

Abstract
Cellular metabolism in hematopoietic stem cells (HSCs) is an area of intense research interest, but the metabolic requirements of HSCs and their adaptations to their niches during development have remained largely unaddressed. Distinctive from other tissue stem cells, HSCs transition through multiple hematopoietic sites during development. This transition requires drastic metabolic shifts, insinuating the capacity of HSCs to meet the physiological demand of hematopoiesis. In this review, we highlight how mitochondrial metabolism determines HSC fate, and especially focus on the links between mitochondria, endoplasmic reticulum (ER), and lysosomes in HSC metabolism.
Introduction
Tissue stem cells operate in a dual system of quiescence and expansion to proficiently generate and regenerate their respective tissue systems (van Velthoven and Rando, 2019). Eukaryotic cells first acquired mitochondria through fusion to α-proteobacteria (Gray et al., 1999) and this metabolic symbiosis allowed cells to replicate (Chandel et al., 2016). However, cells in higher organisms often halt proliferation and become quiescent in order to maintain homeostasis. For example, hematopoietic stem cells (HSCs), mesenchymal stem cells, neural stem cells, epidermal stem cells in the hair follicle, and satellite cells in the skeletal muscle are normally quiescent. The induction of quiescence in stem cells often requires conversion from an active to an inactive cellular metabolism mainly through the suppression of aerobic respiration.
HSCs are rare and versatile cells that sustain life-long hematopoiesis and can generate all lineages of mature hematopoietic cells upon transplantation. Their development is unique among stem cell systems as HSCs originate in different tissues during development. In vertebrates, the initial wave of hematopoiesis occurs in blood islands of the yolk sac, outside of the embryo. Large primitive nucleated erythrocytes, with the occasional presence of primitive macrophages and megakaryocytes, represent the major hematopoietic output of the yolk sac. Hematopoietic cells in the yolk sac may contribute to adult hematopoiesis (Samokhvalov et al., 2007), yet definitive hematopoiesis mainly arises in a region around the ventral wall of the dorsal aorta called the aorta-gonad mesonephros (AGM) at E10.5 in mice. Definitive HSCs, which are serially transplantable and have long-term engraftment capacity, emerge alongside non-self-renewing hematopoietic progenitor cells in the AGM. HSCs next migrate to the fetal liver and spleen and eventually reside in the bone marrow (BM) (Orkin and Zon, 2008; Dzierzak and Bigas, 2018). While embryonic and neonatal HSCs rapidly proliferate and expand to supply the developing hematopoietic system, adult HSCs rarely divide (Crisan and Dzierzak, 2016; Bernitz et al., 2016). These transitions from embryonic/neonatal stage to adult hematopoiesis require drastic alterations in metabolic state (Figure 1).
Figure 1. Schematic Representation of HSC Dynamics during Development.
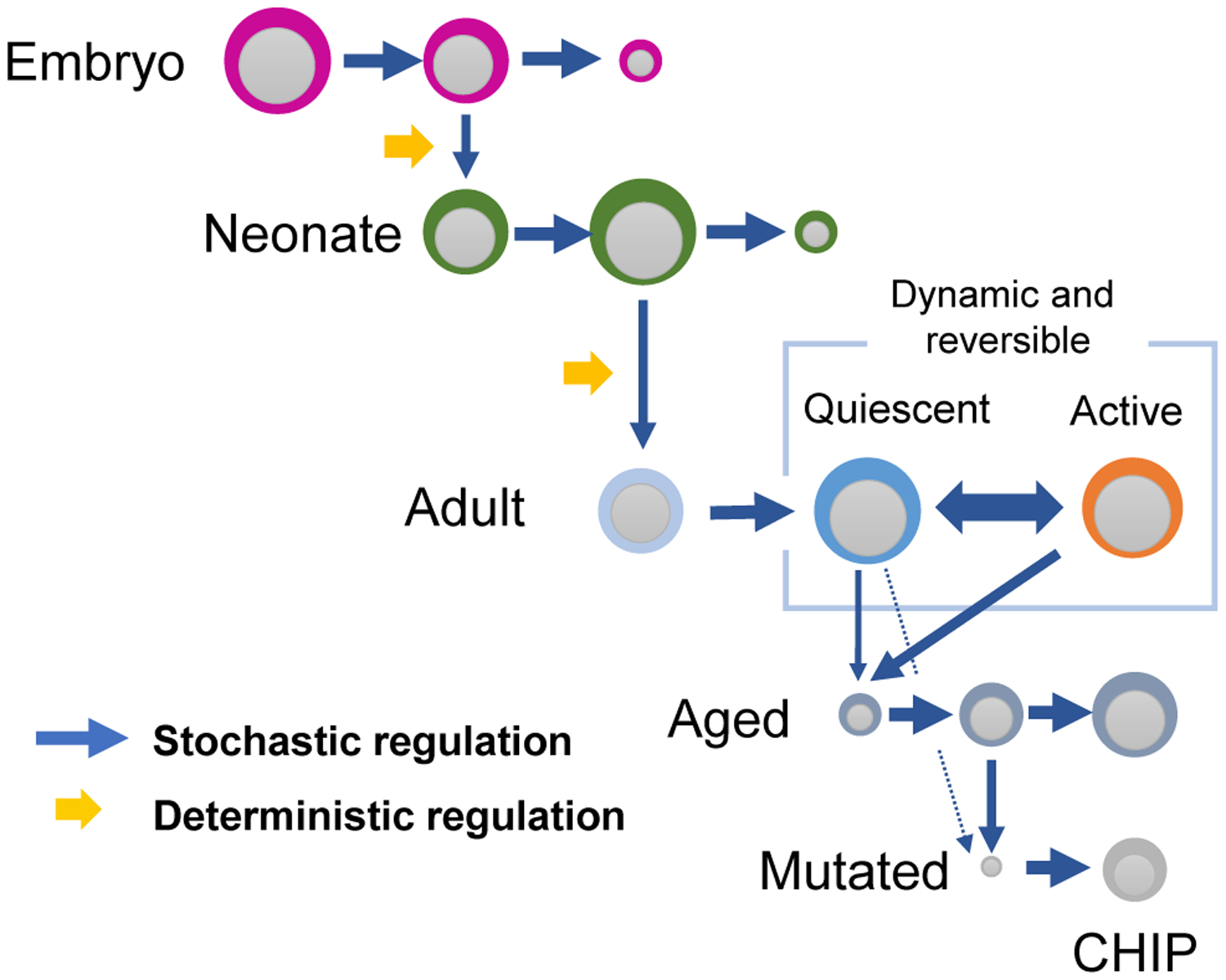
HSCs of different developmental state (embryonic, neonatal, adult, and aged HSCs) clonally expand through stochastic processes. HSCs give rise to differential clones during development through a deterministic process possibly through the modification of hematopoietic environment. Adult HSCs maintain a quiescent state which may be reversed to an active proliferative state upon stress. Aged HSCs eventually accumulate genetic mutations leading to the expansion of abnormal clones (CHIP).
“Metabolism” is derived from the word metaballein (“to change” in Greek), which is fitting, given that the changes in energy production from embryo to the mature organism can be drastic. As a major energy-converting organelle, mitochondria provide ATP for the survival of eukaryotic cells through the tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation (OXPHOS). The adult human body contains approximately 1 × 1016 mitochondria, which accounts for 10% of body weight (Lane, 2005). Mitochondria are also a center for vital cellular processes such as the regulation of reactive oxygen species (ROS) levels, calcium signaling, apoptosis, proteostasis, and heme synthesis (Filippi and Ghaffari, 2019). Cellular metabolism in HSCs has become an area of intense research interest (Ito and Suda, 2014; Chandel et al., 2016). Although metabolic changes during their development have been shown, the metabolic requirements of HSCs in adaptation to their niches have yet to be fully explored. This review focuses on how HSC metabolism transforms and adjusts through hematopoietic ontogenesis, with a special focus on mitochondrial function (Figure 2).
Figure 2. Metabolic Characteristics of Quiescent and Cycling HSCs.
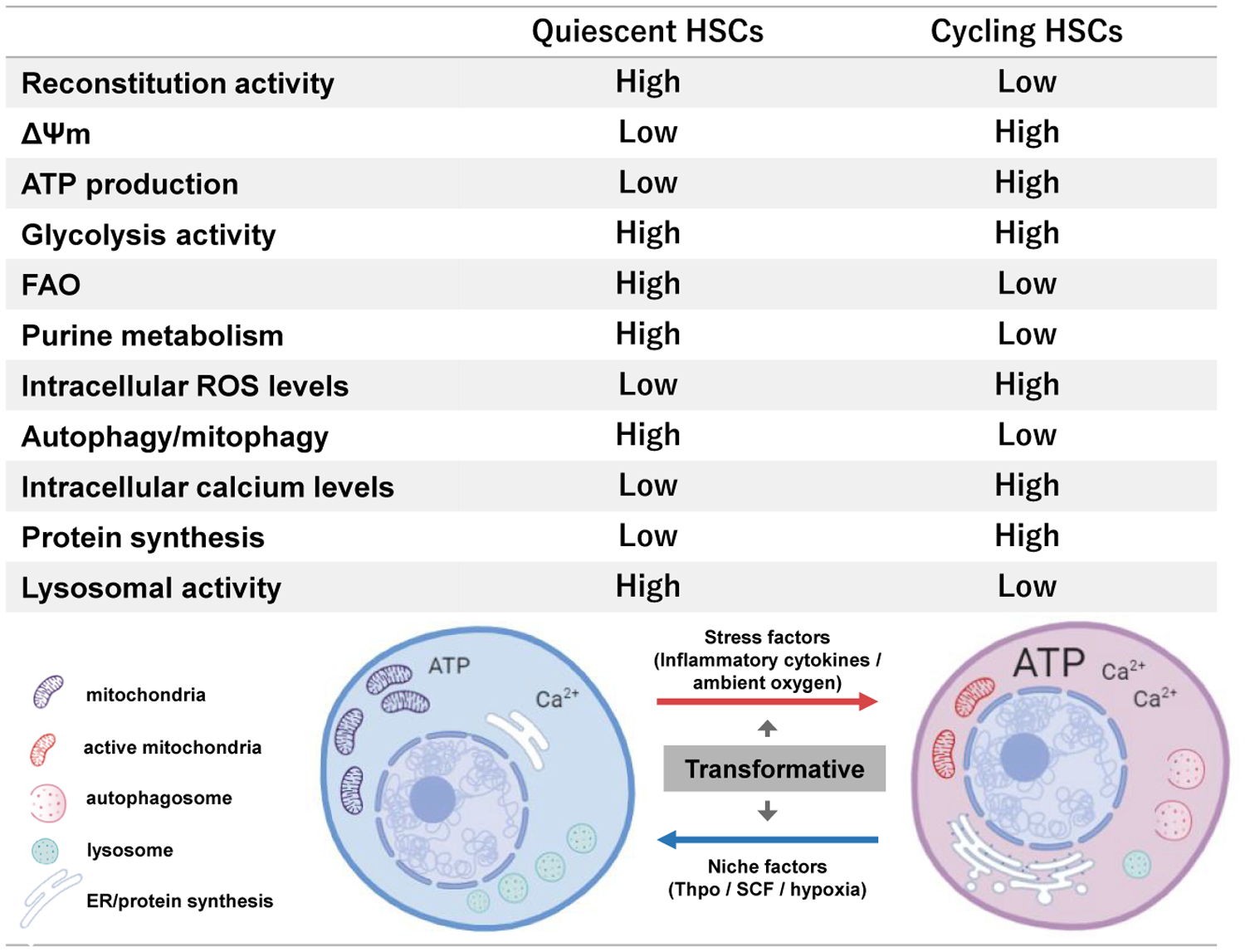
Quiescent adult HSCs exhibit high reconstitution potential and differ in organelle (mitochondria, ER, lysosome, and autophagosome) content compared to cycling HSCs. The difference in organelle activity reflects the overall metabolic state (ΔΨm, ATP production, protein synthesis, autophagy, glycolysis, FAO, purine metabolism, ROS levels, and calcium levels).
Metabolic Transition during HSC Development
During development, definitive HSCs are generated from mesoderm-derived hemogenic endothelium (HE) in the AGM region (Dzierzak and Bigas, 2018). The establishment of arterial identity is a prerequisite for endothelial to hematopoietic transition (EHT) from which definitive HSCs emerge. While transcriptional regulation of EHT has been extensively studied (Zhu et al., 2020; Dzierzak and Bigas, 2018), metabolic changes during EHT have not been fully characterized. HSC expansion during early hematopoiesis involves redox reactions such as the modulation of nitric oxide (NO) and ROS levels (Harris et al., 2013). The EHT process depends on Notch/Wnt/Bmp signaling (Shin et al., 2009), and as both Bmp and Notch regulate NO production, EHT may be influenced through endothelial cell-derived nitric NO metabolism (Nogueira-Pedro et al., 2014). NO is a short-lived free radical implicated in cell proliferation (Villalobo, 2006). NO synthesis in HSCs facilitated by endoglin enhances definitive hematopoiesis (Frame et al., 2017; Manesia et al., 2015). It is also known that prostaglandin E2 contributes to the HSC expansion in zebrafish and mammals during development through the modification of reactive oxygen species (ROS) levels (North et al., 2007; Ludin et al., 2014).
HSCs migrate from the AGM region to the fetal liver for further expansion. Unlike adult HSCs, which require quiescence to maintain stemness, fetal liver HSCs maintain stem cell potential while proliferating (Ema and Nakauchi, 2000). Stem cell transplantation and limiting dilution methods reveal that the absolute number of competitive repopulating units (CRU) per liver increases 38-fold from E12 to 16. Embryonic HSCs largely rely on hepatoblast-expressed Scf, and its receptor, c-Kit, for expansion (Khodadi et al., 2016; Chou and Lodish, 2010). Nestin+ pericytes in the fetal liver also function as a niche through the production of factors such as Scf, Cxcl12, and Spp1 (Khan et al., 2016). While thrombopoietin (Thpo) and its receptor, Mpl, maintains adult HSC quiescence and self-renewal, this signaling pathway is dispensable in fetal HSC maintenance (Qian et al., 2007).
Fetal HSCs are generally thought to contain more mitochondria with higher bioenergetic function than adult HSCs (Manesia et al., 2015; Liu et al., 2010). However, direct comparison of mitochondrial mass between these HSC populations remains technically challenging. This is because adult HSCs express two ABC transporters (Abcg2/Bcrp1and mdr-1a/b), which export the dyes Hoechst33342 and Rhodamine123. This adult HSC-specific dye efflux hinders direct comparison of mitochondria mass between fetal/neonate and adult HSCs using dye-dependent methods (Goodell et al., 1996; Uchida et al., 2004). Careful assessment of mitochondria function using different methodologies is necessary (Snoeck, 2017).
Mitochondrial metabolism is vital for fetal HSC expansion. Fetal HSCs exhibit higher mitochondrial membrane potential (ΔΨmt) and levels of oxygen consumption than adult HSCs (Manesia et al., 2015). Fetal HSCs also exhibit anti-oxidant protection against high mitochondria metabolism, and fetal HSCs defective in manganese superoxide dismutase 2 (Sod2) show defective erythroid hematopoiesis (Friedman et al., 2001). Deletion of components of the TCA cycle and electron transport chain (ETC), fumarate hydratase, and Rieske iron-sulfur protein (RISP) also result in failure of fetal hematopoiesis (Guitart et al., 2017; Anso et al., 2017). Additionally, deletion of an autophagy regulator, Fip200 (200-kDa FAK-family interacting protein), impairs fetal HSC functions through accumulation of mitochondrial mass and ROS (Liu et al., 2010).
Adult HSCs suppress protein synthesis (Signer et al., 2014), while fetal liver HSCs have higher protein synthesis levels (Sigurdsson et al., 2016). In order to counteract ER stress (described later in this review), fetal liver HSCs utilize bile acids as chemical chaperones to withstand unfolded protein response (UPR). Differing UPR levels may also affect metabolic quality control in proliferating fetal/neonatal HSCs versus quiescent adult HSCs. Additionally, mitochondrial UPR (UPRmt) is activated upon adult HSC exit from quiescence (Mohrin et al., 2018; Luo et al., 2019). Thus, the metabolic state of fetal/neonatal HSCs is unique: high mitochondrial metabolism without ROS generation and high protein synthesis without ER stress. However, what triggers HSC metabolic changes during development remains a largely uncultivated field.
Hematopoiesis shifts to the bone marrow (BM) late in gestation and is then maintained there. HSCs acquire cell cycle quiescence around 4 weeks after birth (Bowie et al., 2006). Changes in HSC metabolism associated with emergence of the G0 state have not been analyzed extensively and are an area of interest. Moreover, it is unclear whether neonatal HSCs are similar to adult HSCs, which are genetically and functionally heterogenous (Crisan and Dzierzak, 2016). Single-cell genetic analysis and clonal lineage tracing assays on HSCs should further characterize the shift from neonate to adult hematopoiesis.
Cell Metabolism in Adult HSCs
Glycolysis
Dormant HSCs reside in a hypoxic microenvironment that promotes glycolysis and enables them to retain a low metabolic profile (Spencer et al., 2014). Their quiescence allows them withstand premature exhaustion from cell proliferation. Adult HSCs depend upon discrete genetic and proteomic control in order to utilize anaerobic glycolysis as their main energy source (Unwin et al., 2006; Takubo et al., 2010). HSCs express high levels of hypoxia inducible factor-1 alpha (HIF1α) and Hif1α deletion results in loss of HSC quiescence and stem cell potential (Takubo et al., 2010; Suda et al., 2011). However, using similar conditional deletion mouse models, other studies have described HIF1α and HIF2α as dispensable for HSC maintenance (Vukovic et al., 2016; Guitart et al., 2013), necessitating further analysis of the function of HIFs during steady-state or stress hematopoiesis.
Glycolytic metabolism in HSCs is complex and interlinked with other metabolic pathways such as glutamine metabolism or branched-chain amino acid (BCAA) catabolism (Jin et al., 2018; Taya et al., 2016; Ananieva and Wilkinson, 2018). Alternative polyadenylation regulates the switching of glutaminase isoforms, which is required for HSC activation (Sommerkamp et al., 2020). The enzymes responsible for the early steps of BCAA catabolism include branched-chain aminotransferase 1 (BCAT1), which converts BCAAs into branched-chain ketoacids by transferring the BCAA amino group onto alpha-ketoglutarate (αKG) (generating glutamate). BCAT1 has been implicated in the regulation of leukemic stem cells (LSC) and knockdown of BCAT1 downregulates HIF1α activity and LSC growth (Raffel et al., 2017). BCAA metabolism, which produces αKG and TET protein, is implicated more in the regulation of proliferating HSCs and LSCs than in control of quiescent HSCs. Interestingly, mutations in isocitrate dehydrogenase (IDH) which catalyzes the oxidative decarboxylation of isocitrate into αKG, are also implicated in leukemogenesis (Dang et al., 2009; Lu et al., 2012). Mutant IDH enzymes produce 2-hydroxyglutarate, which leads to aberrant HSC self-renewal and leukemogenesis in part through TET2 inhibition (Rakheja et al., 2013). Ascorbic acid is a TET cofactor and facilitates TET2-mediated DNA hydroxymethylation (Agathocleous et al., 2017; Cimmino et al., 2017). These studies offer seminal insights into how metabolites affect HSC function through the modulation of epigenetic state.
Fatty Acid Metabolism
Fatty acid oxidization (FAO) is the mitochondrial process that breaks down a fatty acid into acetyl-CoA. Acetyl-CoA enters the citric acid cycle while NADH and FADH2, which are co-enzymes, are fueled into the electron transport chain (ETC). Notably, inhibition of the FAO pathway through the deletion of Peroxisome-proliferator activated receptor delta (Ppard), a regulator of fatty acid transport and oxidization that is regulated by promyelocytic leukemia (Pml), results in loss of HSC reconstitution potential (Ito et al., 2012, 2016). Interestingly, inhibition of PPARγ promotes ex vivo expansion of human cord blood hematopoietic stem and progenitor cells (HSPCs), while causing a metabolic shift to glycolysis (Guo et al., 2018).
Beyond their central role in fatty acid metabolism, mitochondria also participate in the biosynthesis of nucleotides, cholesterol, amino acids, as well as heme, and these roles in HSC regulation still need to be investigated.
HSC Expansion through the Modulation of Metabolism
Manipulating HSC metabolism allows the expansion of HSCs in vitro. A study conducted on the role of a glycolytic enzyme, pyruvate dehydrogenase kinase (Pdk), in HSCs revealed the interplay between anaerobic metabolism and aerobic metabolism (Takubo et al., 2013). Pdk inhibits pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl-CoA which fuels aerobic metabolism. Loss of Pdk2 and Pdk4 in HSCs results in loss of stem cell potential. The report highlights that Pdk agonists expand HSCs in vitro.
Murine HSCs in long-term culture can be maintained in quiescent states using hypoxic culture conditions (in 1% O2 conditions), low concentrations of SCF and Thpo with the addition of 4% bovine serum albumin (BSA) as a source of fatty acids (Kobayashi et al., 2019). Although efforts in ex vivo cord blood expansion and preservation have shown considerable success (Chaurasia et al., 2014; Fares et al., 2014; Wagner et al., 2016), the optimal conditions of adult human HSC ex vivo expansion are yet to be determined. Niche scaffolds which mimic the in vivo bone marrow microenvironment have successfully expand HSCs through regulating of mitochondrial activity and ROS production (Bai et al., 2019). Invention of similar methods should follow.
Mitochondrial Energy Production and Synthesis
Mitochondrial Function and the Electron Transport Complex
Mitochondria contain the major enzymes that oxidize carbohydrates, proteins, and lipids to produce ATP. In this process, appropriate substrates are catabolized to acetyl-CoA, which enters the TCA cycle (tricarboxylic acid cycle, or Krebs cycle) housed in the mitochondrial matrix (Baldwin and Krebs, 1981). Subsequent enzymatic steps of the TCA cycle result in the formation of reduced cofactors NADH (nicotinamide adenine dinucleotide) and FADH2 (flavin adenine dinucleotide) and ultimately fuel the ETC on the inner mitochondria membrane (IMM) for ATP production. Electron transfer from NADH to oxygen allows protons to be pumped out of the matrix to generate an electrochemical proton gradient in the IMM. The electron transport ends at complex V (or ATP synthase) where the electrochemical proton gradient couples the conversion of ADP +Pi to ATP (OXPHOS) (Spinelli and Haigis, 2018; Figure 3).
Figure 3. Mitochondria Bioenergetics in HSCs.

Mitochondria bioenergetics pathways implicated in HSC metabolism. Other than ATP production through the TCA cycle and electron transport system, HSC mitochondria produce energy through FAO (beta-oxidation). ROS is generated as a byproduct of oxidative phosphorylation and is detoxified by cystolic redox reaction. Mitochondria also home specific pathways for proteostasis and process unfolded proteins through chaperone proteins and proteases. Intracellular calcium levels are regulated by the mitochondria through crosstalk with ER.
HSCs depend mainly on glycolysis for ATP production, leaving mitochondrial OXPHOS activity relatively inert. Nonetheless, HSCs maintain mitochondrial activity, raising the question of how mitochondrial energy production is involved in HSC maintenance and proliferation. High mitochondrial membrane potential (ΔΨmt) is linked to lower reconstitution capacity and affects proliferation in adult HSCs (Ansó et al., 2017; Vannini et al., 2016; Sukumar et al., 2016). ΔΨmt has traditionally been assayed with dyes. Tetramethylrhodamine methyl ester (TMRM) dye accumulates in the mitochondrial matrix at levels that correlate with ΔΨmt and cellular OXPHOS activity. Long-term HSCs (LT-HSCs) have the lowest level of TMRM staining among all hematopoietic cells (Vannini et al., 2016, 2019). Inhibition or modulation of multidrug resistance (MDR)-mediated efflux did not affect the staining intensity of Rhodamine-123 and TMRE (tetramethylrhodamine, ethyl ester) in HSPCs (Kim et al., 1998; Liang et al., 2020). On the other hand, some studies have shown that verapamil, a wide spectrum inhibitor of efflux pumps, modifies the low retention of Rhodamine-123 in human or murine HSPCs (Zijlmans et al., 1995; Wagner-Souza et al., 2008). Similar results were obtained using a Ca2+-independent MDR inhibitor, Cyclosporin H (Bonora et al., 2018; Morganti et al., 2019a). ΔΨmt is balanced between proton pumping activity by the ETC and proton flow across mitochondrial F1/FO ATP synthase; HSPCs sustain a unique equilibrium of ΔΨmt with a high ETC complex II:complex V ratio (Bonora et al., 2018; Morganti et al., 2019a). ETC complex II maintains complex III in proton pumping, and a low coupling capacity due to minimal F1/FO ATP synthesis results in relatively high ΔΨmt in HSPCs, even though corresponding rates of respiration and phosphorylation are low (Morganti et al., 2019a). Unexpected changes in staining intensity may be detected by flow cytometry when TMRM dye levels have not yet reached perfect equilibrium. Therefore, discrepancies in TMRM staining in HSPCs could depend on staining period, which suggests that staining conditions for measuring ΔΨmt should be standardized (Morganti et al., 2019b). Moreover, as flow cytometry quantifies fluorescence intensity per cell, it is difficult to discern whether mitochondrial volume affects fluorescence intensity values. Combined measurement of ΔΨm and mitochondrial volume/morphology (e.g., 3D image-based content analysis) could help reveal mitochondrial activity in HSPCs. Furthermore, understanding the precise roles of ETC proteins, such as succinate dehydrogenase (SDH; complex II) may clarify the links between ETC complex/TCA cycle, ΔΨmt, and the cell cycle state of HSPCs. Moreover, ATP levels and OXPHOS activity in HSCs can also be quantified through luminescent cell viability assays and Seahorse metabolic flux analyses, respectively (Nakamura-Ishizu et al., 2018; Tan et al., 2019; Ho et al., 2017). However, these methods require a substantial number of cells for accurate analysis and cannot be used for the assessment of individual HSCs. Additional non-dye-based methodologies are needed for the precise evaluation of ΔΨmt in HSCs.
Components of the mitochondria electron transport complex serve as initiators of apoptosis/programmed-cell death. Cytochrome c shuttles electrons between respiratory chain complexes II and IV and is essential for electron flow and ATP synthesis (Santucci et al., 2019). The release of cytochrome c from the electron transport system to the cytoplasm activates caspase signals (Santucci et al., 2019). Cytochrome c-triggered apoptosis is noted in proliferating hematopoietic cells (Garland and Rudin, 1998). During apoptosis, cytochrome c and soluble internal proteins (SIMPs) are released into the cytoplasm and interact with apoptosis protease activating factor (Apaf-1) to form apoptosomes (Marsden et al., 2002). The release of cytochrome c to the cytoplasm requires the peroxidization of cardiolipin, a phospholipid within the mitochondrial membrane (Yin and Zhu, 2012). Thus, cytochrome c is annotated as an “extreme multifunctional protein” which alters its localization, changes its conformation, and determines cell fate (Santucci et al., 2019). However, detailed analysis on how cytochrome c function alters with mitochondria state and whether it regulates HSCs is largely unknown.
Mitochondrial Biogenesis in Adult HSCs
Both excessive and insufficient mitochondrial function is detrimental to adult HSC homeostasis. Abnormal mitochondrial biogenesis affects HSC function (Table 1). Mammalian target of rapamycin (mTOR) complex-1 governs multiple pathways for cellular metabolism and upregulates the transcription factor PPARγ coactivator-1α (PGC-1α), which is a key regulator of mitochondrial biogenesis (Cunningham et al., 2007). mTORC1 activation causes HSC exhaustion and deletion of a component of mTORC1, Raptor, results in pancytopenia and inhibition of HSC regeneration (Kalaitzidis et al., 2012). mTOR activation also leads to HSC exhaustion through the activation of p16Ink4a and p53 (Lee et al., 2010) and is associated with high oxidative stress in HSCs (Jang and Sharkis, 2007).
Table 1.
Mitochondrial Regulators Associated with HSC Function
| Genes | Mitochondria-Related Function | HSC Phenotype in Transgenic Mice | Mitochondria Phenotype | References |
|---|---|---|---|---|
| Mitochondria Biosynthesis Regulators | ||||
| mTOR | upregulates mitochondria synthesis through PGC-1α | activation causes HSC expansion, exhaustion, and leukemogenesis; deletion causes pancytopenia and impairs HSC regeneration | activation results in high oxidative stress | Kalaitzidis et al. (2012); Lee et al. (2010); Jang and Sharkis (2007); Qian et al. (2016) |
| Ppargc1a (Pgc1a) | peroxisome proliferator-activated-γ coactivator-1α; regulator of mitochondrial biogenesis | deletion impairs reconstitution potential of HSCs but does not affect physiological HSCs | deletion reduces MMP and mitochondrial mass in bone marrow cells | Basu et al. (2013) |
| Tsc1 | tuberous sclerosis complex; inhibits mTOR | deletion results in loss of HSC quiescence | deletion causes increased mitochondrial biogenesis and elevated ROS | Chen et al. (2008) |
| Src3 | p160 SRC family of nuclear receptor coactivators; increases Pgc-1α expression | deletion results in defective HSCs | deletion results in high MMP and high ROS; deletion increases Pgc-1α expression | Hu et al. (2018) |
| Lkb1/Stk11 | Lkb1 tumor suppressor; activates AMPK and TSC1 to inhibit mTOR | deletion of Lkb1 causes loss of HSC quiescence and repopulation potential | deletion results in the impairment of mitochondrial function and biogenesis with the reduced MMP and ATP levels | Gan et al. (2010); Gurumurthy et al. (2010); Nakada et al. (2010) |
| Mitochondria-Localized Proteins | ||||
| Ptpmt1 | PTEN-like mitochondrial phosphatase | deletion causes BM failure due to defective HSC differentiation and expansion | deletion causes decrease in oxygen consumption and extracellular acidification rates, indicating defective mitochondrial bioenergetics as well as glycolysis | Yu et al. (2013); Liu et al. (2015) |
| Uqcrfs1 | Rieske iron-sulfur protein (RISP) in mitochondrial complex III | deletion causes loss of HSC quiescence and severe pancytopenia | deletion impairs respiration resulting in a decreased NAD+/NADH ratio; also leads to DNA hypermethylation and histone hypoacetylation through the increases in 2-hydroxyglutarate (2HG) | Ansó et al. (2017) |
| Mortalin (Hspa9) | mitochondria heat shock protein | knock down of Hsp9 led to loss of HSC quiescence and reconstitution potential | knock down of Hsp9 leads to increased ROS in HSCs | Tai-Nagara et al. (2014) |
| Mitochondria Fusion/Fission | ||||
| Mfn2 | mitochondria fusion | deletion causes defective lymphoid lineage differentiation of HSCs | deletion reduces mitochondria tethering to ER and alters intracellular calcium levels | Luchsinger et al. (2016) |
| Drp1 | mitochondria fission | deletion causes loss of reconstitution potential | depletion causes aggregation and depolarization of mitochondria | Hinge et al. (2020) |
| Mitophagy/Autophagy | ||||
| Atad3a | AAA+-ATPase regulator of Pink1-dependent mitophagy | deletion causes HSCs to expand in number with defective lineage differentiation | deficient HSPCs have the reduced mitochondrial content due to hyperactivated mitophagy by Pink1 accumulation | Jin et al. (2018) |
| Atg7 | Autophagy related 7 | Atg7-deficient mice show expansion of HSPCs with defective reconstitution potential | depletion causes aberrant increase in mitochondrial mass, higher mitochondrial ROS level, and increased DNA damage | Mortensen et al. (2011); Gomez-Puerto et al. (2016) |
| Atg5 | Autophagy related 5 | deletion from hematopoietic lineage cells cause lymphopenia and anemia; impaired reconstitution potential | deficiency causes impaired clearance of damaged mitochondria | Jung et al. (2019) |
| Parkin/Pink1 | PTEN-induced putative kinase 1 (Pink1, or Park6), Parkinson protein 2, E3 ubiquitin protein ligase (Parkin, or Park2) | acute silencing causes the impaired self-renewing expansion of Tie2+ HSCs | – | Ito et al. (2016) |
| FIP200 | 200-kDa FAK-family interacting protein | deletion in fetal HSCs increased mitochondrial mass and ROS levels | deletion causes perinatal death due to anemia; fetal HSCs increase proliferation and lose reconstitution potential | Liu et al. (2010) |
| Atg12 | Autophagy related 12 | deletion results in decreased HSC reconstitution potential with myeloid skewing | deficient HSCs present increase of active mitochondria with high MMP | Ho et al. (2017) |
| Other Functions | ||||
| Pml/Ppard | FAO; Pml regulates Ppard, a regulator of fatty acid transport and oxidization | loss of HSC potential | – | Ito et al. (2012) |
| Polg | integrity of mtDNA; proof-reading-deficient version of mitochondrial DNA polymerase gamma | defective Polg causes an accrue of mutations in mtDNA and compromises HSC differentiation in early lymphoid and erythroid lineages | mutations in mtDNA lowered MMP | Norddahl et al. (2011) |
| Pparg | peroxisome proliferator-activated receptor gamma | PPARγ antagonist promotes expansion of human cord blood HSPCs | PPARγ antagonism enhances glycolysis without compromising mitochondrial metabolism | Guo et al. (2018) |
Multiple molecular mechanisms have been implicated on the regulation of mTOR activity in HSCs, and these include non-coding RNAs originating from Dlk1-Gtl2 locus, tuberous sclerosis complex (TSC)-1, and ERK-dependent phosphorylation of MEK1 (pT292) (Qian et al., 2016; Chen et al., 2008; Baumgartner et al., 2018). Mitochondria in HSCs deficient of Lkb-1, which regulates cell metabolism through AMPK, exhibit low ΔΨmt and ATP production (Gurumurthy et al., 2010; Nakada et al., 2010; Gan et al., 2010). However, the deletion of Pgc1a impairs long-term reconstitution potential of HSCs in BM transplantation with minimal effects on physiological HSCs (Basu et al., 2013). Thus, mTOR and related molecules are critical in regulating HSC metabolism.
Dynamics of Mitochondria Mass and Morphology
Mitochondria Structure and Mass in Adult HSCs
Mitochondria dynamically alter their morphology in response to cellular energy demands (Seo et al., 2018). Mitochondria consist of an inner membrane folded into multiple cristae structures that contain OXPHOS machinery. Functionally active mitochondria are oblong or oval and house numerous cristae to maximize inner membrane surface area (Pernas and Scorrano, 2016). Mitochondria mass and shape alters with mitochondria fission, fusion, and mitophagy processes (Youle and van der Bliek, 2012). Mitochondrial shape also changes in response to environmental conditions, such as a hypoxia, changes in energy demand, and metabolites (Westermann, 2010). Additionally, ultrastructural electron microscopic characteristics, including cristae number and electron density, are also related to mitochondria function (Cogliati et al., 2013; Kumar et al., 2014; Hinge et al., 2020; Nakamura-Ishizu et al., 2018). In embryonic stem cells (ESCs), individual mitochondria are small and round with infrequent cristae (Zhang et al., 2011). Although these morphologic features suggest that mitochondrial respiration in ESCs is relatively inactive, ESCs exhibit high ΔΨmt (Folmes et al., 2011). Mitochondrial morphology in quiescent HSCs is similar to that of ESCs, suggesting that ΔΨmt rather than mitochondrial morphology better reflects a primed state for differentiation and proliferation (Schieke et al., 2008). Detailed studies to understand the relationship of mitochondrial morphology to function in HSCs are needed.
HSCs contain higher number and volume of mitochondria compared to hematopoietic progenitor cells. Multiple studies suggest that a dye-based MitoTracker staining underestimates mitochondrial content in HSCs (de Almeida et al., 2017; Takihara et al., 2019; Bonora et al., 2018). In these studies, mitochondrial volume is quantified through mitochondrial DNA levels or through the assessment of mito-dendra2 transgenic mice. Evaluation of immunohistochemical staining of TOM20 (Translocase of outer membrane 20) in HSCs from transgenic mice constitutively expressing GFP in the mitochondria matrix (mtGFP mice) (Shitara et al., 2010) showed lower mitochondrial volumes in HSCs compared to progenitor cells. Further clarification and advancement in techniques such as tomography of electron microscopy are necessary to delineate the exact mitochondrial volume and morphology in HSCs.
Whether HSCs heterogeneously contain different amounts of mitochondria is another interesting topic. Through assessment of mitochondrial volume in HSCs from mito-dendra2 transgenic mice, HSCs can be fractionated into high and low mitochondrial volume subgroups, the former presenting with higher stem cell potential and quiescent characteristics (Takihara et al., 2019). However, highly proliferative and active HSCs upregulate mitochondrial volume (Nakamura-Ishizu et al., 2018; Bonora et al., 2018), indicating that mitochondrial volume does not necessarily correlate with cell cycle state.
Regulation of Mitochondrial Dynamics in Adult HSCs
Mitochondria dynamically alter their shape though fission and fusion processes (Westermann, 2010; Figure 4). Loss of mitofusin2 (Mfn2), an OMM protein which facilitates mitochondrial fusion, results in defective lymphoid lineage differentiation of HSCs (Luchsinger et al., 2016). Although structurally similar to Mfn2, the role of Mfn1 in HSC regulation has not been elucidated (Chen et al., 2003). Deletion of Optic atrophy 1 (Opa1), another fusion regulator located on the IMM (Olichon et al., 2003), lowers the activity of mitochondrial ETC complex I and ATP production in neutrophils (Amini et al., 2018). Further investigation on Mfn1 or Opa1 function may reveal how mitochondrial fusion associates with HSC maintenance.
Figure 4. Mitochondria Dynamics through Fission and Fusion.
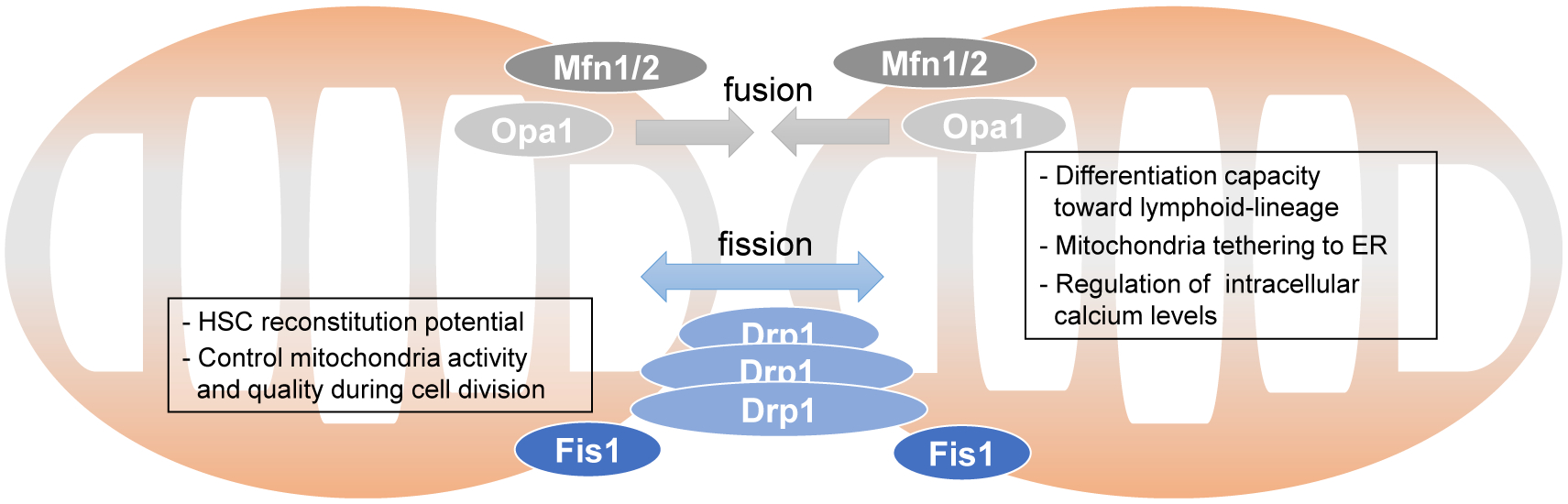
Mfn1/2 located on the OMM and Opa1 on the IMM stimulate mitochondria fusion. Deletion of Mfn2 affect HSC mitochondria function and stem cell potential. Drp1 and Fis1 stimulate mitochondria fission. Fis1 is implicated to affect HSC mitochondria segregation during division and stem cell functions.
Deficiency of the mitochondrial fission regulator Drp1 results in mitochondrial aggregation, depolarization, and subsequently loss of reconstitution potential in HSCs (Hinge et al., 2020). Drp1 activity is regulated through phosphorylation (Ser637, Ser616), small ubiquitin-like modifier proteins, or s-nitrosylation by nitric oxide (Chang and Blackstone, 2007; Foster et al., 2009; Wasiak et al., 2007; Kashatus et al., 2015). Fis1 is located on the outer mitochondria membrane (OMM) and facilitates attachment of Drp1 for mitochondrial fission. Fis1-deficient skeletal muscle cells exhibit swollen mitochondria and a decline in mitochondrial function (Zhang et al., 2019), yet no study has addressed the role of Fis1 in HSCs.
Advances in delineating HSC function and cell fate at the single-cell level has revealed mitochondrial heterogeneity in HSCs. Observation of single asymmetric HSC divisions revealed that mitochondria are distributed in an imbalanced manner to HSC daughter cells (Loeffler et al., 2019). Another study also showed that, under stress conditions (e.g., after transplantation), HSC mitochondria aggregate and depolarize (Hinge et al., 2020). Mitochondrial dynamism and motility were also lost in post-transplantation proliferative HSCs. Mitochondrial patterns were unevenly inherited to daughter cells during asymmetric HSC division, which was frequently observed in post-transplanted HSCs. HSCs with depleted Drp1 have decreased reconstitution potential due to the accumulation of dysfunctional mitochondria by asymmetric segregation. Altered fidelity of mitochondrial morphology implies that HSCs carry dysfunctional mitochondria to track their divisional history and limit their self-renewal capacity (Hinge et al., 2020).
Regulation of ROS in Adult HSCs
ROS are a byproduct of mitochondrial OXPHOS, which at high levels harms HSCs through the generation of reactive free radicals (Bigarella et al., 2014). HSCs acquire various safeguard mechanisms against cytotoxic accumulation of ROS. Loss of Ataxia telangiectasia mutated (Atm) causes accumulation of ROS and loss of quiescence in HSCs through the activation of p16Ink4a or p38MAPK(Ito et al., 2004,2006). Loss of Atm-mediated phosphorylation of Bid (a BH3-only BCL2 family member) along with loss of Mtch2 (Mitochondrial carrier homolog 2) leads to less quiescence and an increase in mitochondrial oxidative stress (Maryanovich et al., 2012). As ROS production mainly derives from OXPHOS, high ΔΨmt is likely associated with high cellular ROS levels. This was highlighted by a study indicating that HSCs exposed to brief periods of ambient oxygen showed a rise in ΔΨmt and ROS levels, which lowered stem cell potential (Mantel et al., 2015). This phenomenon, named “extra physiologic oxygen shock/stress” (EPHOSS), involved oxidative stress-induced opening of mitochondrial permeability transition pore (MPTP).
Cells with damaged mitochondria, which are generally associated with low ΔΨmt, also exhibit high ROS levels. Foxo3a deficiency elevates HSC ROS levels through reduction of ROS detoxification molecules (Miyamoto et al., 2007). Despite presenting high ROS levels, Foxo3a−/− HSCs exhibit low ATP production and low ΔΨmt (Rimmelé et al., 2015). Aged HSCs decrease in stem cell potential but display both low ΔΨmt and ROS levels (Ho et al., 2017). ROS regulation in HSCs is complex, and impaired HSC function may not directly reflect ROS levels, but rather reflect the general integrity of mitochondrial function. These studies collectively support the observation that mitochondrial quality is associated with ROS production, which needs further assessment in HSCs.
Proper levels of intracellular ROS have been implicated in cell survival (Bigarella et al., 2014; Tan and Suda, 2018). ROS mediates the oxidization of target proteins at specific amino acid residues (especially cysteines or methionines) to regulate protein function (Holmström and Finkel, 2014). ROS modulates the activity of protein kinases (c-Kit, Akt, and MAPK) as well as transcriptions factors (p53, HIFs, and FOXOs) which are crucial pathways for HSC maintenance (Paulsen et al., 2011; Murata et al., 2003; Galli et al., 2008; Bigarella et al., 2014). p38MAPK activation leads to upregulation of purine metabolism to fuel HSC proliferation after stress, suggesting the involvement of oxidative stress (Karigane et al., 2016).
Mitochondria participate in anti-oxidant processes which reduce ROS levels. Mitochondrial superoxide dismutase (SOD2) or cytosolic SOD1 converts superoxide radical anion (O2−) to hydrogen peroxide (H2O2) (McCord et al., 1971). While Sod2 deficiency impairs erythroid cell production (Mohanty et al., 2013), no report has indicated its direct involvement in HSC regulation in vivo. Nuclear factor erythroid 2-related factor 2 (Nrf2) is known to regulate HSC quiescence and stem cell potential through modulation of oxidative stress (Murakami et al., 2017; Merchant et al., 2011). Glutathione-dependent enzymes also catalyze biological redox reactions, yet due to their redundancy in function, transgenic mice that lack glutathione-dependent enzymes are often phenotypically intact (Henderson and Wolf, 2011). However, Microsomal glutathione transferase 1 (Mgst1)-deficient HSCs exhibit defective differentiation (Bräutigam et al., 2018). Collectively, while quiescent HSCs generally retain low ROS levels, low ROS levels do not necessarily indicate low mitochondrial function. Neonatal HSCs exhibit active mitochondrial metabolism but low ROS, and more importantly, are capable of expansion in vivo. It should be emphasized that the complexity of HSC response to oxidative stress emphasizes the potential indications of ROS levels on mitochondria and redox state.
Mitochondria and the Endoplasmic Reticulum
Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs)
Mitochondria and endoplasmic reticulum (ER) are essential organelles in eukaryotic cells and coordinate various biological pathways such as maintenance of calcium homeostasis, protein folding, and lipid metabolism. Mitochondria interact with ER to facilitate calcium and lipid exchange between the two membrane systems (Krols et al., 2016). Mitochondria and ER communicate via the formation of mitochondrial-associated membranes (MAM, or mitochondria-associated ER membranes). MAMs are formed through the juxtaposition of ER subdomains to mitochondria and defects in their formation and function alter cell survival and death. MAM proteins either directly connect ER and mitochondria or modulate tethering complexes. Mfn1 and 2 (Mfn1/2) are found in the MAM complex (de Brito and Scorrano, 2009). Mitochondrial ubiquitin ligase Mitol/March5, a MAM protein, ubiquitinates mitochondrial Mfn2, and its deletion impairs MAM domain formation results in decreased tethering of mitochondria to ER (Sugiura et al., 2013). The role of MAMs in the metabolic regulation of HSCs is yet to be explored.
Regulation of Cellular Calcium
Upregulation of mitochondrial Ca2+ in physiological condition activates mitochondrial enzymes, which facilitate the TCA cycle and OXPHOS. α-ketoglutarate dehydrogenase, IDH, and pyruvate dehydrogenase (PDH) are Ca2+-dependent enzymes (McCormack and Denton, 1993). ATP synthase activity also depends on Ca2+ (Das and Harris, 1990). Thus, increased Ca2+ levels enhance electron transport activity in mitochondria, resulting in elevated generation of ATP (Hansford, 1994; Figure 5).
Figure 5. Metabolic Crosstalk between Cellular Organelles in HSCs.

Mitochondria interact with various cellular organelles. These interactions alter the metabolic and cellular fate of HSCs. The nucleus provides nuclear (nc) DNA encoded proteins, while mitochondria possess mitochondria (mt) DNA for the production of mitochondria proteins. Mitochondria also initiate apoptosis. Mitochondria clearance is regulated through a specific form of autophagy, mitophagy. Mitochondria are firmly associated with ER to which they tether and control intracellular calcium levels.
In contrast, prolonged or excessive upregulation of mitochondrial Ca2+ promotes apoptosis. MAMs convey mitochondrial apoptotic signals to ER, especially through the refinement of Ca2+ levels (Iwasawa et al., 2011). Ca2+ flux from ER to mitochondria stimulates Bcl-2-associated X protein (BAX) to oligomerize and permeate the mitochondrial membrane (Rostovtseva et al., 2005). Mitochondrial permeability transition pores (PTPs) are induced by high Ca2 levels (Hunter and Haworth, 1979). PTP opening increases mitochondrial membrane permeability, leading to apoptosis by releasing cytochrome c, apoptosis-inducing factor (AIF), and Smac/DIABLO (Bernardi et al., 2001).
Several reports have highlighted the role of intracellular calcium levels in HSC regulation. Increase of intracellular calcium triggered HSC divisions during stress hematopoiesis induced by 5-fluouracil administration (Umemoto et al., 2018). Dampening of intracellular calcium inhibited mitochondrial functions and prolonged the cell cycle phase of HSCs which resulted in slower recovery of stress hematopoiesis. The suppression of intracellular calcium level by nifedipine, a calcium channel blocker, prolonged the cell division interval and maintained HSCs.
Further association between intracellular calcium levels and mitochondria have been highlighted through the function of Mfn2 (Luchsinger et al., 2016, 2019). Deletion of Mfn2 in HSCs decreased intracellular calcium buffering through ER-mitochondria tethering activity. As intracellular calcium stimulated nuclear translocation and transcriptional activity of Nuclear factor of activated T cells (Nfat), Mfn2-deficient HSCs exhibited loss of lymphoid differentiation potential. Steady-state HSCs were also endowed with low intracellular calcium levels (Luchsinger et al., 2019). HSCs cultured in low concentration of calcium exhibited reduced mitochondrial respiration but not glycolysis. The study also highlighted how intracellular calcium levels regulate calpain and subsequently the activity of TET (Ten-eleven translocation) enzymes for HSC maintenance. However, high concentration of cytoplasmic calcium has also been associated with dormant HSCs (Fukushima et al., 2019).
Mitochondria fission (Drp1) and unfolded protein response have been implicated in regulation of cellular calcium (Wang et al., 2017; Favaro et al., 2019) and these should be further investigated in HSCs. Along with improved methods to accurately measure intracellular calcium in HSCs, detailed monitoring of external and intracellular regulators of calcium levels will further clarify the role of calcium in HSC maintenance. The mechanism of how cytoplasmic calcium levels affect mitochondrial activation should also be investigated.
ER Stress and Mitochondrial UPR
Increased protein synthesis may lead to the accumulation of unfolded and misfolded proteins. This accumulation triggers ER stress as a part of the unfolded protein response (UPR). Since ER stress is a multi-layered, bidirectional cellular response that results in either cell survival or death, proper management of UPR and ER stress signals are crucial for HSCs. It has been suggested that UPRmt is implicated in cellular senescence and aging (Jensen and Jasper, 2014). UPRmt is upregulated upon mitochondrial biogenesis and involves retrograde signaling from the mitochondria to the nucleus to stimulate transcription of mitochondrial chaperones, mediating enhanced mitochondrial function during cell proliferation. Mitochondrial proteostasis has been implicated in HSC regulation. HSCs induced to proliferate with polyinosinic:polycytidylic acid (pI:pC, or poly(I:C)) increased the expression of mitochondrial chaperones and protease-associated genes (Mohrin et al., 2018). The deletion of Sirtuin 7 (Sirt7), a protein deacylase, elevates UPRmt, impairs regenerative potential, and compromises lymphopoiesis of HSCs (Mohrin et al., 2015). Sirt7 represses the activity of Nrf1 (nuclear respiratory factor 1) to suppress mitochondrial metabolism and cell proliferation. Mitochondrial stress may also trigger the formation of aberrant Nlrp3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasomes in HSCs and promote HSC aging (Luo et al., 2019).
Mitochondria Clearance
Autophagy and Mitophagy in Adult HSCs
Autophagy degrades damaged organelles and remove aggregated proteins within cells in response to stress (Shahrabi et al., 2019). Nutrient-sensing pathways, such as the AMPK pathway, activate autophagy-related genes (Atgs) to initiate formation of autophagosomes and clearance of unwanted cellular material. HSCs modulate autophagic flux to control their maintenance, differentiation, and proliferation (Mihaylova and Shaw, 2011; Figure 6). Impairment of autophagy leads to premature aging, while maintenance of autophagy has been implicated in the maintenance of various stem cells including HSCs (Revuelta and Matheu, 2017). For example, deletion of Atg5 in hematopoietic cells causes abnormal clearance of damaged mitochondria and a decline in HSC reconstitution potential (Jung et al., 2019). Conditional knock-out of Atg12 impaired HSC reconstitution potential and resulted in premature HSC aging (Ho et al., 2017). Interestingly, Atg12-deficient HSCs presented an increase in active mitochondria with high ΔΨmt, indicating that Atg12-mediated autophagy is involved in the clearance of healthy but active mitochondria. Similarly, deletion of Atg7 resulted in decrease HSC function and exhaustion accompanied with increased mitochondria numbers and ROS levels (Mortensen et al., 2011).
Figure 6. Regulation of Autophagic Clearance of Active and Damaged Mitochondria in HSCs.
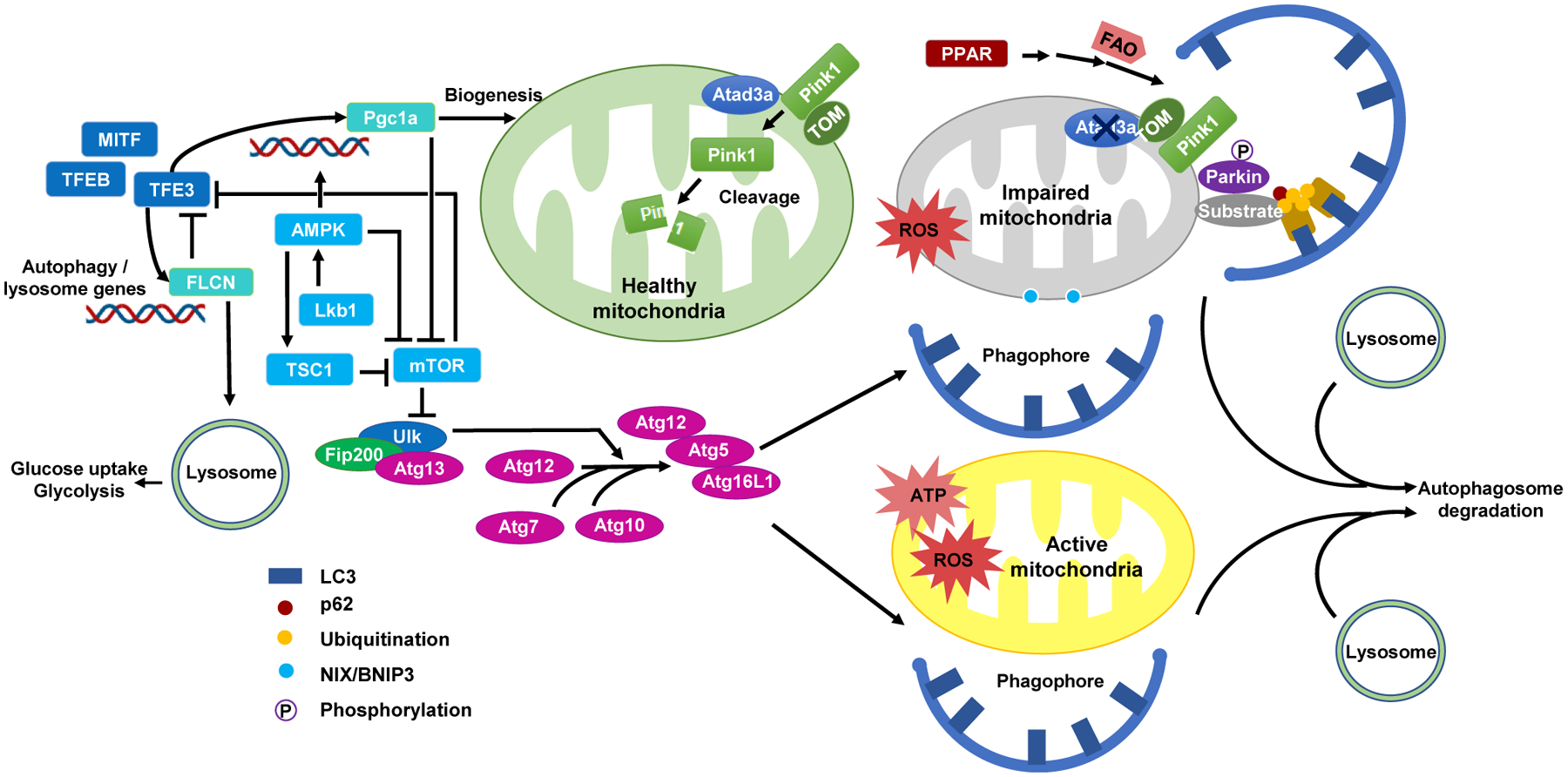
The removal of active and damaged mitochondria through autophagy maintains HSCs in a low metabolic, quiescent state. Active mitochondria with high ATP production and ROS levels are cleared through macroautophagy. Mitochondria with impaired function, such as deletion of Atad3a, may provoke Pink/Parkin pathway for mitophagy. Phosphorylated Parkin promotes the ubiquitination of mitochondrial substrates, recruits p62, and links mitochondria to LC3 for mitophagy. The presence of Atad3a on healthy mitochondria promotes the degradation of Pink1 via protease cleavage in the mitochondria matrix. Autophagy and mitophagy are regulated through a cascade of reactions involving Atg (Atg5, 7, 10, 12, 13, 16L1), Ulk1, FIP200, and other autophagy-related proteins. Autophagic pathways are interlinked with mitochondria biogenesis (mTOR, AMPK, LKB1, TSC2, and PGC1a) along with lysosomal pathways (FLCN, TFEB, TFE3, and MITF).
One of the key regulators of mitophagy, Pten-induced putative kinase 1 (Pink1), interacts with the E3 ubiquitin ligase Parkin (or Park2) and directly facilitates mitochondria aggregation to form autophagosomes (Ito et al., 2016). Pink1 specifically senses depolarized and damaged mitochondria, accumulates on the surface of the mitochondria to recruit and activate Parkin (or Park2), and eventually leads to autophagic removal of mitochondria (Pickrell et al., 2015; Jin et al., 2010). Acute silencing of Parkin/Pink1 in HSCs has been shown to promote enhanced clearance of damaged mitochondria mediated through Ppar-FAO pathway, and defective mitophagy impairs the self-renewal of Tie2+ HSCs. In addition, the studies with Ulk1- and Bnip3l/Nix- deficient mice have found defective mitochondrial clearance during terminal stages of red blood cell maturation, suggesting a potential role of an Atg5/Atg7-independent non-canonical autophagy pathway in hematopoietic cells (Honda et al., 2014).
Enhanced mitophagy also affects HSCs. The deletion of AAA+-ATPase, Atad3a, a regulator of Pink1-dependent mitophagy, hyperactivated mitophagy in HSCs (Jin et al., 2018). Atad3a locates on both IMM and OMM and serves as a mitochondria-ER organizing network protein (Yang and Suda, 2018). Its anchorage into the IMM enables it to interact with ER membranes via N terminus and regulate mitochondria-ER interactions to support steroid and lipid biosynthesis. Atad3a-deficient HSCs exhibited defective lineage differentiation yet expanded in number. Thus, enhanced mitophagy also induces HSC dysfunction.
Lysosomal Function
Interactions between mitochondria and lysosomes have also been implicated in HSC regulation. MITF,TFEB, orTFE3 regulate autophagosomal and lysosomal biogenesis through their interaction with ATG13 and ULK1 (Kennedy and Lamming, 2016). Quiescent HSCs exhibited abundant numbers of active lysosomes which sequestered lysosomal cargo such as mitochondria (Liang et al., 2020). The suppression of lysosomal acidification using concanamycin A (con A) greatly increased ΔΨmt as well as the number and the reconstitution potential of primed-HSCs. Con A-treated, primed-HSCs exhibited enlarged lysosomes with enhanced sequestration of mitochondria as well as low glucose uptake and glycolysis. Lysosomal activity therefore coordinates metabolism for the maintenance of HSCs (Liang et al., 2020). Taken together, low lysosomal activity is critical for the regulation of quiescent HSCs.
HSC Metabolism during Stress and Aging
HSC Heterogeneity and Stress Hematopoiesis
Multipotency, quiescence, and self-renewal capacity are fundamental properties that segregate HSCs from progenitor cells. Yet, HSCs are phenotypically and functionally heterogeneous, presenting variable stem cell properties. Adult HSCs have different depths of cell cycle quiescence: deeply dormant HSCs versus easily activated, primed HSCs. It is postulated that dormant HSCs require a longer time to enter the cell cycle compared to primed HSCs (Cabezas-Wallscheid et al., 2017) and primed HSCs primarily contribute to the restoration of hematopoiesis during stress. Native hematopoiesis assessed with non-transplant models revealed that expansion of multi-potent progenitors (MPPs) establishes steady-state hematopoiesis (Sun et al., 2014; Rodriguez-Fraticelli et al., 2018). These data suggest that long-lived progenitors, rather than HSCs, are the main drivers of physiological hematopoiesis in adults. Lastly, subpopulations of HSCs exhibit bias toward specific lineages, signifying their variability in multi-lineage differentiation capacity (Sanjuan-Pla et al., 2013; Yamamoto et al., 2013; Haas et al., 2015). Lineage-biased HSCs prominently emerge during stress hematopoiesis due to inflammation or recovery from myeloablation. It is questioned whether HSPC heterogeneity (i.e., non-biased HSCs, megakaryocyte-biased HSCs, and long-lived progenitors) associates with HSC metabolic state.
During stress hematopoiesis, HSCs require ATP production for expansion and differentiation. Emergent demands for additional production of mature hematopoietic cells are often relayed through cytokine signaling (Singh et al., 2018). Deletion of the helix-loop-helix transcription factor Id1 attenuated the response of HSCs to cytokine induced proliferation due to a reduction in mitochondrial OXPHOS. We recently reported that the cytokine Thpo upregulates mitochondrial metabolism and skews adult HSCs toward a megakaryocyte lineage-biased differentiation (Nakamura-Ishizu et al., 2018). Administration of recombinant Thpo or Romiplostim (Thpo mimetic drug) to wild-type mice rapidly upregulated HSC ΔΨmt and megakaryocyte differentiation. Furthermore, HSCs with a high ΔΨmt displayed a megakaryocyte-lineage skewed reconstitution pattern upon competitive BM transplantation. These findings suggest that HSC response to stress depends on metabolic state and may be modulated by extrinsic factors such as Thpo.
HSC Aging
Aged HSCs display several distinct phenotypes, including an increase in frequency, reduction of regenerative capacity, and differentiation bias toward myeloid-lineage (de Haan and Lazare, 2018). As described above, metabolic changes such as decrease in autophagy and reduction in proteasomal degradation along with the accumulation of DNA damage can accelerate HSC aging. Aged HSCs generally exhibit enhanced mitochondria OXPHOS and increased ROS production which presumably compromise HSC function (Ito et al., 2006; Mohrin et al., 2015). However, it should be carefully assessed whether high ROS levels are a cause or consequence of HSC dysfunction. Changes in the BM microenvironment influence stem cell aging and indeed, anatomical and functional remodeling of HSC niches accelerates myeloid-lineage cell expansion during aging (Ho et al., 2019). However, changes in the niche that promote HSC aging thorough metabolic stress are yet to be explored.
High mitochondria activity relates to alteration of epigenetic states that impact aging. Mitochondria influence and modify the epigenetic state of cells (Reid et al., 2017). The TCA cycle generates citric acid which modulates histone acetylation and gene expression through its conversion to acetyl-CoA (Reid et al., 2017). Mitochondrial FAO generates acetyl-CoA for histone modification in HSCs (Ito and Suda, 2014). These metabolites moderate histone hypoacetylation and hypermethylation. Although not bound to histones, mitochondrial DNA (mtDNA) is epigenetically regulated through methylation, yet the physiological relevance is still unknown (Matilainen et al., 2017). Nevertheless, the integrity of mtDNA influences HSC aging as exemplified in mice with a proof-reading-deficient version of mitochondrial DNA polymerase gamma (Polg) which causes an accumulation of mutations in mtDNA in HSCs (Norddahl et al., 2011).
Clonal Hematopoiesis of Indeterminate Potential (CHIP)
The presence of age-associated clonal hematopoiesis, CHIP (or age-related clonal hematopoiesis, ARCH), is a seminal finding in the study of HSC aging. A small number of HSC clones that disproportionally contribute to peripheral blood production emerge with aging and are detected in 10% of persons older than 65 years of age (Genovese et al., 2014). The majority of these clones present with mutations principally affecting epigenetic modifiers DNMT3A, ASXL2, and TET2. Among 10%–40% of cases, the clone will progress to meet the diagnostic criteria for CHIP (variant allele frequency [VAF] above 2%), which is associated with an increased risk of hematological cancer. TET2 and DNMT3a enzymes regulate metabolic activation and hematopoietic differentiation. Inactivation of these enzymes allows LSCs to utilize OXPHOs without impairing self-renewal (Sperling et al., 2017; Testa et al., 2016). Proteomic analysis of aged HSPCs revealed significant alterations in the expression of specific proteins involved in DNA repair, cell proliferation, and metabolism (Hennrich et al., 2018). Among these pathways, HSPCs uniquely exhibited the elevation of glucose metabolic enzymes upon aging, which led to the hypothesis that the upregulation of glycolytic enzymes may influence the development of CHIP. Modulation of metabolism to affect CHIP development and transition to hematopoietic malignancy is a topic of considerable therapeutic interest.
Concluding Remarks
HSCs are capable of maintaining homeostasis and meeting demand for hematopoiesis through control of their cell cycle status and metabolism. Interestingly, HSCs in fetal liver cells and neonatal bone marrow are engaged in active cycling, while adult HSCs remain in G0. Over the past decade, an abundance of new genetic models and -omics techniques have expanded our knowledge of both respiratory and non-respiratory functions of the mitochondria in adult HSCs. However, our understanding of metabolic regulation during embryonic hematopoiesis is still limited. This fact holds particularly true for human hematopoiesis; it is difficult to recapitulate the emergence of HSCs from pluripotent stem cells in vitro. The development of HSC capacity begins in the embryo with a series of transient hematopoietic waves. A clear description of how this development occurs, with metabolic adaptation across discrete anatomic sites that shift as the embryo evolves, is of fundamental importance.
Cellular metabolism can be extrinsically modulated. Yet, as the complete recapitulation of bone marrow physiology in vitro is difficult, findings obtained from cultured cells may not reflect in vivo metabolic state. Self-renewing division, which yields two functional HSCs, is believed to be infrequent after birth, as HSCs undergo symmetric self-renewal only 4–6 times across the entire lifespan of a mouse (Wilson et al., 2008). Upon stress, HSCs begin to divide and gradually lose regenerative capacity. This observation leads to many open questions. Why are self-renewing divisions rare in adult HSCs? How do the mitochondria, ER, and lysosomes cooperatively transform the metabolism of HSCs as they exit or re-enter quiescence? New analytical tools that can map spatial distributions of biomolecules (i.e., 4D digital imaging mass spectrometry) will identify novel interactions between HSCs and their niche. This will allow a range of interventions to tailor metabolic events that determine the equilibrium between quiescent and proliferating HSCs. Furthermore, unraveling the mechanism of HSC quiescence may provide insights into rejuvenation and prolonging life of an individual organism. Recently, it was shown that Polycomb complex members (CBX7) are upregulated during diapause state in killifish, a model of short-lived vertebrates (Hu et al., 2020). Diapause state suspends development and helps organisms survive extreme stress. A multi-disciplinary view of dormancy or hibernation may stimulate research and novel concepts in metabolism in organisms and dormant tissue stem cells.
It is unclear whether HSC metabolism during self-renewing cell divisions differs from that of divisions associated with differentiation and cell commitment. Metabolic comparisons between these division patterns provokes high scientific interest; ex vivo HSC expansion from limited donor cells is required for therapeutic application. Although the number of cells necessary for full metabolomics analysis limits analysis of rare populations, recent advances in continuous live imaging and tracking have highlighted an imbalanced apportioning of mitochondrial parameters between HSC daughter cells during asymmetric division (Loeffler et al., 2019). The reliable measurement of single-cell metabolomics in paired daughter cells will contribute to unveil specific metabolic modes to HSC fate decisions (Zenobi, 2013).
Elucidating the mechanisms of HSC fate control is a central goal of ongoing research in the field. However, HSC expansion ex vivo has had only limited success (Wilkinson et al., 2019; Bai et al., 2019) and HSC culture systems must be improved to fully realize the potential of HSC-based therapy. Interestingly, while adult HSCs and embryonic/neonatal HSCs both exhibit low mitochondrial ROS levels, adult HSCs exhibit a low metabolic and embryonic/neonatal HSCs exhibit high mitochondrial activity. Comparative analysis of mitochondrial contributions between adult and embryonic hematopoiesis could help clarify the underlying mechanism behind developmental changes and achieve an in vitro model of the physiological BM microenvironment. This will in turn illuminate the key metabolic requirements of healthy stem cell aging and may be applied to the invention of novel therapies for hematological disorders.
ACKNOWLEDGMENTS
This study was supported by the National Medical Research Council Grant of Singapore Translational Research Investigator Award (NMRC/STaR/18 may-0004/2019-2023, to T.S.), grants from the National Institutes of Health (to K.I.), Research Scholar of the Leukemia and Lymphoma Society (to K.I.), KAKEN Grant-in-Aid for Scientific Research (S) (18H05284 to T.S.), KAKEN Grant-in-Aid for Scientific Research (C) (18K08364 to A.N.-I.), and a grant from Japan Leukemia Research Fund (to A.N.-I.).
REFERENCES
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, Gelman L, Soriano ME, Nuoffer JM, Scorrano L, et al. (2018). Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun 9, 2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananieva EA, and Wilkinson AC (2018). Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 21, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansó E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, Liu X, Zhang Y, Shao Z, Steadman M, et al. (2017). The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol 19, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai T, Li J, Sinclair A, Imren S, Merriam F, Sun F, O’Kelly MB, Nourigat C, Jain P, Delrow JJ, et al. (2019). Expansion of primitive human hematopoietic stem cells by culture in a zwitterionic hydrogel. Nat. Med 25, 1566–1575. [DOI] [PubMed] [Google Scholar]
- Baldwin JE, and Krebs H (1981). The evolution of metabolic cycles. Nature 291,381–382. [DOI] [PubMed] [Google Scholar]
- Basu S, Broxmeyer HE, and Hangoc G (2013). Peroxisome proliferator-activated-γ coactivator-1α-mediated mitochondrial biogenesis is important for hematopoietic recovery in response to stress. Stem Cells Dev. 22, 1678–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner C, Toifl S, Farlik M, Halbritter F, Scheicher R, Fischer I, Sexl V, Bock C, and Baccarini M (2018). An ERK-Dependent Feedback Mechanism Prevents Hematopoietic Stem Cell Exhaustion. Cell Stem Cell 22, 879–892.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Petronilli V, Di Lisa F, and Forte M (2001). A mitochondrial perspective on cell death. Trends Biochem. Sci 26, 112–117. [DOI] [PubMed] [Google Scholar]
- Bernitz JM, Kim HS, MacArthur B, Sieburg H, and Moore K (2016). Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 167, 1296–1309.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigarella CL, Liang R, and Ghaffari S (2014). Stem cells and the impact of ROS signaling. Development 141, 4206–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Ito K, Morganti C, Pinton P, and Ito K (2018). Membrane-potential compensation reveals mitochondrial volume expansion during HSC commitment. Exp. Hematol 68, 30–37.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, and Eaves CJ (2006). Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Invest 116, 2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam L, Zhang J, Dreij K, Spahiu L, Holmgren A, Abe H, Tew KD, Townsend DM, Kelner MJ, Morgenstern R, and Johansson K (2018). MGST1, a GSH transferase/peroxidase essential for development and hematopoietic stem cell differentiation. Redox Biol. 17, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB, Pastor-Flores D, Roma LP, Renders S, Zeisberger P, et al. (2017). Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 169, 807–823.e19. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Jasper H, Ho TT, and Passegué E (2016). Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol 18, 823–832. [DOI] [PubMed] [Google Scholar]
- Chang CR, and Blackstone C (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem 282, 21583–21587. [DOI] [PubMed] [Google Scholar]
- Chaurasia P, Gajzer DC, Schaniel C, D’Souza S, and Hoffman R (2014). Epigenetic reprogramming induces the expansion of cord blood stem cells. J. Clin. Invest 124, 2378–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, and Chan DC (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan K-L, Liu Y, and Zheng P (2008). TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med 205, 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, and Lodish HF (2010). Fetal liver hepatic progenitors are supportive stromal cells for hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 107, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. (2017). Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, and Dzierzak E (2016). The many faces of hematopoietic stem cell heterogeneity. Development 143, 4571–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, and Puigserver P (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AM, and Harris DA (1990). Intracellular calcium as a regulator of the mitochondrial ATP synthase in cultured cardiomyocytes. Biochem. Soc. Trans 18, 554–555. [DOI] [PubMed] [Google Scholar]
- de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, and Snoeck H-W (2017). Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 21, 725–729.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, and Scorrano L (2009). Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9, 222–226. [DOI] [PubMed] [Google Scholar]
- de Haan G, and Lazare SS (2018). Aging of hematopoietic stem cells. Blood 131,479–487. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, and Bigas A (2018). Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell 22, 639–651. [DOI] [PubMed] [Google Scholar]
- Ema H, and Nakauchi H (2000). Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood 95, 2284–2288. [PubMed] [Google Scholar]
- Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, Mayotte N, Csaszar E, Knapp DJHF, Miller P, Ngom M, et al. (2014). Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 345, 1509–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C, Albiero M, Canato M, Gherardi G, De Stefani D, et al. (2019). DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun 10, 2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi MD, and Ghaffari S (2019). Mitochondria in the maintenance of hematopoietic stem cells: new perspectives and opportunities. Blood 133, 1943–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CDL, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, and Terzic A (2011). Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 14, 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, Hess DT, and Stamler JS (2009). Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med 15, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame JM, Lim SE, and North TE (2017). Hematopoietic stem cell development: Using the zebrafish to identify extrinsic and intrinsic mechanisms regulating hematopoiesis. Methods Cell Biol. 138, 165–192. [DOI] [PubMed] [Google Scholar]
- Friedman JS, Rebel VI, Derby R, Bell K, Huang TT, Kuypers FA, Epstein CJ, and Burakoff SJ (2001). Absence of mitochondrial superoxide dismutase results in a murine hemolytic anemia responsive to therapy with a catalytic antioxidant. J. Exp. Med 193, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T, Tanaka Y, Hamey FK, Chang C-H, Oki T, Asada S, Hayashi Y, Fujino T, Yonezawa T, Takeda R, et al. (2019). Discrimination of Dormant and Active Hematopoietic Stem Cells by G0 Marker Reveals Dormancy Regulation by Cytoplasmic Calcium. Cell Rep. 29, 4144–4158.e7. [DOI] [PubMed] [Google Scholar]
- Galli S, Antico Arciuch VG, Poderoso C, Converso DP, Zhou Q, Bal de Kier Joffé E, Cadenas E, Boczkowski J, Carreras MC, and Poderoso JJ (2008). Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PLoS ONE 3, e2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, and Depinho RA (2010). Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 468, 701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland JM, and Rudin C (1998). Cytochrome c induces caspase-dependent apoptosis in intact hematopoietic cells and overrides apoptosis suppression mediated by bcl-2, growth factor signaling, MAP-kinase-kinase, and malignant change. Blood 92, 1235–1246. [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Puerto MC, Folkerts H, Wierenga ATJ, Schepers K, Schuringa JJ, Coffer PJ, and Vellenga E (2016). Autophagy Proteins ATG5 and ATG7 Are Essential for the Maintenance of Human CD34(+) Hematopoietic Stem-Progenitor Cells. Stem Cells 34, 1651–1663. [DOI] [PubMed] [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, and Mulligan RC (1996). Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med 183, 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, and Lang BF (1999). Mitochondrial evolution. Science 283,1476–1481. [DOI] [PubMed] [Google Scholar]
- Guitart AV, Subramani C, Armesilla-Diaz A, Smith G, Sepulveda C, Gezer D, Vukovic M, Dunn K, Pollard P, Holyoake TL, et al. (2013). Hif-2α is not essential for cell-autonomous hematopoietic stem cell maintenance. Blood 122, 1741–1745. [DOI] [PubMed] [Google Scholar]
- Guitart AV, Panagopoulou TI, Villacreces A, Vukovic M, Sepulveda C, Allen L, Carter RN, van de Lagemaat LN, Morgan M, Giles P, et al. (2017). Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J. Exp. Med 214, 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Huang X, Lee MR, Lee SA, and Broxmeyer HE (2018). Antagonism of PPAR-γ signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat. Med 24, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, et al. (2010). The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 468, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, Wurzer S, Prendergast AM, Schnell A, Hexel K, et al. (2015). Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 17, 422–434. [DOI] [PubMed] [Google Scholar]
- Hansford RG (1994). Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr 26, 495–508. [DOI] [PubMed] [Google Scholar]
- Harris JM, Esain V, Frechette GM, Harris LJ, Cox AG, Cortes M, Garnaas MK, Carroll KJ, Cutting CC, Khan T, et al. (2013). Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood 121, 2483–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CJ, and Wolf CR (2011). Knockout and transgenic mice in glutathione transferase research. Drug Metab. Rev 43, 152–164. [DOI] [PubMed] [Google Scholar]
- Hennrich ML, Romanov N, Horn P, Jaeger S, Eckstein V, Steeples V, Ye F, Ding X, Poisa-Beiro L, Lai MC, et al. (2018). Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat. Commun 9, 4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, Sesaki H, Li T, Yu J, Wunderlich M, et al. (2020). Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 26, 420–430.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegue E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YH, Del Toro R, Rivera-Torres J, Rak J, Korn C, García-García A, Macías D, González-Gómez C, Del Monte A, Wittner M, et al. (2019). Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 25, 407–418.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström KM, and Finkel T (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol 15, 411–421. [DOI] [PubMed] [Google Scholar]
- Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, and Shimizu S (2014). Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun 5, 4004. [DOI] [PubMed] [Google Scholar]
- Hu M, Zeng H, Chen S, Xu Y, Wang S, Tang Y, Wang X, Du C, Shen M, Chen F, et al. (2018). SRC-3 is involved in maintaining hematopoietic stem cell quiescence by regulation of mitochondrial metabolism in mice. Blood 132, 911–923. [DOI] [PubMed] [Google Scholar]
- Hu CK, Wang W, Brind’Amour J, Singh PP, Reeves GA, Lorincz MC, Alvarado AS, and Brunet A (2020). Vertebrate diapause preserves organisms long term through Polycomb complex members. Science 367, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DR, and Haworth RA (1979). The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys 195, 453–459. [DOI] [PubMed] [Google Scholar]
- Ito K, and Suda T (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, and Suda T (2006). Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med 12, 446–451. [DOI] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, and Pandolfi PP (2012). A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med 18, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, et al. (2016). Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasawa R, Mahul-Mellier A-L, Datler C, Pazarentzos E, and Grimm S (2011). Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YY, and Sharkis SJ (2007). A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110, 3056–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MB, and Jasper H (2014). Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 20, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, and Youle RJ (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol 191, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Xu C, Zhang X, Long J, Rezaeian AH, Liu C, Furth ME, Kridel S, Pasche B, Bian XW, and Lin HK (2018). Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol 19, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HE, Shim YR, Oh JE, Oh DS, and Lee HK (2019). The autophagy protein Atg5 plays a crucial role in the maintenance and reconstitution ability of hematopoietic stem cells. Immune Netw. 19, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C, Sinha AU, Lane SW, Souza AL, Clish CB, et al. (2012). mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 11,429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karigane D, Kobayashi H, Morikawa T, Ootomo Y, Sakai M, Nagamatsu G, Kubota Y, Goda N, Matsumoto M, Nishimura EK, et al. (2016). p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 19, 192–204. [DOI] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, and Kashatus DF (2015). Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 57, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, and Lamming DW (2016). The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 23, 990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan JA, Mendelson A, Kunisaki Y, Birbrair A, Kou Y, Arnal-Estapé A, Pinho S, Ciero P, Nakahara F, Ma’ayan A, et al. (2016). Fetal liver hematopoietic stem cell niches associate with portal vessels. Science 351, 176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodadi E, Shahrabi S, Shahjahani M, Azandeh S, and Saki N (2016). Role of stem cell factor in the placental niche. Cell Tissue Res. 366, 523–531. [DOI] [PubMed] [Google Scholar]
- Kim M, Cooper DD, Hayes SF, and Spangrude GJ (1998). Rhodamine-123 staining in hematopoietic stem cells of young mice indicates mitochondrial activation rather than dye efflux. Blood 91, 4106–4117. [PubMed] [Google Scholar]
- Kobayashi H, Morikawa T, Okinaga A, Hamano F, Hashidate-Yoshida T, Watanuki S, Hishikawa D, Shindou H, Arai F, Kabe Y, et al. (2019). Environmental Optimization Enables Maintenance of Quiescent Hematopoietic Stem Cells Ex Vivo. Cell Rep. 28, 145–158.e9. [DOI] [PubMed] [Google Scholar]
- Krols M, Bultynck G, and Janssens S (2016). ER-Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J. Cell Biol 214, 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Ciraolo G, Hinge A, and Filippi MD (2014). An efficient and reproducible process for transmission electron microscopy (TEM) of rare cell populations. J. Immunol. Methods 404, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N (2005). Power, sex, suicide: mitochondria and the meaning of life (Oxford University Press; ). [Google Scholar]
- Lee JY, Nakada D, Yilmaz OH, Tothova Z, Joseph NM, Lim MS, Gilliland DG, and Morrison SJ (2010). mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 7, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, et al. (2020). Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 26, 359–376.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, and Guan JL (2010). FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 116, 4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng H, Yu WM, Cooper TM, Bunting KD, and Qu CK (2015). Maintenance of mouse hematopoietic stem cells ex vivo by reprogramming cellular metabolism. Blood 125, 1562–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D, Wehling A, Schneiter F, Zhang Y, Müller-Bötticher N, Hoppe PS, Hilsenbeck O, Kokkaliaris KD, Endele M, and Schroeder T (2019). Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573, 426–429. [DOI] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, and Snoeck H-W (2016). Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger LL, Strikoudis A, Danzl NM, Bush EC, Finlayson MO, Satwani P, Sykes M, Yazawa M, and Snoeck H-W (2019). Harnessing Hematopoietic Stem Cell Low Intracellular Calcium Improves Their Maintenance In Vitro. Cell Stem Cell 25, 225–240.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, and Lapidot T (2014). Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal 21, 1605–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Mu WC, Karki R, Chiang HH, Mohrin M, Shin JJ, Ohkubo R, Ito K, Kanneganti TD, and Chen D (2019). Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 26, 945–954.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manesia JK, Xu Z, Broekaert D, Boon R, van Vliet A, Eelen G, Vanwelden T, Stegen S, Van Gastel N, Pascual-Montano A, et al. (2015). Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem Cell Res. (Amst.) 15, 715–721. [DOI] [PubMed] [Google Scholar]
- Mantel CR, O’Leary HA, Chitteti BR, Huang X, Cooper S, Hangoc G, Brustovetsky N, Srour EF, Lee MR, Messina-Graham S, et al. (2015). Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell 161, 1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DCS, Cecconi F, Kuida K, Tomaselli KJ, et al. (2002). Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Anaf-1/casnase-9 apoptosome. Nature 419, 634–637. [DOI] [PubMed] [Google Scholar]
- Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, Lapidot T, Jung S, and Gross A (2012). The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol 14, 535–541. [DOI] [PubMed] [Google Scholar]
- Matilainen O, Quirós PM, and Auwerx J (2017). Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol. 27, 453–463. [DOI] [PubMed] [Google Scholar]
- McCord JM, Keele BB Jr., and Fridovich I (1971). An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc. Natl. Acad. Sci. USA 68, 1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, and Denton RM (1993). Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev. Neurosci 15, 165–173. [DOI] [PubMed] [Google Scholar]
- Merchant AA, Singh A, Matsui W, and Biswal S (2011). The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood 118, 6572–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, and Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol 13,1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, et al. (2007). Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1, 101–112. [DOI] [PubMed] [Google Scholar]
- Mohanty JG, Nagababu E, Friedman JS, and Rifkind JM (2013). SOD2 deficiency in hematopoietic cells in mice results in reduced red blood cell deformability and increased heme degradation. Exp. Hematol 41, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, and Chen D (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Widjaja A, Liu Y, Luo H, and Chen D (2018). The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell 17, e12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti C, Bonora M, Ito K, and Ito K (2019a). Electron transport chain complex II sustains high mitochondrial membrane potential in hematopoietic stem and progenitor cells. Stem Cell Res. (Amst.) 40, 101573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti C, Bonora M, and Ito K (2019b). Improving the Accuracy of Flow Cytometric Assessment of Mitochondrial Membrane Potential in Hematopoietic Stem and Progenitor Cells Through the Inhibition of Efflux Pumps. J. Vis. Exp 30, 10.3791/60057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SEW, et al. (2011). The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med 208, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Suzuki T, Harigae H, Romeo P-H, Yamamoto M, and Motohashi H (2017). NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells. Mol. Cell. Biol 37, e00086–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Ihara Y, Nakamura H, Yodoi J, Sumikawa K, and Kondo T (2003). Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem 278, 50226–50233. [DOI] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, and Morrison SJ (2010). Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Matsumura T, Stumpf PS, Umemoto T, Takizawa H, Takihara Y, O’Neil A, Majeed ABBA, MacArthur BD, and Suda T (2018). Thrombopoietin Metabolically Primes Hematopoietic Stem Cells to Megakaryocyte-Lineage Differentiation. Cell Rep. 25, 1772–1785.e6. [DOI] [PubMed] [Google Scholar]
- Nogueira-Pedro A, Dias CC, Regina H, Segreto C, Addios PC, Lungato L, D’Almeida V, Barros CC, Higa EMS, Buri MV, et al. (2014). Nitric oxide-induced murine hematopoietic stem cell fate involves multiple signaling proteins, gene expression, and redox modulation. Stem Cells 32, 2949–2960. [DOI] [PubMed] [Google Scholar]
- Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, and Bryder D (2011). Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 8, 499–510. [DOI] [PubMed] [Google Scholar]
- North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, et al. (2007). Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 447, 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, and Lenaers G (2003). Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem 278, 7743–7746. [DOI] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, and Carroll KS (2011). Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol 8, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L, and Scorrano L (2016). Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol 78, 505–531. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, and Youle RJ (2015). Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 87, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Månsson R, Thoren LA, Ekblom M, Alexander WS, and Jacobsen SE (2007). Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 1, 671–684. [DOI] [PubMed] [Google Scholar]
- Qian P, He XC, Paulson A, Li Z, Tao F, Perry JM, Guo F, Zhao M, Zhi L, Venkatraman A, et al. (2016). The Dlk1-Gtl2 Locus Preserves LT-HSC Function by Inhibiting the PI3K-mTOR Pathway to Restrict Mitochondrial Metabolism. Cell Stem Cell 18, 214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, et al. (2017). BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388. [DOI] [PubMed] [Google Scholar]
- Rakheja D, Medeiros LJ, Bevan S, and Chen W (2013). The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front. Oncol 3, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Dai Z, and Locasale JW (2017). The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol 19,1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revuelta M, and Matheu A (2017). Autophagy in stem cell aging. Aging Cell 16, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmelé P, Liang R, Bigarella CL, Kocabas F, Xie J, Serasinghe MN, Chipuk J, Sadek H, Zhang CC, and Ghaffari S (2015). Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 16, 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, and Camargo FD (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, and Colombini M (2005). On the role of VDAC in apoptosis: fact and fiction. J. Bioenerg. Biomembr 37, 129–142. [DOI] [PubMed] [Google Scholar]
- Samokhvalov IM, Samokhvalova NI, and Nishikawa S (2007). Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446, 1056–1061. [DOI] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Bouriez Jones T, et al. (2013). Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502,232–236. [DOI] [PubMed] [Google Scholar]
- Santucci R, Sinibaldi F, Cozza P, Polticelli F, and Fiorucci L (2019). Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol 136, 1237–1246. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Ma M, Cao L, McCoy JP, Liu C, Hensel NF, Barrett AJ, Boehm M, and Finkel T (2008). Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J. Biol. Chem 283, 28506–28512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BJ, Yoon SH, and Do JT (2018). Mitochondrial dynamics in stem cells and differentiation. Int. J. Mol. Sci 19, 3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahrabi S, Paridar M, Zeinvand-Lorestani M, Jalili A, Zibara K, Abdollahi M, and Khosravi A (2019). Autophagy regulation and its role in normal and malignant hematopoiesis. J. Cell. Physiol 234, 21746–21757. [DOI] [PubMed] [Google Scholar]
- Shin M, Nagai H, and Sheng G (2009). Notch mediates Wnt and BMP signals in the early separation of smooth muscle progenitors and blood/endothelial common progenitors. Development 136, 595–603. [DOI] [PubMed] [Google Scholar]
- Shitara H, Shimanuki M, Hayashi J, and Yonekawa H (2010). Global imaging of mitochondrial morphology in tissues using transgenic mice expressing mitochondrially targeted enhanced green fluorescent protein. Exp. Anim 59, 99–103. [DOI] [PubMed] [Google Scholar]
- Signer RAJ, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson V, Takei H, Soboleva S, Radulovic V, Galeev R, Siva K, Leeb-Lundberg LMF, Iida T, Nittono H, and Miharada K (2016). Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 18, 522–532. [DOI] [PubMed] [Google Scholar]
- Singh SK, Singh S, Gadomski S, Sun L, Pfannenstein A, Magidson V, Chen X, Kozlov S, Tessarollo L, Klarmann KD, and Keller JR (2018). Id1 Ablation Protects Hematopoietic Stem Cells from Stress-Induced Exhaustion and Aging. Cell Stem Cell 23, 252–265.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeck HW (2017). Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol 49, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerkamp P, Altamura S, Renders S, Narr A, Ladel L, Zeisberger P, Eiben PL, Fawaz M, Rieger MA, Cabezas-Wallscheid N, and Trumpp A (2020). Differential Alternative Polyadenylation Landscapes Mediate Hematopoietic Stem Cell Activation and Regulate Glutamine Metabolism. Cell Stem Cell 26, 722–738.e7. [DOI] [PubMed] [Google Scholar]
- Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, et al. (2014). Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling AS, Gibson CJ, and Ebert BL (2017). The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 17, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, and Haigis MC (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol 20, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, and Semenza GL (2011). Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310. [DOI] [PubMed] [Google Scholar]
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, et al. (2013). MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell 51, 20–34. [DOI] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, et al. (2016). Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 23, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, and Camargo FD (2014). Clonal dynamics of native haematopoiesis. Nature 514, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai-Nagara I, Matsuoka S, Ariga H, and Suda T (2014). Mortalin and DJ-1 coordinately regulate hematopoietic stem cell function through the control of oxidative stress. Blood 123, 41–50. [DOI] [PubMed] [Google Scholar]
- Takihara Y, Nakamura-Ishizu A, Tan DQ, Fukuda M, Matsumura T, Endoh M, Arima Y, Chin DWL, Umemoto T, Hashimoto M, et al. (2019). High mitochondrial mass is associated with reconstitution capacity and quiescence of hematopoietic stem cells. Blood Adv. 3, 2323–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, and Suda T (2010). Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7, 391–402. [DOI] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. (2013). Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan DQ, and Suda T (2018). Reactive Oxygen Species and Mitochondrial Homeostasis as Regulators of Stem Cell Fate and Function. Antioxid. Redox Signal 29, 149–168. [DOI] [PubMed] [Google Scholar]
- Tan DQ, Li Y, Yang C, Li J, Tan SH, Chin DWL, Nakamura-Ishizu A, Yang H, and Suda T (2019). PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep. 26, 2316–2328.e6. [DOI] [PubMed] [Google Scholar]
- Taya Y, Ota Y, Wilkinson AC, Kanazawa A, Watarai H, Kasai M, Nakauchi H, and Yamazaki S (2016). Depleting dietary valine permits nonmyeloa-blative mouse hematopoietic stem cell transplantation. Science 354, 1152–1155. [DOI] [PubMed] [Google Scholar]
- Testa U, Labbaye C, Castelli G, and Pelosi E (2016). Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol 44, 540–560. [DOI] [PubMed] [Google Scholar]
- Uchida N, Dykstra B, Lyons K, Leung F, Kristiansen M, and Eaves C (2004). ABC transporter activities of murine hematopoietic stem cells vary according to their developmental and activation status. Blood 103, 4487–4495. [DOI] [PubMed] [Google Scholar]
- Umemoto T, Hashimoto M, Matsumura T, Nakamura-Ishizu A, and Suda T (2018). Ca2+-mitochondria axis drives cell division in hematopoietic stem cells. J. Exp. Med 215, 2097–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin RD, Smith DL, Blinco D, Wilson CL, Miller CJ, Evans CA, Jaworska E, Baldwin SA, Barnes K, Pierce A, et al. (2006). Quantitative proteomics reveals posttranslational control as a regulatory factor in primary hematopoietic stem cells. Blood 107, 4687–4694. [DOI] [PubMed] [Google Scholar]
- van Velthoven CTJ, and Rando TA (2019). Stem Cell Quiescence: Dynamism, Restraint, and Cellular Idling. Cell Stem Cell 24, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, and Lutolf MP (2016). Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun 7, 13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, Ragusa S, Stefanidis E, Ryu D, Rainer PY, et al. (2019). The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 24, 405–418.e7. [DOI] [PubMed] [Google Scholar]
- Villalobo A (2006). Nitric oxide and cell proliferation. FEBS J. 273,2329–2344. [DOI] [PubMed] [Google Scholar]
- Vukovic M, Sepulveda C, Subramani C, Guitart AV, Mohr J, Allen L, Panagopoulou TI, Paris J, Lawson H, Villacreces A, et al. (2016). Adult hematopoietic stem cells lacking Hif-1 a self-renew normally. Blood 127, 2841–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JE Jr., Brunstein CG, Boitano AE, DeFor TE, McKenna D, Sumstad D, Blazar BR, Tolar J, Le C, Jones J, et al. (2016). Phase I/II Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell 18, 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner-Souza K, Diamond HR, Ornellas MH, Gomes BE, Almeida-Oliveira A, Abdelhay E, Bouzas LF, and Rumjanek VM (2008). Rhodamine 123 efflux in human subpopulations of hematopoietic stem cells: comparison between bone marrow, umbilical cord blood and mobilized peripheral blood CD34+ cells. Int. J. Mol. Med 22, 237–242. [PubMed] [Google Scholar]
- Wang Y, Subramanian M, Yurdagul A Jr., Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, and Tabas I (2017). Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell 171, 331–345.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasiak S, Zunino R, and McBride HM (2007). Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol 177, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol 11, 872–884. [DOI] [PubMed] [Google Scholar]
- Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, Yamamoto R, Loh KM, Nakamura Y, Watanabe M, et al. (2019). Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 571, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, and Nakauchi H (2013). Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126. [DOI] [PubMed] [Google Scholar]
- Yang C, and Suda T (2018). Hyperactivated mitophagy in hematopoietic stem cells. Nat. Immunol 19, 2–3. [DOI] [PubMed] [Google Scholar]
- Yin H, and Zhu M (2012). Free radical oxidation of cardiolipin: chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radic. Res 46, 959–974. [DOI] [PubMed] [Google Scholar]
- Youle RJ, and van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W-M, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, and Qu C-K (2013). Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 12, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenobi R (2013). Single-cell metabolomics: analytical and biological perspectives. Science 342, 1243259. [DOI] [PubMed] [Google Scholar]
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, et al. (2011). UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 30, 4860–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Sliter DA, Bleck CKE, and Ding S (2019). Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion 49, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Gao P, Tober J, Bennett L, Chen C, Uzun Y, Li Y, Howell ED, Mumau M, Yu W, et al. (2020). Developmental trajectory of pre-hematonoietic stem cell formation from endothelium. Blood. Published online May 11, 2020 10.1182/blood.2020004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlmans JMJM, Visser JWM, Kleiverda K, Kluin PM, Willemze R, and Fibbe WE (1995). Modification of rhodamine staining allows identification of hematopoietic stem cells with preferential short-term or long-term bone marrow-repopulating ability. Proc. Natl. Acad. Sci. USA 92, 8901–8905. [DOI] [PMC free article] [PubMed] [Google Scholar]