

Abstract
Since its discovery in 2007, we have seen the lives of patients diagnosed with advanced anaplastic lymphoma kinase (ALK)‐rearranged non‐small cell lung cancers (NSCLC) transform with the advent of molecular therapies with first‐, second‐, and third‐generation ALK inhibitors now available in the clinic. Despite great gains in patient survival now measured in years and preserved quality of life with targeted therapies, drug resistance is unfortunately inevitably encountered in this rare and unique molecular subset of lung cancer, and patients will eventually succumb to the disease. As these patients are often young, fit, and never smokers, the clinical and scientific communities have aligned to expedite drug development and access. Drug resistance profiling and further strategies are being explored through clinical trials, including the evaluation of specific drug sequencing and combinations to overcome such resistance and promote patient longevity. The cases of this report focus on precision medicine and aim to portray the pertinent aspects to consider when treating ALK‐rearranged NSCLC in 2020, an ever‐shifting space. By way of case examples, this report offers valuable information to the treating clinician, including the evolution of systemic treatments and the management of oligo‐progression and multisite drug resistance. With the maturation of real‐world data, we are fortunate to be experiencing quality and length of life for patients with this disease surpassing prior expectations in advanced lung cancer.
Key Points
This report focuses on the importance of genetic analysis of serial biopsies to capture the dynamic therapeutic vulnerabilities of a patient's tumor, providing a perspective on the complexity of ALK tyrosine kinase inhibitor (ALKi) treatment sequencing.
These case examples contribute to the literature on ALK‐rearranged and oncogene addicted non‐small cell lung cancer (NSCLC), providing a framework for care in the clinic.
In oligo‐progressive disease, local ablative therapy and continuation of ALKi postprogression should be considered with potential for sustained disease control.
ALK G1202R kinase domain mutations (KDM), highly prevalent at resistance to second‐generation ALKi resistances, may emerge in non‐EML4‐ALK variant 3 cases and is sensitive to third‐generation lorlatinib. When in compound with one or more ALK KDMs, resistance to lorlatinib is expected.
In the case of rampantly progressive disease, rebiopsy and redefining biology in a timely manner may be informative.
Short abstract
This report underscores the importance of genetic analysis of serial biopsies to capture the dynamic therapeutic vulnerabilities within a patient's tumor, while providing a perspective on the complexity of ALK tyrosine kinase inhibitor treatment sequencing. Two cases are presented to illustrate the experiences of patients with advanced ALK‐rearranged non‐small cell lung cancer, including the effect on their quality of life and the need for holistic care.
Introduction
Since the discovery of anaplastic lymphoma kinase (ALK) rearrangements as oncogenic drivers in non‐small cell lung cancer (NSCLC), we have seen a rapid emergence of highly effective and tolerable first‐, second‐, and third‐generation ALK tyrosine kinase inhibitors (ALKis), revolutionizing both quality and length of life for the majority of patients presenting with advanced disease [1, 2, 3, 4, 5, 6]. ALK fusions have been detected in 3%–7% of NSCLCs and have been associated with an absence of smoking, younger age, and adenocarcinoma histology [7]. Furthermore, ALK NSCLC has a predilection to the brain, with central nervous system (CNS) disease reported in approximately one‐fifth of patients at diagnosis and up to three‐fourths of patients throughout their disease course in those treated with front‐line first‐generation ALKi crizotinib [2, 8]. Later generation agents have been designed to enable greater CNS penetrability, efficacy, and CNS protection from progressive or relapsing disease [4, 5, 9, 10]. Despite this, those with CNS disease on clinical trials have experienced inferior survival compared with those without. Median overall survival (OS) for patients with baseline brain metastases from the phase III first‐line crizotinib trial PROFILE‐1014 was 23.5 months, whereas it has not been reached for those without brain metastasis, with 56.6% of the entire cohort alive at 4 years [11]. These data are comparable with ASCEND‐3, which demonstrated median OS of 52.1 months overall with front‐line ceritinib [12]. In the updated ALEX results for highly brain penetrant alectinib, progression‐free survival was 27.7 months in those with CNS disease at diagnosis and almost 7 months greater, 34.8 months, in those without [5].
Although OS rates from real‐world ALK cohorts are now also in the order of years, drug resistance and eventual disease relapse unfortunately remains inevitable [2, 13, 14, 15]. Such mechanisms of resistance can be broadly categorized into primary (intrinsic) and secondary (acquired). Primary resistance is rare (~5%) and is currently poorly understood; however, it may be attributed to false positive genotyping, an accompanying genetic “codriver” enabling bypass activation, differing drug sensitivities with different gene fusion variants resulting in varying protein stability, or early phenotypic change such as small cell transformation [2, 16, 17]. The presence of a de novo ALK kinase domain mutation (KDM) is uncommon [18]. Secondary resistance on the contrary is expected, via ALK‐dependent and/or ALK‐independent pathways [14, 19]. Acquired resistance can be characterized by genotyping following rebiopsy of a solid lesion or plasma sampling to inform the optimal next therapy for the individual patient [14, 19]. Heterogeneous disease control is common in patients on ALK‐inhibitor therapy (ALKi), and targeting a progressing or new solid lesion enables the identification of spatial and temporal tumor heterogeneity and polyclonal resistance. However, under current regulations, ALKis are prescribed empirically, without genetic resistance status testing, because testing is not established in routine clinical care.
An intriguing case report involving serial biopsies of a patient progressing through multiple ALKis documented the emergence of compound ALK KDMs, L1198F and C1156Y, at progression on fifth‐line lorlatinib, paradoxically resensitizing the tumor to crizotinib, with which the patient was rechallenged and responded to for a further 6 months [20]. This case highlights the temporal evolution of ALK tumors promoted through clonal selection with ALKis.
With the recent major improvements in the therapeutic paradigm for patients with advanced ALK‐rearranged NSCLC, we look to the real‐world experience to inform our practice, given most patients are managed on multiple lines of therapy not encompassed in clinical trial reports.
This report underscores the importance of genetic analysis of serial biopsies to capture the dynamic therapeutic vulnerabilities within a patient's tumor, while giving a perspective on the complexity of ALKi treatment sequencing. Two cases are presented to illustrate the experience for a person with advanced ALK‐rearranged NSCLC, including the impact on their quality of life and their need for holistic care.
These case examples contribute to the expanding literature on ALK‐rearranged and oncogene‐addicted NSCLC in the current therapeutic landscape, providing a framework to care for these patients in the clinic.
Patient Stories
Case Report 1
In May 2014, Mrs. ND, aged 31 years and of Vietnamese background, developed an uncharacteristic personality change while holidaying with her husband. She was previously well, with no significant medical history and no regular medications. She was born and living in Australia, was an engineer, had never smoked, had no children, and had no family history of malignancy.
Upon return home, she encountered symptoms of raised intracranial pressure and partial seizure activity, heralding emergent medical attention.
A computed tomography scan (CT) of the brain followed by magnetic resonance imaging with gadolinium contrast (MRI‐B) identified a solitary left anterior temporal lobe mass (Fig. 1).
Figure 1.
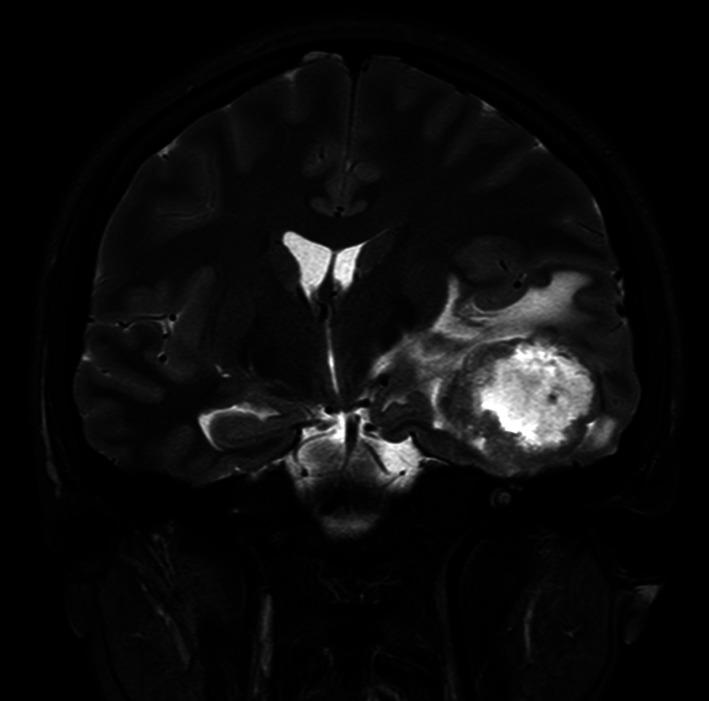
Mrs. ND's presentation magnetic resonance imaging with gadolinium contrast coronal T2 flare image of a left temporal lobe lesion with associated mass effect.
Analysis of the resected tissue confirmed the tumor to be a mucinous adenocarcinoma, thyroid transcription factor‐1 (TTF‐1) positive on immunohistochemistry (IHC), with strong uniform IHC staining for ALK (D5F3 antibody), confirmed via fluorescence in situ hybridization (FISH) with the classic split signal identified in 30% of cells.
A staging CT‐chest, abdomen, and pelvis and subsequent 18F‐fluorodeoxyglucose‐positron emission tomography (FDG‐PET) scan identified a mass in the right lower lobe of the lung with a standardized uptake value maximum (SUVmax) of 12.2 and associated ipsilateral hilar and mediastinal lymph node avidity and no further distant disease.
Following postoperative stereotactic radiosurgery (SRS) to the surgical bed, compassionate crizotinib was initiated, 250 mg twice daily (BD), in June 2014. Crizotinib was not yet government reimbursed in Australia at that time.
Within a few weeks of starting crizotinib, Mrs. ND experienced multiple common adverse events, all low grade (grade 1), including visual disturbance, gastroesophageal reflux, and lower limb edema. Day 12 on the drug, she encountered a grade 4 elevation in her transaminases (AST, 700 U/L). Crizotinib was dose interrupted, and liver enzymes normalized within two weeks; crizotinib was then reinitiated at 200 mg BD.
Restaging imaging with an MRI‐B and CT at 2 months revealed only a very small residual pulmonary scar‐like mass and no CNS disease. After 3 months, Mrs. ND returned to work, and by 6 months she was working full‐time reporting a normal quality of life.
In May 2015, 11 months on crizotinib, a restaging CT was stable; however, an MRI‐B revealed presumed recurrence in the left temporal lobe with multiple new small cortical and subcortical enhancing foci. A repeat FDG‐PET revealed minor systemic progression with avidity within the preexisting thoracic disease.
Disease progression coincided with the opportunity to access the second‐generation ALKi brigatinib, and Mrs. ND ceased crizotinib and commenced brigatinib 180 mg daily in June 2015.
While receiving brigatinib, Mrs. ND experienced low grade (grade 1) fatigue, cramps, unsteadiness, headaches, vertigo, and heart palpitations and was intermittently weak. Neurological and cardiac causes were excluded. Grade 2 edema and rash followed, and she was dose interrupted and reduced to 150 mg daily in August 2015.
The first restaging imaging on brigatinib at 2 months revealed a partial response as per RECIST version 1.1 in her lung via CT, and MRI‐B was stable [21].
In February 2016, with ongoing stable disease, Mrs. ND developed refractory hypertension. She was again dose interrupted and reduced to 120 mg daily. Despite her circumstances, Mrs. ND was still working full‐time and exercising daily, with plans to travel overseas. In June 2016, following more than 1 year on brigatinib, imaging including serial MRI‐Bs remained stable, with multiple, mostly nonenhancing supratentorial and infratentorial intracranial lesions persisting (largest, 8 mm), and her lesion in the right lower lobe remained stable (10 mm).
She continued to manage, with no definite or sustained symptomatic deterioration, until May 2017, when she experienced a possible seizure‐associated aura. Imaging was noted as stable and the episode was considered isolated.
In August 2017, imaging was again stable based on RECIST 1.1 guidelines; brigatinib was re‐escalated to the recommended phase II dose, 180 mg daily. However, over the next month, her blood pressure remained persistently elevated and was managed with antihypertensives and reducing brigatinib to 150 mg daily.
In January 2018, Mrs. ND encountered increasing tiredness and transient vague neurologic symptoms, including altered visual perceptions and balance impairment. Clinically, she had an abnormal heel‐toe test, veering to the left. Repeat brain imaging was stable.
A plasma sample was next obtained to evaluate circulating tumor (ct) DNA via next‐generation sequencing (NGS) (ctDx; Resolution Bioscience); however, this was uninformative, isolating no detectable genetic alteration (supplemental online Appendix 1A) [22]. There was no safe solid organ site to biopsy.
On balance, in January 2018, because of concern for clinical progression with neurological deterioration, Mrs. ND was changed to the second‐generation ALKi alectinib, 600 mg BD, coinciding with its reimbursement in Australia the same month.
With alectinib, she encountered several grade 1 toxicities, manageable with supportive measures, including constipation, mild myalgias, subjectively altered temperature regulation, and occasional visual disturbances.
The first MRI‐B on alectinib in March 2018 was stable. Treatment continued until July 2018, when an MRI‐B revealed minor progression in only a preexisting right vertex lesion, adjacent to the sensorimotor strip without associated edema. The remaining lesions were stable in keeping with oligo‐progression (Fig. 2).
Figure 2.
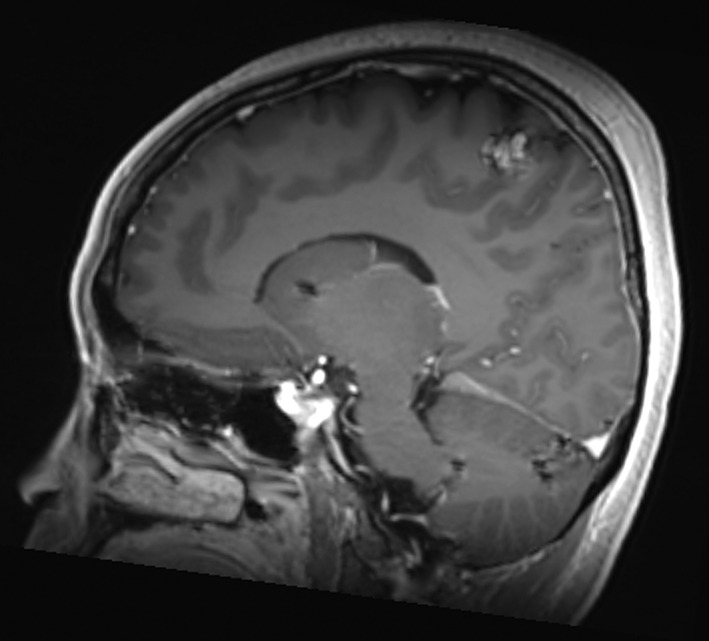
Mrs. ND, July 2018 sagittal T1 FLAIR image with contrast of the 14‐mm right vertex lesion in the sensorimotor strip progressing while on third‐line alectinib. The remaining intraparenchymal lesions were unchanged, and no new lesions were appreciated.
Radiation oncology and surgical opinions were sought while alectinib continued. Disease progression was only present intracranially. To obtain important current biological information, now more than 4 years since diagnosis, the patient‐physician joint decision was to proceed to craniotomy and resection of the progressing now symptomatic lesion, which occurred July 2018.
The postoperative course was complicated by focal seizures and left‐sided parasthesias with associated functional impairment. Anticonvulsants were initiated and titrated with improvement. Recovery continued with inpatient rehabilitation over three weeks.
In August 2018, with alectinib continuing, a repeat MRI‐B revealed stable multifocal low‐volume residual disease.
Mrs. ND's biopsy contained a poorly differentiated lung adenocarcinoma with nests and sheets of epithelioid cells with ample eosinophilic cytoplasm, necrosis, and hemorrhage and scanty intratumoral lymphocytes. This differed from her original resected brain specimen, which contained larger pleomorphic cells with abundant cytoplasm, papillary architecture, prominent fibrovascular cores, and an acute and chronic inflammatory infiltrate (Fig. 3). The specimen from July 2018 was further analyzed by a local targeted amplicon NGS assay and the commercially available FoundationOne CDx Solid Tumor Panel. These identified an Echinoderm Microtubule‐Associated Protein‐Like 4 (EML4)‐ALK variant 3a/b (E6:A20) fusion and the ALK solvent front G1202R KDM with a variant allelic frequency (VAF) of 33.8% (supplemental online Appendix 1B, C). The tumor mutational burden was low (5 mutations per megabase), and a SETD2 splice site c.4715+1G>A mutation was noted (32.9% VAF). The original temporal lobe tumor, resected in May 2014, did not contain the G1202R KDM (confirmed by Sanger sequencing).
Figure 3.
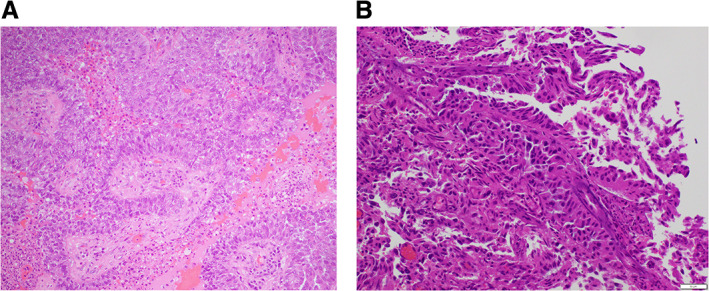
Photomicrographs of H&E‐stained cross‐sectional images of the temporal brain metastases resected from Mrs. ND 2014 (A) and right vertex lesion from 2019 (B). Image in (B) demonstrates a morphological difference from the original (A) resected brain specimen with poorly differentiated nests and sheets of epithelioid cells with ample eosinophilic cytoplasm and scanty intratumoral lymphocytes, necrosis, and hemorrhage. This image has unavoidable changes from intraoperative heat effect. The original (A) was composed of lager pleomorphic cytoplasm abundant cells with a papillary architecture and prominent fibrovascular cores and an acute on chronic inflammatory infiltrate
Based on these findings, a compassionate case was put forward to access the novel third‐generation ALKi lorlatinib, at the time not registered under Therapeutic Goods Administration in Australia, which Mrs. ND began in September 2018, 100 mg daily.
On lorlatinib, Mrs. ND encountered manageable left lower limb paresthesia persisting since her operation. She began encountering mood lability, which was assessed to be multifactorial, and her anticonvulsants were carefully weaned.
The first reimaging in October 2018 confirmed a partial response in her low volume CNS and pulmonary disease, all <7 mm.
As has now been well documented to potentially occur with lorlatinib, Mrs. ND developed hypercholesterolemia (total cholesterol >10 mmol/L) and began rosuvastatin. She also encountered weight gain, bloating, and hair thinning. Her mood continued to be labile as she felt more sensitive and intermittently depressed. The antidepressant mirtazapine was initiated.
Mrs. ND returned to work in late September 2018 and fundraised $25,000 for ALK NSCLC research in her center's partnering research institute. She sees a psychologist on a regular basis and exercises by walking daily.
In February 2019, Mr. and Mrs. ND holidayed overseas.
In August and November 2019, repeat imaging confirmed minimal measurable disease, predominately cystic in the brain, more than 5 years after presenting with symptomatic stage IV CNS disease. Figure 4 illustrates a timeline of Mrs. ND's treatment from diagnosis to the present, including the best radiological response with each therapy and genotyping results informing therapy.
Figure 4.
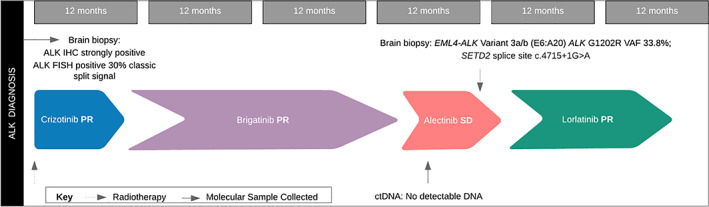
Timeline of Mrs. ND's treatment course from diagnosis to data‐cut December 2019, including the best objective radiological response obtained on each therapy. The timing and results of genotyping informing subsequent treatment are also indicated. Abbreviations: ctDNA, circulating tumor DNA; FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; PR, partial response; SD, stable disease.
Mrs. ND and her husband holidayed in Europe at the end of 2019, and in May 2020, she continues to be well on lorlatinib, with sustained disease control and quality of life.
Case Report 2
Ms. CG, a 33‐year‐old single Chinese Mandarin‐speaking woman, presented in February 2010 with a symptomatic occipital lobe embolic infarct and underwent thrombectomy. She had no prior medical history of note, took no medications, was a lifelong nonsmoker, and was an accountant. Further imaging during the admission, including chest x‐ray, CT‐chest, and FDG‐PET, identified a 5‐cm FDG‐PET avid left lower lobe lung mass with bilateral hilar, left mediastinal, and right cervical lymphadenopathy and a small left pleural effusion (Fig. 5). Percutaneous fine needle aspirate of the lung lesion confirmed a TTF‐1‐positive adenocarcinoma, in keeping with primary NSCLC, at a time premature for more detailed reflex molecular testing.
Figure 5.
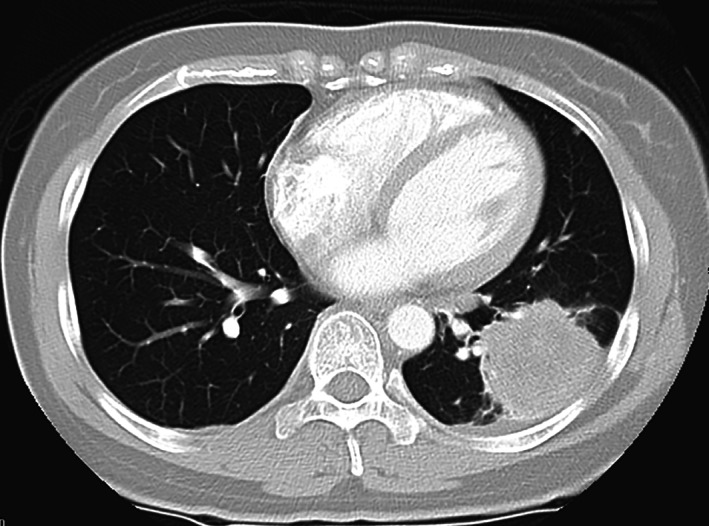
Ms. CG's baseline diagnostic contrast enhanced axial computed tomography‐chest lung window view depicting a large left lower lobe lung primary lesion.
Given a lack of social support in Australia, Ms. CG initially returned to China for treatment. In March 2010, she started erlotinib empirically, with disease progressing radiologically on a 4‐week interval scan. In April 2010, she received cisplatin plus pemetrexed, which was poorly tolerated with nausea and vomiting, and changed to carboplatin plus pemetrexed for a further two cycles; progressive disease was again noted at first reimaging. In July 2010, she was recommended carboplatin plus paclitaxel with bevacizumab, and after one cycle, she returned to Australia with her mother and was screened for a clinical trial investigating the then‐novel ALKi crizotinib [23]. An ALK‐rearrangement was identified on FISH, and in August 2010, she started crizotinib (250 mg BD) and continued on it for almost 7 years until April 2017. Initially, she achieved an excellent partial response in the primary lung tumor and the satellite lung and nodal disease; however, by November 2010, all three targets had increased in size, meeting RECIST 1.1 criteria for progression, although below their baseline measurement [11]. Given her well clinical state, Eastern Cooperative Oncology Group (ECOG) performance status 0, she was permitted to remain on trial beyond progression. In December 2010, following a generalized seizure, left frontal and occipital brain metastases were identified. These were treated with SRS. Remarkably, by April 2011, her lung primary had diminished to a scar after initial progression. Between May 2011 and November 2014, while still on crizotinib, Ms. CG had six episodes of progression in low volume asymptomatic supra‐ and infratentorial brain metastases, including four new lesions, with the largest measuring 7 mm. These were each managed with SRS. Crizotinib was withheld on day (D) −1, D1, and D2 of radiotherapy.
In October 2015, Ms. CG progressed in the chest, with her left lower lobe lung primary now measuring 14 mm. An FDG‐PET confirmed this to be the only metabolically active lesion, and she was again treated beyond progression on crizotinib. Over a year later, December 2016, CT showed that the primary had progressed to 42 mm and the left hilar node had progressed to 29 mm; nevertheless, Ms. CG remained on crizotinib with no focal therapy. By February 2017, these lesions had decreased in size. Although measurable disease was stable in April 2017, two new left upper lobe lung nodules and a hilar node were noted. Via MRI‐B the preexisting lesions were stable, and a new 8‐mm left lateral ventricle lesion was noted. This was treated with SRS. A few years later, Ms. CG admitted her compliance waned at times on crizotinib.
Apart from some grade 1 toxicity, including fluid retention, diarrhea, and the typical visual symptoms associated with changes in ambient lighting, Ms. CG tolerated crizotinib therapy well.
Given now unequivocal progression, Ms. CG changed to ceritinib 600 mg BD in April 2017, continuing for 7 months until November 2017. At the time, this was the only alternative ALKi available in Australia in the clinic. On ceritinib, she experienced grade 1 diarrhea, fatigue, nausea, and vomiting. A confluent erythematous grade 2 eruptive rash required dose interruption and reduction to 450 mg BD June 2017. Although thoracic disease was stable on ceritinib, an MRI‐B in June 2017 revealed two new small lesions. These were monitored, and by November 2017, she had developed 7 new intracranial lesions, disease stable extracranially, and treatment was changed to alectinib 600 mg BD via compassionate access. A partial intracranial response was noted by January 2018, before a complete response by July 2018. Extra‐cranially on alectinib CT staging in January 2018 revealed stable disease; however, by July, further progression at the site of her left lower lobe primary tumor was noted. An FDG‐PET confirmed this was the sole avid site, with an SUVmax of 14.8, and she received five fractions of consolidative SRS to the site while continuing on alectinib. Unfortunately, in October 2018, she had developed new bilateral miliary lung metastases.
Ms. CG encountered grade 2 fatigue, headaches, bloating, and alopecia with alectinib, requiring a dose reduction to 600 mg mane/450 mg nocte initially in December 2017. This was re‐escalated to full dose at later disease progression to no avail prior to it being ceased.
In January 2019, Ms. CG began empiric compassionate lorlatinib 100 mg daily, although by March 2019, she had progression in the contralateral right pleura and hilum. An FDG‐PET in April 2019 identified multiple new bilateral pulmonary sites of FDG‐PET intense avidity as well as subcutaneous chest wall metastases (SUVmax, 20.3). Despite being ECOG 0 prior to 2019, she had now deteriorated to ECOG 1–2 with breathlessness and cough, requiring two hospital admissions to manage superimposed lower respiratory tract infections. During these admissions, she was reviewed by the inpatient palliative care team and linked to the community service. In April 2019, a progressive symptomatic right pleural effusion was drained (900 mL), and cytology was reconfirmed adenocarcinoma. This was then sent for extended analysis with a local solid tumor cancer‐targeted NGS DNA panel and RNA fusion panel, identifying a variant 1 EML4‐ALK fusion (E13:A20) and two ALK KDMs: the solvent front G1202R (VAF, 8.4%) and the F1174C KDM (VAF 8.4%), which sits near the alpha‐C helix domain of the kinase domain (supplemental online Appendix 1D). These two mutations and the variant 1 fusion were confirmed in a ctDNA NGS plasma panel performed in May 2019 (Resolution Bioscience ctDx), although with lower VAFs (ALK fusion, 0.9%; G1202R, 0.5%; F1174C, 0.2%). A RET mutation of uncertain clinical significance, RET A741G (VAF 52.9%) was also reported. Given the high VAF, this was thought likely to be a germline variant (supplemental online Appendix 1A).
Based on updated clinical, radiological, pathological, and molecular reassessment, lorlatinib was ceased and standard dosing 3‐weekly carboplatin plus pemetrexed chemotherapy was initiated in May 2019, with some clinical improvement after cycle 1. Ms. CG was not well enough nor consenting to quadruplet therapy, carboplatin, pemetrexed chemotherapy, atezolizumab (PD‐L1 inhibitor), and bevacizumab (VEGF inhibitor) [24]. Despite only minor radiological progression after four cycles, she encountered ongoing clinical deterioration with worsening breathlessness, requiring supplemental home oxygen therapy.
An MRI‐B in June 2019 revealed nine new lesions since March 2019. These lesions had not been identified on prior imaging, and it was reported that several of the previously observed lesions remained in complete response.
Next‐generation sequencing of a repeat plasma sample in July 2019, after three cycles of chemotherapy, confirmed the previously identified ALK fusion (VAF down to 0.3%), KDMs (G1202R, VAF 0.3%; F1174C, VAF 0.2%), and RET A741G (VAF, 51.8%) and identified a new TP53 splice mutation [(c.919+1G>A), VAF 0.2%].
On a subsequent MRI‐B in August 2019, the small bilateral supratentorial metastases were stable with some response. Lorlatinib was added to continuing maintenance pemetrexed for its nonproven biological potential to augment disease control, particularly in the CNS.
Although a CT pulmonary angiogram performed at hospital readmission for breathlessness in August 2019 excluded a pulmonary embolus, there was marked volume loss and diffuse peribronchial and parenchymal change in the right lung and new interseptal interstitial thickening in keeping with lymphangitis carcinomatosis, consistent with her worsening clinical condition, now ECOG 3 (Fig. 6).
Figure 6.
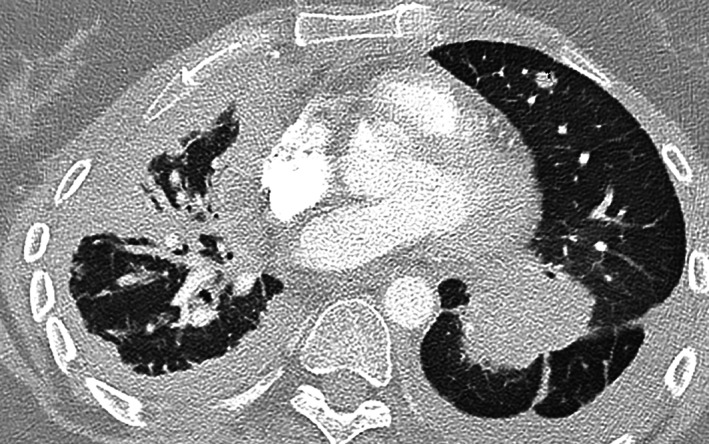
Ms. CG's final computed tomography‐pulmonary angiogram axial view performed through the Emergency Department confirming progressive burdensome disease.
In September 2019, Ms. CG presented to the emergency department with terminal agitation and hallucinations, having self‐discharged a few days prior from the affiliated palliative care unit largely because of her great determination to keep searching for the next potentially effective therapy. During her remarkable course, sequentially managed on four ALKis, informed by genotyping, including early access to first‐generation ALKi crizotinib in Australia on a phase II trial, Ms. CG psychologically grappled with her diagnosis and had great difficulty facing her mortality.
She passed away, almost 10 years from her diagnosis with advanced cancer and over 9 years with CNS disease, with her mother and brother by her side. An autopsy was not carried out. Figure 7 details the timeline from diagnosis to death for Ms. CG, including her treatments, best radiological responses, and molecular profiling timing and results.
Figure 7.
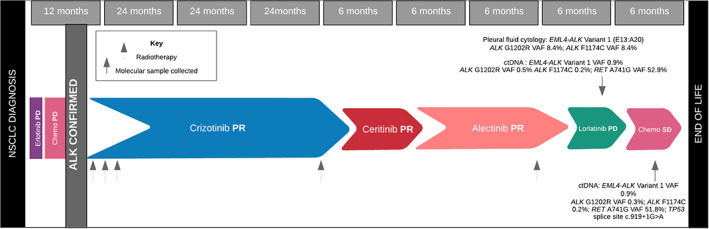
A schematic timeline of Ms. CG's treatment pathway from diagnosis to death, including the best objective radiological response on each therapy. Genotyping and results are detailed, with arrows marking the timing of testing including at primary resistance to last line chemotherapy at clinical deterioration. Abbreviations: ctDNA, circulating tumor DNA; PD, progressive disease; PR, partial response; SD, stable disease.
Discussion
Case 1 offers a remarkable example of a patient with ALK‐rearranged NSCLC presenting with bulky symptomatic CNS disease, remaining in remission over 5 years from diagnosis. Her longevity has been enabled by proactive care and access to drugs early in their clinical evaluation. Now well described in ALK NSCLC, her disease has remained CNS predominant [25]. In the first instance of CNS progression, her dose of brigatinib was uptitrated to enable higher drug exposure in the brain. Challenges were encountered in obtaining genetic information at progression, with plasma negative for ctDNA [22], as Mrs. ND underwent a craniotomy to resect monoprogressive symptomatic disease. Genetic analysis identified EML4‐ALK variant 3 and an ALK G1202R pan‐resistant KDM. This enabled early request for compassionate lorlatinib and explained the minimal benefit of alectinib, post brigatinib [26]. Solvent front mutations, such as G1202R, impair drug binding likely through steric hindrance [27]. Lorlatinib was engineered specifically to overcome such resistance and to be highly brain penetrant, with a reported intracranial objective response rate of 64% when taken after two prior ALKis compared and 37% extracranially [6].
Although G1202R has been reported in leptomeningeal disease through CSF sampling, [28], this is the first report to our knowledge of G1202R in a progressing metastatic intracranial clonal site.
Understanding CNS acquired resistance in an era of highly drug penetrable ALKis is a major emerging focus of ALK NSCLC research. ALK patients are living longer, and therefore patient management requires a critical balance between the biological, radiological, molecular, and psychosocial elements encountered.
Case 2 is an exceptional example of survival with advanced ALK lung cancer. It demonstrates the impact of treating beyond progression, particularly with crizotinib in the case of CNS progression, and with managing oligo‐progression with SRS, an area which remains under some debate given a paucity of randomized data. Retrospective series, however, do demonstrate the ongoing benefit in targeting resistant clones with radiation, and continuing treatment with the same ALKi using this approach [29]. Ms. CG was maintained, clinically well, on crizotinib for 7 years, in an era when later‐generation brain penetrant ALKis were mostly unavailable as they were early in their development phase. Unfortunately, her case also illustrates the rampant progression that may occur at later stage disease. Ms. CG's primary resistance to fourth‐line lorlatinib was explained by the presence of two ALK KDMs, the highly prevalent G1202R and F1174C. No tissue from prior to 2019, including from the primary tumor, was available to document the emergence of the KDMs although the published experience predicts that they develop as a result of selection pressure through sequential earlier generation ALKis. Independently, these KDMs have been shown to be sensitive individually to lorlatinib in vitro and in vivo; however, to our knowledge this is the first time these two particular ALK mutations have been reported together conferring resistance to lorlatinib [19, 30]. A recent case report detailed the presence of eight ALK KDMs in blood at progression on lorlatinib including F1174L and G1202R, further highlighting the complex resistance potential with multiple KDMs impeding drug binding ability and affinity [22]. Case 2 documents the emergence of the solvent front G1202R ALKi resistance mutation in variant 1. Although common in variant 3, this has not been reported previously in vivo, to our knowledge. It has been proposed that upfront monotherapy with lorlatinib for variant 3 may prevent the emergence of the highly prevalent G1202R KDM, whereas for variant 1, trial combination therapy to prevent alternate resistance, more commonly including bypass tracts [17]. Ms. CG's results did not demonstrate ALK‐independent genetic variants.
While on chemotherapy, ctDNA analysis of a repeat plasma specimen identified the persisting KDMs as well as a new TP53 mutation. In ALK‐driven NSCLCs, mutant TP53 is a negative prognostic marker rather than predictive resistance [31]. It however is expected to shed transiently into the blood with chemotherapy, which she was receiving at plasma collection. Although TP53 performs many functions, inhibiting its role in the upregulation of VEGF with anti‐VEGF therapy may be beneficial in combination with an ALKi [32]. With time of the essence, the re‐addition of lorlatinib to pemetrexed chemotherapy was instituted based on both the wish of the patient and recent retrospective reported data supporting the potential biological rationale. This was not based on level 1 evidence [33].
Finally, Ms. CG initially presented with a vertebral artery thromboembolism (TE). TE has been increasingly reported in oncogene‐addicted cancers and in approximately 30% of ALK patients in selected series, predominately venous TE, with the exact mechanism unexplained beyond the expected prothrombotic state of malignancy [34].
Conclusion
Patients with advanced ALK NSCLC are now expected to live years. Their disease is CNS tropic; however, ALK patients are often highly motivated, well, and willing to commit to invasive procedures if it may inform individualized treatment. In oligo‐progressive disease, local ablative therapy and continuation of ALKi postprogression should be considered with potential for sustained disease control. The ALK G1202R KDM, highly prevalent at resistance to second‐generation ALKis, may emerge in non‐EML4‐ALK variant 3 cases and is sensitive to third‐generation lorlatinib. When in compound with one or more ALK KDMs, resistance to lorlatinib is expected. In the case of rampantly progressive disease rebiopsy and redefining biology in a timely manner may be informative. The two case reports contribute to the expanding literature on ALK‐rearranged and oncogene‐addicted NSCLC in the current therapeutic landscape, providing a framework to care for these patients in the clinic.
Glossary of Genomic Terms and Nomenclature
- ALK gene
anaplastic lymphoma kinase gene, mutations of which may increase the growth of cancer cells.
- ALK G1202R kinase domain mutation
ALK resistance mutation.
- ALK inhibitor
a substance used as a targeted therapy to block ALK protein activity and, in the process, also block cancer cells from growing and spreading.
- ALK C1156Y kinase mutation
ALK resistance mutation
- Circulating tumor DNA
tumor DNA in the peripheral blood thought to be released upon tumor cell death.
- Fusion
a hybrid gene formed from two previously separated genes.
- Germline variant
a gene change in a reproductive cell that becomes incorporated into the DNA of every cell in the body of the offspring.
- EML4‐ALK variant
when the echinoderm microtubule‐associated protein‐like 4 (EML4) gene is fused to the anaplastic lymphoma kinase (ALK) gene leading to an oncogene encoding the oncoprotein EML4‐ALK. EML4 fuses at different breakpoints that bind the entire intracellular kinase domain of ALK. The different break points result in different variants.
- ALK L1198F kinase domain mutation
ALK resistance mutation.
- Next‐generation sequencing
sequencing technology that allows for quick and inexpensive sequencing of DNA and RNA with high accuracy.
- Oligo‐progression
the formation of several metastatic lesions from an initial tumor.
- Oncogene addicted cancer
phenomenon of cancer cell dependence on individual oncogenes to sustain growth, resulting in a cancer that is dependent on one specific mutation.
- Oncogenic driver
a molecule that causes the formation or supports the progression of cancer.
- RET point mutation
a single mutation in a gene sequence.
- Sequencing
the process of determining the exact order of the bases in a strand of DNA
- Tumor mutational burden
the number of mutations in the tumor genome, often shown as mutations per megabase.
- Variant allelic frequency
percentage of reads at a site that contain a variant allele.
Author Contributions
Conception/design: Malinda Itchins
Provision of study material or patients: Malinda Itchins, Brandon Lau, Cathy Yi Xia, Michael Rodriguez, Heng Wei, Michael Buckland, Bob T. Li, Mark Li, Vivek Rathi, Anthony J. Gill, Stephen J. Clarke, Michael J. Boyer, Nick Pavlakis
Collection and/or assembly of data: Malinda Itchins, Brandon Lau, Amanda L. Hudson, Helen Westman, Cathy Yi Xia, Sarah A. Hayes, Viive M. Howell, Michael Rodriguez, Anthony J. Gill
Data analysis and interpretation: Malinda Itchins, Amanda L. Hudson, Helen Westman, Wendy A. Cooper, Michael Buckland, Bob T. Li, Mark Li, Vivek Rathi, Stephen B. Fox, Anthony J. Gill, Stephen J. Clarke, Michael J. Boyer, Nick Pavlakis
Manuscript writing: Malinda Itchins, Brandon Lau, Amanda L. Hudson, Viive M. Howell, Nick Pavlakis
Final approval of manuscript: Malinda Itchins, Nick Pavlakis
Disclosures
Bob T. Li: Roche/Genentech, Guardant Health, Eli Lilly & Co. (C/A), Genentech, Eli Lilly & Co., Amgen, Daiichi Sankyo, AstraZeneca, Hengrui Therapeutics, GRAIL, Guardant Health, MORE Health (RF); Memorial Sloan Kettering Cancer Center institutional patents (IP, patent numbers US62/685,057, US62/514,661), Resolution Bioscience, MORE Health (Other: academic travel support). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1. Supporting Information.
Acknowledgments
Dr. Malinda Itchins is funded by an NSW Health PhD Scholarship, the Dwyer Family, and a Sydney Vital Scholar Award.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Soda M, Choi YL, Enomoto M, Takada S et al. Identification of the transforming EML4–ALK fusion gene in non‐small‐cell lung cancer. Nature 2007;448:561–566. [DOI] [PubMed] [Google Scholar]
- 2. Solomon BJ, Mok T, Kim DW et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. New Engl JMed 2014;371:2167–2177. [DOI] [PubMed] [Google Scholar]
- 3. Soria JC, Tan DSW, Chiari R et al. First‐line ceritinib versus platinum‐based chemotherapy in advanced ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐4): A randomised, open‐label, phase 3 study. Lancet 2017;389:917–929. [DOI] [PubMed] [Google Scholar]
- 4. Camidge DR, Kim HR, Ahn MJ et al. Brigatinib versus crizotinib in ALK‐positive non–small‐cell lung cancer. New Engl J Med 2018;379:2027–2039. [DOI] [PubMed] [Google Scholar]
- 5. Camidge DR, Dziadziuszko R, Peters S et al. Updated efficacy and safety data and impact of the EML4‐ALK fusion variant on the efficacy of alectinib in untreated ALK‐positive advanced non‐small cell lung cancer in the global phase III ALEX study. J Thorac Oncol 2019;14:1233–1243. [DOI] [PubMed] [Google Scholar]
- 6. Solomon BJ, Besse B, Bauer TM et al. Lorlatinib in patients with ALK‐positive non‐small‐cell lung cancer: Results from a global phase 2 study. Lancet Oncol 2018;19:1654–1667. [DOI] [PubMed] [Google Scholar]
- 7. Shaw AT, Yeap BY, Mino‐Kenudson M et al. Clinical features and outcome of patients with non–small‐cell lung cancer who harbor EML4‐ALK. J Clin Oncol 2009;27:4247–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaw AT, Felip E, Bauer TM et al. Lorlatinib in non‐small‐cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open‐label, single‐arm first‐in‐man phase 1 trial. Lancet Oncol 2017;18:1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Metro G, Lunardi G, Floridi P et al. CSF concentration of crizotinib in two ALK‐positive non‐small‐cell lung cancer patients with CNS metastases deriving clinical benefit from treatment. J Thorac Oncol 2015;10:e26–e27. [DOI] [PubMed] [Google Scholar]
- 10. Gainor JF, Sherman CA, Willoughby K et al. Alectinib salvages CNS relapses in ALK‐positive lung cancer patients previously treated with crizotinib and ceritinib. J Thorac Oncol 2015;10:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solomon BJ, Kim DW, Wu YL et al. Final overall survival analysis from a study comparing first‐line crizotinib versus chemotherapy: Results from PROFILE 1014. J Clin Oncol 2018;36:2251–2258. [DOI] [PubMed] [Google Scholar]
- 12. Nishio M, Felip E, Orlov S et al. Final overall survival, other efficacy and safety results from ASCEND‐3: Phase II study of ceritinib in ALKi‐naive patients with ALK‐rearranged NSCLC. J Thorac Oncol 2019;15:609–617. [DOI] [PubMed] [Google Scholar]
- 13. Duruisseaux M, Besse B, Cadranel J et al. Overall survival with crizotinib and next‐generation ALK inhibitors in ALK‐positive non‐small‐cell lung cancer (IFCT‐1302 CLINALK): A French nationwide cohort retrospective study. Oncotarget. 2017;8:21903–21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doebele RC, Pilling AB, Aisner DL et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non‐small cell lung cancer. Clin Cancer Res 2012;18:1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itchins M, Hayes SA, Gill AJ et al. Pattern of care and survival of anaplastic lymphoma kinase rearranged non–small cell lung cancer (ALK+NSCLC) in an Australian Metropolitan Tertiary Referral Centre: A retrospective cohort analysis. Asia Pac J Clin Oncol 2018;14:e275–e282. [DOI] [PubMed] [Google Scholar]
- 16. Zhu YC, Liao XH, Wang WX et al. Dual drive coexistence of EML4‐ALK and TPM3‐ROS1 fusion in advanced lung adenocarcinoma. Thorac Cancer 2018;9:324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Früh M, Peters S. EML4‐ALK variant affects ALK resistance mutations. J Clin Oncol 2018;36:1257–1259. [DOI] [PubMed] [Google Scholar]
- 18. Lucena‐Araujo AR, Moran JP, VanderLaan PA et al. De novo ALK kinase domain mutations are uncommon in kinase inhibitor‐naïve ALK rearranged lung cancers. Lung Cancer 2016;99:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaw AT, Solomon BJ, Besse B et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer. J Clin Oncol 2019;20;37:1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaw AT, Friboulet L, Leshchiner I et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med 2016;374:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisenhaur E, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:2228–2247. [DOI] [PubMed] [Google Scholar]
- 22. Swalduz A, Ortiz‐Cuaran S, Avrillon V et al. Fusion detection and longitudinal circulating tumor DNA (ctDNA) profiling in ALK+ non‐small cell lung cancer (NSCLC) patients. J Clin Oncol 2018;36(suppl):e21031a. [Google Scholar]
- 23. Crino L, Kim D, Riely G et al. Initial phase II results with crizotinib in advanced ALK‐positive non‐small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol 2011;29(15 suppl):7514a. [Google Scholar]
- 24. Socinski MA, Jotte RM, Cappuzzo F et al. Atezolizumab for first‐line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–2301. [DOI] [PubMed] [Google Scholar]
- 25. Cao L, Long L, Li M et al. The utilization of next‐generation sequencing to detect somatic mutations and predict clinical prognosis of Chinese non‐small cell lung cancer patients. Onco Targets Ther 2018;11:2637–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gainor JF, Dardaei L, Yoda S et al. Molecular mechanisms of resistance to first‐and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov 2016;6:1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ignatius Ou SH, Azada M, Hsiang DJ et al. Next‐generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high‐level resistance to alectinib (CH5424802/RO5424802) in ALK‐rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol 2014;9:549–553. [DOI] [PubMed] [Google Scholar]
- 28. Zheng MM, Li YS, Jiang BY et al. Clinical utility of cerebrospinal fluid cell‐free DNA as liquid biopsy for leptomeningeal metastases in ALK‐rearranged NSCLC. J Thorac Oncol 2019;14:924–932. [DOI] [PubMed] [Google Scholar]
- 29. Weickhardt AJ, Scheier B, Burke JM et al. Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene‐addicted non‐small‐cell lung cancer. J Thorac Oncol 2012;7:1807–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gainor JF, Dardaei L, Yoda S et al. Molecular mechanisms of resistance to first‐and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov 2016;6:1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang WX, Xu CW, Chen YP et al. TP53 mutations predict for poor survival in ALK rearrangement lung adenocarcinoma patients treated with crizotinib. J Thoac Dis 2018;10:2991–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwaederlé M, Lazar V, Validire P et al. VEGF‐A expression correlates with TP53 mutations in non–small cell lung cancer: Implications for antiangiogenesis therapy. Cancer Res 2015;75:1187–1190. [DOI] [PubMed] [Google Scholar]
- 33. Lin J, Schoenfeld A, Zhu VW et al. Efficacy of plarinum/pemetrexed combination chemotherapy in ALK‐positive NSCLC refractory to second‐generation ALK‐inhibitors. J Thorac Oncol 2020;15:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zer A, Moskovitz M, Hwang DM et al. ALK‐rearranged non‐small‐cell lung cancer is associated with a high rate of venous thromboembolism. Clin Lung Cancer 2017;18:156–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1. Supporting Information.