

Summary
Protein kinases are essential for signal transduction and control of most cellular processes, including metabolism, membrane transport, motility, and cell cycle. Despite the critical role of kinases in cells and their strong association with diseases, good coverage of their interactions is available for only a fraction of the 535 human kinases. Here, we present a comprehensive mass-spectrometry-based analysis of a human kinase interaction network covering more than 300 kinases. The interaction dataset is a high-quality resource with more than 5,000 previously unreported interactions. We extensively characterized the obtained network and were able to identify previously described, as well as predict new, kinase functional associations, including those of the less well-studied kinases PIM3 and protein O-mannose kinase (POMK). Importantly, the presented interaction map is a valuable resource for assisting biomedical studies. We uncover dozens of kinase-disease associations spanning from genetic disorders to complex diseases, including cancer.
Keywords: protein kinases, interaction network, disease module, signaling regulation, protein complexes, proteomics, systems biology, kinome, cancer
Graphical Abstract
Highlights
-
•
AP-MS confirms over 1,300 known and reveals over 5,000 kinase protein interactions
-
•
Functional contexts and regulatory circuits for poorly studied kinases
-
•
Kinases are biochemically linked to a broad range of Mendelian disease modules
-
•
Biochemical context for kinase complexes containing human cancer proteins
In this issue of Molecular Cell, Buljan et al. present a comprehensive account of human kinase complexes for inferring new kinase functions, kinase signaling modules, and kinase complexes linked to genetic diseases.
Introduction
Reversible phosphorylation of serine, threonine, and tyrosine residues represents a central molecular mechanism to control key properties of proteins, including their enzyme activity, half-life, complex formation, and subcellular localization. Phosphosites have been detected in almost all human proteins (Hornbeck et al., 2015), and protein regulation by kinases is essential for orchestrating the majority of biological processes in eukaryotes. In the human genome, the protein kinase lineage tree encompasses more than 500 evolutionarily related proteins (Manning et al., 2002; Wilson et al., 2018) (Table S1). These are organized in 10 large families and more than 100 subgroups. While a subset of the kinases has been extensively characterized (e.g., those belonging to the CMGC, STE, and RTK families), for many kinases, we still only have a limited understanding of their functional roles.
Kinases are strongly associated not only with cancer development but also with Mendelian developmental disorders, metabolic conditions (Lahiry et al., 2010), and different multifactorial diseases. Together with GPCRs, they represent the main group of current drug targets (Wang and Gray, 2015a, 2015b; Wu et al., 2015). For the understanding of cellular processes kinases participate in, and consequently for the charting of associated disease-relevant signaling pathways, it is critical to map kinase interaction networks (Gstaiger and Aebersold, 2013). However, at present, good coverage of protein interaction information is available only for a subset of human kinases. A well-established method for capturing stable protein interactions is affinity purification coupled to mass spectrometry (AP-MS). Through orthogonal data integration and statistical analyses, AP-MS-generated interaction data can be used to identify proteins that are involved in the same cellular processes, protein complexes, or functional and/or disease modules. In this context, we refer to a protein complex as an assembly of proteins that stably interact with each other as part of a single macromolecular entity and to a module as interactors of the same bait protein that also share a functional and/or disease association but that do not need to be part of a single assembly (Chen et al., 2014).
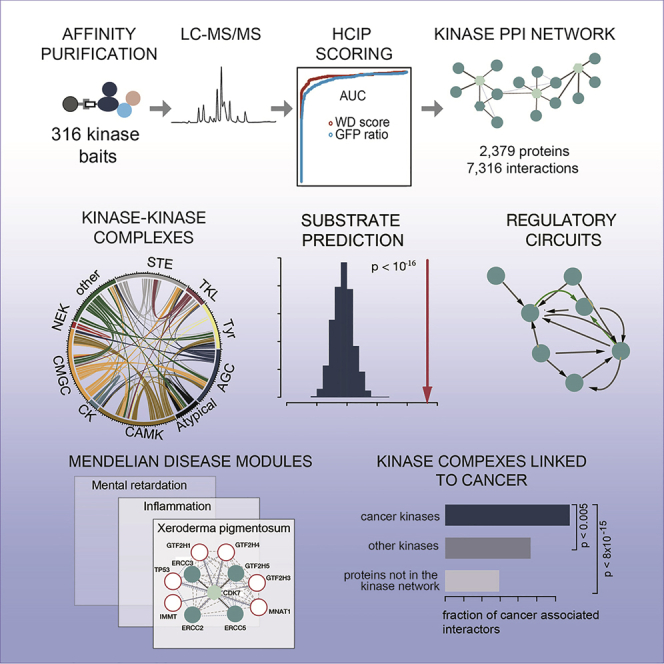
Here, we performed a systematic AP-MS analysis of interaction partners of human kinases. Our study has an unprecedented depth and includes more than 300 kinase baits representing all kinase families, many of which have not been included in previously published large-scale AP-MS studies (Hein et al., 2015; Huttlin et al., 2015, 2017; Varjosalo et al., 2013), as well as more than 5,000 previously unreported kinase interactions. This allowed us to extensively characterize kinase regulation of physiological functions and to define the biochemical context for several, as yet poorly investigated protein kinases. This is exemplified by PIM3 and POMK kinases for which the identified interaction partners predicted central roles in cytoskeleton regulation and the processing of glycosylated proteins, respectively. For these two kinases, we also validated interactions identified by AP-MS using orthogonal assays with BioID-MS and reciprocal co-purification. Further, using high-content imaging experiments, we could demonstrate that some of the poorly characterized kinases (NEK9 and PKMYT1) are required for cell shape control, which is consistent with their preferred binding to proteins with roles in cytoskeletal organization. In addition, we found multiple instances where interaction neighborhoods of individual kinases were significantly enriched in particular disease terms, including both Mendelian disorders and multifactorial diseases. These findings are exemplified by the nephronophthisis module associated with the NEK7 kinase and proteins linked to schizophrenia around the PAK5 kinase. Overall, this study represents the most comprehensive systematic analysis of human kinase interactions to date and demonstrates that the obtained network data are a highly valuable resource for assisting functional studies of kinase complexes in health and disease. Interaction data can be searched via the following website: https://sec-explorer.shinyapps.io/Kinome_interactions/.
Results
Systematic Analysis of Human Kinase Interaction Networks: Study Design and Scope
Building on an experimental workflow described previously, we generated 316 cell lines, each expressing a specific epitope-tagged kinase family member from a single locus under an inducible promoter (Glatter et al., 2009) (Figure 1A). Selection of the proteins used in the study was based on the feasibility to detect the expressed kinase construct from each generated cell line by AP-MS, and this excluded most membrane kinases. To enhance detectability of sub-stoichiometric or weakly interacting proteins, we used relatively large amounts of cellular starting material (>108 cells) and a rapid single-step affinity purification protocol (Figure 1A). We used a large set (n = 94) of negative controls (i.e., cell lines expressing GFP; see STAR Methods ) to facilitate subsequent data filtering. By comparing receiver operating characteristic (ROC) curves of several established filtering methods, we identified the combination of weighted D (WD) score (Sowa et al., 2009) (see STAR Methods) and protein abundance ratio compared to GFP control purifications as best performing, and we set the cutoff values such that the number of false-positive interactions was kept below 1% (see STAR Methods for details and additional data filtering). Only 6% of the initial interactions passed these filtering criteria (Figures S1A and S1B), and the filtering strategy also removed a large number of typical AP-MS contaminant proteins from the CRAPome database. The kinase baits used here cover 55 to 75% of kinase families members (Figure 1B) with the tyrosine (Tyr) kinase family being the only exception. Overall, we identified 7,316 high-confidence interactions involving 2,379 unique proteins, with prey proteins being evenly distributed across kinase families (Figure 1C; Table S2). Across all kinase families, we identified between 66% and 93% novel interactions that were not deposited in public protein-protein interaction (PPI) databases (Figure 1D). The number of identified interactors varied significantly across individual kinases, but it was comparable across kinase families (Figure 1C).
Figure 1.

Systematic Mapping of Kinase Interactions by AP-MS
(A) Workflow used to generate the kinase interaction network. This includes generation of more than 300 cell lines stably expressing doxycycline-inducible kinases, single-step pull-downs, duplicate runs on a hybrid linear ion trap-Orbitrap mass spectrometer, peptide identification with X!Tandem, and statistical evaluation.
(B) Coverage of kinases across different families. Dark blue represents included and orange not included kinases.
(C) Distribution of the number of identified interactors per kinase.
(D) Novel versus known interactions for each kinase family are plotted against the kinome evolutionary tree. Each kinase family is represented with a different color, and the same coloring scheme is used in all figures.
Data Quality and Novelty
The number of protein interactions reported in public databases for individual kinases varies dramatically (Figure 2A). These differences likely represent research bias toward heavily studied kinases and proteins in general (Edwards et al., 2011). To assess this, we investigated if the number of interaction partners per kinase correlated with the number of studies that involved the respective kinase. Even though poorly studied kinases (as determined by the number of citations in PubMed; Table S1) had on average a significantly lower number of PPIs annotated in the public databases compared to frequently studied kinases (Figure 2A), we observed no such differences in the connectivity in the here-generated systematic AP-MS dataset, supporting the unbiased character of our study. Of note, 90 of the kinases used as baits here had 30 or fewer citations (Table S1).
Figure 2.

Assessment of the Network Scope and Data Quality
(A) Barplot depicting the number of interactions per kinase found in public databases (http://dcv.uhnres.utoronto.ca/iid/; gray bars) and in the presented study (blue bars). A running average of the number of citations per kinase is shown as a red line. Bars are ordered by the decreasing number of interactions based on IID.
(B) Venn diagram shows an overlap between kinase baits used in our study and baits used in two previous large-scale AP-MS studies: Hein et al. (2015) and BioPlex 2 (Huttlin et al., 2017).
(C) Fraction of all protein interactions detected in different AP-MS studies that were also reported by additional studies deposited in the BioGRID database (top). The number of citations associated with the recapitulated interactions is also shown (bottom).
(D) Protein pairs that were almost exclusively purified together with different kinase baits (p value < 10−10) are shown.
(E) Stacked barplots show the fraction of the HSP90 chaperone complex members found here as interactors of kinases that were previously classified as its strong or weak clients or were not identified as HSP90 clients (Taipale et al., 2012).
(F) Representative kinase-containing CORUM complexes that were recapitulated in the generated network. Conditions for this were more than two subunits and more than 75% of complex subunits covered.
(G) Contacts between CORUM protein pairs in the kinase network are enriched in a comparison to 500 reshuffled networks of the same composition and topology.
(H) Distribution of co-elution correlation values for all protein pairs measured in HEK293 SEC analysis (Heusel et al., 2019) (gray area). Average correlation values for protein pairs found in the same CORUM complex (dark blue area) or here in the kinase network (dark green area) were higher than those for other protein pairs.
In total, more than 150 baits included in this study have not been assessed by previous large-scale AP-MS studies (Figure 2B), and ∼15% of the interactions have been already reported in the PPI database Biological General Repository for Interaction Datasets (BioGRID) (Figure 2C, top) (Stark et al., 2006), a value that is comparable to other AP-MS studies. As a measure of data robustness, we analyzed the number of independent reports supporting the set of already known interactions. We found that the fraction of known interactions reported by at least two independent publications was greater than 50% (Figure 2C, bottom). Overall, our repository confirms, as a single resource, 1,236 PPIs reported in more than 800 publications.
Identification of Known and Putative Novel Protein Modules and Complexes
Several previous interaction studies have shown that a strong co-purification or joint detection of proteins across multiple baits can indicate physical or functional association (Drew et al., 2017; Knight et al., 2017; Youn et al., 2018). Using this principle, we applied a hypergeometric test to identify protein pairs co-purifying at significantly high frequency (see STAR Methods). We found that many of the frequently co-purifying protein pairs (1,718 protein pairs, i.e., 1% of all pairs, with adjusted p value < 0.01 and with at least five shared kinase baits) were indeed part of larger protein complexes or functional modules (Table S3). Pairs identified with a highly stringent cutoff (adjusted p value < 10−10) are shown in Figure 2D. Several of these proteins belonged to complexes with well-annotated roles in the regulation of kinase stability and activity, such as HSP90-CDC37 and striatin-interacting phosphatase and kinase (STRIPAK) complexes, respectively. Among others, the analysis highlighted several groups of co-purifying proteins with distinct roles in carbon metabolism or rRNA and mRNA processing, as well as proteins from stable complexes. The latter included mitochondrial complex composed of HADHA and HADHB subunits, the TRiC chaperonin complex, and the less well-studied BCLAF1-THRAP3 protein complex with a role in DNA damage response (Vohhodina et al., 2017) (Figure 2D). The identified modules of co-purifying proteins can also assist protein functional assignments. An example is the less well-studied FTSJ3 protein (Ringeard et al., 2019) that is annotated as a putative rRNA methyltransferase. We found that FTSJ3 co-purified (p value < 10−5) with several proteins involved in rRNA processing (Figure 2D; Table S3), which corroborated its suggested function.
The strongest hit in the co-purification analysis was the HSP90 chaperone complex (composed of HSP90A, HSP90B, and CDC37 proteins). This complex is highly expressed and its core components are often considered contaminants in AP-MS studies. However, ∼60% of all kinases are known to be clients of the heat shock protein (HSP) complex (Taipale et al., 2012; Verba et al., 2016). We reasoned that we could use retention/exclusion patterns of this complex across the baits in this study to evaluate the specificity of our pull-downs and filtering. For this, we compared our results with the extensive biochemical characterization of HSP90-kinase interactions carried out by Taipale and colleagues (Taipale et al., 2012). In their work, they could distinguish between kinases that are strongly or weakly associated with HSP90. We found that our data were in very good agreement with their results; the majority of the kinases previously classified as strong interactors of the HSP90 complex members were present in our pull-downs. In contrast, this was the case only for a smaller percentage of the weak interactors and a minor fraction of the noninteractors (Figure 2E; Table S4). These results thus support the filtering strategy employed here.
Next, we benchmarked the kinase interaction network against the manually annotated core protein complexes deposited in the comprehensive resource of mammalian protein complexes (CORUM) database (core complexes, release 02.02.2017) and in this way assessed the integrity of kinase-containing complexes. Overall in our set, we could retrieve 50%–100% of the components for about 50% of the kinase-containing CORUM complexes, with several instances of kinases bound to their adaptors/accessory subunits (e.g., PKA, CK2, and IKK), or within their stable complexes, such as the EVDP, as illustrated in Figures 2F and S1C. In a comparison to randomized kinase networks (see STAR Methods), the overlap between the CORUM and our interactions was highly significant (Figure 2G). To further validate that we are able to capture stable interactions among kinases and associated protein complexes, we compared PPIs from the kinase interaction network to correlation profiling data generated from size-exclusion chromatography (SEC) experiments in our laboratory (Heusel et al., 2019). Our analysis indeed showed that the here-identified interaction pairs had significantly higher SEC co-eluting values than random protein pairs (i.e., they were often found physically associated in an independent assay) (Figure 2H).
Together, these results support the organization of the kinase interaction network in well-defined functional units and additionally show that co-purification patterns can be effectively used to suggest new functional associations.
Functional Landscape of the Human Kinase Interaction Network
To explore the functional space occupied by different kinases, we first analyzed Gene Ontology (GO) terms associated with kinases and their interaction partners. GO analysis at the kinase family level lacked specificity, so we focused on the analysis of GO term enrichment among the interactors of evolutionarily more strongly related kinase subgroups. The thus-identified significant GO terms defined subgroup-specific functional fingerprints and indicated that processes could be broadly partitioned in those controlled by several different kinase subfamilies (such as cell cycle, protein transport, and apoptosis) and those primarily regulated by a single or low number of kinase subfamilies (such as a mRNA splicing, circadian rhythm, and specialized signaling pathways; Figures 3A and S2A). To determine to what extent our data recapitulated already established functional knowledge and revealed novel functional associations, we used a semantic similarity score (Yu et al., 2010). Using this score, which measures similarity between groups of GO terms, we compared the most significant GO terms obtained in this analysis with the GO terms of the known interaction partners of the same kinase (obtained from the public repository Integrated Interaction Database [IID]; see STAR Methods). For several kinase subfamilies (e.g., casein kinase I, mitogen-activated protein kinase [MAPK], IKK, Aurora, Polo, and STE20), we found a strong functional match between prior knowledge and our results, suggesting that the newly discovered interactions confirm and expand our knowledge of the kinase activities in previously defined contexts (Figure 3B).
Figure 3.

Functional Landscape of the Kinase Interaction Network
(A) Dotplot of the most significant terms associated with kinase subgroups covered in this study.
(B) Semantic similarity between the top GO terms based on the interactions from this study and interaction partners deposited in the IID.
(C) Main associations in the PIM3 interaction network include the actin cytoskeletal and G proteins and SRC modules.
(D) Cellular phenotyping after gene silencing. Volcano plot depicts siRNA gene targets that displayed the strongest changes in the representative cell area and shape phenotypes relative to the negative siRNA control. Targets that had a −log10 (p value) greater than 2.5 are labeled with their gene names. Colors represent different features measured, as defined by the CellProfiler software tool. Different siRNAs for the same gene are indicated with −1 or −2, and combinations of siRNAs are denoted by the symbol “|”. Heatmaps summarize the information on how many siRNA and siRNA combinations per gene were observed as significant.
(E) Circos plot of all kinase-kinase interactions. Coloring scheme is the same as in Figure 1.
(F) Ratio between intra-family and inter-family connections.
(G) Number of kinases versus total number of interactors per kinase bait.
(H) WNK3 kinase hub with its interactors and known associations with the endocytosis proteins.
In addition, our GO analysis suggested a better definition of the biochemical roles for several less well-studied subfamilies and specific kinases. This is illustrated by the proteins interacting with PIM3 kinase (Figure 3C). PIM3 is the least-studied member of the PIM subfamily of CAMK kinases. As of January 2020, only four interactions were reported in the IID PPI database (Kotlyar et al., 2016) and only one in BioGRID (Stark et al., 2006). The PIM family has been implicated in the progression of several malignancies, possibly through regulating cell motility. PIM3 itself was linked to a decreased survival in prostate cancer patients (Santio and Koskinen, 2017). In line with these observations, the PIM3 interactors identified in this study included five different proto-oncogenic SRC kinases (FYN, LYN, SRC, YES1, and FRK) as well as subunits of heterotrimeric G proteins. In addition, our study revealed a strong association of PIM3 with cytoskeletal proteins, in particular several actin-regulating modules, with an interaction pattern comparable to that observed for the cytoskeletal kinase DAPK1. To confirm the PIM3 association with cytoskeletal proteins, we tagged separately N- and C- terminus of PIM3 with the biotin ligase Flag-BirA∗ and carried out a proximity labeling experiment, BioID coupled to MS, in an orthogonal cell line, HeLa CC2, using a doxycycline-inducible system. We found that cytoskeleton-related GO terms were also overrepresented in the thus-defined PIM3 proximal proteome (Figure S2B). While the overlap between AP-MS and BioID-MS was relatively low (as expected from the different chemistries and lysis conditions the two methods use and in accordance with previous reports; Lambert et al., 2015), the large majority of the proteins found by both AP-MS and BioID-MS were indeed cytoskeletal, thus independently confirming the initial finding (Figure S2C). By this means, our data also suggest possible routes for the PIM3 control of cell shape and motility.
Besides PIM3, we found that several other poorly studied kinases (with less than 50 citations) preferentially associated with proteins linked to cell shape related GO terms (Table S5). To test whether these kinases are indeed functionally linked to cell shape control, we subjected them to small interfering RNA (siRNA)-mediated knockdown and subsequent high-content image-based profiling of cell shape phenotypes in two cell lines (LN229 and SKOV3). Our results revealed that several of the tested kinases (in particular NEK9 and PKMYT1/MYT1) were indeed necessary for controlling cell shape (Figures 3D, S2D, and S2E). Both genes, as well as several other kinases with positive phenotypes, are known to play a role in cell-cycle regulation. Therefore, additional experiments are needed to determine whether the identified cell shape changes in the kinase-knockdown cells result from a direct involvement of these kinases in controlling cell shape or indirectly via affecting cell-cycle control.
The Kinase-Kinase Interaction Network
Consistent with the notion that kinases preferentially bind other kinases (Breitkreutz et al., 2010; Colinge et al., 2014), we found that “kinase” was the most significantly enriched domain among the proteins associated with the tested baits. This corresponds to 454 kinase-kinase contacts (i.e., 5% of all observed interactions) (Figure S2F; see STAR Methods). Overall, more than 250 of these kinase-kinase interactions were not reported previously. To better understand the architecture of these associations, we represented all kinase-kinase contacts as a circos plot (Figure 3E). This highlighted a strong interconnectivity among kinases that belong to the same families. The highest degree of intra-family connectivity was among kinases from small kinase families, such as CK1 and NEK, and among kinases from the larger STE family (Figure 3F). At the other end of the spectrum, the highest frequency of inter-family connections was observed for the tyrosine like-kinases (TKLs) and for kinases not assigned to any of the families and therefore classified as “other.” Of note, the number of kinase-kinase interactions did not correlate with the total number of interactions per kinase (Figure 3G).
This analysis also highlighted several new kinase hubs (defined here are kinases that interacted with three or more other kinases). An example for this is the WNK3 kinase (Figure 3H). The WNK3 interactors included its two homologs, WNK2 and WNK1, as well as four other kinases, STK39, PIK3, GAK, and BIKE. WNKs have been previously shown to regulate the surface expression of ion transporters by a variety of means, including endocytosis (WNK1/4) and activation of STK39. Besides the known interaction with STK39 (McCormick and Ellison, 2011), WNK3 was associated with several components of the endocytic machinery. Interestingly, BIKE, GAK, and PIK3 have also been previously reported to interact with endocytosis regulators, and the activation of PIK3 was shown to be required for the WNK1-mediated regulation of the potassium channel ROMK endocytosis (Cheng and Huang, 2011), suggesting that, similar to WNK1, WNK3 may also be involved in the endocytosis of ion transporters.
Importantly, besides interactions among kinases, hundreds of new interactions with other proteins involved in cellular regulation, such as phosphatases, E3 ubiquitin ligases, DUB enzymes, and epigenetic factors, were identified in the study (Figures S2G–S2J). In particular, a large fraction of the kinase interactions with epigenetic factors was not previously deposited (Figure S2H). In summary, our analysis provides the first systematic insight into the global organization of inter-kinase relationships in human cells and suggests wide a biochemical context for kinase regulatory activity in controlling protein phosphorylation, ubiquitylation, and gene expression.
The Kinase Interaction Network as a Valuable Resource for Mapping Kinase-Substrate Relationships
By now, more than 200,000 phosphoresidues in the human proteome have been cataloged (Hornbeck et al., 2015). However, for only a small fraction of these are the kinases catalyzing the phosphorylation event known (Wagih et al., 2016). We therefore investigated if the interaction dataset generated here could be used to nominate kinase-substrate assignments. We found that 75 of the physical interactions in our dataset were previously reported as kinase-substrate pairs. To determine the significance of this observation, we generated a set of 1,000 random networks that mirrored the kinase interaction network size and topology and assessed the number of known kinase-substrate interactions in these networks (see STAR Methods). There, the mean number of interactions documented as a regulatory event was 16 (Figure 4A). Hence, the observed 75 kinase-substrate interactions represented a strong enrichment (p value < 10−53) and indicated that the generated network likely included instances of kinases bound to their substrates.
Figure 4.

Kinase Interaction Network Assists Assignments of Regulatory Interactions
(A) In the generated kinase network, 75 interactions (red arrow) were known kinase-substrate pairs, which corresponds to a significant enrichment compared to random networks (shown as a histogram on the left).
(B) Criteria for the prediction of novel kinase-substrate relationships.
(C) In total, 534 interactions in the kinase network were predicted as possible kinase-substrate interaction (depicted with red arrow). This is higher than the average number of predicted pairs in random networks (histogram on the left).
(D) Several substrate proteins predicted to be regulated by casein kinase 2 form stable protein complexes. Known and predicted regulatory interactions are depicted with red and gray arrows, respectively. Dashed lines indicate connections between proteins from the same CORUM complex (Ruepp et al., 2010). CK2 holoenzyme is a tetramer with two CK2b regulatory subunits and CK2a1 and CK2a2 catalytically active subunits.
(E) CK2 predicted substrates with more than five CK2 phosphomotifs are shown. Number of predicted phosphomotifs is shown for each protein. Proteins that were reported as CK2 substrates in previous studies are indicated in dark red.
(F) Kinase-kinase regulatory interactions occur within and across kinase families. The full set of predicted regulatory events is shown in Figure S3B. Arrows colored in green represent activation loop phosphorylation. Kinase families are colored according to the scheme in Figure 1.
Next, we aimed to predict novel kinase-substrate relationships in our dataset (Figure 4B). For this, we used the NetPhorest tool (Miller et al., 2008) and considered thus-identified kinase motifs around the experimentally confirmed phosphoresidues (i.e., sites identified in five or more large-scale studies or a single targeted small-scale study; Hornbeck et al., 2015)). We restricted our analysis to the top three kinase families predicted to be able to recognize each phosphosite (see STAR Methods). In total, we found 550 instances where computationally predicted kinase-substrate directional interactions were also supported by a physical contact in the generated kinase interaction network (Figure 4C; Table S6). This overlap, which consisted of 534 nondirectional protein interactions (16 interactions corresponded to reciprocal kinase-substrate relationships), represented a significant enrichment when compared to a set of 1,000 random networks of the same size and topology (mean of 394, p < 10−16; Figure 4C). Collectively, for the phosphosites included in the analysis, this increases the fraction of sites with an upstream kinase assignment from 15% to 26%.
Since AP-MS largely captures stable interactions, we assessed if any of the bait kinases were predicted to regulate two or more proteins from the same CORUM complex (Ruepp et al., 2008). This highlighted the CK2 α2/β2 holoenzyme (Figure 2F) as a possible regulator of multiple subunits from epigenetic complexes and the splicing machinery (Figure 4D; see also Figure S2J). Importantly, several of these proteins were already known substrates of the CK2 CSNK2A1/CK2a1 kinase. In addition to this, CK1 kinases were identified as possible regulators of smaller complexes involved in transcriptional elongation (Figure S3A).
Many of the proteins annotated as known CK2 substrates contain multiple phosphosites that can be recognized by this complex (Shi et al., 2009; Wise et al., 1997; Yang et al., 2013). In line with this, 20 of the proteins that we predicted as CSNK2A1/CK2a1 and/or CSNK2A2/CK2a2 substrates had six or more confident phosphosites (defined as above) that mapped within sequence motifs that can be recognized by CK2 (Figure 4E). None of these 20 proteins were among the known CK2 substrates deposited in PhosphoSitePlus (Hornbeck et al., 2015). However, CK2 kinase regulation of several chromatin modifiers, which we predict here, has actually been reported by previous individual studies. These instances include HIRIP3 (Assrir et al., 2007), DEK (Kappes et al., 2004), SMARCA4 (Padilla-Benavides et al., 2017), and TCOF1 (Ciccia et al., 2014; Wise et al., 1997) proteins. In addition to CK2 substrates, predicted substrates of CLK2 and CLK3 splicing regulators also had multiple phosphomotifs, which could be recognized by these kinases (Figure S3A).
In particular, kinase-kinase phosphorylations are an important element of regulatory networks and are known to modulate signal amplification and duration (Breitkreutz et al., 2010; Garrington and Johnson, 1999). Kinase-kinase regulatory interactions reflected to a large extent intra- and inter-family connectivity observed in the nondirectional interaction network (Figures 3E and S3B). For instance, members of the SRC and protein kinase C subfamilies interacted almost exclusively with other members of the same subfamily. The majority of inter-family regulatory circuits involved kinase-substrate interactions between CMGC and CAMK families as well as CMGC and AGC families (Figure S3B). In addition, CMGC kinase CDK1/CDC2 was predicted to regulate kinases that belonged to different kinase families but that had similar roles in mitosis (NEK4 and PKMYT1/MYT1) or in MAPK signaling (RAF1, IRAK1, and MAP2K2), as depicted in Figure 4F. Collectively, these analyses present the kinase interaction network as a resource that, via integration of orthogonal data, can be used to identify novel regulatory relationships with an effect on a broad range of cellular functions.
Kinase Complexes Linked to Genetic Diseases
Next, we investigated if any of the kinase neighborhoods associated with particular genetic diseases. For this, we compared occurrences of specific disease terms associated with the interactors of an individual kinase to the frequency of these terms in the background human proteome (i.e., among all other proteins not present in the kinase network). For this, we used gene-disease annotations from the (1) Online Mendelian Inheritance in Man (OMIM) database (Hamosh et al., 2005) and (2) DisGeNET database (Piñero et al., 2015). OMIM provides curated and confident annotations with an emphasis on Mendelian diseases, while DisGeNET additionally includes predicted disease associations for both Mendelian and complex diseases. The overall number of gene-disease associations in the latter resource is significantly higher, but nonspecific associations can also prevent true relationships from being detected as significant. In this analysis, we excluded cancer-associated (CA) terms, as these are a focus of a separate analysis described below, and we excluded terms with a very limited support in DisGeNET (see STAR Methods). In total, 15 of the studied bait kinases had interaction neighborhoods where at least one OMIM disease term was strongly overrepresented (adjusted p value < 0.05, Fisher’s exact test; Table S7; see STAR Methods), and 137 kinases had interaction neighborhoods in which at least one of the DisGeNET disease annotations was strongly overrepresented (adjusted p value < 0.05, Fisher’s exact test; Table S7; see STAR Methods). The most significant disease modules identified with either of the two resources are shown in Figure 5. In nearly half of these instances, the bait kinase was also annotated with the highlighted disease term. The obtained kinase-disease associations cover a broad range of medical conditions. These, among others, include congenital disorders, psychiatric conditions, and diseases of specific organs. Below, we describe in detail some of these associations.
Figure 5.

Kinase Modules Associated with Genetic Diseases
Kinase interaction modules in which interactors with the same disease term were present at a statistically significant frequency are shown. Shapes colored in light green (significant baits) or gray (other bait and prey proteins) indicate that the protein lacked the respective annotation. p values indicate enrichment significance.
Among the most strongly enriched instances in the OMIM analysis was a known module involved in DNA repair centered on the CDK7 kinase (p value < 8.1 × 10−5). Mutation in any of these proteins that are part of the well-studied TFIIH transcription factor complex (Figure 2F) can lead to the development of the skin condition xeroderma pigmentosum (Singh et al., 2015). The same disease module was also recovered in the DisGeNET analysis (p value < 1.2 × 10−6; Table S7) extended with few additional proteins from the same complex (Figure 5). In addition, the OMIM analysis highlighted a known cluster of proteins around the SRPK2 kinase annotated with the disease term retinitis pigmentosa (p value < 0.013), a serious eye disorder leading to blindness. Further, significant DisGeNET disease modules also included the AMPK holoenzyme, which is composed of PRKAG2 and PRKAG3γ noncatalytic subunits and PRKAA1/AMPKa1 and PRKAA2/AMPKa2 catalytic subunits and is known to be linked to the Wolff-Parkinson-White syndrome, a condition related to heart arrhythmia (p value < 3 × 10−6). In addition, the DisGeNET annotations suggested an association of the module around the PAK5 kinase with schizophrenia (13 out 20 interactors, p value < 10−4). The kinase itself lacked this annotation (Figure 5). However, PAK5 was recently linked to psychosis (Morris et al., 2014), and the kinase is predominantly expressed in the brain. Furthermore, both approaches identified several significant disease clusters around the POMK kinase, which we discuss in more detail below.
The analysis of OMIM Mendelian disease annotations further reported the enrichment of the “nephronophthisis” term around the NEK8 kinase. Nephronophthisis is a genetic disorder of kidneys that affects children. The two proteins involved in the disease development (i.e., NEK8 and ANKS6) also interacted with the NEK7 kinase. NEK7’s physical association with this disease module is supported by previous experimental work (Hoff et al., 2013). In addition, we found here that two other NEK kinases (NEK6 and NEK9), as well as the adaptor protein ANKS3, all interacted with NEK7 and with each other. ANKS3 associates with ciliary disorders (Shamseldin et al., 2016) that are etiologically related to nephronophthisis, and both ANKS3 and NEK7 were suggested to be important in development of renal ciliopathies (Viau et al., 2018).
In addition to these proteins, NEK7 also interacted with proteins associated with other Mendelian diseases, and some of these interactions were not reported previously. This included interactions with the TMPO protein (linked to cardiomyopathy) and with the PGK1 kinase (with a role in hemolytic anemia and neurological dysfunction). We performed reciprocal purification of TMPO and PGK1 proteins in a different cell line (A549) and using antibodies against NEK7. The experiment validated these interactions at NEK7 endogenous expression levels (Figure S4A). Overall, disease modules identified here tend to associate with pathologies of diverse organs, thus further signifying the broad functional roles of protein kinases. Significant modules warrant further investigations on the exact mechanisms of action and the role of the associated kinases in the respective diseases.
POMK Frequently Associates with Membrane Proteins Involved in Glycan Biosynthesis
Within the interaction network generated in this study, the POMK kinase stood out as a kinase with a high number of interaction partners (171 proteins), where the majority (i.e., 92%) of the interactions were as yet not reported in PPI databases. Until recently, POMK was considered a pseudokinase, and it had a generic name SGK196 (Yoshida-Moriguchi et al., 2013). The kinase localizes within the endoplasmic reticulum (ER) membrane (Figure 6A) and has a role in adding a phosphate to the O-mannose sugar moieties (Ogawa et al., 2015; Yoshida-Moriguchi et al., 2013). One of its known substrates is dystroglycan, a receptor that connects cytoskeleton to the extracellular matrix (Jae et al., 2013). Using the above-described analysis of Mendelian disease terms, we found that the interaction partners of POMK were strongly enriched in proteins involved in muscular dystrophy (p value < 0.003) or in the related “congenital disorder of glycosylation” (p value < 0.001). Diseases phenotypes of the same class were also linked to the loss-of-function mutations in POMK, which prevent phosphorylation of mannose on dystroglycan (von Renesse et al., 2014). Importantly, in the here-identified POMK disease module, only the interaction with the POMGNT1 protein was previously cataloged in PPI databases (Figure 5).
Figure 6.

POMK Kinase Frequently Interacts with Proteins Involved in Glycan Metabolism
(A) Protein sequence of the POMK kinase is depicted. The kinase is embedded in the ER membrane with the largest fraction of the protein residing within the ER lumen.
(B) Functional GO terms and KEGG pathway annotations overrepresented in the set of POMK interaction proteins are shown.
(C) Interaction partners of the POMK kinase that according to KEGG annotations are members of metabolic pathways (53 proteins in total) are grouped based on the KEGG pathway assignments. In the instances where the same protein belonged to several pathways, it was assigned to the larger one.
We further characterized POMK interaction partners using GO functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations in the DAVID database (Dennis et al., 2003). This showed that POMK interaction partners were strongly enriched in membrane proteins, and in proteins involved in metabolic pathways (p value < 3.6 × 10−31 and p value < 3.5 × 10−12, respectively; Figures 6B and 6C). This agrees with the reports that many proteins associated with the same congenital disorder phenotypes as POMK tend to function in the glycan biosynthesis pathway and are often categorized as membrane and metabolic proteins (Freeze et al., 2014; Yoshida-Moriguchi et al., 2013; Zhang et al., 2018). Of note, several of the here-identified POMK interaction partners (i.e., DDOST, STT3A, STT3B, RPN1/OST1, and RPN2 proteins) are subunits of the core N-oligosaccharyltransferase complex (OST), which is located in the ER membrane. Even though O-linked glycosylation predominantly occurs in the Golgi, O-mannosylation is initiated in the ER, and the Pmt1-Pmt2 protein complex, which mediates mannose transfer, is in a physical proximity of the OST complex (Bai et al., 2019). To further validate these results, we tested POMK interactions with several of these proteins by reciprocal co-purifications in another cell line (A549) and used POMGNT1 as a positive control. The data confirmed interactions with STT3A, RPN1, and RPN2 proteins and thus supported POMK’s association with proteins involved in N-glycosylation (Figure S4B). Overall, the POMK interaction partners we identified using AP-MS should help in further delineating the exact roles of this kinase in developmental and metabolic disorders (Freeze et al., 2014).
Kinase Modules Linked to Cancer
Kinases are the most overrepresented class in the Cancer Gene Census (CGC), i.e., a set of genes, whose mutations have been causally implicated in cancer (Fleuren et al., 2016). Using the generated kinase interaction network, we assessed if CA kinases indeed interacted more often with other CA proteins. To this end, we annotated as CA those proteins that are already classified as cancer drivers (i.e., included in the CGC) or were reported to be mutated at a significant frequency in cancer patients (Davoli et al., 2013; Futreal et al., 2004; Lawrence et al., 2014; Vogelstein et al., 2013). We found that among the interaction partners of CA kinases, a significantly higher fraction of proteins were themselves annotated as CA, both when compared to the interaction partners of non-CA kinases used as baits here or to human proteins that were not in the kinase network (Figure 7A; p value < 0.005 and p value < 8 × 10−15, respectively, chi-square test). This observation hence further supports the usage of kinase physical interactions for the study of their CA roles (Fessenden, 2017).
Figure 7.

Kinase Network Associated with Cancer
(A) Interactors of CA kinases are often themselves associated with cancer.
(B) Individual kinases whose direct protein interaction neighborhoods were strongly enriched in CA proteins (p value < 0.025) are listed together with their respective p values.
(C) Interaction network represents kinases from (B) together with their associated proteins that are annotated as CA. Coloring scheme is the same as in (B). Significant bait kinases are highlighted with bold edges. When several of the interaction partners share the same GO term, this is indicated with the colored background around the kinase name (most common terms are shown).
(D) Volcano plot indicates the enrichment of Dyrk2 interaction partners identified by BioID-MS compared to the GFP control (BirA-tagged GFP). Proteins enriched with a log2FC (Dyrk2/GFP) ≥ 1 (adjusted p value ≤ 0.05) were considered as high-confidence interactors (thresholds are indicated with dashed lines). Interactors identified by both BioID-MS and AP-MS measurements are presented as green dots (11 were detected with a high confidence).
(E) Quantitative changes in the Dyrk2 interactions after the treatment with Adriamycin (ADR) are shown. Only interaction partners identified with both AP-MS and BioID are shown. Significant changes (log2FC (ADR/ctr) ≥ 1 and adjusted p value ≤ 0.05) are indicated with an asterisk.
Next, we searched for individual kinase modules that were enriched in CA proteins (see STAR Methods). We identified 33 kinase modules where kinase interacting proteins were significantly enriched in CA proteins (adjusted p value < 0.05, Fisher’s exact test; Figure S5A). In addition to kinases with well-established cancer driver roles, such as CDK4 and CDK7, this highlighted several other kinases linked to cellular processes relevant for cancer development (Figures 7B, S5A, and 7C). For instance, the PLK1 kinase, an important regulator of mitosis (Petronczki et al., 2008), which is also frequently overexpressed in cancer (Liu et al., 2017); JNK1/MAPK8, which is known to regulate proliferation and apoptosis (Chen et al., 1996); and Tribbles pseudokinase 2 (TRIB2/Trb2), which, when overexpressed, is able to induce acute myelogenous leukemia in mice (Keeshan et al., 2010). Of note, more than half of protein-protein interactions from the subnetwork depicted in Figure 7C were newly identified in this study. Jointly, these analyses strongly supported the notion that better mapping of CA protein modules could add links to cancer pathways and nominate gene candidates of interest while also providing a cellular context for their activity.
DYRK2 Kinase Changes Affinity to Its Interactors during DNA Damage Response
Kinases are known to play a central role in the regulation of DNA damage response during cancer development (Arcas et al.,2014). We therefore annotated proteins with a role in this process (see STAR Methods for the definition of this list) and looked for the modules with multiple DNA-damage-associated proteins. Among the kinases that had five or more interaction partners linked to DNA damage repair were SRPK kinases, CK2 kinases, and the DYRK2 kinase (Table S8). DYRK2 is a less well-studied kinase that can translocate to the nucleus and phosphorylate p53 upon the DNA damage (Taira et al., 2007). The kinase is known to form a stable E3 ligase complex with DDB1 (DNA-damage-binding protein 1), UBR5, and VPRBP, and it was hypothesized that DYRK2’s role in the DNA damage response is likely to depend on its stable interaction with DDB1 (Bensimon et al., 2011). Here, with AP-MS, we additionally identified a new interaction between DYRK2 and BIRC6, a scaffold protein that assists recruitment of DNA damage factors to DNA breaks (Ge et al., 2015a, 2015b). To validate the AP-MS interactions and additionally assess if DYRK2 interactors change during the DNA damage response, we performed BioID-MS experiments in HEK293 cells under normal conditions and upon the treatment with the genotoxic agent Adriamycin (ADR). The BioID-MS analysis confirmed 11 DYRK2 interactions initially identified by AP-MS (Figure 7D). Among these were the above mentioned BIRC6 and proteins from the stable E3 ligase complex (DDB1, UBR5, and VPRBP). However, after ADR treatment, DYRK2 interactions with both BIRC6 protein and the associated E3 ligase complex were reduced (Figure 7E), thus suggesting that DYRK2 is likely to act independently during DNA repair. In contrast, ADR treatment increased interactions between DYRK2 and several proteins that can promote phase transition, such as SRRM2 (Rai et al., 2018). In addition to interaction partners that overlapped with AP-MS, BioID-MS showed that ADR treatment induced stronger binding of DYRK2 to P53, GTSE1, and several other DNA-damage-associated proteins (Figures S5B–S5D). Overall, the observed quantitative changes among DYRK2 interaction partners suggest that DYRK2 could have a role in the DNA damage response that is independent of the DDB1 protein and the core E3 ligase complex.
Discussion
Here, we present the first comprehensive interaction map for the human kinome. Understanding the biochemical context of protein kinase activity is crucial to decode the architecture of cellular signaling and ultimately interpret disease-associated genomic variation. Notwithstanding the high level of comprehensiveness of the interactome mapping performed here, the study has several known limitations that are inherent to large-scale AP-MS experiments. The results did not capture many physiologically relevant interactions, such as for instance cell-line-specific interactions or interactions that occur only upon specific stimuli. Similarly, mutated kinases may have distinct interactomes and some of the interactions crucial for disease development will not be observed with wild-type isoforms used here. In addition, AP-MS is likely to miss many transient but physiologically relevant interactions, and the protocol we used here is not tailored towards large-scale identification of interactions that occur among membrane or nuclear proteins. Furthermore, overexpressed or misfolded kinases will have a stronger interaction with Hsp90 and other chaperones, and some of the interactions could occur only after cell lysis. It is also important to note that 20 of the studied kinases are not endogenously expressed in the HEK293 cell line used here and 37 other kinases are expressed only at very low levels (Table S9). Finally, the kinase constructs used in the study may not reflect the exact isoforms expressed in human cells (Table S9), and the performed N-terminal tagging of the constructs could have affected protein stability, localization, and binding. These caveats are important to bear in mind when assigning new roles to kinases based on their interaction neighborhoods.
Kinases tend to have a relatively high number of interaction partners and pleiotropic roles in the cell (Huttlin et al., 2017; Lahiry et al., 2010), so it is not uncommon that the same kinase family associates with different disease phenotypes (Lahiry et al., 2010). For instance, DYRK2 is also involved in the regulation of proteasome-mediated protein degradation and cell proliferation through phosphorylation of the Rpt3 19S proteasome subunit (Guo et al., 2016). Here, we predict a number of novel modules where interaction partners of the studied kinases share the same disease associations (Figure 5). Better mapping of such modules can aid understanding of kinase roles in the cell and in disease development (Cheng et al., 2014; Csermely et al., 2013; Goh and Choi, 2012), as we suggest here for NEK7, PAK5, and other kinases. Furthermore, mutations that drive cancer development often affect kinase regulation and their interaction properties (Buljan et al., 2018). Combined with the rich cancer genomics data, context information from the kinase interaction network can be of a high value for identifying kinase-linked cancer drivers and establishing a link to cancer pathologies, which is not readily evident from mutation patterns alone. Importantly, there are currently more than 50 approved kinase inhibitors and more than 130 in clinical trials (Wilson et al., 2018). This emphasizes the direct relevance of the mapped disease and functional modules. Moreover, these interactions shed a new light on the biology of several under-investigated kinases, such as POMK and PIM3. Furthermore, the generated interaction network is able to assist kinase-substrate predictions, as illustrated here by epigenetic complexes that we predict to be controlled by CK2 holoenzyme (Figure 4D).
Overall, well-defined interaction modules can be a valuable resource for integrating incomplete functional and disease data on human genes. In this regard, the kinase interaction network represents a stepping stone toward a systemic understanding of kinases biochemical context, function, and regulation.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NEK7 Antibody | Abcam | ab133514 |
| V5 Antibody | Life technologie Europe BV | R960-25 |
| Anti-HA Antibody | Lucerna Chemie AG | HA-11, 901513 |
| secondary anti-rabbit IgG HRP-conjugated antibody | Cell Signaling | #7074S; RRID:AB_2099233 |
| Alexa Fluor® 488 anti-Vimentin | Biolegend | #677809; RRID:AB_2650955 |
| Alexa Fluor® 647 anti-Tubulin Beta 3 | Biolegend | #657406; RRID:AB_2563610 |
| DAPI | Sigma Aldrich | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Strep-Tactin Sepharose | IBA | 2-1201-010 |
| X-Treme Gene 9 DNA Transfection Reagent | Roche | 06365787001 |
| Hygromycin B | Invitrogen | 10687010 |
| BlasticidinS Hcl, 50 mg | Life Technologies | R21001 |
| Biotin | Sigma | B4501-1G |
| Doxycycline Hyclate | Sigma | D9891-5G |
| Critical Commercial Assays | ||
| jetPRIME transfection Reagent | Chemie Brunschwig AG | 114-15 |
| Lipofectamine RNAiMAX | Invitrogen | 13778075 |
| Adriamycin | Sigma | D1515 |
| Deposited Data | ||
| Processed Protein-Protein Interaction data | This paper | Table S2 |
| Raw Mass Spectrometry Data and Peptide Identifications | This paper | Peptide Atlas: PASS01469 https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01469. |
| Experimental Models: Cell Lines | ||
| Human: Flp-Ln T-Rex 293 Cell Line | Thermo Fisher Scientific (Invitrogen) | R78007 |
| Human: A549 Cell Line | ATCC | ATCC® CCL-185 |
| Human: T-REx-HeLa CCL2 Flp-In cells | Thermo Fisher Scientific (Invitrogen) | R71407 |
| Human: LN-229 (glioblastoma cell line) | ATCC | ATCC® CRL-2611 |
| Human: SKOV3 (ovarian cancer cell line) | ATCC | ATCC® HTB-77 |
| Oligonucleotides | ||
| Silencer select GSG2 siRNA | ThermoFisher | s38320 |
| Silencer select PIM3 siRNA | ThermoFisher | s53946 |
| Silencer select PIM3 siRNA | ThermoFisher | s53947 |
| Silencer select PKMYT1 siRNA | ThermoFisher | s194986 |
| Silencer select PKMYT1 siRNA | ThermoFisher | s194985 |
| Silencer select CSNK1G1 siRNA | ThermoFisher | s28823 |
| Silencer select CSNK1G1 siRNA | ThermoFisher | s28822 |
| Silencer select WNK3 siRNA | ThermoFisher | s35278 |
| Silencer select WNK3 siRNA | ThermoFisher | s35279 |
| Silencer select POMK siRNA | ThermoFisher | s534531 |
| Silencer select POMK siRNA | ThermoFisher | s534532 |
| Silencer select NEK7 siRNA | ThermoFisher | s44315 |
| Silencer select NEK7 siRNA | ThermoFisher | s44316 |
| Silencer select NEK6 siRNA | ThermoFisher | s21184 |
| Silencer select NEK6 siRNA | ThermoFisher | s21185 |
| Silencer select RIOK2 siRNA | ThermoFisher | s622 |
| Silencer select SCYL3 siRNA | ThermoFisher | s32780 |
| Silencer select SCYL3 siRNA | ThermoFisher | s32778 |
| Silencer select NEK9 siRNA | ThermoFisher | s40772 |
| Silencer select NEK9 siRNA | ThermoFisher | s40771 |
| siRNA Positive control: Silencer Cy3-labeled GAPDH siRNA | ThermoFisher | Catalog #: AM4649 |
| siRNA Negative control: Silencer Select Negative Control No. 2 siRNA | ThermoFisher | Catalog #: 4390846 |
| Recombinant DNA | ||
| hORFeome V5.1 | Horizon Discovery/ Dharmacon | Open Biosystem |
| hORFeome V8.1 | Horizon Discovery/ Dharmacon | The CCSB ORFeome Collection |
| pTOSH-GW-FRT-HA-Strep | Glatter et al., 2009 | N/A |
| pOG44 Flp recombinase expression vector | Invitrogen | V600520 |
| Kinome Constructs | Varjosalo et al., 2008 | N/A |
| Software and Algorithms | ||
| Cytoscape 3.6.0 | Kohl et al., 2011 | https://www.cytoscape.org/ |
| X!Tandem | Craig & Beavis, 2004 | https://www.thegpm.org/tandem/ |
| Abacus | Fermin et al., 2011 | http://abacustpp.sourceforge.net/ |
| ProteinProphet | Deutsch et al., 2010 | Trans Proteomic Pipeline (TPP, v.4.6.0) |
| R package GOSemSim | Yu et al., 2010 | Bioconductor (v. 2.6.2) |
| R package RDAVIDWebService | Fresno and Fernández, 2013 | Bioconductor |
| NetPhorest | Miller et al., 2008 | http://netphorest.info |
| CellProfiler 2.2.0 | McQuin et al., 2018 | https://cellprofiler.org |
| MATLAB R2019a | https://www.mathworks.com/ | |
| CompPASS | Sowa et al., 2009 | http://besra.hms.harvard.edu/ipmsmsdbs/cgi-bin/tutorial.cgi |
| Other | ||
| Calculation of co-purification p values | This paper | N/A |
| Opera Phenix automated spinning-disk confocal microscope at 20x magnification | Perkin Elmer | HH14000000 |
| Upon acceptance, we will share all images generated for the Manuscript through Mendeley Data | N/A | |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Matthias Gstaiger (matthias.gstaiger@imsb.biol.eth.ch).
Materials Availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
Raw data generated during this study is available at: https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01469.
Kinase network is accessible via the following website: https://sec-explorer.shinyapps.io/Kinome_interactions/.
The code is available upon request.
Experimental Model and Subject Details
Expression constructs
To generate expression vectors for doxycycline-inducible expression of N-terminal Strep-HA-tagged bait proteins, human ORFs provided as pDONR223 vectors were selected either from a Gateway® compatible human orfeome collections horfeome v5.1, horfeome v8.1 and ORFeome Collaboration Clones (OpenBiosystems, Horizon Discovery) or from the collection of kinome constructs (Varjosalo et al., 2008) for LR recombination with the destination vector pcDNA5/FRT/TO/SH/GW (Glatter et al., 2009). Information on this is provided in the Table S9.
Stable cell line generation
HEK Flp-In 293 T-Rex cells (Invitrogen) containing a single genomic FRT site and stably expressing the tet repressor were cultured in DMEM medium (4.5 g/l glucose, 2 mM L-glutamine, Invitrogen) supplemented with 10% FCS, 50 μg/ml penicillin, 50 μg/ml streptomycin, 100 μg/ml zeocin and 15 μg/ml blasticidin. The medium was exchanged with DMEM medium (10% FCS, 50 μg/ml penicillin, 50 μg/ml streptomycin) before transfection. For cell line generation, Flp-In HEK293 cells were co-transfected with the corresponding expression plasmids and the pOG44 vector (Invitrogen) for co-expression of the Flp-recombinase using the Fugene6 transfection reagent (Roche). Two days after transfection, cells were selected in hygromycin-containing medium (100 μg/ml) for 2–3 weeks. Of note, HEK293 used here are transformed by adenovirus and they express adenoviral oncoproteins E1A/B.
Method Details
Protein purification
Following hygromycin selection, stable isogenic HEK293 cell pools were grown in eight 14 cm Nunclon dishes to 80% confluency, 1.3 μg/ml doxycycline (Sigma) was added for 24h to induce the expression of SH-tagged bait proteins and harvested with PBS containing 10 mM EDTA. Cells were collected, frozen in liquid nitrogen and stored at −80°C prior to protein complex purification. Effectively, > 1x108 cells were used per pulldown.
The frozen cell pellets were resuspended in 4 mL HNN lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 50 mM NaF, 0.5% Igepal CA-630 (Nonidet P-40 Substitute), 200 μM Na3VO4, 1 mM PMSF, 20 μg/ml Avidin and 1x Protease Inhibitor mix (Sigma) and incubated on ice for 10 min. Insoluble material was removed by centrifugation. Cleared lysates were incubated on a rotating wheel at 4°C with 50 μl pre-equilibrated Strep-Tactin Sepharose beads (IBA Biotagnology) for 15min and loaded on a spin column (Bio-Rad). The beads were washed two times with 1 mL HNN lysis buffer and three times with HNN buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 50 mM NaF). Bound proteins were eluted with 600 ul 0.5 mM Biotin in the HNN buffer. To remove the biotin, eluted samples were incubated for 1 h in presence of 25% TCA on ice, washed with acetone, air-dried and re-solubilized in 50 μl 8 mM Urea in 50 mM NH4HCO3 pH 8.8. Cysteine bonds were reduced with 5 mM TCEP for 30 min at 37°C and alkylated in 10 mM iodoacetamide for 30 min at room temperature in the dark. Samples were diluted with NH4HCO3 to 1.5 M Urea and digested with 1 μg trypsin (Promega) overnight at 37°C. The peptides were purified using C18 microspin columns (The Nest Group Inc.) according to the protocol of the manufacturer and eluted with 0.1% formic acid, 3% acetonitrile for mass spectrometry analysis.
Affinity purification and western blotting
For Co-AP and western blotting analysis shown in Figure S4B 5x 105 A549 cells were co-transfected with plasmids for the transient expression of the V5-tagged kinase and SH-tagged proteins using jetPRIME transfection Reagent (114-15, Chemie Brunschwig AG). The transfected cells were lysed 24 hr after transfection in 1 mL of HNN lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 50 mM NaF, 0.5% Igepal CA-630 (NP-40 substitute), 200 uM Na3VO4, 1 mM PMSF, and 1x Protease Inhibitor mix (sigma)). The cleared lysate was incubated with 15 μL equilibrated Streptactin beads (Streptactin superflow 50%, IBA) o/n at 4°C on a rotation shaker. The beads were washed three times with the lysis buffer and after final wash the complexes were eluted in 50 μL of 3X Laemmli buffer. The samples were boiled for 5 min, separated by PAGE and transferred to nitrocellulose membranes (Whatman GmbH, Germany). Immunoprecipitated proteins were detected either with anti-HA (HA-11, 901513, Lucerna Chemie AG)) or anti-V5 (R960-25, Life technologie Europe BV) primary and secondary horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-mouse, labforce) by enhanced chemiluminescence. For the detection of complexes with endogenous NEK7 kinase (Figure S4A), A549 cells were transfected with vectors expressing SH-tagged proteins, lysed, lysates purified and subjected to western blotting as described above. Endogenous NEK7 protein was detected by western blotting of the Streptactin purified samples using primary anti NEK7 antibodies (Abcam, ab133514) and secondary anti-rabbit IgG HRP-conjugated antibody (Cell Signaling, #7074S).
Mass spectrometry
LC-MS/MS analysis was performed on a Thermo Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Peptide separation was carried out by a Thermo Easy-nLC 1000 HPLC system using a 15 cm long, 75 μm diameter ID PepMap column (Thermo, particle size 2 μm) with a 45 min gradient from 5% B to 35% B at a flow rate of 300 nl/min. Solvents were A: 3% acetonitrile, 0.1% formic acid in water; B: 3% water, 0.1% formic acid in acetonitrile. The data acquisition mode was set to obtain one high resolution MS scan in the Orbitrap (240,000@400 m/z). The 10 most abundant ions from the first MS scan were fragmented by collision induced fragmentation (CID) and MS/MS fragment ion spectra were acquired in the linear ion trap at normal scan rate. Charge state screening was enabled and unassigned or singly charged ions were rejected. Precursor isolation width was 2 m/z in all cases and dynamic exclusion was enabled for 30 s. After every technical replicate set, a peptide reference sample containing 200 fmol of human [Glu1]-Fibrinopeptide B (Glufib) (Sigma-Aldrich) was analyzed to monitor the LC-MS/MS systems performance.
Carry-over was systematically controlled through all the MS runs. Each subsequent control (Glufib) measurement and bait replicate set were manually screened for the presence of previous bait and most abundant interaction candidate peptides. Samples with carried-over interacting proteins were re-measured using cleaned LC columns.
Kinase classification
To classify kinases in families, we used as primary reference the Uniprot annotation (reported either in individual entries or in the summary file https://www.uniprot.org:443/docs/pkinfam.txt, released July 2016). For kinases that were not classified in Uniprot and for Atypical kinases, we used the classification from (Manning et al., 2002) (http://kinase.com/web/current/kinbase/). For three kinases for which the classification was “other” in Uniprot, we followed the more specific classification by Manning and colleagues (Q96S38 and Q9Y6S9 classified as AGC; P49842 classified as Atypical). The following proteins were not considered in the classification but were kept in the interactome: 1) two putative kinases: Q9UJY1 and Q12792. 2) Non-catalytic subunits of the 5′-AMP-activated protein kinase: Q9UGI9, Q9UGJ0, O43741, P54619. Similarly, subfamily classification was primarily based on Uniprot and integrated with information reported in Manning et al., 2002. The Manning classification contains up to three classes (Group, Family, Subfamily); for consistency, we refer to the highest Manning class as “Family” and the first subordinate class as “Subfamily,” even if the first class is in some instances, e.g., AGC, defined as Group. The classification is reported in Table S1.
Citation analysis
Citation data for kinome baits were downloaded (July 2017) from ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2pubmed.gz using their geneIDs. Pubmed ID counts for entries with multiple geneIDs were summed up (kinase P57078 was not considered for analysis, as it doesn’t have an associated geneID). The number of unique publicly available interactions was derived from the Integrated Interaction Database (IID; version 2017-04). The full data is reported in Table S1. Red line in Figure 2A that represents a running average of the citation number was calculated using R package zoo (k = 5).
Reference databases and comparison with HT AP-MS studies
We used as a reference database for published and deposited interactions the integrated interaction database (IID) (Kotlyar et al., 2016) (version 2017-04, unless stated otherwise). BioGrid was used for the calculation of the fraction of the identified interactions that have been already deposited. For protein complexes, we used the list of core CORUM complexes (release: 02.02.2017; http://mips.helmholtz-muenchen.de/corum/#download) and removed residual duplicates complexes as well as complexes with only a single protein. Baits from (Hein et al., 2015) were identified from column ‘expected.bait.reference.uniprot.id’ column in the ‘bait cell line’ sheet, supplemental data. Baits GeneIDs from Huttlin et al. (2015) and Huttlin et al. (2017) were mapped to UniprotIDs using Uniprot Retrieve/ID Mapping function (Uniprot, December 2018) and reviewed entries were used for the comparison (17 and 30 unmapped GeneID identifiers, respectively).
GO analysis
As input for the GO analysis, we used the combined (non-redundant) interactome from the kinome/IID (version 2018-05) for all subgroups with at least 3 proteins among our baits, including the baits and excluding chaperones. The analysis was carried out using the R package RDAVIDWebService (Fresno and Fernández, 2013), using as annotation category “GOTERM_BP_DIRECT” and selecting all the entries with an adjusted (Benjamini-Hochberg) p value < 0.05, or up to the top 5 entries in the instances when less than 5 entries satisfied the significance threshold. Specific GO terms were manually collapsed into more general ones; first, by clustering terms together based on semantic similarity and then by choosing shared terms among the inferred trees (AmiGO 2 version 2.5.12) of those GO terms. When multiple terms for the same subfamily were mapped to the same general term, the lower p value value has been represented in Figure 3A. To measure semantic similarity between Kinome network and IID interaction networks, we compared GO terms associated with the Kinome and IID for the indicated subgroups (as defined above). The comparison was carried out with R packages GOSemSim (v. 2.6.2)(Yu et al., 2010), using the Wang similarity measure.
To classify interactors among phosphatases, ubiquitin ligases, DUB enzymes, transcription factors and epigenetic factors, and to define protein complexes belonging to the latter class, we relied on curated information included in, or associated with, the following publications (Li et al., 2008, Medvedeva et al., 2015, Nijman et al., 2005, Sacco et al., 2012, Wingender et al., 2013). To evaluate the fraction of novel interactions including interactors belonging to these classes, we used as a reference the IID (Kotlyar et al., 2016) (version 2017-04).
Network visualization
Protein Interaction data was visualized with Cytoscape 3.6.0 (https://www.cytoscape.org/) (Kohl et al., 2011).
Protein Domains
In order to assess the overrepresentation of functional domains, domain assignments were downloaded from the Pfam database (Pfam 31.0, March 2017) (Finn et al., 2016). Proteins identified as interaction partners of protein kinases were compared to all other proteins in the reference human proteome (Uniprot-all table from the Uniprot database obtained in June 2018, containing 20,361 entries in total) (UniProt Consortium, 2015). Only proteins with at least one annotated domain were considered. For each domain, a fraction of proteins with the domain was compared between the kinase interactors and background proteins. Significantly overrepresented domains were identified with one-sided Fisher’s exact test in R and the obtained p values were corrected using the Benjamini-Hochberg method.
Prediction of kinase-substrate interactions
In order to predict the phosphosites and the possible upstream kinases, NetPhorest software tool (distribution November 2013) (Miller et al., 2008) was locally installed and ran on the fasta sequences of all proteins in the kinase interaction network. The output of this analysis is a position of each Serine, Threonine or Tyrosine that is found within a motif which could be recognized by one or more kinase groups and a posterior probability for each group. As the predicted phosphosites we kept only residues that were experimentally found to be phosphorylated: either by a small-scale study or by five or more large-scale studies deposited in the PhosphositePlus database (release November 2018). For each individual site, we considered only the top three predicted kinase groups and required a minimum posterior probability of 0.035, as well as the posterior to be higher than the prior (Freschi et al., 2014; So et al., 2015; Tan et al., 2009). Individual kinase group members were assigned to each upstream kinase group based on the file provided by the NetPhorest support team and based on the kinase family or subfamily name match to the NetPhorest groups. Finally, the kinase-substrate relationships predicted from the sequence features were considered further only when they were additionally supported by the physical interaction in the filtered AP-MS interaction data obtained here.
A list of the known kinase-substrate pairs was obtained from the PhosphoSitePlus database (data extracted in Nov 2018), (Hornbeck et al., 2015). This dataset was overlapped with the Kinase interaction network and the number of interactions that agreed with the annotated regulatory events was noted. In order to assess if this was higher than expected, random networks were composed. These preserved the size and topology of the kinase network, i.e., the distribution of the number of interactors per individual kinase, and interactors were randomly sampled from the pool of all prey proteins identified in the study. This was repeated 1,000 times and each time the total number of interaction partners that were also known kinase-substrate pairs was noted. Distribution of the values observed for the random networks was compared to the corresponding value for the original Kinase network using the pnorm test in R. Reciprocal regulation, i.e., instances where two kinases interacted and both were known to phosphorylate each other, were counted as a single interaction event. Next, we evaluated the observed number of predicted kinase-substrate regulatory events. Analogous to the approach used for known substrates, random networks were generated in the same way and the number of predicted kinase-substrate events in the kinase network was compared to the distribution of these values for random networks with the pnorm test in R. The subnetwork with kinase-kinase regulatory interactions was obtained by asking that both the kinase and the substrate are listed as kinases according to Manning annotations included in Table S1 (Manning et al., 2002). Additionally, the number of predicted phosphosites in each substrate protein was counted and proteins with more than five phosphosites that are predicted to be regulated by the same kinase were noted. Kinases that had more than five substrate proteins with multiple, i.e., six or more, phosphosites, were then investigated in more detail.
Many kinases that belong to the AGC, CAMK, CMGC or Tyrosine Kinase families have conserved sequence segments around the more variable active loop region (located between the Mg binding and P+1 loops). Phosphosites that map within the active loop are often associated with specific roles and they can function as regulatory switches for kinase activity. In order to identify active loop coordinates in the predicted substrate kinases, all bait kinases were aligned using the multiple sequence alignment tool Clustal Omega (Sievers et al., 2011). For this, we noted positions of activation loops in exemplary well-annotated kinases and used the sequence alignment to infer positions of these in other kinases. Based on differences in sequence motifs we used as reference kinases CDK9 from CMGC family and BRAF from TLK family. Amino acid (aa) positions before and after the activation loop were noted in both CDK9 and BRAF kinase. For all other bait kinases it was assessed if the regions before and after the less-conserved activation loops in CDK9 (first choice) or BRAF shared a sequence similarity with the aligned regions in these proteins (for instance, kinases from CMGC family had an excellent alignment with CDK9 annotated regions). To consider that there was a sequence conservation in the regions surrounding the active loop, the predicted active loop sequence segment had to be between 5 and 50 amino acids long and at least half of the amino acids in sequence segments around the active loop had to be similar and aligned. The length of the surrounding regions was estimated from sequence annotations and alignment with other kinases and it was 9 and 7 amino acids for CDK9 and 11 and 9 amino acids for BRAF for the segment preceding and after the activation loop, respectively. Finally, when applicable, phosphosites in the predicted substrate proteins were mapped to the active loop coordinates defined in this way. This highlighted 25 predicted substrate kinases in which one or more regulated phosphosite was within an active loop (this information is included in the Table S6).
In addition to phosphosites, sequences of predicted substrates were searched for the presence of docking motifs that could be recognized by protein kinase domains. For this, annotations in the annotated eukaryotic linear motifs were downloaded from the ELM database (ELM 2016) (Dinkel et al., 2016).
Detection of disease-associated kinase modules
To obtain confident annotations for human gene-disease relationships, “morbidmap” table with the curated data on known Mendelian disorders was retrieved from the Online Mendelian Inheritance in Man (OMIM) catalog (a dataset generated in January 2017) (Hamosh et al., 2005). OMIM associates diseases to gene names, so all gene names assigned to individual Uniprot identifiers were searched. Because similar disease phenotypes can have slightly different OMIM terms, each disease term was comma separated, unspecific character symbols were removed, and only the expression before the first comma was assigned to the associated protein. Additionally, as a control, all proteins in the kinase network were annotated with individual words occurring in OMIM terms. Six expressions that were not captured with the above approach, but were re-occurring in the kinase network, were separately evaluated as regular expression terms (these terms were: “glycogen storage disease,” “mental retardation,” “thalassemia,” “deafness” and “ventricular”). Cancer-associated terms were skipped as these were analyzed separately. Only terms that were shared among three or more proteins in the kinase network were considered in the further analyses. Finally, to reduce the unspecific enrichments, actin and myosin proteins as well as chaperone proteins (defined as having HSP or chaperone in the gene name, or HSP, CPN60 or FKBP domain in the protein sequence) were excluded from the analysis. To identify disease modules, frequency of these terms in each individual kinase neighborhood - composed of the kinase itself and its direct interactors – was compared to the frequency of the terms in the background set. Background set was composed of Uniprot proteins in the representative human proteome (version August 2016 with 20,197 entries in total) after excluding proteins identified in the kinase network. In addition, only proteins with at least one OMIM annotation were included in the analysis. To identify disease terms overrepresented around individual kinases, Fisher’s exact test followed with Benjamini-Hochberg correction was applied. Significant terms were visualized and at this step three mitochondrial ribosomal proteins around the MAST1 kinase were removed. The kinase is located within cytoplasm and has a role in cytoskeleton regulation, so there is a risk that these could be false identifications.
Functions associated with the POMK interaction partners were investigated separately. Information on the GO terms and KEGG pathways that were most strongly overrepresented in a comparison with all other human proteins was obtained using the annotations in DAVID database (release 6.8). Terms that were highly significant (p value < 0.0001, Benjamini Hochberg correction) and where gene sets annotated with the particular term were not subsets of the more significant terms were visualized (i.e., overlapping terms are not reported in the figure).
In order to expand the list of gene-disease associations, DisGeNET v5.0 table with all available associations was retrieved (Piñero et al., 2015). This included annotations with different confidence levels and gene-disease relationships that were of a lower confidence were skipped. These were the terms supported only (i) either by the BeFree text mining system, or (ii) Human Phenotype Ontology, or (iii) Genetic Association Database or (iv) only by Comparative Toxicogenomics Database. Analogously to the OMIM analysis, only disease terms annotated to at least three proteins from the kinase network were considered, and kinase interaction partners that were chaperones, actin or myosin proteins, were omitted from the analysis. Additionally, all cancer-associated terms were skipped as cancer was studied separately. Again, occurence of each term was counted within each kinase neighborhood, and significant overrepresentation compared to the background human proteins (i.e., UniProt reference proteome after excluding proteins identified in this study) was assessed with the Fisher’s exact test, followed with the Benjamini Hochberg correction. For the consistency in calculations, chaperones, actins and myosin proteins were also excluded from the background set and only proteins with at least one DisGeNET annotation were considered.
Cellular phenotype assay
In order to identify kinases whose interaction partners are enriched in roles associated with cell shape regulation we searched for the interactors annotated with Uniprot GO terms that contained the words ‘ciliary’, ‘cytoskeleton’, ‘polarity’, ‘kinasin’, ‘microtubule’, ‘focal adhesion’, ‘cell adhesion’, ‘lamellipodium’, ‘mitotic’ or ‘actin filament’. To increase the specificity of the identified kinases, enrichment was calculated by excluding (i) chaperones (i.e., interactors with HSP, Cpn60 or FKBP domain, or those that contained HSP or chaperon in their name), (ii) cytoskeletal proteins that frequently copurify in AP-MS with different baits (this was done with text-mining of protein names, and those that contained MYH, ACTIN, or started with CCT, ACT, ACL or ARP were skipped), and (iii) proteins that interacted with more than 10 different kinases. Enrichment was calculated by comparing interactors of individual kinases to the background of all Uniprot proteins. Significant kinases that already had strong support and a confident GO term that indicated their role in the cellular shape regulation were excluded. In addition to kinases selected in this way, we included in the screen kinases from the NEK7 interaction neighborhood (NEK6 and NEK9) and, based on preliminary literature links, also the SCYL3 pseudokinase. Finally, positive and negative control siRNAs were also included in the screen. Whenever possible, validated ThermoFisher siRNA reagents were used in the experiments.
Two human cell lines LN-229 (glioblastoma cell line) and SKOV3 (ovarian cancer cell line) were used for the siRNA silencing experiments. LN-229 cells were cultured in DMEM media supplemented with 2% FBS, 1% Pen-strep, and 25mM HEPES while SKOV3 cells were cultured in RPMI-1640 media supplemented with 10% FBS and 1% Antibiotic-Antimycotic (all products from GIBCO). Cells were passaged at approximately 70% confluency with 0.25% Trypsin-EDTA (GIBCO, 25200056) and seeded at 0.7-1.0x103 cells/well into clear-bottom, tissue-culture treated, CellCarrier-384 Ultra Microplates (Perkin Elmer, 6057300) with three replicate wells per condition. Silencer Select Pre-Designed siRNAs (Table S5) were transfected at 0.66 pmol/well using Lipofectamine RNAiMAX (Invitrogen, 13778075). Both the siRNAs and Lipofectamine transfection reagent were dispensed using a Labcyte Echo liquid handler in a randomized plate layout to control for plate effects. For each gene target, between 1-3 siRNAs were tested for both cell lines. Cells were incubated at 37C, 5% CO2 for 48 hours following siRNA transfection. Cells were subsequently fixed with 4% PFA in PBS, blocked for 1 hour at room temperature in blocking solution (PBS containing 5% FBS and 0.1% Triton). Cells were stained overnight at 4C in blocking solution with the following antibodies: Alexa Fluor® 488 anti-Vimentin (1:1000, Biolegend, #677809), Alexa Fluor® 647 anti-Tubulin Beta 3 (1:1000, Biolegend, #657406) and DAPI (10mg/ml, 1:1000, Sigma Aldrich). High-content imaging was performed with an Opera Phenix automated spinning-disk confocal microscope at 20x magnification (Perkin Elmer, HH14000000). To measure cell area shape features, single-cells were segmented using CellProfiler 2.2.0 (McQuin et al., 2018). Nuclei segmentation relied on the DAPI channel, while LN-229 cytoplasmic segmentation utilized the Tubulin Beta 3 (TUBB3) channel and the SKOV3 cytoplasmic segmentation utilized the Vimentin (VIM) channel. CellProfiler module ‘MeasureObjectSizeShape’ based on the cytoplasmic segmentation was used to derive 18 different cell area shape features. Area shape features that were not dependent on cell density were chosen for plotting. Downstream image analysis and data visualization was performed with MATLAB R2019a where single cell feature data was averaged across each well/condition and compared to the negative siRNA control. Student’s paired t test was performed to calculate statistical significance.
Identification of kinase modules associated to cancer
Kinase modules with overrepresentation of CA proteins were identified in a similar approach as other disease modules. The list of known CA proteins was compiled from the known cancer drivers deposited in the Cancer Gene Census (version January 2017) (Futreal et al., 2004) and from a prominent review on the roles of cancer proteins (Vogelstein et al., 2013). Additionally, the list of CA proteins was extended with those identified as frequently mutated in two recent comprehensive analyses of pan-cancer patient mutation data (Davoli et al., 2013; Lawrence et al., 2014). In this and the following analyses, heat shock proteins were excluded due to their broad scope of kinase interaction partners. In order to assess if the interactome of CA kinases was enriched in other CA proteins, a set of all kinase interaction partners was divided into those that interacted with one or more of the CA kinase baits and all other interactors, and a fraction of CA proteins was noted in each set. Additionally, a background fraction of CA proteins was calculated in the set of all other Uniprot proteins in the representative human proteome after excluding proteins in the kinase network (UniProt Consortium, 2015) (version August 2016). A tendency of CA kinases to interact with proteins that are also associated with cancer was assessed with the chi-square test in the comparison to two other sets.
Next, a fraction of CA proteins in each kinase neighborhood, i.e., a set of proteins formed by each kinase and its interactors, was calculated and compared to the fraction of CA proteins in the background set of all other Uniprot proteins, when excluding those in the kinase network. Modules with at least two CA proteins were assessed and significant clusters were identified using the Fisher’s test and Benjamini Hochberg correction for the obtained p values. DYRK1A/B kinases were excluded from the final list of significant kinase clusters due to their known interaction with adenoviral E1A protein expressed in HEK cell line (Komorek et al., 2010). Furthermore, the interaction network for the most significant modules (adjusted p value < 0.025) was visualized using the Cytoscape software (Kohl et al., 2011). In order to functionally annotate the significant modules, KEGG, Biocarta and GO biological process annotations were retrieved for all proteins in the network using DAVID service (Dennis et al., 2003). Terms that were most frequently occurring within each module were listed, and manually checked in order to select the ones that are most frequent, and when possible, that occur also in other modules in the network.
BioID experiments
The expression of FLAG-BirA∗-tagged Dyrk2 (T-REx-HEK293 Flp-In cells) or Pim3 (T-REx-HeLa CCL2 Flp-In cells) was induced by addition of 4ug/ml doxycycline for 24h in one 150 mm tissue culture plate. Then the media was replaced by fresh media supplemented with 50 μm biotin and cells were incubated for additional 24h. Cells were harvested and snap frozen in liquid nitrogen followed by cell lysis in 1 mL RIPA buffer (50mM Tris-HCl (pH 8), 150 mM NaCL, 1% Triton X-100, 1 mM EDTA, 0.1% SDS supplemented with 1 mM PMSF and protease inhibitor cocktail (Sigma)) and Benzonase (Sigma) treatment (250U) at 10°C for 30 min. Cell debris was removed by centrifugation (20000 x g, 20 min at 4°C) and the cleared cell lysate was incubated with disuccinimidyl suberate (DSS) (Sigma) crosslinked Strep-Tactin beads (IBA LifeSciences) for 1h on a rotation shaker at 4°C. Then beads were washed three times with RIPA buffer, three times with HNN buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 50 mM NaF) and two times with 100 mM NH4CO3. For protein denaturation the beads were incubated with 8 M urea followed by reduction with 5 mM Tris(2-carboxyethyl)phosphine TCEP and protein alkylation with 10 mM iodoacetamide. The sample was diluted to 4 M urea with 100 mM NH4CO3 and proteins were digested on the beads with 0.5 μg LysC (Wako) for 3h followed by dilution to 1 M urea and digestion by 0.8 μg trypsin overnight. In order to stop protein digestion 5% formic acid was added and the peptides were purified by C18 UltraMicroSpin columns (The NestGroup) and dried in a speedvac. The dried peptides were dissolved in 2% acetonitrile and 0.1% formic acid.
BioID-MS data analysis
In general, BioID-MS experiments were performed in triplicates (n = 3). Acquired MS/MS scans were searched against the UniProtKB/Swiss-Prot protein database (31.03.2016) using MaxQuant v1.5.2.8 (Cox and Mann, 2008) with default parameters. High confidence interactors of FLAG-BirA∗-Dyrk2 were determined by filtering against proteins identified in FLAG-BirA∗-GFP control experiments. Proteins significantly enriched (log2FC ≥ 1, adj. p value ≤ 0.05) compared to the GFP control were considered as high confident interaction partners. The fold change of interactors was calculated based on precursor MS1 values (LFQ intensities) determined by MaxQuant v1.4.2.8. The statistical analysis was performed by customized R scripts. Briefly, MS1 intensities were median-normalized and missing values were imputed using random sampling from a distribution based on the 5th lowest quantile. Changes in the Dyrk2 BioID interaction network upon perturbation were quantified using bait-normalized precursor MS1 intensities (LFQ intensity). The statistical analysis was performed as described above.
For the identification of high confident interaction partners of FLAG-BirA∗-Pim3 expressed in HeLa CCL2 cells the BioID-MS interaction dataset was filtered with SAINTexpress (Mellacheruvu et al., 2013) using default parameters and sixteen control experiments (FLAG-BirA∗ and FLAG-BirA∗-GFP expressed in HeLa CCL2 cells) of the CRAPome database (http://crapome.org/). IDs assigned with a SAINT score = 1 were considered as high confidence interactors.
DNA damage induction
To generate a reference list of DNA repair proteins, we obtained a set of proteins which are confidently annotated with the process (Arcas et al.,2014) and we extended it with proteins that were shown to physically migrate to the foci of DNA damage after UV laser microirradiation (Izhar et al., 2015). In order to confirm BIRC6 as new Dyrk2 interaction partner and to investigate Dyrk2 interactome remodeling in response to DNA damage we performed BioID-MS interaction analysis with BirA-tagged Dyrk2 under normal conditions and upon induction of DNA damage by Adriamycin (ADR). In contrast to AP-MS, proximity labeling by BioID allows stringent lysis conditions that disrupt the nuclear membrane and facilitates the identification of nuclear interaction partners. To induce genotoxic stress HEK293 cells were treated with 2 μg/ml ADR for 24h and the quantification of changes in the Dyrk2 interactome was based on precursor MS1 intensities obtained from MaxQuant v1.5.2.8. Interaction partners were accurately quantified with a replicate CV < 9% and the triplicates of untreated and ADR treated cells emerge as individual cluster (Figure S5B).
Quantification and Statistical Analysis
Protein identification
Acquired spectra were searched with X!Tandem (Craig and Beavis, 2004) (release 2011.12.01) against the canonical human proteome reference dataset (http://www.uniprot.org/), extended with reverse decoy sequences for all entries. The search parameters were set to include semi-tryptic peptides (KR/P) containing up to two missed cleavages. Carbamidomethyl (+57.021465 amu) on Cys was set as static peptide modification. Oxidation (+ 15.99492 amu) on Met and phosphorylation (+79.966331 amu) on Ser, Thr, Tyr were set as dynamic peptide modifications. The precursor mass tolerance was set to 25 ppm, the fragment mass error tolerance to 0.5 Da. Obtained peptide spectrum matches were statistically evaluated using PeptideProphet and protein inference by ProteinProphet, both part of the Trans Proteomic Pipeline (TPP, v.4.6.0) (Deutsch et al., 2010). A minimum protein probability of 0.9 was set to match a false discovery rate (FDR) of < 1%. The resulting pep.xml and prot.xml files were used as input for the spectral counting software tool Abacus to calculate spectral counts and values (Fermin et al., 2011) (release 2013.02.14). Raw MS data is available at the following site: https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01469. The site also includes peptide identifications from all MS runs (all_pep_xml_together.txt), and provides the script and instructions for filtering that corresponds to Abacus criteria and generates peptide identifications used in this study (processingPepXml.pl and NOTES_ON_PEP_XML).
Evaluation of high confidence interacting proteins (HCIP)
To identify the best performing strategy to filter the obtained AP-MS raw data we compared the receiver operator characteristics of several established computational methods using published kinase protein interactions (reported by at least two references) and the corresponding random set of protein interactions. We found that the weighted D (WD) score (Sowa et al., 2009) as well as spectral count ratios corrected for false positive detection based on data from a large set of GFP control purifications outperformed the other scores tested with respect to sensitivity and specificity (Figure S1A). We therefore combined WD score and spectral count ratios for generating the final high-confidence protein interaction dataset. The chosen cutoff for the WD score and the spectral count ratios was set to keep the number of false positive interactions below one percent as determined from the ROC curve while maximizing the recovery of already known PPI in the filtered data.
A custom CompPASS implementation was used to compute the WD-score derived from the original method (Sowa et al., 2009):
k = total number of baits
p = number of replicate runs in which the interactor is present
r = number of replicate runs per bait
The GFP-ratio was computed using all control measurements and defined as the differential expression of the interactors over the mean of all controls:
, where was set to the minimum value > 0 of all interactors, if .
Total spectral count values of identified co-purified proteins were compared to a control AP dataset consisting of 94 Strep HA-GFP purification experiments, from which 68 GFP control datasets have been measured on an LTQ Orbitrap XL mass spectrometer and deposited in the CRAPome contaminant repository for affinity purification (Mellacheruvu et al., 2013).
A custom R script was used to select an enrichment cut off over the control dataset (GFP-ratio) and a WD-score threshold, candidate bait-prey interactions were looked-up for literature references in the IntAct database (release date: 31.03.2015). Matches with at least two publications were considered to be true positive hits. A corresponding number of true false hits was generated by permutation of the prey and bait identifiers in the input Table 20 times with replacement and random sampling. The WD-score and GFP-ratio thresholds of 73.6 and 18.4, respectively, were selected by computation of receiver operating characteristic and selection of a FPR threshold of 1%.
Only candidate interactions above the WD-score and GFP-ratio threshold were considered to be of high confidence. Additional filtering criteria applied included removal of Keratins, adenoviral proteins, self-bait interactions, iRT peptides, obsolete entries and interactions that had a total number of spectral count lower than 2. Finally, interactors present in more than 70% of the 411 CRAPome human control runs (v1.1) as well as residual carry-over proteins were removed from the filtered table.
Co-purification analysis
A local perl script was used to apply the hypergeometric test and calculate significance of co-purification for each of the protein pairs that were purified together with the same kinase baits. In the formula for hypergeometric test the variables were:
m = Number of times two proteins are seen with the same kinase bait
k = Number of kinase baits protein 1 was purified with
l = Number of kinase baits protein 2 was purified with
N = Total number of kinase baits
The obtained values were adjusted for multiple testing using Benjamini-Hochberg correction. The most significant terms are depicted as a network and shown in Figure 2. Only the pair NUP188 – GCN1 is left out from the illustration. The reason for this is that NUP18 is present in 25% of the studies in the CRAPome database and there is a risk it could be a non-specific binder.
Additional resources
Interaction data was deposited onto the following website:
Acknowledgments
The project was supported by SystemsX.ch (project PhosphoNetX PPM to R.A.), the SNSF (grant 3100A0-688 107679 to R.A.), and the ERC (grant ERC-20140AdG 670821 to R.A.). M.G. and R.C. acknowledge support by the IMI project ULTRA-DD (FP07/2007-2013, grant 115766). M.B. was funded by an SNSF SystemsX.ch fellowship (TPdF 2013/135). M.M. was supported by an EMBO fellowship (ALTF 928-2014). The authors are grateful to M. Claassen, M. Altmeyer, M. Aebi, L. Gillet., J. Mena, and C. Li for their advice on the literature and methodological approaches.
Author Contributions
M.B. and R.C. analyzed the data, prepared the figures, and wrote the manuscript with input from R.A. and M.G. A.V. and A.D. generated recombinant cell lines and performed MS experiments. M.V. provided constructs and assisted generation of recombinant cell lines. G.R., A.V., and L.E.P. performed initial data filtering and preliminary data analyses. M.M. performed BioID and DNA damage studies. S.L. and B.S. performed siRNA assays. V.S. assisted with DNA damage analyses. R.A. and M.G. designed and led the study.
Declaration of Interests
The authors declare no competing interests.
Published: July 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.07.001.
Contributor Information
Ruedi Aebersold, Email: aebersold@imsb.biol.ethz.ch.
Matthias Gstaiger, Email: matthias.gstaiger@imsb.biol.eth.ch.
Supplemental Information
References
- Arcas A., Fernández-Capetillo O., Cases I., Rojas A.M. Emergence and evolutionary analysis of the human DDR network: implications in comparative genomics and downstream analyses. Mol. Biol. Evol. 2014;31:940–961. doi: 10.1093/molbev/msu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assrir N., Filhol O., Galisson F., Lipinski M. HIRIP3 is a nuclear phosphoprotein interacting with and phosphorylated by the serine-threonine kinase CK2. Biol. Chem. 2007;388:391–398. doi: 10.1515/BC.2007.045. [DOI] [PubMed] [Google Scholar]
- Bai L., Kovach A., You Q., Kenny A., Li H. Structure of the eukaryotic protein O-mannosyltransferase Pmt1-Pmt2 complex. Nat. Struct. Mol. Biol. 2019;26:704–711. doi: 10.1038/s41594-019-0262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A., Aebersold R., Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Breitkreutz A., Choi H., Sharom J.R., Boucher L., Neduva V., Larsen B., Lin Z.-Y., Breitkreutz B.-J., Stark C., Liu G. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M., Blattmann P., Aebersold R., Boutros M. Systematic characterization of pan-cancer mutation clusters. Mol. Syst. Biol. 2018;14:e7974. doi: 10.15252/msb.20177974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.R., Wang X., Templeton D., Davis R.J., Tan T.H. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J. Biol. Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- Chen B., Fan W., Liu J., Wu F.-X. Identifying protein complexes and functional modules--from static PPI networks to dynamic PPI networks. Brief. Bioinform. 2014;15:177–194. doi: 10.1093/bib/bbt039. [DOI] [PubMed] [Google Scholar]
- Cheng C.-J., Huang C.-L. Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J. Am. Soc. Nephrol. 2011;22:460–471. doi: 10.1681/ASN.2010060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Jia P., Wang Q., Zhao Z. Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget. 2014;5:3697–3710. doi: 10.18632/oncotarget.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Huang J.-W., Izhar L., Sowa M.E., Harper J.W., Elledge S.J. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA. 2014;111:18631–18636. doi: 10.1073/pnas.1422488112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinge J., César-Razquin A., Huber K., Breitwieser F.P., Májek P., Superti-Furga G. Building and exploring an integrated human kinase network: global organization and medical entry points. J. Proteomics. 2014;107:113–127. doi: 10.1016/j.jprot.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Craig R., Beavis R.C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Csermely P., Korcsmáros T., Kiss H.J.M., London G., Nussinov R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol. Ther. 2013;138:333–408. doi: 10.1016/j.pharmthera.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T., Xu A.W., Mengwasser K.E., Sack L.M., Yoon J.C., Park P.J., Elledge S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:R60. [PubMed] [Google Scholar]
- Deutsch E.W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel H., Van Roey K., Michael S., Kumar M., Uyar B., Altenberg B., Milchevskaya V., Schneider M., Kühn H., Behrendt A. ELM 2016--data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 2016;44(D1):D294–D300. doi: 10.1093/nar/gkv1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K., Lee C., Huizar R.L., Tu F., Borgeson B., McWhite C.D., Ma Y., Wallingford J.B., Marcotte E.M. Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 2017;13:932. doi: 10.15252/msb.20167490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A.M., Isserlin R., Bader G.D., Frye S.V., Willson T.M., Yu F.H. Too many roads not taken. Nature. 2011;470:163–165. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
- Fermin D., Basrur V., Yocum A.K., Nesvizhskii A.I. Abacus: a computational tool for extracting and pre-processing spectral count data for label-free quantitative proteomic analysis. Proteomics. 2011;11:1340–1345. doi: 10.1002/pmic.201000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessenden M. Protein maps chart the causes of disease. Nature. 2017;549:293–295. doi: 10.1038/549293a. [DOI] [PubMed] [Google Scholar]
- Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleuren E.D.G., Zhang L., Wu J., Daly R.J. The kinome ‘at large’ in cancer. Nat. Rev. Cancer. 2016;16:83–98. doi: 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]
- Freeze H.H., Chong J.X., Bamshad M.J., Ng B.G. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am. J. Hum. Genet. 2014;94:161–175. doi: 10.1016/j.ajhg.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freschi L., Osseni M., Landry C.R. Functional divergence and evolutionary turnover in mammalian phosphoproteomes. PLoS Genet. 2014;10:e1004062. doi: 10.1371/journal.pgen.1004062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno C., Fernández E.A. RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics. 2013;29:2810–2811. doi: 10.1093/bioinformatics/btt487. [DOI] [PubMed] [Google Scholar]
- Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrington T.P., Johnson G.L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Ge C., Che L., Ren J., Pandita R.K., Lu J., Li K., Pandita T.K., Du C. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc. Natl. Acad. Sci. USA. 2015;112:E1210–E1219. doi: 10.1073/pnas.1418335112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C., Che L., Du C. The UBC domain is required for bruce to promote BRIT1/MCPH1 function in DSB signaling and repair post formation of BRUCE-USP8-BRIT1 complex. PLoS ONE. 2015;10:e0144957. doi: 10.1371/journal.pone.0144957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter T., Wepf A., Aebersold R., Gstaiger M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol. 2009;5:237. doi: 10.1038/msb.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh K.-I., Choi I.-G. Exploring the human diseasome: the human disease network. Brief. Funct. Genomics. 2012;11:533–542. doi: 10.1093/bfgp/els032. [DOI] [PubMed] [Google Scholar]
- Gstaiger M., Aebersold R. Genotype-phenotype relationships in light of a modular protein interaction landscape. Mol. Biosyst. 2013;9:1064–1067. doi: 10.1039/c3mb25583b. [DOI] [PubMed] [Google Scholar]
- Guo X., Wang X., Wang Z., Banerjee S., Yang J., Huang L., Dixon J.E. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 2016;18:202–212. doi: 10.1038/ncb3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A., Scott A.F., Amberger J.S., Bocchini C.A., McKusick V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein M.Y., Hubner N.C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I.A., Weisswange I., Mansfeld J., Buchholz F. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Heusel M., Bludau I., Rosenberger G., Hafen R., Frank M., Banaei-Esfahani A., van Drogen A., Collins B.C., Gstaiger M., Aebersold R. Complex-centric proteome profiling by SEC-SWATH-MS. Mol. Syst. Biol. 2019;15:e8438. doi: 10.15252/msb.20188438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff S., Halbritter J., Epting D., Frank V., Nguyen T.-M.T., van Reeuwijk J., Boehlke C., Schell C., Yasunaga T., Helmstädter M. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013;45:951–956. doi: 10.1038/ng.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E.L., Ting L., Bruckner R.J., Gebreab F., Gygi M.P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E.L., Bruckner R.J., Paulo J.A., Cannon J.R., Ting L., Baltier K., Colby G., Gebreab F., Gygi M.P., Parzen H. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545:505–509. doi: 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhar L., Adamson B., Ciccia A., Lewis J., Pontano-Vaites L., Leng Y., Liang A.C., Westbrook T.F., Harper J.W., Elledge S.J. A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep. 2015;11:1486–1500. doi: 10.1016/j.celrep.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jae L.T., Raaben M., Riemersma M., van Beusekom E., Blomen V.A., Velds A., Kerkhoven R.M., Carette J.E., Topaloglu H., Meinecke P. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science. 2013;340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappes F., Damoc C., Knippers R., Przybylski M., Pinna L.A., Gruss C. Phosphorylation by protein kinase CK2 changes the DNA binding properties of the human chromatin protein DEK. Mol. Cell. Biol. 2004;24:6011–6020. doi: 10.1128/MCB.24.13.6011-6020.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K., Bailis W., Dedhia P.H., Vega M.E., Shestova O., Xu L., Toscano K., Uljon S.N., Blacklow S.C., Pear W.S. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood. 2010;116:4948–4957. doi: 10.1182/blood-2009-10-247361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J.D.R., Choi H., Gupta G.D., Pelletier L., Raught B., Nesvizhskii A.I., Gingras A.-C. ProHits-viz: a suite of web tools for visualizing interaction proteomics data. Nat. Methods. 2017;14:645–646. doi: 10.1038/nmeth.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl M., Wiese S., Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol. Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- Komorek J., Kuppuswamy M., Subramanian T., Vijayalingam S., Lomonosova E., Zhao L.-J., Mymryk J.S., Schmitt K., Chinnadurai G. Adenovirus type 5 E1A and E6 proteins of low-risk cutaneous beta-human papillomaviruses suppress cell transformation through interaction with FOXK1/K2 transcription factors. J. Virol. 2010;84:2719–2731. doi: 10.1128/JVI.02119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyar M., Pastrello C., Sheahan N., Jurisica I. Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res. 2016;44(D1):D536–D541. doi: 10.1093/nar/gkv1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry P., Torkamani A., Schork N.J., Hegele R.A. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010;11:60–74. doi: 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- Lambert J.-P., Tucholska M., Go C., Knight J.D.R., Gingras A.-C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteomics. 2015;118:81–94. doi: 10.1016/j.jprot.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Bengtson M.H., Ulbrich A. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Sun Q., Wang X. PLK1, a potential target for cancer therapy. Transl. Oncol. 2017;10:22–32. doi: 10.1016/j.tranon.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- McCormick J.A., Ellison D.H. The WNKs: atypical protein kinases with pleiotropic actions. Physiol. Rev. 2011;91:177–219. doi: 10.1152/physrev.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva Y.A., Lennartsson A., Ehsani R., Kulakovskiy I.V., Vorontsov I.E., Panahandeh P., Khimulya G., Kasukawa T., Drabløs F., FANTOM Consortium EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database (Oxford) 2015;2015:bav067. doi: 10.1093/database/bav067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.-P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.L., Jensen L.J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S.A., Bordeaux J., Sicheritz-Ponten T. Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 2008;1:ra2. doi: 10.1126/scisignal.1159433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris D.W., Pearson R.D., Cormican P., Kenny E.M., O’Dushlaine C.T., Perreault L.-P.L., Giannoulatou E., Tropea D., Maher B.S., Wormley B., International Schizophrenia Consortium, SGENE+ Consortium. Wellcome Trust Case Control Consortium 2 An inherited duplication at the gene p21 Protein-Activated Kinase 7 (PAK7) is a risk factor for psychosis. Hum. Mol. Genet. 2014;23:3316–3326. doi: 10.1093/hmg/ddu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman S.M., Luna-Vargas M.P., Velds A. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Ogawa M., Sawaguchi S., Furukawa K., Okajima T. N-acetylglucosamine modification in the lumen of the endoplasmic reticulum. Biochim. Biophys. Acta. 2015;1850:1319–1324. doi: 10.1016/j.bbagen.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Padilla-Benavides T., Nasipak B.T., Paskavitz A.L., Haokip D.T., Schnabl J.M., Nickerson J.A., Imbalzano A.N. Casein kinase 2-mediated phosphorylation of Brahma-related gene 1 controls myoblast proliferation and contributes to SWI/SNF complex composition. J. Biol. Chem. 2017;292:18592–18607. doi: 10.1074/jbc.M117.799676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M., Lénárt P., Peters J.-M. Polo on the rise-from mitotic entry to cytokinesis with Plk1. Dev. Cell. 2008;14:646–659. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Piñero J., Queralt-Rosinach N., Bravo À., Deu-Pons J., Bauer-Mehren A., Baron M., Sanz F., Furlong L.I. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A.K., Chen J.-X., Selbach M., Pelkmans L. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature. 2018;559:211–216. doi: 10.1038/s41586-018-0279-8. [DOI] [PubMed] [Google Scholar]
- Ringeard M., Marchand V., Decroly E., Motorin Y., Bennasser Y. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature. 2019;565:500–504. doi: 10.1038/s41586-018-0841-4. [DOI] [PubMed] [Google Scholar]
- Ruepp A., Brauner B., Dunger-Kaltenbach I., Frishman G., Montrone C., Stransky M., Waegele B., Schmidt T., Doudieu O.N., Stümpflen V., Mewes H.W. CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008;36:D646–D650. doi: 10.1093/nar/gkm936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A., Waegele B., Lechner M., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Mewes H.W. CORUM: the comprehensive resource of mammalian protein complexes--2009. Nucleic Acids Res. 2010;38:D497–D501. doi: 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco F., Perfetto L., Castagnoli L., Cesareni G. The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012;586:2732–2739. doi: 10.1016/j.febslet.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santio N.M., Koskinen P.J. PIM kinases: From survival factors to regulators of cell motility. Int J Biochem Cell Biol. 2017;93:74–85. doi: 10.1016/j.biocel.2017.10.016. [DOI] [PubMed] [Google Scholar]
- Shamseldin H.E., Yakulov T.A., Hashem A., Walz G., Alkuraya F.S. ANKS3 is mutated in a family with autosomal recessive laterality defect. Hum. Genet. 2016;135:1233–1239. doi: 10.1007/s00439-016-1712-4. [DOI] [PubMed] [Google Scholar]
- Shi Y., Han G., Wu H., Ye K., Tian Z., Wang J., Shi H., Ye M., Zou H., Huo K. Casein kinase 2 interacts with human mitogen- and stress-activated protein kinase MSK1 and phosphorylates it at multiple sites. BMB Rep. 2009;42:840–845. doi: 10.5483/bmbrep.2009.42.12.840. [DOI] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A., Compe E., Le May N., Egly J.-M. TFIIH subunit alterations causing xeroderma pigmentosum and trichothiodystrophy specifically disturb several steps during transcription. Am. J. Hum. Genet. 2015;96:194–207. doi: 10.1016/j.ajhg.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So J., Pasculescu A., Dai A.Y., Williton K., James A., Nguyen V., Creixell P., Schoof E.M., Sinclair J., Barrios-Rodiles M. Integrative analysis of kinase networks in TRAIL-induced apoptosis provides a source of potential targets for combination therapy. Sci. Signal. 2015;8:rs3. doi: 10.1126/scisignal.2005700. [DOI] [PubMed] [Google Scholar]
- Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M., Krykbaeva I., Koeva M., Kayatekin C., Westover K.D., Karras G.I., Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira N., Nihira K., Yamaguchi T., Miki Y., Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell. 2007;25:725–738. doi: 10.1016/j.molcel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Tan C.S.H., Bodenmiller B., Pasculescu A., Jovanovic M., Hengartner M.O., Jørgensen C., Bader G.D., Aebersold R., Pawson T., Linding R. Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci. Signal. 2009;2:ra39. doi: 10.1126/scisignal.2000316. ra39. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M., Björklund M., Cheng F., Syvänen H., Kivioja T., Kilpinen S., Sun Z., Kallioniemi O., Stunnenberg H.G., He W.-W. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell. 2008;133:537–548. doi: 10.1016/j.cell.2008.02.047. [DOI] [PubMed] [Google Scholar]
- Varjosalo M., Keskitalo S., Van Drogen A., Nurkkala H., Vichalkovski A., Aebersold R., Gstaiger M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3:1306–1320. doi: 10.1016/j.celrep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Verba K.A., Wang R.Y.-R., Arakawa A., Liu Y., Shirouzu M., Yokoyama S., Agard D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science. 2016;352:1542–1547. doi: 10.1126/science.aaf5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viau A., Bienaimé F., Lukas K., Todkar A.P., Knoll M., Yakulov T.A., Hofherr A., Kretz O., Helmstädter M., Reichardt W. Cilia-localized LKB1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney. EMBO J. 2018;37:e98615. doi: 10.15252/embj.201798615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohhodina J., Barros E.M., Savage A.L., Liberante F.G., Manti L., Bankhead P., Cosgrove N., Madden A.F., Harkin D.P., Savage K.I. The RNA processing factors THRAP3 and BCLAF1 promote the DNA damage response through selective mRNA splicing and nuclear export. Nucleic Acids Res. 2017;45:12816–12833. doi: 10.1093/nar/gkx1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Renesse A., Petkova M.V., Lützkendorf S., Heinemeyer J., Gill E., Hübner C., von Moers A., Stenzel W., Schuelke, M POMK mutation in a family with congenital muscular dystrophy with merosin deficiency, hypomyelination, mild hearing deficit and intellectual disability. J. Med. Genet. 2014;51:275–282. doi: 10.1136/jmedgenet-2013-102236. [DOI] [PubMed] [Google Scholar]
- Wagih O., Sugiyama N., Ishihama Y., Beltrao P. Uncovering phosphorylation-based specificities through functional interaction networks. Mol. Cell. Proteomics. 2016;15:236–245. doi: 10.1074/mcp.M115.052357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Gray N.S. SnapShot. Mol. Cell. 2015;58:708.e1. doi: 10.1016/j.molcel.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Wang J., Gray N.S. SnapShot: kinase inhibitors II. Mol. Cell. 2015;58:710.e1. doi: 10.1016/j.molcel.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Wilson L.J., Linley A., Hammond D.E., Hood F.E., Coulson J.M., MacEwan D.J., Ross S.J., Slupsky J.R., Smith P.D., Eyers P.A., Prior I.A. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018;78:15–29. doi: 10.1158/0008-5472.CAN-17-2291. [DOI] [PubMed] [Google Scholar]
- Wingender E., Schoeps T., Dönitz J. TFClass: an expandable hierarchical classification of human transcription factors. Nucleic Acids Res. 2013;41:D165–D170. doi: 10.1093/nar/gks1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise C.A., Chiang L.C., Paznekas W.A., Sharma M., Musy M.M., Ashley J.A., Lovett M., Jabs E.W. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl. Acad. Sci. USA. 1997;94:3110–3115. doi: 10.1073/pnas.94.7.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P., Nielsen T.E., Clausen M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Yang X.-L., Li Q.-R., Ning Z.-B., Zhang Y., Zeng R., Wu J.-R. Identification of complex relationship between protein kinases and substrates during the cell cycle of HeLa cells by phosphoproteomic analysis. Proteomics. 2013;13:1233–1246. doi: 10.1002/pmic.201200357. [DOI] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T., Willer T., Anderson M.E., Venzke D., Whyte T., Muntoni F., Lee H., Nelson S.F., Yu L., Campbell K.P. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science. 2013;341:896–899. doi: 10.1126/science.1239951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J.-Y., Dunham W.H., Hong S.J., Knight J.D.R., Bashkurov M., Chen G.I., Bagci H., Rathod B., MacLeod G., Eng S.W.M. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell. 2018;69:517–532.e11. doi: 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- Yu G., Li F., Qin Y., Bo X., Wu Y., Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26:976–978. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- Zhang H., Zhu Q., Cui J., Wang Y., Chen M.J., Guo X., Tagliabracci V.S., Dixon J.E., Xiao J. Structure and evolution of the Fam20 kinases. Nat. Commun. 2018;9:1218. doi: 10.1038/s41467-018-03615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data generated during this study is available at: https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01469.
Kinase network is accessible via the following website: https://sec-explorer.shinyapps.io/Kinome_interactions/.
The code is available upon request.