

Abstract
Hypertrophic cardiomyopathy (HCM), the most common inherited cardiac disease, is caused by several mostly heterozygous mutations in sarcomeric genes. Hallmarks of HCM are cardiomyocyte and myofibrillar disarray and hypertrophy and fibrosis of the septum and the left ventricle. To date, a pathomechanism common to all mutations remains elusive. We have proposed that contractile imbalance, an unequal force generation of neighboring cardiomyocytes, may contribute to development of HCM hallmarks. At the same calcium concentration, we found substantial differences in force generation between individual cardiomyocytes from HCM patients with mutations in β-MyHC (β-myosin heavy chain). Variability among cardiomyocytes was significantly larger in HCM patients as compared with donor controls. We assume that this heterogeneity in force generation among cardiomyocytes may lead to myocardial disarray and trigger hypertrophy and fibrosis. We provided evidence that burst-like transcription of the MYH7-gene, encoding for β-MyHC, is associated with unequal fractions of mutant per wild-type mRNA from cell to cell (cell-to-cell allelic imbalance). This will presumably lead to unequal fractions of mutant per wild-type protein from cell to cell which may underlie contractile imbalance. In this review, we discuss molecular mechanisms of burst-like transcription with regard to contractile imbalance and disease development in HCM.
Keywords: Hypertrophic cardiomyopathy, Contractile imbalance, Burst-like transcription, Cell-to-cell allelic imbalance
Introduction
Hypertrophic cardiomyopathy (HCM) is characterized by asymmetric hypertrophy of the interventricular septum and the left ventricular wall. At the cellular level, often pronounced disarray of cardiomyocytes and myofibrils and increased fibrosis is found in the myocardium (Maron and Maron 2013). Clinically, the disease is highly heterogeneous, ranging from mild and almost asymptomatic disease courses to severe diastolic dysfunction or heart failure. HCM is the most common cause of sudden cardiac death in young athletes (Maron and Maron 2013). The prevalence of HCM is generally assumed to be 1:500 (Maron et al. 2012); recent publications even estimate a prevalence of 1:200 (Semsarian et al. 2015).
Several different mutations in an increasing number of genes have been associated with HCM. Almost all patients are heterozygous for the respective mutation; the disease is transmitted autosomal dominant (Maron et al. 2012). According to the European Society for Cardiology, 40–60% of the HCM patients encode for mutations that affect sarcomeric proteins (Authors/Task Force et al. 2014). Among these, MYH7, encoding for the β-myosin heavy chain (β-MyHC) and MYBPC3, encoding for the cardiac myosin binding protein C (cMyBP-C) account for more than 80%. Another 10% is covered by TNNT2 and TNNI3, encoding for cardiac troponin T (cTnT) and troponin I (cTnI), respectively (Walsh et al. 2017). HCM is mostly a monogenic disorder; patients usually encode for one causative mutation. However, rare cases of patients encoding for two distinct mutations in the same gene on different alleles (compound heterozygous) or two mutations in different genes (double heterozygous) have been reported. These patients often exhibit a much more severe course of disease (Richard et al. 2003; Van Driest et al. 2004). In addition, the emergence of high throughput sequencing methods enables the comprehensive analysis of single nucleotide variants (SNV). This led to the detection of SNVs that are frequent in the healthy population but may trigger susceptibility to HCM if patients cumulate two or more of these SNVs (Burns et al. 2017; Thomson et al. 2019).
To date, no common mechanism has been determined to explain how different mutations in different genes can lead to the same phenotype of HCM. Patients with missense mutations, the most common type of HCM mutations (Maron et al. 2012), express both mutant and wild-type mRNA and protein. According to the poison peptide hypothesis, HCM mutations alter the physiological function of the respective proteins, thereby affecting the force generating mechanism in cardiomyocytes (Bonne et al. 1998). Depending on the affected gene and on the localization within the gene, mutations can alter calcium sensitivity, isometric force levels, acto-myosin ATPase activity, shortening velocity, relaxation properties, and/or cross-bridge cycle kinetics (reviewed in (Ashrafian et al. 2011; Marian and Braunwald 2017; Moore et al. 2012)). Interestingly, alterations can be highly divergent. MYH7-mutations R719W and R723G e.g. decrease calcium sensitivity (Kirschner et al. 2005) whereas MYH7-mutation I736T (Kirschner et al. 2005) or TnT-mutation R92Q (Robinson et al. 2002) increase calcium sensitivity. ATPase rates can either be increased (MYH7-mutation R719W (Seebohm et al. 2009)) or reduced (MYH7-mutation R453C (Bloemink et al. 2014; Sommese et al. 2013)). This exemplary listing may depict the complexity of HCM mutation effects on sarcomeric function. In addition, the poison peptide mechanism likely does not apply to mutations that lead to a truncated protein, as many mutations in MYBPC3 do. In most patients, the truncated isoform is not incorporated into sarcomeres, the total level of functional cMyBP-C is reduced in cardiomyocytes and originates from the wild-type allele only (haploinsufficiency) (Vignier et al. 2009). The cMyBP-C is a structural protein that interacts with the thick and the thin filament and seems to act as an inhibitor of cross-bridge interactions (van Dijk et al. 2012). Reduced levels of cMyBP-C in HCM patients with haploinsufficiency presumably lead to alterations in force generation, especially altered cross-bridge cycle kinetics (reviewed in (Marian and Braunwald 2017; Schlossarek et al. 2011)). Taken together, a common mechanism of HCM development caused by different mutations remains elusive.
Evidence from our own previous work, however, led us to suggest a concept for a potential common mechanism that contributes to HCM development for heterozygous mutations which alter parameters of cardiomyocyte force generation. When we analyzed the effects of MYH7 mutations in single M. soleus fibers, we detected a large variability in calcium-dependent force generation among individual fibers from the HCM patients, respectively (Kirschner et al. 2005). The variability was substantially larger than between fibers from control individuals. We also observed a similarly large variability among individual cardiomyocytes from HCM patients with different mutations in the MYH7 gene, respectively (Montag et al. 2018). We hypothesized that different fractions of mutated and wild-type protein from cell to cell might be the reason for the observed highly variable function of individual muscle cells (Brenner et al. 2014; Kirschner et al. 2005; Kraft et al. 2016; Montag et al. 2018). A mosaic of stronger and weaker cells may thus lead to contractile imbalance between individual cardiomyocytes (Brenner et al. 2014; Kraft et al. 2016; Montag et al. 2018). This hypothesis was supported by the finding that fractions of mutant and wild-type mRNA varied substantially among individual HCM cardiomyocytes from the same cardiac tissue which had been used in functional studies (Kraft et al. 2016; Montag et al. 2018). As underlying mechanism that may lead to the observed unequal allelic expression of MYH7, we identified stochastic, burst-like expression which is independent for mutant and wild-type alleles (Montag et al. 2018). The resulting contractile imbalance may disrupt the functional syncytium of the myocardium and contribute to development of myocardial disarray, hypertrophy, and fibrosis. In this review, we aim to further elucidate the mechanisms that underlie contractile imbalance and how it may affect disease development in HCM.
Stochastic gene expression from cell to cell
Several years ago, researchers detected in bacteria and yeast that cells, which originate from the same genetic background, express divergent levels of specific proteins. In a culture of clonal cells, some cells expressed low levels, some expressed high levels, and some expressed medium levels of the identical protein. This evoked a phenotypic variability between individual cells (Blake et al. 2003; Elowitz et al. 2002) that resulted in a mixture of cells with different functional properties. Interestingly, over time, protein expression in individual cells changed and likewise did the functional activity (Cai et al. 2006). Such a phenotypic heterogeneity was subsequently also shown in cultured mammalian cells (Lo et al. 2015; Sigal et al. 2006) and cancer cell lines (Roumeliotis et al. 2017).
The variable protein expression from cell to cell and over time was attributed to the so-called burst-like transcription. This model of gene expression assumes that transcription occurs in stochastic pulses (Blake et al. 2003; Elowitz et al. 2002). Transcription of a gene starts, pauses, and restarts again in a stochastic manner over time (Fig. 1). Burst-like transcription has been determined in bacteria, yeast, and in mammalian cells, including cells within tissues (Blake et al. 2003; Elowitz et al. 2002; Montag et al. 2018; Raj et al. 2006; Raj and van Oudenaarden 2008). In a tissue, this means that one cell actively transcribes a respective gene while in a neighboring cell transcription can pause at the same time (Fig. 1). Even though accumulation of mRNA from recent bursts within the cells will partly equalize total mRNA and protein levels from cell to cell, a distinct variability between cells has been reported to remain (Blake et al. 2003; Cai et al. 2006; Elowitz et al. 2002; Symmons et al. 2019). To minimize functional differences between cells, the stochasticity of on and off switches appears low for structural proteins to gain comparable levels of proteins from cell to cell and allow for concerted actions of the tissue (Rajapakse and Smale 2017; Sun and Zhang 2020). In contrast, it seems higher for secreted proteins which do not only affect a single cell (Sun and Zhang 2020).
Fig. 1.
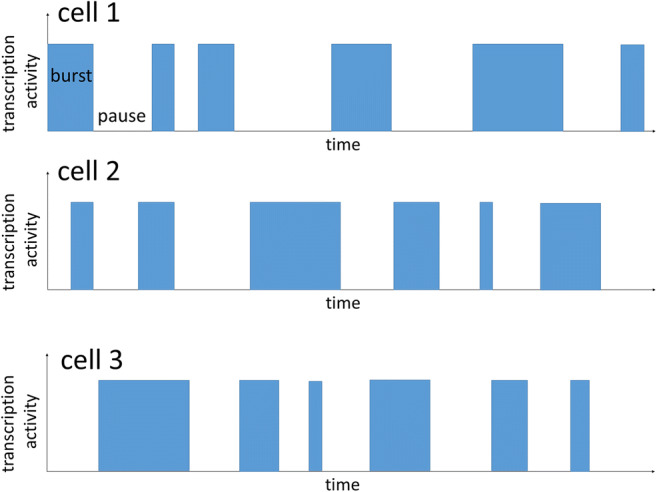
The transcriptional activity of three individual cells is schematically depicted. A burst of transcription is indicated by a blue bar, transcriptional pauses are indicated by the absence of bars. At a given time point, some cells are in a bursting phase whereas in other cells transcription is pausing. This will lead to different mRNA and protein amounts in the cells, which may cause phenotypic variability between the individual cells
Determinants and regulators of burst-like transcription
The mechanisms that underlie gene expression and burst-like transcription shall be discussed here in more detail. Permanent inactivation of specific genes or alleles is exerted by DNA methylation at CpG islands (CG-rich regions in promoters and in coding regions of genes). This epigenetic mechanism controls e.g. tissue specific inactivation of specific genes. DNA methyltransferases add methyl groups to cytosines within CpG islands. These methyl groups interfere with transcription factor binding to the DNA and thereby inhibit transcription. In addition, methylcytosine binding proteins (MBP) recruit histone-deacetylases and other co-repressors, finally increasing the chromosomal density and thereby forming transcriptionally inactive chromosomal structures (reviewed in (Attwood et al. 2002)). These structures do not take part in transcriptional bursting.
Adjustable regulation of gene expression shapes the bursts of transcription and occurs via alterations in the occupancy of chromosomes with nucleosomes, also called nucleosomal density. Nucleosomes are defined as chromosomal DNA which is wrapped around a histone octamer (Kornberg 1974). Genes with a high nucleosomal density are associated with numerous histones especially at the promoter sites and show lower transcription rates. In contrast, promoters of active genes show a lower nucleosomal density (reviewed in (Nicolas et al. 2017)). Key regulators of nucleosome density are histone modifications. Specific residues of histones can be methylated, acetylated, phosphorylated, and/or ubiquitinated (for a detailed review about modifications see (Bannister and Kouzarides 2011)). In brief, most kinds of acetylation, ubiquitination, and phosphorylation lead to reduced nucleosomal density and thereby to a higher transcriptional activity (Bannister and Kouzarides 2011). Histone methylation mainly increases the nucleosomal density, reducing transcription activity (Bannister and Kouzarides 2011).
Genes or alleles with a low nucleosomal density are accessible for transcription factors (TF) and RNA polymerase II (RNA-Pol II) that enable transcription (Li et al. 2007). Binding and release of TFs and RNA-Pol II occurs stochastically (Larsson et al. 2019; Urban and Johnston Jr. 2018), based on the thermodynamics of DNA and TF/RNA-Pol II interaction in the nuclear surrounding (Blake et al. 2006). The probability of TF and RNA-Pol II-binding increases with a decreasing nucleosomal density. In line with this, activated genes with a lower nucleosomal density show a higher burst frequency and burst size (duration) (Brown et al. 2013; Hornung et al. 2012b).
Based on this background, generation of transcriptional bursts can be described in a simplified way: to activate a gene, the nucleosomal density on the promoter is reduced. Random binding events of TFs and RNA-Pol II to the DNA lead to bursts of transcription; stochastic unbinding terminates the burst; rebinding will start another burst.
Burst-like transcription can be governed at different levels. We can distinguish between control of basal burst rates for specific genes and flexible modification of burst size and frequency to regulate gene expression. Basal burst rates seem to be determined mainly by regulatory sequences. Specific gene elements such as TATA boxes in the promoter recruit TFs and RNA polymerases. Genes, which encode for TATA boxes, show higher burst frequencies (Blake et al. 2006; Hornung et al. 2012a) and burst frequency and size depend on specific TATA box sequences (Hornung et al 2012a). Additionally, RNA-Pol II was shown to pause during transcription of a particular mRNA molecule and to continue after a certain time. This pausing and restarting was shown to underlie transcriptional bursts (Dobrzynski and Bruggeman 2009). It seems to be stochastic and can presumably be influenced by specific DNA sequences to affect burst size and frequency (Fujita et al. 2016). Flexible modifiers of bursts adjust gene expression to specific circumstances. Upregulated genes show e.g. decreasing nucleosomal density which will lead to a higher accessibility for TF and RNA polymerase and thereby a higher burst frequency and size (Kalo et al. 2015). The nucleosome and deacetylation remodeling complex (NuDR) was shown to fine tune gene expression by controlling the accessibility of TF and RNA-Pol II to the DNA (Bornelov et al. 2018). In addition, the availability of TFs and RNA-Pol II will shape the bursts. Higher levels of TFs may cause a higher burst frequency, as shown for cFos (Senecal et al. 2014). Moreover, binding probabilities of TFs on the DNA and thereby the bursts can be affected by trans-acting factors that modify the affinity of the TF for a particular promoter (Boettiger 2013).
Assays to determine burst-like transcription
Different methods can be used to determine burst-like transcription. In fixed cells or tissues, single-molecule RNA fluorescence in situ hybridization (smRNA-FISH) can visualize actively transcribed alleles (Levesque et al. 2013). Probe sets that can bind to the pre-mRNA at the chromosomal locus and to the nascent mRNA are hybridized in fixed cells or tissue. Co-localization of the probe sets in the nucleus indicates an active transcription site (aTS), an actively transcribed allele (Levesque et al. 2013). Burst-like transcription, which is independent for the two alleles, is indicated by the occurrence of cells without, cells with one, and cells with two aTS in the same population. This method was used to determine active transcription sites in cultured cells (Levesque et al. 2013) and tissue sections from mouse kidney (Symmons et al. 2019). Our group provided evidence for burst-like transcription of the MYH7 gene in cardiac tissue from HCM patients and non-transplanted donor hearts (Montag et al. 2018). Preliminary data indicate that MYBPC3 and TNNI3 are also expressed burst-like (Montag et al. 2019).
In living cells, fluorescently tagged mRNAs can be used to examine kinetics of burst-like transcription over time. Here, specific stem loop sequences are inserted by genome editing to the 3′- or 5′-end of the mRNA of interest. Fluorescently labeled bacterial proteins that can bind to the specific stem loop sequences are co-expressed in these cells and fluorescent signals indicate whether the respective RNA is transcribed. Live cell imaging then directly visualizes transcription of the mRNA molecules in the nuclei and the stochastic on and off switch of the gene of interest (Darzacq et al. 2007; Yunger et al. 2010).
The activity of a respective gene is most likely correlated with the number of aTS per nucleus. In highly active genes, the bursts will occur more frequently and have a longer duration (Dar et al. 2012). This will result in a higher percentage of cells that contain one or two aTS. In contrast, genes with a low activity will show high percentage of cells without aTS and more cells with only one aTS (Fig. 2a). This will also apply to polyploid cells that contain more than 2n of chromosome sets. In two independent studies where the ploidy of healthy adult human cardiomyocytes was determined, 19 or 22% of the nuclei were diploid, 45 or 60% tetraploid, 2% hexaploid, 15% or 23% octoploid, 1 or 11% 16-ploid, and 0 or 1% were 32-ploid (Herget et al. 1997; Montag et al. 2018). Increases in ploidy are found in cardiac diseases such as myocardial infarction and HCM (Herget et al. 1997; Montag et al. 2018; Shliakhto et al. 2007; Sukhacheva et al. 2015). In line with this, we have determined nuclei with more than two MYH7-aTS for both donor and HCM patients (Montag et al. 2018). Genes with a low activity will show mostly nuclei without aTS and few nuclei with more than two aTS. In contrast, most nuclei from highly active genes will show more than two aTS. For cardiomyocytes, where most cells are tetraploid, we expect the maximum at 4 aTS per nucleus (Fig. 2b).
Fig. 2.

Burst-like transcription of alleles leads to stochastic distribution of active transcription sites (aTS) per nucleus. During phases with high activity, the probability for each allele to be active increases; thus, more nuclei will show more aTS. The graphs depict estimated fractions of aTS per nuclei in highly and lowly expressed genes. a In diploid cells, genes with a low activity will provide mostly nuclei without aTS and in genes with high activity most nuclei will show two aTS. b In polyploidy cells, low activity genes will again show mostly nuclei without aTS. Highly active genes in polyploid cells will also show a higher number of nuclei with more than one aTS. According to our results and published data on the ploidy of cardiomyocytes (Herget et al. 1997; Montag et al. 2018), we assume that most cells are tetraploid; therefore, the maximum of nuclei will show four aTS
Burst-like transcription leads to allelic imbalance from cell to cell
So far, we have discussed burst-like transcription at the gene level. Importantly, the described mechanisms that underlie burst-like transcription also lead to a stochastic expression of the alleles over time, meaning each allele is switched on and off independently from the other one. Therefore, in cells from the same population, at a given time point, either allele A alone can be active, allele B alone can be active, both alleles can be active, or no allele can be active (Fig. 3a). This induces an allelic imbalance from cell to cell (Fig. 3b); in the same population, some cells contain high fractions of mRNA from allele A, some contain high fractions of mRNA from allele B, and others contain mRNA from both alleles in different ratios (Montag et al. 2018; Raser and O’Shea 2005; Symmons et al. 2019).
Fig. 3.

Burst-like transcription of MYH7 leads to allelic imbalance from cell to cell. a Schematic of cardiomyocytes within cardiac tissue with active transcription sites (aTS) for allele A (wild-type; light gray star) and allele B (mutant; dark gray star). The tissue contains cells that at the given time point transcribe both alleles, no allele, only allele A, and only allele B. b Accumulation of mRNA molecules from bursting of the two alleles leads to different ratios of wild-type vs. mutant mRNA from cell to cell, ranging from cells with mainly wild-type allele A (light gray waves), mainly mutant allele B (dark gray waves), and mixtures of both alleles. c Using smRNA-FISH, we visualized active transcription sites (aTS) in nuclei from an HCM patient with the MYH7 mutation R723G. We found nuclei without aTS (top), cells with one aTS (middle), and cells with more than one aTS (here, two aTS, bottom). This indicates burst-like, independent transcription of the two MYH7 alleles. d In individual cardiomyocytes isolated by laser-microdissection from cryosections of myocardial samples from the same patient, we detected highly variable fractions of mutant MYH7 mRNA from cell to cell as revealed by single-cell RT-PCR and allele-specific restriction analysis (top). Densitometric quantification of mutant and wild-type mRNA fractions showed substantial allelic imbalance from cell to cell, with cells that express essentially only mutant mRNA, cells that express almost only wild-type mRNA, and cells with mRNA from both alleles in different ratios (bottom). Figures from c and d are reprinted and modified from (Montag et al. 2018), with permission from Frontiers
Initial studies termed this phenomenon random monoallelic expression. They could show that the expression of the alleles can differ between the cells from the same population (Reinius and Sandberg 2015). However, in contrast to the association that may be evoked by the term “monoallelic,” the authors also detected cells, which expressed both the maternal and the paternal allele. This detection of mono- and biallelic mRNA expression led to the assumption of a dynamic on and off switch, a stochastic choice of the two alleles (Deng et al. 2014; Levesque et al. 2013; Reinius and Sandberg 2015). With the emergence of single-cell RNA sequencing techniques, the unequal allelic expression from cell to cell is determined in more and more different cell types (Borel et al. 2015; Deng et al. 2014; Sun and Zhang 2020).
Importantly, stochastic on- and off-switching of the alleles over time may lead to accumulation of mRNA from previous bursts depending on the particular mRNA turnover rates. Thus, cells without aTS may contain mRNA molecules of this gene and cells with only allele A being active still may contain mRNA molecules from allele B. In addition, the allelic expression within one cell will change over time. Therefore, a cell with mainly mRNA from allele B can express high levels of mRNA from allele A at another time point. The mechanisms described for regulation of bursts will also affect unequal allelic mRNA distribution from cell to cell. Higher burst rates will lead to a higher activation rate of both alleles and may thus reduce the allele-specific variability between individual cells from one population (Kalo et al. 2015).
For the MYH7 gene, we have recently provided evidence that burst-like transcription of mutant and wild-type alleles could lead to allelic imbalance from cell to cell. In cardiomyocytes from HCM patients with different MYH7 mutations, we observed burst-like transcription of the two MYH7 alleles by smRNA-FISH (Fig. 3c) and highly variable fractions of mutant and wild-type mRNA from cell to cell (Fig. 3d) (Montag et al. 2018). Model calculations were used to test our hypothesis that burst-like transcription underlies the observed allelic imbalance from cell to cell (Montag et al. 2018). Indeed, using the fraction of cardiomyocytes with active transcription sites from our experiments and published MYH7-mRNA turnover rates as an input for the model, the calculated variability of allelic expression of mutant and wild-type β-MyHC among individual cardiomyocytes was comparable to our experimental data. Thus, we assume that the MYH7 transcriptional bursts underlie the observed unequal allelic expression from cell to cell (Montag et al. 2018). In line with our research, a recent study shows burst-like transcription of maternal and paternal alleles for different genes in mouse kidney and relates this to cell-to-cell allelic imbalance of these genes (Symmons et al. 2019).
Translation of unequal allelic mRNA fractions from cell to cell into unequal fractions of mutated and wild-type protein
To date, quantification of mutant and wild-type β-MyHC at the single-cell level is not possible due to technical limitations. Model calculations showed comparable variable fractions of mutant and wild-type alleles from cell to cell at mRNA and protein level (Montag et al. 2018). The assumption that variable fractions of mutated mRNA from cell to cell correspond to a similar variability also at the protein level is strongly supported by the large functional variability among individual HCM cardiomyocytes (Kraft et al. 2016; Montag et al. 2018). Additional evidence for variable allele-specific protein distribution from cell to cell is provided by independent studies that showed a patchy distribution of wild-type cMyBP-C protein from cell to cell and even within individual cardiomyocytes of HCM patients with cMyBP-C truncation mutations (Aldag-Niebling et al. 2018; Kraft and Montag 2019; Parbhudayal et al. 2018; Theis et al. 2009). The truncations lead to haploinsufficiency; thus, the remaining protein in the sarcomeres originates just from the wild-type allele. Only periods with bursts from the wild-type allele will therefore lead to (overall reduced) expression of functional protein, which may underlie the patchy distribution of cMyBP-C from cell to cell. In addition, a recent publication shows that MYBPC3-mRNA is transported to the Z-disc where it is translated and incorporated into the sarcomeres (Lewis et al. 2018). If the mRNA from a burst of one allele is transported to a certain area of a cell, this may lead to the observed uneven distribution of wild-type cMyBP-C within individual cardiomyocytes (Aldag-Niebling et al. 2018; Kraft and Montag 2019; Theis et al. 2009). Taken together, model calculations as well as uneven distribution of wild-type cMyBP-C and functional variability from cell to cell strongly suggest that allelic mRNA-imbalance translates into imbalance of wild-type and mutated protein from cell to cell.
Consequences of allelic imbalance from cell to cell: the contractile imbalance hypothesis and HCM pathogenesis
In heterozygous individuals where mutant and wild-type alleles encode for proteins with different functional properties, it will affect the function of the cell from which allele the protein is translated at a specific time point. For mutations in secreted proteins, the impact on each individual cell may be small since all cells in the vicinity will be exposed to a comparable mixture of wild-type and mutated proteins (Sun and Zhang 2020). In HCM patients with mutations that alter force generation as shown for MYH7 mutations (Bloemink et al. 2014; Kirschner et al. 2005; Seebohm et al. 2009; Sommese et al. 2013), different fractions of mutant and wild-type sarcomeric proteins from cell to cell may lead to different phenotypes of individual cells. Such a phenotypic heterogeneity could increase disease severity in addition to primary functional effects of the mutation itself (Deng et al. 2014).
To characterize functional effects of HCM mutations in β-MyHC, we analyzed calcium-dependent force generation in isolated, permeabilized single cardiomyocytes from HCM patients with mutations A200V and R723G compared with donor controls (Kraft et al. 2016; Montag et al. 2018). Mutation A200V is located in the myosin head domain, close to the nucleotide-binding pocket. Mutations in this region are assumed to affect the ATPase activity of β-MyHC (Colegrave and Peckham 2014). Mutation R723G is located in the converter region of β-MyHC. The mutation leads to an increased stiffness of the converter and thereby increased force generation (Seebohm et al. 2009). Taking together all analyzed cells, respectively, both mutations lead to a right shift of the force-pCa-curve, indicating calcium desensitization. However, when looking at the individual force-pCa curves of the HCM cardiomyocytes for each mutation, we found a rather large variability in calcium sensitivity and thus in force generation at physiological calcium concentrations (Fig. 4). This variability was much larger in HCM cardiomyocytes than in donor cells. Whereas some HCM cardiomyocytes showed relative force generation comparable to donor cells, other cardiomyocytes from the very same patient showed a 10–20-fold reduced force at the identical calcium concentration (Fig. 4). We named this functional variability among individual cardiomyocytes contractile imbalance. Preliminary data suggest that a missense mutation in cTnI and a truncation mutation in cMyBP-C also lead to contractile imbalance (Montag et al. 2019).
Fig. 4.

Contractile imbalance in HCM patients with mutations in β-MyHC. Single permeabilized cardiomyocytes, isolated from heart tissue of HCM patients (red) with the mutation A200V (left) or R723G (right), respectively, and from donor myocardium (blue) as controls were adjusted for phosphorylation levels. Cardiomyocytes were subjected to different calcium concentrations to measure isometric force generation. a Forces of each individual cardiomyocyte were normalized to maximum force and plotted against the respective calcium concentration (force-pCa-relations). Each symbol and curve represents a different individual cell. b Relative forces at physiological calcium concentrations during twitch (indicated by boxes in a) were plotted for individual cardiomyocytes from patients and controls. Whereas for controls, force generation varied no more than twofold, the patient’s cardiomyocytes showed a much larger variability of 10 to 20-fold. Data were published in Montag et al. (2018); a is reprinted and modified from (Montag et al. 2018), with permission from Frontiers
Implications and limits of the contractile imbalance hypothesis
The finding that individual cardiomyocytes generate different forces at the same calcium concentration may have a severe impact on the physiological function of the myocardium. Our results suggest that the coordinated contraction of neighboring cardiomyocytes within the functional cardiac syncytium may be disturbed in the heart of HCM patients. During each twitch, contracting neighboring cardiomyocytes will generate different forces. This will render them unable to act uniformly. Stronger cells in the myocardium may over-contract, whereas weaker cells may be over-stretched, finally disrupting the myocardial network. Over the years while the phenotype develops, contractile imbalance may thereby exacerbate or lead to the typical, HCM-associated myocardial disarray.
As derived from model calculations, burst-like transcription which is independent for the two alleles will lead to varying fractions of mutated and wild-type mRNA and protein within each cell over time. Thus, cardiomyocytes with high fractions of wild-type protein and therefore almost normal function can convert into cells with high fractions of mutant protein (or low levels of functional protein in case of truncation mutations) and highly altered function at another time point. Repeatedly altering function of individual cardiomyocytes may further contribute to disruption of the syncytium and thereby to development of cardiomyocyte and myofibrillar disarray.
Increased stretch in cultured cardiomyocytes induces release of TGF-β, angiotensin II, and endothelin-1 (Ruwhof et al. 2000; van Wamel et al. 2001) and expression of hypertrophy marker genes (van Wamel et al. 2002). In a HCM mouse model, increased strain due to the mutation was associated with increased TGF-β expression and activated pro-fibrotic pathways and hypertrophic remodeling (Teekakirikul et al. 2010). In other mouse models, expression of TGF-β activated fibroblasts and myofibroblasts and thereby induced cardiac fibrosis and hypertrophy (Khalil et al. 2017). Accordingly, we assume that contractile imbalance in HCM patients may lead to myocardial disarray and release of TGF-β, finally triggering hypertrophy and fibrosis (Brenner et al. 2014).
Notably, substantial alteration of sarcomere function and the force generating mechanism by HCM mutations is pathologic for the myocardium per se, as can be seen from very rare homozygous HCM patients where a mutation-induced contractile imbalance cannot be expected. Interestingly, these patients show a different and mostly more severe course of disease compared with their heterozygous relatives (Nishi et al. 1994). In homozygous HCM patients, the disease may be aggravated due to a gene dosage effect, as discussed in more detail in Brenner et al. (2014). Another hypothesis suggests a mechanism which may underlie a non-uniformity between cardiomyocytes in heterozygous and homozygous patients. It assumes that the mutation-induced disturbed protein function may affect the development of the contractile system in early stages of life and induce different degrees of myofibrillar disarray from cell to cell early on (Mansson 2014).
Conclusions
We have shown that the alleles of the MYH7 gene are transcribed in bursts, most likely causing allelic imbalance from cell to cell. The unequal fractions of mutated and wild-type β-MyHC are associated with contractile imbalance among the individual cardiomyocytes; at the same calcium concentration, cardiomyocytes generate highly different forces. This may disrupt the myocardial syncytium and trigger cardiomyocyte disarray, a hallmark of HCM. In addition, the variable forces may induce pro-hypertrophic and pro-fibrotic pathways in HCM patients. We assume that this mechanism may apply for all HCM mutations that alter force generation, irrespective of the kind of alteration or the underlying mechanism.
Acknowledgments
We thank Joachim Meißner for critically reading the manuscript.
Authors’ contributions
JM and TK had the idea for the article, JM performed literature search and drafted the manuscript, and TK critically revised the work.
Funding information
Open Access funding provided by Projekt DEAL. This work was supported by a HiLF-Grant (Hochschulinterne Leistungsförderung) of Hannover Medical School to JM and grants of the Deutsche Forschungsgemeinschaft to TK (KR1187/19-1 and KR1187/22-1).
Availability of data and material (data transparency)
Not applicable.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval (include appropriate approvals or waivers)
Not applicable.
Consent to participate (include appropriate statements)
Not applicable.
Consent for publication (include appropriate statements)
Not applicable.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aldag-Niebling D, Radocaj A, Hilfigker-Kleiner D, Dos Remedios C, Brenner B, Kraft T. Hypertrophic cardiomyopathy: variable expression of myosin-binding protein C from cell-to-cell and functional imbalance among individual cardiomyocytes. Biophys J. 2018;114:312A. doi: 10.1016/j.bpj.2017.11.1762. [DOI] [Google Scholar]
- Ashrafian H, McKenna WJ, Watkins H, Robbins J (2011) Disease Pathways and Novel Therapeutic Targets in Hypertrophic Cardiomyopathy. Circ Res 109:86–96 [DOI] [PubMed]
- Attwood JT, Yung RL, Richardson BC. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci. 2002;59:241–257. doi: 10.1007/s00018-002-8420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Authors/Task Force m et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) Eur Heart J. 2014;35:2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake WJ, KAErn M, Cantor CR, Collins JJ (2003) Noise in eukaryotic gene expression. Nature 422:633–637. 10.1038/nature01546 [DOI] [PubMed]
- Blake WJ, et al. Phenotypic consequences of promoter-mediated transcriptional noise. Mol Cell. 2006;24:853–865. doi: 10.1016/j.molcel.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Bloemink M, Deacon J, Langer S, Vera C, Combs A, Leinwand L, Geeves MA. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac beta-myosin. J Biol Chem. 2014;289:5158–5167. doi: 10.1074/jbc.M113.511204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN. Analytic approaches to stochastic gene expression in multicellular systems. Biophys J. 2013;105:2629–2640. doi: 10.1016/j.bpj.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonne G, Carrier L, Richard P, Hainque B, Schwartz K. Familial hypertrophic cardiomyopathy: from mutations to functional defects. Circ Res. 1998;83:580–593. doi: 10.1161/01.RES.83.6.580. [DOI] [PubMed] [Google Scholar]
- Borel C, et al. Biased allelic expression in human primary fibroblast single cells. Am J Hum Genet. 2015;96:70–80. doi: 10.1016/j.ajhg.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornelov S, et al. The nucleosome remodeling and deacetylation complex modulates chromatin structure at sites of active transcription to fine-tune gene expression. Mol Cell. 2018;71(56-72):e54. doi: 10.1016/j.molcel.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner B, Seebohm B, Tripathi S, Montag J, Kraft T. Familial hypertrophic cardiomyopathy: functional variance among individual cardiomyocytes as a trigger of FHC-phenotype development. Front Physiol. 2014;5:392. doi: 10.3389/fphys.2014.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Mao C, Falkovskaia E, Jurica MS, Boeger H. Linking stochastic fluctuations in chromatin structure and gene expression. PLoS Biol. 2013;11:e1001621. doi: 10.1371/journal.pbio.1001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C, Bagnall RD, Lam L, Semsarian C, Ingles J (2017) Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ Cardiovasc Genet:10. 10.1161/CIRCGENETICS.116.001666 [DOI] [PubMed]
- Cai L, Friedman N, Xie XS. Stochastic protein expression in individual cells at the single molecule level. Nature. 2006;440:358–362. doi: 10.1038/nature04599. [DOI] [PubMed] [Google Scholar]
- Colegrave M, Peckham M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken) 2014;297:1670–1680. doi: 10.1002/ar.22973. [DOI] [PubMed] [Google Scholar]
- Dar RD, et al. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci U S A. 2012;109:17454–17459. doi: 10.1073/pnas.1213530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- Dobrzynski M, Bruggeman FJ. Elongation dynamics shape bursty transcription and translation. Proc Natl Acad Sci U S A. 2009;106:2583–2588. doi: 10.1073/pnas.0803507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Fujita K, Iwaki M, Yanagida T. Transcriptional bursting is intrinsically caused by interplay between RNA polymerases on DNA. Nat Commun. 2016;7:13788. doi: 10.1038/ncomms13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herget GW, Neuburger M, Plagwitz R, Adler CP. DNA content, ploidy level and number of nuclei in the human heart after myocardial infarction. Cardiovasc Res. 1997;36:45–51. doi: 10.1016/s0008-6363(97)00140-5. [DOI] [PubMed] [Google Scholar]
- Hornung G, Bar-Ziv R, Rosin D, Tokuriki N, Tawfik DS, Oren M, Barkai N. Noise-mean relationship in mutated promoters. Genome Res. 2012;22:2409–2417. doi: 10.1101/gr.139378.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung G, Oren M, Barkai N. Nucleosome organization affects the sensitivity of gene expression to promoter mutations. Mol Cell. 2012;46:362–368. doi: 10.1016/j.molcel.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalo A, et al. Cellular levels of signaling factors are sensed by beta-actin alleles to modulate transcriptional pulse intensity. Cell Rep. 2015;13:1284–1285. doi: 10.1016/j.celrep.2015.10.053. [DOI] [PubMed] [Google Scholar]
- Khalil H, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner SE, et al. Hypertrophic cardiomyopathy-related beta-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am J Physiol Heart Circ Physiol. 2005;288:H1242–H1251. doi: 10.1152/ajpheart.00686.2004. [DOI] [PubMed] [Google Scholar]
- Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- Kraft T, Montag J. Altered force generation and cell-to-cell contractile imbalance in hypertrophic cardiomyopathy. Pflugers Arch. 2019;471:719–733. doi: 10.1007/s00424-019-02260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft T, Montag J, Radocaj A, Brenner B. Hypertrophic cardiomyopathy: cell-to-cell imbalance in gene expression and contraction force as trigger for disease phenotype development. Circ Res. 2016;119:992–995. doi: 10.1161/CIRCRESAHA.116.309804. [DOI] [PubMed] [Google Scholar]
- Larsson AJM, et al. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565:251–254. doi: 10.1038/s41586-018-0836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque MJ, Ginart P, Wei Y, Raj A. Visualizing SNVs to quantify allele-specific expression in single cells. Nat Methods. 2013;10:865–867. doi: 10.1038/nmeth.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis YE, Moskovitz A, Mutlak M, Heineke J, Caspi LH, Kehat I. Localization of transcripts, translation, and degradation for spatiotemporal sarcomere maintenance. J Mol Cell Cardiol. 2018;116:16–28. doi: 10.1016/j.yjmcc.2018.01.012. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Lo CA, Kays I, Emran F, Lin TJ, Cvetkovska V, Chen BE. Quantification of protein levels in single living cells. Cell Rep. 2015;13:2634–2644. doi: 10.1016/j.celrep.2015.11.048. [DOI] [PubMed] [Google Scholar]
- Mansson A. Hypothesis and theory: mechanical instabilities and non-uniformities in hereditary sarcomere myopathies. Front Physiol. 2014;5:350. doi: 10.3389/fphys.2014.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:749–770. doi: 10.1161/CIRCRESAHA.117.311059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–715. doi: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- Montag J, et al. Burst-like transcription of mutant and wildtype MYH7-alleles as possible origin of cell-to-cell contractile imbalance in hypertrophic cardiomyopathy. Front Physiol. 2018;9:359. doi: 10.3389/fphys.2018.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag J, et al. Contractile imbalance as trigger for HCM pathogenesis: evidence from mutations in different sarcomeric proteins. European Heart Journal. 2019;40:ehz748.0376. doi: 10.1093/eurheartj/ehz748.0376. [DOI] [Google Scholar]
- Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas D, Phillips NE, Naef F. What shapes eukaryotic transcriptional bursting? Mol BioSyst. 2017;13:1280–1290. doi: 10.1039/c7mb00154a. [DOI] [PubMed] [Google Scholar]
- Nishi H, Kimura A, Harada H, Adachi K, Koga Y, Sasazuki T, Toshima H. Possible gene dose effect of a mutant cardiac beta-myosin heavy chain gene on the clinical expression of familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 1994;200:549–556. doi: 10.1006/bbrc.1994.1483. [DOI] [PubMed] [Google Scholar]
- Parbhudayal RY, et al. Variable cardiac myosin binding protein-C expression in the myofilaments due to MYBPC3 mutations in hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2018;123:59–63. doi: 10.1016/j.yjmcc.2018.08.023. [DOI] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse I, Smale S. Emergence of function from coordinated cells in a tissue. Proc Natl Acad Sci U S A. 2017;114:1462–1467. doi: 10.1073/pnas.1621145114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O’Shea EK. Noise in gene expression: origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B, Sandberg R. Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat Rev Genet. 2015;16:653–664. doi: 10.1038/nrg3888. [DOI] [PubMed] [Google Scholar]
- Richard P, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- Robinson P, et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem. 2002;277:40710–40716. doi: 10.1074/jbc.M203446200. [DOI] [PubMed] [Google Scholar]
- Roumeliotis TI, et al. Genomic determinants of protein abundance variation in colorectal cancer cells. Cell Rep. 2017;20:2201–2214. doi: 10.1016/j.celrep.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwhof C, van Wamel AE, Egas JM, van der Laarse A. Cyclic stretch induces the release of growth promoting factors from cultured neonatal cardiomyocytes and cardiac fibroblasts. Mol Cell Biochem. 2000;208:89–98. doi: 10.1023/A:1007046105745. [DOI] [PubMed] [Google Scholar]
- Schlossarek S, Mearini G, Carrier L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol. 2011;50:613–620. doi: 10.1016/j.yjmcc.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Seebohm B, et al. Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys J. 2009;97:806–824. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. doi: 10.1016/j.jacc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Senecal A, et al. Transcription factors modulate c-Fos transcriptional bursts. Cell Rep. 2014;8:75–83. doi: 10.1016/j.celrep.2014.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shliakhto EV, et al. Cellular aspects of hypertrophic cardiomyopathy pathogenesis: the role of cardiomyocyte polyploidy and activation of the proliferating cell nuclear antigen in the myocardium. Tsitologiia. 2007;49:817–823. [PubMed] [Google Scholar]
- Sigal A, et al. Variability and memory of protein levels in human cells. Nature. 2006;444:643–646. doi: 10.1038/nature05316. [DOI] [PubMed] [Google Scholar]
- Sommese RF, et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc Natl Acad Sci U S A. 2013;110:12607–12612. doi: 10.1073/pnas.1309493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhacheva TV, Chudinovskikh YA, Eremeeva MV, Serov RA. Age-related features of cardiomyocyte ploidy in patients with hypertrophic obstructive cardiomyopathy. Bull Exp Biol Med. 2015;159:95–99. doi: 10.1007/s10517-015-2899-4. [DOI] [PubMed] [Google Scholar]
- Sun M, Zhang J. Allele-specific single-cell RNA sequencing reveals different architectures of intrinsic and extrinsic gene expression noises. Nucleic Acids Res. 2020;48:533–547. doi: 10.1093/nar/gkz1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons O, et al. Allele-specific RNA imaging shows that allelic imbalances can arise in tissues through transcriptional bursting. PLoS Genet. 2019;15:e1007874. doi: 10.1371/journal.pgen.1007874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teekakirikul P, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis JL, et al. Expression patterns of cardiac myofilament proteins: genomic and protein analysis of surgical myectomy tissue from patients with obstructive hypertrophic cardiomyopathy. Circ Heart Fail. 2009;2:325–333. doi: 10.1161/CIRCHEARTFAILURE.108.789735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson KL, et al. Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet Med. 2019;21:1576–1584. doi: 10.1038/s41436-018-0375-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban EA, Johnston RJ., Jr Buffering and amplifying transcriptional noise during cell fate specification. Front Genet. 2018;9:591. doi: 10.3389/fgene.2018.00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk SJ, et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- van Wamel AJ, Ruwhof C, van der Valk-Kokshoom LE, Schrier PI, van der Laarse A. The role of angiotensin II, endothelin-1 and transforming growth factor-beta as autocrine/paracrine mediators of stretch-induced cardiomyocyte hypertrophy. Mol Cell Biochem. 2001;218:113–124. doi: 10.1023/A:1007279700705. [DOI] [PubMed] [Google Scholar]
- van Wamel AJ, Ruwhof C, van der Valk-Kokshoorn LJ, Schrier PI, van der Laarse A. Stretch-induced paracrine hypertrophic stimuli increase TGF-beta1 expression in cardiomyocytes. Mol Cell Biochem. 2002;236:147–153. doi: 10.1023/A:1016138813353. [DOI] [PubMed] [Google Scholar]
- Vignier N, et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–248. doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- Walsh R, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J. 2017;38:3461–3468. doi: 10.1093/eurheartj/ehw603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunger S, Rosenfeld L, Garini Y, Shav-Tal Y. Single-allele analysis of transcription kinetics in living mammalian cells. Nat Methods. 2010;7:631–633. doi: 10.1038/nmeth.1482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.