

Abstract
We reported previously that, in budding yeast, transcription rate affects both the efficiency and fidelity of pre-mRNA splicing, especially of ribosomal protein transcripts. Here, we report that the majority of ribosomal protein transcripts with non-consensus 5′ splice sites are spliced less efficiently when transcription is faster, and more efficiently with slower transcription. These results support the “window of opportunity” model, and we suggest a possible mechanism to explain these findings.
Keywords: introns, RNA polymerase speed, RNA polymerase, yeast
INTRODUCTION
Cotranscriptional splicing
The coding sequences of most eukaryotic genes are interrupted by introns (mainly non-coding sequences) that are also present in the nascent transcripts. Pre-mRNA splicing is the process that removes the introns and joins the flanking sequences (exons) to produce mature mRNAs. Evidence accumulating during the last two decades suggests that many, and possibly the majority of, splicing events occur cotranscriptionally, that is before transcription termination (Ameur et al. 2011; Khodor et al. 2011; Carrillo Oesterreich et al. 2016). This raises the intriguing possibility of functionally significant interactions between splicing, chromatin, transcription and other RNA processing events, when they occur in close proximity (Li and Manley 2006; Aitken et al. 2011; Neugebauer 2019). Two models were proposed to explain how transcription and splicing are coupled, referred to as the recruitment coupling and the kinetic coupling models, which are not mutually exclusive (for review, see Dujardin et al. 2013).
Recruitment coupling
The term “recruitment coupling” refers to the ability of the transcription machinery to promote the recruitment of RNA processing factors to the site of transcription. In particular, the carboxyterminal domain (CTD) of the largest subunit of RNA polymerase II (RNAPII) acts as a “landing pad” for cotranscriptional recruitment of capping, splicing and 3′ end processing factors to nascent RNA (Bentley 2014). Strong support for this model was obtained by a study with human cell lines showing CTD-dependent inhibitory action by serine/arginine-rich (SR) splicing factor SRSF3 (SRp20) in inclusion of fibronectin cassette exon 33 (E33) (de la Mata and Kornblihtt 2006).
Kinetic coupling
Early evidence for coupling between transcription and splicing was reported by Eperon et al. (1988), who showed that, in HeLa cells, the use of an alternative splice site positioned within a potential stem–loop structure depended on a “window” of availability of the splice site in the nascent RNA for splicing factors or hnRNP proteins to bind before the inhibitory stem–loop formed. A prediction of their model was that the rate of transcription elongation could control alternative splicing by determining the duration of the window. Indeed, de la Mata et al. (2003) showed that, in cultured human cells, a slow RNAPII promoted inclusion of the fibronectin EDI (extra domain I) exon, and they proposed that slower transcription elongation expanded the “window of opportunity” for recognition of the weak 3′ splice site (3′SS) upstream of the exon before transcription of a competing 3′SS downstream. In this model, the rate of transcript elongation could affect the extent and even the order in which splicing factors assemble cotranscriptionally on a nascent transcript, although it need not affect splicing catalysis, which may occur post-transcriptionally on cotranscriptionally assembled spliceosomes.
Evidence of kinetic coupling was obtained also in budding yeast by Howe et al. (2003), who observed enhanced inclusion of the second exon of modified DYN2 transcripts in a slow RNAPII mutant or when cells were treated with chemical inhibitors of transcription. In striking contrast, slower elongation caused skipping of human CFTR exon 9 in a minigene construct due to enhanced recruitment of ETR-3, a negative splicing factor, to the 3′SS of the exon (Dujardin et al. 2014). This demonstrates that slower transcription can also expand the window of opportunity for recruitment of factors that block splicing of a newly transcribed exon, reducing its inclusion rate. Overall, it seems that transcription rate determines the temporal window of opportunity for selection or rejection of an upstream sequence before a competing downstream sequence is transcribed. However, Fong et al. (2014) found that, in human cells, both faster and slower elongating RNA polymerase mutants may disrupt splicing in the same way, which seems contrary to the “window of opportunity” model. They proposed that an optimal rate of transcriptional elongation is required for normal cotranscriptional pre-mRNA splicing. Therefore, for the vast majority of genes, it is unclear what determines the splicing outcome of altering transcription elongation rate or how changes in transcription rate are regulated locally at alternatively spliced exons. It has been proposed that mechanisms may exist to slow or pause transcription downstream from introns in budding yeast, thereby stretching the window of opportunity for cotranscriptional spliceosome assembly to occur (Alexander et al. 2010; Oesterreich et al. 2010).
Transcription rate affects splicing efficiency
To investigate the effect of transcription elongation rate on splicing efficiency (the fraction of transcripts that get spliced) in budding yeast (Aslanzadeh et al. 2018), we previously used RNAPII trigger loop mutants that elongate, on average, four times faster (Rpb1-G1097D) or eight times slower (Rpb1-H1085Y) than wild-type RNAPII (∼12 nt) in vitro (Kaplan et al. 2012). In all these strains, UPF1 was deleted to reduce degradation caused by nonsense mediated decay. Both 4-thio-uracil labeling for analysis of newly synthesized RNA and Native Elongating Transcript (NET)-RTqPCR to analyze RNAPII-associated transcripts for several genes showed that more splicing occurs cotranscriptionally with the slow mutant, and less with the fast mutant compared with wild-type RNAPII. Importantly, these differences were not simply due to there being more or less unspliced pre-mRNA as a consequence of different decay rates in the mutants, as the level of spliced mRNA was elevated in the slow mutant and reduced in the fast mutant compared with the WT. These results are compatible with both recruitment coupling and kinetic coupling; slower elongation allows more time for cotranscriptional recruitment of splicing factors to the nascent transcript, whereas faster elongation has the opposite effect. Moreover, analysis of total RNA (as opposed to nascent RNA analyzed by NET-RTqPCR) revealed that overall splicing efficiency was also decreased with the fast RNAPII mutant and slightly improved with the slow mutant compared to wild-type. These results indicate that post-transcriptional splicing in the fast mutant does not compensate for its reduced cotranscriptional splicing (Aslanzadeh et al. 2018).
Interestingly, transcriptome-wide analysis of splicing efficiency by RNA sequencing of total RNA revealed that the effect of the fast RNAPII mutant was mainly to reduce splicing efficiency with ribosomal protein (RP) coding transcripts (seen as increased accumulation of unspliced pre-mRNAs; Fig. 1C in Aslanzadeh et al. 2018), whereas the slow mutant increased the splicing efficiency with both RP and non-RP transcripts.
FIGURE 1.
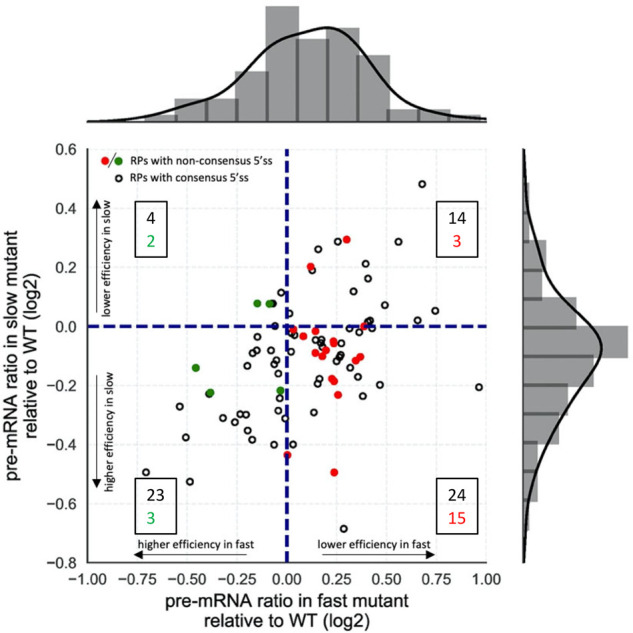
Faster transcription reduces and slower transcription increases the efficiency of splicing RP transcripts with non-canonical 5′ splice sites. Splicing efficiencies were calculated (Supplemental Table S1) as RNA-seq read counts from pre-mRNA divided by total reads (pre-mRNA + mRNA) for each transcript (Aslanzadeh et al. 2018). Transcripts that splice significantly more or less efficiently in the fast mutant compared to WT lie to the left and right of the vertical dashed line, respectively. Those that splice more or less efficiently in the slow mutant compared to WT lie below or above the horizontal dashed line, respectively. RP transcripts with non-consensus 5′SS are represented by green (more efficiently spliced in the fast mutant) or red (less efficiently spliced in the fast mutant) dots. The boxed numbers indicate the number of genes represented in each quadrant, consensus in black, non-consensus in colored font. Histograms show the distribution of pre-mRNA ratios relative to WT in the fast (top) and slow (right) mutants. In the fast mutant there are more genes with increased pre-mRNA ratio (reduced splicing efficiency) relative to WT and in the slow mutant there are more genes with reduced pre-mRNA ratio (increased splicing efficiency) relative to WT.
We previously reported that RP transcripts are spliced faster and more cotranscriptionally (Barrass et al. 2015; Wallace and Beggs 2017) and that they tend to be spliced with greater efficiency and higher fidelity (Aslanzadeh et al. 2018). Taken together with the greater sensitivity to transcription speed, these observations indicate that the splicing of RP transcripts is more functionally coupled to transcription than that of non-RP transcripts, and that this coupling is highly beneficial. Yet, it remains largely unknown what determines the extent to which splicing of a transcript is more or less coupled to transcription. Notably, many RP transcripts are spliced more efficiently with slower transcription and less efficiently with faster transcription. Here, we investigate whether this can be explained by variation in splice site sequences.
RESULTS AND DISCUSSION
Figure 1 shows the effects of the fast and slow RNAPII mutants on RP splicing efficiencies plotted relative to wild-type (which normalizes for effects of expression level in the data analysis) for 88 RP transcripts, using data (Supplemental Table S1) from Aslanzadeh et al. (2018). For 65 RP introns (74% of the 88 RP introns analyzed) splicing efficiencies are higher in the slow RNAPII mutant compared to wild-type (Fig. 1, all dots below the horizontal dashed line) and for 56 RP introns (64%) splicing efficiencies are lower in the fast mutant (all dots right of the vertical dashed line). The quadrant containing data from the largest number of genes is bottom right, with 39 introns (44% of the 88 RP introns analyzed) whose splicing efficiency is both reduced with the fast RNAPII mutant and increased with the slow mutant. Therefore, for 44% of the RP genes the effect of RNAPII elongation rate on efficiency of splicing fits the window of opportunity model.
It was noted that 23 of the 88 RP transcripts have non-consensus 5′ splice sites (5′SS) (i.e., do not match the consensus sequence GUAUGU), and that 18 (78%) of these were spliced more efficiently in the slow strain (colored dots below the horizontal dashed line in Fig. 1; Supplemental Table S2). The same number (18) with non-consensus 5′SS were spliced less efficiently in the fast strain (red dots right of the vertical dashed line in Fig. 1). Conceivably, the reduced splicing efficiency observed for these RP transcripts in the fast mutant could be explained by a combination of delayed splicing due to the non-consensus 5′SS and the shorter window of opportunity for splicing to occur cotranscriptionally due to the faster transcription elongation, but which is compensated by slow transcription. Consistent with this explanation, 15 of these introns with non-consensus 5′SS, splicing was both less efficient in the fast RNAPII strain and more efficiently in the slow strain (red dots in the bottom right quadrant in Fig. 1; Supplemental Table S1). Therefore, the splicing efficiencies for 65% of the RP transcripts with non-consensus 5′SS fit the window of opportunity model. This compares with 24 of 65 introns (37%) with consensus 5′SS that fit this model, which is itself notable (expect ∼25% if the effect of transcription speed was random). It appears, therefore, that the splicing of RP introns with non-consensus 5′SS is more highly coupled to transcription.
Interestingly, genome-wide studies in mammalian cells also found that specific transcript features, such as suboptimal splice sites, may make a particular splicing event more sensitive to transcription rate, especially in transcripts that encode RNA binding proteins, such as ribosomal proteins, or RNA processing factors (Ip et al. 2011; Fong et al. 2014).
For five RP introns with non-consensus 5′SS (RPS14B, RPS19A, RPS21B, RPL30, and RPL43B) splicing was more efficient rather than less efficient in the fast RNAPII strain (green dots in Fig. 1), suggesting that, for these transcripts, other factors have a greater influence on splicing efficiency than 5′SS. In the case of two of these transcripts, RPS14B and RPL30, their protein products Rps14p and Rpl30p, when in excess, bind to their respective precursor transcripts and inhibit splicing (Fewell and Woolford 1999; Macías et al. 2008). Therefore, in these two cases at least, competition between cotranscriptional spliceosome assembly and feedback inhibition of splicing by the protein product, may explain the different response to changes in transcription speed, for example if faster transcription through the intron permits spliceosome assembly or optimal secondary structure in the transcript before binding of the inhibitory protein. It would be interesting to investigate whether splicing of the other three transcripts in this category is also subject to negative regulation.
Curiously, the same trends are not apparent among non-RP transcripts with non-consensus 5′SS, but many of these have other atypical intron features that may affect their splicing kinetics.
Our observation that a non-consensus 5′SS correlates with sensitivity to transcription elongation rate supports both the recruitment and kinetic coupling models and suggests that, at least for RP transcripts, a non-consensus 5′SS may reduce or slow spliceosome assembly and/or splicing catalysis in a manner that is compensated by slower transcription elongation. The most frequently occurring non-consensus 5′SS in the RP genes has the sequence GUACGU (Supplemental Table S2). Carrillo Oesterreich et al. (2016) showed, using a reporter gene, that this sequence delayed spliceosome assembly and cotranscriptional splicing compared with a consensus 5′SS, which they proposed could be due to weakened 5′SS base-pairing with U1 snRNA delaying spliceosome assembly. However, it is questionable whether replacing U with C as the base at position +4 of a 5′SS significantly alters the stability of the 5′SS:U1 snRNA interaction. The opposing base at position +5 in U1 snRNA is pseudoU (Ψ) (Plaschka et al. 2018), which can pair weakly with either U or C, with relative stabilities depending on the context of the surrounding sequence (Kierzek et al. 2014) and contacting proteins. Interestingly, in the study by Carrillo Oesterreich et al. (2016), ChIP analysis showed U1 snRNP apparently recruited equally efficiently to reporter transcripts containing either GUAUGU or GUACGU at the 5′SS, whereas the U1 snRNP ChIP signal was lost more slowly from the GUACGU reporter. A possible explanation for this delayed departure of U1 snRNP from the cotranscriptionally assembling spliceosomes is the reduced ability of CGU in the non-consensus 5′SS to pair with ACA in the ACAGAGA motif of U6 snRNA, an interaction that is coupled with U1 snRNP displacement from the 5′SS by Prp28 (Fig. 2; Staley and Guthrie 1999; Chen et al. 2001). In this case, slower transcription elongation would allow more time for the assembling spliceosome to proceed through this checkpoint cotranscriptionally and/or for cotranscriptional recruitment of factors required to form catalytically active spliceosomes, providing a rational explanation for the effect of transcription speed on splicing efficiency for this subset of transcripts.
FIGURE 2.

A potential mechanism to explain the observed effects of transcription speed with the suboptical 5′ splice site GUACGU. Gray boxes, exons. (Upper) Base-pairing of a canonical 5′SS with U1 snRNA, and with U6 snRNA following Prp28 function. (Lower) Likely base-pairing of the non-canonical 5′SS, GUACGU, with U1 snRNA, and with U6 snRNA following Prp28 function. Vertical bars do not indicate the strength of base-pairing.
Overall, the work discussed here supports the window of opportunity model as an explanation for the effects of transcription speed on splicing efficiency of many RP transcripts, especially those with non-consensus 5′SS. We propose a possible mechanistic explanation for this as being mediated by effects on cotranscriptional spliceosome assembly. For a small number of RP introns feedback regulation by their gene products may explain why splicing is more efficient with the faster RNAPII. The work by Fong et al. (2014) challenged the window of opportunity model, finding that for many human genes both faster and slower elongating RNAPII mutants seemed to disrupt splicing in the same way. However, whereas their slow mutant elongated 3.4 times slower than normal RNAPII, their fast RNAPII mutant elongated only 12% faster than normal, resulting in quantitatively smaller changes in splicing, which may not be very revealing. In comparison, the yeast mutants used in this study elongate on average eight times slower or four times faster than wild-type RNAPII (Kaplan et al. 2012), which may provide a more robust comparison. Perhaps more importantly, Fong et al. measured changes in alternative splicing events, whereas this yeast study monitored changes in splicing efficiency. Conceivably, the effects of transcription speed on alternative splicing may be more complex.
What other features of RP transcripts might cause their splicing to be more coupled to transcription? Promoter structure was shown to be important for alternative splicing in human cells (Cramer et al. 1997) and yeast (Kawashima et al. 2014), and it was shown that RP transcripts in budding yeast have distinct promoter architectures, with an exclusive pattern of DNA binding proteins that enhance their transcription (Knight et al. 2014). Indeed, considering that RP genes are more highly expressed than most non-RP genes, expression level could be an underlying factor in determining the extent of coupling. Chromatin structure and modifications also affect both transcription and splicing (Gunderson et al. 2011; Neves et al. 2017; Venkataramanan et al. 2017). One informative future approach would be to study the effect on cotranscriptional splicing of swapping the promoters and different intron features of yeast RP and non-RP transcripts.
MATERIALS AND METHODS
For details of the data generation and analysis methods, see Aslanzadeh et al. (2018).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Edward Wallace for helpful comments about the manuscript. This work was supported by Wellcome Trust funding to J.D.B. (104648) and a Darwin Trust of Edinburgh studentship to V.A. Work in the Wellcome Centre for Cell Biology was supported by Wellcome Trust core funding (092076).
Author contributions: The original experimental work to produce the data analyzed here was performed by V.A., supervised by J.D.B. Data analysis and manuscript preparation were done by V.A. and J.D.B.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.075895.120.
Freely available online through the RNA Open Access option.
REFERENCES
- Aitken S, Alexander RD, Beggs JD. 2011. Modelling reveals kinetic advantages of co-transcriptional splicing. PLoS Comput Biol 7: e1002215 10.1371/journal.pcbi.1002215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander RD, Innocente SA, Barrass JD, Beggs JD. 2010. Splicing-dependent RNA polymerase pausing in yeast. Mol Cell 40: 582–593. 10.1016/j.molcel.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, Feuk L. 2011. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat Struct Mol Biol 18: 1435–1440. 10.1038/nsmb.2143 [DOI] [PubMed] [Google Scholar]
- Aslanzadeh V, Huang Y, Sanguinetti G, Beggs JD. 2018. Transcription rate strongly affects splicing fidelity and cotranscriptionality in budding yeast. Genome Res 28: 203–213. 10.1101/gr.225615.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrass JD, Reid JEA, Huang Y, Hector RD, Sanguinetti G, Beggs JD, Granneman S. 2015. Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biol 16: 282 10.1186/s13059-015-0848-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. 2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175. 10.1038/nrg3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo Oesterreich F, Herzel L, Straube K, Hujer K, Howard J, Neugebauer KM. 2016. Splicing of nascent RNA coincides with intron exit from RNA polymerase II. Cell 165: 372–381. 10.1016/j.cell.2016.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY-F, Stands L, Staley JP, Jackups RR, Latus LJ, Chang T-H. 2001. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol Cell 7: 227–232. 10.1016/S1097-2765(01)00170-8 [DOI] [PubMed] [Google Scholar]
- Cramer P, Pesce CG, Baralle FE, Kornblihtt AR. 1997. Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci 94: 11456–11460. 10.1073/pnas.94.21.11456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Kornblihtt AR. 2006. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol 13: 973–980. 10.1038/nsmb1155 [DOI] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. 2003. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12: 525–532. 10.1016/j.molcel.2003.08.001 [DOI] [PubMed] [Google Scholar]
- Dujardin G, Lafaille C, Petrillo E, Buggiano V, Gómez Acuña LI, Fiszbein A, Godoy Herz MA, Nieto Moreno N, Muñoz MJ, Alló M, et al. 2013. Transcriptional elongation and alternative splicing. Biochim Biophys Acta 1829: 134–140. 10.1016/j.bbagrm.2012.08.005 [DOI] [PubMed] [Google Scholar]
- Dujardin G, Lafaille C, de la Mata M, Marasco LE, Muñoz MJ, Le Jossic-Corcos C, Corcos L, Kornblihtt AR. 2014. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 54: 683–690. 10.1016/j.molcel.2014.03.044 [DOI] [PubMed] [Google Scholar]
- Eperon LP, Graham IR, Griffiths AD, Eperon IC. 1988. Effects of RNA secondary structure on alternative splicing of pre-mRNA: Is folding limited to a region behind the transcribing RNA polymerase? Cell 54: 393–401. 10.1016/0092-8674(88)90202-4 [DOI] [PubMed] [Google Scholar]
- Fewell SW, Woolford JL. 1999. Ribosomal protein S14 of Saccharomyces cerevisiae regulates its expression by binding to RPS14B pre-mRNA and to 18S rRNA. Mol Cell Biol 19: 826–834. 10.1128/MCB.19.1.826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu X-D, Bentley DL. 2014. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 28: 2663–2676. 10.1101/gad.252106.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson FQ, Merkhofer EC, Johnson TL. 2011. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc Natl Acad Sci 108: 2004–2009. 10.1073/pnas.1011982108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KJ, Kane CM, Ares M. 2003. Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces cerevisiae. RNA 9: 993–1006. 10.1261/rna.5390803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, Blencowe BJ. 2011. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 21: 390–401. 10.1101/gr.111070.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Jin H, Zhang IL, Belyanin A. 2012. Dissection of Pol II trigger loop function and Pol II activity–dependent control of start site selection in vivo. PLoS Genet 8: e1002627 10.1371/journal.pgen.1002627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Douglass S, Gabunilas J, Pellegrini M, Chanfreau GF. 2014. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae. PLoS Genet 10: e1004249 10.1371/journal.pgen.1004249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodor YL, Rodriguez J, Abruzzi KC, Tang C-HA, Marr MT, Rosbash M. 2011. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev 25: 2502–2512. 10.1101/gad.178962.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierzek E, Malgowska M, Lisowiec J, Turner DH, Gdaniec Z, Kierzek R. 2014. The contribution of pseudouridine to stabilities and structure of RNAs. Nucleic Acids Res 42: 3492–3501. 10.1093/nar/gkt1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight B, Kubik S, Ghosh B, Bruzzone MJ, Geertz M, Martin V, Dénervaud N, Jacquet P, Ozkan B, Rougemont J, et al. 2014. Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev 28: 1695–1709. 10.1101/gad.244434.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Manley JL. 2006. Cotranscriptional processes and their influence on genome stability. Genes Dev 20: 1838–1847. 10.1101/gad.1438306 [DOI] [PubMed] [Google Scholar]
- Macías S, Bragulat M, Tardiff DF, Vilardell J. 2008. L30 binds the nascent RPL30 transcript to repress U2 snRNP recruitment. Mol Cell 30: 732–742. 10.1016/j.molcel.2008.05.002 [DOI] [PubMed] [Google Scholar]
- Neugebauer KM. 2019. Nascent RNA and the coordination of splicing with transcription. Cold Spring Harb Perspect Biol 11: a032227 10.1101/cshperspect.a032227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves LT, Douglass S, Spreafico R, Venkataramanan S, Kress TL, Johnson TL. 2017. The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae. Genes Dev 31: 702–717. 10.1101/gad.295188.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterreich FC, Preibisch S, Neugebauer KM. 2010. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell 40: 571–581. 10.1016/j.molcel.2010.11.004 [DOI] [PubMed] [Google Scholar]
- Plaschka C, Lin P-C, Charenton C, Nagai K. 2018. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature 559: 419 10.1038/s41586-018-0323-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1999. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell 3: 55–64. 10.1016/S1097-2765(00)80174-4 [DOI] [PubMed] [Google Scholar]
- Venkataramanan S, Douglass S, Galivanche AR, Johnson TL. 2017. The chromatin remodeling complex Swi/Snf regulates splicing of meiotic transcripts in Saccharomyces cerevisiae. Nucleic Acids Res 45: 7708–7721. 10.1093/nar/gkx373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace EWJ, Beggs JD. 2017. Extremely fast and incredibly close: cotranscriptional splicing in budding yeast. RNA 23: 601–610. 10.1261/rna.060830.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.