

Abstract
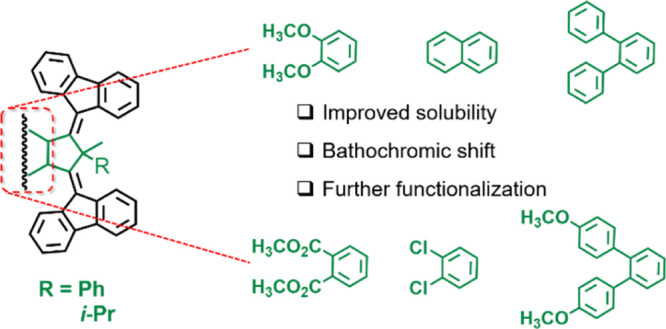
The synthesis and characterization of a series of light-driven third-generation molecular motors featuring various structural modifications at the central aromatic core are presented. We explore a number of substitution patterns, such as 1,2-dimethoxybenzene, naphthyl, 1,2-dichlorobenzene, 1,1′:2′,1″-terphenyl, 4,4″-dimethoxy-1,1’:2′,1″-terphenyl, and 1,2-dicarbomethoxybenzene, considered essential for designing future responsive systems. In many cases, the synthetic routes for both synthetic intermediates and motors reported here are modular, allowing for their post-functionalization. The structural modifications introduced in the core of the motors result in improved solubility and a bathochromic shift of the absorption maxima. These features, in combination with a structural design that presents remote functionalization of the stator with respect to the fluorene rotors, make these novel motors particularly promising as light-responsive actuators in covalent and supramolecular materials.
Introduction
The field of molecular machines and motors has experienced amazing developments enabling a transition from molecules to dynamic molecular systems.1−15 One reason for this development relies on the promise that the integration of these artificial molecular tools into functional materials provides the opportunity for dynamic, responsive, and adaptive properties.16−18 Moreover, the possibility to perform work upon applying an external stimulus in the form of chemical energy or light7,19 represents an appealing perspective for materials science. In this context, light-driven rotary molecular motors represent promising candidates for further investigation because of their ability to undergo 360° unidirectional rotary motion upon irradiation.20−25 Recent examples of molecular motors embedded in light-responsive polymer networks,26,27 surfaces,28 and metal organic frameworks29−31 have shown some of the future directions of stimuli-responsive materials. In order to advance the field and envision further opportunities to explore, fundamental work on the design and synthesis of novel molecular motor structures is paramount.
The light-driven molecular motors developed in our group are based on overcrowded alkenes that undergo 360° unidirectional rotary motion thanks to a unique interplay between point and helical chirality.32−34 These molecular systems are classified into three generations, depending on the number of stereogenic centers, which control the unidirectionality of the rotary motion.34 First- and second-generation molecular motors possess two35 and one stereogenic center,36 respectively, while the recently developed third-generation molecular motors37,38 are mesostructures. General chemical structures of first-, second-, and third-generation motors are shown in Figure 1.
Figure 1.
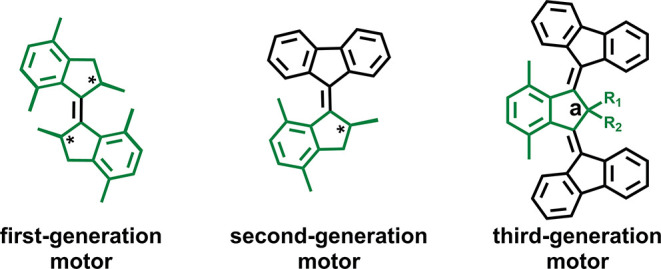
General chemical structures of first- (left), second- (center), and third-generation (right) molecular motors.
Third-generation molecular motors can be considered as a combination of two second-generation motors with opposite helicity.37,38 They feature one center of pseudo-asymmetry (denoted by the letter a in Figure 1) that allows for the unidirectional rotary motion of the parallel rotors with respect to the central aromatic core, implying that one rotor rotates clockwise and the other anti-clockwise.37,38 Drawing a parallel with the macroscopic world, the fluorene rotors rotate similarly to the left and right wheels of a car from the perspective of the driver. This peculiar feature makes this generation of motors particularly appealing for locomotion and cargo transport applications because the combination of the unidirectional rotary motions of the two rotors should result in unidirectional translation, as conceptually demonstrated with the nanocar.39
Previous investigations on light-driven third-generation motors carried out in our group focused on (i) the introduction of the concept of unidirectional rotary motion in achiral light-driven molecular motors37 and (ii) the consequences of structural variations on speed and unidirectionality.38 Concerning (ii), particular attention was dedicated to the role of the substituents at the pseudoasymmetric carbon atom, while no synthetic work was carried out on the central aromatic core that presented either a benzene or p-xylene moiety. Reliable procedures for the modification of the core of these motors are highly desirable as it would greatly facilitate their incorporation into functional materials. We now report the synthesis and characterization of a library of core-modified third-generation light-driven molecular motors (Chart 1). Most of the new motors (2–7 and 9) present either the methyl, phenyl (Me,Ph) or methyl, iso-propyl (Me,i-Pr) combination of substituents at the pseudo-asymmetric carbon atom. These combinations of substituents have been reported to lead to 70 and 100% unidirectionality, respectively.38 The octadecyl, phenyl (C18,Ph) combination was installed in motor 8 instead. The replacement of the Me substituent with the more sterically hindered C18 should result in an increase in unidirectionality from 70 to 78%.38 Despite this modest effect, our major motivation for the C18,Ph combination was its potential use in facilitating the deposition of third-generation motors onto solid supports because long alkyl chains are typically responsible for more favorable molecule–surface interactions. We focused on a number of motifs for the central aromatic core, such as 1,2-dimethoxybenzene (2), naphthyl (3), 1,2-dichlorobenzene (4), 1,1′:2′,1″-terphenyl (5), 4,4″-dimethoxy-1,1′:2′,1″-terphenyl (6), and 1,2-dicarbomethoxybenzene (9). Therefore, what distinguishes these motors from previous examples is the presence of substituents on the central aromatic cores, which results in (1) bathochromic shifts of the absorption maxima and (2) ample possibilities for further functionalization/integration into light-responsive systems/materials and surface anchoring.
Chart 1. Chemical Structures of the Third-Generation Motors Presented Here.

Results and Discussion
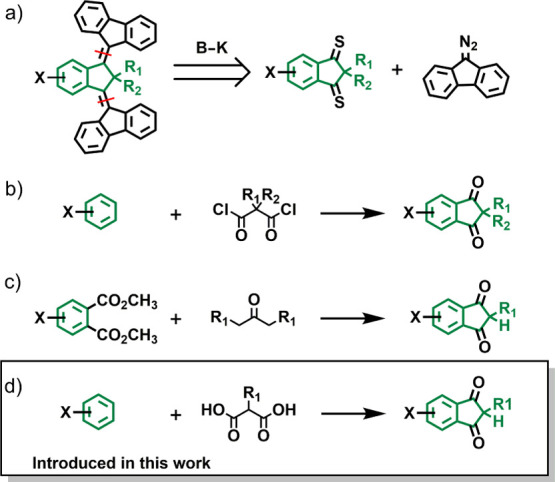
The key step in the synthesis of overcrowded alkene-based third-generation molecular motors is the Barton–Kellogg olefination (B–K reaction).37,38 This approach is shown in Scheme 1a, where disconnection at the level of the overcrowded alkene moieties results in two building blocks: an indanedithione and a diazo compound. The high stability of indanedithiones deriving from the 1,3-indanedione skeleton with a quaternary carbon atom in the α-position to the two thiocarbonyl moieties, as well as that of 9-diazofluorene, makes this combination of reactants particularly convenient. Both can be stored as stable synthetic intermediates without the necessity to generate them in situ or use them immediately after purification. The use of indanedithiones which can be directly prepared from the diketone precursors shifts the attention to the synthesis of 1,3-indanedione derivatives. In previous studies,37,38 we have successfully prepared these compounds using Friedel–Crafts acylation of electron-rich aromatic compounds (Scheme 1b) or NaH-induced Claisen condensation between a dimethyl phthalate-type derivative and symmetrical ketones with two methylene groups at the α-position (Scheme 1c).40,41 In the present study, we predominantly applied the Claisen condensation approach in view of its more widely compatible reaction conditions. However, we also adopted a third synthetic strategy consisting of Friedel–Crafts acylation with malonic acids in polyphosphoric acid41 (PPA) (Scheme 1d). Although less general than the Claisen condensation,40 the PPA strategy can save many synthetic steps with certain core designs.
Scheme 1. (a) Retrosynthetic Approach for Third-Generation Motors and Approaches for the Synthesis of 1,3-Indanedione Derivatives Based on (b) Friedel–Crafts Acylation with Acyl Chlorides, (c) NaH-Induced Claisen Condensation, and (d) PPA-Induced Friedel–Crafts Acylation with Malonic Acids.
Synthesis of Motor Cores
As indicated, in most cases, we followed the Claisen condensation approach40 for preparing the required 1,3-indanediones. Starting from the corresponding dimethyl phthalate derivatives 10–18, the NaH-induced cyclization reaction with commercially available 1,3-diphenyl-2-propanone or 2,6-dimethyl-4-heptanone in toluene at 110 °C was performed (Scheme 2a, i). The reaction mixtures turned into deep red suspensions at different reaction times (1–5 d) depending on the dimethyl phthalate starting material used. The red precipitates corresponded to the sodium salts of the cyclized products (intermediates between brackets in Scheme 2a), which were not soluble in toluene. Different from our previous reports on third-generation motors,37,38 in the present procedure, we did not isolate the neutral compounds by treating the Na+ salts with hydrochloric acid (HCl). Simple filtration of the red suspensions allowed for obtaining the crude Na+ salts, which were then subjected to alkylation with methyl iodide (MeI) in acetone at 60 °C (Scheme 2a, ii). These reactions were conducted in the presence of mixtures of Aliquat 336 and potassium fluoride (KF) on celite, as their combination had previously been shown to favor C-alkylation of the enolates over the competing O-alkylation.37,38 Core structures 19–27 were obtained in good yields (from 30 to 76%), with the only exceptions of 22 and 25 (11 and 18%, respectively), after chromatographic purification on silica (Scheme 2a).
Scheme 2. Synthesis of Motor Cores Applying the (a) Claisen Condensation Approach, Followed by Filtration of the Crude Na+ Salts and Immediate Methylation; (b) PPA-Induced Friedel–Crafts Acylation, Followed by Alkylation of the Isolated Intermediate.

Reaction conditions: (i) diester (1 equiv), 1,3-diphenyl-2-propanone or 2,6-dimethyl-4-heptanone (1.5 equiv), NaH 60 wt % (2 equiv), toluene, 110 °C, 1–5 d depending on the starting material; (ii) MeI (2 equiv), Aliquat 336 (0.05 equiv), KF on celite (0.05 equiv), acetone, 60 °C, o/n; (iii) phenylmalonic acid (1.5 equiv), PPA (115% H3PO4 basis), 90 °C, mechanical stirring, o/n; and (iv) MeI or 1-iodooctadecane (2 equiv), Aliquat 336 (0.05 equiv), KF on celite (0.05 equiv), acetone, 60 °C, o/n.
Given the laborious synthetic routes to obtain starting diester 11 (four steps),42,43 we attempted the direct PPA-induced double Friedel–Crafts acylation of commercially available 1,2-dimethoxybenzene 28 (Scheme 2b). Encouraged by a related functionalization with iso-propylmalonic acid,44 we slightly adapted the reaction conditions to phenylmalonic acid. Gratifyingly, we successfully obtained 29 in 31% yield after a simple extraction–recrystallization sequence. Alkylation of 29 with MeI afforded motor core unit 20 after chromatographic purification (Scheme 2b, iv). It must be emphasized that applying this route is particularly convenient to access 20 because it shortens the synthesis from six steps (four steps account for the preparation of 11) to only two steps. Finally, alkylation of 2-phenyl-1H-indene-1,3(2H)-dione38 with 1-iodooctadecane afforded 30 in 83% yield (Scheme 2b, iv).
A particularly convenient aspect of some of our synthetic routes to these core structures is their modularity as additional substituents can be introduced via both dimethyl phthalate and core building blocks. Bromo-substituted dimethyl phthalate 14 or 1,3-indanedione 23 could be converted into derivatives 15–17 or 24–26, respectively, by means of the Suzuki–Miyaura cross-coupling reaction with phenylboronic acids catalyzed by palladium-tetrakis(triphenylphosphine) [Pd(PPh3)4]. Details on the preparation of 14 can be found in the Experimental Section, while here, we briefly discuss the post-modification of 23 (Scheme 3). Applying the Suzuki–Miyaura cross-coupling on this core structure, 24–26 were obtained in 50–75% yield (Scheme 3). While the same reaction on 14 consistently afforded the corresponding esters 15–17 in higher yields (see the Experimental Section), the post-modification of 23 has the additional advantage of diversifying these syntheses after the NaH-induced Claisen condensation, which represents the critical step in the preparation of these core units. Hence, direct modification of 14 is the best option if the target is one molecular design only; alternatively, post-modification of 23 is more convenient when the screening of multiple molecular designs is desired.
Scheme 3. Suzuki–Miyaura Cross Coupling Reaction on Indanedione Core 23 with Phenylboronic Acid, 4-Methoxyphenylboronic Acid, and 4-(tert-Butyldimethylsilyloxy)phenylboronic Acid Affording 24, 25, and 26, Respectively.
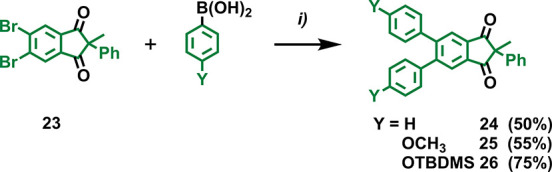
Reaction conditions: (i) boronic acid (2.2 equiv), Pd(PPh3)4 (10 mol %), toluene, 110 °C, 18 h.
Synthesis and Characterization of the Motors
With 1,3-indanedione derivatives 19–27 and 30 in hand, we then focused on their conversion into indanedithiones, which in some cases was not straightforward. Previous work on third-generation motors highlighted the importance of using mixtures of phosphorous pentasulfide (P2S5) and Lawesson’s reagent (LR) in boiling toluene.37,38 We found that this mixture was necessary in most cases, while treating 20 with only LR sufficed to achieve full conversion (Scheme 4a). This behavior was attributed to the electron-donating effect of the methoxy substituents. Unfortunately, core unit 23 could not be successfully converted into its corresponding indanedithione, probably because of the electron-withdrawing effect of the two bromine substituents. Besides electronic effects, steric hindrance may also play a role in the 1,3-indanedione → indanedithione conversion. For example, when 30 was subjected to the reaction conditions of Scheme 4a, we obtained a mixture of unreacted 30 as well as mono- and di-substituted (38) products that could not be fully separated by chromatography. tert-Butyldimethylsilyl protecting groups, which are of interest for potential post-functionalization of the motors because of their ease of removal, were found to be particularly inert and survive indanedithione formation. Finally, core 27 could not be converted to indanedithione 39 with the reaction conditions reported in Scheme 4a. We could, however, obtain 39 by reacting 27 in the presence of LR under microwave irradiation for 4 h, albeit in a very low yield (8%) (Scheme 4b). These conditions were unsuccessful with 23 as we only recovered unreacted starting material. Thioketones 31–39 were characterized by 1H NMR to ensure full conversion and were directly (without isolation) used in the subsequent Barton–Kellogg olefination (B–K reaction).
Scheme 4. Preparation of the Indanedithiones Starting from the Corresponding Substituted 1,3-Indanediones, Applying (a) Conventional Heating and (b) Microwave Irradiation.
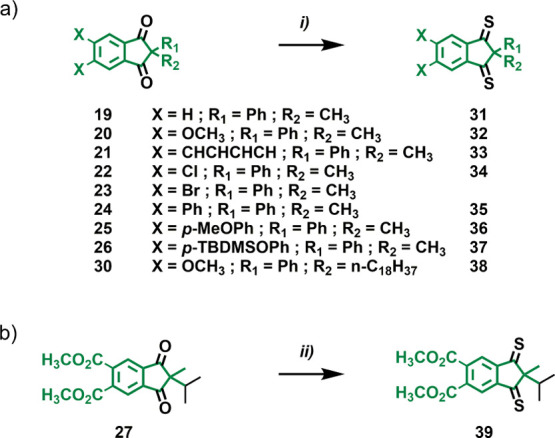
Reaction conditions: (i) P2S5 (4 equiv), LR (4 equiv), toluene, 110 °C, o/n; (ii) LR (8 equiv), toluene, 110 °C, 4 h, microwave irradiation.
B–K reactions were run starting from toluene solutions of indanedithiones 31–39 at room temperature, followed by the addition of solid diazofluorene (Scheme 5a). The typical green-blue toluene solutions of 31–39 turned red immediately upon addition of diazofluorene, and the progress of these reactions was monitored by thin-layer chromatography (TLC). The desulfurizing agent hexamethylphosphorous triamide (HMPT) was added when no traces of starting indanedithione were detected, and the resulting mixtures were heated up to 90 °C and stirred overnight (Scheme 5a). Motors 1–9 (1 had previously been reported) were obtained after chromatographic purification. Following the same strategy of post-functionalization previously discussed for core 23, we highlight the possibility to also access motor 5via post-modification of 4 by direct two-fold catalytic cross-coupling with phenyllithium (Ph-Li) (Scheme 5b). Attempts at preparing 6via similar postmodification of 4 resulted in negligible conversions as judged by crude product 1H NMR analysis. This justified the choice of the higher yielding B–K reaction to obtain 6 and 7.
Scheme 5. Synthesis of Core-Modified Third-Generation Molecular Motors via (a) Barton–Kellogg Olefination and (b) Direct Catalytic Cross-Coupling Postmodification of Motor 4 with Phenyllithium To Yield Motor 5.
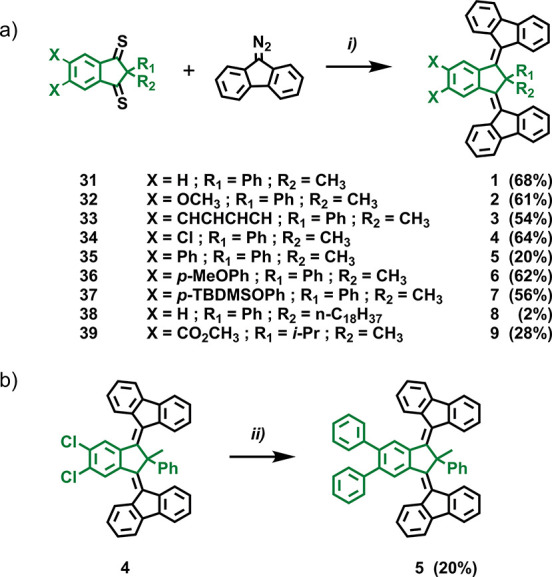
Reaction conditions: (i) diazofluorene (2.5 equiv), toluene, room temperature, 4–18 h. HMPT (2 equiv), 90 °C, 18 h; (ii) dichloro[1,3-bis(2,6-di-3-pentylphenyl)imidazol-2-ylidene](3-chloropyridyl)palladium(II) (Pd-PEPPSI-IPent catalyst, 5 mol %), dry toluene, 40 °C, 5 min. Slow addition (1 h) of PhLi (2.5 equiv in 1 mL dry toluene), 40 °C.
All newly synthesized motors were obtained as powders with colors ranging from orange to deep red depending on the nature of the core substitution. Core-modified light-driven third-generation motors 2–9 are significantly more soluble than reference motor 1 in halogenated solvents and a number of other organic solvents (acetone, THF, diethyl ether, and ethyl acetate). The presence of substituents at distal positions in the molecular structures with respect to the overcrowded alkene moieties probably results in favored solvation.
Previous work on third-generation molecular motors revealed that the substituents at pseudo-asymmetric carbon atom a (Figure 1) define the number of isomers potentially accessible from one motor structure.38 The steric hindrance of these substituents was found to be the key structural parameter.38 The two substituents at carbon a are placed one each in a pseudo-equatorial and pseudo-axial orientation and can interconvert their positions through thermal conformational isomerization. Depending on the steric hindrance exerted by the two substituents, the two conformational isomers are not degenerate, with one being energetically preferred. The more stable isomer for reference motor 1 was reported to be the one with the phenyl ring in the pseudo-axial position (s-isomer), more remote from the two fluorene moieties.38 This allows the phenyl ring to have more spatial freedom. The ratio between the two isomers with the phenyl ring placed pseudo-axially (s-isomer) and pseudo-equatorially (r-isomer), respectively, was determined to be 2:1 at room temperature by integrating the two different singlets corresponding to the methyl substituent in the 1H NMR spectrum of 1.38 The singlet of the methyl group of the s-isomer was more downfield-shifted compared to that of the r-isomer.38 The two isomers also possess different local asymmetries with respect to the two fluorene moieties, and, as a result, third-generation motors with this Me,Ph substitution pattern at carbon a are not fully unidirectional. The 2:1 isomer distribution of reference motor 1 implies that roughly 67% of the motor population rotate in one direction, while the remaining 33% rotate in the opposite direction, resulting in 34% net unidirectional rotation. The thermal interconversion between the two isomers was confirmed by the observation of signal coalescence in temperature-dependent (TD) NMR (TD-NMR) studies.38 Replacing the phenyl substituent with the more sterically demanding iso-propyl group resulted in exclusive formation of one isomer only. As a result, third-generation motors featuring Me,i-Pr substitution on carbon a are fully unidirectional; the NMR spectra are characterized by much sharper signals, and no coalescence phenomena are observed in TD-NMR studies.37,38 The newly synthesized third-generation motors 2–9 perfectly fit into this qualitative description. The 1H NMR spectra of motors 2–7, with the Me,Ph substitution pattern, show two almost overlapping singlets for the methyl substituent at room temperature. Moreover, these motors show signal coalescence upon heating/cooling of NMR samples (see the Supporting Information for 1H NMR spectra measured at 90 and −45 °C). Measuring 1H NMR spectra at −45 °C for motors 2–7 allows to separate the singlets of the methyl group of the r- and s-isomers and ultimately determine their degree of unidirectionality at the specific temperature (Table S1). The s-isomer, with the methyl group in the pseudoequatorial position, was the more stable isomer for 2–7, as suggested by the more intense downfield-shifted signal (vide supra).38 A similar analysis was performed on motor 8, bearing the C18,Ph substituents. Hence, motors 2–8 show preferred directional rotary motion, in line with previous findings.37,38 Motor 9, with the Me,i-Pr combination, instead, possesses a 1H NMR spectrum characterized by sharp signals and only one resonance for both the methyl and iso-propyl groups (see the Supporting Information) and hence should undergo fully unidirectional rotary motion.
Third-generation molecular motors possess the highest speed of rotation among the three generations of artificial molecular motors (ultrafast rotary motion). The unidirectional rotary cycle is composed of a photochemical E–Z isomerization (PEZ), followed by subsequent thermal helix inversion (THI).34 This sequence covers the first 180° rotation, while another PEZ–THI combination completes the rotary cycle.34 Assuming a high enough photon flux, the THI is considered the rate-limiting step of the rotary cycle and thus defines the rotary speed of our molecular motors. The ultrafast rotary motion of third-generation molecular motors implies very low thermal activation barriers for THI.38 Although the unidirectionality of the rotary motion is controlled by the substituents on carbon a, the speed of rotation is mainly governed by the central aromatic core. The limited steric hindrance exerted by the central aromatic core on the fluorene moieties, especially in the case of the unsubstituted benzene core, is the main reason for the high rotary speed. Earlier studies confirmed the ultrafast rotary motion looking at motor structures that featured the p-xylene core and desymmetrized fluorine rotors.37 The lack of readily available p-xylene derivatives simultaneously functionalized with two ester moieties in the 1,2-positions and two identical substituents in the 4,5-positions prevented direct measurement of the ultrafast rotary motion. The temperatures required for studying the rotation of motors 2–9 (below −110 °C) are instrumentally inaccessible and/or not compatible with the melting point of many organic solvents. Congruously, any attempt at studying the unidirectional rotary motions of motors 2–9 by 1H NMR at −80 °C via in situ irradiation with 365, 395, or 405 nm light did not afford any change in their 1H NMR spectra (see the Supporting Information). Hence, we anticipate that 2–9 undergo ultrafast rotary motion by their close structural resemblance to previous third-generation molecular motors.37,38 On one hand, this represents a challenge to address in the future for a rigorous physicochemical understanding of the behavior of these molecules using transient spectroscopy methods, which is a part of an upcoming study. On the other hand, it also makes this class of photochemically driven compounds particularly promising for the preparation of light-responsive materials and actuators because their use should result in a much faster actuation of the polymeric or supramolecular ensembles they will be embedded in.
Study of the single-crystal X-ray structures of some of the newly synthesized motors highlighted that the core-modifications introduced in the design of 2–9 resulted in significant variations compared to the previous systems. We obtained single crystals of motors 2, 4, and 5 by slow diffusion of hexane (antisolvent) into a concentrated 1,2-dichloroethane (solvent) solution of 2, 4, and 5.45 The crystal structures of motors 4 and 5 are shown in Figure 2a,b, respectively, while that of 2 is shown in Figure S88 because of its similarity to the X-ray structure of 4. Regardless of the presence of both possible isomers (Me,Ph substituents) in solution, all motors crystallized as one single isomer. The same behavior had also been observed in previous investigations,38 with reference 1 crystallizing with the phenyl substituent in the pseudo-axial orientation. Although in the single crystals of 4, the phenyl substituent is placed pseudo-axial and has more spatial freedom (Figure 2a), in the crystals of 5, we observed the opposite isomer featuring a pseudo-equatorial phenyl ring (Figure 2b). Interestingly, the two aromatic rings of the 1,1′:2′,1″-terphenyl system in 5 do not possess the same dihedral angle [45.54(17)° and 58.57(16)°] with respect to the central aromatic core, which removes the symmetry plane of the molecule in the single crystal.
Figure 2.
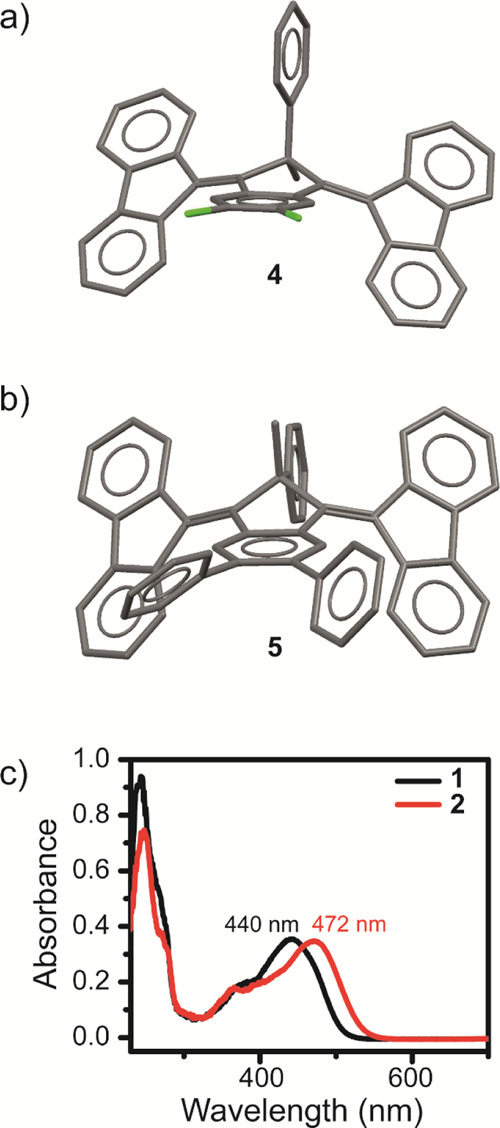
(a) Single-crystal X-ray structure of motor 4. (b) Single-crystal X-ray structure of motor 5. Hydrogen atoms are omitted for clarity in the crystal structures. Carbon atoms are depicted in gray and chlorine atoms in green. (c) UV–vis spectrum of reference motor 1 (black trace) and motor 2 (red trace) in CH2Cl2 (25 °C, 5 × 10–6 m, an optical path of 1 cm).
Finally, further shifting of the absorption spectra of light-triggered molecular motors toward the visible and near-IR regions is also highly desirable and of particular attention in recent motor designs.31,46−49 The introduction of two methoxy groups in the core of motor 2 resulted in a bathochromic shift of 32 nm compared to 1 (Figure 2c). This was already well visible in the appearance of the two motors in the solid state: while 1 is an orange powder, 2 is deeply red colored. However, although increasing the electron density by means of electron-donating substituents proved to be beneficial in this respect, preliminary studies toward electron-withdrawing substituents showed only a modest or no effect (see the Supporting Information).
Conclusions
In conclusion, the synthesis and characterization of several new light-driven third-generation molecular motors with different designs of the central aromatic core and functionalities are reported. Although in previous efforts, the focus was exclusively on benzene- and p-xylene-type cores, in this investigation, a number of moieties were explored, such as 1,2-dimethoxybenzene (2), naphthyl (3), 1,2-dichlorobenzene (4), 1,1′:2′,1″-terphenyl (5), 4,4″-dimethoxy-1,1′:2′,1″-terphenyl (6), and 1,2-dicarbomethoxybenzene (9). We present modular and scalable synthetic routes, which further offer the possibility to post-functionalize synthetic intermediates and motors. This aspect is particularly beneficial in case screening of different molecular designs is required to optimize the structure for a specific task. The structural modifications introduced in the newly synthesized motors resulted in an improved solubility compared to reference motor 1 as well as a bathochromic shift of the absorption spectra. Hence, the core modifications presented here offer ample opportunity for application/incorporation of these motors in light-responsive materials. In particular, the functionalization of the cores at the distal side with respect to the fluorene rotors makes the design of model motors 2–9 highly promising for ongoing research toward cargo transport and locomotion along tracks.
Experimental Section
Instrumentation
Microwave reactions were performed on a Discover SP Microwave Synthesizer.
Column chromatography was performed using a Grace Reveleris instrument.
1H NMR and 13C NMR spectra were recorded on a Varian Mercury Vx 400 MHz (100 MHz for 13C), Varian Oxford AS 500 MHz (125 MHz for 13C), or Bruker 600 MHz (150 MHz for 13C) NMR spectrometer. Chemical shifts are given in ppm (δ) values relative to the solvent (for CDCl3: 1H δ: 7.26 and 13C δ: 77.16; for Cl2DCCDCl2: 1H δ: 6.00 and 13C δ: 73.78). Splitting patterns are labeled s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; h, heptet; and m, multiplet.
UV–vis absorption spectra were recorded on a Jasco V-630 or a Hewlett-Packard 8453 spectrometer.
Infrared spectra were recorded on a PerkinElmer Spectrum One 1600 Fourier transform infrared (FT-IR) spectrometer or a PerkinElmer Spectrum Two FT-IR spectrometer, equipped with a PerkinElmer Universal ATR Sampler Accessory.
High-resolution mass spectra were recorded on an LTQ Orbitrap XL.
Single crystals were mounted on a cryoloop and placed in the nitrogen stream (100 K) of a Bruker-AXS D8 Venture diffractometer. Data collection and processing was carried out using the Bruker APEX3 software suite.50 A multiscan absorption correction was applied based on the intensities of symmetry-related reflections measured at different angular settings (SADABS).50 The structure was solved using either SHELXS51 (LP18006) or SHELXT52 (18005, 18011), and refinement was performed using SHELXL.51 The hydrogen atoms were generated by geometrical considerations, constrained by idealized geometries and allowed to ride on their carrier atoms with an isotropic displacement parameter related to the equivalent displacement parameter of their carrier atoms.
Materials
Compound 10 was purchased from TCI. Compound 28 was purchased from Sigma-Aldrich. All other commercially available products were purchased from TCI, Combi Blocks, or Sigma-Aldrich and used as received.
General Method A: Procedure for Esterification
The desired dicarboxylic acid (1 equiv) was loaded in a round-bottom flask and dissolved/suspended in MeOH (2.2 mL per mmol of acid) at 0 °C. Then, SOCl2 (4 equiv) was added dropwise via a syringe. The reaction mixture was heated under reflux and left overnight. After being cooled to room temperature, the volatiles were removed. The crude mixture was dissolved in dichloromethane (DCM) and washed with water (2 × 100 mL). The organic phase was then dried over MgSO4, filtered, and evaporated to achieve the desired esters. All products were used in the next step without further purification.
General Method B: NaH-Induced Procedure for the Core Preparation
The appropriate diester (1 equiv) and ketone (2 equiv) were weighed in a round-bottom flask and dissolved in toluene (2 mL per mmol of diester). Sodium hydride on oil (60 wt %, 2 equiv) was added in small portions with a spatula. The resulting suspension was stirred for 5 min at room temperature under nitrogen and then heated at reflux for 1–5 days. The starting white-gray suspensions turned deep red upon reacting. The mixtures were cooled down to room temperature. The precipitate was filtered, washed with pentane, and dried. The dried solid (powder) was loaded with KF on celite (50 wt %, 0.02 equiv) and methyl-N,N,N-trioctan-1-ammonium (0.02 equiv) in a round-bottom flask. Acetone (the same volume as toluene) was added, and the mixture was stirred for 5 min at room temperature under nitrogen. Methyl iodide (2 equiv) or 1-iodooctadecane (2 equiv) was added, and the reaction mixture was heated at reflux overnight. The color of the reaction mixture changed from deep red to yellow upon reacting. The solvent was removed, and the crude material was dissolved in CH2Cl2 and H2O. The organic phase was washed (×3) with H2O, dried over MgSO4, and filtered. The solvent was removed, and the crude material was purified by column chromatography (SiO2, gradient from 100% pentane to pentane/EtOAc mixtures).
General Method D: Procedure for Indanedithione Preparation with Conventional Heating
The appropriate indanedione (1 equiv) was added to an oven-dried round-bottom flask under nitrogen. Dry toluene (5 mL per mmol) was added, followed by the addition of P4S10 (4 equiv) and LR (4 equiv). The resulting suspension was heated overnight at reflux and monitored with TLC. When full conversion was not achieved overnight, additional P4S10 (4 equiv) was added and the reaction was kept under reflux for additional 24 h. The reaction mixture was cooled down to room temperature. The suspension was filtered over a short pad of celite, and the filtered solution (green in most cases) was evaporated. The crude product was immediately purified with column chromatography (SiO2, gradient from 100% pentane to 90:10 pentane/EtOAc). In many cases, the desired indanedithione was not obtained in 100% purity because of the presence of other products deriving from noncomplete conversion or unknown impurities. Using these slightly impure indanedithiones in the following Barton–Kellogg olefinations never affected the outcome of the reactions; hence, they did not undergo further purification.
General Method E: Procedure for Preparation of Indanedithione 39 with Microwave Heating
Indanedione 27 (400 mg; 1.26 mmol) was loaded into a 25 mL microwave vial and dissolved in toluene (10 mL). LR (8 equiv; 4.06 g; 10.05 mmol) was added, and the vial was sealed. The resulting suspension was sonicated for 5 min. The reaction mixture was heated at 110 °C for 4 h under microwave irradiation. The suspension was filtered over a short pad of celite, and the filtered solution was evaporated. The crude product was immediately purified with column chromatography (SiO2, gradient from 100% pentane to 95:5 pentane/EtOAc). Indanedithione 39 was almost always obtained in mixtures with unreacted 27 and the product of single conversion. Prolonged reaction times resulted in the degradation of 39 and/or no further conversion.
General Method F: Procedure for the Barton–Kellogg Olefination
The appropriate indanedithione (1 equiv) was dissolved in toluene (5 mL per mmol of indanedithione). Next, 9-diazo-9H-fluorenone53 (2.5 equiv) was added, and the resulting mixture was stirred at room temperature under a nitrogen atmosphere for 8 h. Hexamethylphosphanetriamine (2 equiv) was added. The mixture was heated at 90 °C overnight. The volatiles were evaporated under reduced pressure. The residue was adsorbed on celite and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%).
General Method G: Procedure for the Suzuki Couplings
The appropriate dibromo derivative (1 equiv) and boronic acid (2.5 equiv) were weighed in a round-bottom flask and dissolved/suspended in toluene (30 mL per mmol of dibromo derivative). An 8 M aqueous solution of K2CO3 (21 equiv) was added to the organic phase. The mixture was degassed for 15 min by bubbling nitrogen. Palladium(0)tetrakis(triphenylphospine) (0.1 equiv) was added. The reaction mixture was heated at reflux overnight. The reaction mixture was cooled down and loaded into a separatory funnel. The toluene layer was separated, and the aqueous layer was washed with CH2Cl2 (×3). All the organic phases were dried over MgSO4 and filtered. After solvent removal, the crude materials were purified by column chromatography (SiO2, gradient pentane/EtOAc; 0–50%).
Dimethyl 4,5-Dimethoxyphthalate (11)
Compound 11 was synthesized according to literature procedures.42,431H NMR (CDCl3, 600 MHz, δ): 7.15 (s, 2H), 3.90 (s, 6H), 3.84 (s, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 167.9, 150.7, 125.2, 111.4, 56.2, 52.6. FT-IR (dry powder) (cm–1): 3018 (C–H), 2955 (C–H), 1709 (C=O). High-resolution mass spectrometry (HR-MS) (m/z): [M + Na]+ calcd for C12H14O6Na, 276.0683; found, 276.0680 (0.8 ppm). mp 86.2–87.1 °C.
Dimethyl 4,5-Dichlorophthalate (13)
Compound 13 was synthesized with general method A (white solid; 15.8 g; 60.3 mmol; 91% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.78 (s, 2H), 3.88 (s, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 166.0, 135.8, 131.4, 131.0, 53.1. FT-IR (dry powder) (cm–1): 3096 (C–H), 3037 (C–H), 2956 (C–H), 1720 (C=O). HR-MS (m/z): [M + H]+ calcd for C10H9Cl2O4, 262.9872; found, 262.9873 (0.2 ppm). mp 44.5–46 °C.
Dimethyl 4,5-Dibromophthalate (14)
1,2-Dibromo-4,5-dimethyl-benzene (5 g; 18.94 mmol) was weighed in a custom-made Teflon beaker equipped with a Teflon cap. The compound was suspended in 35 mL of a 30 wt % HNO3 aqueous solution. A stirring bar was added, and the Teflon beaker was sealed with the Teflon cap. The Teflon reactor was inserted into an autoclave. The autoclave was placed on top of a hot plate previously set at 170 °C. The reaction mixture was left overnight, after which it was cooled down to room temperature. The Teflon reactor was extracted from the autoclave, and the reaction mixture was poured into a pH 14 aqueous solution (200 mL). The resulting solution was filtered (folded paper filter). The filtered solution was acidified with concentrated HCl (37 wt %) to induce precipitation (addition of HCl significantly developed heat; the mixture was cooled down during acidification). The precipitate was filtered (Buchner filter) and dried. The obtained white solid (1,2-dibromophthalic acid) was then subjected to esterification following general method A (white solid; 4.3 g; 12.3 mmol; 65% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.96 (s, 2H), 3.91 (s, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 166.1, 134.1, 132.0, 128.4, 53.2. FT-IR (dry powder) (cm–1): 2953 (C–H), 2923 (C–H), 2852 (C–H), 1730 (C=O). HR-MS (m/z): [M + H]+ calcd for C10H9Br2O4, 352.8842; found, 352.8841 (0.2 ppm). mp 72.6–74.3 °C.
Dimethyl [1,1′:2′,1″-Terphenyl]-4′,5′-dicarboxylate (15)
Compound 15 was synthesized with general method G and purified by column chromatography (SiO2, gradient pentane/EtOAc; 0–50%) (off-white solid; 2.12 g; 6.12 mmol; 63% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.80 (s, 2H), 7.24–7.23 (m, 6H), 7.15–7.13 (m, 4H), 3.94 (s, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 168.0, 143.6, 139.7, 131.4, 130.9, 129.8, 128.3, 127.5, 52.8. FT-IR (dry powder) (cm–1): 2955 (C–H), 1721 (C=O). HR-MS (m/z): [M + Na]+ calcd for C22H18O4Na, 369.1097; found, 369.1094 (0.9 ppm). mp 108.8–110.1 °C.
Dimethyl 4,4″-Dimethoxy-[1,1′:2′,1″-terphenyl]-4′,5′-dicarboxylate (16)
Compound 16 was synthesized with general method G and purified by column chromatography (SiO2, gradient pentane/EtOAc; 0–50%) (off-white solid; 7.05 g; 17.3 mmol; 81% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.74 (s, 2H), 7.07–7.06 (AA′BB′ system, 4H), 6.79–6.77 (AA′BB′ system, 4H), 3.93 (s, 6H), 3.78 (s, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 168.1, 159.1, 143.0, 132.2, 131.3, 130.9, 130.5, 113.8, 55.3, 52.8. FT-IR (dry powder) (cm–1): 2955 (C–H), 2841 (C–H), 1722 (C=O). HR-MS (m/z): [M + Na]+ calcd for C24H22O6Na, 429.1309; found, 429.1303 (1.4 ppm). mp 113.6–115.1 °C.
Dimethyl 4,4″-Bis((tert-butyldimethylsilyl)oxy)-[1,1′:2′,1″-terphenyl]-4′,5′-dicarboxylate (17)
Compound 17 was synthesized with general method G and purified by column chromatography (SiO2, gradient pentane/EtOAc; 0–30%) (transparent oil; 6.7 g; 11.03 mmol; 97% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.75 (s, 2H), 6.99–6.98 (AA′BB′ system, 4H), 6.71–6.69 (AA′BB′ system, 4H), 3.93 (s, 6H), 0.97 (s, 18H), 0.18 (s, 12H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 168.2, 155.3, 143.2, 132.9, 131.2, 130.9, 130.4, 120.8, 120.0, 116.0, 52.8, 25.8, 25.8, 18.4, −4.28. FT-IR (dry liquid) (cm–1): 2954 (C–H), 2930 (C–H), 2858 (C–H), 1728 (C=O), 1244 (Si–O). HR-MS (m/z): [M + H]+ calcd for HR-MS C34H47O6Si2, 607.2906; found, 607.2887 (3.1 ppm).
Tetramethyl Benzene-1,2,4,5-tetracarboxylate (18)
Compound 18 was synthesized with general method A (white solid; 25 g; 75.1 mmol; 95% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.07 (s, 2H), 3.94 (s, 12H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 166.5, 134.4, 129.8, 53.2. FT-IR (dry powder) (cm–1): 2956 (C–H), 1720 (C=O). HR-MS (m/z): [M + Na]+ calcd for C14H14O8Na, 333.0581; found, 333.0579 (0.6 ppm). mp 137.1–138.9 °C.
5,6-Dimethoxy-2-methyl-2-phenyl-1H-indene-1,3(2H)-dione (20)
Compound 20 was synthesized with general method B and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 882 mg; 2.98 mmol; 76% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.32 (s, 2H), 7.28–7.15 (m, 5H), 3.96 (s, 6H), 1.63 (s, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 201.0, 156.4, 138.4, 136.5, 129.4, 128.8, 128.8, 128.8, 127.6, 126.7, 103.9, 57.6, 56.8, 20.0. FT-IR (dry powder) (cm–1): 3012 (C–H), 2979 (C–H), 2959 (C–H), 1688 (C=O). HR-MS (m/z): [M + H]+ calcd for C18H18O4, 297.1121; found, 297.1120 (0.5 ppm). mp 168.2–170.7 °C.
2-Methyl-2-phenyl-1H-cyclopenta[b]naphthalene-1,3(2H)-dione (21)
Compound 21 was synthesized with general method B and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 1.24 g; 4.33 mmol; 40% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.58 (s, 2H), 8.12–8.11 (m, 2H), 7.73–7.71 (m, 2H), 7.39–7.38 (m, 2H), 7.31–7.23 (m, 3H), 1.78 (s, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 202.3, 138.2, 136.9, 136.3, 130.7, 129.8, 128.9, 127.7, 126.9, 125.2, 59.6, 20.3. FT-IR (dry powder) (cm–1): 2979 (C–H), 2934 (C–H), 2867 (C–H), 1688 (C=O). HR-MS (m/z): [M + H]+ calcd for C20H16O2, 287.1067; found, 287.1065 (0.5 ppm). mp 113.1–115.6 °C.
5,6-Dichloro-2-methyl-2-phenyl-1H-indene-1,3(2H)-dione (22)
Compound 22 was synthesized with general method B and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 1.01 g; 3.31 mmol; 11% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.07 (s, 2H), 7.28–7.21 (m, 5H), 1.66 (s, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 199.7, 141.6, 140.1, 137.2, 129.1, 128.1, 126.7, 125.7, 58.5, 20.3. FT-IR (dry powder) (cm–1): 2929 (C–H), 1709 (C=O). HR-MS (m/z): [M + H]+ calcd for C16H12Cl2O2, 305.0131; found, 305.0129 (0.4 ppm). mp 104.9–106.8 °C.
5,6-Dibromo-2-methyl-2-phenyl-1H-indene-1,3(2H)-dione (23)
Compound 23 was synthesized with general method B and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 4.0 g; 10.15 mmol; 62% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.25 (s, 2H), 7.27–7.21 (m, 5H), 1.66 (s, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 199.8, 140.5, 137.1, 134.5, 129.1, 129.1, 129.0, 128.1, 126.7, 58.4, 20.2. FT-IR (dry powder) (cm–1): 3072 (C–H), 2934 (C–H), 1707 (C=O). HR-MS (m/z): [M + H]+ calcd for C16H12Br2O2, 392.9120; found, 392.9189 (0.4 ppm). mp 145.6–147.1 °C.
2-Methyl-2,5,6-triphenyl-1H-indene-1,3(2H)-dione (24)
Compound 24 was synthesized with general method B and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 1.26 g; 3.25 mmol; 61% yield). Compound 24 was also synthesized with general method G and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 900 mg; 2.32 mmol; 50% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.07 (s, 2H), 7.41–7.39 (m, 2H), 7.34–7.31 (m, 2H), 7.27–7.23 (m, 7H), 7.16–7.14 (m, 4H), 1.76 (s, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 201.8, 149.2, 140.3, 139.7, 138.1, 129.7, 129.0, 128.4, 128.1, 127.8, 126.9, 125.9, 58.6, 20.3. FT-IR (dry powder) (cm–1): 3060 (C–H), 1707 (C=O). HR-MS (m/z): [M + H]+ calcd for C22H22O2, 389.1536; found, 389.1534 (0.4 ppm). mp 195.3–197.2 °C.
5,6-Bis(4-methoxyphenyl)-2-methyl-2-phenyl-1H-indene-1,3(2H)-dione (25)
Compound 25 was synthesized with general method B (off-white solid; 2.07 g; 0.53 mmol; 18% yield) and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture). Compound 25 was also synthesized with general method G and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (off-white solid; 950 mg; 2.12 mmol; 55% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.02 (s, 2H), 7.40–7.39 (m, 2H), 7.34–7.31 (m, 2H), 7.27–7.25 (m, 1H), 7.10–7.09 (AA′BB′ system, 4H), 6.82–6.80 (AA′BB′ system, 4H), 3.80 (s, 6H), 1.75 (s, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 201.9, 159.5, 148.7, 140.0, 138.2, 132.2, 131.0, 129.0, 127.7, 126.9, 125.7, 114.0, 58.5, 55.4, 20.2. FT-IR (dry powder) (cm–1): 3059 (C–H), 2961 (C–H), 1701 (C=O). HR-MS (m/z): [M + H]+ calcd for C30H25O4, 449.1747; found, 449.1738 (2.0 ppm). mp 196.2–198.7 °C.
5,6-Bis(4-((tert-butyldimethylsilyl)oxy)phenyl)-2-methyl-2-phenyl-1H-indene-1,3(2H)-dione (26)
Compound 26 was synthesized with general method G and purified by column chromatography (SiO2, gradient from 100% pentane to 8:2 pentane/EtOAc mixture) (off-white solid; 622 mg; 0.96 mmol; 75% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.02 (s, 2H), 7.40–7.39 (m, 2H), 7.34–7.31 (m, 2H), 7.27–7.25 (m, 1H), 7.02–7.01 (AA′BB′ system, 4H), 6.74–6.73 (AA′BB′ system, 4H), 1.75 (s, 3H), 0.98 (s, 18H), 0.19 (s, 12H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 201.9, 155.8, 148.9, 140.0, 138.2, 132.8, 131.0, 129.6, 129.0, 128.9, 127.7, 126.9, 125.6, 120.2, 58.5, 25.8, 20.2, 18.4, −4.3. FT-IR (dry powder) (cm–1): 2931 (C–H), 2858 (C–H), 1703 (C=O), 1266 (Si–O). HR-MS (m/z): [M + H]+ calcd for C40H49O4Si2, 649.3164; found, 649.3147 (2.6 ppm). mp 134.2–136.5 °C.
Dimethyl 2-Isopropyl-2-methyl-1,3-dioxo-2,3-dihydro-1H-indene-5,6-dicarboxylate (27)
Compound 27 was synthesized with general method B and purified by column chromatography (SiO2, gradient from 100% pentane to 7:3 pentane/EtOAc mixture) (yellow solid; 3.07 g; 9.65 mmol; 30% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.25 (s, 2H), 3.96 (s, 6H), 2.16 (h, J = 6 Hz, 1H), 1.27 (s, 3H), 0.91 (d, J = 6 Hz, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 203.5, 166.4, 142.7, 138.6, 123.9, 57.5, 53.4, 34.7, 18.1, 17.4. FT-IR (dry powder) (cm–1): 2957 (C–H), 2876 (C–H), 1729 (C=O), 1710 (C=O). HR-MS (m/z): [M + H]+ calcd for C17H19O6, 319.1176; found, 319.1175 (0.4 ppm). mp 92.2–94.6 °C.
5,6-Dimethoxy-2-phenyl-1H-indene-1,3(2H)-dione (29)
Compound 29 was synthesized adapting a literature procedure.44 (off-white solid; 7.78 g; 27.5 mmol; 31% yield). 1H NMR (CDCl3, 600 MHz, δ): 7.40 (s, 2H), 7.34–7.28 (m, 3H), 7.18–7.16 (m, 2H), 4.20 (s, 1H), 4.04 (s, 6H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 197.4, 156.3, 138.0, 133.9, 129.1, 128.8, 127.9, 103.8, 59.6, 56.9. FT-IR (dry powder) (cm–1): 3006 (C–H), 2946 (C–H), 1686 (C=O). HR-MS (m/z): [M + H]+ calcd for C17H15O4, 283.0965; found, 283.0961 (1.3 ppm). mp 101.2–103.5 °C.
2-Octadecyl-2-phenyl-1H-indene-1,3(2H)-dione (30)
Compound 30 was synthesized with general method B applying only the alkylation part on 2-phenyl-1H-indene-1,3(2H)-dione38 and purified by column chromatography (SiO2, gradient from 100% pentane to 9:1 pentane/EtOAc mixture) (off-white solid; 2.89 g; 6.09 mmol; 83% yield). 1H NMR (CDCl3, 600 MHz, δ): 8.03–8.02 (m, 2H), 7.86–7.85 (m, 2H), 7.42–7.40 (m, 2H), 7.30–7.28 (m, 2H), 7.24–7.22 (m, 1H), 2.25 (t, J = 9 Hz, 2H), 1.30–1.13 (m, 35H), 0.88 (t, J = 6 Hz, 3H). 13C NMR {1H} (CDCl3, 150 MHz, δ): 202.2, 142.2, 137.4, 136.0, 128.9, 127.7, 127.0, 123.7, 62.5, 36.5, 32.1, 30.1, 29.8, 29.8, 29.8, 29.7, 29.7, 29.6, 29.5, 29.3, 25.4, 22.8, 14.3. FT-IR (dry powder) (cm–1): 2949 (C–H), 2916 (C–H), 2850 (C–H), 1705 (C=O). HR-MS (m/z): [M + H]+ calcd for C33H47O2, 475.3571; found, 475.3557 (2.9 ppm). mp 63.2–64.3 °C.
In some cases, indanedithiones 31–39 were not obtained in 100% purity. Hence, we only report their 1H NMR spectra. However, the impurities present in 31–39 did not affect the subsequent B–K olefinations to obtain motors 1–9.
5,6-Dimethoxy-2-methyl-2-phenyl-1H-indene-1,3(2H)-dithione (32)
Compound 32 was synthesized with general method D and purified by column chromatography (SiO2, gradient from 100% pentane to 9:1 pentane/EtOAc) (green solid; 180 mg; 0.55 mmol; 33% yield). 1H NMR (CDCl3, 300 MHz, δ): 7.40 (s, 2H), 7.21–7.17 (m, 5H), 4.09 (s, 6H), 1.92 (s, 3H).
2-Methyl-2-phenyl-1H-cyclopenta[b]naphthalene-1,3(2H)-dithione (33)
Compound 33 was synthesized with general method D and purified by column chromatography (SiO2, gradient from 100% pentane to 90:10 pentane/EtOAc) (green solid; 576 mg; 1.81 mmol; 52% yield). 1H NMR (CDCl3, 300 MHz, δ): 8.63 (s, 2H), 8.15–8.12 (m, 2H), 7.72–7.69 (m, 2H), 7.22–7.12 (m, 5H), 2.00 (s, 3H).
5,6-Dichloro-2-methyl-2-phenyl-1H-indene-1,3(2H)-dithione (34)
Compound 34 was synthesized with general method D and purified by column chromatography (SiO2, gradient from 100% pentane to 9:1 pentane/EtOAc) (green solid; 275 mg; 0.81 mmol; 32% yield). 1H NMR (CDCl3, 400 MHz, δ): 8.15 (s, 2H), 7.24–7.15 (m, 5H), 1.91 (s, 3H).
2-Methyl-2,5,6-triphenyl-1H-indene-1,3(2H)-dithione (35)
Compound 35 was synthesized with general method D and purified by column chromatography (SiO2, gradient from 100% pentane to 9:1 pentane/EtOAc). The compound was not obtained in high purity (green solid; 120 mg; 2.85 mmol; 35% yield). 1H NMR (CDCl3, 400 MHz, δ): 8.14 (s, 2H), 7.31–7.29 (m, 10H), 7.25–7.22 (m, 5H), 2.01 (s, 3H).
5,6-Bis(4-methoxyphenyl)-2-methyl-2-phenyl-1H-indene-1,3(2H)-dithione (36)
Compound 36 was synthesized with general method D and purified by column chromatography (SiO2, gradient from 100% pentane to 9:1 pentane/EtOAc) (green solid; 113 mg; 0.23 mmol; 45% yield). 1H NMR (CDCl3, 400 MHz, δ): 8.05 (s, 2H), 7.28–7.21 (m, 5H), 7.16–7.14 (AA′BB′ system, 4H), 6.83–6.80 (AA′BB′ system, 4H), 3.81 (s, 6H), 1.96 (s, 3H).
5,6-Bis(4-((tert-butyldimethylsilyl)oxy)phenyl)-2-methyl-2-phenyl-1H-indene-1,3(2H)-dione (37)
Compound 37 was synthesized with general method D and purified by column chromatography (SiO2, gradient from 100% pentane to 95:5 pentane/EtOAc) (green solid; 200 mg; 0.29 mmol; 48% yield). 1H NMR (CDCl3, 400 MHz, δ): 8.06 (s, 2H), 7.25–7.20 (m, 5H), 7.08–7.06 (AA′BB′ system, 4H), 6.75–6.73 (AA′BB′ system, 4H), 1.97 (s, 3H), 0.98 (s, 18H), 0.19 (s, 12H).
Dimethyl 2-Isopropyl-2-methyl-1,3-dioxo-2,3-dihydro-1H-indene-5,6-dicarboxylate (27)
Compound 27 was synthesized with general method E and purified by column chromatography (SiO2, gradient from 100% pentane to 9:1 pentane/EtOAc). However, this procedure afforded a singly converted product in all attempts, with the only exception of one case in which desired 39 was obtained (green solid; 181 mg; 0.51 mmol; 33% yield). 1H NMR (CDCl3, 400 MHz, δ): 8.27 (s, 2H), 3.96 (m, 6H), 2.41 (h, J = 7 Hz, 1H), 1.52 (s, 3H), 0.86 (d, J = 7 Hz, 6H).
Motor 2
Motor 2 was synthesized with general method F and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%) (deep red solid; 200 mg; 0.34 mmol; 61% yield). 1H NMR (Cl2DCCDCl2, 500 MHz, 90 °C, δ) (signals of the main isomer): 8.41 (d, J = 6 Hz, 2H), 7.80 (d, J = 6 Hz, 2H), 7.72 (d, J = 6 Hz, 2H), 7.68 (d, J = 6 Hz, 2H), 7.65 (s, 2H), 7.30 (t, J = 6 Hz, 3H), 7.27 (t, J = 6 Hz, 3H), 7.18 (t, J = 6 Hz, 3H), 7.13 (t, J = 6 Hz, 3H), 3.86 (s, 6H), 2.44 (s, 3H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 160.7, 157.2, 154.3, 152.2, 150.2, 149.7, 142.2, 140.9, 140.4, 140.1, 139.6, 139.5, 138.9, 137.6, 137.4, 137.3, 132.9, 130.4, 128.7, 128.6, 128.0, 127.5, 127.3, 127.2, 127.0, 126.8, 126.7, 126.7, 126.6, 126.2, 126.0, 125.8, 123.6, 119.9, 119.6, 119.5, 119.4, 110.7, 105.4, 71.4, 68.7, 56.9, 56.7, 56.6, 23.4, 19.4. HR-MS (m/z): [M + H]+ calcd for C44H33O2, 593.2475; found, 593.2461 (2.3 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 247 (149,200), 471 (69,400). mp 260–262 °C. Single crystals for X-ray diffraction (XRD) were obtained from slow diffusion of hexane (antisolvent) into a saturated solution of 1,2-dichloroethane (solvent).
Motor 3
Motor 3 was synthesized with general method F and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%) (orange solid; 395 mg; 0.678 mmol; 54% yield). 1H NMR (Cl2DCCDCl2, 500 MHz, 90 °C, δ): 8.72 (s, 2H), 8.64 (d, J = 6 Hz, 2H), 7.84–7.82 (m, 4H), 7.80–7.78 (m, 2H), 7.71 (d, J = 6 Hz, 2H), 7.68–7.67 (m, 2H), 7.57–7.55 (m, 4H), 7.73 (t, J = 6 Hz, 3H), 7.29–7.26 (m, 3H), 7.15–7.12 (m, 5H), 2.48 (s, 3H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 159.5, 156.6, 144.0, 141.8, 141.6, 140.6, 140.2, 140.1, 140.00, 139.6, 139.4, 137.5, 135.6, 134.3, 134.0, 133.5, 133.5, 132.0, 130.9, 129.5, 129.0, 128.9, 128.8, 128.6, 128.0, 127.8, 127.7, 127.5, 127.3, 127.2, 126.5, 126.4, 126.2, 125.2, 125.0, 123.1, 122.9, 119.6, 119.5, 119.4, 118.8, 70.0, 67.6, 20.1, 19.7. HR-MS (m/z): [M + H]+ calcd for C46H31, 583.2420; found, 583.2410 (1.8 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 244 (239,352), 400 (94,064). mp > 300 °C.
Motor 4
Motor 4 was synthesized with general method F and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%) (orange solid; 313 mg; 0.52 mmol; 64% yield). 1H NMR (Cl2DCCDCl2, 500 MHz, 90 °C, δ) (signals of the main isomer): 8.35 (d, J = 6 Hz, 2H), 8.32 (s, 2H), 7.73–7.68 (m, 5H), 7.63 (d, J = 6 Hz, 2H), 7.34 (t, J = Hz, 3H), 7.26 (t, J = 6 Hz, 3H), 7.21 (t, J = 6 Hz, 3H), 7.10 (t, J = 6 Hz, 3H), 2.42 (s, 3H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 157.1, 154.4, 154.2, 147.2, 144.6, 140.8, 140.4, 140.3, 140.2, 139.8, 139.6, 138.5, 138.4, 137.1, 135.3, 135.0, 134.4, 134.0, 133.1, 132.7, 130.7, 130.4, 129.6, 128.8, 128.3, 128.2, 128.1, 127.9, 127.7, 127.5, 126.6, 126.4, 125.4, 125.3, 124.4, 123.5, 123.3, 122.8, 120.3, 120.2, 119.8, 119.6, 119.5, 119.0, 70.7, 68.5, 19.7, 18.9. HR-MS (m/z): [M + H]+ calcd for C42H27Cl2, 601.1484; found, 601.1481 (0.5 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 241 (156,766), 441 (59,518). mp 269–271 °C. Single crystals for XRD were obtained from slow diffusion of hexane (antisolvent) into a saturated solution of 1,2-dichloroethane (solvent).
Motor 5
Motor 4 (108 mg; 0.18 mmol) and the Pd-PEPPSI-IPent complex (5 mol %) were dissolved in toluene (4 mL) in a dried Schlenk flask under N2. The mixture was stirred at 40 °C for 5 min. Subsequently, a toluene solution (1 mL) of PhLi (2.5 equiv) was added over 1 h by the use of a syringe pump. After complete addition, MeOH (1 mL) was added to quench the remaining PhLi. The reaction mixture was transferred to a round-bottom flask, celite was added, and the solvents were evaporated in vacuo. Purification with column chromatography (SiO2, elution in gradient from 100% pentane to 100% DCM) afforded motor 5 (red solid; 25 mg; 0.036 mmol; 20% yield). Motor 5 was also synthesized with general method F (50 mg; 0.072 mmol; 20% yield from 24). 1H NMR (Cl2DCCDCl2, 500 MHz, 90 °C, δ) (signals of the main isomer): 8.63 (d, J = 6 Hz, 2H), 8.30 (s, 2H), 7.80 (d, J = 6 Hz, 3H), 7.69–7.64 (m, 6H), 7.31–7.11 (m, 20H), 2.50 (s, 3H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 159.8, 156.6, 147.0, 144.6, 141.5, 141.3, 140.5, 140.2, 140.1, 140.0, 139.9, 139.8, 139.6, 139.1, 137.5, 135.6, 133.2, 132.7, 131.9, 131.4, 129.8, 129.8, 129.3, 128.9, 128.7, 128.5, 128.3, 128.0, 127.7, 127.6, 127.5, 127.4, 127.3, 127.0, 126.9, 126.6, 126.3, 126.2, 126.1, 125.1, 123.7, 123.3, 119.6, 119.4, 118.9, 70.7, 68.6, 19.9, 19.4. HR-MS (m/z): [M + H]+ calcd for C54H37, 685.2890; found, 685.2881 (1.3 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 246 (209192), 391 (63390), 449 (86394). mp > 300 °C. Single crystals for XRD were obtained from slow diffusion of hexane (antisolvent) into a saturated solution of 1,2-dichloroethane (solvent).
Motor 6
Motor 6 was synthesized with general method F and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%) (red solid; 108 mg; 0.14 mmol; 62% yield). 1H NMR (Cl2DCCDCl2, 500 MHz, 90 °C, δ) (signals of the main isomer): 8.62 (d, J = 6 Hz, 2H), 8.25 (s, 2H), 7.79 (d, J = 6 Hz, 2H), 7.68 (d, J = 6 Hz, 2H), 7.64 (d, J = 6 Hz, 2H), 7.29 (t, J = Hz, 3H), 7.25 (t, J = 6 Hz, 3H), 7.18 (t, J = 6 Hz, 3H), 7.15 (d, J = 6 Hz, 4H), 7.12 (t, J = 6 Hz, 3H), 6.80 (d, J = 6 Hz, 4H), 3.82 (s, 6H), 2.49 (s, 3H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 160.0, 158.1, 158.0, 156.9, 146.6, 144.2, 141.4, 141.0, 140.8, 140.6, 139.9, 139.7, 139.5, 139.2, 137.5, 135.6, 132.8, 132.7, 132.6, 132.4, 131.7, 131.0, 130.9, 130.5, 128.7, 127.6, 127.4, 127.2, 126.7, 126.3, 126.2, 125.1, 123.7, 123.3, 119.5, 119.4, 113.2, 70.6, 68.5, 55.3, 19.8, 19.5. HR-MS (m/z): [M + H]+ calcd for C56H41O2, 745.3101; found, 745.3088 (1.7 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 244 (206,000), 395 (69,400), 450 (84,200). mp 268–270 °C.
Motor 7
Motor 7 was synthesized with general method F and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%) (red solid; 120 mg; 0.13 mmol; 56% yield). 1H NMR (Cl2DCCDCl2, 500 MHz, 90 °C, δ) (signals of the main isomer): 8.62 (d, J = 6 Hz, 2H), 8.26 (s, 2H), 7.79 (d, J = 6 Hz, 2H), 7.68 (d, J = 6 Hz, 2H), 7.65 (d, J = 6 Hz, 2H), 7.30 (t, J = Hz, 3H), 7.25 (t, J = 6 Hz, 3H), 7.18 (t, J = 6 Hz, 3H), 7.12 (t, J = 6 Hz, 3H), 7.09 (d, J = 6 Hz, 4H), 6.72 (d, J = 6 Hz, 4H), 2.49 (s, 3H), 1.03 (s, 18 H), 0.23 (s, 12H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 160.1, 156.9, 154.4, 154.3, 146.6, 144.2, 141.3, 141.1, 140.6, 139.9, 139.9, 139.7, 139.5, 139.2, 137.5, 135.6, 133.5, 133.3, 132.8, 132.3, 131.5, 131.0, 130.9, 128.7, 127.6, 127.4, 127.2, 126.6, 126.2, 123.4, 119.7, 119.5, 119.4, 70.7, 68.5, 29.9, 25.6, 19.8, 19.6, 18.2, −4.4. HR-MS (m/z): [M + H]+ calcd for C66H65O2Si2, 945.4518; found, 945.4505 (1.4 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 246 (178,420), 396 (59,400), 451 (72,400). mp 248–251.6 °C.
Motor 8
Motor 8 was synthesized with general method F, after column chromatography (SiO2, gradient pentane/DCM; 0–100%) and preparative TLC (SiO2, pentane/EtOAc 98:2) (orange solid; 32 mg; 0.041 mmol; 2% yield from 30). 1H NMR (CDCl3, 500 MHz, −45 °C, δ): 8.45 (d, J = 8.0 Hz, 2H), 8.24–8.22 (m, 2H), 7.75 (d, J = 8.0 Hz, 2H), 7.71–7.67 (m, 4H), 7.39–7.25 (m, 11H), 7.15 (t, J = 8 Hz, 2H), 7.19 (t, J = 8 Hz, 2H), 3.04 (m, 2H), 1.23 (m, 20H), 1.01 (m, 2H), 0.87 (m, 5H), 0.74 (m, 2H), 0.67 (m, 2H), 0.56 (m, 2H), 0.44 (m, 2H), 0.34 (m, 2H). 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 155.1, 148.7, 141.0, 140.0, 139.8, 138.8, 137.4, 133.3, 130.5, 129.1, 128.5, 127.5, 127.4, 127.4, 126.8, 126.1, 126.0, 123.5, 119.4, 119.3, 72.06, 32.1, 30.7, 29.9, 29.9, 29.9, 29.8, 29.6, 29.6, 29.5, 29.0, 28.9, 27.9, 24.7, 22.9, 14.5. HR-MS (m/z): [M + H]+ calcd for C59H63, 771.4924; found, 771.4912 (1.5 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 241 (105,922), 453 (40,224). mp 194.2–196.6 °C.
Motor 9
Motor 9 was synthesized with general method F and purified by column chromatography (SiO2, gradient pentane/CH2Cl2; 0–100%) (orange solid; 90 mg; 0.146 mmol; 28% yield). 1H NMR (CDCl3, 600 MHz, 25 °C, δ): 8.44 (s, 2H), 8.20 (d, J = 6 Hz, 2H), 8.01 (d, J = 6 Hz, 2H), 7.77 (d, J = 6 Hz, 2H), 7.73 (d, J = 6 Hz, 2H), 7.39 (t, J = 6 Hz, 2H), 7.34–7.30 (m, 6H), 7.13 (t, J = 6 Hz, 2H), 3.89 (s, 6H), 3.01 (h, J = 6 Hz, 1H), 2.39 (s, 3H), 1.07 (d, J = 6 Hz, 6H) [hexamethylphosphoramide (HMPA) present in the sample due to a strong interaction with the compound; the doublet at 2.65 in the 1H NMR spectrum belongs to HMPA]. 13C NMR {1H} (CDCl3, 125 MHz, −45 °C, δ): 167.8, 155.8, 150.2, 145.2, 140.9, 139.7, 139.6, 138.1, 136.1, 134.3, 131.1, 129.1, 128.9, 128.3, 128.2, 127.8, 127.3, 126.7, 126.4, 125.2, 123.5, 120.0, 119.7, 119.6, 75.0, 70.0, 53.3, 39.9, 28.8, 24.2. HR-MS (m/z): [M + H]+ calcd for C43H35O4, 615.2530; found, 615.25174(2.0 ppm). UV–vis (CH2Cl2) λmax, nm (ε): 238 (196,366), 374 (49,228), 440 (65,830). mp > 300 °C.
Acknowledgments
Renze Sneep (University of Groningen) is acknowledged for HR-MS measurements, and Pieter van der Meulen (University of Groningen) is acknowledged for assistance during NMR experiments. This work was supported financially by the European Research Council (ERC, advanced grant no. 694345 to B.L.F.), the Dutch Ministry of Education, Culture and Science (Gravitation Program no. 024.001.035), and the European Commission (MSCA-IF no. 793082 to L.P.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01235.
Crystallographic data of 2 (CIF)
Crystallographic data of 4 (CIF)
Crystallographic data of 5 (CIF)
List of all synthesized compounds, 1H NMR and 13C NMR spectra, UV–vis spectra of motors 1–9, FT-IR spectra, crystal structure of motors 2, 4, and 5, TD-1H NMR experiments with motor 6, and 1H NMR in situ irradiation experiments at −80 °C with motor 6 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sauvage J.-P. Transition Metal-Containing Rotaxanes and Catenanes in Motion: Toward Molecular Machines and Motors. Acc. Chem. Res. 1998, 31, 611–619. 10.1021/ar960263r. [DOI] [Google Scholar]
- Balzani V.; Credi A.; Raymo F. M.; Stoddart J. F. Artificial Molecular Machines. Chem. Rev. 2000, 39, 3348–3391. . [DOI] [PubMed] [Google Scholar]
- Molecular Machines and Motors; Sauvage J.-P., Ed.; Springer-Verlag Berlin Heidelberg, 2001. [Google Scholar]
- Stoddart J. F. Molecular Machines. Acc. Chem. Res. 2001, 34, 410–411. 10.1021/ar010084w. [DOI] [PubMed] [Google Scholar]
- Browne W. R.; Feringa B. L. Making Molecular Machines Work. Nat. Nanotechnol. 2006, 1, 25–35. 10.1038/nnano.2006.45. [DOI] [PubMed] [Google Scholar]
- Champin B.; Mobian P.; Sauvage J.-P. Transition Metal Complexes as Molecular Machine Prototypes. Chem. Soc. Rev. 2007, 36, 358–366. 10.1039/b604484k. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Bergamini G.; Ceroni P. From the Photochemistry of Coordination Compounds to Light-Powered Nanoscale Devices and Machines. Coord. Chem. Rev. 2008, 252, 2456–2469. 10.1016/j.ccr.2007.11.009. [DOI] [Google Scholar]
- Kay E. R.; Leigh D. A.; Zerbetto F. Synthetic Molecular Motors and Mechanical Machines. Angew. Chem., Int. Ed. 2007, 46, 72–191. 10.1002/anie.200504313. [DOI] [PubMed] [Google Scholar]
- Coskun A.; Banaszak M.; Astumian R. D.; Stoddart J. F.; Grzybowski B. A. Great Expectations: Can Artificial Molecular Machines Deliver on Their Promise?. Chem. Soc. Rev. 2012, 41, 19–30. 10.1039/c1cs15262a. [DOI] [PubMed] [Google Scholar]
- Erbas-Cakmak S.; Leigh D. A.; McTernan C. T.; Nussbaumer A. L. Artificial Molecular Machines. Chem. Rev. 2015, 115, 10081–10206. 10.1021/acs.chemrev.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay E. R.; Leigh D. A. Rise of the Molecular Machines. Angew. Chem., Int. Ed. 2015, 54, 10080–10088. 10.1002/anie.201503375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassem S.; van Leeuwen T.; Lubbe A. S.; Wilson M. R.; Feringa B. L.; Leigh D. A. Artificial Molecular Motors. Chem. Soc. Rev. 2017, 46, 2592–2621. 10.1039/c7cs00245a. [DOI] [PubMed] [Google Scholar]
- Moulin E.; Faour L.; Carmona-Vargas C. C.; Giuseppone N. From Molecular Machines to Stimuli-Responsive Materials. Adv. Mater. 2020, 32, 1906036. 10.1002/adma.201906036. [DOI] [PubMed] [Google Scholar]
- Baroncini M.; Silvi S.; Credi A. Photo- And Redox-Driven Artificial Molecular Motors. Chem. Rev. 2020, 120, 200–268. 10.1021/acs.chemrev.9b00291. [DOI] [PubMed] [Google Scholar]
- Feringa B. L. The Art of Building Small: From Molecular Switches to Molecular Motors. J. Org. Chem. 2007, 72, 6635–6652. 10.1021/jo070394d. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Credi A.; Venturi M. Light Powered Molecular Machines. Chem. Soc. Rev. 2009, 38, 1542. 10.1039/b806328c. [DOI] [PubMed] [Google Scholar]
- Baroncini M.; Groppi J.; Corra S.; Silvi S.; Credi A. Light-Responsive (Supra)Molecular Architectures: Recent Advances. Adv. Opt. Mater. 2019, 7, 1900392. 10.1002/adom.201900392. [DOI] [Google Scholar]
- Dattler D.; Fuks G.; Heiser J.; Moulin E.; Perrot A.; Yao X.; Giuseppone N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. 10.1021/acs.chemrev.9b00288. [DOI] [PubMed] [Google Scholar]
- Silvi S.; Venturi M.; Credi A. Light Operated Molecular Machines. Chem. Commun. 2011, 47, 2483–2489. 10.1039/c0cc03829f. [DOI] [PubMed] [Google Scholar]
- Greb L.; Lehn J.-M. Light-Driven Molecular Motors: Imines as Four-Step or Two-Step Unidirectional Rotors. J. Am. Chem. Soc. 2014, 136, 13114–13117. 10.1021/ja506034n. [DOI] [PubMed] [Google Scholar]
- Gerwien A.; Mayer P.; Dube H. Photon-Only Molecular Motor with Reverse Temperature-Dependent Efficiency. J. Am. Chem. Soc. 2018, 140, 16442–16445. 10.1021/jacs.8b10660. [DOI] [PubMed] [Google Scholar]
- Filatov M.; Paolino M.; Min S. K.; Kim K. S. Fulgides as Light-Driven Molecular Rotary Motors: Computational Design of a Prototype Compound. J. Phys. Chem. Lett. 2018, 9, 4995–5001. 10.1021/acs.jpclett.8b02268. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Schildhauer M.; Rott F.; Huber L. A.; Guentner M.; Thumser S.; Hoffmann K.; Oesterling S.; de Vivie-Riedle R.; Riedle E.; et al. Complete Mechanism of Hemithioindigo Motor Rotation. J. Am. Chem. Soc. 2018, 140, 5311–5318. 10.1021/jacs.8b02349. [DOI] [PubMed] [Google Scholar]
- Nikiforov A.; Gamez J. A.; Thiel W.; Filatov M. Computational Design of a Family of Light-Driven Rotary Molecular Motors with Improved Quantum Efficiency. J. Phys. Chem. Lett. 2016, 7, 105–110. 10.1021/acs.jpclett.5b02575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C.-Y.; Lu H.-F.; Chao I.; Yang J.-S. A Rotary Molecular Motor Gated by Electrical Energy. Org. Lett. 2014, 16, 6100–6103. 10.1021/ol502946v. [DOI] [PubMed] [Google Scholar]
- Li Q.; Fuks G.; Moulin E.; Maaloum M.; Rawiso M.; Kulic I.; Foy J. T.; Giuseppone N. Macroscopic Contraction of a Gel Induced by the Integrated Motion of Light-Driven Molecular Motors. Nat. Nanotechnol. 2015, 10, 161–165. 10.1038/nnano.2014.315. [DOI] [PubMed] [Google Scholar]
- Foy J. T.; Li Q.; Goujon A.; Colard-Itté J.-R.; Fuks G.; Moulin E.; Schiffmann O.; Dattler D.; Funeriu D. P.; Giuseppone N. Dual-Light Control of Nanomachines That Integrate Motor and Modulator Subunits. Nat. Nanotechnol. 2017, 12, 540–545. 10.1038/nnano.2017.28. [DOI] [PubMed] [Google Scholar]
- Chen K.-Y.; Ivashenko O.; Carroll G. T.; Robertus J.; Kistemaker J. C. M.; London G.; Browne W. R.; Rudolf P.; Feringa B. L. Control of Surface Wettability Using Tripodal Light-Activated Molecular Motors. J. Am. Chem. Soc. 2014, 136, 3219–3224. 10.1021/ja412110t. [DOI] [PubMed] [Google Scholar]
- Danowski W.; van Leeuwen T.; Abdolahzadeh S.; Roke D.; Browne W. R.; Wezenberg S. J.; Feringa B. L. Unidirectional Rotary Motion in a Metal–Organic Framework. Nat. Nanotechnol. 2019, 14, 488–494. 10.1038/s41565-019-0401-6. [DOI] [PubMed] [Google Scholar]
- Martinez-Bulit P.; Stirk A. J.; Loeb S. J. Rotors, Motors, and Machines Inside Metal–Organic Frameworks. Trends Chem. 2019, 1, 588–600. 10.1016/j.trechm.2019.05.005. [DOI] [Google Scholar]
- Danowski W.; Castiglioni F.; Sardjan A. S.; Krause S.; Pfeifer L.; Roke D.; Comotti A.; Browne W. R.; Feringa B. L. Visible Light Driven Rotation of Molecular Motors in a Dual-Function Metal Organic Framework Enabled by Energy Transfer. J. Am. Chem. Soc. 2020, 142, 9048–9056. 10.1021/jacs.0c03063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feringa B. L. The Art of Building Small: From Molecular Switches to Molecular Motors. J. Org. Chem. 2007, 72, 6635–6652. 10.1021/jo070394d. [DOI] [PubMed] [Google Scholar]
- Feringa B. L. In Control of Motion: From Molecular Switches to Molecular Motors. Acc. Chem. Res. 2001, 34, 504–513. 10.1021/ar0001721. [DOI] [PubMed] [Google Scholar]
- Roke D.; Wezenberg S. J.; Feringa B. L. Molecular Rotary Motors: Unidirectional Motion around Double Bonds. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 9423–9431. 10.1073/pnas.1712784115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumura N.; Zijlstra R. W. J.; van Delden R. A.; Harada N.; Feringa B. L. Light-Driven Monodirectional Molecular Rotor. Nature 1999, 401, 152–155. 10.1038/43646. [DOI] [PubMed] [Google Scholar]
- Koumura N.; Geertsema E. M.; Van Gelder M. B.; Meetsma A.; Feringa B. L. Second Generation Light-Driven Molecular Motors. Unidirectional Rotation Controlled by a Single Stereogenic Center with near-Perfect Photoequilibria and Acceleration of the Speed of Rotation by Structural Modification. J. Am. Chem. Soc. 2002, 124, 5037–5051. 10.1021/ja012499i. [DOI] [PubMed] [Google Scholar]
- Kistemaker J. C. M.; Štacko P.; Visser J.; Feringa B. L. Unidirectional Rotary Motion in Achiral Molecular Motors. Nat. Chem. 2015, 7, 890–896. 10.1038/nchem.2362. [DOI] [PubMed] [Google Scholar]
- Kistemaker J. C. M.; Štacko P.; Roke D.; Wolters A. T.; Heideman G. H.; Chang M.-C.; Van Der Meulen P.; Visser J.; Otten E.; Feringa B. L. Third-Generation Light-Driven Symmetric Molecular Motors. J. Am. Chem. Soc. 2017, 139, 9650–9661. 10.1021/jacs.7b04412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudernac T.; Ruangsupapichat N.; Parschau M.; Maciá B.; Katsonis N.; Harutyunyan S. R.; Ernst K.-H.; Feringa B. L. Electrically Driven Directional Motion of a Four-Wheeled Molecule on a Metal Surface. Nature 2011, 479, 208–211. 10.1038/nature10587. [DOI] [PubMed] [Google Scholar]
- Mosher W. A.; Soeder R. W. Reactions of Some Methylene Ketones with Dimethyl Phthalate. A New Route to 2-Substituted 1,3-Indandiones. J. Org. Chem. 1971, 36, 1561–1563. 10.1021/jo00810a028. [DOI] [Google Scholar]
- Murthy A. R.; Wyrick S. D.; Hall I. H. Hypolipidemic Activity of Indan-l,3-Dione Derivatives in Rodents. J. Med. Chem. 1985, 28, 1591–1596. 10.1021/jm00149a008. [DOI] [PubMed] [Google Scholar]
- Anderson D. R.; Koch T. H. 2,3-Bis(Trimethylsilyloxy)-l,3-Butadiene as a Useful Reactive Diene in the Diels-Alder Reaction. J. Org. Chem. 1978, 43, 2726–2728. 10.1021/jo00407a048. [DOI] [Google Scholar]
- Yang Q.; Ma W.; Wang G.; Bao W.; Dong X.; Liang X.; Zhu L.; Lee C.-S. Tunable Cyclization Strategy for the Synthesis of Zizaene-, Allo -Cedrane-, Seco -Kaurane-, and Seco -Atesane-Type Skeletons. Org. Lett. 2017, 19, 5324–5327. 10.1021/acs.orglett.7b02610. [DOI] [PubMed] [Google Scholar]
- Vile J.; Carta M.; Bezzu C. G.; Mckeown N. B. Tribenzotriquinacene-Based Polymers of Intrinsic Microporosity. Polym. Chem. 2011, 2, 2257–2260. 10.1039/c1py00294e. [DOI] [Google Scholar]
- CCDC 2005072 [2], 20050723 [4], and 2005074 [5] Contain the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge from The Cambridge Crystallographic Data Center.
- van Leeuwen T.; Pol J.; Roke D.; Wezenberg S. J.; Feringa B. L. Visible-Light Excitation of a Molecular Motor with an Extended Aromatic Core. Org. Lett. 2017, 19, 1402–1405. 10.1021/acs.orglett.7b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roke D.; Feringa B. L.; Wezenberg S. J. A Visible-Light-Driven Molecular Motor Based on Pyrene. Helv. Chim. Acta 2019, 102, e1800221 10.1002/hlca.201800221. [DOI] [Google Scholar]
- Roke D.; Sen M.; Danowski W.; Wezenberg S. J.; Feringa B. L. Visible-Light-Driven Tunable Molecular Motors Based on Oxindole. J. Am. Chem. Soc. 2019, 141, 7622–7627. 10.1021/jacs.9b03237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer L.; Scherübl M.; Fellert M.; Danowski W.; Cheng J.; Pol J.; Feringa B. L. Photoefficient 2nd Generation Molecular Motors Responsive to Visible Light. Chem. Sci. 2019, 10, 8768–8773. 10.1039/c9sc02150g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruker . APEX3 (V2016.1-0), SAINT (Version 8.37A) and SADABS (Version 2014/5); Bruker AXS Inc.: Madison, Wisconsin, USA, 2016. [Google Scholar]
- Sheldrick G. M. A short history ofSHELX. Acta Crystallogr Sect. A 2008, 64, 112–122. 10.1107/s0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. SHELXT- Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. 10.1107/s2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M.; He Z.; Gao B.; Li L.; Ni C.; Hu J. Copper-Catalyzed Gem -Difluoroolefination of Diazo Compounds with TMSCF 3 via C–F Bond Cleavage. J. Am. Chem. Soc. 2013, 135, 17302–17305. 10.1021/ja409941r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.