

Summary
Heavy‐chain‐only antibodies (HCAbs), which are devoid of light chains, have been found naturally occurring in various species including camelids and cartilaginous fish. Because of their high thermostability, refoldability and capacity for cell permeation, the variable regions of the heavy chain of HCAbs (VHHs) have been widely used in diagnosis, bio‐imaging, food safety and therapeutics. Most immunogenetic and functional studies of HCAbs are based on case studies or a limited number of low‐throughput sequencing data. A complete picture derived from more abundant high‐throughput sequencing (HTS) data can help us gain deeper insights. We cloned and sequenced the full‐length coding region of VHHs in Alpaca (Vicugna pacos) via HTS in this study. A new pipeline was developed to conduct an in‐depth analysis of the HCAb repertoires. Various critical features, including the length distribution of complementarity‐determining region 3 (CDR3), V(D)J usage, VJ pairing, germline‐specific mutation rate and germline‐specific scoring profiles (GSSPs), were systematically characterized. The quantitative data show that V(D)J usage and VHH recombination are highly biased. Interestingly, we found that the average CDR3 length of classical VHHs is longer than that of non‐classical ones, whereas the mutation rates are similar in both kinds of VHHs. Finally, GSSPs were built to quantitatively describe and compare sequences that originate from each VJ pair. Overall, this study presents a comprehensive landscape of the HCAb repertoire, which can provide useful guidance for the modeling of somatic hypermutation and the design of novel functional VHHs or VHH repertoires via evolutionary profiles.
Keywords: antibody diversity, high‐throughput sequencing, immune repertoire, nanobody, protein design
A pipeline was developed to conduct an in‐depth analysis of the next‐generation sequencing data repertoire of the heavy‐chain‐only antibodies (HCAbs) in Vicugna pacos. Both naive and immunized samples were sequenced and analyzed to provide a comprehensive landscape of the HCAb repertoire.
Abbreviations
- AAs
amino acids
- ASR
average substitution rat
- CDR
complementarity‐determining region
- GSSPs
germline‐specific scoring profiles
- HCAbs
heavy‐chain‐only antibodies
- HTS
high‐throughput sequencing
- MSAs
multiple sequence alignments
- PCR
polymerase chain reaction
- SR
substitution rat
- VHHs
the variable regions of heavy chain of HCAbs
Introduction
The antigen‐binding domain of functional heavy‐chain‐only antibodies (HCAbs) discovered in camelids and sharks is composed of a single variable domain. 1 , 2 The variable regions of heavy chain of HCAbs (VHHs), also known as nanobodies, have attracted growing interest in various applications, as they are more soluble and stable than canonical antibodies (VHs). 3 , 4 , 5 , 6 In camels, the ratio of HCAbs to total IgG can reach more than 80%, which indicates that HCAbs play a significant role in immune protection. 7 However, it is obvious that the diversity of HCAbs is dramatically lower than that of canonical antibodies because of the lack of variable heavy chain and variable light chain (VH/VL) combinational diversification. This raises a question of how HCAbs can compete with canonical antibodies. Several hypotheses and observations have been proposed to address the problem of diversity reduction inherent to HCAbs. One hypothesis is that the complementarity‐determining region 3 (CDR3) of VHHs contains longer loops than canonical antibody VHs (18 amino acids versus 13 amino acids), which helps to compensate for the lack of diversity. 8 Evidently, longer CDR3 length increases the paratope size, as well as the three‐dimensional structural diversity and contact surface area with antigens. 9 Another explanation, inferred from a structural study that compared two independently generated anti‐lysozyme nanobodies, is that the in vivo maturation and selection systems are strong enough to compensate for the decrease in the VHHs primary repertoire. 10
High‐throughput sequencing (HTS) technology enables scientists to evaluate millions of sequences in parallel, resulting in the collection of more complete and comprehensive information for target samples. This capability makes HTS suitable for the characterization of immune repertoires that are highly plastic and diverse. Although HTS is now routinely applied in studies of human adaptive immunity, 11 vaccine development 12 and diagnostic research, 13 only a few studies were tried on VHHs. Fridy et al. developed a pipeline combining HTS and proteomics to identify specific VHHs. 14 Similarly, Turner et al. demonstrated that HTS can be used as a complementary tool for phage‐display bio‐panning to rapidly obtain additional clones from an immune VHH library. 15 For the first time, Li et al. compared the repertoires of classical antibodies and HCAbs of Bactrian camels, with analysis data including CDR3 length distribution, mutation rate, characteristic amino acids, the distribution of cysteine codons, and the non‐classical VHHs. 8 Nevertheless, the features of HCAbs, such as the germline usage and mutation preferences, remain unknown. Like classical immunoglobulin heavy chains, VHHs are encoded by recombined V(D)J genes that are formed from sets of Variable (V), Diversity (D) and Joining (J) genes (IGHV, IGHD, IGHJ) on the genome. An in‐depth analysis of the origin and mutation profiles of VHHs would help us to better understand the diversity of the HCAb repertoire, as well as the diversity compensation. Furthermore, appropriate interpretation of the information is important to guide the design of novel functional VHHs. 16 , 17
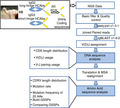
This study is mainly focused on the HCAb repertoire. First, the coding sequences of VHHs from long‐hinge HCAbs (IgG2) and short‐hinge HCAbs (IgG3) were amplified from the non‐immunized and the antigen‐immunized antibody repertoires of Vicugna pacos, where full‐length coding sequences of VHHs were obtained by an Illumina MiSeq System (2 × 300) under the paired‐end module. Next, a new pipeline combined with multiple software tools was developed to characterize the diversity and evolutionary features of the VHHs, including CDR3 length distribution, V(D)J usage, VJ pairing, DJ pairing, germline‐specific mutation rate and germline‐specific scoring profiles (GSSPs) (Fig. 1). Considering that the diversity of antibody repertoires is position, chain, and species‐dependent, 18 , 19 , 20 comparative studies are also made on amino acid sequences derived from different germline genes.
Figure 1.
Workflow for the analysis of heavy‐chain‐only antibodies (HCAbs) repertoire. The coding sequences of variable regions of the heavy chain of HCAbs (VHHs) are amplified from long‐ and short‐hinge HCAbs, respectively, where the next‐generation sequencing (NGS) data are processed using fastq‐join to merge paired‐end sequences after discarding the low‐quality reads. Next, IgBLAST is used to assign V(D)J genes for each transcript. Coding sequence (CDS) length distribution, V(D)J usage, VJ pairing and DJ pairing usage are based on DNA sequences, other analyses are based on amino acid sequences.
Materials and methods
RNA extraction and reverse transcription
Peripheral blood mononuclear cells were separated from peripheral blood by Ficoll‐1.077 (Sangon, Shanghai, China) gradient centrifugation, separately. Three naive blood samples were collected from three non‐immunized healthy male alpaca (Vicugna pacos). To collect immunized blood samples, one donor was immunized by subcutaneous, lower‐back injections every 2 weeks. Samples of fresh blood were collected 1 week after the fifth and seventh immunizations. For each blood sample, RNA was purified from approximately 2 × 107 peripheral blood mononuclear cells using RNAprep Kit (Tiangen, Beijing, China), following the manufacturer’s instruction. First‐strand complementary DNA (cDNA) was synthesized with random hexamer primers using a PrimeScript™ RT‐PCR Kit (TAKARA, Dalian, China), and then stored at −80°C. 21
Library construction and Illumina sequencing
The VHH coding region was amplified from cDNA by a nested polymerase chain reaction (PCR) as described before. 22 , 23 In brief, the variable region was first amplified by primers AlpVh‐LD and AlpVHH, which anneal to the conserved region of the leading sequence and CH2 region, respectively. Next, the PCR products were diluted as a template for the second round of PCR, which employed primer pairs AlpVHH‐F/AlpVHH‐R1 and AlpVHH‐F/AlpVHH‐R2 to amplify coding sequences of short‐ and long‐hinge heavy‐chain antibodies, respectively. The PCR products that encoded VHHs (~450 bp) were purified using TAKARA gel extraction kits, and then subjected to next‐generation sequencing by the Beijing Genomics Institute sequencing center. Sequences were generated with a MiSeq System using a 2 × 300 paired‐end module.
Basic data processing
Adapter sequences were first checked and removed from the reads. Then, the reads that bases of ‘N’ were >10% or have >50% bases with quality values ≤5 were discarded, resulting in 14·13 × 2 million paired‐end reads. The pairwise reads were joined using the fastq‐join tool (version 1.3.1). 24 The main parameters were the maximum difference percentage (8%) and the minimum base overlap (6 bp). Phylogenetic trees of V germline genes were built using MEGA version X. 25
V(D)J assignment and numbering
The V(D)J germline gene sequences were obtained from the international ImMunoGeneTics information system (IMGT) antibody repertoire database. 26 The out‐frame bases in the 3′ end of J gene sequences were manually deleted, where the elaborated germline gene sequences were used to build an IgBLAST database. The resulting five 85 million joined sequences were subjected to IgBLAST1.8.0 with default parameters. 27 The origin of each sequence, either from long‐hinge or short‐hinge IgGs, was identified by BLAST 2.7.1+ according to the E‐value and sequence identity of the alignments. 28 The V(D)J germline genes on the top of the resulting list of IgBLAST were assigned to the sequences. An in‐house Python script was used to analyze VHH length distribution, CDR3 length distribution, V/D/J germline gene usage, VJ pairing, DJ pairing and amino acid substitution. The IMGT numbering system was adopted for the coding sequences of VHHs.
Construction and comparison of GSSPs
The sequences were translated and aligned to all alleles of the gene. Sequences with more than one stop codon or no amino acid substitution were discarded. Sequences belonging to the same VJ germline gene were parsed from IgBLAST output to build multiple sequence alignments (MSAs); redundant amino acid sequences were removed in each MSA. To improve the accuracy of sequence alignment, the V and J segments of each amino acid sequence in an MSA were re‐aligned with corresponding IMGT numbered germline sequences using an in‐house NW‐align program (Y. Zhang, https://zhanglab.ccmb.med.umich.edu/NW‐align/) before GSSP construction. The GSSPs were built and compared as described in previous work. 20 In brief, the MSAs whose number of sequences was greater than a given threshold (e.g. 100, 500 and 1000) were used to build GSSPs. A divergence matrix between GSSPs was calculated; each element in the matrix was the Jensen–Shannon divergence calculated between each pair of sequences from the MSAs. The R function cmdscale was used for multidimensional scaling and generating coordinates for plotting. The logo plot of MSA was drawn using a stand‐alone version of weblogo 3.6. 29
Calculation of substitution frequencies for the 20 amino acids
The GSSPs were used to calculate the substitution frequencies. The substitution rate (SR) from each GSSP was calculated by
| (1) |
where fi is the mutation frequency of sequence i to the corresponding germline genes. L is the length of the GSSP, and N is the total sequences in the MSA. The average substitution rate (ASR) for the 20 AAs of a GSSP is calculated by
| (2) |
where ASR(a, b) is the average substitution rate of amino acid a in germline gene substituted by observed amino acid b in MSA, fi(a, b) is the frequency of amino acid a in germline gene substituted by amino acid b at the position i of an MSA, f(a) is the frequency of amino acid a in germline sequence, L is the length of the MSA, and N is the total sequences in the MSA.
Statistical analysis
To investigate the likelihood of pairing preference between germline segments, we used an in silico simulation protocol as described in a previous study. 30 Briefly, in each simulation, an equal number of real data sequences was constructed using the same individual frequencies of V, D and J segments observed in the real data. After 2000 simulation steps, the DJ and VJ pairing that appeared in each simulation were counted. The relative deviation (RD) of minimum, maximum and real frequencies of each kind of pairing were calculated by
| (3) |
where x is the minimum or maximum frequencies of simulation, or frequencies of real sequence data, and is the average frequency of each pairing in the 2000 simulation steps.
We used the function spearmanr in the python module scipy to calculate the Spearman’s rank correlation coefficient to evaluate the statistical dependence of the germline usage, VJ and DJ pairings, and the substitution preference between samples.
Results
Sequence data filtration and formation
A summary of the sequencing data sets processed in this study is shown in Table 1. The MiSeq sequencing of the non‐immune and antigen‐experienced HCAb repertoires yielded a total of 38·25 × 2 million reads. As the sample Naive‐1 generated the most sequencing reads (14·13 × 2 million reads), it was used to build and test the pipeline. A number of 2 550 856 unique DNA sequences were subjected to IgBLAST to identify the germline gene origination of each sequence, after the redundant DNA sequences of the joined paired‐end reads were removed. Both V and J germline genes are found in more than 97% of the non‐redundant DNA sequences. Following these filtrations, a total of 2 490 298 unique DNA sequences with VJ assignment hits were used to determine the coding sequence (CDS) distribution, V(D)J usage, VJ pairing and DJ pairing. Briefly, the CDS length distribution centers around 375 bp and follows an approximately normal distribution, where the maximum CDS length is 438 bp in the data set (see Fig. S1). A number of 1 973 186 unique amino acid sequences deduced from this data set were used to construct multiple sequence alignments (MSAs), to analyze CDR3 length distribution, and to calculate substitution rates and construct GSSPs. VHHs from long‐hinge and short‐hinge HCAbs were identified and analyzed for comparison.
Table 1.
Summary of sequencing data sets in this work
| Sample 1 | Data 3 (counts) | Joined data 3 (counts) | Unique DNA data (counts) | Unique amino acid data (counts) |
|---|---|---|---|---|
| Naive‐1 | 14 130 770 × 2 | 5 850 191 | 2 550 586 | 1 973 186 |
| Naive‐2 | 9 783 739 × 2 | 6 381 831 | 2 317 312 | 1 717 133 |
| Naive‐3 | 7 939 987 × 2 | 5 285 912 | 1 763 675 | 1 271 695 |
| Immune‐1 | 2 935 298 × 2 | 1 841 952 | 1 270 186 | 782 529 |
| Immune‐2 | 3 459 366 × 2 | 2 270 196 | 1 653 673 | 1 149 386 |
Naive‐1, Naive‐2 and Naive‐3 were collected from three healthy donors; Immune‐1 and Immune‐2 were collected from one donor after fifth and seventh immunizations, respectively.
Data are the total sequences of paired‐end reads after filter and quality control.
Joined data are the number of sequences that were generated from paired‐end reads.
Germline gene usage
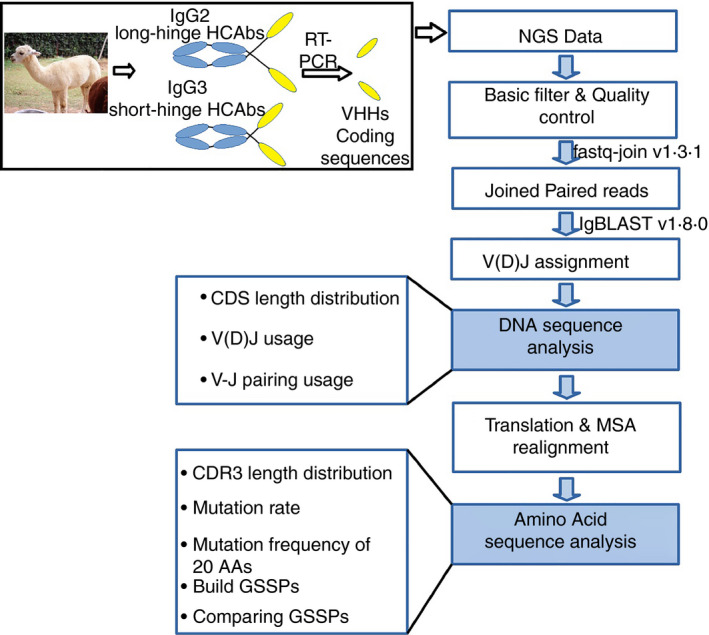
Studies of canonical antibody repertoires have demonstrated that specific V, D and J germline genes have very different frequencies in humans and mice. 30 , 31 , 32 , 33 Meanwhile, HCAbs and canonical IgGs in the alpaca (Vicugna pacos) genome have been shown to originate from the same IgH locus, which is composed of 88 V genes (including four pseudogenes), eight D genes and seven J genes. 34 Here, we used the tool IgBLAST to determine the origination of V, D and J of each clone. The 84 functional V genes, eight D genes and seven J genes were employed to create a reference database for IgBLAST. The IgBLAST results showed that the V, D and J segment usages have strong preferences for specific germline genes (Fig. 2).
Figure 2.
V, D and J germline gene usage of variable regions of the heavy chain of heavy‐chain‐only antibodies (VHHs) repertoire is highly biased. (a) Usage of V germline genes. (b) Usage of D germline genes. (c) Usage of J germline genes.
The V segments of all clones were generated from the subgroups of IgHV3. The V segments IGHV3S65*01, IGHV3S3‐3*01 and IGHV3S53*01 are used by more than 10% of all the clones, whereas the top 11 V germline genes are used by more than 95% of all the clones (Fig. 2a). All the 17 V germline genes, which contain at least two framework region 2 (FR2) hallmark residues, F37, E44, R45 and G47 in the Kabat numbering system, 35 are in a sub‐cluster of IgHV3 (see Fig. S2). Germline genes from this sub‐cluster contribute more than 85% of V gene usage (Fig. 2a). These hallmark residues are considered to be important for the solubility and stability of VHHs, as well as the VH/VL association of conventional VHs. A novel promiscuous class of VHHs that do not have any FR2 imprints was reported in Sanger sequencing studies. 36 , 37 It is now clear that sequences that lack FR2 imprints are generated from other V germline genes, in which IGHV3S39*01, IGHV3S41*01, IGHV3S25*01, IGHV3‐1*01, IGHV3S9*01 and IGHV3S1*01 constitute the top six contributors. These hallmark‐free V segments are responsible for about 10% of V gene usage in the data set.
The usage of D segments was relatively evenly distributed across the germline genes, where six out of eight D germline genes have above 10% usage (Fig. 2b). Similar to the V gene usage, the J germline gene usage was also highly biased (Fig. 2c). For instance, the germline gene IGHJ4*01 was used by two‐thirds of the J segments. As only a few sequences were assigned to IGHJ5*01 and IGHJ1*01 (0·15% and 0·09%, respectively), we manually checked the DNA and corresponding amino acid sequences. The IGHJ5*01 hits of J segments were correctly assigned by IgBLAST. However, because of the defects in the 3′ sequences, all the IGHJ1*01 assignments were false positives, indicating that VHHs never use IGHJ1*01. These sequences were therefore discarded in the subsequent analyses.
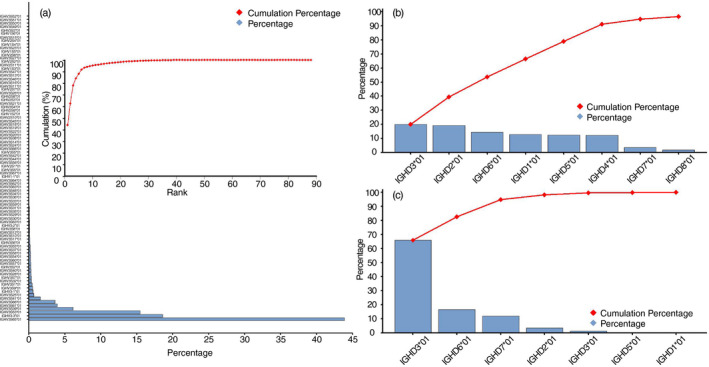
V(D)J recombination preferences
VJ pairing data showed that more than 90% of the VJ pairs are composed of genes from the top 21 most used VJ germline gene combinations (Fig. 3a), indicating that VJ pairing is biased. Theoretically, the combination of VJ pairing should be much more than 21, even though the V and J usage is highly biased toward specific germline genes. To evaluate whether V(D)J pairing exhibits bias, simulated antibody repertoires were employed to test statistical preference. As the V(D)J recombination occurs in two steps to assemble a complete variable region in vivo, we first analyzed the DJ pairing, and then the VJ pairing. Although most relative deviation of the real data was <100%, DJ pairing showed a preference (Fig. 3b). The VJ pairing results indicated a stronger bias, as the relative deviations of the six types of VJ combination were more than 200% (Fig. 3c). Notably, all the highly biased VJ pairings were from FR2 hallmark‐free V germline genes, which were IGHV3‐1*01, IGHV3S1*01, IGHV3S25*01 and IGHV3S39*01.
Figure 3.
V(D)J pairing of variable regions of the heavy chain of heavy‐chain‐only antibodies (VHHs) is biased. (a) VJ pairing of germline genes in the naive VHH repertoire is highly biased to pairs of certain germline genes. The top 21 VJ pair made up >90% of the clones in the repertoire. (b, c) In silico simulation of DJ and VJ combination. The error bars illustrate the relative deviation of the 2000 steps of simulation, while the columns represent the relative deviation of the VHHs repertoire.
CDR3 length and distribution
The CDR3 length of VHHs from the HTS data mainly ranged from 4 to 34 amino acids, according to the IMGT numbering system (Fig. 4). The overall average length of HCAbs CDR3 is 18 amino acids, consistent with previous studies. 8 We found that the shortest and longest CDR3 lengths were 2 and 39 amino acids, respectively, although they were quite rare. Interestingly, VHHs derived from various germline genes showed different CDR3 length distributions (see Table S1), indicating a bias of insertion during the process of in vivo V(D)J recombination. Hence, we further compared the sequences derived from the top 11 V germline genes (Fig. 4). Notably, the results showed that the average CDR3 length of clones derived from hallmark germline genes is longer than that of hallmark‐free germline genes, except IGV3S9*01.
Figure 4.
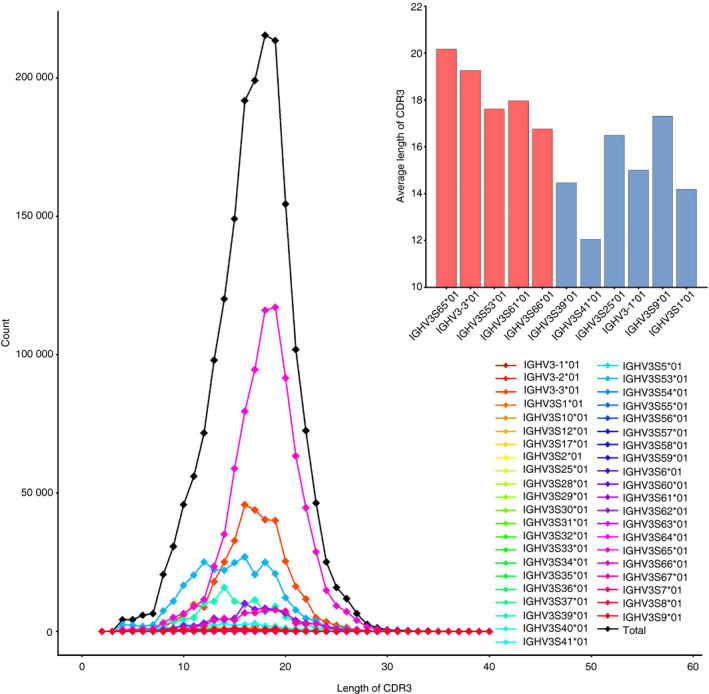
Distribution of CDR3 amino acid length is dependent on germline gene families. The average CDR3 lengths of the top 11 V germline genes are presented in the upper right panel. Hallmark and non‐hallmark residue V genes are shown in red and blue bars, respectively.
Substitution and insertion analysis
As CDR3 contains random insertions and is highly diverse, only the VJ paired segments were used for substitution analysis. The SR, which represents the mutation strength of a VJ pair lineage, ranged from 12% to 22% (see Table S2). To analyze the substitution preference of each amino acid, we calculated the ASR of the VJ pairing that comprises >1000 lineages, and then overall ASR. The results demonstrated that partial substitutions tended to be biased and most types of mutations were rare (Fig. 5). As to the overall ASR, 79 out of 441 substitution types were higher than 1%. Insertion of glycine (20·78%) and alanine (12·18%) were preferred at the tip of the CDR1 and CDR2 loop. Meanwhile, we found that each germline VJ pair showed various substitution patterns. Therefore, we further calculated the GSSPs to quantify and compare the diversity clustered by each germline VJ pair.
Figure 5.
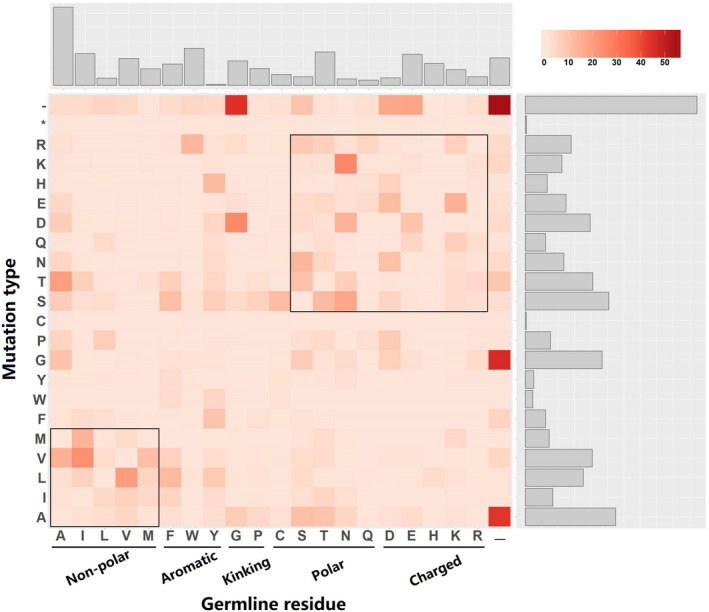
Most substitutions are rare, and partial mutations are preferred. Most observed types of substitution occur rarely because of mutation bias. The histograms in the upper and right are the sums of the cases in the corresponding column or row, respectively. The asterisk in mutation type means stop codon. Dashes mean gaps in both germline residues and mutation types.
Construction and comparison of VHH profiles
A GSSP that captures the frequency at which each amino acid appears at every position in an MSA is an N‐by‐L matrix, where N is the number of residue types and L is the alignment length. The weighted average of the Jensen–Shannon divergence between GSSPs was calculated to quantitatively compare different profiles and then visualized using multidimensional scaling. To test the robustness of this quantification method for GSSPs, we calculated Jensen–Shannon divergence of VJ pairing types that have more than 100, 500 or 1000 lineages, respectively. The results confirmed that lineages from common V genes tend to be clustered, no matter what cut‐off values were used (Fig. 6). Moreover, plotting Jensen–Shannon divergence of the top 11 V germline family showed that some classes are close to each other, indicating that mutation patterns are similar between clustered families (Fig. 6b, d, and f).
Figure 6.
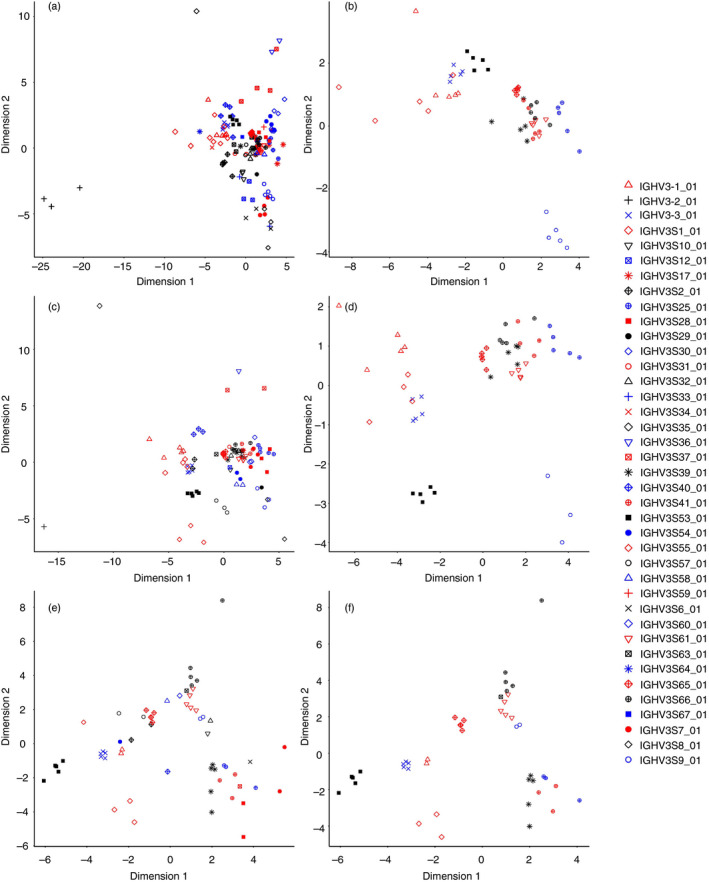
The similarity of germline‐specific scoring profiles (GSSPs) between VJ germline gene families. Jensen–Shannon divergence was used to compare GSSPs, and the distance matrix was visualized using multidimensional scaling. The VJ pair types that have >100 (a and b), >500 (c and d), or >1000 (e and f) lineages were calculated and plotted respectively. GSSPs of the same V gene tend to be clustered together. Meanwhile, the distance of partial VJ pair is close, which indicates sharing similar mutation patterns. VJ pairs from the top 11 V genes are shown in the right column (b, d and f).
Comparison of long‐hinge and short‐hinge HCAbs
Specific primers were designed to amplify IgG2 and IgG3, which enabled the identification of each clone type. The IgG‐specific primer sequences were found in 5 674 954 sequences (97% of all unique sequences). Comparison of IgG2 and IgG3 showed that the ranks of J gene usage were the same, but ranks of V and D usage were different, indicating a different preference of V and D segments (Fig. 7). Notably, a bias of gene rearrangement was observed for these two types of HCAbs. The top five hallmark V germline genes contribute 90·91% of long‐hinge (IgG2) clones, but only 75·30% of short‐hinge (IgG3).
Figure 7.
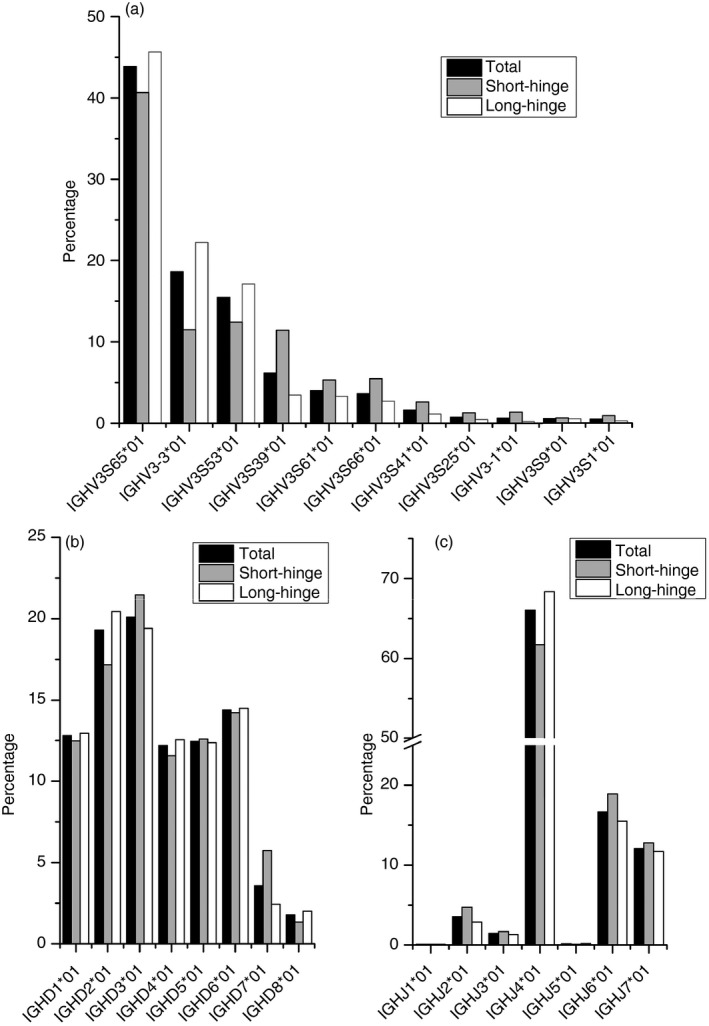
Comparison of V(D)J usage of long‐hinge (IgG2) and short‐hinge (IgG3) heavy‐chain‐only antibodies (HCAbs). Sequences belonging to IgG2 or IgG3 were identified using specific primers for BLAST. (a–c) are the V, D and J gene usage of both types of HCAbs, respectively.
Comparison of VHHs from different donors
To test the robustness of the pipeline, the HTS sequences from four other peripheral blood samples (Naive‐2, Naive‐3, Immune‐1 and Immune‐2), which were collected from the non‐immunized and immunized donors (Table 1), were processed following the same pipeline, respectively. The V and J germline usage, VJ pairing, DJ pairing and the substitution preference were highly correlated between the five samples (see Table S3). Interestingly, the correlations of the D germline usage are low between samples (see Table S3), especially between the naive and the immunized samples (Spearman rank correlation coefficients: Immune‐1 and Naive‐1, ρ = 0·683, P = 0·042; Immune‐1 and Naive‐3, ρ = 0·467, P = 0·205).
Discussion
HCAbs occur naturally in various species, such as camelids (e.g. camels and llamas) and cartilaginous fish (e.g. sharks). 38 This remarkable evolutionary convergence implies the advantages of functional HCAbs. Hence, systematic investigation of HCAb repertoires is important to reveal the mystery of evolutionary conservation as well as to understand the compensation for the lack of diversity in HCAbs. In this study, we developed a novel pipeline to analyze the full coding sequences of variable domains. In order to automatically process data and maintain its reliability, we tried to avoid using arbitrary filters in the workflow when possible, and checked the intermediate results from each step. As MSAs are crucial for calculating substitution rates and building GSSPs, a classic NW‐align algorithm was employed to re‐align MSAs from IgBLAST. To mitigate effects from noise in the data, we set 1000 as the minimum number of lineages to calculate ASR. The pipeline can be easily extended to analyze the HTS data of antibody repertoires from other species.
V(D)J recombination is one of the mechanisms of antibody diversity. A previous study confirmed that the V germline genes of HCAbs and conventional IgGs were located in the same IgH locus on the genome. 34 The hallmark residues in FR2 regions have been shown to be characteristic of VHHs. A previous study reported the presence of novel hallmark‐free variable domains that can be rearranged to both camelid classical antibodies and HCAbs. 36 Here, we found that more than 10% of hallmark‐free sequences are in the non‐immunized HCAb repertoire, indicating an increase in the HCAbs diversity by sharing V germline genes with tetrameric IgGs. Interestingly, the germline gene IGHV3S39*01 contributes about 60% of V segment usage among all non‐hallmark V germlines. The biological mechanism of how hallmark‐free HCAbs are developed is still unknown. It is well accepted that imunoglobulin heavy chains are selected at the pre‐B‐cell receptor checkpoint. Martin et al. found specific structural requirements (CDR3 length and amino composition) to select immunoglobulin μ heavy chains during maturation of the pre‐B stage. 39 In our data set, the hallmark residues (F37, E44 and G47), which usually form a contact interface with the CDR3 to stabilize the structure, show greater diversity than the others in FR2 except for those near CDR regions (see Fig. S3). Based on our observations, we infer that partial VH germline genes, if not all, are capable of rearranging to HCAbs, but only a small portion (~10%) pass the pre‐B checkpoint. Nevertheless, our data confirm that germline gene usage shows a high preference for specific genes. Five out of 17 hallmark‐containing germline V segments are responsible for 85·54% of V gene usage in the data set (Fig. 2a).
Studies of antibody repertoires from humans and mice demonstrated that germline gene usage is dynamic during vaccination or infection. Hence, we investigated non‐immunized samples from three individuals and two samples from one antigen‐injected animal with a 2‐week interval. The results show that the V and J germline usage, VJ pairing, DJ pairing and amino acid substitution preference are highly correlated whether antigen immunized or not (see Table S3). This high similarity is in accordance with a recent work that revealed the high prevalence of shared clonotypes in human B‐cell repertoires. 40 In contrast, the D germline usage shows poor correlations, especially between the naive and immunized samples, indicating that the D fragment seems to be the main driving force for in vivo antibody maturation.
The CDR3 is the most polymorphic region of IgGs and the main contributor to antigen binding. 41 , 42 The CDR3 loop in VHHs of dromedary is longer than the loop in VHs from humans or mice (average of 14 or 13 amino acids, respectively). 43 Longer loop lengths increase the paratope size and consequently help compensate for the diversity loss that occurs when light chains are absent. 9 Analysis of 114 conventional camel antibodies showed that CDR3 length is dependent on the germline gene family. 44 Our HTS data demonstrated that the distribution of CDR3 length varies on V germline families, indicating that the length of CDR3 loops is determined by the usage of the germline gene. This finding is in accordance with studies of T‐cell receptor, whose repertoire distribution patterns depend on the use of the germline genes. 45 , 46
Somatic hypermutation has long been known as a key process for increasing diversity and improving the affinity of antibodies. Comparative analysis of the immune repertoire between the conventional antibodies and the HCAbs from the Bactrian camel showed that the nucleotide mutation rate of HCAbs is higher than that of canonical antibodies. 8 In contrast, the calculation of the amino acid mutation rate shows no significant difference in the substitution rate between hallmark and non‐hallmark germline gene families (see Table S2). However, the substitution patterns of each VJ family did not converge (Fig. 6). Quantitative analysis of GSSPs also confirmed the diversity of the lineages that originated from various VJ germline genes.
A recent study on antibody maturation showed that antibodies that respond to the same antigen to a large extent share similar amino acid substitutions. 47 It appears that the conserved antibody structures that drive adaptive immune responses are highly limited and selected. 31 This is consistent with the results of dominant mutations in HCAbs, suggesting the existence of some preferred mutation patterns. For the result of overall ASR, we observed that non‐polar amino residues tend to mutate to non‐polar amino acids; polar and charged residues are more likely to be substituted by polar and charged amino acids (Fig. 5, a boxed region in heat‐map). Phenylalanine (F), alanine (A), serine (S) and aspartic acid (D) are the germline residues that are most preferred to be substituted (Fig. 5, upper histogram), while alanine (A), serine (S), glycine (G) and aspartic acid (D) are the residues that are most likely to be mutated (Fig. 5, right histogram).
The new pipeline developed in this work has revealed novel and detailed features of the HCAbs repertoire, which is important for VHH engineering or design. The GSSPs built in this work can describe the mutation sequence space of variable domains of antibodies. 20 In previous studies, we have shown that coupling with appropriate evolutionary profile information, our evolution‐based protein design protocol, evodesign, exhibits a high accuracy in designing protein folds 48 , 49 and protein–protein interactions. 16 , 17 The preference of germline usage and mutation of HCAbs can be very useful to reduce the effective searching space of amino acid sequences and increase the accuracy of protein design.
Disclosures
The authors have declared no competing interests.
Authors’ contributions
ZT and YZ conceived and designed the study. ZT developed the computer code, carried out the statistical analysis and wrote the initial draft. ZT, XH and YZ reviewed and edited the manuscript. JF, NH and YL conducted the experiments of sample collection and deep sequencing. WZ participated in the data visualization. All authors read and approved the final manuscript.
Supporting information
Figure S1. Distribution of CDS length of VHHs.
Figure S2. Phylogenetic tree of V germline genes in the IgH locus of Vicugna pacos. The phylogenetic tree is built using MEGA version X. The germline genes, which contain at least two hallmark residues, are shown in the rectangular box.
Figure S3. Position specific score matrix of sample Naive‐1. The logo plot was built using weblogo version 3.6. The hallmark residues are labelled with asterisk, corresponding to F37, E44, R45 and G47 in the Kabat numbering system.
Table S1. Distribution of CDR3 amino acid length of top 11 V germline genes
Table S2. Substitution rates of VHHs from top 11 V germline genes
Table S3. Spearman’s rank correlation of germline usage, pairing and substitution preference for the five samples
Acknowledgements
This work was supported in part by the National Key R&D Program of China (Grants 2018YFC1602203), the National Natural Science Foundation of China (Nos 31860260, 31301479), the Science and Technology Innovation Platform Project of Jiangxi Province (No. 20192BCD40001), the Research Program of State Key Laboratory of Food Science and Technology (No. SKLF‐ZZA‐201912, SKLF‐ZZB‐201925), the National Institute of General Medical Sciences (GM083107, GM116960), the National Institute of Allergy and Infectious Diseases (AI134678) and the National Science Foundation (DBI1564756, IIS1901191). The authors gratefully acknowledge financial support from the China Scholarship Council. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation grant number ACI‐1548562. 50
Contributor Information
Yanping Li, Email: liyanping@ncu.edu.cn.
Yang Zhang, Email: zhng@umich.edu.
References
- 1. Hamers‐Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB et al Naturally‐occurring antibodies devoid of light‐chains. Nature 1993; 363:446–48. [DOI] [PubMed] [Google Scholar]
- 2. Greenberg AS, Avila D, Hughes M, Hughes A, McKinney EC, Flajnik MF. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995; 374:168–73. [DOI] [PubMed] [Google Scholar]
- 3. Herce HD, Schumacher D, Schneider AFL, Ludwig AK, Mann FA, Fillies M et al Cell‐permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nat Chem 2017; 9:762–71. [DOI] [PubMed] [Google Scholar]
- 4. Bruce VJ, McNaughton BR. Evaluation of nanobody conjugates and protein fusions as bioanalytical reagents. Anal Chem 2017; 89:3819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bannas P, Hambach J, Koch‐Nolte F. Nanobodies and nanobody‐based human heavy chain antibodies as antitumor therapeutics. Front Immunol 2017; 8:1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discov Today 2016; 21:1076–13. [DOI] [PubMed] [Google Scholar]
- 7. Blanc MR, Anouassi A, Ahmed Abed M, Tsikis G, Canepa S, Labas V et al A one‐step exclusion‐binding procedure for the purification of functional heavy‐chain and mammalian‐type gamma‐globulins from camelid sera. Biotechnol Appl Biochem 2009; 54:207–12. [DOI] [PubMed] [Google Scholar]
- 8. Li X, Duan X, Yang K, Zhang W, Zhang C, Fu L et al Comparative analysis of immune repertoires between Bactrian camel's conventional and heavy‐chain antibodies. PLoS One 2016; 11:e0161801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muyldermans S, Baral TN, Retamozzo VC, De Baetselier P, De Genst E, Kinne J et al Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol 2009; 128:178–83. [DOI] [PubMed] [Google Scholar]
- 10. De Genst E, Silence K, Ghahroudi MA, Decanniere K, Loris R, Kinne J et al Strong in vivo maturation compensates for structurally restricted H3 loops in antibody repertoires. J Biol Chem 2005; 280:14114–21. [DOI] [PubMed] [Google Scholar]
- 11. Rubelt F, Busse CE, Bukhari SAC, Burckert JP, Mariotti‐Ferrandiz E, Cowell LG et al Adaptive immune receptor repertoire community recommendations for sharing immune‐repertoire sequencing data. Nat Immunol 2017; 18:1274–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galson JD, Pollard AJ, Truck J, Kelly DF. Studying the antibody repertoire after vaccination: practical applications. Trends Immunol 2014; 35:319–31. [DOI] [PubMed] [Google Scholar]
- 13. Ye B, Smerin D, Gao Q, Kang C, Xiong X. High‐throughput sequencing of the immune repertoire in oncology: applications for clinical diagnosis, monitoring, and immunotherapies. Cancer Lett 2018; 416:42–6. [DOI] [PubMed] [Google Scholar]
- 14. Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, Scheid JF et al A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods 2014; 11:1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turner KB, Naciri J, Liu JL, Anderson GP, Goldman ER, Zabetakis D. Next‐generation sequencing of a single domain antibody repertoire reveals quality of phage display selected candidates. PLoS One 2016; 11:e0149393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pearce R, Huang X, Setiawan D, Zhang Y. evodesign: designing protein–protein binding interactions using evolutionary interface profiles in conjunction with an optimized physical energy function. J Mol Biol 2019; 431:2467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shultis D, Mitra P, Huang X, Johnson J, Khattak NA, Gray F et al Changing the apoptosis pathway through evolutionary protein design. J Mol Biol 2019; 431:825–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen RM, Kleinstein SH, Louzoun Y. Somatic hypermutation targeting is influenced by location within the immunoglobulin V region. Mol Immunol 2011; 48:1477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui A, Di Niro R, Vander Heiden JA, Briggs AW, Adams K, Gilbert T et al A model of somatic hypermutation targeting in mice based on high‐throughput Ig sequencing data. J Immunol 2016; 197:3566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheng Z, Schramm CA, Kong R, Program NCS, Mullikin JC, Mascola JR et al Gene‐specific substitution profiles describe the types and frequencies of amino acid changes during antibody somatic hypermutation. Front Immunol 2017; 8:537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tu Z, Xu Y, He QH, Fu JH, Liu X, Tao Y. Isolation and characterisation of deoxynivalenol affinity binders from a phage display library based on single‐domain camelid heavy chain antibodies (VHHs). Food Agric Immunol 2012; 23:123–31. [Google Scholar]
- 22. Tu Z, Xu Y, Liu X, He Q, Tao Y. Construction and biopanning of camelid naive single‐domain antibody phage display library. China Biotechnol 2011; 31:31–6. [Google Scholar]
- 23. Liu X, Xu Y, Xiong YH, Tu Z, Li YP, He ZY et al VHH phage‐based competitive real‐time immuno‐polymerase chain reaction for ultrasensitive detection of ochratoxin A in cereal. Anal Chem 2014; 86:7471–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aronesty E. Comparison of sequencing utility programs. Open Bioinform Journal 2013; 7:1–8. [Google Scholar]
- 25. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018; 35:1547–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lefranc MP, Giudicelli V, Duroux P, Jabado‐Michaloud J, Folch G, Aouinti S et al IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 2015; 43:D413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013; 41:W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al BLAST+: architecture and applications. BMC Bioinformatics 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res 2004; 14:1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arnaout R, Lee W, Cahill P, Honan T, Sparrow T, Weiand M et al High‐resolution description of antibody heavy‐chain repertoires in humans. PLoS One 2011; 6:e22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henry Dunand CJ, Wilson PC. Restricted, canonical, stereotyped and convergent immunoglobulin responses. Philos Trans R Soc Lond B Biol Sci 2015; 370:20140238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen L, Kutskova YA, Hong F, Memmott JE, Zhong S, Jenkinson MD et al Preferential germline usage and VH/VL pairing observed in human antibodies selected by mRNA display. Protein Eng Des Sel 2015; 28:427–35. [DOI] [PubMed] [Google Scholar]
- 33. Jayaram N, Bhowmick P, Martin AC. Germline VH/VL pairing in antibodies. Protein Eng Des Sel 2012; 25:523–29. [DOI] [PubMed] [Google Scholar]
- 34. Achour I, Cavelier P, Tichit M, Bouchier C, Lafaye P, Rougeon F. Tetrameric and homodimeric camelid IgGs originate from the same IgH locus. J Immunol 2008; 181:2001–9. [DOI] [PubMed] [Google Scholar]
- 35. Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity‐determining regions to binding of antibody‐combining sites. J Immunol 1991; 147:1709–19. [PubMed] [Google Scholar]
- 36. Deschacht N, De Groeve K, Vincke C, Raes G, De Baetselier P, Muyldermans S. A novel promiscuous class of camelid single‐domain antibody contributes to the antigen‐binding repertoire. J Immunol 2010; 184:5696–704. [DOI] [PubMed] [Google Scholar]
- 37. Monegal A, Olichon A, Bery N, Filleron T, Favre G, de Marco A. Single domain antibodies with VH hallmarks are positively selected during panning of llama (Lama glama) naive libraries. Dev Comp Immunol 2012; 36:150–56. [DOI] [PubMed] [Google Scholar]
- 38. Flajnik MF, Deschacht N, Muyldermans S. A case of convergence: why did a simple alternative to canonical antibodies arise in sharks and camels? PLoS Biol 2011; 9:e1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin DA, Bradl H, Collins TJ, Roth E, Jack HM, Wu GE. Selection of Ig µ heavy chains by complementarity‐determining region 3 length and amino acid composition. J Immunol 2003; 171:4663–71. [DOI] [PubMed] [Google Scholar]
- 40. Soto C, Bombardi RG, Branchizio A, Kose N, Matta P, Sevy AM et al High frequency of shared clonotypes in human B cell receptor repertoires. Nature 2019; 566:398–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Genst E, Saerens D, Muyldermans S, Conrath K. Antibody repertoire development in camelids. Dev Comp Immunol 2006; 30:187–98. [DOI] [PubMed] [Google Scholar]
- 42. Miqueu P, Guillet M, Degauque N, Dore JC, Soulillou JP, Brouard S. Statistical analysis of CDR3 length distributions for the assessment of T and B cell repertoire biases. Mol Immunol 2007; 44:1057–64. [DOI] [PubMed] [Google Scholar]
- 43. Kaplinsky J, Li A, Sun A, Coffre M, Koralov SB, Arnaout R. Antibody repertoire deep sequencing reveals antigen‐independent selection in maturing B cells. Proc Natl Acad Sci USA 2014; 111:E2622–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Griffin LM, Snowden JR, Lawson AD, Wernery U, Kinne J, Baker TS. Analysis of heavy and light chain sequences of conventional camelid antibodies from Camelus dromedarius and Camelus bactrianus species. J Immunol Methods 2014; 405:35–6. [DOI] [PubMed] [Google Scholar]
- 45. Nishio J, Suzuki M, Nanki T, Miyasaka N, Kohsaka H. Development of TCRB CDR3 length repertoire of human T lymphocytes. Int Immunol 2004; 16:423–31. [DOI] [PubMed] [Google Scholar]
- 46. Gomez‐Tourino I, Kamra Y, Baptista R, Lorenc A, Peakman M. T cell receptor beta‐chains display abnormal shortening and repertoire sharing in type 1 diabetes. Nat Commun 2017; 8:1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tian M, Cheng C, Chen X, Duan H, Cheng HL, Dao M et al Induction of HIV neutralizing antibody lineages in mice with diverse precursor repertoires. Cell 2016; 166:1471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brender JR, Shultis D, Khattak NA, Zhang Y. An evolution‐based approach to de novo protein design In: Samish Ilan, (ed.). Methods in Molecular Biology. Clifton: Humana Press, 2017; 1529:243–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mitra P, Shultis D, Zhang Y. EvoDesign: de novo protein design based on structural and evolutionary profiles. Nucleic Acids Res 2013; 41:W273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A et al XSEDE: accelerating scientific discovery. Comput Sci Eng 2014; 16:62–4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of CDS length of VHHs.
Figure S2. Phylogenetic tree of V germline genes in the IgH locus of Vicugna pacos. The phylogenetic tree is built using MEGA version X. The germline genes, which contain at least two hallmark residues, are shown in the rectangular box.
Figure S3. Position specific score matrix of sample Naive‐1. The logo plot was built using weblogo version 3.6. The hallmark residues are labelled with asterisk, corresponding to F37, E44, R45 and G47 in the Kabat numbering system.
Table S1. Distribution of CDR3 amino acid length of top 11 V germline genes
Table S2. Substitution rates of VHHs from top 11 V germline genes
Table S3. Spearman’s rank correlation of germline usage, pairing and substitution preference for the five samples