

Abstract
Cell-free DNA in plasma has been used for noninvasive prenatal testing and cancer liquid biopsy. The physical properties of cell-free DNA fragments in plasma, such as fragment sizes and ends, have attracted much recent interest, leading to the emerging field of cell-free DNA fragmentomics. However, one aspect of plasma DNA fragmentomics as to whether double-stranded plasma molecules might carry single-stranded ends, termed a jagged end in this study, remains underexplored. We have developed two approaches for investigating the presence of jagged ends in a plasma DNA pool. These approaches utilized DNA end repair to introduce differential methylation signals between the original sequence and the jagged ends, depending on whether unmethylated or methylated cytosines were used in the DNA end-repair procedure. The majority of plasma DNA molecules (87.8%) were found to bear jagged ends. The jaggedness varied according to plasma DNA fragment sizes and appeared to be in association with nucleosomal patterns. In the plasma of pregnant women, the jaggedness of fetal DNA molecules was higher than that of the maternal counterparts. The jaggedness of plasma DNA correlated with the fetal DNA fraction. Similarly, in the plasma of cancer patients, tumor-derived DNA molecules in patients with hepatocellular carcinoma showed an elevated jaggedness compared with nontumoral DNA. In mouse models, knocking out of the Dnase1 gene reduced jaggedness, whereas knocking out of the Dnase1l3 gene enhanced jaggedness. Hence, plasma DNA jagged ends represent an intrinsic property of plasma DNA and provide a link between nuclease activities and the fragmentation of plasma DNA.
There are a lot of recent research efforts at investigating the nascent field of fragmentomics of cell-free DNA (cfDNA) in plasma. The nonrandom fragmentation patterns of cfDNA, related to the tissues of origin, were first revealed by studying fetally and maternally derived cfDNA molecules in the plasma of pregnant women (Chan et al. 2004; Lo et al. 2010). The fetal DNA fragments were demonstrated to be generally shorter than the maternal DNA molecules (Chan et al. 2004; Lo et al. 2010). The size profile of cfDNA exhibiting a 166-bp major peak and smaller peaks with 10-bp intervals suggested that the biology of cfDNA might be associated with nucleosomal organization (Lo et al. 2010). Similar patterns were also observed in plasma DNA in patients with cancer (Jiang et al. 2015; Underhill et al. 2016), organ transplantation recipients (Zheng et al. 2012), and patients with autoimmune diseases (Chan et al. 2014).
In the light of the realization of the nonrandomness of cfDNA fragmentation, the nucleosome footprints of cfDNA have been demonstrated to be able to inform the tissue of origin (Snyder et al. 2016; Sun et al. 2019). Recently, a catalog of genomic locations were reported to be preferentially cleaved during the generation of plasma DNA molecules, referred to as plasma DNA preferred ends (Chan et al. 2016; Jiang et al. 2018). Such preferred ends were also shown to be related to the tissue of origin of plasma DNA (Chan et al. 2016; Jiang et al. 2018). Furthermore, certain motifs were preferentially present at the ends of plasma DNA molecules (Serpas et al. 2019; Jiang et al. 2020). Such end motif analysis revealed that DNASE1L3 (deoxyribonuclease 1-like 3), an endonuclease, plays an important role in the fragmentation of plasma DNA (e.g., the forming of the “CCCA” end motif of plasma DNA molecules), as elucidated by a Dnase1l3 deletion mouse model (Serpas et al. 2019). The down-regulation of DNASE1L3 in hepatocellular carcinoma (HCC) tumor tissues coincided with the reduction of the CCCA end motif in plasma of patients with HCC (Jiang et al. 2020), resembling the consequence of Dnase1l3-deficient mice. In addition, different nucleases such as DNASE1 and DNASElL3 may be responsible for different cutting patterns with regard to the ends of cell-free DNA (Han et al. 2020). The discovery of new biological properties is a catalyst for new diagnostic approaches. For example, by leveraging the size difference between fetal and maternal DNA, a noninvasive approach for detecting fetal chromosomal aneuploidies was developed (Yu et al. 2014, 2017).
In this work, we investigated whether plasma DNA might consist of double-stranded DNA molecules with nonflush or, in other words, single-stranded protruding ends. We refer to such protruding ends as jagged ends. However, the jagged ends could not be revealed in previous studies because the process of DNA end repair which commonly precedes library construction for massively parallel sequencing would convert the jagged ends into blunt ends. If such jagged ends exist, then we would also explore whether their characteristics might be related to the tissue of origin of the respective cfDNA molecules.
Results
Principle for detecting jagged ends
A double-stranded DNA molecule is assumed to carry 5′ protruding or 3′ protruding ends, termed jagged ends. We envisioned that the jagged ends could be traceable if we could extend those jagged ends with nucleotides with a detectable characteristic. For instance, the detectable characteristics could be a methylated or unmethylated base. The length of the jagged ends could then be deduced from the methylation density of the resultant DNA molecule.
Figure 1 shows the principle as to how the 5′ jagged ends could be detected by introducing unmethylated cytosines (together with three other unmodified nucleotides) for DNA polymerase-mediated extension. All cytosines present in the CpG dinucleotides of a newly synthesized strand would be completely unmethylated. On the other hand, the CpG cytosines present in the original DNA molecule would reflect the methylation status of the DNA molecule. As the genome-wide methylation levels range from 69% to 80% across different tissues (Supplemental Table S1; Roadmap Epigenomics Consortium et al. 2015), it is expected that the methylation level of the CpG sites (the proportion of sequenced cytosines at CpG sites) on the original DNA molecule would be much higher than that of the newly synthesized strand. The blunt-end molecules were subjected to bisulfite treatment by which unmethylated cytosines were converted into uracils but leaving methylated cytosines unchanged. The difference in the measured methylation levels between a segment proximal to the 5′ end (e.g., read1) and the 3′ end (e.g., read2) of a DNA molecule depends on the length of the newly synthesized DNA on the 3′ end and would hence reflect the jaggedness of the original DNA molecule (Fig. 1). To quantify the jaggedness based on CG methylation signals, the jag index (denoted by JI-U which stands for Jagged Index-Unmethylated) is defined by the formula below:
where M1 represents the methylation density of read1, and M2 represents the methylation density of read2.
Figure 1.
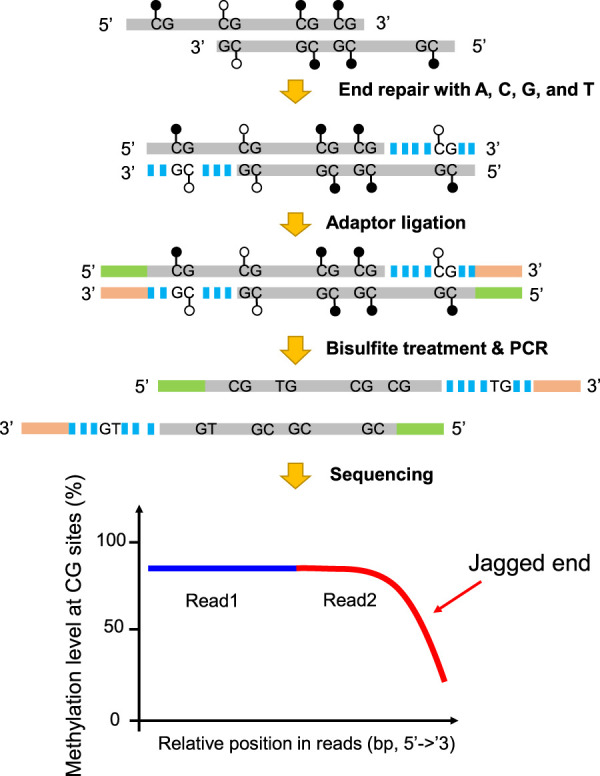
Schematic illustration of plasma DNA jagged end detection using methylation levels at CG sites. A DNA molecule carrying 5′ protruding ends (i.e., jagged ends) would be filled up by the unmethylated nucleotides (i.e., A, C, G, and T) with the presence of T4 DNA polymerase and the Klenow fragment, thus turning into blunt ends. The CG sites present in the newly generated strand were expected to be unmethylated, whereas the CG sites in the original sequence of the same molecule had, on average, a 70% chance of being methylated. The resultant blunted molecules were subjected to bisulfite treatment and PCR amplification. If a molecule contained jagged ends with a 5′ protruding single strand, the methylation levels at CG sites proximal to the 3′ end (e.g., read2) would be lower than that close to the 5′ end (e.g., read1). Filled lollipops represent methylated Cs, and unfilled lollipops represent unmethylated Cs. The dashed blue lines represent newly filled-up nucleotides.
Jaggedness in sonicated tissue DNA and plasma DNA
We used previously published bisulfite sequencing data from 10 DNA samples from blood cells (Adams et al. 2012) and 30 plasma DNA samples (Jiang et al. 2017), with a median of 205 million paired-end reads per sample (range: 65.2–384.3 million) using massively parallel bisulfite sequencing. In the sonicated DNA samples, the mean methylation level dropped from 79.1% to 1.1% within the five positions proximal to the 3′ end (Fig. 2A). In the plasma DNA samples, a progressive reduction of methylation levels was observed in a stretch of at least 30 nucleotides proximal to the 3′ end (Fig. 2B). These results suggested that plasma DNA molecules might carry longer jagged ends in comparison with sonicated DNA molecules. The JI-U of plasma DNA (mean: 22.1; range: 19.6–25.4) was significantly higher than that of sonicated DNA (mean: 2.1; range: 0.7–3.4; P-value < 0.0001, Mann–Whitney U test) (Fig. 2C).
Figure 2.

Presence of jagged ends in sonicated DNA and plasma DNA. (A) Methylation levels in white blood cell DNA across different CG sites in read1 and read2 after pooling all aligned paired-end reads. The blue dots represent the pooled methylation signal contributed by sequenced CGs at the relative positions in read1. The red dots represent the pooled methylation signal at the relative positions in read2. The methylation signal was calculated by the percentage of sequenced CGs with respect to the total sequenced CGs and TGs. (B) Methylation levels in white blood cell DNA across different CG sites in read1 and read2 after pooling all aligned paired-end reads. (C) JI-U between sonicated DNA and plasma DNA samples. The JI-U represents the jagged index deduced from unmethylated signals at CG sites.
Overall patterns of plasma DNA jagged ends
Bisulfite sequencing results from a previously published cohort of 30 pregnant women, with 10 from each of the first (12–14 wk), second (20–24 wk), and third (38–40 wk) trimesters, were re-analyzed for DNA jaggedness (Jiang et al. 2017). The JI-U varied with fragment sizes of plasma DNA (Fig. 3A), showing multiple peaks at 226 bp, 405 bp, and 556 bp. The average distance between two adjacent major peaks in Figure 3A was found to be 165 bp, suggesting that the generation of plasma DNA jagged ends might be associated with nucleosome arrays in the human genome. This characteristic pattern could be consistently observed in all samples (Supplemental Fig. S1A–AE).
Figure 3.

Jagged ends in plasma DNA of pregnant women. (A) The profile of JI-U across different sizes of plasma DNA fragments, ranging from 50 bp to 600 bp. (B) The JI-U between shared DNA molecules (mainly of maternal origin) and fetal-specific DNA molecules without size selection. (C) The JI-U between shared DNA molecules and fetal-specific DNA molecules within a range of 130 to 160 bp. (D) JI-U across different sizes for shared and fetal-specific plasma DNA molecules. The JI-U represents the jagged index deduced from unmethylated signals at CG sites.
Difference in jaggedness between fetal- and maternal-derived plasma DNA molecules
To assess whether there is any difference in the jaggedness between fetal- and maternal-derived plasma DNA molecules, we analyzed the plasma DNA molecules covering informative single nucleotide polymorphism (SNP) loci (Jiang et al. 2017). Informative SNPs refer to SNPs in which the mother was homozygous (i.e., genotype AA) and the fetus was heterozygous (i.e., genotype AB). In this situation, the B allele would be fetal-specific whereas the A allele would be shared by both the fetus and the mother. In a maternal plasma sample, most of the DNA was derived from the mother, as the median fetal DNA fraction among those samples was 17.1%. Hence, the JI-U of the DNA molecules carrying an A allele would reflect the jaggedness of maternally derived DNA.
Figure 3B shows that the JI-U of fetally derived molecules (median: 23.8; range: 21.4–27.7) was significantly higher than that of molecules carrying the shared alleles (median: 22.1; range: 19.2–24.7; P-value < 0.0001, Mann–Whitney U test). The difference in the jaggedness was observed for plasma DNA between 130 and 160 bp (Fig. 3C). There was a 28.4% increase in the JI-U of fetal DNA for molecules within a range of 130 to 160 bp, whereas there was only a 7.5% increase for all molecules without size selection. The fetal-specific DNA molecules bore similar patterns in the variation of JI-U against size compared with DNA molecules carrying shared alleles (Fig. 3D). However, the JI-U of fetal-specific DNA was higher than that of shared sequences (Fig. 3D).
High-resolution jagged end analysis
The aforementioned approach for detecting jagged ends was informative only at CpG dinucleotides, which accounted for only 1% of dinucleotides in the human genome. Such a low prevalence of CpG sites in the genome would largely limit the resolution of jagged ends analysis, making it difficult to infer the exact jagged end length for a DNA molecule. In contrast to CpG cytosines, cytosines at non-CpG sites (i.e., CH sites, where H represents A, C, or T) are generally unmethylated in the human genome (the methylation level at CH sites <1%) (Supplemental Table S1). As shown in Figure 4A, if we used methylated cytosines (mCs) instead of Cs in the process of end repair, the jagged ends would be filled in by mCs. Therefore, methylation signals at CH sites would be used for differentiating jagged ends from the original double-stranded DNA in such a molecule. Similar to the CpG-based method, the DNA molecules carrying jagged ends after the end repair using the enzyme mix with mCs would be filled up to generate the blunt molecules. The end-repaired molecules were subjected to bisulfite treatment. The unconverted cytosines (i.e., mCs) at CH sites in the newly generated strand would indicate the presence of jagged ends, whereas the converted cytosines (i.e., Ts) at CH sites would indicate the original cytosines in the double-stranded DNA. In this context of CH, the jaggedness index (denoted by JI-M which stands for Jagged Index-Methylated) is defined as the proportion of sequenced mCs at CH sites in read2. As CH sites account for 19.8% of the human genome compared with 1% of CpG sites, the resolution and accuracy of jagged end analysis would be greatly improved.
Figure 4.
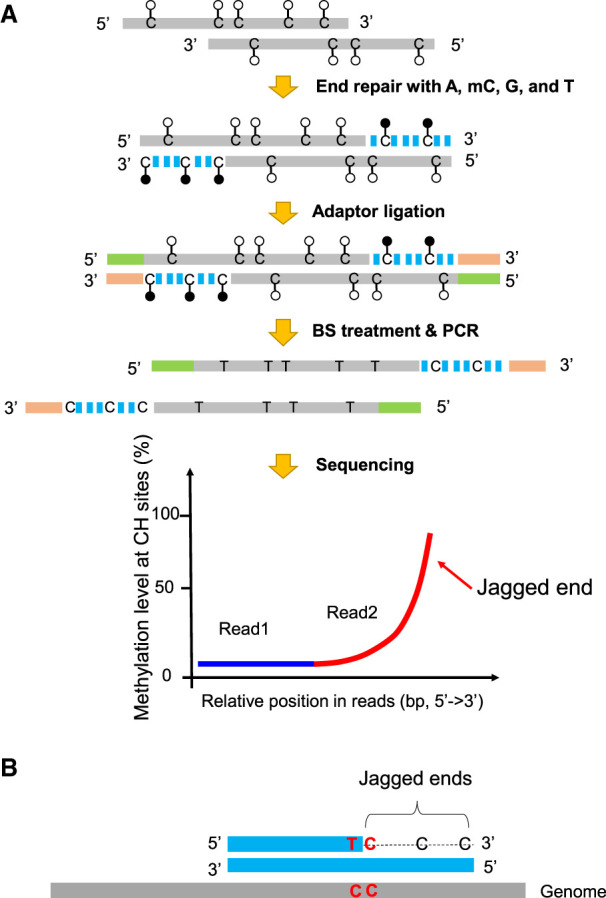
Schematic illustration of plasma DNA jagged end detection using methylation levels at CH sites. (A) The general principle for detecting the presence of jagged ends. In the process of end repair, a DNA molecule carrying 5′ protruding ends (i.e., jagged ends) would be filled up by A, T, mC (methylated C), G with the Klenow fragment without 3′ → 5′ exonuclease activity. The CH (H: A, C, or T) sites present in the newly generated strand were expected to be methylated, whereas the CH sites in the original sequence of the same molecule were generally unmethylated. After the end repair of plasma DNA, the resultant blunted molecules were subjected to bisulfite treatment and PCR amplification. If a molecule contained jagged ends with a 5′ protruding single strand, the methylation levels at CH sites proximal to the 3′ ends (e.g., read2) would be higher than that close to the 5′ end (e.g., read1). Filled lollipops represent methylated Cs, and unfilled lollipops represent unmethylated Cs. The dashed blue lines represent newly filled-up nucleotides. (B) The exact jagged end length deduction. A molecule containing two consecutive cytosines (i.e., “CC” tag) would provide a possibility to directly determine the exact length of a jagged end, when the below criteria are fulfilled: The first one is located within the double-stranded DNA and the other just corresponds to the first nucleotide of the jagged end. Such a “CC” tag would be converted into a “TC” tag after the bisulfite conversion followed by PCR amplification. Therefore, the dashed line indicates the exact jagged end length deduced by CC-tag strategy.
In addition, this approach provides a strategy for accurately deducing the exact length of a jagged end, as illustrated in Figure 4B. Considering a situation where there are two consecutive Cs (i.e., “CC”) in a stretch of DNA, the first one is located within the double-stranded DNA and the other just corresponds to the first nucleotide of jagged end involving the incorporation of mC during the end-repair process. Such a “CC” tag would be converted into a “TC” pattern in bisulfite sequencing, because after the bisulfite treatment and PCR amplification, the originally unmethylated Cs in a double-strand are converted to Ts, whereas the newly incorporated mC at the first position of the jagged end remains unchanged. The length from the site of “TC” to the 3′ end in a molecule would indicate the exact length of the jagged end. We named this method the “CC-tag” strategy for detecting the exact length of a jagged end (Fig. 4B).
Evaluation of the incorporation of methylated cytosines in the end-repair step
To test if mCs were able to be incorporated into the newly generated strand complementary to the single strand of a jagged end during the process of DNA end repair, we added two synthetic double-stranded DNA fragments carrying jagged ends with known lengths as internal controls, namely 13 nt and 22 nt in size, respectively. Each of the two double-stranded synthetic DNAs consisted of a target sequence providing the annealing site for the P7 adapter (Illumina), unmethylated double-stranded DNA, a molecular barcode indicating the length of the synthetic jagged end, and a single-stranded fragment. As shown in Supplemental Figure S2, A and B, on average, 96% (range: 90%–99%) of cytosines within the jagged ends (lowercase letters) were observed to be unchanged in the plasma DNA of pregnant subjects, suggesting the successful incorporation of mCs along jagged ends. In contrast, none of Cs within a double-stranded stretch was found to be methylated, suggesting that nearly all Cs in the original double-stranded stretch had been converted to Ts. These results suggested that the use of mC in the end-repair mix would allow us to detect the jagged ends. On the other hand, we further performed an assay related to a synthetic oligonucleotide carrying a 14-nt jagged end that contained “CC” tags. Supplemental Figure S2C shows that the preponderance of cytosines at the 32nd position (84.1%), the sites immediately preceding the first base in jagged ends, were converted to thymine, whereas the preponderance of cytosines at the 33rd position (98.3%), the starting site of the synthetic jagged end, were kept unchanged. These results demonstrated the analytical validity of the CC-tag strategy.
To further validate this approach based on methylation signals at CH sites, we carried out the incorporation of nucleotides to jagged ends for two plasma DNA aliquots from one pregnant sample using mCs and Cs, respectively, in the enzyme mix of DNA end repair. For the DNA portion end-repaired with mCs, we observed the progressive increase in the proportion of sequenced mC at CH sites across a stretch of at least 30 nt proximal to the 3′ end, from 7.5% up to 79.9%, but we did not see this pattern in the DNA close to 5′ end (Supplemental Fig. S2D). In contrast, such an increase in methylation levels at CH sites disappeared in the DNA portion that was end-repaired with unmethylated Cs (Supplemental Fig. S2D). This result provided extra evidence supporting the specific incorporation of mCs along jagged ends and further confirmed the existence of jagged ends in plasma DNA.
CH sites in the human genome (∼271 million loci) are much more prevalent than CpG sites (∼28 million loci), leading to a much higher resolution analysis of jagged ends. As shown in Supplemental Table S2, we analyzed the percentage of fragments carrying cytosines that could be used for inferring the presence of jagged ends, termed informative Cs in jagged ends. The method using the enzyme mix with mCs for end repair could detect a much higher proportion of fragments carrying informative Cs, compared with the method using the enzyme mix with unmethylated Cs. For example, the approach using the methylation signals at CH sites revealed 59.8% of fragments carrying at least one informative C in jagged ends for a molecule, whereas only 8.4% of fragments would be informative for the approach using the methylation signals at CpG sites.
Analysis for the length of jagged ends in plasma DNA
The jaggedness of plasma DNA from 15 pregnant women was analyzed using the “CC-tag” approach (Fig. 5A). A mean of 12.3% (range: 8.1%–17.3%) of fragments had blunt ends, that is, no jaggedness. In other words, the fragments with jagged ends accounted for 87.8% (range: 83.7%–91.9%). A mean of 11.2% (range: 8.3%–13.1%) molecules had a jagged end of only 1 nt and a mean of 26.6% (range: 23.3%–29.9%) of molecules have a jagged end of 5 nt or less. As shown in Figure 5A, the longer the jagged ends, the lower the relative frequency would be observed in plasma DNA. Such general patterns were reproducible across other samples (Supplemental Fig. S3). Furthermore, the shorter jagged ends were accompanied with a higher GC content (Supplemental Fig. S4), which was consistent with the fact that plasma DNA ends were enriched with the “CC” motif (Chandrananda et al. 2015; Serpas et al. 2019; Jiang et al. 2020).
Figure 5.

The profile of exact jagged end lengths of plasma DNA. (A) Jagged end length distribution deduced by the CC-tag strategy. (B) Averaged jagged end lengths across different sizes of plasma DNA fragments based on CC-tag strategy.
Figure 5B shows that the mean lengths of jagged ends varied according to different sizes of plasma DNA fragments. The major peaks occurred at 238 bp, 419 bp, and 583 bp, with a distance between two neighboring peaks which was reminiscent of the size of one nucleosome unit. Below 160 bp, a series of small peaks occurred in a 10-bp periodicity (Fig. 5B).
Difference in jaggedness between fetal and maternal DNA molecules
A median of 201,352 informative SNPs (range: 178,623–208,552) for which the mother was homozygous (AA) and the fetus was heterozygous (AB) were used to explore the difference in jagged end length between fetal and maternal DNA molecules with the use of the high-resolution analysis method. Both fetal-specific and shared sequences possessed jagged ends, as the increase of methylation levels at CH sites proximal to the 3′ end was present in both the fetal-specific and shared sequences (Fig. 6A).
Figure 6.

Differential jagged ends between fetal and maternal DNA molecules revealed by the incorporation of methylation at CH sites. (A) Methylation level at CH sites in the relative positions of read1 and read2 for shared and fetal-specific DNA molecules. (B) Averaged jagged end lengths between shared and fetal-specific DNA molecules. (C) Relationship between JI-M deduced by methylation at CH sites in read2 and plasma DNA fragment sizes. (D) The cumulative JI-M between fetal-specific and shared DNA molecules. (E) The cumulative difference in JI-M, from 130 bp to 160 bp, between fetal and shared (mainly of maternal origin) DNA sequences. The black line represents the cumulative difference in jaggedness index between the shared (mainly of maternal origin) and fetal-specific DNA molecules pooled from all pregnant subjects. The positive values indicate relative higher jaggedness for molecules below a particular size cutoff. (F) The correlation between the average jagged end length within 130–160 bp and fetal DNA fraction. The JI-M represents the jagged index deduced from the methylated signals at CH sites.
The average lengths of jagged ends present in the fetal-specific DNA molecules (mean: 21.0; range: 18.7–23.1) were significantly higher than those from molecules carrying shared alleles (mean: 19.1; range: 17.4–20.8; P-value < 0.0001, Mann–Whitney U test) (Fig. 6B). The longer jagged ends in the fetal-specific DNA molecules were consistent with the fact that higher JI-M values were observed in fetal DNA molecules across the different fragment sizes (Fig. 6C). Supplemental Figure S5 shows the plot of the difference in methylation levels at CH sites (i.e., ΔJ), across different sizes from short to long molecules, between the fetal and maternal DNA molecules in relation to the different sizes of plasma DNA fragments. The positive and gradually rising values of ΔJ within the size range of 130 bp to 160 bp indicated that the longer jagged ends were present in fetal-specific DNA across this size range (Supplemental Fig. S5). The positive ΔJ values were consistently observed in different plasma DNA samples of pregnant women (Supplemental Fig. S6).
To further illustrate the trend of aggregated differences in jaggedness of plasma DNA between the fetal and maternal DNA molecules, the cumulative JI-M values within the size range of 130 to 160 bp for fetal and maternal DNA were plotted, respectively (Fig. 6D). The cumulative JI-M was calculated by the proportion of methylated cytosines at CH sites in those progressively accumulated DNA molecules from short to long sizes. The cumulative curve of the fetal JI-M was above that of the maternal DNA (Fig. 6D), thus leading to a positive value in the cumulative difference in JI-M for all pregnant subjects (Fig. 6E). These results thus suggested that the fetal DNA bore more molecules with longer jagged ends.
Correlation between jagged ends and fetal DNA fractions
As fetal DNA molecules had longer jagged ends compared with maternal DNA molecules (Fig. 6D), we hypothesized that the overall jaggedness would correlate with the fetal DNA fractions. To test this hypothesis, we calculated the mean of jagged end lengths regarding the DNA molecules within the size range from 130 to 160 bp, when the difference in jaggedness between fetal and maternal DNA achieved a maximal value (Supplemental Fig. S5). The mean jagged end lengths correlated with the fetal DNA fractions which were calculated based on informative SNPs (Pearson's r: 0.7; P-value: 0.004) (Fig. 6F).
Difference in jaggedness between plasma DNA molecules carrying mutant and wild-type alleles in patients with hepatocellular cancer
Three plasma DNA samples of patients with HCC were sequenced to a median of 12.5× human haploid genome coverage (range: 10.3–35.2×). Paired white blood cell DNA and tumor DNA were sequenced to a median of 23.5× coverage (range: 15.3–50.2×). The single nucleotide variants which were present in tumor DNA but absent in the matched white blood cell DNA were identified as previously described (Jiang et al. 2018) and were considered as somatic mutations. The plasma DNA carrying the mutant alleles was of tumoral origin, whereas the plasma DNA carrying the wild-type alleles was mainly nontumoral. There was a median of 21,657 tumor-derived DNA molecules (range: 3899–31,234). The jaggedness of tumor-derived DNA was observed to be higher than that of sequences carrying wild-type alleles in three plasma DNA samples of patients with HCC, as the cumulative difference in JI-M between the tumor-derived DNA molecules and wild-type molecules was found to be positive (Fig. 7A–C; Supplemental Fig. S7A–C).
Figure 7.

The cumulative difference in JI-M between plasma DNA molecules carrying mutant (tumor-derived DNA) and wild-type alleles (mainly nontumoral DNA). The patterns of cumulative difference in JI-M for three plasma DNA samples of patients with HCC, including HCC 01, 02, and 03, are shown in (A), (B), and (C), respectively.
Diagnostic applications of plasma DNA jaggedness for HCC detection
As tumor-derived DNA molecules contained higher jaggedness, we further explored if it was feasible to use plasma DNA jaggedness for detecting patients with HCC. We analyzed a cohort in a previously published study (Jiang et al. 2020), consisting of healthy control subjects (n = 8), patients infected with chronic hepatitis B virus (HBV, n = 17), and patients with hepatocellular carcinoma (HCC, n = 34), using bisulfite sequencing. This experimental protocol involved a step of end repair with the use of unmethylated cytosines, allowing for calculating JI-U for each sample. As shown in Figure 8A, JI-U for those fragments within a range of 130 to 160 bp was significantly elevated in patients with HCC (mean JI-U: 15.2; range: 13.2–17.3), compared with subjects without HCC (mean JI-U: 14.0; range: 12.2–15.6; P-value < 0.0001, Mann–Whitney U test). The area-under-the-curve (AUC) of the ROC (receiver operating characteristic curve) was 0.87 (Fig. 8B). These results suggested the diagnostic potential of jaggedness of plasma DNA for detecting patients with HCC. Furthermore, the AUC of the jaggedness index for those fragments between 130 and 160 bp was much higher than that for all fragments without size selection (0.54) (Supplemental Fig. S8A,B).
Figure 8.

Diagnostic performance. (A) Jaggedness indices across healthy control subjects (CTR), patients infected with chronic hepatitis B virus (HBV), and patients with hepatocellular carcinoma (HCC). The y-axis indicates the jaggedness index based on the filling of unmethylated cytosine (JI-U). (B) ROC of jaggedness index between subjects with and without HCC.
To test whether jagged ends would provide an independent diagnostic marker, we performed logistic regression analysis using the mean size of plasma DNA and jaggedness index between subjects with and without HCC. In univariate logistic regression analysis, the odds ratio of the mean size of plasma DNA was 0.86 (95% confidence interval [CI]: 0.77–0.93; P-value: 0.0013). The odds ratio of JI-U was 6.06 (95% CI: 2.68–18.61; P-value: 0.0002). Thus, both the mean size of plasma DNA and jaggedness index were statistically significant as predictors in differentiating between patients with and without HCC. In multivariate logistic regression analysis, only the odds ratio of JI-U (4.70; 95% CI: 1.92–15.2) was found to be significant (P-value: 0.0029), whereas the odds ratio of the mean size of plasma DNA (0.93; 95% CI: 0.82–1.03) did not show statistical significance (P-value: 0.19). This analysis suggested that these two metrics might interact with one another, but jaggedness was shown to be a relatively stronger predictor in this cohort.
Biological implications of plasma DNA jaggedness
As DNA nucleases have been reported to be responsible for plasma DNA end motifs (Serpas et al. 2019; Han et al. 2020; Jiang et al. 2020), we studied whether jaggedness of plasma DNA, as a new fragmentomic marker, could be used as an indicator reflecting DNASE1L3 and DNASE1 activities using mouse models with the deletion of different nucleases. To this end, we measured the jaggedness index in wild type (n = 12), Dnase1−/− (n = 7), and Dnase1l3−/− (n = 5), with the use of the filling of methylated cytosines. The median number of mapped paired-end reads was 126 million (range: 31–228 million).
As shown in Figure 9, an increase of jaggedness was observed in mice with the deletion of Dnase1l3 (Dnase1l3−/−) compared with wild-type mice, whereas a decreasing trend was seen in mice with deletion of Dnase1 (Dnase1−/−) (P-value: 0.0109, Kruskal–Wallis test). These results suggested that nucleases may play a role in the production of jagged ends.
Figure 9.
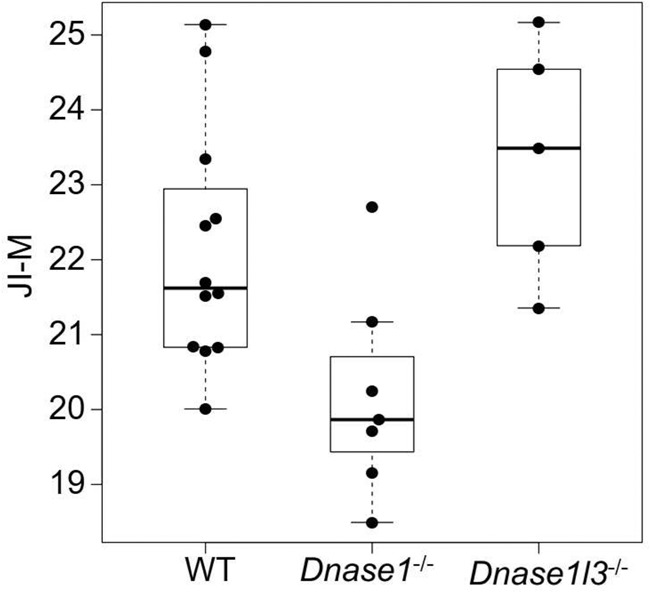
Jaggedness indices of plasma DNA in mice across different genotypes including wild type, Dnase1−/−, and Dnase1l3−/−. The y-axis indicates the jaggedness index based on the filling of methylated cytosines (JI-M).
The higher jaggedness index was also observed in both fetal DNA molecules and tumoral DNA molecules compared with background DNA molecules of mainly hematopoietic origin. Such an increase of jaggedness of plasma DNA might be related to DNASE1 activity in corresponding tissues (i.e., placental and HCC tumor tissues). In this regard, the gene expression of DNASE1 was higher in placenta tissues than white blood cells (Illumina Body Map data set) (Supplemental Fig. S9A; Cabili et al. 2011). Furthermore, the gene expression of DNASE1 was also higher in HCC tumor tissues (n = 34) when compared with normal liver tissues (n = 18) (data from The Cancer Genome Atlas [TCGA] Research Network) (Supplemental Fig. S9B).
Discussion
We have demonstrated a generic approach for revealing the existence of plasma DNA jagged ends and quantitatively analyzing the jagged ends at single-base resolution, by introducing nucleotides with detectable characteristics (e.g., methylation) into the jagged ends. The majority of the plasma DNA molecules were found to bear jagged ends. The jaggedness was found to be associated with different sizes of plasma DNA. The periodic patterns of the degree of jaggedness across different fragment sizes appeared to be consistent with the size of one nucleosome unit, suggesting that the generation of jagged ends might be associated with nucleosome structures. Furthermore, on the basis of results concerning mouse models with deletions of nuclease genes, our data suggested that the jaggedness was associated with nuclease activities including DNASE1 and DNASE1L3. However, the impact on jaggedness caused by DNASE1 and DNAS1L3 appeared to exhibit contrasting effects, for which DNASE1 enhanced jaggedness whereas DNASE1L3 might create fragments with less jaggedness. Such contrasting effects of DNASE1 and DNASE1L3 might be related to their unique enzymatic characteristics. It was reported that DNASE1 and DNASE1L3 displayed different substrate specificities. For example, DNASE1L3 is more efficient at cleaving chromatin, in contrast to DNASE1 preferentially cleaving naked regions (Mizuta et al. 2006; Napirei et al. 2009).
A number of aspects regarding the nonrandomness of plasma DNA fragmentation have been elucidated in previous studies. For example, the characteristic size profile of cell-free DNA is reminiscent of nuclease-cleaved nucleosomes (Lo et al. 2010). It has subsequently been demonstrated that the existence of preferentially cleaved genomic locations might inform the tissue of origin of plasma DNA (Chan et al. 2016; Jiang et al. 2018). Plasma DNA was reported to comprise the nucleosome footprints (Snyder et al. 2016; Sun et al. 2018). In addition, plasma DNA end motifs were recently studied, unlocking the linkage between nuclease biology and cell-free DNA fragmentation (Serpas et al. 2019) as well as diagnostic potential (Jiang et al. 2020). However, the jaggedness of plasma DNA molecules could not be revealed in the previous studies because the sequencing protocols involved the process of DNA end repair, which would transform the jagged ends into blunt ends and the blunted molecules could not be traceable back to the original end patterns. As reported in this study, we made use of the fact that the differential methylation states were present between the original strand and the new strand synthesized during the process of end repair, allowing the detection of jagged ends of plasma DNA.
There were two versions of the approach present in this study for deciphering the jagged ends of nucleic acids. The first version using the methylation patterns at CpG sites could be conveniently adopted in the analysis of many existent bisulfite sequencing data sets, because it was common to use unmethylated Cs in the mix of DNA end repair prior to DNA library preparation for bisulfite sequencing. Even though the low prevalence of CpG sites in the human genome would limit the resolution of the jagged end analysis, the general characteristic patterns of jagged index were able to be actually uncovered. This was supported by the fact that the periodic patterns in the distribution of jaggedness and a higher extent of jaggedness in the fetal DNA were by and large, consistent between the two approaches present in this study. One limitation for these two approaches for detecting DNA jagged ends was that only the 5′ protruding ends (5′ jagged ends) could be detected in the present version since the DNA polymerase-mediated DNA synthesis in the end-repair step only occurs in the 5′ to 3′ direction. Other approaches would need to be developed to analyze the 3′ protruding ends, such as a hybridization-based approach published recently (Harkins et al. 2020). The presence of 5′ protruding forms of double-stranded plasma DNA was hinted in a previous study, using [α-32P]dATP-mediated end-labeling of plasma DNA (Suzuki et al. 2008). However, in contrast to the qualitative nature of [α-32P]dATP-mediated end-labeling, the approaches for jagged end detection in this study would lead to a quantitative measurement.
The second version with using methylation signals at CH sites created new possibilities to measure the exact length of jagged ends for a subset of DNA molecules. It further demonstrated that the increased jaggedness was found in fetal DNA molecules in comparison with maternal DNA molecules. The difference in jagged end length between the fetal and maternal DNA molecules would suggest that the plasma DNA jagged ends would be potentially linked to the tissue of origin. Such speculation was further evidenced by the fact that (1) there was a generally positive correlation between the overall jaggedness and fetal DNA fraction in the plasma DNA of pregnant women, and (2) the tumor-derived DNA molecules were associated with an increase of jaggedness compared with the nontumoral DNA molecules. Compared with patients without HCC, a significant increase of jaggedness could be observed in the DNA pool with all fragments for patients with HCC, suggesting that the jagged ends of plasma DNA, as a new biomarker, would be useful for differentiating HCC and non-HCC subjects. It would be also intriguing to investigate the jaggedness in patients with different cancers in a large-scale cohort for future exploration in this new avenue. Further large-scale use of plasma DNA jaggedness might reveal how gender, age, and other biological factors would affect the inter-/intra-individual variations of such new biomarker. The use of plasma DNA jaggedness as a surrogate for monitoring different nuclease activities would be another possible application.
In summary, we have developed new methods that allow the detection and characterization of the jagged ends of plasma DNA molecules. Using these methods, we revealed that the jagged ends of plasma DNA may serve as a new class of physical properties of cell-free DNA fragmentation. It may open up new possibilities for plasma DNA fragmentomics-based molecular diagnostics in noninvasive prenatal testing, organ transplantation, oncology, and autoimmune diseases.
Methods
Mouse sample collection and processing
Mice with deletion of the Dnase1 gene (Dnase1−/−) were obtained from the Knockout Mouse Project Repository of the University of California at Davis. Mice with deletion of the Dnase1l3 gene (Dnase1l3−/−) were obtained from The Jackson Laboratory. The mice involved a third-party distribution agreement and were nontransferable. The mice were maintained in the Laboratory Animal Center of The Chinese University of Hong Kong (CUHK). All experimental procedures were approved by the Animal Experimentation Ethics committee of CUHK and performed in compliance with the Guide for the Care and Use of Laboratory Animals (8th ed., 2011) established by the National Institutes of Health.
Plasma from the blood samples was collected through centrifugation at 1600g for 10 min, followed by 16,000g for 10 min at 4°C. Plasma DNA was extracted from ∼0.5 mL plasma with the use of the QIAamp Circulating Nucleic Acid kit (Qiagen).
Human sample collection and processing
Pregnant women in the first, second, or third trimester were recruited from the Department of Obstetrics and Gynecology of the Prince of Wales Hospital, Hong Kong. Patients with HCC were recruited from the Department of Surgery and the Department of Medicine and Therapeutics of the Prince of Wales Hospital, Hong Kong. Healthy subjects were recruited as controls. The demographic and clinical information for these HCC patients are listed in Supplemental Table S3. The study was approved by the Joint Chinese University of Hong Kong–Hospital Authority New Territories East Cluster Clinical Research Ethics Committee, with written informed consent.
Plasma from the blood samples was collected through centrifugation at 1600g for 10 min, followed by16,000g for 10 min at 4°C. Plasma DNA was extracted from ∼4 mL plasma with the use of a QIAamp Circulating Nucleic Acid kit (Qiagen).
Sequencing library preparation
Plasma DNA libraries were constructed with the modified Paired-end Sequencing Sample Preparation kit (Illumina) as illustrated in Supplemental Figure S10. First, Exo T was used to remove 3′ protruding ends. Subsequently, Klenow Fragment (exo-) together with dATP (A), dGTP (G), dTTP (T), and methylated dCTP (5mC) were applied to fill in 5′ protruding ends, forming blunt ends. In this filling step, the Klenow fragment of DNA polymerase I (Escherichia coli) facilitated the A-tailing by which a nontemplated A was added to the 3′ blunt ends. Next, PNK was used for 5′ phosphorylation. Lastly, T4 DNA ligase was used to ligate methylated sequencing adapters to plasma DNA after end repair.
Adapter-ligated DNA was treated with two rounds of bisulfite conversion by the EpiTect Plus DNA Bisulfite kit (Qiagen) according to the manufacturer's instructions. Bisulfite-converted products were amplified 8–11 cycles with KAPA HiFi HotStart Uracil + ReadyMix (Roche). The quality of each DNA library was assessed on an Agilent 4200 TapeStation. The libraries were run on D1000 ScreenTape (Agilent) for DNA size and quantity assessment prior to sequencing. A peak observable at around 320 bp indicated readiness for sequencing. The amplified DNA libraries were sequenced on a HiSeq 4000 system (Illumina) in a 75-bp × 2 paired-end format.
Sequencing alignment
After base-calling, the sequencing reads were preprocessed by removing the adapter sequences and low-quality bases (i.e., quality score of <20). The trimmed reads in a FASTQ format were aligned to the human (hg19) or mouse (mm9) reference genome using BSMAP using the “unique pair” output option and the other parameters with default settings (Xi and Li 2009). Only paired-end reads with both ends uniquely aligned to the same chromosome with the correct orientation, spanning an insert size of ≤1000 bp were used for downstream analysis. For human sequence data, re-analysis of the data using the GRCh38 (UCSC hg38) human reference genome would not affect the results significantly, as the major difference between these two versions of the human reference genome is the sequence representation for centromeres (i.e., repetitive regions), and the short sequencing reads derived from those regions were generally not able to be aligned uniquely, thus not being included for downstream analysis. Similarly, for mouse sequence data, re-analysis of the data using the GRCm38 (UCSC mm10) mouse reference genome would not affect the results significantly because only uniquely aligned reads were included for downstream analysis.
Data access
All raw sequencing data generated in this study have been submitted to the European Genome–Phenome Archive (EGA; https://ega-archive.org/) under accession number EGAS00001004080. The bioinformatics pipeline for detecting the jagged ends was written in Perl language and is available as Supplemental Code.
Competing interest statement
K.C.A.C., R.W.K.C., and Y.M.D.L. hold equities in DRA, Take2 Holdings Limited, and Grail. K.C.A.C., R.W.K.C., and Y.M.D.L. are consultants to Grail. K.C.A.C., R.W.K.C., and Y.M.D.L. receive research funding from Grail. Y.M.D.L. is a scientific cofounder of and serves on the scientific advisory board of Grail. R.W.K.C. is a consultant to Illumina. H.L.Y.C. received an honorarium from Grail. H.L.Y.C. is a consultant of Roche Diagnostics. P.J. holds equities in Grail. P.J. is a Director of KingMed Future. P.J., S.H.C., K.C.A.C., R.W.K.C., and Y.M.D.L. have filed patent applications based on the data generated from this work. Patent royalties are received from Grail, Illumina, Sequenom, DRA, Take2 Health, and Xcelom.
Supplementary Material
Acknowledgments
This work was supported by the Research Grants Council of the Hong Kong SAR Government under the Theme-based Research Scheme (T12-403/15-N and T12-401/16-W), a collaborative research agreement from Grail, and the Vice Chancellor's One-Off Discretionary Fund of The Chinese University of Hong Kong (VCF2014021). Y.M.D.L. is supported by an endowed chair from the Li Ka Shing Foundation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.261396.120.
Freely available online through the Genome Research Open Access option.
References
- Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, Bock C, Boehm B, Campo E, Caricasole A, et al. 2012. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol 30: 224–226. 10.1038/nbt.2153 [DOI] [PubMed] [Google Scholar]
- Cabili M, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927. 10.1101/gad.17446611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KCA, Zhang J, Hui ABY, Wong N, Lau TK, Leung TN, Lo K-W, Huang DWS, Lo YMD. 2004. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem 50: 88–92. 10.1373/clinchem.2003.024893 [DOI] [PubMed] [Google Scholar]
- Chan RWY, Jiang P, Peng X, Tam L-S, Liao GJW, Li EKM, Wong PCH, Sun H, Chan KCA, Chiu RWK, et al. 2014. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci 111: E5302–E5311. 10.1073/pnas.1421126111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KCA, Jiang P, Sun K, Cheng YKY, Tong YK, Cheng SH, Wong AIC, Hudecova I, Leung TY, Chiu RWK, et al. 2016. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc Natl Acad Sci 113: E8159–E8168. 10.1073/pnas.1615800113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrananda D, Thorne NP, Bahlo M. 2015. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med Genomics 8: 29 10.1186/s12920-015-0107-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DSC, Ni M, Chan RWY, Chan VWH, Lui KO, Chiu RWK, Lo YMD. 2020. The biology of cell-free DNA fragmentation and the roles of DNASE1, DNASE1L3, and DFFB. Am J Hum Genet 106: 202–214. 10.1016/j.ajhg.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkins KM, Schaefer NK, Troll CJ, Rao V, Kapp J, Naughton C, Shapiro B, Green RE. 2020. A novel NGS library preparation method to characterize native termini of fragmented DNA. Nucleic Acids Res 48: e47 10.1093/nar/gkaa128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VW-S, Wong GLH, Chan SL, Mok TSK, Chan HLY, et al. 2015. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci 112: E1317–E1325. 10.1073/pnas.1500076112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Tong YK, Sun K, Cheng SH, Leung TY, Chan KCA, Chiu RWK, Lo YMD. 2017. Gestational age assessment by methylation and size profiling of maternal plasma DNA: a feasibility study. Clin Chem 63: 606–608. 10.1373/clinchem.2016.265702 [DOI] [PubMed] [Google Scholar]
- Jiang P, Sun K, Tong YK, Cheng SH, Cheng THT, Heung MMS, Wong J, Wong VWS, Chan HLY, Chan KCA, et al. 2018. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc Natl Acad Sci 115: E10925–E10933. 10.1073/pnas.1814616115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Sun K, Peng W, Cheng SH, Ni M, Yeung PC, Heung MMS, Xie T, Shang H, Zhou Z, et al. 2020. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov 10: 664–673. 10.1158/2159-8290.CD-19-0622 [DOI] [PubMed] [Google Scholar]
- Lo YMD, Chan KCA, Sun H, Chen EZ, Jiang P, Lun FMF, Zheng YW, Leung TY, Lau TK, Cantor CR, et al. 2010. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2: 61ra91 10.1126/scitranslmed.3001720 [DOI] [PubMed] [Google Scholar]
- Mizuta R, Mizuta M, Araki S, Shiokawa D, Tanuma S, Kitamura D. 2006. Action of apoptotic endonuclease DNase γ on naked DNA and chromatin substrates. Biochem Biophys Res Commun 345: 560–567. 10.1016/j.bbrc.2006.04.107 [DOI] [PubMed] [Google Scholar]
- Napirei M, Ludwig S, Mezrhab J, Klöckl T, Mannherz HG. 2009. Murine serum nucleases—contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J 276: 1059–1073. 10.1111/j.1742-4658.2008.06849.x [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpas L, Chan RWY, Jiang P, Ni M, Sun K, Rashidfarrokhi A, Soni C, Sisirak V, Lee W-S, Cheng SH, et al. 2019. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc Natl Acad Sci 116: 641–649. 10.1073/pnas.1815031116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. 2016. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164: 57–68. 10.1016/j.cell.2015.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Jiang P, Wong AIC, Cheng YKY, Cheng SH, Zhang H, Chan KCA, Leung TY, Chiu RWK, Lo YMD. 2018. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc Natl Acad Sci 115: E5106–E5114. 10.1073/pnas.1804134115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Jiang P, Cheng SH, Cheng THT, Wong J, Wong VWS, Ng SSM, Ma BBY, Leung TY, Chan SLY, et al. 2019. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res 29: 418–427. 10.1101/gr.242719.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Kamataki A, Yamaki J, Homma Y. 2008. Characterization of circulating DNA in healthy human plasma. Clin Chim Acta 387: 55–58. 10.1016/j.cca.2007.09.001 [DOI] [PubMed] [Google Scholar]
- Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP, Shendure J. 2016. Fragment length of circulating tumor DNA. PLoS Genet 12: e1006162 10.1371/journal.pgen.1006162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Li W. 2009. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10: 232 10.1186/1471-2105-10-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SCY, Chan KCA, Zheng YWL, Jiang P, Liao GJW, Sun H, Akolekar R, Leung TY, Go ATJI, van Vugt JMG, et al. 2014. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci 111: 8583–8588. 10.1073/pnas.1406103111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SCY, Jiang P, Chan KCA, Faas BHW, Choy KW, Leung WC, Leung TY, Lo YMD, Chiu RWK. 2017. Combined count- and size-based analysis of maternal plasma DNA for noninvasive prenatal detection of fetal subchromosomal aberrations facilitates elucidation of the fetal and/or maternal origin of the aberrations. Clin Chem 63: 495–502. 10.1373/clinchem.2016.254813 [DOI] [PubMed] [Google Scholar]
- Zheng YWL, Chan KCA, Sun H, Jiang P, Su X, Chen EZ, Lun FMF, Hung ECW, Lee V, Wong J, et al. 2012. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: a transplantation model. Clin Chem 58: 549–558. 10.1373/clinchem.2011.169318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.