

Abstract
The immune system is a fascinating world of cells, soluble factors, interacting cells, and tissues, all of which are interconnected. The highly complex nature of the immune system makes it difficult to view it as a whole, but researchers are now trying to put all the pieces of the puzzle together to obtain a more complete picture. The development of new specialized equipment and immunological techniques, genetic approaches, animal models, and a long list of monoclonal antibodies, among many other factors, are improving our knowledge of this sophisticated system. The different types of cell subsets, soluble factors, membrane molecules, and cell functionalities are some aspects that we are starting to understand, together with their roles in health, aging, and illness. This knowledge is filling many of the gaps, and in some cases, it has led to changes in our previous assumptions; e.g., adaptive immune cells were previously thought to be unique memory cells until trained innate immunity was observed, and several innate immune cells with features similar to those of cytokine-secreting T cells have been discovered. Moreover, we have improved our knowledge not only regarding immune-mediated illnesses and how the immune system works and interacts with other systems and components (such as the microbiome) but also in terms of ways to manipulate this system through immunotherapy. The development of different types of immunotherapies, including vaccines (prophylactic and therapeutic), and the use of pathogens, monoclonal antibodies, recombinant proteins, cytokines, and cellular immunotherapies, are changing the way in which we approach many diseases, especially cancer.
Keywords: Vaccines, CAR T cells, Trained immunity, Microbiota, Cancer, Monoclonal antibodies
Subject terms: Immunization, Tumour immunology
Introduction
The knowledge of human immunology has improved exponentially in recent years, and more advances in the near future are certainly imminent. The immune system is extremely complex, but we are now developing new tools and skills to study it. Several factors have been involved in these advancements, and the most important ones include the development of thousands of different monoclonal antibodies that allow the identification of a large variety of cell subpopulations and the functional analysis of immune cells. These tools, together with new and sophisticated technologies, such as single-cell analysis, imaging techniques, omics (including massive DNA-RNA sequencing, proteomics, and metabolomics data and new tools for processing these data, such as artificial intelligence and machine learning approaches, mathematical modeling, etc.), newly designed animal models (using conventional transgenic/knockout/knock-in mice or new technologies such as CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats–CRISPR-associated protein 9), are increasing our knowledge about how our immune system functions. The study of the interaction between the immune system and other systems, such as the nervous and endocrine systems or the microbiome, in several illnesses has produced interesting results with important clinical applications.
All of these advances can be applied to several immune-mediated pathologies, but overall, the success achieved with some types of immunotherapies in recent years is revealing new ways to explore and manipulate the immune system for our benefit.
Writing a review about human immunology is a significant challenge, but we have attempted to bring together recent knowledge about the immune system, immune-mediated illnesses and types of immunotherapies.
New findings in fundamental immunology
The last two decades have witnessed a major revolution in the field of immunology. The traditional classification of the immune system into two different arms, namely, innate and adaptive components that collaborate to respond to foreign antigens or to perform self-/nonself-discrimination, has become much more complex. The development and application of new technologies have provided new findings and created a new landscape in which the immune system establishes cross talk, not only between immune components but also with commensal microorganisms1,2 and other important systems, such as the endocrine and nervous systems3–5. These developments have forced immunologists to reformulate the immunological architecture that confers protection, which has made the study of the immune system especially attractive. Moreover, these advances have led to an increased interest in better understanding, managing, and manipulating the immune response in both health and disease.
Cell subsets
The characterization of new immune cell subsets has been a constant feature in the immunology field. This evolution is clearly reflected in the discovery of an innate counterpart of T lymphocytes, collectively named innate lymphoid cells (ILCs)6, and in the identification of different types of effector CD4 and regulatory T cells7.
Innate lymphoid cells (ILCs)
ILCs are lymphocytes, but in contrast to adaptive immune cells, they can colonize lymphoid and barrier tissue sites during fetal development, do not undergo somatic recombination and do not express antigen-specific receptors8,9. In addition to lymphoid organs, ILCs are enriched in barrier tissues, such as the gastrointestinal tract, airways, and skin10,11. These innate cells have been considered to be tissue-resident cells, but recent studies suggest that ILCs can migrate through the lymphatic system during homeostasis or enter into the circulation upon infection and inflammation6,12. Currently, five different ILCs are defined on the basis of their transcription factor expression, different cytokine production and/or developmental patterns6: natural killer (NK) cells (discussed below), lymphoid tissue inducer cells (LTis) and three subsets of helper-like ILCs (ILC1s, ILC2s, and ILC3s), which are considered to be the innate counterparts of T helper (Th) 1, Th2, and Th17 cells, respectively. The main focus of this review is ILCs.
ILC1s are dependent on the T-box transcription factor T-bet and produce interferon gamma (INF-γ), but they differ in the expression of eomesodermin transcription factor13. ILC1s express CD127 in humans and CD200R in mice, but the natural cytotoxicity receptor NKp46 (also known as NCR1) is expressed in both species14,15.
ILC2s constitute the most homogeneous class of ILCs; they are dependent on GATA3 and RORα, and they produce type 2 cytokines, mainly interleukin 5 (IL-5) and IL-13. ILC2s are involved in immune responses to parasite infection, and in humans, they express chemoattractant receptor-homologous molecule expressed in TH2 cells (CRTH2) and high levels of CD161, whereas most mouse ILC2s express ST2 (a member of the IL-1 receptor family)14,15.
The development and function of ILC3s depend on the transcription factor RORγt. Both human and mouse ILC3s can produce granulocyte macrophage colony-stimulating factor (GM-CSF), IL-17, and/or IL-2216,17. In humans, two major ILC3 subsets can be distinguished on the basis of the expression of the natural cytotoxicity receptor NKp44 (also known as NCR2)14,15. Both types can produce IL-17, but the production of IL-22 is mainly confined to NKp44+ ILC3s.
Extensive research has focused on deciphering the role of ILCs to ensure the maintenance of tissue homeostasis and immune protection11,18. ILCs express particular sets of receptors in a tissue-specific manner, and these allow the detection of host-derived signals (including those from alarmins, neuronal mediators, microbia, and the diet)19. The integration of these endogenous signals is essential for the maintenance of tissue homeostasis, but dysregulation of ILC responses leads to inflammation and disorder12,20. ILC are mainly involved in early protection against viruses and bacteria13,21, but their response to dysregulated local proinflammatory cytokine production in adipose tissues leads to the development of metabolic disorders and obesity20. IL-5 and IL-13 produced by ILC2s induce goblet cell differentiation and the recruitment of eosinophils, basophils, and mast cells22, which are involved in protection against infection by helminths and viruses, but when uncontrolled, these cells drive allergic responses and metabolic disorders. Moreover, the depletion of ILC2s in animal models suggests a role for these cells in atopic dermatitis and asthma23.
ILC3s are abundant in mucosal tissues, and NCR2+ ILC3s have been proven to be essential for regulating the balance between commensal and pathogenic bacteria through the production of IL-2224. In contrast, NCR2− ILC3s can promote colitis in a model of inflammatory bowel disease25. The lack of immunodeficiency in ILC-deficient patients led to the proposal that ILCs are dispensable in the presence of functional T cells and B cells26. However, recent studies support the idea that ILCs cannot be considered to have functions that only duplicate those of the adaptive immune system.
In addition to those showing the essential role of LTi cells in the formation of secondary lymphoid organs during embryogenesis and the postnatal development of intestinal lymphoid clusters, recent studies also provide evidence that subsets of ILCs express multiple factors that modulate the adaptive immune response in health and disease27,28. In particular, ILC2s and ILC3s modulate the T-cell response. Studies in mice suggest that in healthy intestine, ILC3s express major histocompatibility complex (MHC) class II molecules but lack the expression of costimulatory molecules; therefore, they inhibit microbiota-specific T-cell responses, thus preventing intestinal inflammation29. It seems that the interaction between ILC3s and Tfh cells limits IL-4 secretion and the production of IgA by mucosal B cells30.
Studies with murine models have significantly contributed to the classification and understanding of the role of ILCs in the immune system, especially since similarities have been observed between ILCs identified in mice and humans15. However, the differences between these two species present real challenges15,31 because human ILCs have unique attributes that are only now being elucidated, with further work required in this exciting field. The roles of ILCs in immunity and their cross talk with other components of the immune response await further analysis. Detailed coverage of this topic is beyond the scope of this review, and we refer the reader to recent reviews that provide more information on the biology of human32 and mouse33,34 ILCs.
T cells and plasticity
T cells are categorized as Tα/β and Tγ/δ cells, depending on the type of T-cell receptor (TCR) that they express35. Human Tγ/δ cells, similar to their murine counterparts, are a minor population (1–10% of nucleated cells) in peripheral blood, but are especially abundant in barrier tissues such as the epidermis35–37.
The three main subsets of T cells carrying α/β receptor are the CD4+T helper cells and CD8+cytotoxic and CD4+ CD25+ regulatory T cells38.
New effector CD4+ helper T-cell subsets (initially classified as Th1 and Th2)39,40 have been recently described, and at least six human Th cell subsets have been identified to date: Th1, Th2, Th17, Tfh, Th9, and Th22 cells38,41. All of these cells recognize foreign peptides presented by class II MHC molecules on antigen-presenting cells (dendritic cells, macrophages, and B lymphocytes).
Th1 cells are required to activate macrophages and cell-mediated immunity to kill intracellular pathogens42, whereas Th2 cells are important in facilitating eosinophils to fight against parasitic helminths and B cells for antibody production and antibody class-switching to generate IgA or IgE43. Th17 cells are required to mobilize neutrophils for the clearance of fungi and extracellular bacteria, and they are also involved in mucosal protection44. Th9 and Th22 cells are also involved in mucosal immunity; Th9 cells protect against parasites45,46, and Th22 cells prevent microbial translocation across epithelial surfaces and promote wound healing47,48. As mentioned in the introduction to ILCs, studies on human Th cells isolated from lymphoid organs and blood samples, along with recent observations on the developmental mechanism of distinct Th cell subsets, have revealed both similarities and differences of human and mouse Th cells41,49,50.
Tfh cells are very important for germinal center reactions, antibody class switching, affinity maturation, and the development of high affinity antibodies and memory B cells51,52. At the surface marker level, Tfh cells are generally characterized by the expression of CXCR5, the chemokine receptor for CXCL13, which is highly expressed on B-cell follicles for expressing inducible T-cell costimulator (ICOS) and programmed death protein 1 (PD-1)53,54, which enable their involvement in the interaction of Tfh cells and B cells55.
The definition of a given T cell lineage is based on its ability to sense different inductive cytokines, to produce particular cytokines or to express a lineage-specifying transcription factor. Th1 cells produce IFN-γ and express T-bet56; Th2 cells are characterized by IL-4, IL-5, and IL-13 production and GATA-3 expression57,58; pTregs, which are induced in the periphery from naïve precursors, produce TGF-β and express Foxp3 (Tr1 cells are IL-10-secreting Tregs that do not express Foxp3)59. Th17 cells produce IL-17A, IL-17F, and IL-22 and express RORγt60,61, and Tfh cells produce IL-4 and IL-21 and express the BCL6 transcription factor. In addition, Th22 cells, which produce IL-22 and express the aryl hydrocarbon receptor (AHR)47,62, and Th9 cells, are characterized by the expression of IL-9 and the transcription factor PU.163. Additional levels of regulation, such as the differential expression of microRNAs, long noncoding RNAs (lncRNAs), and protein stability and function, have been found to control various aspects of Th cell differentiation and effector function64,65.
CD8+ cytotoxic T cells express the dimeric CD8 marker and have specific lytic capacity to target cells through several mechanisms, including the release of cytotoxic granules, secretion of cytokine tumor necrosis factor alpha (TNFa) and interferon gamma, and the induction of cell death through the interactions of Fas and the Fas ligand38,66. Their TCRs are restricted to interactions with peptides presented by class I MHCs.
Regulatory T cells (Tregs) include thymically derived and peripherally induced regulatory T cells (tTregs and pTregs, respectively), and they produce either IL10, TGF-beta, IL-35 or combinations of these proteins67. tTregs express the transcription factor Foxp3 and secrete IL10 and TGF-β; pTregs, which are induced in the periphery from naïve precursors, can also be subdivided into IL-10-induced Tregs [Tr1 cells] (which secrete large amounts of IL-10 and moderate levels of TGFβ), TH3 cells (which produce IL-10 and TGF-β), and TGFβ-induced Tregs [iTregs], which may or may not express Foxp3.
Moreover, new subsets of regulatory T cells have been described. They include follicular regulatory T cells (which express Foxp3 and Bcl-6 and CXCR5), which modulate the function of Tfh cells and fine-tune the germinal center response68–70, and a IL-35-dependent regulatory population of cells (referred to as iTr35 cells), which show potent suppressive potential in several mouse disease models71. Other regulatory populations have also been described, including Bregs and CD8+ Tregs, which are the analogous counterparts of Tregs72–74.
Recent studies have revealed the capacity of differentiated T cells, particularly Th17 cell and pTreg subsets, to change their phenotype in response to changing contexts75–79. Becattini et al.78 found that human memory CD4 T cells primed in vivo by pathogens (e.g., Candida albicans and Mycobacterium tuberculosis) or vaccines (Tetanus toxoid) are highly heterogeneous, both at the population and clonal levels. With respect to studies on human arthritis, Nistala et al.79 proposed that Th17 cells are recruited to the joint and converted to Th17/1 or Th1 cells in response to local IL-12 levels. This plasticity has also been observed with in vitro assays under conditions that mimic a disease site, namely, low TGF-β and high IL-12 levels79. These results are inconsistent with the original idea of Th lineage stability and provide new possibilities for disease treatment aimed at inducing particular Th subsets to modulate the immune response against pathogens or to control detrimental immunity76,77,80.
Trained and adaptive immune memory
Other classical concepts in fundamental immunology, such as immune memory, are also changing. The specificity and the capacity to generate long-lived memory cells are two properties that have been classically used to distinguish innate immunity from adaptive immunity. Adaptive immunity is clearly based on the specific recognition of antigenic determinants by somatically diversified receptors (B cell and T cell receptors (BCR and TCRs, respectively)) and on its capacity to respond more effectively to restimulation with the same antigen. In contrast, innate immune responses have traditionally been considered nonspecific and without the capacity to adapt81. However, the discovery of germline-encoded pattern recognition receptors (PRRs) and the “trained innate” immunity (or innate immune memory) have provoked a shift in our understanding of the immune response. In 1997, Medzhitov et al. demonstrated that pattern recognition receptors (PRRs) expressed on innate cells recognize invariant molecular structures expressed by invading pathogens82. After the interaction, PRRs trigger the expression of costimulatory molecules and activate important signaling pathways to induce the activation of innate and adaptive immune cells. PRRs mainly belong to four families: Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), and peptidoglycan recognition proteins (PGRPs)83,84. The profiles of PRRs expressed by innate cells can lead to partially specific recognition of a type of microorganism; e.g., innate cells can distinguish between gram-negative and gram-positive bacteria and modulate the immune response based on this recognition, although they cannot differentiate between bacterial species85.
The idea that only jawed vertebrates developed immunological memory has also been challenged by the observation of resistance to reinfection in organisms that lack an adaptive immune response, such as plants86 and invertebrates87,88. Recent studies have shown that monocytes and macrophages exposed to Candida albicans or β-glucans exhibited an enhanced secondary response89. In addition, immunization of mice with bacillus Calmette-Guérin (BCG, the tuberculosis vaccine) induces T cell-independent protection against secondary infections by Candida albicans, Schistosoma mansoni or influenza virus90–93. Thus, organisms are protected not only against the original microorganism but also to unrelated pathogens.
The mechanisms underlying the establishment of this innate immune memory differ from those involved in adaptive immune memory81. After infection or vaccination, innate immune cells (such as monocytes and macrophages) display long-term functional changes through epigenetic and metabolic reprogramming, including histone acetylation, methylation and modulation of noncoding RNAs94–96. In turn, the faster and more pronounced reactivity of adaptive immune cells (T and B lymphocytes) upon reinfection is characterized by permanent changes in the genome of cells, such as mutations, gene rearrangement, clonal expansions, as well as epigenetic modifications, all of which ensure a more persistent effect than is endowed by trained immunity81,94,95.
Other cells for which immunological memory has been described include Tγ/δ cells97 and innate lymphoid cells98. Recently, some authors have proposed that NK cells are also capable of immunological memory99–102. Antigen-specific recall responses by human NK cells were observed by Nikzad et al.103 in humanized mice and in varicella zoster virus (VZV)-exposed adult human volunteers, in which cytotoxic NK cells were recruited to sites of an VZV test antigen challenge on the skin. Sensitization with haptens using mice lacking T cells and B cells led to the generation of hapten-specific memory NK cells99. The recall response persisted for more than four months after priming, and was adoptively transferred to naïve mice100. Interestingly, NK cells exhibit memory that is not only specific to a given virus, such as cytomegalovirus101,102, but that is also induced in the absence of a defined antigen104,105.
Furthermore, new studies suggest that trained immunity is not a phenomenon that is restricted to immune cells, because epithelial stem cells also retain memory of previous inflammatory challenges by displaying an enhanced wound-healing capacity upon skin damage106. Given the data outlined above, immunological memory is now recognized to be highly diverse and not restricted to B cell- or T cell-mediated adaptive immunity. Much remains to be learned in this field, but the different manifestations of immunological memory described above offer an important basis for clinical applications, such as the development of novel vaccination strategies107 or new therapies for pathological situations in which immunological memory can be detrimental, such as allergies or autoimmune diseases94,108,109.
Interaction of the immune system and the microbiome
The immune system has evolved in the presence of commensal microorganisms that colonize barrier surfaces of vertebrates and invertebrates1,110. The cross talk between the natural host microbiome and immune system is particularly interesting in the gastrointestinal tract, where the density and diversity of indigenous bacteria, viruses and fungi are greatest compared to those of other anatomical sites111. In the literature, reports of observed changes in microbial community composition during diseases are diverse and include those in inflammatory bowel disease (IBD), obesity, metabolic syndrome, and multiple sclerosis112–116. However, the microbiome can be influenced by different factors, such as the specific niche that it occupies, diet, stress, environmental factors, and host genetics, and a specific correlation does not necessarily infer causation. The presence of these commensals in mucosal tissues has been known since before Metchnikoff, but the current knowledge on the role of the microbiome in shaping the immune system throughout life came mostly from the development of next-generation sequencing (in particular, the reduction in the cost of 16S ribosomal RNA gene sequencing) and the use of germ-free animal models, which can be colonized even with human microbiota117.
Germ-free mice are characterized by atrophy of Peyer’s patches with few germinal centers and isolated lymphoid follicles, a lower number of B, T, and dendritic cells and a decreased level of immunoglobulins, particularly IgA and IgG118. These effects are observed at the mucosal and systemic levels, and they can be reversed within weeks after the colonization of germ-free mice with commensal bacteria119. Moreover, colonization with commensal Bacteroides fragilis revealed the immunomodulatory effect of bacterial polysaccharides in restoring systemic cells and the differentiation of CD4+ T cells into regulatory T cells (Foxp3+ Tregs), which in turn favor mucosal immunomodulation120. The induction of Th17 cell maturation by segmented filamentous bacteria has also been reported121. These important examples emphasize the major roles of the commensal microbiome in the maturation of mucus-associated lymphoid tissue and the systemic immune system. The development of new technologies to better track the locations and activities of distinct microbial populations is essential to elucidate host-microbe interactions, through which other systems, such as the nervous system, seem to play important roles2,122–125.
The better characterization of some immune cell subsets, trained immunity, and host-microbiome interactions provides a few very good examples that prove the maturation of immunology in the last few decades. In this sense, studies with mouse models have significantly contributed to the increase in our fundamental knowledge; however, the differences between murine and human immunology are notable, and conclusions drawn from mouse studies are sometimes not fully translated to humans31. If we want to fully exploit the power of the immune system for human health, greater effort is required for understanding human immunology. Immunologists, in cooperation with experts from other fields, have developed a variety of protocols and tools to achieve greater selectivity in the identification and analysis of human cell subsets, types of cytokines and receptors, chemokines, etc. These tools range from biological approaches that rely on next-generation sequencing, mass spectrometry, and bioinformatics to immune monitoring technologies based on multiparameter flow cytometry and single-cell gene expression analysis. Although not without limitations, these techniques provide a much better picture of the whole immune system than individual and independent approaches.
Immune-mediated illnesses
Immune-mediated illnesses comprise a wide variety of diseases characterized by the dysregulation of a normal immune response. Most of these illnesses are complex disorders believed to arise from a combination of genetic and environmental factors126.
Infectious diseases
Infectious diseases are caused by pathogens (viruses, bacteria, fungi or parasites that infect the host body), and they remain a leading cause of mortality worldwide. Prominent examples include illnesses produced by Mycobacterium tuberculosis, human immunodeficiency virus (HIV), Plasmodium falciparum or the current coronavirus disease 2019 (COVID-19) outbreak caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which has already infected millions of people and produced thousands of deaths in many countries.
For a number of years, many people believed Koch’s postulates, which implied that virulence traits reside solely in the pathogen. However, recent advances in molecular biology have shown that host genes play major roles in infection, together with a wide range of environmental variables127.
To date, six gene products endowing infectious disease susceptibility have been validated in the literature: (1) hemoglobin subunit beta; (2) band 3-anion transport protein; (3) Duffy antigen/receptor, which is associated with Plasmodium spp. infections; (4) the prion protein associated with Creutzfeldt–Jakob disease; (5) fucosyltransferase 2 and 3, which is associated with Norwalk virus infections; and (6) C-C motif chemokine receptor 5 (CCR5) coreceptor, encoded by an immune-related gene and leads to the impairment of the entry of the human immunodeficiency virus (HIV) into helper T cells, thus avoiding/decreasing the progression to acquired immunodeficiency syndrome128.
Another gene associated with infectious disease and the immune system is the natural-resistance-associated macrophage protein (NRAMP1), which encodes an integral membrane protein expressed exclusively in the lysosomal compartment of monocytes and macrophages. It is a susceptibility locus for increased ratios of infection with Leishmania spp. parasites and certain strains of Salmonella spp., Mycobacterium bovis and Mycobacterium tuberculosis129,130. In addition, it has been suggested that functional variants of immunoglobulin Fc gamma RIIa (CD32) are related to the development of invasive encapsulated bacterial infections131.
Moreover, because of recently acquired genomic data, new human polymorphisms have been discovered, some of which play roles in changing immunoglobulin levels, seroconversion rates or the intensity of antigen-specific immune responses. In addition, they also contribute to human susceptibility to infection by viruses such as influenza, rhinovirus and respiratory syncytial virus132. These polymorphisms are mapped within the MHC (HLA-DQB1*03, HLA-DRβ1, or HLA-DPβ1), natural killer cell immunoglobulin-like receptors 1 and 4 (KIR3DL1 and KIR2DS4) and natural killer lectin-like receptor D1 (KLDR-1)133.
Several recent studies available as preprints have analyzed certain genes that may explain the differences in the variable expression of and susceptibility to COVID-19 by patients, either by affecting the host receptor for the virus (angiotensin I converting enzyme 2 (ACE-2))134, immune genes (TLR7 and others) or blood groups (group O seems to be the most protective)135, and more extensive omics studies are now underway with larger numbers of patients.
Autoimmune diseases
In 1901, the physician Paul Ehrlich first used the term “Horror autotoxicus” to describe the way autoimmunity contradicts the natural aversion to self-injury (“Living with the Enemy”, reviewed in136). Currently, according to the American Autoimmune Related Disorders Association, more than 100 autoimmune diseases have been identified. Historically, these diseases were considered to be rare, but current epidemiological data have shown that they affect approximately 3–5% of the population worldwide. Some of the most common autoimmune diseases include type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus, and inflammatory bowel disease (https://www.aarda.org/diseaselist/). Although significant progress has been made in understanding the mechanisms of autoimmune diseases and the nature of self-tolerance, these disease remain major burdens on health systems around the world.
Autoimmune diseases arise when the immune system attacks normal components of the body137. The concept of immune tolerance is defined as the ability of the immune system to prevent the targeting of self-molecules, self-cells or self-tissues. On the other hand, the failure to distinguish self from nonself is often termed a break of tolerance, and it is the basis for an autoimmune disease138.
What are the mechanisms that lead to a break in tolerance? Autoimmune diseases are complex disorders that are believed to arise from a combination of genetic (mutations and higher inheritance frequency of some types of major histocompatibility complex alleles), epidemiological (age and sex) and environmental (infections, microbiota, tobacco, chemicals and pharmaceutical drugs). factors These factors trigger a break in self-tolerance with the activation of self-reactive lymphocytes through several mechanisms, such as molecular mimicry, the overexpression and abnormal expression of MHC class II molecules in peripheral tissues, thymic aging, and immunodeficiencies (discussed below) and many others. Some lymphocytes escape control due to polymorphisms in several genes that affect the routes of lymphocyte activation. Other causes may include defective antigen presentation by some MHC variants with specific polymorphisms. Therefore, the self-reactive lymphocytes that have escaped control and react against self-constituents initiate the autoimmune process139.
Although a large number of genome-wide association studies (GWAS) have led to the identification of hundreds of polymorphisms associated with the development of different autoimmune diseases, it has proven difficult to define the role of most of these polymorphisms in the breakdown of tolerance to a self-antigen139–145. It is worth highlighting, however, that the MHC remains the main genetic factor associated with human autoimmunity138,139.
Other gene variants identified are common to many autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, type I diabetes, ulcerative colitis, autoimmune hepatitis and numerous other autoimmune diseases. For example, the protein tyrosine phosphatase nonreceptor type 22 (PTPN22) gene encodes a protein that inhibits T-cell activation in the adaptive immune system, whereas it promotes myeloid cell activation; interferon regulatory factor 5–transportin 3 (IRF5–TNPO3) is involved in the accumulation of lymphocytes within lymphoid organs and failed elimination of autoreactive naïve T cells; BTB domain and CNC homolog 2 (BACH2) has a critical role in immunoglobulin class-switching recombination, somatic hypermutation of immunoglobulin encoding genes and the activation of tissue macrophages. A more complete list of genes associated with autoimmunity can be found in the review by Wang et al.138
Researchers are currently looking for the missing heritability in autoimmune diseases by focusing on the study of methylome profiles, genetic cargos in extracellular vesicles, genetic alterations, and ways in which the microbiome may affect these diseases.
Rejection of transplants
Immune-mediated rejection of tissue allografts was first described in 1945 by the British immunologist Peter Medawar146,147. Only three years later, George Snell described the MHC, which carries the histocompatibility genes, and one decade later, Jean Dausset described the human leukocyte antigen (HLA); each of these scientists was recognized with the Nobel Prize in Physiology and Medicine148. Since its discovery, MHC has emerged as the most polymorphic gene locus in eukaryotes with 24093 HLA and related alleles, more than 362709 nucleotide variants reported in the Individual-Participant Data–International ImMunoGeneTics/Human Leukocyte Antigen (IPD–IMGT/HLA) work group database (https://www.ebi.ac.uk/ipd/imgt/hla/), release 3.39.0, 2020/01/20149.
Although the main barrier for long-term organ and tissue grafting is driven by HLA incompatibilities, other important players play roles in transplant rejection. In particular, minor histocompatibility antigens, which are peptides derived from allelic variants of normal cellular proteins, presented by class I or II MHC antigens induce cellular immune responses in HLA-matched individuals who lack the same allelic variant150.
Natural killer (NK) cells also play important roles in transplantation through their killer cell immunoglobulin-like receptors (KIRs), which are receptors for HLA class I molecules. NK cells expressing an inhibitory KIR-binding self-HLA can be activated when exposed to allografts that lack a ligand for the inhibitory receptor151. The locus that codifies these receptors displays a considerable degree of polymorphism, with 1110 alleles reported in the Individual-Participant Data–International/Killer Cell Immunoglobulin-Like Receptors (IPD/KIR) work group database, release 2.9.0, 2019/12/11149.
More recently, we have begun to appreciate the importance of non-HLA genetic factors in the development of transplant rejection; examples include polymorphisms in the genes encoding cytokines, such as tumor necrosis factors (TNF), interleukins (IL-1, IL-6 and IL-10), interferon gamma (IFN-γ), and transforming growth factor-β3 (TGF-β3). Other genes encode pathogen recognition receptors, with nucleotide-binding oligomerization domain-containing 2 (NOD2 (CARD15)) being the most widely studied, although conclusive data have not been obtained to date148.
Immunodeficiencies
Primary immunodeficiencies (PIDs) comprise a heterogeneous group of more than 400 genetic disorders that result in defects in the immune response152. PIDs are considered Mendelian disorders because they are mainly autosomal recessive disorders that often display incomplete penetrance, which affects the severity and onset of the disease. With the exception of immunoglobulin A (IgA) deficiency, PIDs are considered to be rare disorders, as their prevalence worldwide ranges from 1 to 9 among 100,000 people153. Unsurprisingly, these types of diseases are not uncommon in highly consanguineous populations such as those in the Middle East/Northern Africa (MENA) region. The incidence of consanguinity marriage in these areas ranges between 20 and 56%, which leads to a unique population in which autosomal recessive diseases arise, with the prevalence of PID in these countries as high as 30 in 100,000 people154.
Although more than 400 genes have been described for PIDs, approximately 60% of the causal genes remain unknown, and next-generation sequencing studies performed in MENA populations are contributing to the search for currently unknown genes that cause PIDs155. A complete and updated list of PID-causing genes and diseases can be found at the European Society for Immunodeficiencies (ESID) webpage (https://esid.org)156.
Clinical manifestations of PIDs are highly variable; many disorders involve an increased susceptibility to several types of infections, but some patients develop autoimmune diseases. Patients usually present recurrent sinus or ear infections or pneumonia within a one-year period; other indicators are failure to thrive, poor response to prolonged use of antibiotics, and persistent thrush or skin abscesses153.
Depending on the affected pathway, PIDs are associated with varying levels of severity, times of onset, and risks of infection by certain groups of microorganisms. According to the International Union of Immunological Societies (IUIS) (https://iuis.org/committees/iei/), 430 inborn errors of immunity can be classified as follows: (a) immunodeficiencies that affect cellular and humoral immunity; (b) combined immunodeficiency (CID) with associated or syndromic features; (c) predominant antibody deficiencies; (d) diseases of immune dysregulation; (e) congenital defects of phagocyte number, function, or both; (f) defects in intrinsic and innate immunity; (g) autoinflammatory disorders; (h) complement deficiencies; and (i) phenocopies of a PID156,157.
However, PIDs are broadly classified as follows according to the component of the immune system affected:
T-cell immunodeficiency, e.g., defects in the IFN-γ/IL-12 pathway and mutations in the autoimmune regulator (AIRE) gene.
B-cell (antibody-mediated) immunodeficiency: gamma-globulinemia, X-linked common variable immunodeficiency (CVID), selective IgA deficiency, specific antibody deficiency, and IgG subclass deficiency.
Combined immunodeficiency: Wiskott–Aldrich syndrome, ataxia telangiectasia, DiGeorge syndrome and severe combined immunodeficiency (SCID).
Phagocyte defects: chronic granulomatous disease, hyperimmunoglobulin E (IgE) syndrome and leukocyte adhesion deficiency.
Complement defects (deficiency in early, late or regulatory complement components)158.
Autoinflammatory diseases
Systemic autoinflammatory diseases (AIDs) are characterized by recurrent acute inflammatory episodes secondary to a dysregulated inflammatory process that typically develops during childhood, with recurrent episodes of fever, rashes, and disease-specific patterns of organ inflammation. Genetically speaking, these are hereditary disorders, andto date, more than 40 genes (Table 1) have been identified as causes of AIDs, which can be grouped according to the pathway that is altered159.
Inflammasome. The inflammasome is a multiprotein intracellular complex that detects pathogenic microorganisms and stressors and activates the highly pro-inflammatory cytokines IL-1β and IL-18. Genes affected in this group are MEFV (Mediterranean fever pyrin innate immunity regulator), which is related to familial Mediterranean fever (FMF); NLRC4 (NLR family CARD domain-containing 4); NLRP1 (NLR family pyrin domain-containing 1) and WDR1 (WD repeat domain 1)159.
Type-I interferon (IFN)-mediated disorders. These disorders are characterized by the upregulated expression of genes induced by IFN. The gain of function by variants of TMEM173 (transmembrane protein 173) is the core manifestation of this disorder group, but other genes have been identified, including DDX58 (DExD/H-box helicase 58), DNASE2 (lysosomal deoxyribonuclease 2), POLA1 (DNA polymerase alpha 1 subunit) and USP18 (ubiquitin-specific peptidase 18)159,160.
Ubiquitination disorders. Ubiquitination is a process that marks proteins for degradation via the proteasome, which is required for the processing of intracellular antigens (such as virus proteins or mutated tumor proteins) and their presentation by class I HLA molecules. Ubiquitination involves three main steps: activation, conjugation and ligation, which are performed by ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s). Ubiquitination disorders are caused by variants of the PSMB8, PSMB9, PSMA3 and PSM4 genes (proteasome 20S subunit beta 8, subunit beta 9, subunit alpha 3 and subunit alpha 4, respectively), affecting the proteasome subunits, proteasome maturation protein gene (POMP) and/or proteasome assembly chaperone 2 (PSMG2), by encoding proteasome assembly molecules161. In addition, other genes in this group, such as OTULIN (OTU deubiquitinase with linear linkage specificity), encode ubiquitin peptidases, i.e., proteins involved in ubiquitination assembly complexes, such as HOIL-1 (heme-oxidized IRP2 ubiquitin ligase 1) and HOIP (NHP2-like protein 1 homolog). Finally, the loss of function due to variants of the TNFAIP3 (TNF-alpha-induced protein 3, also known as A20) gene, which encodes a protein with ubiquitin ligase and ubiquitinase activity, has also been described159.
Inflammatory or innate immune regulators. A large number of genes have been found to affect the pathways/mechanisms involved in macrophage and B-cell differentiation and lymph node development, among many functions. Genes in this group include ADA2 (adenosine deaminase 2), TNFRSF11A (TNF receptor superfamily member 11a), ADGRE2 (adhesion G protein-coupled receptor E2), TRNT1 (tRNA nucleotidyltransferase 1), LACC1 (laccase domain-containing 1) and AP1S3 (adaptor related protein complex 1 subunit sigma 3)159.
Table 1.
Genes involved in autoinflammatory diseases and their pattern of inheritance
| Gene | Protein | Disorder | Mode of inheritance |
|---|---|---|---|
| ADA2 | ADA2 | Deficiency of adenosine deaminase 2 (DADA2) | AR |
| ADGRE2 | ADGRE2 | Vibratory urticaria | AD |
| AP1S3 | AP1S3 | AP1S3-mediated psoriasis (AMPS) | AD |
| CARD14 | CARD14 | CARD14-mediated psoriasis (CAMPS)/psoriasis susceptibility locus 2 (PSORS2) | AD |
| DDX58 | DDX58 | Singleton–Merten Syndrome (SMS) | AR |
| DNASE2 | DNASE2 | Interferon pathology | Unknown |
| HLA-B*51 | HLA-B | Behçet disease (present epistasis over ERAP1) | Complex |
| HLA-DRB1*11 | HLA-DRB1 | Systemic juvenile idiopathic arthritis | Complex |
| IFIH1 | MDA5 | Singleton–Merten Syndrome (SMS) | AD |
| IL10 | IL-10 | IL-10 deficiency (IL-10D) | AR |
| IL10RA, IL10RB | IL-10 receptor | Very early-onset inflammatory bowel disease (VEOIBD) | AR |
| IL1RN | IL-1 receptor antagonist | Deficiency of IL-1 receptor antagonist (DIRA) | AR |
| IL36 RN | IL-36 receptor antagonist | Deficiency of IL-36 receptor antagonist (DITRA) | AR |
| LACC1 | LACC1/FAMIN | Monogenic form of systemic juvenile idiopathic arthritis (sJIA) | AR |
| LPIN2 | Lipin 2 | Majeed syndrome | AR |
| MEFV | Pyrin/marenostrin | Familial Mediterranean fever (FMF) | AR |
| MEFV | Pyrin/marenostrin | Pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND) | AD |
| MVK | Mevalonate kinase | Hyperimmunoglobulinemia D with periodic fever syndrome (HIDS)/mevalonate kinase deficiency (MKD); porokeratosis 3 (POROK3)/disseminated superficial actinic porokeratosis (DSAP) | AR |
| NLRC4 | NLRC4 | NLRC4-macrophage activation syndrome (NLRC4-MAS) | AD - mosaicism |
| NLRP12 | NLRP12 | Familial cold autoinflammatory syndrome 2 (FACS2)/NLRP12-associated periodic syndrome (NAPS12) | AD |
| NLRP12 | NLRP12 | Multiple self-healing palmoplantar carcinoma (MSPC); familial keratosis lichenoides chronica (FKLC)/NLRP1-associated autoinflammation with arthritis and dyskeratosis (NAIAD) | AD |
| NLRP3 | Cryopyrin/NLRP3 | Cryopyrin-associated periodic syndrome (CAPS) | AD |
| NLRP3 | Cryopyrin/NLRP3 | Schnitzler syndrome | AD - mosaicism |
| NLRP7 | NLRP7 | Hydatidiform mole | AR |
| NOD2 | NOD2 | Blau syndrome/early-onset sarcoidosis | AD |
| OUTLIN | OUTLIN | Otulipenia/otulin-related autoinflammatory syndrome (ORAS) | AR |
| PLCG2 | PLCγ2 | Autoinflammatory PLCγ2-associated antibody deficiency and immune dysregulation (APLAID) | AD |
| POLA1 | POLA1 | X-linked reticulate pigmentary disorder (XLPDR) | X-linked |
| POMP | POMP | POMP-related autoinflammation and immune dysregulation disease (PRAID) | AD |
| PSMA3 PSMB4 PSMB9 POMP | Immunoproteasome subunits | PRASS | AR - digenism |
| PSMB8 | Immunoproteasome β5i subunit | Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) | AR |
| PSMB8 | Immunoproteasome β5i subunit | Proteasome-associated autoinflammatory syndrome (PRASS) | AR - digenism |
| PSMG2 | PSMG2 | PRASS | AR |
| PSTPIP1 | PSTPIP1 | Pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) | AD |
| RBCK1 | HOIL-1 | HOIL-1 deficiency | AR |
| RNF31 | HOIP | HOIP deficiency | AR |
| SH3BP2 | SH3BP2 | Cherubism | AD |
| SLC29A3 | hENT3 | Histiocytosis-lymphadenopathy plus syndrome | AR |
| TMEM173 | STING | STING-associated vasculopathy with onset in infancy (SAVI) | AD - mosaicism |
| TNFAIP3 | A20 | A20 haploinsufficiency (HA20) | AD |
| TNFRSF11A | TNFRSF11A | TNFRSF11A-associated periodic syndrome (TRAPS11) | AD |
| TNFRSF1A | TNFR1 | Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) | AD |
| TRNT1 | TRNT1 | Sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD) | AR |
| UPS18 | UPS18 | TORCH Syndrome | AD |
| WDR1 | WDR1 | Periodic fever, immunodeficiency, and thrombocytopenia (PFIT) | AR |
Allergy
Allergic diseases can be termed complex diseases that involve both genetic and environmental factors, and they influence not only the development of IgE-mediated sensitivity in the case of hypersensitivity type I allergies but also the subsequent development of clinical symptoms in a range of tissues, including skin, nose, and lung tissue162.
Since the first report of a link between chromosome 11q12 and atopy in 1989163, knowledge about the common risk variants for allergic diseases has increased exponentially, mainly because of GWAS. Most allergic diseases have allergy-related traits such as asthma, with the strongest association mapped to chromosome 17q21. However, the disease-associated gene at this locus remains unclear; one of the candidate genes is ORMDL3 (sphingolipid biosynthesis regulator 3) due to its role in sphingolipid synthesis and the regulation of eosinophils. Other genes associated with asthma are interleukin 33 (IL33) and its receptor, IL1RL1 (interleukin 1 receptor-like 1), HLA region, SMAD3 (SMA- and MAD-related protein 3) and IL2RB (interleukin 2 receptor subunit beta)164.
As asthma and other allergic-associated traits could be present in patients without allergies, some researchers performed GWAS analysis on cohorts of patients who had high levels of allergen-specific immunoglobulin E (IgE) or a positive skin prick test. As a result, 18 loci were identified, and the strongest association was on chromosome 11q13. This locus has been associated with two genes: C11orf30 (EMSY transcriptional repressor, BRCA2 interacting), a potential regulator of interferon-stimulated gene, and LRRC32 (leucine rich repeat-containing 32), which is involved in Transforming Growth Factor Beta (TGFβ)-signaling in T regulatory cells.
The rest of the associated loci involved in the pathogenesis of allergy highlight the importance of the Th2 responses (STAT6 (signal transducer and activator of transcription 6), TSLP (thymic stromal lymphopoietin), BCL6 (B-cell lymphoma 6 protein), IL1RL1 (interleukin 1 receptor-like 1), IL33 (interleukin 33), GATA3 (trans-acting t-cell-specific transcription factor binding protein 3)); innate immunity (TLR1/6/10 (Toll-like receptor 1/6/10)); TGFβ-signaling (LRRC32 (leucine rich repeat-containing 32), SMAD3 (mothers against decapentaplegic homolog 3)); T-cell (IL2 (interleukin 2), PTGER4 (Prostaglandin E Receptor 4)) and T regulatory box (LRRC32 (leucine rich repeat-containing 32), IL-2, NFATC2 (nuclear factor of activated T cells 2), FOXA1 (forkhead box A1))164.
In the last two years, researchers have focused on epigenome-wide association study (EWAS) of allergy processes. The epigenetic landscape is specific for a given cell; thus, EWAS requires careful selection of the relevant cell type for a given biomedical condition. For allergies, EWAS has mainly been performed on nasal mucosal cells and whole blood (although the result was later normalized by the number of circulating eosinophils). Nasal mucosal cells comprise CD8+ T cells, CD4+ T cells, myeloid cells, innate lymphoid cells, B cells, double-negative T cells, granulocytes, CD117+ cells, and plasma cell populations165. In all of these studies, 36 CpG-associated regions were identified, from which the SMAD3 gene, coding for an important regulator of T-cell differentiation, was replicated in three independent cohorts166. Of all of the genes in whole blood identified using EWAS, only the ACOT7 (acyl-CoA thioesterase 7), EPX (eosinophil peroxidase), GJA4 (gap junction protein alpha 4) and METTL1 (methyltransferase-like 1) genes were confirmed in the nasal cell populations167.
Cancer immunology
In 1909, Ehrlich proposed the idea that mutant cells arise continuously and that the immune system scans for and eradicates these mutant cells before they manifest clinically168. However, immune surveillance remained a controversial topic until its acceptance in the 1990s169.
Immune surveillance is the recognition and elimination of cancerous cells by lymphocytes, which act as sentinels that recognize transformed cells. Ultimately, during tumor progression, cancer cells show low immunogenicity and resistance to immune effector cells, thus expanding and escaping immune control. The way in which cancer cells modify the immune system has been called immune editing169.
The key of immunosurveillance is cancerous cell expression of tumor antigens that can activate various immune cell phenotypes; for simplicity, any overexpressed, mutated, dysregulated, or rearranged gene product expressed by a cancerous cell may be considered a tumor antigen. It is critical to consider that most of these proteins, except those derived from virus-infected cancer cells, are primarily self-proteins, but they are expressed with mutation(s) or minor changes in their antigenic structure170.
One mechanism by which cancer cells escape from immune recognition is antigenic modulation. For example, the loss of MHC class I molecule expression leads to aberrant antigen masking, which is one of the mechanisms described for tumor cells that escape specific antitumor T-cell immune responses171. In addition, the MHC-peptide-T cell receptor complex elicited by a tumor antigen shows weak stability, since high-affinity T-cells tend to be rendered tolerant to these antigens172.
Another mechanism is the direct inhibition induced by cancer cells due to their interaction with surface regulatory molecules, also called checkpoint molecules. These molecules include programmed cell death-1 (PD1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which induce the inhibition of host T cells. Although these checkpoints usually help conventional immune responses control immune activation, they can also be used by tumor cells to inhibit antitumoral T-cell responses173.
PD1) is a transmembrane protein expressed on T, B, and NK cells, and it binds to PD1 ligands (PD-L1 and PD-L2) on target cells. When it binds to its ligand on tumor cells, PD1 inhibits tumor cell apoptosis, causes peripheral effector T-cell exhaustion, and promotes the conversion of effector T cells into regulatory T cells172,174.
CTLA4 is also a physiological negative regulator of T-cell activation. The interaction with CD80/CD86 in the tumor leads to the inhibition of T-cell function and suppressed effector activity175. Knowledge of these two checkpoint inhibitors has opened the door to new antitumoral therapeutic approaches, such as the use of monoclonal antibodies that block the aforementioned interactions (anti-PD1, anti-PD-L1, or anti-CTLA-4), which are called checkpoint inhibitors176.
In addition, tumor cells create an inhibitory microenvironment around them. Malignant cells can recruit other cells, such as immune cells and fibroblasts, which can be corrupted by tumor cells. The interaction between tumor and nontumor cells creates the tumor microenvironment, which is mostly driven by the dynamics of the tumor promoting the proliferation/expansion of cancer cells. For example, tumor and stromal cells release multiple factors, such as the chemokine CCL28 (C-C motif chemokine ligand 28), which inhibits effector T-cell functions and attracts Tregs to the microenvironment172.
Tumor cells use different mechanisms to promote cancer progression and further metastasis. The complete immunological eradication of cancer is the goal of antitumoral immunotherapy and is discussed later in this review.
Immunosenescence and inflammaging
Aging is accompanied by the decline and dysregulation of immune efficacy, which results in an increased vulnerability to infectious diseases, diminished responses to vaccination, and reduced tumor clearance. Immune alterations mainly manifest as a reduction in the number of naïve peripheral blood cells and a relative increase in some types of memory cells177.
Natural aging causes progressive atrophy of the thymus, which is called thymic involution. The endpoint is a significant decrease in naïve T cells, which reduces the diversity of the T-cell antigen receptor (TCR) repertoire and culminates in disrupted T-cell homeostasis178. The cellular and molecular hallmarks of aging have been described as genomic instability, telomere attrition, epigenetic alterations, sarcopenia, changes in intracellular communications, cellular senescence, immunosenescence and mitochondrial dysfunction179.
The process of aging alters the innate and adaptive immune systems. In terms of innate immunity, aging results in a decreased number of circulating monocytes and dendritic cells, reduced phagocytic properties of macrophages and neutrophils, and impaired antigen presentation by dendritic cells179. As mentioned above, aging also generates a reduction in the T-cell and B-cell receptor repertoire due to the accumulation of senescent or exhausted lymphocytes, together with a decrease in the number of circulating naïve T and B cells178,179. On the other hand, NK cell cytotoxicity is maintained in centenarians, and an increase in the number of these cells is observed in healthy aging people177. Moreover, CD4+ T cells exhibit cytotoxic features in centenarians; this is an acquired characteristic for CD4+ T cells that usually have helper, but not cytotoxic functions under physiological conditions180.
In addition to these features, chronic inflammation is considered the key that underlies the phenomenon called ‘inflammaging’, which is related to elevated self-reactivity and results in the typical chronic low-grade, systemic inflammatory phenotype observed in the elderly in the absence of acute infection. Currently, it is believed that self-reactive T cells are the main contributors to this process. It has been proposed that this basal inflammatory state contributes to the development of some diseases, such as Type II diabetes, Alzheimer’s disease and atherosclerosis178. Understanding the mechanisms of age-related disorders in immune regulation is important for identifying more efficient strategies of immune rejuvenation and for the effective induction of vaccination-mediated immunity in older individuals177.
Immunotherapy
Immunotherapy includes the use of certain components of the immune system (antibodies, cells, cytokines, etc.) for the treatment of various cancers and autoimmune diseases and the manipulation of the immune system through vaccines for the prevention and treatment of infectious and allergic diseases (Fig. 1).
Fig. 1.
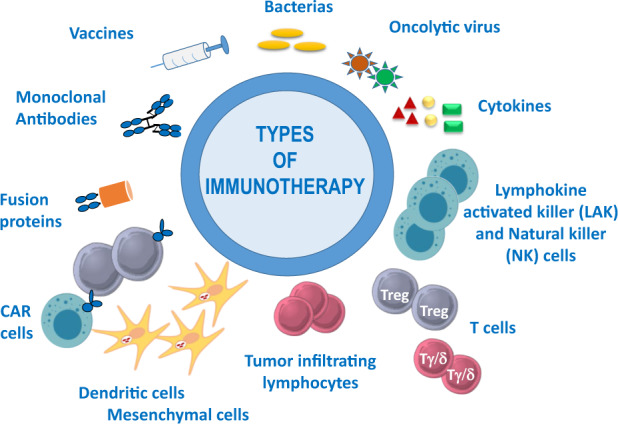
Examples of immunotherapy, including the use of vaccines, monoclonal antibodies, fusion proteins, bacteria, oncolytic viruses, cytokines, and different types of cellular immunotherapy: chimeric antigen receptor (CAR) T cells, dendritic and mesenchymal cells, tumor-infiltrating lymphocytes, regulatory (Treg) and gamma/delta (Tγ/δ) T cells, lymphocyte activated killer (LAK) and natural killer (NK) cells
Immunotherapy using microorganisms or their components in vaccines was first practiced centuries ago; soluble substances such as poly- and monoclonal antibodies, as well as cytokines, have been used for many years, but recently, cellular immunotherapy has emerged in clinical practice. Although immunotherapy can be used for many diseases (infections, autoimmune diseases, macular degeneration, allergic diseases, etc.), it is being used most expansively in the cancer field. The main goal is to destroy the tumor, either directly or indirectly (by enhancing the patient’s immune system), while offering greater specificity and fewer side effects than conferred by conventional therapies.
Pathogens and vaccines for infectious diseases
Immunotherapy associated with pathogens was first linked to the prevention of infectious diseases, starting from variolization (in the X century), followed by Edward Jenner’s vaccination against smallpox (in the XVIII century) and subsequently many other preventive vaccines for infectious diseases. The great advances in the knowledge about infectious diseases took place in the nineteenth century, but the XX and XXI centuries are clearly the vaccination centuries, as many new successful vaccines (with attenuated or dead pathogens, subunits, recombinant proteins, carbohydrates or DNA) introduced against a variety of pathogens. Currently, vaccines are among the factors that, together with hygiene, antibiotics and surgery, save the most lives181. Vaccination enabled the eradication of smallpox infection worldwide in 1980, and we are quite close to eradicating polio182. However, new and better vaccines are urgently needed; e.g., a vaccine against the new coronavirus 2019, SARS-Cov-2; prevalent pathogens, such as human immunodeficiency virus (HIV); parasites, such as Plasmodium spp., which produce malaria; and bacteria, such as Mycobacterium tuberculosis. However, anti-vaccine groups in more affluent countries are putting society at risk for a return of the serious illnesses that had almost been forgotten, such as diphtheria and tetanus183, with an increase in measles in unvaccinated people at epidemic levels, thus negating many of the advances made over many years.
Therapy with microorganisms
Bacteria
Whole pathogens or their products can also be used in human therapy for some types of cancer. At the end of the XIX century, the father of immunotherapy, Dr. Coley, popularized the use of extracts from cultures of Streptococcus pyogenes and Serratia marcescens184 (called Coley’s toxin) for the treatment of patients with sarcoma, lymphoma, testis cancer, etc., but because of variable results and, indeed, cases of death, these treatments were discontinued. Later, because of the research on cancer performed by Dr. Lloyd J. Old with Mycobacteria, bacillus Calmette-Guérin (BCG) was approved by the American Food and Drug Administration (FDA) in 1976 for use in a therapeutic procedure for bladder cancer —a treatment that is still in use today185,186.
More recently, and with the increased knowledge of the human microbiome, the use of microorganisms in therapy has seen a resurgence. Some intestinal infections, such as those produced by Clostridium difficile, can be cured with the transfer of intestinal bacteria from healthy people (feces transplantation)187. Numerous other attempts to use microorganisms to cure inflammatory illnesses (Crohn’s disease, ulcerative colitis, etc.) have met with limited success188, which indicates that this type of therapy is much more complex than initially anticipated. As a consequence, many more studies are required to ensure that this approach can be used for curative immunotherapy. Researchers are also working on genetically modified or artificial bacteria (e.g., based on Salmonella enterica, Listeria monocytogenes or Lactobacillus lactis), but only limited effects have been observed to date189.
Oncolytic viruses (OVs)
Although the use of bacteria in antitumoral therapy has been largely restricted, the use of therapeutic viruses is increasing. Virus-based therapy was introduced in the 1990s with the use of adenovirus, but only in recent years has it been used in practice in the clinic. Oncologic viruses190 have the capacity to attack tumor cells in a preferential manner and induce immunogenic cell death (ICD) and host antitumor immunity (Fig. 2).
Fig. 2. Effect of oncolytic virus on cancer cells.
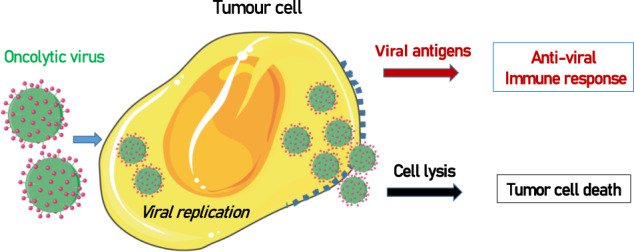
Oncolytic viruses replicate inside tumor cells, which causes cell lysis. In addition, the expression of viral antigens induces an antiviral immune response that helps destroy tumor cells
The first virus approved for use in therapy was a recombinant oncolytic adenovirus named H101, which was licensed in 2005 by the China Food and Drug Administration (CFDA) for treating head and neck carcinoma in combination with chemotherapy191. Ten years later, the oncolytic attenuated-modified virus herpes simplex I-talimogene laherparepvec (T-VEC, Imlygic®) was approved by both European (EMEA) and American (FDA) agencies for the treatment of melanoma192. The virus is modified by the insertion of human GM-CSF and deletion of the ICP47 gene. Since the approval of T-VEC, a new era has dawned on the use of OVs in cancer therapy193,194.
Currently, oncolytic viruses from the Adenoviridae, Herpesviridae, Picornaviridae, Reoviridae and Poxviridae families are in different phases of clinical studies for several types of tumors194,195. For example, reovirus against brain tumors (alone or combined with other therapies)196 or Maraba virus against triple-negative breast tumors197,198 offer some hope to patients with these types of cancer.
Viral sequences can be modified by genetic engineering techniques, thus making the virus more prone to infect some cells and enhancing viral infiltration and tumor tropism. Combinations with other components (immunomodulators, drugs, and cytokines) are also being explored to suppress antiviral immunity and enhance antitumoral cytotoxicity199.
Other vaccines
Vaccines for cancer prevention
It is clear that certain viruses and bacteria play roles in cancer development. Viruses such as genital herpes, hepatitis B, Epstein Barr or human papilloma and bacteria such as Helicobacter pylori have been associated with cancers of the uterus and liver, in Burkitt’s lymphoma, and oral/genital and stomach cancers, respectively200. Therefore, immunization against these pathogens offer protection not only from infection but also from cancer.
Therapeutic vaccines
Once an illness has developed, the intention of a therapeutic vaccine is to eliminate or decrease its pathology. Thus, vaccines are used for cases of allergies, cancers and autoimmune diseases.
Allergy (Type 1)
Allergen-specific immunotherapy (AIT) aims to modulate the immune system against an allergen, thus modifying the natural course of the allergic disease and conferring long-lasting benefits201. The basic AIT involves the introduction of repeated doses of allergen (either injectable or sublingual allergen extract tablets) and often in escalating doses in a controlled manner, followed by a maintenance phase. In cases for which long-lasting tolerance is acquired, therapy may be discontinued. Allergen extracts can be obtained from different sources, such as cat hair and pelt, mites, different types of pollen, venom protein, foods, etc. Allergy vaccines are currently the only effective therapy that can stop the progression of the illness because treatment with anti-inflammatory drugs, such as anti-histaminic drugs or corticoids, mitigates the symptoms of the allergic processes but does not modify the natural course of the disease202,203.
AIT has been shown to induce the activation of antigen-specific Tregs and IL-10-producing Bregs (Br1) subtype cells, which is combined with anergy caused by Th2 cells201 and the production of allergen-specific IgG antibodies that can compete with IgE for binding to allergens204.
In the past, most vaccines were developed using natural allergen extracts. However, significant progress has been made in recent years to correctly characterize the allergen at the molecular level, and some of these allergens are now being produced by recombinant technologies, nucleic acid-based strategies, or synthetic peptide chemistry205.
Cancer
Another therapeutic approach for vaccines is in the field of cancer. Therapeutic cancer vaccines that contain self- or nonself-patient tumor lysates, viral vectors, mutated tumor proteins or peptides, among other types206 administered in the presence of adjuvants can activate the immune system to induce antitumoral responses207. The goal is to activate the Th and Tc cell compartments to expand specific cytotoxic T and NK cells directed against tumor cells.
Some vaccines are more immunogenic than others, and this effect can be related to several factors, such as the types/numbers of genetic mutations in the tumor, expression of neoantigens, production of viral proteins, an immunosuppressive environment, lack of expression of histocompatibility complex molecules, etc., which together may explain the large variability in tumor elimination208. Therapeutic cancer vaccines are generally very safe, and major secondary effects have not been observed, although large differences in patient responses are detected. Moreover, this strategy may be used in conjunction with other complementary therapies209, such as monoclonal antibodies, chemotherapy or cellular therapy209,210. Several patients are currently taking part in clinical trials and are receiving therapeutic cancer vaccines against different types of tumors, such as lung (ClinicalTrials.gov Identifier: NCT04397926), prostate (ClinicalTrials.gov Identifier: NCT03525652) or pancreas (ClinicalTrials.gov Identifier: NCT04161755), using individual or combined therapies.
Autoimmunity
In the case of therapeutic vaccines for autoimmune diseases, such as multiple sclerosis, diabetes, Myasthenia gravis or Guillain Barré syndrome, the intention is to induce tolerance to self-antigens through the activation of regulatory cells (Tregs and Bregs) and tolerogenic dendritic cells, thus avoiding the immune response to self-components211. Due to the large variety of autoimmune diseases, the different etiologies and extensive variability, even in the same type of disease, designing a vaccine that can be useful for a wide range of patients is very difficult.
However, several researchers are obtaining good results in animal models with nanostructures and peptides that induce specific tolerance, and it is predicted that, in the near future, these types of therapies will be applied to patients suffering from autoimmune diseases (reviewed by Serra and Santamaria212).
Polyclonal antibodies (pAbs)—serotherapy
The discovery of antibodies by Dr. E. von Behring and Kitasato213 at the end of the XIX century highlighted the potential of antibodies to neutralize tetanus and diphtheria toxins. This discovery opened the way to exploring the potential clinical applications of conventional antiserum-containing polyclonal antibodies from immunized animals/humans214. This “serotherapy” was initiated by Dr. Roux and Dr. Yersin, who used anti-diphtheria serum to treat several children215. After this initial success, the use of serotherapy was increased for use against diphtheria and other diseases but also led to the identification of problems, such as immunogenicity with the formation of immune complexes (Arthus reaction), the variability and limitation of the antibody batches, the content of a mixture of classes and subclasses of antibodies with different biological activities, and their temporal effects. For all of these reasons, therapy with polyclonal antibodies was very much restricted to special cases, such as the use of gamma-globulins for the prevention of Rhesus (RH) maternal-fetal incompatibility and tetanus or snake venom toxicity216.
With the identification of gamma-globulin-deficient patients by Dr. Bruton in 1952217, the use of immunoglobulins as therapeutic molecules for the treatment of humoral immunodeficiencies was initiated. However, some problems were encountered in the initial phases, mostly related to the serum preparation and aggregation/fragmentation of antibodies. Since their initial use, several efforts have been made to avoid impurities and to improve the purification process, and several commercial products are now available (as intravenous or subcutaneous preparations). Currently, many patients with humoral immunodeficiencies are successfully being treated to prevent them from catching infectious diseases. More recently, the therapeutic applications of immunoglobulins have expanded to other diseases, such as against COVID-19 caused by SARS-Cov-2 infection (see below), autoimmune disorders and Kawasaki syndrome in children218. The beneficial effects seem to be mediated by several immunological mechanisms, including viral neutralization, inhibition of inflammatory cells and activation of immune regulators214.
Monoclonal antibodies (mAbs)
The development of monoclonal antibodies (mAbs) by C. Milstein and G. Köhler in 1975219 (Nobel Prize winners for Physiology/Medicine in 1984) changed medicine and immunology completely, along with many other disciplines. Monoclonal antibodies are produced from the fusion of two cells to generate a hybrid cell or hybridoma with two characteristics, i.e., the production of one specific antibody and immortality. Dr. Milstein is considered to be the father of modern immunology for his crucial contribution220. The development of many different mAbs has enabled the identification of new molecules and the development of more accurate diagnostic approaches; specific, fast and inexpensive technologies; processes for the purification/concentration of compounds; and better and more specific therapy. mAbs can now be used against specific targets according to the concept of the “magic bullet”, a term coined by Dr. Paul Ehrlich at the beginning of the XX century (reviewed in ref. 221).
Numerous different mouse and rat mAbs were produced against several molecules, but due to their murine origin, patients treated with these mAbs suffered from hypersensitivity and immune responses222,223. Thus, most mAbs currently used in clinical applications are linked to radioactive elements and used for diagnostic purposes (Table 2).
Table 2.
List of some approved monoclonal antibodies for clinical applications
| FDA | Antibody | Commercial name | Target antigen | Clinical application |
|---|---|---|---|---|
| Murine | ||||
| 1996 | Arcitumomab | CEA-scan Murine-(99mTc) | CEA (carcinoembryonic antigen) | Diagnostic imaging of colorectal cancers |
| 2014 | Blinatumomab | Blincyto | CD19 | B-cell precursor acute lymphoblastic leukemia |
| 1996 | Capromab pendetide | ProstaScint Murine-(111In) | PSMA (prostate specific membrane antigen) | Detection of prostate tumor |
| 2004 | Fanolesomab | NeutroSpec Murine-(99mTc) | CD15 | Diagnosis of appendicitis |
| 2002 | Ibritumomab tiuxetan | Zevalin Murine-(90Y) | CD20 | Non-Hodgkin lymphoma therapy |
| 1996 | Imciromab-Pentetate | Myoscint Murine-(111In) | Heart myosin | Cardiac imaging |
| 1986 | Muromonab-CD3 | OKT3 Murine | CD3 | Prevention of rejection of kidney, heart and liver allografts. No longer in production. |
| 1996 | Nofetumomab merpentan | Verluma Murine Fab—(99mTc) | CAA (carcinoma-associated antigen) | Diagnosis of several cancers (lung, gastrointestinal, breast, ovary, pancreas, etc.) |
| 1992 | Satumomab pendetide | OncoScint Murine-(111In) | TAG-72 | Ovarian and colorectal cancer diagnosis (radioimaging) |
| 2003 | Tositumomab/Iodine 131 Tositumomab | Murine-(131I) | CD20 | Non-Hodgkin follicular lymphoma |
| Chimeric | ||||
| 1994 | Abciximab | ReoPro | Platelet glycoprotein | High-risk angioplasty |
| 1998 | Basiliximab | Simulect | CD25 | Immunosuppressant agent to prevent rejection in organ transplantation |
| 2011 | Brentuximab vedotin | Adcetris | CD30 + drug | Anaplastic and cutaneous large cell lymphomas; Hodgkin lymphoma |
| 2005 | Catumaxomab | Proxinium | EpCAM | Malignant ascites with EpCAM-positive carcinomas |
| 2004 | Cetuximab | Erbitux | EGFR | Colorectal, head and neck cancer |
| 2015 | Dinutuximab, Dinutuximab beta | Unituxin/Isquette | GD2 | Neuroblastoma |
| 1998 | Infliximab | Remicade | TNF-α | Psoriasis, Crohn’s disease, ankylosing spondylitis, psoriatic arthritis, rheumatoid arthritis and ulcerative colitis. |
| 2016 | Infliximab | Inflectra and other biosimilars | TNF -α | Psoriasis, Crohn’s disease, ankylosing spondylitis, psoriatic arthritis, rheumatoid arthritis and ulcerative colitis. |
| 2016 | Obiltoxaximab | Anthim | B. anthracis toxin | Bacillus anthracis toxin |
| 1997 | Rituximab | Rituxan, and other biosimilars | CD20 | Non-Hodgkin lymphoma, rheumatoid arthritis, chronic lymphocytic leukemia, and others |
| 2014 | Siltuximab | Sylvant | IL-6 | Multiple myeloma and other tumors |
| Humanized | ||||
| 2001 | Alemtuzumab | Campath | CD52 | Non-Hodgkin lymphoma, chronic lymphocytic leukemia, multiple sclerosis. |
| 2016 | Atezolizumab | Tecentriq | PD-L1 | Urothelial carcinoma, small cell lung cancer, triple-negative breast cancer |
| 2010 | Atlizumab or tocilizumab | Actemra/RoActemra | IL-6R | Rheumatoid arthritis, systemic juvenile idiopathic arthritis, giant cell arteritis, cytokine release syndrome. |
| 2004 | Bevacizumab | Avastin | VEGF-A | Colon, lung, glioblastoma, renal-cell carcinoma; age-related macular degeneration. |
| 2008 | Certolizumab pegol | Cimzia | TNF-α | Morbus Crohn, rheumatoid arthritis |
| 1997 | Daclizumab | Zenapax | CD25 | Prevention of allograft rejection |
| 2016 | Daclizumab | Zinbryta | CD25 | Multiple sclerosis. Withdrawn from the market in 2018 |
| 2007 | Eculizumab | Soliris | C5-complement factor | Paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome and neuromyelitis optica |
| 2003 | Efalizumab | Raptiva | CD11a | Psoriasis treatment. Withdrawn from the market in 2009 |
| 2015 | Elotuzumab | Empliciti | SLAMF7-CD319 | Multiple myeloma |
| 2000 | Gemtuzumab ozogamicin | Mylotarg | CD33-drug | Relapsed acute myeloid leukemia. |
| 2015 | Idarucizumab | Praxbind | Dabigatran etexilate | Reversal of anticoagulant effects of dabigatran |
| 2016 | Ixekizumab | Taltz | IL-17A | Moderate to severe plaque psoriasis, active ankylosing spondylitis |
| 2006 | Natalizumab | Tysabri | α4β1 | Multiple sclerosis, Crohn’s disease |
| 2013 | Obinutuzumab | Gazyva | CD20 | Follicular lymphoma |
| 2017 | Ocrelizumab | Ocrevus | CD20 | Immunosuppressive drug, relapsing forms of multiple sclerosis (MS) |
| 2003 | Omalizumab | Xolair | Ig E | Severe asthma |
| 1998 | Palivizumab | Synagis | Protein F | Prevention of respiratory syncytial virus infections |
| 2014 | Pembrolizumab/Lambrolizumab | Keytruda | PD-1 | Several types of cancer (metastatic solid tumors) |
| 2012 | Pertuzumab | Perjeta | HER2 | HER2-positive metastatic breast cancer and as neoadjuvant |
| 2006 | Ranibizumab | Lucentis | VEGF-α | Age-related macular degeneration |
| 2016 | Reslizumab | Cinqair | IL5 | Eosinophil-meditated inflammation (asthma) |
| 2010 | Tocilizumab or atlizumab | Actemra/RoActemra | IL-6R | Rheumatoid arthritis, systemic juvenile idiopathic arthritis, giant cell arteritis, cytokine release syndrome |
| 1998 | Trastuzumab | Herceptin | HER2/neu | HER2-positive breast and stomach cancer |
| 2013 | Trastuzumab-emtansine | Kadcyla | HER2/neu | HER2-positive metastatic breast cancer |
| 2014 | Vedolizumab | Entyvio | Integrin-α4β7 | Ulcerative colitis and Crohn’s disease |
| Human | ||||
| 2002 | Adalimumab | Humira, Trudexa | TNF-α | Rheumatoid arthritis, psoriasis, Crohn’s disease |
| 2006 | Panitumumab | Vectibix | EGFR | Metastatic colorectal carcinoma |
| 2016 | Adalimumab | Amjevita and other biosimilars | TNF-α | Arthritis rheumatoid, psoriasis, Crohn’s disease |
| 2015 | Alirocumab | Praluent | PCSK9 | High levels of LDL cholesterol |
| 2017 | Avelumab | Bavencio | PD-L1 | Gastric cancer, Merkel-cell carcinoma |
| 2011 | Belimumab | Benlysta | BAFF | Systemic lupus erythematosus |
| 2016 | Bezlotoxumab | Zinplava | C. difficile B toxin | Prevention of recurrence of Clostridium difficile infection |
| 2017 | Brodalumab | Siliq/Kyntheum | IL-17RA | Severe plaque psoriasis |
| 2009 | Canakinumab | Ilaris/ACZ885 | IL-1b | Cryopyrin-associated periodic syndromes, autoinflammatory syndromes |
| 2015 | Daratumumab | Darzalex | CD38 | Multiple myeloma |
| 2010 | Denosumab | Prolia/Xgeva | RANKL | Osteoporosis at high risk of fractures |
| 2017 | Dupilumab | Dupixent | IL4Rα | Allergic diseases, atopic dermatitis, asthma and nasal polyps |
| 2017 | Durvalumab | Imfinzi | PD-L1 | Bladder and lung cancer; other tumors |
| 2015 | Evolocumab | Repatha | LDL-C/PCSK9 | Hyperlipidemia |
| 2009 | Golimumab | Simponi | TNFα | Rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis |
| 2011 | Ipilimumab | Yerboy | CTLA-4 | Melanoma, renal cell carcinoma |
| 2015 | Necitumumab | Portrazza | EGFR | Squamous non-small cell lung carcinoma |
| 2015 | Nivolumab | Opdivo | PD-1 | Several types of cancer (melanoma, lung, renal, colon, liver, etc.)□ |
| 2009 | Ofatumumab | Arzerra | CD20 | Chronic lymphocytic leukemia |
| 2016 | Olaratumab | Lartruvo | PDGFR-α | Soft-tissue sarcoma |
| 2014 | Ramucirumab | Cyramza | VEGFR2 | advanced gastric cancer, gastro-esophageal junction adenocarcinoma; other tumors (non-small cell lung carcinoma, colorectal cancer, hepatocellular carcinoma) |
| 2012 | Raxibacumab | ABthrax | Bacillus anthracis, Anthrax toxin | Prophylaxis and treatment of inhaled Bacillus anthracis |
| 2015 | Secukinumab | Cosentyx | IL-17A | Psoriasis, ankylosing spondylitis, psoriatic arthritis |
| 2009 | Ustekinumab | Stelara | IL-12/IL23 p40 | Psoriasis, Crohn’s disease, ulcerative colitis |
In an effort to avoid immunogenicity, mAbs were subsequently modified by genetic engineering approaches to carry mostly sequences of human origin. Several research groups and companies developed chimeric and humanized mAbs (Table 2), and these mAbs included additional modifications, such as changes in the carbohydrates (glycosylation) and/or antibody regions, with the aim of improving their therapeutic action224–228. Moreover, fragments of recombinant antibodies (Fabs, single-chain Fvs, different V regions, fusion proteins, smaller antibodies, etc.) increased the variability of these potential therapeutic agents.
The generation of fully human mAbs took more time due to technical difficulties and ethical issues; therefore, researchers sought alternative methods to conventional approaches, such as the development of transgenic animals carrying human immunoglobulin genes using minilocus vectors, artificial yeast/human chromosomes or P1 vectors. The generation of knockout mice (in which mice lack Ig genes) and further crosses with transgenic mice carrying human antibody sequences led to the generation of mouse strains that were able to produce fully human mAbs229,230. Other initiatives, such as the generation of immunodeficient mice in which human bone marrow or libraries of recombinant phages carrying human variable genes were reconstituted, allowed the development of more fully human antibodies (Table 2). Sir Greg Winter, Nobel Prize winner in Chemistry in 2018231,232, became the pioneer of mAb humanization through the genetic engineering of an antibody (Campath 1), later developing a fully human antibody (antitumor necrosis factor alfa, TNF-a) using recombinant phage technology225,233,234. Several companies are currently developing human antibodies using these and new technologies (reviewed in225,227,233,234).
Since 1975, the list of approved mAbs for human therapy has continued to increase (Table 2), and many more mAbs are in clinical trials235–237. The versatility of mAbs is based on a different mechanism of action238:
Neutralization/blocking of soluble elements. For example, the neutralization of cytokines (TNF-α) and growth factors (vascular endothelium growth factor) prevents the exhibition of their effects, i.e., inflammatory and angiogenic effects, respectively239,240.
Complement activation. IgG/IgM antibodies activate the complement cascade by the classical route, which leads to the death of the target cell241,242.
Cytotoxicity mediated by NK cells. NK cells can facilitate mAb killing of target cells. The mAb, after binding to a target cell, can attach to Fc receptors on the surface of NK cells to trigger the release of granzymes and perforin, thus inducing cell target death243,244.
Induction of cell death by apoptosis. Certain mAbs directed against some membrane molecules can directly activate apoptosis243.
Blocking activation signals. Antibodies can block some membrane receptors and avoid cell activation/proliferation activation/proliferation243,245.
Carriers of toxins, pro-drugs, enzymes, and radioactive elements. mAbs are able to concentrate select compounds around target cells, providing a much more selective therapy than conventional chemo- or radiotherapy244.
Check point inhibitors. Leading to a recent revolution in cancer therapy, the identification of several inhibitory molecules can be blocked by mAbs, thus leading to the activation and proliferation of antitumoral T cells. Molecules such as CTLA-4 and PD1 and its ligand PDL-1, maintain immune cells under controlled conditions. However, it is possible to reactivate the antitumoral immune responses by blocking some of these molecules with mAbs, either directed to only one of them or by using various antibodies in combination (for example, against CTLA-4 and PD1)246.
The results obtained with these therapeutic mAbs against checkpoint inhibitors in some types of cancer have been amazing. For their contribution to the understanding of the roles of CTLA-4247 and PD-1248, the Swedish academy gave the Nobel Prize in 2018 to Dr. J.P. Allison and Dr. T. Honjo, respectively249. However, this therapy is not efficacious in all types of cancers for several reasons, such as the expression of these and other checkpoint inhibitors in immune cells, the number of antitumoral cells in each patient, an immunosuppressant microenvironment, the rate of cancer mutations, and the expression of histocompatibility molecules.
Recombinant proteins
There is a large list of recombinant proteins that are currently being used for human therapy, including interleukin 2 (IL-2), interferons (IFNs) and GM-CSF.
IL-2 was identified in 1976 as a growth factor for T lymphocytes, and soon after Dr. Rosenberg started to use it in antitumoral therapy250,251. Years later, in 1991, IL-2 was approved by the FDA for patients with metastatic renal cancer and in 1998 for the treatment of metastatic melanoma251.
Interferon (IFN) was described in 1957 by Isaacs and Lindenmann252. The interferon family is the largest family of cytokines and is classified into three different types (I, II, and III). Type I IFNs (including IFN-α and IFN-β) exhibit several molecular actions that may be very relevant for use in therapy for a range of pathologies (such as autoimmune diseases and cancers)253. In 1986, the FDA approved human IFN-α2a and IFN-α2b for patients with hairy cell leukemia and later on for patients with multiple sclerosis. Since their initial use, these interferon species have been approved for many other diseases, including chronic hepatitis B and C, lymphoma, advanced melanoma, and as adjuvants together with other therapies for several types of cancers254,255.
Another cytokine is GM-CSF, which activates the production of granulocytes and monocytes from bone marrow myeloid progenitors and has shown adjuvant antitumoral effects256,257. Other cytokines, such as IL-5, IL-7, IL-12, IL-15, IL-18, and IL-21258,259, are being tested in several clinical trials, and it is expected that some of them, either alone or in combination, can be used in future antitumoral therapy.
Other recombinant proteins are already on the market, some of which are derived from antibodies, with some advantages such as small size, low immunogenicity and general ease of production. Examples are etanercept and abatacept (CTLA-4 Ig), which were approved by the European Medicines Agency in 2000 and 2007, respectively. The former is a chimeric protein that carries the external portion of the tumor necrosis factor (TNF) receptor linked to the IgG Fc region, which captures soluble TNF to block its inflammatory effects260. The latter example is a fusion protein that combines the extracellular portion of human CTLA-4 and IgG1 Fc. Abatacept is a competitive inhibitor that blocks T-cell activation and can be used in the treatment of inflammatory illnesses such as rheumatoid arthritis261.
Cellular immunotherapy
Natural killer (NK) and lymphokine-activated killer (LAK) cells
Natural killer (NK) cells were described in the 1970s based on their capacity to eliminate tumor cells without prior sensitization, with differences observed compared with specific cytotoxic T cells (which are activated based on the recognition of the target cells)262,263. In 1985, Piontek et al. reported that NK cells have the ability to preferentially kill cells that had lost the expression of the major histocompatibility complex class I molecules264,265.
Lymphokine-activated killer (LAK) cells are a heterogeneous population that includes not only NK but also NKT and T cells, which can be generated in an in vitro culture of peripheral blood mononuclear cells (PBMCs) in the presence of IL-2266. Dr. Rosenberg and collaborators carried out studies using these cells in the presence of IL-2 (reviewed by Rosemberg251). These LAK cells showed good antitumoral responses in 22% of the melanoma patients who received them as therapy250. However, secondary effects such as liver toxicity and the expansion of the Treg population limited their therapeutic effect. Researchers started to design new recombinant IL-2 with some mutations to avoid the activation of Tregs267, with linking it to polyethylene glycol (PEG) to increase its half-life and efficacy268.
Another cytokine described later, IL-15, showed similarities to IL-2 in many respects269, and it had some unique advantages, such as the capacity to activate NK and cytotoxic T cells (Tc) but not Tregs. IL-15 is being used in different versions (alone, as a heterodimer with receptor IL-15/IL15Ra or IL15Rα IgFc, or in an agonist complex with ALT-803)269 and in combination with other therapies in several clinical trials (examples: NCT01021059, NCT03905135, and NCT03759184).
More recently, researchers have focused their attention on other cytokines and combinations (such as IL-15, IL-12, and IL-18)270, which are able to activate NK cells in vitro and induce a good responses in animal models. In some human clinical trials, remission has been observed for patients with acute myeloid leukemia271,272, which broadens the options for the use of NK cells in the treatment of this pathology.
The properties of NK cells reveal their versatility as treatments against tumors. NK cells are able to kill tumors through several mechanisms, including receptor-mediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity (ADCC) and death receptor-mediated apoptosis, but they also secrete cytokines such as interferon gamma that enhance the antitumoral adaptive immune response. NK cell adoptive transfer (either autologous or allogenic NKs) is currently being tested in clinical trials for hematological diseases and solid tumors, and numerous research groups have recognized their potential in other situations, such as transplant rejection and pregnancy. NK cell lines, memory-like NK cells and stem cell-derived NK cells are additional types of cells that can be used for tumor immunotherapy273.
Regarding other cellular therapies, NK cells as substitutes for T cells for use upon transformation with an chimeric antibody receptor (CAR) are being explored (see below).
Dendritic cells
Paul Langerhans identified dendritic cells in human skin in 1868274, but these cells were not named until 1973 by Dr. Ralph M. Steinman (Nobel Prize in 2011) and Dr. Zanvil A. Cohn, who chose the term because the cell morphology, with long extensions, resembles that of neuronal dendrites275. In humans, dendritic cells are obtained from different sources that vary in origin, maturation state and tissue distribution (skin, lymphoid tissue, circulating cells). Among the main types of dendritic cells, plasmocytoids are conventional myeloid DC1 and DC2, pre-DC and monocyte-derived dendritic cells. In the epidermis, there are three types: Langerhans cells (LC), monocyte-derived LC-like cells and inflammatory dendritic epidermal cells (IDECS)276. As indicated above, DCs are antigen-presenting cells and are the only cells that are able to activate naïve T lymphocytes. A subpopulation of DCs also carries out a process known as cross-presentation. In this way, they facilitate the activation of both helper and cytotoxic T lymphocytes277. In addition to their participation in the immune response, they can be used in antitumoral therapeutic vaccines277,278.
It is possible to generate a type of blood monocyte-derived dendritic cell in the presence of a mixture of cytokines in culture279—a process that induces their subsequent maturation and activation in the presence of tumor antigens (cell lysates, recombinant or purified antigens, peptides, RNA, DNA, and viral vectors280). These cells can also be obtained from bone marrow hematopoietic CD34 + progenitor cells281. Other sources, such as circulating or skin dendritic cells, are relatively scarce and are therefore not usually used.
After their differentiation and activation in vitro278,282, DCs are exposed to tumor antigens and infused back into the patient (either by blood infusion or injected into areas near the lymph nodes or even directly into them) to reach the secondary lymphoid organs as soon as possible, at which point they can present antigens to the T cells. This approach is a type of individualized therapy and is therefore expensive.
The first approved vaccine in which autologous dendritic cells were used was Sipuleucel-T (Provenge)283, which was a treatment for prostate cancer refractory to hormonal treatment. Immunotherapy with dendritic cells is currently being tested in more than 200 clinical trials for various tumors: brain, pancreas, mesothelioma, melanoma and many others (ClinicalTrials.gov Identifiers: NCT01204684, NCT02548169, NCT02649829, and NCT03300843, respectively). The data indicate that the therapy is well tolerated and has led to increased patient survival in some trials. Furthermore, complete cure and partial remission outcomes have also been observed. The lack of efficacy on other tests was probably due to the presence of immunosuppressive factors in the tumor environment.
Another therapeutic use of dendritic cells involves their induction of immunosuppression both in transplants and in autoimmune diseases284. In an autoimmune pathology such as multiple sclerosis, the intention is to achieve stable tolerogenic dendritic cells that can act against some autoantigens (such as myelin peptides) in the presence of vitamin D3, dexamethasone, or other agents285. Phase I clinical trials have generally shown good tolerance to this therapy without serious adverse effects286.
However, greater control of this treatment is necessary in several respects to obtain the best therapeutic results284; e.g., the type of dendritic cells and ex vivo differentiation, the antigens used, and the injection route are important considerations.
Gamma/delta T cells (Tγ/δ)
Human T cells expressing γ/δ TCR cells have interesting properties, including the capacity to kill a broad range of tumor cells. The advantages of these cells in cancer therapy are based on their independence from MHC expression on tumor cells and that their relative insensitivity to some inhibitor molecules (such as PD-1). The initial clinical application, with the adoptive transfer of autologous Vδ2+ cells after ex vivo expansion, showed only sporadic responses287, and different exploratory studies are currently being carried out to increase their clinical therapeutic use. Allogeneic Vδ2+ cells are also being explored in cancer therapy; e.g., they are being used against refractory hematological malignancies288 and advanced cholangiocarcinoma289.
Regulatory T cells (Tregs)
Although the basis of immune regulation was suggested centuries ago, regulatory T cells were described by Sakaguchi et al. as CD4+ CD25+ natural regulatory T cells290 that expressed the forkhead box P3 transcription factor (foxp3)291. Later, induced or adaptive regulatory T cells were also identified, including different subsets that carry several phenotypic markers and express various cytokine secretion profiles292. All of these factors play crucial roles in the maintenance of immunological self-tolerance by suppressing autoreactive T cells.
The manipulation of Tregs to achieve therapeutic outcomes is a field of great interest, because of both their expansion and activation in diseases, such as allergic and autoimmune diseases, and as a potential targets for cancer immunotherapy293.
Tumor-infiltrating lymphocytes (TILs)
Lymphocytes that infiltrate solid tumors are called tumor-infiltrating lymphocytes (TILs). In 1957, Thomas and Burnet proposed that the immune system performs tumor immune vigilance, with lymphocytes as sentinel cells leading to the elimination of somatic cells transformed by spontaneous mutations294,295.
Since the end of the 1980s, Dr. Rosenberg has been trying to prove and improve the effective use of TILs. The process starts with surgery and the isolation of TILs from a tumor, followed by TIL activation in culture in the presence of cytokines, cellular expansion and, finally reinfusion into the patient. Since its inception, this therapy has been improved markedly, with an increase in optimal responses from less than 30% to the current 50–75%, in some cases. These higher success rates are due, in particular, to the prior preparation of the patient, including the depletion of lymphoid tissues, to avoid an expansion of regulatory cells296, myeloid suppressor cells and other cells that can compete with the transferred TILs.
Currently, there are more than 200 trials in which TILs are being used alone or in combination with other immunotherapies on several tumors, such as melanoma, metastatic colorectal cancer, glioblastoma, pancreatic cancer, hepatobiliary cancer, ovarian cancer and breast cancer. This individualized therapy has limitations; it can only be used on solid tumors, and the number and specificity of the TILs and the type of tumor and microenvironment make standardizing this therapy difficult.
Chimeric antigen receptor (CAR)
Since TILs include a variety of T lymphocytes with different specificities, the next step was to obtain T cells of a single type (monoclonal cells) carrying a clonal receptor capable of recognizing tumor antigens. This effort was carried out for the first time in mice and subsequently, in 2006, in humans with a transgenic TCR against the MART-1 melanoma antigen297,298. These types of receptors are known as tTCRs, but their ability to recognize antigens is restricted since they can only identify the peptides presented by antigen-presenting cells on self-histocompatibility molecules.
This situation changed because of one of the latest revolutions in antitumor therapy, the development of T lymphocytes that carry a chimeric antigen receptor (CAR) based on a specific antibody directed to a target surface molecule299,300. These modified T cells can directly recognize tumor cells without required antigen processing or presentation by professional antigen-presenting cells. Moreover, the CAR includes all of the necessary elements for intracellular signaling and activation of helper and cytotoxic T lymphocytes.
CAR therapy was developed by one of its pioneers, Dr. Carl June at the University of Pennsylvania in the United States300, who used modified T lymphocytes that carried a chimeric antigen receptor to target CD19+ leukemic B cells. After interacting with CD19+ cells, these modified CAR T cells were activated and able to proliferate and exert cytotoxic functions against target cells. In this case, both tumor and healthy B cells were affected. Although bone marrow continues to produce B lymphocytes, in cases of severe B lymphopenia, it is possible to provide exogenous immunoglobulins periodically.
The whole process of the current CAR T-cell therapy begins with blood donation, from which lymphocytes are purified and genetically modified in vitro by a viral vector, which carries the genes coding for the chimeric antigen receptor. The cells are expanded in the presence of cytokines in culture and are subsequently reinfused into the patient. This type of cellular immunotherapy is individualized for each patient, with his/her CAR T cells ultimately destroying the tumor.
Since the first generation of CARs appeared, namely, a chimeric receptor composed of an anti-CD19-specific single-chain variable fragment linked to a transmembrane domain and intracellular signaling domain of the T cell receptor (CD3 ζ chain), researchers started to modify the original design. New generations of CARs, including the CD3 ζ subunit together with other signaling domains, such as CD28, CD134, CD137 (4–1BB), CD27, and ICOS, or combinations (CD3 ζ, CD28, and CD134)301, have been developed in the second and third generations of CARs to improve several aspects, such as the activation, proliferation and survival of CAR T cells. The fourth generation of CARs show improved the antitumoral effects by carrying additional molecules (such as cytokines or drugs), improvements to the safety of CAR T-cell therapy through the use of suicide genes301 and many new designs, such as dual CARs or the so-called split universal and programmable (SUPRA) CAR system302.
In addition to T cells, other types of cells, such as NK cells, are now being explored for use in antitumoral responses303. In an effort to avoid using personalized treatment, researchers are now working on universal CARs that may be used on many different patients without inducing the problem of rejection304–307.
The encouraging results obtained with this therapy have led to interest from companies, and some commercialized examples are available, although many more “in-house” or academia-produced CARs are in clinical trials. CAR T-cell therapy was initially designed for use against hematological cancers (leukemia and lymphomas), but many new opportunities have been opened for its use against solid tumors308, infectious diseases (such as HIV)309, allotransplantation, autoimmune diseases310 and severe allergies311. China and the USA are the leading countries in producing CAR T-cell therapy, and numerous clinical trials are underway.
Immunotherapy for COVID-19 patients
Coronavirus disease 2019 (COVID-19), which is produced by severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2), affects millions of people in many countries. Most of the infected patients (80–85%) are asymptomatic or have mild symptoms, but the disease in some patients progresses to a moderate or severe illness that requires hospitalization in intensive care units because of respiratory distress, multiorgan failure, and/or other pathologies, and more than one-half million fatal cases have been reported worldwide. The most vulnerable population includes aging patients and those with comorbidities such as hypertension, diabetes and cardiovascular diseases.
There are several aspects of the COVID-19 pathogeny that suggest an overreaction of the immune system in severely ill patients, with increased levels of inflammatory cytokines such as IL-6, IL-1 and others (creating the so-called “cytokine storm”), together with blood lymphopenia and CD8 T cell and NK cell exhaustion. Special therapies have not yet been identified to cure these patients, and preventive vaccines are not yet available, but some immunotherapies have been proposed as adjunct therapies, and some of these are currently in different phases of clinical trials312.
The immunotherapeutic strategies include the following:
Targeting inflammatory molecules. To attenuate the cytokine storm (IL-6 receptor, IL-6, IL-1, GM-CSF, VEGF, etc.), monoclonal antibodies against receptors and/or cytokines, receptor antagonists and/or inhibitors are proposed.
Passive immunotherapy. Patients who were infected and recovered, but developed neutralizing antibodies against the SARS-Cov-2 virus, can donate plasma to treat severe/critical patients. Some reports have indicated promising results in a low number of patients who received convalescent plasma313,314, but conclusions cannot be drawn until several randomized studies and more patients are analyzed. In addition to the use of convalescence plasma, hyperimmune globulin therapy or monoclonal antibodies directed against the virus have also been proposed, and clinical assays are ongoing.
Immunomodulation therapy. Intravenous immunoglobulins are aimed at blocking inflammation and preventing secondary infections312. This approach is being used with success in cases of Kawasaki syndrome in children.
Cellular immunotherapy. To date, very little attention has been paid to the cellular immunotherapy approach in treatments of COVID-19, but several attempts may include the use of SARS-Cov-2-specific T and NK cells to trigger antiviral responses and autologous or allogenic Tregs to modulate inflammatory processes.
Future challenges in immunotherapy
Immunotherapy has been used for centuries, but only in recent years has this area expanded rapidly in several respects, mostly by the use of soluble elements (monoclonal antibodies and cytokines) and, more recently, with immune cells (cellular immunotherapy). There are many fields in which immunotherapy faces a range of challenges:
Vaccines
1. Researchers are working on reducing the number of injections by employing a combination of vaccines. Several current vaccines contain components from 3–6 pathogens in a single injection, and these are able to provide adequate protection against all of these pathogens315.
2. Researchers are developing more stable and durable vaccines. Improvements in the half-lives of vaccines, for example, by lyophilization, while maintaining immunogenicity is expected to reduce current problems, especially those involved in the transportation of vaccines to remote areas316. In this respect, nanotechnology can help in the design of more stable vaccines that lead to slow antigen release and improved immunogenicity317.
3. Researchers are working on vaccines that confer protection against all serotypes of a specific pathogen (universal). This outcome is especially important for pathogens with high variability (such as the influenza virus). Researchers are designing vaccines that can protect against several variants by using common regions that can induce protective immune responses to all or most of the variants318.
4. Researchers are developing alternative routes of administration (e.g., oral, inhaled, intranasal, skin, rectum, vagina) as substitutes for intramuscular or subcutaneous injections. One of the problems to be solved is the immune tolerance developed to elements delivered by the oral route, but some vaccines are already effectively administered by this route (such as the oral polio, cholera, typhoid fever and rotavirus vaccines). The intranasal route has also proven effective for some vaccines (nasal influenza vaccine), and vaccines administered through other routes are under investigation.
5. Researchers are seeking the early protection of newborns319. Newborns are very susceptible to infections due to their immature immune system320. Moreover, the protection exerted by maternal antibodies transferred through the placenta during pregnancy against some pathogens interferes with the development of the newborn’s own immune response. Greater knowledge on ways to activate the immature immune system early will enable the development of vaccines for newborns. Moreover, immunization of pregnant women may help to enhance neonatal protection against several pathogens321.
6. Researchers are developing new and more effective vaccines. This effort is crucial for very prevalent pathogens such as Mycobacteria tuberculosis, HIV virus or plasmodium falciparum. Although there are treatments against these pathogens, most are not curative—as in the case of HIV; prevention is the best way to stop their spread.
7. Researchers are working to address emerging pandemics. In the case of new pathogens, such as SARS-Cov-2, which has produced a recent global outbreak, effective vaccines are urgently required322. New technologies for vaccine formulations and routes of administration, the identification of immune-related factors of protection and modifications to the governmental regulatory and approval process for vaccines for emerging pathogens are challenges that must be faced to achieve a rapid vaccination procedure for outbreaks. Hundreds of vaccines against SARS-Cov-2 (using different strategies such as live attenuated or inactivated pathogens, viral vector-based, viral RNA, DNA, recombinant proteins, peptides, etc.)323 are now under development, and some are in clinical trials. However, the need to develop a new vaccine in a short period of time should not negate the main principles of vaccination use: safety and immune protection.
8. Researchers are working on genetic (RNA, DNA) vaccines because they have great advantages, including no requirement for growing a pathogen. Genetic vaccines can be obtained in a much shorter time, with much faster and safer production processes, and can be transported much more easily. However, the immunogenicity of these vaccines must be improved, and other problems need be avoided, such as the potential deleterious effects of integrating vaccine sequences into cells324.
9. Researchers are developing safer and more powerful adjuvants. Many years ago, the only adjuvant authorized for vaccines was aluminum hydroxide (alum), but currently, several adjuvants are on the market325. The use of ligands that activate the innate immune response, such as those linked to TLRs or nanostructures with adjuvant effects, is currently under study.
10. Researchers are boosting trained immunity, a new concept related to the innate immune memory-like described for NK cells (expansion) and macrophages (epigenetic modifications). Knowledge of how to handle trained immunity will enable better vaccine design and more effective secondary responses326.
11. Researchers are seeking to eradicate diseases from the earth. The greatest challenge, eradicating disease is possible in the short term for some pathogens, such as poliovirus. Very few cases of polio have been recently reported, and these reports came from only three countries; therefore, it is feasible that this disease can be eradicated in the near future.
12. Advances are challenged by the anti-vaccine movement. Paradoxically, there are people who doubt the beneficial effects of vaccines, and they are putting the health of their own children and society in general at risk327. The effectiveness of community protection conferred through vaccinated people is disrupted by decreased numbers of immunized persons. This lesser coverage enables pathogens to infect the most susceptible people, such as small children, elderly patients and those who cannot be vaccinated due to certain pathologies or because they are undergoing immunosuppression treatment. Thus, news about the return of illnesses that were nearly forgotten, such as tetanus in Italy (the first case in 30 years), the death of a child in Catalonia from diphtheria, or the exponential increase of measles cases (already counted in the thousands) worldwide328, should make parents think carefully about the risks of not protecting children by vaccines. The World Health Organization (https://www.who.int/topics/vaccines/en/) argues that anti-vaccine movements can roll back all the achievements thus far in this field and have cited this issue as one of the main challenges to be resolved. Addressing the anti-vaccine movement requires a coordinated effort of professionals to inform parents adequately and perhaps other types of coercive measures that some countries are already applying (financial fines, denial of access to public assistance in childcare units, removal of authorization to travel/live in some countries, new laws, and so on).
Antibodies and cytokines
Immunotherapy with monoclonal antibodies has been a true revolution for many pathologies, as has the use of certain cytokines and recombinant fusion proteins. It is therefore predicted that these approaches may have a bright future, and regulatory agencies are expected to authorize many more mAb-based therapies in the coming years, especially given the good results obtained in clinical trials. Complete antibodies or those modified to increase their functionality or decrease their immunogenicity, combinations of antibodies and cytokines, antibody fragments, etc., are only some of the many possibilities for this type of product, which will expand the range of therapeutic options.
One of the main problems regarding the use of antibodies in therapy, especially in cancer, is based on their often unpredictable efficacy. Large variability in terms of remission and durable clinical benefits between patients is observed (for example, in the antitumoral responses by antibodies directed to the checkpoint inhibitors). Thus, the main challenge is to understand the situations in which an antibody will have the desired effect. It is crucial to find validated biomarkers (with predictive and/or prognostic value) that can help to stratify or select patients for the best immunotherapy. A better understanding is also required for tumor heterogeneity, resistance to some drugs and immunosuppressive microenvironments329. An in-depth immunological study, together with a personalized approach, is certainly the way to improve the success of these types of therapies.
In combination with conventional therapies (radiotherapy, chemotherapy, and surgery), other immunotherapeutic drugs or cellular immunotherapies can also help to maximize the efficacy of this immunotherapy, but increases in toxicity will be another challenge to face.
Pathogens
The use of oncolytic viruses (OVs), bacteriophages that selectively infect bacteria, modified pathogens for vaccines or for antitumor immuno-activation, and the manipulation/ modification of the microbiota are some of the therapies that are being considered.
OVs are designed to kill tumor cells and to activate the immune system against those cells. However, many of OVs have shown limited therapeutic effects when applied in monotherapy; therefore, much more work is required to improve their systemic antitumor effects and avoid the attenuation of the virus, which limits the viral replication. Several obstacles, such as low viral delivery and spread, resistance to therapy and antiviral immunity, have been observed330. Thus, the main challenges with oncolytic viruses are addressed by improving their antitumoral efficacy, including the optimization of viral delivery, the development of OVs engineered to activate the immune system (e.g., by releasing cytokines), and their use as adjuvant therapies or in combination with other immunotherapeutic agents, such as immunomodulators331.
Regarding gut microbiota manipulation as a therapeutic approach, fecal microbiota transplantation is an effective therapy for recurrent Clostridium difficile infection332 and is now being investigated for other indications, such as inflammatory bowel disease and cancer. Some of the challenges facing microbiome transplantation are the lack of precise knowledge about the complete microbiome and the mechanisms of action involved in its therapeutic capacity, the large variability of its effectiveness and the external factors that affect it. More studies are centered on understanding how to manipulate bacterial colonies, the discovery of therapeutic molecules, nutrient competitions, etc., that are required for successful application. The best type of therapy (either individual or the combination of bacteria) is also under debate, along with how to reach the market by translating this individualized therapy into commercial scale products. The safety and potential adverse long-term effects are also being assessed.
Other components (nanomaterials and small molecules)
Nanomaterials. To obtain approval for the use of other elements from incipient fields, such as the use of different types of nanostructures, either alone or in combination with other immunotherapies, it is important to resolve certain issues. In the case of nanoparticle use, a better understanding of the interaction between nanomaterials and biological media; nanoparticle biodistribution, metabolism and biocompatibility; and the reproducibility of the synthesis and scaled up production of nanomaterials are among the issues to address.
Small molecules. A greater knowledge of several molecules involved in the immune system has led to the development of new therapeutic agents, which have been synthesized by traditional chemistry and block or activate intracellular signaling. The low cost of production of these molecules, along with the scaling and reproducibility of small-molecule batches, has attracted the attention of pharmaceutical companies interested in a whole set of new immunomodulatory drugs. A better understanding of the mechanism of action of small-molecule-based drugs and proof that they offer higher efficacy than existing therapies, either in monotherapy or in combination therapy, are challenges that face those seeking to engineer new types of targeting molecules.
Cellular immunotherapy
To date, cellular immunotherapy has been an individualized therapy with high production costs, and it requires the involvement of multidisciplinary groups in hospitals. A real challenge in the field of cellular immunotherapy is the acquisition of universal off-the-shelf cell therapies to replace those currently made to order in a very personalized manner. The development of universal cells, for example, in the case of CAR T-cell therapy, would increase the number of patients who could benefit from this treatment at thus reduce the costs.
Other challenging aspects of cellular immunotherapy are the life-threatening toxicity of induced and their lack of effect on solid tumors, which is mostly due to the immunosuppressive tumor microenvironment. This approach requires new strategies to overcome these difficulties. In addition to cancer, cellular immunotherapy has a long history of use against autoimmunity, infectious diseases, allergies and transplantation rejection. It is also important to find biomarkers for prognosis/prediction that can help to optimize this method. Other therapies that involve the use of activated NK cells, tumor-infiltrating lymphocytes, vaccination with dendritic cells, etc., are having partial clinical success. Similar to other treatments, these approaches require further study, but it is feasible that they may become reality in the near future.
Conclusions
Greater knowledge of the immune system, especially concerning the variety of cellular and humoral components and the close regulation among them, the interaction with other systems or with elements such as the microbiota, will allow the development of new types of therapies that may be safer, more effective and specific but with much lower toxicity than found in current therapies. This long journey has been possible due to the efforts of numerous researchers (throughout the centuries), who have contributed with their work, creativity, successes and failures to advance our knowledge of the immune system, cellular components, membrane markers, interactions, signaling pathways and many more aspects. This great combined effort has paved the way for the achievements that are currently being realized.
Acknowledgements
This work was financially supported by the Ministerio de Economía y Competitividad (BIO2017-84974-R), Xunta de Galicia “Grupo Referencia Competitiva 2016” (ED431C 2016/041) and the European Union (European Regional Development Fund, Ref. ED431G2019/06). JV and SM acknowledge contracts from Programa INTERREG V-A España-Portugal (POCTEP) 2014-2020 and Retención de Talento Investigador from the University of Vigo, respectively.
Author contributions
A.G-F conceptualized the study and conceived the project, and all the authors participated in writing the paper.
Competing interests
A.G-F is a copromoter of the spin-off Nanoimmunotech. There are no competing financial interests in relation to the work described.
Footnotes
These authors contributed equally: Jezabel Varadé, Susana Magadán, África González-Fernández
References
- 1.Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535:75–84. doi: 10.1038/nature18848. [DOI] [PubMed] [Google Scholar]
- 2.Yoo BB, Mazmanian SK. The enteric network: interactions between the immune and nervous systems of the gut. Immunity. 2017;46:910–926. doi: 10.1016/j.immuni.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mate I, Madrid J, Fuente M. Chronobiology of the neuroimmunoendocrine system and aging. Curr. Pharm. Des. 2014;20:4642–4655. doi: 10.2174/1381612820666140130201131. [DOI] [PubMed] [Google Scholar]
- 4.Dantzer R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol. Rev. 2018;98:477–504. doi: 10.1152/physrev.00039.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taams LS. Neuroimmune interactions: how the nervous and immune systems influence each other. Clin. Exp. Immunol. 2019;197:276–277. doi: 10.1111/cei.13355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vivier E, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Sallusto F. Heterogeneity of human CD4+ T cells against microbes. Annu. Rev. Immunol. 2016;34:317–334. doi: 10.1146/annurev-immunol-032414-112056. [DOI] [PubMed] [Google Scholar]
- 8.Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 9.Cherrier DE, Serafini N, Di Santo JP. Innate lymphoid cell development: a T cell perspective. Immunity. 2018;48:1091–1103. doi: 10.1016/j.immuni.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricardo-Gonzalez RR, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018;19:1093–1099. doi: 10.1038/s41590-018-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebbo M, Crinier A, Vély F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat. Rev. Immunol. 2017;17:665–678. doi: 10.1038/nri.2017.86. [DOI] [PubMed] [Google Scholar]
- 13.Abt MC, et al. Innate immune defenses mediated by two ilc subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe. 2015;18:27–37. doi: 10.1016/j.chom.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simoni Y, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. 2017;46:148–161. doi: 10.1016/j.immuni.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guia S, Narni-Mancinelli E. Helper-like innate lymphoid cells in humans and mice. Trends Immunol. 2020;41:436–452. doi: 10.1016/j.it.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Melo-Gonzalez F, Hepworth MR. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology. 2017;150:265–275. doi: 10.1111/imm.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shikhagaie MM, et al. Neuropilin-1 is expressed on lymphoid tissue residing LTi-like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep. 2017;18:1761–1773. doi: 10.1016/j.celrep.2017.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulenouar S, et al. Adipose type one innate lymphoid cells regulate macrophage homeostasis through targeted cytotoxicity. Immunity. 2017;46:273–286. doi: 10.1016/j.immuni.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Jacquelot N, Luong K, Seillet C. Physiological regulation of innate lymphoid cells. Front. Immunol. 2019;10:405. doi: 10.3389/fimmu.2019.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Sullivan TE, et al. Adipose-Resident Group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. 2016;45:428–441. doi: 10.1016/j.immuni.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weizman O-E, et al. ILC1 confer early host protection at initial sites of viral infection. Cell. 2017;171:795–808.e12. doi: 10.1016/j.cell.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nussbaum JC, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502:245–248. doi: 10.1038/nature12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scanlon ST, McKenzie ANJ. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr. Opin. Immunol. 2012;24:707–712. doi: 10.1016/j.coi.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Rankin LC, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat. Immunol. 2016;17:179–186. doi: 10.1038/ni.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirchberger S, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vély F, et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016;17:1291–1299. doi: 10.1038/ni.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019;19:599–613. doi: 10.1038/s41577-019-0194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hepworth MR, Sonnenberg GF. Regulation of the adaptive immune system by innate lymphoid cells. Curr. Opin. Immunol. 2014;27:75–82. doi: 10.1016/j.coi.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hepworth MR, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498:113–117. doi: 10.1038/nature12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melo-Gonzalez F, et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J. Exp. Med. 2019;216:728–742. doi: 10.1084/jem.20180871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J. Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 32.Hazenberg MD, Spits H. Human innate lymphoid cells. Blood. 2014;124:700–709. doi: 10.1182/blood-2013-11-427781. [DOI] [PubMed] [Google Scholar]
- 33.Walker JA, Barlow JL, McKenzie ANJ. Innate lymphoid cells-how did we miss them? Nat. Rev. Immunol. 2013;13:75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 34.Sonnenberg GF, Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity. 2012;37:601–610. doi: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandre D, Lefranc MP. The human γ/δ+ and α/β+ T cells: a branched pathway of differentiation. Mol. Immunol. 1992;29:447–451. doi: 10.1016/0161-5890(92)90001-E. [DOI] [PubMed] [Google Scholar]
- 36.Cruz MS, Diamond A, Russell A, Jameson JM. Human αβ and γδ T cells in skin immunity and disease. Front. Immunol. 2018;9:1. doi: 10.3389/fimmu.2018.01304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beetz S, Marischen L, Kabelitz D, Wesch D. Human γδ T cells: candidates for the development of immunotherapeutic strategies. Immunol. Res. 2007;37:97–111. doi: 10.1007/BF02685893. [DOI] [PubMed] [Google Scholar]
- 38.Brummelman J, Pilipow K, Lugli E. The single-cell phenotypic identity of human CD8 + and CD4 + T cells. Int. Rev. Cell Mol. Biol. 2018;341:63–124. doi: 10.1016/bs.ircmb.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 40.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 41.Schmitt N, Ueno H. Regulation of human helper T cell subset differentiation by cytokines. Curr. Opin. Immunol. 2015;34:130–136. doi: 10.1016/j.coi.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fietta P, Delsante G. The effector T helper cell triade. Riv. Biol. 2009;102:61–74. [PubMed] [Google Scholar]
- 43.Romagnani S. Regulation of the development of type 2 T-helper cells in allergy. Curr. Opin. Immunol. 1994;6:838–846. doi: 10.1016/0952-7915(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 44.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 45.Veldhoen M, et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 46.Licona-Limón P, Arias-Rojas A, Olguín-Martínez E. IL-9 and Th9 in parasite immunity. Semin. Immunopathol. 2017;39:29–38. doi: 10.1007/s00281-016-0606-9. [DOI] [PubMed] [Google Scholar]
- 47.Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nat. Immunol. 2009;10:864–871. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- 48.Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009;10:857–863. doi: 10.1038/ni.1767. [DOI] [PubMed] [Google Scholar]
- 49.Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol. 2015;16:142–152. doi: 10.1038/ni.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Jong E, Suddason T, Lord GM. Translational mini-review series on Th17 cells: development of mouse and human T helper 17 cells. Clin. Exp. Immunol. 2010;159:148–158. doi: 10.1111/j.1365-2249.2009.04041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hams E, et al. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J. Immunol. 2011;186:5648–5655. doi: 10.4049/jimmunol.1003161. [DOI] [PubMed] [Google Scholar]
- 52.Good-Jacobson KL, et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 2010;11:535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kroenke MA, et al. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J. Immunol. 2012;188:3734–3744. doi: 10.4049/jimmunol.1103246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edholm E-S, et al. Nonclassical MHC class I-dependent invariant T cells are evolutionarily conserved and prominent from early development in amphibians. Proc. Natl Acad. Sci. U.S.A. 2013;110:14342–14347. doi: 10.1073/pnas.1309840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaerli P, et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 2000;192:1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szabo SJ, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/S0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 57.Lantelme E, Mantovani S, Palermo R, Campanelli B, Sallusto F, Giachino C. Kinetics of GATA-3 gene expression in early polarizing and committed human T cells. Immunology. 2001;102:123–130. doi: 10.1046/j.1365-2567.2001.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng WP, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/S0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 59.Vieira PL, et al. IL-10-secreting regulatory T cells do not express foxp3 but have comparable regulatory function to naturally occurring CD4 + CD25 + regulatory T cells. J. Immunol. 2004;172:5986–5993. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 60.Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 61.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 62.Eyerich S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 2009;119:3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jia L, Wu C. Differentiation, regulation and function of Th9 cells. Adv. Exp. Med. Biol. 2014;841:181–207. doi: 10.1007/978-94-017-9487-9_7. [DOI] [PubMed] [Google Scholar]
- 64.Pagani M, et al. Role of microRNAs and long-non-coding RNAs in CD4+ T-cell differentiation. Immunol. Rev. 2013;253:82–96. doi: 10.1111/imr.12055. [DOI] [PubMed] [Google Scholar]
- 65.Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat. Rev. Immunol. 2013;13:666–678. doi: 10.1038/nri3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woodland DL, Dutton RW. Heterogeneity of CD4+ and CD8+ T cells. Curr. Opin. Immunol. 2003;15:336–342. doi: 10.1016/S0952-7915(03)00037-2. [DOI] [PubMed] [Google Scholar]
- 67.Sawant DV, Hamilton K, Vignali DAA. Interleukin-35: expanding its job profile. J. Interf. Cytokine Res. 2015;35:499–512. doi: 10.1089/jir.2015.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Linterman MA, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maceiras AR, Fonseca VR, Agua-Doce A, Graca L. T follicular regulatory cells in mice and men. Immunology. 2017;152:25–35. doi: 10.1111/imm.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lim HW, Hillsamer P, Kim CH. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell–driven B cell responses. J. Clin. Investig. 2004;114:1640–1649. doi: 10.1172/JCI200422325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Collison LW, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010;11:1093–1101. doi: 10.1038/ni.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Machicote A, Belén S, Baz P, Billordo LA, Fainboim L. Human CD8+ HLA-DR+ regulatory T cells, similarly to classical CD4+ Foxp3+ cells, suppress immune responses via PD-1/PD-L1 axis. Front. Immunol. 2018;9:2788. doi: 10.3389/fimmu.2018.02788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakagawa H, Wang L, Cantor H, Kim HJ. New insights into the biology of CD8 regulatory T cells. Adv. Immunol. 2018;140:1–20. doi: 10.1016/bs.ai.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 74.Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607–612. doi: 10.1016/j.immuni.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 75.Mazzoni A, et al. Demethylation of the RORC2 and IL17A in human CD4 + T lymphocytes defines Th17 origin of nonclassic Th1 cells. J. Immunol. 2015;194:3116–3126. doi: 10.4049/jimmunol.1401303. [DOI] [PubMed] [Google Scholar]
- 76.Maggi L, et al. Brief report: etanercept inhibits the tumor necrosis factor α-driven shift of Th17 lymphocytes toward a nonclassic Th1 phenotype in juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66:1372–1377. doi: 10.1002/art.38355. [DOI] [PubMed] [Google Scholar]
- 77.Duhen T, Campbell DJ. IL-1β promotes the differentiation of polyfunctional human CCR6 + CXCR3 + Th1/17 cells that are specific for pathogenic and commensal microbes. J. Immunol. 2014;193:120–129. doi: 10.4049/jimmunol.1302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Becattini S, et al. Functional heterogeneity of human memory CD4+ T cell clones primed by pathogens or vaccines. Science. 2015;347:400–406. doi: 10.1126/science.1260668. [DOI] [PubMed] [Google Scholar]
- 79.Nistala K, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl Acad. Sci. U.S.A. 2010;107:14751–14756. doi: 10.1073/pnas.1003852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dupage M, Bluestone JA. Harnessing the plasticity of CD4+ T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016;16:149–163. doi: 10.1038/nri.2015.18. [DOI] [PubMed] [Google Scholar]
- 81.Pradeu T, Du Pasquier L. Immunological memory: what’s in a name? Immunol. Rev. 2018;283:7–20. doi: 10.1111/imr.12652. [DOI] [PubMed] [Google Scholar]
- 82.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 83.Gulati A, Kaur D, Krishna Prasad GVR, Mukhopadhaya A. PRR function of innate immune receptors in recognition of bacteria or bacterial ligands. Adv. Exp. Med. Biol. 2018;1112:255–280. doi: 10.1007/978-981-13-3065-0_18. [DOI] [PubMed] [Google Scholar]
- 84.Dolasia K, Bisht MK, Pradhan G, Udgata A, Mukhopadhyay S. TLRs/NLRs: shaping the landscape of host immunity. Int. Rev. Immunol. 2018;37:3–19. doi: 10.1080/08830185.2017.1397656. [DOI] [PubMed] [Google Scholar]
- 85.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 86.Durrant WE, Dong X. Systemic acquired resistance. Annu. Rev. Phytopathol. 2004;42:185–209. doi: 10.1146/annurev.phyto.42.040803.140421. [DOI] [PubMed] [Google Scholar]
- 87.Pham LN, Dionne MS, Shirasu-Hiza M, Schneider DS. A specific primed immune response in Drosophila is dependent on phagocytes. PLoS Pathog. 2007;3:e26. doi: 10.1371/journal.ppat.0030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kurtz J. Specific memory within innate immune systems. Trends Immunol. 2005;26:186–192. doi: 10.1016/j.it.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 89.Quintin J, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wout JW, Poell R, Furth R. The rof BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice. Scand. J. Immunol. 1992;36:713–720. doi: 10.1111/j.1365-3083.1992.tb03132.x. [DOI] [PubMed] [Google Scholar]
- 91.Spencer JC, Ganguly R, Waldman RH. Nonspecific protection of mice against influenza virus infection by local or systemic immunization with Bacille Calmette-Guérin. J. Infect. Dis. 1977;136:171–175. doi: 10.1093/infdis/136.2.171. [DOI] [PubMed] [Google Scholar]
- 92.Angelidou A, et al. BCG as a case study for precision vaccine development: lessons from vaccine heterogeneity, trained immunity, and immune ontogeny. Front. Microb. 2020;11:332. doi: 10.3389/fmicb.2020.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Netea MG, Van Crevel R. BCG-induced protection: effects on innate immune memory. Sem. Immunol. 2014;26:512–517. doi: 10.1016/j.smim.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 94.Netea MG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:6284. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Netea MG, et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saeed S, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ramírez-Valle F, Gray EE, Cyster JG. Inflammation induces dermal Vγ4+ γδT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc. Natl Acad. Sci. U.S.A. 2015;112:8046–8051. doi: 10.1073/pnas.1508990112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang X, Peng H, Tian Z. Innate lymphoid cell memory. Cell Mol. Immunol. 2019;16:423–429. doi: 10.1038/s41423-019-0212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 2006;7:507–516. doi: 10.1038/ni1332. [DOI] [PubMed] [Google Scholar]
- 100.Paust S, et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 2010;11:1127–1135. doi: 10.1038/ni.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lopez-Vergès S, et al. Expansion of a unique CD57+ NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl Acad. Sci. U.S.A. 2011;108:14725–14732. doi: 10.1073/pnas.1110900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Foley B, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood. 2012;119:2665–2674. doi: 10.1182/blood-2011-10-386995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nikzad R, et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 2019;4:eaat8116. doi: 10.1126/sciimmunol.aat8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cooper MA, et al. Cytokine-induced memory-like natural killer cells. Proc. Natl Acad. Sci. U.S.A. 2009;106:1915–1919. doi: 10.1073/pnas.0813192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cooper MA, Yokoyama WM. Memory-like responses of natural killer cells. Immunol. Rev. 2010;235:297–305. doi: 10.1111/j.0105-2896.2010.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Larsen SB, Cowley CJ, Fuchs E. Epithelial cells: liaisons of immunity. Curr. Opin. Immunol. 2020;62:45–53. doi: 10.1016/j.coi.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.de Bree LCJ, et al. Non-specific effects of vaccines: current evidence and potential implications. Semin. Immunol. 2018;39:35–43. doi: 10.1016/j.smim.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 108.Locht C, Mielcarek N. Live attenuated vaccines against pertussis. Exp. Rev. Vaccines. 2014;13:1147–1158. doi: 10.1586/14760584.2014.942222. [DOI] [PubMed] [Google Scholar]
- 109.Muramatsu D, et al. β-Glucan derived from Aureobasidium pullulans is effective for the prevention of influenza in mice. PLoS ONE. 2012;7:e41399. doi: 10.1371/journal.pone.0041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. 2017;46:562–576. doi: 10.1016/j.immuni.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abrahamsson TR, et al. Low diversity of the gut microbiota in infants with atopic eczema. J. Allergy Clin. Immunol. 2012;129:434–440. doi: 10.1016/j.jaci.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 113.Abrahamsson TR, et al. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin. Exp. Allergy. 2014;44:842–850. doi: 10.1111/cea.12253. [DOI] [PubMed] [Google Scholar]
- 114.Chen J, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016;6:28484. doi: 10.1038/srep28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scher JU, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67:128–139. doi: 10.1002/art.38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Round JL, Palm NW. Causal effects of the microbiota on immune-mediated diseases. Sci. Immunol. 2018;3:eaao1603. doi: 10.1126/sciimmunol.aao1603. [DOI] [PubMed] [Google Scholar]
- 117.Arrieta M-C, Walter J, Finlay BB. Human microbiota-associated mice: a model with challenges. Cell Host Microbe. 2016;19:575–578. doi: 10.1016/j.chom.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 118.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hapfelmeier S, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–1709. doi: 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 121.Tanoue T, Umesaki Y, Honda K. Immune responses to gut microbiota-commensals and pathogens. Gut Microbes. 2010;1:224–233. doi: 10.4161/gmic.1.4.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Braniste V, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014;6:263ra158. doi: 10.1126/scitranslmed.3009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hoban AE, et al. Regulation of prefrontal cortex myelination by the microbiota. Transl. Psychiatry. 2016;6:e774. doi: 10.1038/tp.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yano JM, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161:264–276. doi: 10.1016/j.cell.2015.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hsiao EY, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.David T, Ling SF, Barton A. Genetics of immune-mediated inflammatory diseases. Clin. Exp. Immunol. 2018;193:3–12. doi: 10.1111/cei.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ferris MT, Hood DW. Host genetic regulation of immune-based and infectious diseases: introduction to mammalian genome special issue: genetics of infectious disease. Mamm. Genome. 2018;29:365–366. doi: 10.1007/s00335-018-9779-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klebanov N. Genetic predisposition to infectious disease. Cureus. 2018;10:e3210. doi: 10.7759/cureus.3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bellamy R, et al. Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N. Engl. J. Med. 1998;338:640–644. doi: 10.1056/NEJM199803053381002. [DOI] [PubMed] [Google Scholar]
- 130.Vidal SM, Malo D, Vogan K, Skamene E, Gros P. Natural resistance to infection with intracellular parasites: isolation of a candidate for BCG. Cell. 1993;73:469–485. doi: 10.1016/0092-8674(93)90135-D. [DOI] [PubMed] [Google Scholar]
- 131.Platonov AE, et al. Association of human Fc gamma RIIa (CD32) polymorphism with susceptibility to and severity of meningococcal disease. Clin. Infect. Dis. 1998;27:746–750. doi: 10.1086/514935. [DOI] [PubMed] [Google Scholar]
- 132.Bongen E, Vallania F, Utz PJ, Khatri P. KLRD1-expressing natural killer cells predict influenza susceptibility. Genome Med. 2018;10:45. doi: 10.1186/s13073-018-0554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scepanovic P, et al. Human genetic variants and age are the strongest predictors of humoral immune responses to common pathogens and vaccines. Genome Med. 2018;10:1–13. doi: 10.1186/s13073-018-0568-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benetti, E., et al. ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur. J. Hum. Genet. (2020). 10.1038/s41431-020-0691-z. [DOI] [PMC free article] [PubMed]
- 135.Zhao, J. et al. Relationship between the ABO Blood Group and the COVID-19 susceptibility. medRxiv (2020). 10.1101/2020.03.11.20031096.
- 136.Silverstein AM. Autoimmunity versus horror autotoxicus: the struggle for recognition. Nat. Immunol. 2001;2:279–281. doi: 10.1038/86280. [DOI] [PubMed] [Google Scholar]
- 137.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 138.Wang L, Wang F-S, Gershwin ME. Human autoimmune diseases: a comprehensive update. J. Intern. Med. 2015;278:369–395. doi: 10.1111/joim.12395. [DOI] [PubMed] [Google Scholar]
- 139.Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J. Clin. Investig. 2015;125:2228–2233. doi: 10.1172/JCI78088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.González-Serna D, et al. Analysis of the genetic component of systemic sclerosis in Iranian and Turkish populations through a genome-wide association study. Rheumatology. 2019;58:289–298. doi: 10.1093/rheumatology/key281. [DOI] [PubMed] [Google Scholar]
- 141.López-Isac E, et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 2019;10:4955. doi: 10.1038/s41467-019-12760-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Westra H-J, et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 2018;50:1366–1374. doi: 10.1038/s41588-018-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Inshaw JRJ, Cutler AJ, Burren OS, Stefana MI, Todd JA. Approaches and advances in the genetic causes of autoimmune disease and their implications. Nat. Immunol. 2018;19:674–684. doi: 10.1038/s41590-018-0129-8. [DOI] [PubMed] [Google Scholar]
- 144.Márquez A, et al. Meta-analysis of Immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. 2018;10:97. doi: 10.1186/s13073-018-0604-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Catalina MD, Owen KA, Labonte AC, Grammer AC, Lipsky PE. The pathogenesis of systemic lupus erythematosus: harnessing big data to understand the molecular basis of lupus. J. Autoimmun. 2019;110:102359. doi: 10.1016/j.jaut.2019.102359. [DOI] [PubMed] [Google Scholar]
- 146.Medawar PB. A second study of the behaviour and fate of skin homografts in rabbits: a report to the War Wounds Committee of the Medical Research Council. J. Anat. 1945;79:157–176. [PubMed] [Google Scholar]
- 147.Medawar PB. Immunity to homologous grafted skin; the relationship between the antigens of blood and skin. Br. J. Exp. Pathol. 1946;27:15–24. [PMC free article] [PubMed] [Google Scholar]
- 148.Carreras, E., Dufour, C., Mohty, M. & Kröger, N. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. (2019). NBK554031. [PubMed]
- 149.Robinson J, et al. IPD-IMGT/HLA database. Nucleic Acids Res. 2020;48:948–955. doi: 10.1093/nar/gkz950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Spellman S, et al. Effects of mismatching for minor histocompatibility antigens on clinical outcomes in HLA-matched, unrelated hematopoietic stem cell transplants. Biol. Blood Marrow Transpl. 2009;15:856–863. doi: 10.1016/j.bbmt.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rajalingam R. The impact of HLA class I-specific killer cell immunoglobulin-like receptors on antibody-dependent natural killer cell-mediated cytotoxicity and organ allograft rejection. Front. Immunol. 2016;7:585. doi: 10.3389/fimmu.2016.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gruber C, Bogunovic D. Incomplete penetrance in primary immunodeficiency: a skeleton in the closet. Hum. Genet. 2020;139:745–757. doi: 10.1007/s00439-020-02131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin. Immunol. 2018;14:61. doi: 10.1186/s13223-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Al-Mousa H, Al-Saud B. Primary immunodeficiency diseases in highly consanguineous populations from Middle East and North Africa: epidemiology, diagnosis, and care. Front. Immunol. 2017;8:678. doi: 10.3389/fimmu.2017.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Abolhassani H, Hammarström L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood. 2020;135:656–667. doi: 10.1182/blood.2019000929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tangye SG, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020;40:24–64. doi: 10.1007/s10875-019-00737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bousfiha A, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J. Clin. Immunol. 2018;38:129–143. doi: 10.1007/s10875-017-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McCusker C, Warrington R. Primary immunodeficiency. Allergy Asthma Clin. Immunol. 2011;7:S11. doi: 10.1186/1710-1492-7-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jéru I. Update on the genetics of autoinflammatory disorders. Curr. Allergy Asthma Rep. 2019;19:41. doi: 10.1007/s11882-019-0874-2. [DOI] [PubMed] [Google Scholar]
- 160.Davidson S, Steiner A, Harapas CR, Masters SL. An update on autoinflammatory diseases: interferonopathies. Curr. Rheumatol. Rep. 2018;20:38. doi: 10.1007/s11926-018-0748-y. [DOI] [PubMed] [Google Scholar]
- 161.de Jesus AA, et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019;143:1939–1943.e8. doi: 10.1016/j.jaci.2018.12.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Holloway JW, Yang IA, Holgate ST. Genetics of allergic disease. J. Allergy Clin. Immunol. 2010;125:S81–S94. doi: 10.1016/j.jaci.2009.10.071. [DOI] [PubMed] [Google Scholar]
- 163.Cookson WO, Sharp PA, Faux JA, Hopkin JM. Linkage between immunoglobulin E responses underlying asthma and rhinitis and chromosome 11q. Lancet. 1989;1:1292–1295. doi: 10.1016/S0140-6736(89)92687-1. [DOI] [PubMed] [Google Scholar]
- 164.Bønnelykke K, Sparks R, Waage J, Milner JD. Genetics of allergy and allergic sensitization: common variants, rare mutations. Curr. Opin. Immunol. 2015;36:115–126. doi: 10.1016/j.coi.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jochems SP, et al. Innate and adaptive nasal mucosal immune responses following experimental human pneumococcal colonization. J. Clin. Investig. 2019;130:4523–4538. doi: 10.1172/JCI128865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.DeVries A, et al. Epigenome-wide analysis links SMAD3 methylation at birth to asthma in children of asthmatic mothers. J. Allergy Clin. Immunol. 2017;140:534–542. doi: 10.1016/j.jaci.2016.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kabesch M, Tost J. Recent findings in the genetics and epigenetics of asthma and allergy. Semin. Immunopathol. 2020;42:43–60. doi: 10.1007/s00281-019-00777-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ehrlich P. Ueber den jetzigen stand der karzinomforschung. Ned. Tijdschr. Geneeskd. 1909;5:73–209. [Google Scholar]
- 169.Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52:55–81. doi: 10.1016/j.immuni.2019.12.018. [DOI] [PubMed] [Google Scholar]
- 171.Garrido F, et al. Natural history of HLA expression during tumour development. Immunol. Today. 1993;14:491–499. doi: 10.1016/0167-5699(93)90264-L. [DOI] [PubMed] [Google Scholar]
- 172.Sukari A, Nagasaka M, Al-Hadidi A, Lum LG. Cancer immunology and immunotherapy. Anticancer Res. 2016;36:5593–5606. doi: 10.21873/anticanres.11144. [DOI] [PubMed] [Google Scholar]
- 173.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front. Oncol. 2018;8:86. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129:474–481. doi: 10.1111/j.1365-2567.2010.03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immun. 2017;66:1609–1617. doi: 10.1007/s00262-017-2053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee HT, Lee SH, Heo YS. Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules. 2019;24:1190. doi: 10.3390/molecules24061190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Aiello A, et al. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 2019;10:2247. doi: 10.3389/fimmu.2019.02247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Thomas R, Wang W, Su DM. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun. Ageing. 2020;17:2. doi: 10.1186/s12979-020-0173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Oh SJ, Lee JK, Shin OS. Aging and the immune system: the impact of immunosenescence on viral infection, immunity and vaccine immunogenicity. Immune Netw. 2019;19:e37. doi: 10.4110/in.2019.19.e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hashimoto K, et al. Single-cell transcriptomics reveals expansion of cytotoxic CD4 T cells in supercentenarians. Proc. Natl Acad. Sci. USA. 2019;116:24242–24251. doi: 10.1073/pnas.1907883116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rappuoli R, Pizza M, Del Giudice G, De Gregorio E. Vaccines, new opportunities for a new society. Proc. Natl Acad. Sci. USA. 2014;111:12288–12293. doi: 10.1073/pnas.1402981111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Meyer H, Ehmann R, Smith GL. Smallpox in the post-eradication era. Viruses. 2020;12:138. doi: 10.3390/v12020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Greenwood B. The contribution of vaccination to global health: past, present and future. Philos. Trans. R. Soc. L. B. Biol. Sci. 2014;369:20130433. doi: 10.1098/rstb.2013.0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Decker WK, Safdar A. Bioimmunoadjuvants for the treatment of neoplastic and infectious disease: Coley’s legacy revisited. Cytokine Growth Factor Rev. 2009;20:271–281. doi: 10.1016/j.cytogfr.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 185.Fuge O, Vasdev N, Allchorne P, Green JS. Immunotherapy for bladder cancer. Res. Rep. Urol. 2015;7:65–79. doi: 10.2147/RRU.S63447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Old LJ, Clarke DA, Benacerraf B. Effect of Bacillus Calmette–Guerin infection on transplanted tumours in the mouse. Nature. 1959;184:291–292. doi: 10.1038/184291a0. [DOI] [PubMed] [Google Scholar]
- 187.You, J. H. S., Jiang, X., Lee, W. H., Chan, P. K. S. & Ng, S. C. Cost-effectiveness analysis of fecal microbiota transplantation for recurrent Clostridium difficileinfection in patients with inflammatory bowel disease. J Gastroenterol. Hepatol. (2020). 10.1111/jgh.15002. [DOI] [PubMed]
- 188.Tariq R, et al. Efficacy of fecal microbiota transplantation for recurrent c. difficile infection in inflammatory bowel disease. Inflamm. Bowel Dis. 2019;2019:izz299. doi: 10.1093/ibd/izz299. [DOI] [PubMed] [Google Scholar]
- 189.Sedighi M, et al. Therapeutic bacteria to combat cancer; current advances, challenges, and opportunities. Cancer Med. 2019;8:3167–3181. doi: 10.1002/cam4.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Davola ME, Mossman KL. Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? Oncoimmunology. 2019;8:e1581528. doi: 10.1080/2162402X.2019.1596006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Malhotra A, Sendilnathan A, Old MO, Wise-Draper TM. Oncolytic virotherapy for head and neck cancer: current research and future developments. Oncolytic Virother. 2015;4:83–93. doi: 10.2147/OV.S54503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Greig SL. Talimogene laherparepvec: first global approval. Drugs. 2016;76:147–154. doi: 10.1007/s40265-015-0522-7. [DOI] [PubMed] [Google Scholar]
- 193.Eissa IR, et al. The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers. Cancers. 2018;10:356. doi: 10.3390/cancers10100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kaufman HL, Bommareddy PK. Two roads for oncolytic immunotherapy development. J. Immunother. Cancer. 2019;7:26. doi: 10.1186/s40425-019-0515-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat. Rev. Microbiol. 2014;12:23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Samson A, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Martin NT, et al. Pre-surgical neoadjuvant oncolytic virotherapy confers protection against rechallenge in a murine model of breast cancer. Sci. Rep. 2019;9:1865. doi: 10.1038/s41598-018-38385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bourgeois-Daigneault M-C, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018;10:eaao1641. doi: 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 199.Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol. Ther. - Oncolytics. 2019;15:234–247. doi: 10.1016/j.omto.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.De Flora S, Bonanni P. The prevention of infection-associated cancers. Carcinogenesis. 2011;32:787–795. doi: 10.1093/carcin/bgr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gutowska-Owsiak D, Ogg GS. Therapeutic vaccines for allergic disease. npj Vaccines. 2017;2:12. doi: 10.1038/s41541-017-0014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wu AY. Immunotherapy—vaccines for allergic diseases. J. Thorac. Dis. 2012;4:198–202. doi: 10.3978/j.issn.2072-1439.2011.07.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Valenta R, Campana R, Focke-Tejkl M, Niederberger V. Vaccine development for allergen-specific immunotherapy based on recombinant allergens and synthetic allergen peptides: lessons from the past and novel mechanisms of action for the future. J. Allergy Clin. Immunol. 2016;137:351–357. doi: 10.1016/j.jaci.2015.12.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Larché M, Akdis CA, Valenta R. Immunological mechanisms of allergen-specific immunotherapy. Nat. Rev. Immunol. 2006;6:761–771. doi: 10.1038/nri1934. [DOI] [PubMed] [Google Scholar]
- 205.Zhernov Y, Curin M, Khaitov M, Karaulov A, Valenta R. Recombinant allergens for immunotherapy. Curr. Opin. Allergy Clin. Immunol. 2019;19:402–414. doi: 10.1097/ACI.0000000000000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Schlom J, et al. Therapeutic cancer vaccines. Adv. Cancer Res. 2014;121:67–124. doi: 10.1016/B978-0-12-800249-0.00002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. npj Vaccines. 2019;4:1–10. doi: 10.1038/s41541-019-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Servín-Blanco R, Zamora-Alvarado R, Gevorkian G, Manoutcharian K. Antigenic variability: obstacles on the road to vaccines against traditionally difficult targets. Hum. Vaccin. Immunother. 2016;12:2640–2648. doi: 10.1080/21645515.2016.1191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018;18:168–182. doi: 10.1038/nri.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sivori S, et al. Human NK cells: surface receptors, inhibitory checkpoints and translational applications. Cell. Mol. Immunol. 2019;16:430–441. doi: 10.1038/s41423-019-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pannemans K, Hellings N, Stinissen P. Therapeutic vaccines for autoimmune diseases. Drug Discov. Today. Ther. Strateg. 2009;6:39–44. doi: 10.1016/j.ddstr.2009.11.001. [DOI] [Google Scholar]
- 212.Serra P, Santamaria P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur. J. Immunol. 2018;48:751–756. doi: 10.1002/eji.201747059. [DOI] [PubMed] [Google Scholar]
- 213.Von Behring E, Zustandekommen KS. der Diphtherie-Immunitat and der Tetanus-Immunität bei Thieren. Dtsch. Med. Wochenschr. 1890;16:1113–1114. doi: 10.1055/s-0029-1207589. [DOI] [Google Scholar]
- 214.João C, Negi VS, Kazatchkine MD, Bayry J, Kaveri SV. Passive serum therapy to immunomodulation by IVIG: a fascinating journey of antibodies. J. Immunol. 2018;200:1957–1963. doi: 10.4049/jimmunol.1701271. [DOI] [PubMed] [Google Scholar]
- 215.Roux EYA. Contribution à l’étude de la diphthérie. 3e mémoire. Ann. Inst. Pasteur. 1890;4:384–426. [Google Scholar]
- 216.Galeotti Caroline, Kaveri SriniV, Bayry J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int. Immunol. 2017;29:491–498. doi: 10.1093/intimm/dxx039. [DOI] [PubMed] [Google Scholar]
- 217.Bruton OC. Agamma-globulinemia. Pediatrics. 1952;9:722–728. doi: 10.1542/peds.9.6.722. [DOI] [Google Scholar]
- 218.Perez EE, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J. Allergy Clin. Immunol. 2017;139:S1–S46. doi: 10.1016/j.jaci.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 219.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 220.Springer TA. César milstein, the father of modern immunology. Nat. Immunol. 2002;3:501–503. doi: 10.1038/ni0602-501. [DOI] [PubMed] [Google Scholar]
- 221.Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 222.Shawler DL, Bartholomew RM, Smith LM, Dillman RO. Human immune response to multiple injections of murine monoclonal IgG. J. Immunol. 1985;135:1530–1535. [PubMed] [Google Scholar]
- 223.Kuus-Reichel K, et al. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin. Diagn. Lab. Immunol. 1994;1:365–372. doi: 10.1128/cdli.1.4.365-372.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2017;122:2–19. doi: 10.1016/j.addr.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 225.Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol. 2008;20:450–459. doi: 10.1016/j.coi.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 226.Chester KA, et al. Phage libraries for generation of clinically useful antibodies. Lancet. 1994;343:455–456. doi: 10.1016/S0140-6736(94)92695-6. [DOI] [PubMed] [Google Scholar]
- 227.Frenzel A, Schirrmann T, Hust M. Phage display-derived human antibodies in clinical development and therapy. MAbs. 2016;8:1177–1194. doi: 10.1080/19420862.2016.1212149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front. Immunol. 2017;8:1751. doi: 10.3389/fimmu.2017.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Magadán, S. et al. Production of antigen-specific human monoclonal antibodies: comparison of mice carrying IgH/κ or IgH/κ/λ transloci. Biotechniques 33, (2002). [DOI] [PubMed]
- 230.Mompó SM, González-Fernández Á. Antigen-specific human monoclonal antibodies from transgenic mice. Methods Mol. Biol. 2019;1904:253–291. doi: 10.1007/978-1-4939-8958-4_11. [DOI] [PubMed] [Google Scholar]
- 231.Barderas R, Benito-Peña E. The 2018 Nobel Prize in chemistry: phage display of peptides and antibodies. Anal. Bioanal. Chem. 2019;411:2475–2479. doi: 10.1007/s00216-019-01714-4. [DOI] [PubMed] [Google Scholar]
- 232.Winter G. Harnessing evolution to make medicines (Nobel Lecture) Angew. Chem. Int. Ed. Engl. 2019;58:14438–14445. doi: 10.1002/anie.201909343. [DOI] [PubMed] [Google Scholar]
- 233.Almagro JC, Pedraza-Escalona M, Arrieta HI, Pérez-Tapia SM. Phage display libraries for antibody therapeutic discovery and development. Antibodies (Basel) 2019;8:44. doi: 10.3390/antib8030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 235.Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs. 2020;12:1703531. doi: 10.1080/19420862.2019.1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11:219–238. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. 2018;10:183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Jiang XR, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011;10:101–110. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- 239.Mitoma H, Horiuchi T, Tsukamoto H, Ueda N. Molecular mechanisms of action of anti-TNF-α agents - Comparison among therapeutic TNF-α antagonists. Cytokine. 2018;101:56–63. doi: 10.1016/j.cyto.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 240.Buurman DJ, et al. Quantitative comparison of the neutralizing capacity, immunogenicity and cross-reactivity of anti-TNF-α biologicals and an Infliximab-biosimilar. PLoS ONE. 2018;13:e0208922. doi: 10.1371/journal.pone.0208922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Taylor RP, Lindorfer MA. Cytotoxic mechanisms of immunotherapy: harnessing complement in the action of anti-tumor monoclonal antibodies. Semin. Immunol. 2016;28:309–316. doi: 10.1016/j.smim.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 242.Rogers LM, Veeramani S, Weiner GJ. Complement in monoclonal antibody therapy of cancer. Immunol. Res. 2014;59:203–210. doi: 10.1007/s12026-014-8542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Singh S, et al. Monoclonal antibodies: a review. Curr. Clin. Pharmacol. 2018;13:85–99. doi: 10.2174/1574884712666170809124728. [DOI] [PubMed] [Google Scholar]
- 244.Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017;16:315–337. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 245.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018;17:197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 246.Fritz JM, Lenardo MJ. Development of immune checkpoint therapy for cancer. J. Exp. Med. 2019;216:1244–1254. doi: 10.1084/jem.20182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 248.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 249.Altmann DM. A Nobel Prize-worthy pursuit: cancer immunology and harnessing immunity to tumour neoantigens. Immunology. 2018;155:283–284. doi: 10.1111/imm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yron I, Wood TA, Spiess PJ, Rosenberg SA. In vitro growth of murine T cells. V. The isolation and growth of lymphoid cells infiltrating syngeneic solid tumors. J. Immunol. 1980;125:238–245. [PubMed] [Google Scholar]
- 251.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J. Immunol. 2014;192:5451–5458. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.A I, J L. Virus interference. I. interferon Proc. R. Soc. Lond. Ser. B - Biol. Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 253.Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007;282:20047–20051. doi: 10.1074/jbc.R700004200. [DOI] [PubMed] [Google Scholar]
- 254.Borden EC. Interferons α and β in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019;18:219–234. doi: 10.1038/s41573-018-0011-2. [DOI] [PubMed] [Google Scholar]
- 255.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer. 2016;16:131–144. doi: 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- 256.Bhattacharya P, et al. Dual role of GM-CSF as a pro-inflammatory and a regulatory cytokine: implications for immune therapy. J. Int. Soc. Interface Cytokine Res. 2015;35:585–599. doi: 10.1089/jir.2014.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Chen P, Chen F, Zhou B. Comparisons of therapeutic efficacy and safety of ipilimumab plus GM-CSF versus ipilimumab alone in patients with cancer: a meta-analysis of outcomes. Drug Des. Devel. Ther. 2018;12:2025–2038. doi: 10.2147/DDDT.S154258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. 2019;39:6–21. doi: 10.1089/jir.2018.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Nicholas C, Lesinski GB. Immunomodulatory cytokines as therapeutic agents for melanoma. Immunotherapy. 2011;3:673–690. doi: 10.2217/imt.11.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hassett B, et al. Manufacturing history of etanercept (Enbrel®): consistency of product quality through major process revisions. MAbs. 2018;10:159–165. doi: 10.1080/19420862.2017.1388483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ruderman EM, Pope RM. The evolving clinical profile of abatacept (CTLA4-lg): a novel co-stimulatory modulator for the treatment of rheumatoid arthritis. Arthritis Res. Ther. 2005;7:S21. doi: 10.1186/ar1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer. 1975;16:230–239. doi: 10.1002/ijc.2910160205. [DOI] [PubMed] [Google Scholar]
- 263.Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975;5:117–121. doi: 10.1002/eji.1830050209. [DOI] [PubMed] [Google Scholar]
- 264.Piontek GE, et al. YAC-1 MHC class I variants reveal an association between decreased NK sensitivity and increased H-2 expression after interferon treatment or in vivo passage. J. Immunol. 1985;135:4281–4288. [PubMed] [Google Scholar]
- 265.Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H–2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 266.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J. Exp. Med. 1982;155:1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Sun Z, et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat. Commun. 2019;10:1–12. doi: 10.1038/s41467-018-07882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Charych DH, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res. 2016;22:680–690. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- 269.Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol. Res. 2015;3:219–227. doi: 10.1158/2326-6066.CIR-15-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Miller JS, Lanier LL. Natural killer cells in cancer immunotherapy. Annu. Rev. Cancer Biol. 2019;3:77–103. doi: 10.1146/annurev-cancerbio-030518-055653. [DOI] [Google Scholar]
- 271.Liu Y, Bewersdorf JP, Stahl M, Zeidan AM. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: the dawn of a new era? Blood Rev. 2019;34:67–83. doi: 10.1016/j.blre.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 272.Mavers M, Bertaina A. High-risk leukemia: past, present, and future role of NK cells. J. Immunol. Res. 2018;2018:1586905. doi: 10.1155/2018/1586905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy. Cancer J. 2015;21:486–491. doi: 10.1097/PPO.0000000000000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Langerhans P. Ueber die Nerven der menschlichen Haut. Arch. für Pathol. Anat. und Physiol. und für. Klin. Med. 1868;44:325–337. [Google Scholar]
- 275.Volchenkov R, Sprater F, Vogelsang P, Appel S. The 2011 Nobel Prize in physiology or medicine. Scand. J. Immunol. 2012;75:1–4. doi: 10.1111/j.1365-3083.2011.02663.x. [DOI] [PubMed] [Google Scholar]
- 276.Otsuka M, Egawa G, Kabashima K. Uncovering the mysteries of langerhans cells, inflammatory dendritic epidermal cells, and monocyte-derived Langerhans cell-like cells in the epidermis. Front. immunol. 2018;9:1768. doi: 10.3389/fimmu.2018.01768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.van Spriel AB, de Jong EC. Dendritic cell science: more than 40 years of history. J. Leukoc. Biol. 2013;93:33–38. doi: 10.1189/jlb.0512263. [DOI] [PubMed] [Google Scholar]
- 278.Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95. doi: 10.1038/cr.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Posch W, Lass-Flörl C, Wilflingseder D. Generation of human monocyte-derived dendritic cells from whole blood. J. Vis. Exp. 2016;118:54968. doi: 10.3791/54968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Fong L, Engleman EG. Dendritic cells in cancer immunotherapy. Annu. Rev. Immunol. 2000;18:245–273. doi: 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- 281.Romani N, et al. Proliferating dendritic cell progenitors in human blood. J. Exp. Med. 1994;180:83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. 2016;37:855–865. doi: 10.1016/j.it.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Handy CE, Antonarakis ES. Sipuleucel-T for the treatment of prostate cancer: novel insights and future directions. Future Oncol. 2018;14:907–917. doi: 10.2217/fon-2017-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Phillips BE, Garciafigueroa Y, Trucco M, Giannoukakis N. Clinical tolerogenic dendritic cells: exploring therapeutic impact on human autoimmune disease. Front. Immunol. 2017;8:1279. doi: 10.3389/fimmu.2017.01279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic cells as gatekeepers of tolerance. Semin. Immunopathol. 2017;39:153–163. doi: 10.1007/s00281-016-0583-z. [DOI] [PubMed] [Google Scholar]
- 286.Prietl B, et al. High-dose cholecalciferol supplementation significantly increases peripheral CD4+ Tregs in healthy adults without negatively affecting the frequency of other immune cells. Eur. J. Nutr. 2014;53:751–759. doi: 10.1007/s00394-013-0579-6. [DOI] [PubMed] [Google Scholar]
- 287.Pauza CD, et al. Gamma delta T cell therapy for cancer: it is good to be local. Front. Immunol. 2018;9:1305. doi: 10.3389/fimmu.2018.01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Wilhelm M, et al. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014;12:45. doi: 10.1186/1479-5876-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Alnaggar M, et al. Allogenic Vγ9Vδ2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J. Immunother. Cancer. 2019;7:36. doi: 10.1186/s40425-019-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 291.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. J. Immunol. 2017;198:981–985. [PubMed] [Google Scholar]
- 292.Holm TL, Nielsen J, Claesson MH. CD4+ CD25+ regulatory T cells: I. Phenotype and physiology. APMIS. 2004;112:629–641. doi: 10.1111/j.1600-0463.2004.apm1121001.x. [DOI] [PubMed] [Google Scholar]
- 293.Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. U.S.A. 2018;1417:104–115. doi: 10.1111/nyas.13625. [DOI] [PubMed] [Google Scholar]
- 294.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 295.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 296.Xu L, et al. Depletion of CD4+CD25 high regulatory T cells from tumor infiltrating lymphocytes predominantly induces Th1 type immune response in vivo which inhibits tumor growth in adoptive immunotherapy. Cancer Biol. Ther. 2009;8:66–72. doi: 10.4161/cbt.8.1.7131. [DOI] [PubMed] [Google Scholar]
- 297.Chodon T, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014;20:2457–2465. doi: 10.1158/1078-0432.CCR-13-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Uckert W, Schumacher TNM. TCR transgenes and transgene cassettes for TCR gene therapy: status in 2008. Cancer Immunol. Immunother. 2009;58:809–822. doi: 10.1007/s00262-008-0649-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Casucci M, Bondanza A. Suicide gene therapy to increase the safety of chimeric antigen receptor-redirected T lymphocytes. J. Cancer. 2011;2:378–382. doi: 10.7150/jca.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426–1438.e11. doi: 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Liu E, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Adachi K, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 305.Gomes-Silva D, Ramos CA. Cancer immunotherapy using CAR-T cells: from the research bench to the assembly line. Biotechnol. J. 2018;13:1700097. doi: 10.1002/biot.201700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ruella M, Kenderian SS. Next-generation chimeric antigen receptor T-cell therapy: going off the Shelf. BioDrugs. 2017;31:473–481. doi: 10.1007/s40259-017-0247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol. Ther. 2017;25:1769–1781. doi: 10.1016/j.ymthe.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Knochelmann HM, et al. CAR T cells in solid tumors: blueprints for building effective therapies. Front. immunol. 2018;9:1740. doi: 10.3389/fimmu.2018.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Kuhlmann AS, Peterson CW, Kiem HP. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS. 2018;13:446–453. doi: 10.1097/COH.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018;18:605–616. doi: 10.1038/s41577-018-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Skuljec J, et al. Chimeric antigen receptor-redirected regulatory T cells suppress experimental allergic airway inflammation, a model of asthma. Front. Immunol. 2017;8:1125. doi: 10.3389/fimmu.2017.01125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Bonam SR, Kaveri SV, Sakuntabhai A, Gilardin L, Bayry J. Adjunct immunotherapies for the management of severely Ill COVID-19 patients. Cell Rep. Med. 2020;1:100016. doi: 10.1016/j.xcrm.2020.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ahn JY, et al. Use of convalescent plasma therapy in two covid-19 patients with acute respiratory distress syndrome in Korea. J. Korean Med. Sci. 2020;35:e149. doi: 10.3346/jkms.2020.35.e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Duan K, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl Acad. Sci. U.S.A. 2020;117:9490–9496. doi: 10.1073/pnas.2004168117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Klein NP, Abu-Elyazeed R, Cheuvart B, Janssens W, Mesaros N. Immunogenicity and safety following primary and booster vaccination with a hexavalent diphtheria, tetanus, acellular pertussis, hepatitis B, inactivated poliovirus and Haemophilus influenzae type b vaccine: a randomized trial in the United States. Hum. Vaccines Immunother. 2019;15:809–821. doi: 10.1080/21645515.2018.1549449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kartoglu U, Milstien J. Tools and approaches to ensure quality of vaccines throughout the cold chain. Exp. Rev. Vaccines. 2014;13:843–854. doi: 10.1586/14760584.2014.923761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Peleteiro M, et al. Polymeric nanocapsules for vaccine delivery: influence of the polymeric shell on the interaction with the immune system. Front. Immunol. 2018;9:791. doi: 10.3389/fimmu.2018.00791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Jazayeri SD, Poh CL. Development of universal influenza vaccines targeting conserved viral proteins. Vaccines. 2019;7:169. doi: 10.3390/vaccines7040169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Thompson KM, Gellin BG, Hinman AR, Orenstein WA. The National Vaccine Advisory Committee at 30: impact and opportunity. Vaccine. 2018;36:1330–1344. doi: 10.1016/j.vaccine.2018.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Saso A, Kampmann B. Vaccine responses in newborns. Semin. Immunopathol. 2017;39:627–642. doi: 10.1007/s00281-017-0654-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Switzer C, D’Heilly C, Macina D. Immunological and clinical benefits of maternal immunization against pertussis: a systematic review. Infect. Dis. Ther. 2019;8:499–541. doi: 10.1007/s40121-019-00264-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Lurie N, Saville M, Hatchett R, Halton J. Developing Covid-19 vaccines at pandemic speed. N. Engl. J. Med. 2020;382:1969–1973. doi: 10.1056/NEJMp2005630. [DOI] [PubMed] [Google Scholar]
- 323.Amanat F, Krammer F. Perspective SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Ulmer JB, Wahren B, Liu MA. Gene-based vaccines: recent technical and clinical advances. Trends Mol. Med. 2006;12:216–222. doi: 10.1016/j.molmed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 325.Petrovsky N. Comparative safety of vaccine adjuvants: a summary of current evidence and future needs. Drug Saf. 2015;38:1059–1074. doi: 10.1007/s40264-015-0350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Sánchez-Ramón S, et al. Trained immunity-based vaccines: a new paradigm for the development of broad-spectrum anti-infectious formulations. Front. Immunol. 2018;9:2936. doi: 10.3389/fimmu.2018.02936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Pluviano S, Watt C, Della Sala S. Misinformation lingers in memory: failure of three pro-vaccination strategies. PLoS ONE. 2017;12:e0181640. doi: 10.1371/journal.pone.0181640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Sanyaolu A, et al. Measles outbreak in unvaccinated and partially vaccinated children and adults in the United States and Canada (2018-2019): a narrative review of cases. Inquiry. 2019;56:004695801989409. doi: 10.1177/0046958019894098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Lee Ventola C. Cancer immunotherapy, part 3: challenge and future trends. P T. 2017;42:514–521. [PMC free article] [PubMed] [Google Scholar]
- 330.Martinez-Quintanilla J, Seah I, Chua M, Shah K. Oncolytic viruses: overcoming translational challenges. J. Clin. Investig. 2019;129:1407–1418. doi: 10.1172/JCI122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother. Cancer. 2018;6:140–153. doi: 10.1186/s40425-018-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Samarkos M, Mastrogianni E, Kampouropoulou O. The role of gut microbiota in Clostridium difficile infection. Eur. J. Intern. Med. 2018;50:28–32. doi: 10.1016/j.ejim.2018.02.006. [DOI] [PubMed] [Google Scholar]