


Abstract
In mammalian cells, eight cytoplasmic aminoacyl-tRNA synthetases (AARS), and three non-synthetase proteins, reside in a large multi-tRNA synthetase complex (MSC). AARSs have critical roles in interpretation of the genetic code during protein synthesis, and in non-canonical functions unrelated to translation. Nonetheless, the structure and function of the MSC remain unclear. Partial or complete crystal structures of all MSC constituents have been reported; however, the structure of the holo-MSC has not been resolved. We have taken advantage of cross-linking mass spectrometry (XL-MS) and molecular docking to interrogate the three-dimensional architecture of the MSC in human HEK293T cells. The XL-MS approach uniquely provides structural information on flexibly appended domains, characteristic of nearly all MSC constituents. Using the MS-cleavable cross-linker, disuccinimidyl sulfoxide, inter-protein cross-links spanning all MSC constituents were observed, including cross-links between eight protein pairs not previously known to interact. Intra-protein cross-links defined new structural relationships between domains in several constituents. Unexpectedly, an asymmetric AARS distribution was observed featuring a clustering of tRNA anti-codon binding domains on one MSC face. Possibly, the non-uniform localization improves efficiency of delivery of charged tRNA’s to an interacting ribosome during translation. In summary, we show a highly compact, 3D structural model of the human holo-MSC.
INTRODUCTION
The essential function of the aminoacyl tRNA synthetases (AARSs) is mRNA decoding during protein synthesis. They catalyze ATP-dependent charging of tRNA with cognate amino acids for delivery to the A-site of the ribosome. Nearly all cytoplasmic AARSs exhibit extra-ordinary regulatory activities distinct from their canonical function in protein synthesis (1,2). These non-canonical functions are almost exclusively mediated by domains appended during evolution to the catalytic domains, and not present in early AARS forms such as the bacterial enzymes. Dysregulation or mutation of AARSs are implicated in pathological conditions including neurodegeneration, tumorigenesis, and metabolic disease (3–6), and there is expanding interest in AARS as therapeutics and therapeutic targets (7). Nine of the 20 AARS activities (in eight proteins since GluProRS contains two synthetase activities in a single polypeptide chain) reside in a cytoplasmic multi-tRNA synthetase complex (MSC) with three non-synthetase proteins termed AARS complex-interacting multifunctional proteins (AIMP)-1, -2 and -3 (8,9). Smaller MSC forms with fewer constituents are present in archaea and fungi (10).
Following the discovery of a mammalian MSC (11), much effort has been expended toward its structural elucidation by low-resolution imaging (12–16), X-ray crystallography (17–20), and biochemical approaches (15,21,22). Structures of the catalytic and anti-codon binding domains of the AARSs are highly conserved from bacteria to vertebrates, and have been elucidated by X-ray crystallography. However, appended domains unique to complex animals are largely absent from reported structures, most likely due to dynamic, disordered linkages to the catalytic domains (Figure 1A), and as a consequence, the structure of the mammalian MSC has remained elusive (23,24). The molecular size of the MSC, ∼1.0–1.2 MDa (14,22,25,26), is less than the ∼2 MDa, 60S eukaryotic large ribosomal subunit, however, given that RNA comprises at least half of the ribosome mass, the protein mass of the MSC is comparable to that of the 60S ribosome. Based on stoichiometric studies of the purified complex, and structural studies of purified constituents, the MSC is suggested to be a super-complex of two identical, symmetrically arranged sub-units, each containing a single copy of the constituents, with the exception of LysRS which is present as a dimer in each sub-unit (Figure 1B, adapted from (27,28)). The sub-units are proposed to be joined by dimers of AspRS and the ProRS domain of GluProRS, and possibly by LysRS tetramers (20). Four AARSs containing GST-like domains important in protein-protein interactions form a MetRS-AIMP3–GluProRS–AIMP2 core of the complex (27,29). These proteins, together with AspRS, and possibly LeuRS and IleRS (30), form a distinct sub-complex denoted as sub-complex I (27). Sub-complex II consists of AIMP1, GlnRS, ArgRS, a dimer of LysRS, and AIMP2 (which is shared by both sub-complexes).
Figure 1.
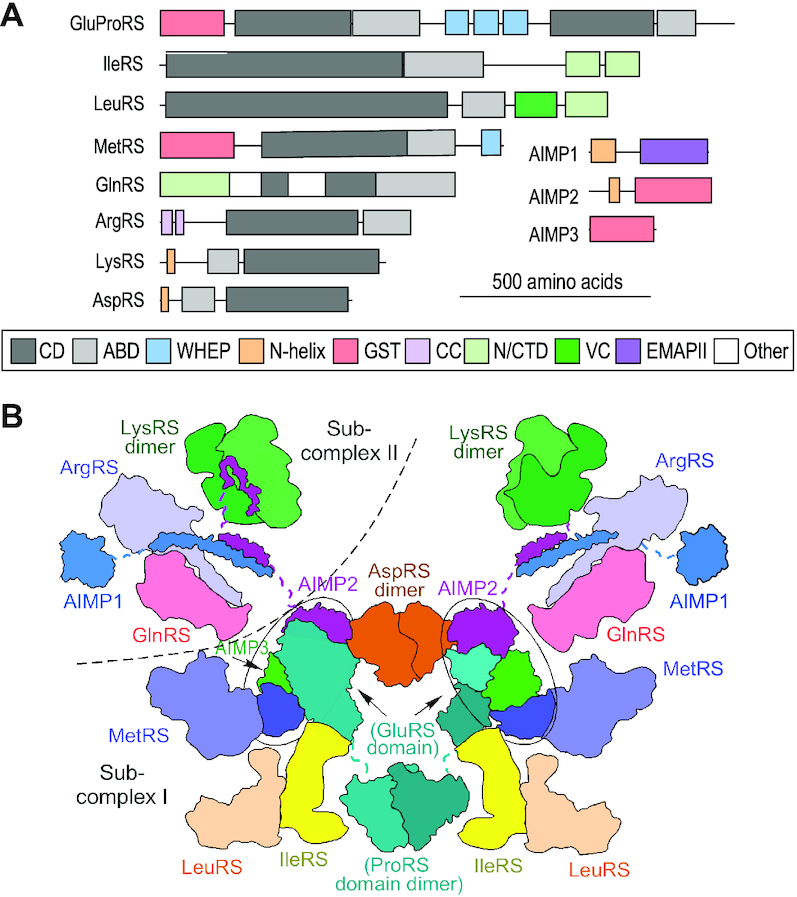
Constituents and proposed architecture of mammalian MSC. (A) Schematic of protein domain arrangements of the nine AARSs and three AIMPs in the MSC. The highly conserved catalytic (dark gray) and anticodon (light gray) binding domains are highlighted. Likewise, the vertebrate-specific appended domains, including GST-like domains (pink), WHEP domains (cyan), lysine-rich N-helical domains (orange), among others, are indicated in box at bottom. Constituents and domains are drawn to scale (scale indicated). (B) Proposed bisymmetrical model of the MSC. Each symmetrical side is a monomeric complex consisting of one copy of each of the eleven proteins with the exception of LysRS, which is present as a dimer. Each unit is sub-divided into sub-complexes I and II (dashed curve). Hubs containing four GST-like domains are highlighted in ovals.
We propose to interrogate MSC structure using XL-MS (31–34). The recent introduction of MS-cleavable cross-linkers has greatly enhanced structural analysis of protein-protein interactions in complex, multi-component systems. MS-mediated cleavage of cross-linkers by collisional activation separates cross-linked peptides into two linear peptides with diagnostic signatures, and thus gives a high confidence of cross-link assignment (34). The method reports specific amino acid linkages in proximate peptides within (intra-protein) and between (inter-protein) constituents. The most commonly used MS-cleavable cross-linker, DSSO, has a 10-Å spacer between the reactive groups, and primarily cross-links pairs of Lys residues at a distance up to ∼27 Å, although limited reactivity towards hydroxyl-bearing amino acids has been reported (35,36). XL-MS tolerates sample heterogeneity, providing information on protein interactions and conformations of recombinant proteins in solution, in crude cell lysates, and even in live cells. Based on its complexity, including proteins with unstructured and possibly dynamic domains, XL-MS is an ideal platform for structural analysis of the MSC.
MATERIALS AND METHODS
Protein cross-linking for XL-MS
Human HEK293T cells (ATCC) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 1% penicillin–streptomycin solution. Cells (30 × 106 cells) were pelleted, washed twice with ice-cold PBS, and lysed in buffer containing 100 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, supplemented with protease inhibitor cocktail (ThermoFisher). Following incubation in an end-to-end rotator for 40 min at 4°C, debris was cleared by centrifugation at 15 000 RPM for 20 min at 4°C. The lysate was pre-cleared with protein A/G magnetic beads (Pierce) pre-blocked with buffer containing 100 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X-100 and 0.02% bovine serum albumin for 1 h. For pull-downs, lysates were incubated with rabbit polyclonal anti-human GluProRS linker (Leu753 to Thr956; BioSynthesis, TX, USA) (37) or IgG control antibody, followed by incubation with protein A/G magnetic beads overnight in an end-to-end rotator at 4°C. The antibody was selected for pull-down because of its very high affinity and specificity for GluProRS. Moreover, under these cellular conditions, gel filtration of lysates shows all GluProRS co-elutes with other MSC constituents, e.g., AIMP1 and AIMP2, in high-molecular weight fractions consistent with the MSC (Supplementary Figure S1). The pull-down preceded cross-linker addition to minimize linkage with non-interacting proteins in the protein-rich cytoplasmic environment. The beads were collected with a magnet (ThermoFisher), and washed four times with ice-cold 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X-100. The beads were further washed twice with cross-linking buffer consisting of 20 mM HEPES, pH 7.8 and 150 mM NaCl, and re-suspended in the same buffer. The MSC complex was cross-linked on the beads by incubation with 1 mM of the MS-cleavable cross-linker disuccinimidyl sulfoxide (DSSO, ThermoFisher) dissolved in DMSO for 45 min at room temperature, and the reaction quenched by addition of 20 mM Tris–HCl (pH 7.8) for 30 min. For in situ, live cell cross-linking, 30 × 106 HEK293T cells were pelleted at 1000 rpm and suspended in 2 ml of hypotonic buffer consisting of 10 mM HEPES, pH 8.0, 10 mM NaCl, 1 mM MgCl2, 0.5 mM DTT, and protease inhibitor cocktail. Suspended cells were incubated with DSSO (3 mM) for 60 min at 4°C, and the reaction quenched with 25 mM Tris–HCl, pH 7.8. The cells were lysed and subjected to pull-down as above.
Sample preparation and analysis of proximate proteins by XL-MS
Immunoprecipitated material was subjected to on-bead tryptic digestion (38). Briefly, trypsin (10 μl, 10 ng/μl) in 100 mM ammonium bicarbonate was added to washed beads, and samples vortexed for 15 s every 2–3 min for 15 min at room temperature. Digestion was continued overnight at 37°C. A second 10-μl aliquot of protease was added for 4 h at 37°C. The supernatant was collected on a magnetic rack and diluted with formic acid (5% v/v, final concentration). The digests were cleaned using PepClean C-18 spin columns (ThermoFisher) according to manufacturer's instructions, the samples dried in a vacuum concentrator, and reconstituted in 1% acetic acid.
Samples were analyzed by LC–MS using an Orbitrap Fusion Lumos Tribrid MS and XlinkX software (ThermoScientific). The instrument was equipped with a Dionex Ultimate 3000 nano UHPLC system, and a Dionex (25 cm × 75 μm id) Acclaim Pepmap C18, 2-μm, 100-Å reversed-phase capillary chromatography column. Peptide digests (5 μl, ∼1–2 μg protein) were injected and eluted with an acetonitrile/0.1% formic acid gradient at a flow rate of 0.3 μl/min. Experiments were analyzed using three standard LC–MS/MS methods (31,39). The first method took advantage of data-dependent acquisition utilizing MS1 scans to identify peptide molecular weights, and collision-induced dissociation (CID)-based MS2 scans to identify peptide sequences. Two XL-MS methods were applied. The first XL-MS approach is a CID-MS2-MS3 method that utilized an initial MS1 scan which identifies 375–1500 Da ions, and measures their mass-to-charge (m/z) ratios, permitting determination of peptide molecular weights and charge states. To filter out the majority of non-cross-linked moieties, peptides with charge state ≥4 were isolated and subjected to MS2 fragmentation using low-energy (25%) CID with high-resolution detection in the Orbitrap (30 000 resolution). MS2 fragmentation dissociated the DSSO-cross-linked peptides to yield ions with a diagnostic mass difference of 31.9721 Da. These fragments were selected for further fragmentation by an MS3 experiment in the ion trap that determined the sequence of both peptides, including identification of cross-linked Lys residues. The second XL-MS method, CID-MS2-electron-transfer/higher-energy collision dissociation (EThcD)-MS2 utilized MS1 to identify peptide molecular weights and charge state selecting only those with charge state ≥4 for MS2 analysis in the Orbitrap. The CID MS2 experiments identified the diagnostic 31.9721 Da mass difference, and the EThcD experiment determined peptide sequence, including cross-linked Lys residues.
Data and error analysis
For the data-dependent XL-MS analysis, collected data were queried against human SwissProtKB databases (Uniprot.org) using Sequest bundled into Proteome Discoverer 2.2 (ThermoScientific) (34). Parameters included methionine oxidation and DSSO modification of lysine, protein mass tolerance of 10 ppm, and fragment ion mass tolerance of 0.6 Da. Protein and peptide validation was performed using Scaffold 4.9.0 (40) with application of a 1% false discovery rate (FDR) to identify high-confidence proteins (41,42). XL-MS data were analyzed using the XLinkX 2.2 node from Proteome Discoverer 2.2 (34). Parameters were as above, specifically selecting DSSO as cross-linker and three missed cleavages. This data was searched against the human SwissProtKB database and a database generated from proteins identified in the data-dependent analysis. Validation of cross-linked peptides was done using Percolator (Matrix Science) with a FDR set at 1% (43,44). A consensus workflow was also applied for the statistical arrangement; de-isotope and TopX filters were used to determine m/z error with selectivity set to ∼10% FDR. XL-MS spectra designated as high confidence were inspected for the following criteria. For the CID–MS2–MS3 experiment, selection of MS3 spectra required three of the four possible fragments contain sequence-specific y and b ions consistent with the amino acid composition of the peptide. For the CID–MS2–EThcD–MS2 method multiple sequence-specific ions needed to be present from each peptide involved in cross-links.
Modeling and visualization of the MSC
Most constituents used to assemble the MSC model have published crystal structures for truncated or incomplete human proteins. Full-length or near-full length models were constructed by appending the crystal structures with three-dimensional models of missing domains obtained by homology modeling using SWISS-MODEL (45,46). For some proteins, crystal structures were available only from non-human species, and these were subjected to homology modeling using SWISS-MODEL. The MSC was step-wise assembled by docking constituents to partially assembled complexes, subject to distance constraints corresponding to Lys–Lys intermolecular crosslinks, using SWISS-MODEL and the protein-protein docking program PatchDock (47) (see Supporting Information for details). Models of MSC constituents with intra-molecular cross-links (Supplemental Figure S2) and assembled complexes were visualized with PyMOL (48).
RESULTS
Summary of inter- and intra-protein cross-links
Lysates of human HEK293T cells were subjected to pull-down with anti-GluProRS or IgG control antibody, and then cross-linked with 1 mM DSSO, an MS-cleavable cross-linker with an amine-reactive ester at each end of a seven-carbon spacer. Bead-bound protein was subjected to cleavage and peptide detection by an Orbitrap Fusion Tribrid MS3 instrument. Cross-linked peptides were analyzed by Thermo XlinkX software, and the data was queried against the human UniProtKB database. The experiment was repeated three times with cross-linker added on-bead, and once in which DSSO was incubated directly with cells, and the results combined. We identified 19 unique inter-protein cross-links pairwise connecting all 11 MSC constituents (Figure 2A, Supplementary Table S1), and 118 unique intra-protein cross-links (Figure 2B, Supplementary Table S2). The latter can provide insights into the spatial relationship of well-defined structural domains connected by dynamic or disordered spacers characteristic of appended domains of nearly all human MSC constituents. All cross-links were between pairs of Lys residues, with the single exception of a heterofunctional cross-link between AIMP2 Tyr35 and LysRS Lys249. The results were validated by determination of cross-link distances in proteins and multi-protein complexes with known crystal structure. For example, intra-protein cross-links in human LysRS, AspRS and GluRS (human structure is modeled based on sequence homology with GluRS from Methanothermobacter thermautotrophicus) were generally consistent with the reported ∼27-Å limit (Figure 3A–C). Likewise, the inter-protein cross-link between the N-terminus of AIMP2 and LysRS satisfied the distance constraint (Figure 3D). Summarizing all intra-protein cross-links found in reported structures, ∼80% are smaller than 30 Å; larger cross-links in ProRS and GlnRS are informative exceptions and discussed below (Figure 3E).
Figure 2.
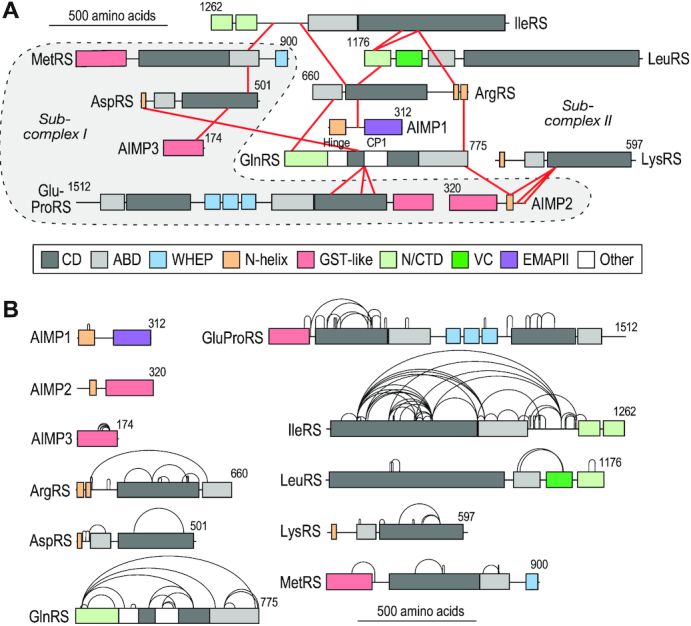
XL-MS-derived cross-links in the MSC. (A) Linkage map generated by xiView depicting inter-protein cross-links determined by XL-MS analysis of HEK293T cells (49). Subcomplex I constituents are highlighted (gray regions surrounded by dashed outline). (B) Linkage map showing intra-protein cross-links obtained by XL-MS.
Figure 3.
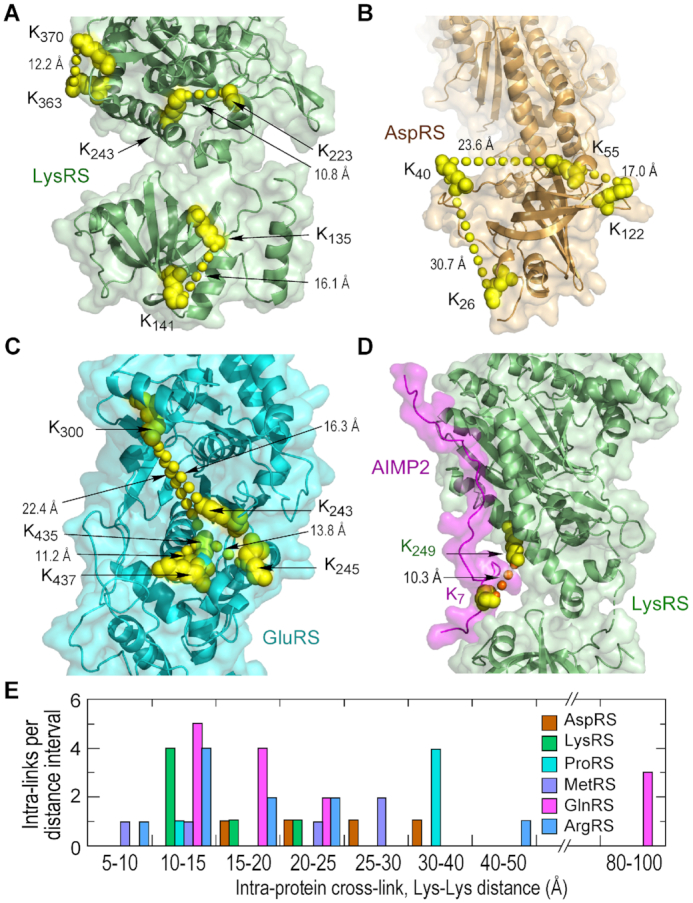
Validation of XL-MS-derived cross-links. (A, B) XL-MS-derived cross-links shown in X-ray structure of (A) LysRS (PDB ID: 6ILD) and (B) AspRS (PDB ID: 4J15). Cross-linked Lys residues are shown as atom-level structures (yellow), and intra-protein cross-links shown as connecting line of yellow spheres. (C) The structure of human GluRS was modeled based on homology to the archaebacterium Methanothermobacter thermautotrophicus GluRS (PDB ID: 3AII). (D) XL-MS-derived crosslinks between LysRS and AIMP2 are shown in the X-ray structure (PDB ID: 6ILD). Inter-protein cross-links are indicated by connecting line of orange spheres. (E) Number of cross-links as a function of cross-link distance within the reported structures.
XL-MS-derived structural model of MSC sub-complex I
Previous X-ray crystallography data established a core of four GST-like domains that was not challenged by our results, and was used as a starting point in our model development (27,29). Our data showed AspRS cross-linked to two core constituents, namely, AIMP3 and MetRS – an unexpected result since previous biochemical and structural studies showed AspRS binding AIMP2 (Figure 1B) (18,27). However, a recent report showed a 58-amino acid degradation fragment of AspRS (AspRS336–393) resides in a crevice formed by AIMP2 and AIMP3 in the GST-like core (29). The cross-link observed by XL-MS, AspRS Lys374 to AIMP3 Lys138, was within this AspRS fragment, confirming this structure in the context of the MSC (Figure 4A). A model of full-length human MetRS was generated from separate crystallographic structures of human MetRS GST-like and catalytic domains, joined by a 14-aa linker modeled as an α-helix (the derived structure of MetRS and all other MSC constituents, with intra-protein cross-links, are displayed in Supplemental Figure S2). Full-length MetRS was situated to satisfy the AspRS Lys451 to MetRS Lys729 cross-link (Figure 4B). The human GluRS catalytic domain of GluProRS was modeled on the crystal structure of Methanothermobacter thermautotrophicus (50) and joined to the human GST-like domain based on XL-MS-derived intra-protein cross-links (Figure 4C). The GluRS catalytic domain was included in the model to complete the pentameric core (Figure 4D). Unfortunately, cross-links between ProRS and GluRS, or with any other MSC constituent, were not observed, thus preventing placement of ProRS, or the linker that joins it, to GluRS.
Figure 4.

Assembly of the sub-complex I pentamer. (A) Schematic of inter-protein cross-links between MetRS, AspRS and AIMP3 (right). Reported crystal structure (PDB ID: 5Y6L REF) of the four GST-like domains with the AspRS peptide (orange). The crystal structure (PDB ID: 4J15) of human AspRS (brown) was used to model and dock full-length AspRS on the GST tetramer. (B) The MetRS catalytic domain (PDB ID: 5GL7) was docked on the pentameric structure using inter-protein distance constraints given by XL-MS. (C) Schematic of intra-protein cross-links in GluRS with cross-links between GST-like and catalytic domains highlighted (red, right). Structure of human catalytic domain determined by homology modeling with archael GluRS (PDB ID: 3AII). (D) Ribbon (left) and space-filled (right) structures of the pentameric complex comprising sub-complex I.
XL-MS-based structural model of MSC sub-complex II
The XL-MS-derived cross-links between MSC constituents outside of sub-complex I were investigated to elucidate their spatial relationships in the MSC. A high-resolution crystal structure of full-length human GlnRS has been reported (51). A 35-aa spacer in the hinge region connecting the N-terminal domain (NTD, Met1-Glu182) to the catalytic domain is disordered, consistent with a flexible or dynamic structure (51). XL-MS revealed three intra-protein cross-links between these domains (Figure 5A, right). According to the crystal structure, the distances corresponding to these cross-links are >80 Å, and thus well beyond the ∼27-Å limit of DSSO (Figure 5A, left). The NTD can be repositioned to give cross-links between 14 and 31 Å, while not inducing spatial conflict (Figure 5A, center). In the altered position, the NTD abuts the catalytic and anti-codon binding domains, and supports the reported regulation of tRNA binding and catalytic activity by the NTD, there by eliminating the need to hypothesize long-distance communication between domains (51).
Figure 5.

Application of XL-MS-derived cross-links to amend GlnRS monomer and GlnRS-ArgRS dimer structures. (A) Schematic (right) of XL-MS-derived intra-links in GlnRS; cross-links between N-terminal domain (NTD) and catalytic domain are highlighted (red). Crystal structure (PDB ID: 4YE6) of the NTD (red) and catalytic domain (pink) of GlnRS (left). Position of K586 is estimated from A584, the nearest neighbor in the crystal structure. Amended model of GlnRS obtained by conformance with XL-MS-derived intra-protein distance constraints (middle). (B) Schematic of inter-links between GlnRS and ArgRS obtained by XL-MS (right, bottom). Crystal structure (PDB ID: 4R3Z) of ArgRS-GlnRS-AIMP1 trimer (left). Structure of ArgRS complexed with GlnRS containing the N-terminus (PDB ID: 4YE6) (middle). Improved model of ArgRS-GlnRS satisfying ArgRS intra-protein and ArgRS-GlnRS inter-protein cross-links (right, top).
Our finding of cross-links between GlnRS and ArgRS confirms the interaction observed in the crystal structure of ArgRS-GlnRS-AIMP1 (17). A major feature of the triad is a split α-helical motif at the ArgRS N-terminus featuring limited interactions with GlnRS and AIMP1 (Figure 5B, left). However, the ∼40-Å lengths of the XL-MS-derived intra- and inter-protein cross-links suggest the crystal structure does not reflect the conformation within the MSC. This possibility is supported by inclusion of the GlnRS NTD in the structure, which introduces an additional 70.8 Å cross-link between ArgRS Lys557 and GlnRS Lys25 (Figure 5B, center). To accommodate the structure of GlnRS with the re-positioned NTD, and satisfy the observed cross-links, the split N-terminal α-helix of ArgRS was adjusted to form a compact helix-turn-helix structure, and the position of ArgRS relative to GlnRS shifted (Figure 5B, right). The adjacency of GlnRS and LysRS is determined by a cross-link of both proteins with Lys64 in the N-helical domain of AIMP2 (Figure 6A). The relative spatial positions of IleRS and LeuRS in the MSC have been elusive, but an association with GluProRS and MetRS in sub-complex I has been proposed (Figure 1B) (52). In contrast, the XL-MS data shows cross-links joining a triad of IleRS, LeuRS, and ArgRS in sub-complex II (Figure 6B).
Figure 6.
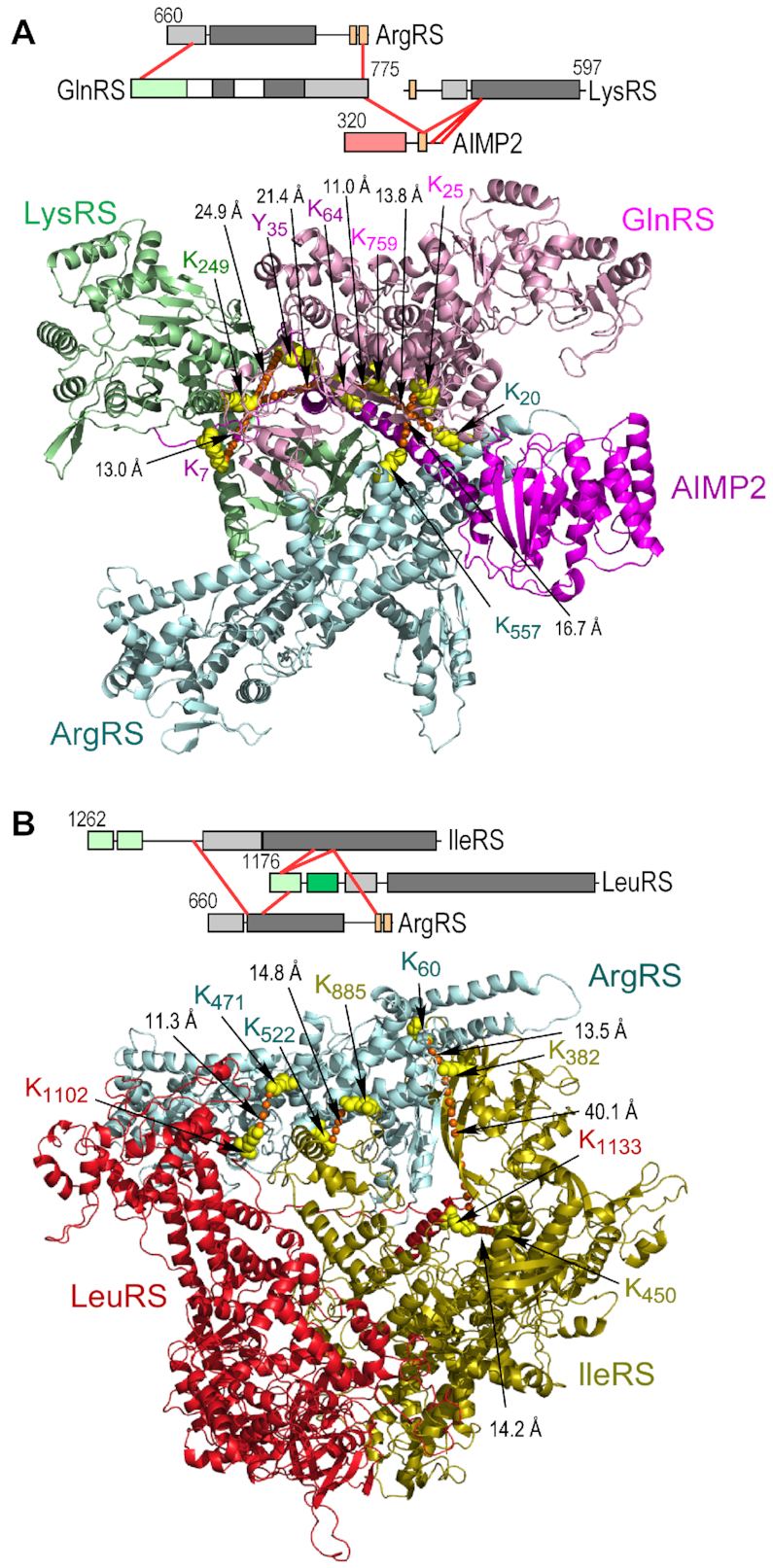
Modeling of protein-protein interactions within sub-complex II. (A) Schematic of inter-protein cross-links between ArgRS, GlnRS, LysRS and AIMP2 (top). Ribbon model of tetrameric complex satisfying distance constraints of all inter-protein cross-links (bottom). (B) Schematic of inter-protein cross-links between ArgRS, IleRS and LeuRS (top). Ribbon model of trimeric ArgRS–IleRS–LeuRS complex satisfying distance constraints of all inter-protein cross-links (bottom).
Interactions of the major MSC sub-complexes
The constituents and structures joining the major sub-complexes of the MSC are unclear; however, AIMP2 binding to GluProRS in sub-complex I and to AIMP1 and LysRS in subcomplex II are likely to contribute (Figure 1B) (53,54). The XL-MS data reveal inter-protein cross-links between five pairs of constituents connecting the two sub-complexes, namely, IleRS–MetRS, AspRS–GlnRS, GlnRS–GluProRS, GlnRS–AIMP2 and AIMP2–LysRS (Figure 7, top, right). GlnRS appears to be central to the joining of the sub-complexes as cross-links with three proteins in sub-complex I are observed, all ≤30 Å (Figure 7, bottom, left and outsets). The model reveals integration of the sub-complexes despite a clear segregation of the constituents, with the exception of AIMP2 that resides in both sub-complexes in accordance with previous models.
Figure 7.

Connections between sub-complexes I and II. Schematic of inter-protein cross-links between five protein pairs joining sub-complexes I and II (top, right). Structural model comprising LysRS, GlnRS, GluRS, MetRS, AspRS, IleRS and AIMP2 satisfying XL-MS-derived inter-protein distance constraints (bottom, left). Red dashed line delineates sub-complexes (SC) I and II; AIMP2 spans both sub-complexes. Expanded outsets show details of GlnRS–GluRS (bottom, right) and AIMP2–GlnRS–LysRS interactions (top, left).
Evidence for dimers of MSC constituents
Biochemical and crystallographic data suggest several MSC constituents are present as dimers including AspRS, LysRS and the ProRS domain of GluProRS (55–61). However, the dimerization requirement for these AARSs for activity might be evaded in the MSC by alternative interactions with neighboring AARSs. Consistent with this idea, molecular modeling shows that AspRS monomer and GlnRS, interacting AARSs in our model, can form a sterically compatible pair (62). Intra-protein cross-links can provide evidence for the presence of homodimers, particularly in anti-parallel structures. For example, an ‘intra-protein’ cross-link between amino acids very distant from each other in the monomeric structure, e.g., >40 Å, but near each other in the dimer, would provide evidence for a homodimer arranged in a head-to-tail configuration. XL-MS data for LysRS shows six distinct intra-protein cross-links ranging from 10.7 to 20.9 Å if both are within the same monomer, and from 50.0 to 101.2 Å if the residues span the dimeric structure (Table 1). The preponderance of cross-links consistent only with the monomeric form does not provide compelling evidence for either structure since the region comprising the dimer interface is relatively small compared to the total volume. Thus, a preponderance of cross-links consistent with a monomer is likely to be observed even in a dimer. For example, a manual count showed only 16 potential Lys–Lys cross-links less than 27 Å spanning the LysRS dimer interface, compared to ∼125 potential intra-protein cross-links within each of the monomers. Thus, assuming equivalent accessibility to the cross-linker, there is a probability of ∼0.06 (16/266) that a given cross-link will provide evidence for a dimeric form. Notably, the vast majority of nearby Lys pairs in LysRS were not observed to be cross-linked, possibly due to spatial obstruction of the cross-linker, weak reactivity of primary amine groups, or inefficient MS detection of cross-linked peptides. Of the four intra-protein cross-links in AspRS, none are suggestive of a dimeric structure. Five intra-protein cross-links were detected in ProRS; the monomeric cross-link distances in three range from 30.9 to 38.7 Å, whereas the same cross-links in dimeric structures range from 16.1 to 23.8 Å, suggestive of a dimeric structure. However, the lengths in the monomer are not much greater than the distance limit of the cross-linker, and might be consistent with an altered conformation of PRS in the MSC compared to the crystal structure. It is noteworthy that a cross-link between identical residues is independent evidence for a homodimer (63); however, such cross-links were not observed in any MSC component.
Table 1.
Summary of Lys–Lys cross-link distances in crystal structures of AARS dimers
| Cross-link | Cross-link length in dimer denoted A–B (Å) | |||
|---|---|---|---|---|
| Both aa's in A | Both aa's in B | aa1 in A; aa2 in B | aa1 in B; aa2 in A | |
| AspRS (PDB ID: 4J15 (64)) | ||||
| K26–K40 | 32.1 | 30.7 | 80.8 | 76.2 |
| K40–K55 | 23.6 | 24.4 | 53.3 | 53.8 |
| K55–K122 | 17.0 | 16.8 | 60.8 | 61.3 |
| K241–K451 | 28.4 | 27.3 | 27.3 | 22.3 |
| LysRS (PDB ID: 6ILD (59)) | ||||
| K135–K141 | 16.1 | 17.6 | 101.2 | 100.0 |
| K223–K243 | 10.8 | 10.7 | 50.1 | 50.0 |
| K305–K479 | 20.9 | 20.7 | 56.2 | 55.9 |
| K363–K370 | 12.2 | 12.7 | 50.0 | 49.7 |
| K402–K479 | 14.0 | 12.4 | 77.6 | 77.6 |
| K407–K479 | 12.9 | 11.1 | 82.4 | 81.2 |
| ProRS (PDB ID: 4HVC (65)) | ||||
| K1089–K1109 | 36.6 | 30.9 | 22.6 | 18.4 |
| K1091–K1109 | 38.7 | 35.3 | 19.5 | 16.1 |
| K1109–K1156 | 34.9 | 36.9 | 18.5 | 23.8 |
| K1143–K1156 | 31.7 | 31.8 | 42.6 | 42.3 |
| K1156–K1213 | 11.6 | 11.5 | 51.8 | 53.6 |
Structural model of the human MSC
A holo-MSC model was constructed satisfying inter- and intra-protein cross-link lengths, and without spatial interference of any domains (Figure 8A, B). All constituents and domains are included except for the ProRS domain of GluProRS and the linker connecting it to the GluRS domain, as no informative XL-MS data were obtained. The overall maximum dimensions are about 198 × 170 × 150 Å. The asymmetric structure is relatively compact and globular, but studded with surface crevices, possibly to facilitate interaction with substrate tRNAs. Our findings reveal the first atomistic, 3D model of the human MSC.
Figure 8.

Three-dimensional models of human holo-MSC. Ribbon (A) and space-filled (B) models of front (left), side (middle) and rear (right) views of the MSC. (C) Space-filled model showing front (left) and back (right) views of the MSC with anticodon binding domains highlighted.
DISCUSSION
Several conclusions can be inferred from our structural model of the MSC. This compactness of the model is due in part to inter-protein crosslinks connecting all MSC constituents. Utilization of the high density of intra-protein cross-links permitted improved modeling of appended domains generally attached to catalytic domains by disordered spacers. For example, the GST-like domain of GluRS, the N-terminus of GlnRS, and the N-helix domain of ArgRS, all were found to be closely associated with the catalytic domain, further contributing to the compactness of the overall structure. In several cases, the structure derived from XL-MS was at variance with the crystal structure, most likely due to the difference in experimental conditions; XL-MS interrogates conformation in situ within the intact MSC, an environment very different from the densely packed condition established during crystallization of purified components. The model also shows tight integration of sub-complexes I and II that can be more accurately considered as interacting hemispheres rather than discrete mini-complexes. Importantly, no cross-links between MSC constituents and the other eleven AARSs were observed in our XL-MS experiments. This finding not only supports cross-link specificity, but also rejects the possibility that the MSC contains all twenty AARSs, but some are reproducibly lost during isolation. Our structural model does not include the ProRS domain of GluProRS, nor the linker joining the catalytic domains, since the XL-MS experiments did not reveal inter-protein cross-links with these domains, nor intra-protein cross-links between the catalytic domains. The absence of cross-links between ProRS with other MSC constituents was not likely due to artifactual interference by the anti-GluProRS linker antibody used in immunoprecipitation since mono-links, i.e. mono-functional reactions of DSSO with single Lys residues, were detected in the linker and ProRS domains (not shown). These results suggest that ProRS might be loosely tethered to GluRS, and to the rest of the MSC, and thus an exception to the observed compactness of the complex. Alternatively, ProRS might be tightly associated with the bulk mass of the complex, but there are not any proximal Lys-Lys pairs available for cross-linking.
The size and stoichiometry of the MSC have not been established with certainty. Our model is comparable in compactness and volume to the MSC defined by early, low-resolution negative stain and cryo-EM images (12) (Figure 9A, B). In contrast, small-angle X-ray scattering (SAXS) suggested a highly elongated structure (16) (Figure 9C). The discrepancy between the low-resolution structures has not been resolved, but might be related to technical issues, for example, solid- versus solution-phase image acquisition. Also, differences in purification protocols can result in altered composition, including tRNA content, and proteolysis (66). Not requiring highly purified material, the XL-MS approach has the advantage of a one-step affinity purification procedure that is less susceptible to artifactual modification during sample preparation. According to a recent structural model, the MSC is a supramolecular complex consisting of two identical units rotated 180° around an axis through AspRS and ProRS dimers, and joined by these dimers (Figure 1B) (27). Each unit is proposed to contain a single copy of each of the 11 constituents, except for LysRS present as a dimer in each. Evidence for the overall shape shown is limited, but possibly derives from early, low-resolution, negative-stain electron microscopy, suggested to reveal a ‘cup’ or ‘U-shape’ structure (13). However, substantial evidence does not support this model. Importantly, the calculated molecular mass of the complex in Figure 1 is ∼2.0 MDa, whereas mass determinations by sucrose gradient centrifugation or gel filtration, done in multiple independent laboratories, consistently report 1.0–1.2 MDa (14,22,26,67,68). The calculated molecular size of the XL-MS-derived MSC with monomeric constituents is ∼930 kDa, increasing to ∼1.2 MDa upon inclusion of the second monomer of the three proposed dimeric constituents, AspRS, LysRS and GluProRS (56,57,69,70). These calculated sizes are consistent with the reported mass determinations, even taking into account the presence of about four ∼25 kDa tRNAs per MSC particle (14). Thus, we suggest the holo-MSC is a unitary structure consisting primarily of constituent monomers, with the exception of limited homodimers.
Figure 9.
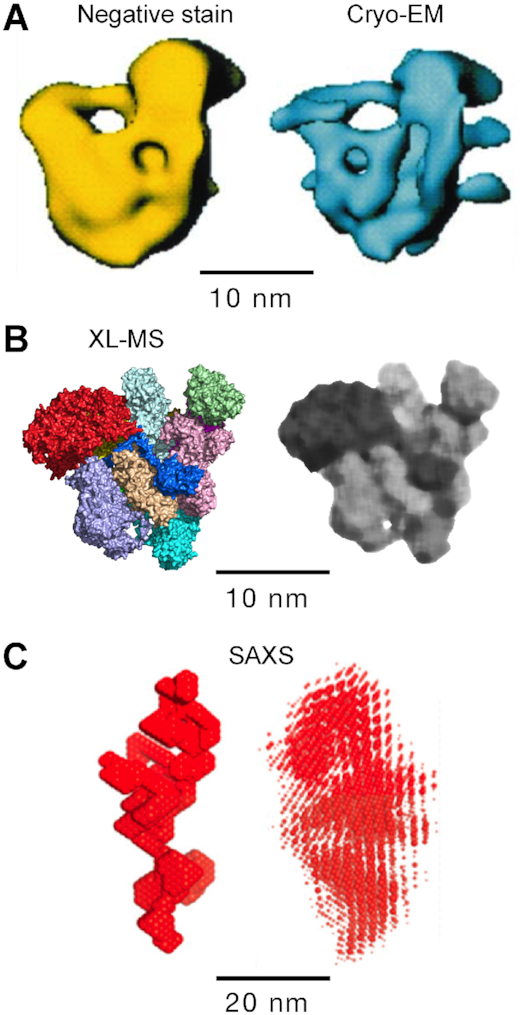
Volumetric comparison of MSC structures. Structures derived from (A) negative stain (left) and cryo-EM (right), (B) XL-MS, before (left) and after (right) application of median low-pass filter (Canvas Draw 4, median filter setting = 5) and (C) low-resolution SAXS bead model (left) and superposition of SAXS bead models (right).
The XL-MS data did not provide evidence for AspRS and LysRS dimers, and only weakly supported a ProRS dimer. Nonetheless, taking advantage of the known crystal structures of the dimers, we have expanded our model to include them (Supplementary Figure S3). The LysRS B chain was inserted into the model without spatial interference with other constituents, but there was limited spatial overlap of AspRS B chain with ArgRS. Inclusion of GluProRS dimer required construction of a monomer that was hampered by the absence of structural information on the relationship between the GluRS, linker, and ProRS domains. The structure of the linker joining the catalytic domains has not been resolved and contains unstructured spacers joining three well-structured, helix-turn-helix WHEP domains. The GluProRS dimer was arbitrarily positioned without spatial constraints. The lack of interaction of the ProRS domain with the main MSC structure is consistent with a report showing specific proteolytic release of ProRS from the MSC (71).
The function of the MSC in vertebrate cells remains uncertain, but interaction with ribosomes has been described, suggesting a ‘channeling’ mechanism in which charged tRNAs are released from MSC AARSs to elongation factor 1α (EF1α) for delivery to the ribosome A-site. This mechanism could improve translation efficiency by recycling tRNA without diffusive release into the cytoplasmic pool (8,72–75). The roughly equimolar stoichiometry of MSC and ribosomes, about 107 copies per cell, provides circumstantial evidence for a role of the MSC/ribosome interaction in translation (74). Several AARSs not in the MSC, most notably ValRS and PheRS, directly bind the EF1α subunit of elongation factor complex, EF1 (76,77). However, evidence for binding of MSC constituents with EF1α is limited (78) and contrasts with reports that fail to show co-purification of the MSC with elongation factors (79). Likewise, our XL-MS experiments did not reveal cross-linking, or even enrichment, of EF1α with the MSC. These data are consistent with a processive pathway in which a tRNA cycles from an MSC AARS to EF1α, then to the ribosome, and back to the MSC, without requiring a high affinity interaction between the AARS and EF1α (80). Intriguingly, AIMP3 (also known as EEF1E1 or eukaryotic translation elongation factor 1 ϵ1) has sequence homology with the β and γ subunits of EF1 required for recycling EF1α-GDP to EF1α-GTP (79). Conceivably, a partnership of AIMP3 and EF1α represents a common mechanism of charged tRNA delivery to elongating ribosomes shared by all MSC AARSs (81,82). Our structural model reveals a topological asymmetry that potentially sheds light on this proposed mechanism. A ‘side-view’ reveals a relatively flat surface opposing a bulged surface (Figure 8A, B, center panel). Interestingly, the anti-codon binding domains of nearly all AARSs are localized on the flat surface, with mechanistic implications (Figure 8C). This asymmetric distribution might suggest that all synthetases are positioned for optimal delivery of charged tRNAs to the ribosome A-site via EF1α, and possibly AIMP3. Alternatively, these sites might face away from the ribosome to facilitate binding of uncharged tRNAs.
At least 9 of the 11 MSC constituents exhibit non-canonical functions implicated in pathology. Phosphorylation of GluProRS contributes to inflammation, aging, and adiposity (83,84). LysRS phosphorylation induces mast cell activation (85) and priming of human immunodeficiency virus type 1 (86). MetRS phosphorylation induces MSC release of AIMP3 that can contribute to tumor suppression, or in excess induces accelerated aging in transgenic mice (87,88). LeuRS acts a leucine sensor that regulates mTORC1 activity (89,90). Finally, mutations in five genes encoding MSC-resident proteins – RARS, DARS, EPRS, AIMP1 and AIMP2 – cause forms of child-onset hypomyelinating leukodystrophy (91–96). In several cases, the constituent escapes from the MSC, e.g. GluProRS and LysRS are released following stimulus-dependent phosphorylation, and a fragment of AIMP1 is released by proteolysis during apoptosis (83,84,97,98). Our structural model can form a basis for understanding the dynamics of the MSC and its constituents. MSC release of GluProRS and LysRS does not adversely influence cell viability, but their effects on MSC structure have not been investigated in depth. Although AspRS has been proposed to undergo stimulus-dependent release from the MSC (18), its central position suggests release might profoundly influence MSC structure. Consistent with previous electron microscopy showing a central location of AIMP1 (99), AIMP1 in our model forms a central belt-like structure interacting with multiple AARSs suggesting that release of intact AIMP1 might be detrimental to MSC structure. Almost no information is available concerning assembly of the MSC, and its potential regulation. Assembly factors, such as those required for ribosome assembly, might likewise be essential for ordered assembly of the MSC. Overall, our data provides the first comprehensive structural model of the mammalian MSC that will provide a foundation for understanding the canonical role of the MSC in translation, the noncanonical functions of its constituents, and potential contributions of the MSC to pathology.
DATA AVAILABILITY
We deposited the PDB file for the model of the multi-tRNA synthetase complex in the publicly accessible database ModelArchive.
Supplementary Material
ACKNOWLEDGEMENTS
Computational resources were provided by the Ohio Supercomputer Center and the Extreme Science and Engineering Discovery Environment of the National Center for Supercomputing Applications, University of Illinois. Permission to reproduce Figure 9C was obtained, Copyright 2002 Published by Elsevier Science B.V. on behalf of the Federation of European Biochemical Societies.
Contributor Information
Krishnendu Khan, Department of Cardiovascular and Metabolic Sciences, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH 44195, USA.
Camelia Baleanu-Gogonea, Department of Chemistry, Cleveland State University, Cleveland, OH 44115, USA.
Belinda Willard, Lerner Research Institute Proteomics and Metabolomics Core, Cleveland Clinic Foundation, Cleveland, OH 44195, USA.
Valentin Gogonea, Department of Chemistry, Cleveland State University, Cleveland, OH 44115, USA.
Paul L Fox, Department of Cardiovascular and Metabolic Sciences, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH 44195, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [P01 HL029582, P01 HL076491 to P.L.F.]; National Heart, Lung, and Blood Institute [R01 HL128300, R01 HL128268 (sub-contracts to V.G.)]; National Institute of General Medical Sciences [R01 GM086430 to P.L.F.]; National Institute of Diabetes and Digestive and Kidney Diseases [R01 DK124203, R01 DK123236 to P.L.F.]; Research Accelerator Program Grant from the Lerner Research Institute, Cleveland Clinic (to P.L.F.); the Fusion Lumos instrument was purchased via National Institutes of Health shared instrument grant [S10 OD023436 to B.W.]. Funding for open access charge: NIH [P01 HL029582].
Conflict of interest statement. None declared.
REFERENCES
- 1. Guo M., Schimmel P.. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013; 9:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yao P., Fox P.L.. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013; 5:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim S., You S., Hwang D.. Aminoacyl-tRNA synthetases and tumorigenesis: more than housekeeping. Nat. Rev. Cancer. 2011; 11:708–718. [DOI] [PubMed] [Google Scholar]
- 4. Guo M., Yang X.L., Schimmel P.. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010; 11:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wei N., Zhang Q., Yang X.L.. Neurodegenerative Charcot-Marie-Tooth disease as a case study to decipher novel functions of aminoacyl-tRNA synthetases. J. Biol. Chem. 2019; 294:5321–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ognjenovic J., Simonovic M.. Human aminoacyl-tRNA synthetases in diseases of the nervous system. RNA Biol. 2018; 15:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kwon N.H., Fox P.L., Kim S.. Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discov. 2019; 18:629–650. [DOI] [PubMed] [Google Scholar]
- 8. Kaminska M., Havrylenko S., Decottignies P., Le Marechal P., Negrutskii B., Mirande M.. Dynamic organization of aminoacyl-tRNA synthetase complexes in the cytoplasm of human cells. J. Biol. Chem. 2009; 284:13746–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee S.W., Cho B.H., Park S.G., Kim S.. Aminoacyl-tRNA synthetase complexes: beyond translation. J. Cell Sci. 2004; 117:3725–3734. [DOI] [PubMed] [Google Scholar]
- 10. Eswarappa S.M., Fox P.L.. Citric acid cycle and the origin of MARS. Trends Biochem. Sci. 2013; 38:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bandyopadhyay A.K., Deutscher M.P.. Complex of aminoacyl-transfer RNA synthetases. J. Mol. Biol. 1971; 60:113–122. [DOI] [PubMed] [Google Scholar]
- 12. Norcum M.T., Boisset N.. Three-dimensional architecture of the eukaryotic multisynthetase complex determined from negatively stained and cryoelectron micrographs. FEBS Lett. 2002; 512:298–302. [DOI] [PubMed] [Google Scholar]
- 13. Norcum M.T. Isolation and electron microscopic characterization of the high molecular mass aminoacyl-tRNA synthetase complex from murine erythroleukemia cells. J. Biol. Chem. 1989; 264:15043–15051. [PubMed] [Google Scholar]
- 14. Wolfe C.L., Warrington J.A., Treadwell L., Norcum M.T.. A three-dimensional working model of the multienzyme complex of aminoacyl-tRNA synthetases based on electron microscopic placements of tRNA and proteins. J. Biol. Chem. 2005; 280:38870–38878. [DOI] [PubMed] [Google Scholar]
- 15. Kerjan P., Cerini C., Sémériva M., Mirande M.. The multienzyme complex containing nine aminoacyl-tRNA synthetases is ubiquitous from Drosophila to mammals. Biochim. Biophys. Acta. 1994; 1199:293–297. [DOI] [PubMed] [Google Scholar]
- 16. Dias J., Renault L., Perez J., Mirande M.. Small-angle X-ray solution scattering study of the multi-aminoacyl-tRNA synthetase complex reveals an elongated and multi-armed particle. J. Biol. Chem. 2013; 288:23979–23989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fu Y., Kim Y., Jin K.S., Kim H.S., Kim J.H., Wang D., Park M., Jo C.H., Kwon N.H., Kim D. et al.. Structure of the ArgRS-GlnRS-AIMP1 complex and its implications for mammalian translation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15084–15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hahn H., Park S.H., Kim H.J., Kim S., Han B.W.. The DRS-AIMP2-EPRS subcomplex acts as a pivot in the multi-tRNA synthetase complex. IUCrJ. 2019; 6:958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ofir-Birin Y., Fang P., Bennett S.P., Zhang H.M., Wang J., Rachmin I., Shapiro R., Song J., Dagan A., Pozo J. et al.. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell. 2013; 49:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo M., Ignatov M., Musier-Forsyth K., Schimmel P., Yang X.L.. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J.Y., Kang Y.S., Lee J.W., Kim H.J., Ahn Y.H., Park H., Ko Y.G., Kim S.. p38 is essential for the assembly and stability of macromolecular tRNA synthetase complex: implications for its physiological significance. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7912–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson D.L., Yang D.C.. Stoichiometry and composition of an aminoacyl-tRNA synthetase complex from rat liver. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:4059–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang X.L. Structural disorder in expanding the functionome of aminoacyl-tRNA synthetases. Chem. Biol. 2013; 20:1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo M., Yang X.L.. Architecture and metamorphosis. Top. Curr. Chem. 2014; 344:89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Dang C., Yang D.C.. Disassembly and gross structure of particulate aminoacyl-tRNA synthetases from rat liver. Isolation and the structural relationship of synthetase complexes. J. Biol. Chem. 1979; 254:5350–5356. [PubMed] [Google Scholar]
- 26. Mirande M., Cirakoglu B., Waller J.P.. Seven mammalian aminoacyl-tRNA synthetases associated within the same complex are functionally independent. Eur. J. Biochem. 1983; 131:163–170. [DOI] [PubMed] [Google Scholar]
- 27. Cho H.Y., Maeng S.J., Cho H.J., Choi Y.S., Chung J.M., Lee S., Kim H.K., Kim J.H., Eom C.Y., Kim Y.G. et al.. Assembly of multi-tRNA synthetase complex via heterotetrameric glutathione transferase-homology domains. J. Biol. Chem. 2015; 290:29313–29328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hyeon D.Y., Kim J.H., Ahn T.J., Cho Y., Hwang D., Kim S.. Evolution of the multi-tRNA synthetase complex and its role in cancer. J. Biol. Chem. 2019; 294:5340–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho H.Y., Lee H.J., Choi Y.S., Kim D.K., Jin K.S., Kim S., Kang B.S.. Symmetric assembly of a decameric subcomplex in human multi-tRNA synthetase complex via interactions between glutathione transferase-homology domains and aspartyl-tRNA synthetase. J. Mol. Biol. 2019; 431:4475–4496. [DOI] [PubMed] [Google Scholar]
- 30. Rho S.B., Lee J.S., Jeong E.J., Kim K.S., Kim Y.G., Kim S.. A multifunctional repeated motif is present in human bifunctional tRNA synthetase. J. Biol. Chem. 1998; 273:11267–11273. [DOI] [PubMed] [Google Scholar]
- 31. Yu C., Huang L.. Cross-linking mass spectrometry: An emerging technology for interactomics and structural biology. Anal. Chem. 2018; 90:144–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O’Reilly F.J., Rappsilber J.. Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat. Struct. Mol. Biol. 2018; 25:1000–1008. [DOI] [PubMed] [Google Scholar]
- 33. Iacobucci C., Gotze M., Ihling C.H., Piotrowski C., Arlt C., Schafer M., Hage C., Schmidt R., Sinz A.. A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein-protein interactions. Nat. Protoc. 2018; 13:2864–2889. [DOI] [PubMed] [Google Scholar]
- 34. Liu F., Rijkers D.T., Post H., Heck A.J.. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods. 2015; 12:1179–1184. [DOI] [PubMed] [Google Scholar]
- 35. Sinz A. Divide and conquer: cleavable cross-linkers to study protein conformation and protein-protein interactions. Anal. Bioanal. Chem. 2017; 409:33–44. [DOI] [PubMed] [Google Scholar]
- 36. Iacobucci C., Piotrowski C., Aebersold R., Amaral B.C., Andrews P., Bernfur K., Borchers C., Brodie N.I., Bruce J.E., Cao Y. et al.. First community-wide, comparative cross-linking mass spectrometry study. Anal. Chem. 2019; 91:6953–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jia J., Arif A., Ray P.S., Fox P.L.. WHEP domains direct noncanonical function of glutamyl-prolyl tRNA synthetase in translational control of gene expression. Mol. Cell. 2008; 29:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohammed H., Taylor C., Brown G.D., Papachristou E.K., Carroll J.S., D'Santos C.S.. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat. Protoc. 2016; 11:316–326. [DOI] [PubMed] [Google Scholar]
- 39. Liu F., Lossl P., Scheltema R., Viner R., Heck A.J.R.. Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 2017; 8:15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Searle B.C. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010; 10:1265–1269. [DOI] [PubMed] [Google Scholar]
- 41. Holding A.N. XL-MS: Protein cross-linking coupled with mass spectrometry. Methods. 2015; 89:54–63. [DOI] [PubMed] [Google Scholar]
- 42. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Forster F., Beck M., Aebersold R.. False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods. 2012; 9:901–903. [DOI] [PubMed] [Google Scholar]
- 43. Kall L., Canterbury J.D., Weston J., Noble W.S., MacCoss M.J.. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007; 4:923–925. [DOI] [PubMed] [Google Scholar]
- 44. The M., MacCoss M.J., Noble W.S., Kall L.. Fast and accurate protein false discovery rates on large-scale proteomics data sets with Percolator 3.0. J. Am. Soc. Mass Spectrom. 2016; 27:1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schwede T., Kopp J., Guex N., Peitsch M.C.. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003; 31:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. et al.. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J.. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleiic Acids Res. 2005; 33:W363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DeLano W.L. DeLano Scientific. 2003; Palo Alto, CA. [Google Scholar]
- 49. Graham M.J., Combe C., Kolbowski L., Rappsilber J.. xiView: a common platform for the downstream analysis of crosslinking mass spectrometry data. 2019; bioRxiv doi:26 February 2019, preprint: not peer reviewed 10.1101/561829. [DOI]
- 50. Moen S.O., Edwards T.E., Dranow D.M., Clifton M.C., Sankaran B., Van Voorhis W.C., Sharma A., Manoil C., Staker B.L., Myler P.J. et al.. Ligand co-crystallization of aminoacyl-tRNA synthetases from infectious disease organisms. Sci. Rep. 2017; 7:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ognjenovic J., Wu J., Matthies D., Baxa U., Subramaniam S., Ling J., Simonovic M.. The crystal structure of human GlnRS provides basis for the development of neurological disorders. Nucleic Acids Res. 2016; 44:3420–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaminska M., Havrylenko S., Decottignies P., Gillet S., Le Marechal P., Negrutskii B., Mirande M.. Dissection of the structural organization of the aminoacyl-tRNA synthetase complex. J. Biol. Chem. 2009; 284:6053–6060. [DOI] [PubMed] [Google Scholar]
- 53. Robinson J.C., Kerjan P., Mirande M.. Macromolecular assemblage of aminoacyl-tRNA synthetases: quantitative analysis of protein-protein interactions and mechanism of complex assembly. J. Mol. Biol. 2000; 304:983–994. [DOI] [PubMed] [Google Scholar]
- 54. Quevillon S., Robinson J.C., Berthonneau E., Siatecka M., Mirande M.. Macromolecular assemblage of aminoacyl-tRNA synthetases: identification of protein-protein interactions and characterization of a core protein. J. Mol. Biol. 1999; 285:183–195. [DOI] [PubMed] [Google Scholar]
- 55. Merritt E.A., Arakaki T.L., Larson E.T., Kelley A., Mueller N., Napuli A.J., Zhang L., Deditta G., Luft J., Verlinde C.L. et al.. Crystal structure of the aspartyl-tRNA synthetase from Entamoeba histolytica. Mol. Biochem. Parasitol. 2010; 169:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Agou F., Waller J.P., Mirande M.. Expression of rat aspartyl-tRNA synthetase in Saccharomyces cerevisiae. Role of the NH2-terminal polypeptide extension on enzyme activity and stability. J. Biol. Chem. 1996; 271:29295–29303. [DOI] [PubMed] [Google Scholar]
- 57. Eriani G., Cavarelli J., Martin F., Dirheimer G., Moras D., Gangloff J.. Role of dimerization in yeast aspartyl-tRNA synthetase and importance of the class II invariant proline. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:10816–10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fang P., Zhang H.M., Shapiro R., Marshall A.G., Schimmel P., Yang X.L., Guo M.. Structural context for mobilization of a human tRNA synthetase from its cytoplasmic complex. Proc. Natl Acad. Sci. U.S.A. 2011; 108:8239–8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hei Z., Wu S., Liu Z., Wang J., Fang P.. Retractile lysyl-tRNA synthetase-AIMP2 assembly in the human multi-aminoacyl-tRNA synthetase complex. J. Biol. Chem. 2019; 294:4775–4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yaremchuk A., Cusack S., Tukalo M.. Crystal structure of a eukaryote/archaeon-like protyl-tRNA synthetase and its complex with tRNAPro(CGG). EMBO J. 2000; 19:4745–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Larson E.T., Kim J.E., Napuli A.J., Verlinde C.L., Fan E., Zucker F.H., Van Voorhis W.C., Buckner F.S., Hol W.G., Merritt E.A.. Structure of the prolyl-tRNA synthetase from the eukaryotic pathogen Giardia lamblia. Acta Crystallogr. D. Biol. Crystallogr. 2012; 68:1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ribas de Pouplana L., Schimmel P.. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001; 104:191–193. [DOI] [PubMed] [Google Scholar]
- 63. Haupt C., Hofmann T., Wittig S., Kostmann S., Politis A., Schmidt C.. Combining chemical cross-linking and mass spectrometry of intact protein complexes to study the architecture of multi-subunit protein assemblies. J. Vis. Exp. 2017; 28:56747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim K.R., Park S.H., Kim H.S., Rhee K.H., Kim B.G., Kim D.G., Park M.S., Kim H.J., Kim S., Han B.W.. Crystal structure of human cytosolic aspartyl-tRNA synthetase, a component of multi-tRNA synthetase complex. Proteins. 2013; 81:1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhou H., Sun L., Yang X.L., Schimmel P.. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2013; 494:121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mirande M. The aminoacyl-tRNA synthetase complex. Subcell. Biochem. 2017; 83:505–522. [DOI] [PubMed] [Google Scholar]
- 67. Dang C.V., Ferguson B., Burke D.J., Garcia V., Yang D.C.. Interactions of aminoacyl-tRNA synthetases in high-molecular-weight multienzyme complexes from rat liver. Biochim. Biophys. Acta. 1985; 829:319–326. [DOI] [PubMed] [Google Scholar]
- 68. Godar D.E., Garcia V., Jacobo A., Aebi U., Yang D.C.. Structural organization of the multienzyme complex of mammalian aminoacyl-tRNA synthetases. Biochemistry. 1988; 27:6921–6928. [DOI] [PubMed] [Google Scholar]
- 69. Dewan V., Wei M., Kleiman L., Musier-Forsyth K.. Dual role for motif 1 residues of human lysyl-tRNA synthetase in dimerization and packaging into HIV-1. J. Biol. Chem. 2012; 287:41955–41962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kerjan P., Triconnet M., Waller J.P.. Mammalian prolyl-tRNA synthetase corresponds to the ∼ 150 kDa subunit of the high-Mr aminoacyl-tRNA synthetase complex. Biochimie. 1992; 74:195–205. [DOI] [PubMed] [Google Scholar]
- 71. Shalak V., Kaminska M., Mitnacht-Kraus R., Vandenabeele P., Clauss M., Mirande M.. The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J. Biol. Chem. 2001; 276:23769–23776. [DOI] [PubMed] [Google Scholar]
- 72. Barbarese E., Koppel D.E., Deutscher M.P., Smith C.L., Ainger K., Morgan F., Carson J.H.. Protein translation components are colocalized in granules in oligodendrocytes. J. Cell Sci. 1995; 108:2781–2790. [DOI] [PubMed] [Google Scholar]
- 73. Negrutskii B.S., Deutscher M.P.. Channeling of aminoacyl-tRNA for protein synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:4991–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. David A., Netzer N., Strader M.B., Das S.R., Chen C.Y., Gibbs J., Pierre P., Bennink J.R., Yewdell J.W.. RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J. Biol. Chem. 2011; 286:20688–20700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Netzer N., Goodenbour J.M., David A., Dittmar K.A., Jones R.B., Schneider J.R., Boone D., Eves E.M., Rosner M.R., Gibbs J.S. et al.. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009; 462:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Motorin Y.A., Wolfson A.D., Orlovsky A.F., Gladilin K.L.. Mammalian valyl-tRNA synthetase forms a complex with the first elongation factor. FEBS Lett. 1988; 238:262–264. [DOI] [PubMed] [Google Scholar]
- 77. Petrushenko Z.M., Budkevich T.V., Shalak V.F., Negrutskii B.S., El'skaya A.V.. Novel complexes of mammalian translation elongation factor eEF1A.GDP with uncharged tRNA and aminoacyl-tRNA synthetase. Implications for tRNA channeling. Eur. J. Biochem. 2002; 269:4811–4818. [DOI] [PubMed] [Google Scholar]
- 78. Sarisky V., Yang D.C.. Co-purification of the aminoacyl-tRNA synthetase complex with the elongation factor eEF1. Biochem. Biophys. Res. Commun. 1991; 177:757–763. [DOI] [PubMed] [Google Scholar]
- 79. Quevillon S., Mirande M.. The p18 component of the multisynthetase complex shares a protein motif with the beta and gamma subunits of eukaryotic elongation factor 1. FEBS Lett. 1996; 395:63–67. [DOI] [PubMed] [Google Scholar]
- 80. Mirande M. Processivity of translation in the eukaryote cell: role of aminoacyl-tRNA synthetases. FEBS Lett. 2010; 584:443–447. [DOI] [PubMed] [Google Scholar]
- 81. Yang X.L., Otero F.J., Ewalt K.L., Liu J., Swairjo M.A., Kohrer C., RajBhandary U.L., Skene R.J., McRee D.E., Schimmel P.. Two conformations of a crystalline human tRNA synthetase-tRNA complex: implications for protein synthesis. EMBO J. 2006; 25:2919–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kang T., Kwon N.H., Lee J.Y., Park M.C., Kang E., Kim H.H., Kang T.J., Kim S.. AIMP3/p18 controls translational initiation by mediating the delivery of charged initiator tRNA to initiation complex. J. Mol. Biol. 2012; 423:475–481. [DOI] [PubMed] [Google Scholar]
- 83. Arif A., Terenzi F., Potdar A.A., Jia J., Sacks J., China A., Halawani D., Vasu K., Li X., Brown J.M. et al.. EPRS is a critical mTORC1-S6K1 effector that influences adiposity in mice. Nature. 2017; 542:357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sampath P., Mazumder B., Seshadri V., Gerber C.A., Chavatte L., Kinter M., Ting S. M., Dignam J.D., Kim S., Driscoll D.M., Fox P.L.. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004; 119:195–208. [DOI] [PubMed] [Google Scholar]
- 85. Lee Y.N., Nechushtan H., Figov N., Razin E.. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FceRI-activated mast cells. Immunity. 2004; 20:145–151. [DOI] [PubMed] [Google Scholar]
- 86. Stark L.A., Hay R.T.. Human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) interacts with Lys-tRNA synthetase: implications for priming of HIV-1 reverse transcription. J. Virol. 1998; 72:3037–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kwon N.H., Kang T., Lee J.Y., Kim H.H., Kim H.R., Hong J., Oh Y.S., Han J.M., Ku M.J., Lee S.Y. et al.. Dual role of methionyl-tRNA synthetase in the regulation of translation and tumor suppressor activity of aminoacyl-tRNA synthetase-interacting multifunctional protein-3. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:19635–19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Oh Y.S., Kim D.G., Kim G., Choi E.C., Kennedy B.K., Suh Y., Park B.J., Kim S.. Downregulation of lamin A by tumor suppressor AIMP3/p18 leads to a progeroid phenotype in mice. Aging Cell. 2010; 9:810–822. [DOI] [PubMed] [Google Scholar]
- 89. Han J.M., Jeong S.J., Park M.C., Kim G., Kwon N.H., Kim H.K., Ha S.H., Ryu S.H., Kim S.. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012; 149:410–424. [DOI] [PubMed] [Google Scholar]
- 90. Yoon M.S., Son K., Arauz E., Han J.M., Kim S., Chen J.. Leucyl-tRNA synthetase activates Vps34 in amino acid-sensing mTORC1 signaling. Cell Rep. 2016; 16:1510–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nafisinia M., Sobreira N., Riley L., Gold W., Uhlenberg B., Weiss C., Boehm C., Prelog K., Ouvrier R., Christodoulou J.. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus-Merzbacher disease. Eur. J. Hum. Genet. 2017; 25:1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Taft R.J., Vanderver A., Leventer R.J., Damiani S.A., Simons C., Grimmond S.M., Miller D., Schmidt J., Lockhart P.J., Pope K. et al.. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 2013; 92:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mendes M.I., GutierrezSalazar M., Guerrero K., Thiffault I., Salomons G.S., Gauquelin L., Tran L.T., Forget D., Gauthier M.S., Waisfisz Q. et al.. Bi-allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2018; 102:676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Feinstein M., Markus B., Noyman I., Shalev H., Flusser H., Shelef I., Liani-Leibson K., Shorer Z., Cohen I., Khateeb S. et al.. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010; 87:820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wolf N.I., Salomons G.S., Rodenburg R.J., Pouwels P.J., Schieving J.H., Derks T.G., Fock J.M., Rump P., van Beek D.M., van der Knaap M.S. et al.. Mutations in RARS cause hypomyelination. Ann. Neurol. 2014; 76:134–139. [DOI] [PubMed] [Google Scholar]
- 96. Shukla A., Das Bhowmik A., Hebbar M., Rajagopal K.V., Girisha K.M., Gupta N., Dalal A.. Homozygosity for a nonsense variant in AIMP2 is associated with a progressive neurodevelopmental disorder with microcephaly, seizures, and spastic quadriparesis. J. Hum. Genet. 2018; 63:19–25. [DOI] [PubMed] [Google Scholar]
- 97. Yannay-Cohen N., Carmi-Levy I., Kay G., Yang C.M., Han J.M., Kemeny D.M., Kim S., Nechushtan H., Razin E.. LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol. Cell. 2009; 34:603–611. [DOI] [PubMed] [Google Scholar]
- 98. Shalak V., Guigou L., Kaminska M., Wautier M.P., Wautier J.L., Mirande M.. Characterization of p43(ARF), a derivative of the p43 component of multiaminoacyl-tRNA synthetase complex released during apoptosis. J. Biol. Chem. 2007; 282:10935–10943. [DOI] [PubMed] [Google Scholar]
- 99. Norcum M.T., Warrington J.A.. The cytokine portion of p43 occupies a central position within the eukaryotic multisynthetase complex. J. Biol. Chem. 2000; 275:17921–17924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We deposited the PDB file for the model of the multi-tRNA synthetase complex in the publicly accessible database ModelArchive.