

Abstract
End-stage renal disease (ESRD) is described by four primary diagnoses, diabetes, hypertension, glomerulonephritis, and cystic kidney disease, all of which have viruses implicated as causative agents. Enteroviruses, such as coxsackievirus (CV), are a common genus of viruses that have been implicated in both diabetes and cystic kidney disease; however, little is known about how CVs cause kidney injury and ESRD or predispose individuals with a genetic susceptibility to type 1 diabetes (T1D) to kidney injury. This study evaluated kidney injury resulting from coxsackievirus B4 (CVB4) inoculation of non-obese diabetic (NOD) mice to glean a better understanding of how viral exposure may predispose individuals with a genetic susceptibility to T1D to kidney injury. The objectives were to assess acute and chronic kidney damage in CVB4-inoculated NOD mice without diabetes. Results indicated the presence of CVB4 RNA in the kidney for at least 14 days post-CVB4 inoculation and a coordinated pattern recognition receptor response, but the absence of an immune response or cytotoxicity. CVB4-inoculated NOD mice also had a higher propensity to develop an increase in mesangial area 17 weeks post-CVB4 inoculation. These studies identified initial gene expression changes in the kidney resulting from CVB4 exposure that may predispose to ESRD. Thus, this study provides an initial characterization of kidney injury resulting from CVB4 inoculation of mice that are genetically susceptible to developing T1D that may one day provide better therapeutic options and predictive measures for patients who are at risk for developing kidney disease from T1D.
Keywords: coxsackievirus, NOD, TLR3, kidney
Introduction
End-stage renal disease (ESRD) is the ninth leading cause of death in the United States and can result from four primary diagnoses: diabetes, hypertension, glomerulonephritis, and cystic kidney disease (38). While viruses are thought to cause kidney diseases (10,21,28,35,40), little is known about the role viruses play in their onset and progression. Coxsackievirus (CV) is one virus that has been identified in clinical reports to cause both diabetes and acute kidney injury (AKI) (4,27,48); however, the impact of CV on the kidney and its contribution to ESRD in each of these diagnoses (diabetes and AKI) are yet unknown.
Coxsackievirus B4 (CVB4), a common childhood virus causing hand, foot, and mouth disease, was first associated with AKI in a 1973 case report of a 9-year-old boy with acute glomerulonephritis, proteinuria, and high blood urea nitrogen (8). Since then, other cases of CVB infections resulting in AKI and sometimes death have been reported (27); however, it remains elusive why some individuals lack kidney injury from exposure to CV, while others experience severe kidney damage.
Some insight into how viruses lead to kidney injury has come from studies using enteroviruses. For example, it was found that CVs have the capacity to infect both human and mouse kidneys and cause injury through direct and indirect mechanisms (10,20) [reviewed in Pasch and Frey (27)]. Direct mechanisms include viral-mediated cell lysis, cellular repair, cell death, or activation/suppression of other gene or signaling pathways in response to the virus in the cell (7,13). Indirect mechanisms include immune cell-mediated injury by the bystander effect or immunoglobulin deposition from normal kidney filtration, which elicits a kidney injury response (16,20,22,47).
While multiple studies identified pathological changes resulting from CV infection (20,22,47), Conaldi et al. first characterized some of the functional changes in vitro (10). They demonstrated that infection of human cells with CVB1–6 reduced the contractile and phagocytic properties of mesangial cells. Furthermore, CVB1, 3, 4, and 5 infection resulted in a persistent mesangial cell infection, while glomerular epithelial cells (podocytes) and proximal tubular epithelial cells quickly died. While Conaldi's studies provided promising data on what could be expected with CVB infection and some functional changes, their studies do not provide details on how whole kidney function is affected by the presence of virus in the kidney, the role of the immune system, or why individuals have differing responses.
Some insight may be gleaned by considering the initiating factors of ESRD, such as diabetes. While both type 1 diabetes (T1D) and type 2 diabetes (T2D) can be triggered by genetic and environmental factors, obesity is the primary trigger of T2D, whereas environmental factors such as viruses can trigger T1D in genetically susceptible individuals (3,5,6,12,15,18,19,23,30,33,34,41,46). We hypothesize that environmental factors (i.e., viruses) may also trigger kidney injury in genetically susceptible individuals. For these reasons, we first sought to describe the kidney phenotype in response to CVB4 inoculation before the development of T1D in mice that are prone to develop T1D, in an effort to identify a mechanism for predisposal to future kidney disease.
This study evaluated the effects of a one-time CVB4 inoculation on the kidneys of non-obese diabetic (NOD) mice, a strain of mice that are genetically vulnerable to CVB4-induced acceleration of T1D (12,26,33,34), before onset of T1D, to define the viral-mediated component of kidney injury in the context of T1D susceptibility. Characterizing the short- and long-term effects of CVB4 on the kidneys of NOD mice may contribute to a better understanding of how otherwise “innocent” viruses such as CVs may have a more important impact on the development of chronic kidney disease (CKD) and ESRD than previously thought, as well as provide insight into why some individuals develop CV-induced kidney damage while others do not in the face of other diseases that lead to kidney disease, such as T1D.
Methods
Ethics statement
All experiments were performed in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines and standards set by federal, state, and local authorities and approved by the Ohio University Institutional Animal Care and Use Committee (IACUC) under protocol numbers 16-H-008, 13-H-043, and 12-H-001. Ohio University's animal care program is maintained in accordance with the Public Health Service (PHS) policy and meets the standards for care and housing set by the “Guide for the Care and Use of Animals” published by the National Research Council. The program also maintains accordance with all regulations of the United States Department of Agriculture. The animal care program is fully accredited by the AAALAC, International.
Mice
Female Tlr3+/+ (9,12,25,26,33,34,36) and Tlr3−/− (1,43) NOD/ShiLtJ mice were used for this study due to their susceptibility to CVB4-accelerated T1D (12,26,33,37). Tlr3+/+ and Tlr3−/− NOD mice received either intraperitoneal inoculation of 500,000 plaque forming units of unpurified CVB4 Edwards strain (GenBank: S76772.1) (46) or sterile phosphate-buffered saline (PBS) as a stress (noninoculated) control at 8 weeks of age. Mice were randomly selected for euthanasia at 0, 2, 3, 5, 7, 9, 10, and 14 days postinoculation (n = 4–8). Time points were based on the natural progression of known viral titers in renal cells (10) and pancreas (26). Additional Tlr3+/+ NOD mice were inoculated with either CVB4 or PBS control at 10 weeks of age and euthanized at 3, 7, 12, and 17 weeks after CVB4 inoculation (n = 6–14).
A target sample size of six was used for all time points of this study; however, variability in the number of mice that either developed T1D (and were thus removed from the study) or died from virus inoculation is represented by the variation in mouse numbers at each time point. Moreover, based on previous studies using NOD mice, sample sizes ranging from 3 to 13 were deemed acceptable (25,32,44). Mice were housed under sterile/pathogen-free conditions in a temperature-controlled (18–22°C) vivarium with a 14/10-h light/dark cycle and fed standard laboratory chow ad libitum for the course of the study (#D12450B; Research Diets).
Body weight and blood glucose were measured once a week for the course of the study. Tail vein blood glucose was measured via a handheld glucometer (FreeStyle Freedom Lite Blood Glucose Monitoring System). All mice with a nonfasting blood glucose level ≥250 mg/dL, confirmed by measurement of glycosuria on Diastix (#2806; Bayer) before euthanasia, were excluded from this study. After euthanasia, both kidneys were collected, the capsule was removed by gently rubbing with a Kimwipe (#06-666; Kimberly-Clark), and the left kidney weight was recorded. Left kidney poles were removed, flash frozen with the whole right kidney in liquid nitrogen, and stored at −80°C. The remainder of the left kidney was prepared for histology as described below.
Histological analysis
Kidney tissue was fixed in 10% neutral buffered formalin (#SF98-20; Fisher) for 24 h and embedded in paraffin (#EG1160; Leica Biosystems). Formalin-fixed paraffin-embedded (FFPE) kidney sections (4 μm) were stained with periodic acid-Schiff (PAS). Images of five glomeruli per sample were obtained at 400 × magnification on a Nikon Eclipse 80i microscope (Nikon) using Image Pro software (Media Cybernetics). Individual glomeruli were isolated from images and total area measured using a freehand selection tool using ImageJ 1.48v software (31). Total PAS-positive area was measured as previously described (31).
In situ hybridization
FFPE kidney sections (4 μm) were probed for (+) CVB4 RNA (#481341; Advanced Cell Diagnostics). In situ hybridization (ISH) was performed using the RNAscope Intro Pack 2.5 HD Reagent Kit Brown-Mm (#322371; Advanced Cell Diagnostics) according to the manufacturer's protocol with 10-min peroxidase, 8-min retrieval, and 15-min protease treatment. Slides were evaluated for (+) CVB4 RNA location and presence/absence over time.
Immunohistochemistry/fluorescence
FFPE tissue sections (4 μm) from noninoculated day 3 and CVB4-inoculated days 3, 7, 10, and 14 were subjected to heat-mediated antigen retrieval at high pH (proliferating cell nuclear antigen [PCNA], cCAS3) or proteinase K (#ab64220; Abcam) (TLR3). Fluorescent sections were blocked 1 min at room temperature with TrueBlack lipofuscin autofluorescence quencher (#32007; Biotium) diluted to 1 × in 70% ethanol. Sections were blocked with 10% goat serum (#ab138478; Abcam) and 1% bovine serum albumin (BSA, (#A7906; Sigma-Aldrich) in PBS 30 min at room temperature.
Primary antibodies to cCAS3 (#9661; Cell Signaling Technology), PCNA (#sc-56; Santa Cruz), and TLR3 (#ab59918; Abcam) were diluted to 1.3, 4, and 5 μg/mL, respectively, with 1% BSA in PBS and incubated overnight at 4°C in a humidified chamber. Secondary antibodies were diluted in 1% BSA in PBS for 1 h at room temperature. Fluorescent slides were counterstained with 1:1,000 4′,6-diamidino-2-phenylindole (DAPI, #62248; Thermo-Fisher) diluted in PBS for 5 min at room temperature and mounted with ProLong Gold Antifade (#P36934; Thermo-Fisher). The 3,3′ diaminobenzidine (DAB) substrate (#D3939; Sigma-Aldrich) was incubated for 3 min on nonfluorescent slides, dehydrated in reverse order as hydration, and mounted with Permount (#SP15; Thermo-Fisher).
To evaluate proliferation in kidney sections, three images per sample were taken at 200 × magnification. PCNA-positive cells were counted using ImageJ cell counter and expressed as a percent relative to total cells (counted as DAPI positive) (31). Apoptosis was evaluated by visually scanning each kidney section and counting the total cCAS3-positive cells. Representative fluorescent images were taken at 600 × magnification on a Nikon A1R confocal microscope (Nikon, Tokyo, Japan). Since no TLR3 was detected, no quantification was performed.
Western blot analysis
Total protein was isolated from the cortex of flash frozen kidney using the Tissue Protein Extraction Reagent (TPER, #78510; Fisher) plus protease/phosphatase inhibitors (#05892791001, #04906837001; Sigma) and Bullet Blender homogenizer with 0.5 mm ZrO2 beads (Next Advance). Proteins were immunoprecipitated (IP) with either phosphoserine or phosphothreonine (#sc-81514, #sc-5267; Santa Cruz) antibodies.
One-third of IP protein samples (200 μg starting) and 20 μg total protein from the same samples were separated by SDS-PAGE on 4–12% Bis-Tris polyacrylamide gels (#NP0323; Fisher) and transferred to nitrocellulose membranes (#LC2009; Fisher). Membranes were blocked with Odyssey blocking buffer (#927-40000; LI-COR) and immunoblotted for PKR (1:1,000, #sc-708; Santa Cruz), IRF1, IRF3, IRF5 (1:1,000, #8478, 4302, 4950; Cell Signaling Technology), and IRF7 (1:1,000, #ab109255; Abcam) followed by anti-mouse or rabbit secondary antibodies (1:15,000, #926-32211 and #827-08364; LI-COR). Blots were imaged on an Odyssey Infrared Imager (LI-COR). Image Studio v5.2 software was used to quantify the signal intensity of each protein band (LI-COR).
Normalization was performed by dividing the signal intensity of experimental bands by the signal intensity of the loading control, either ACTB or TUBA1A. IP quantification was evaluated by dividing the signal intensity of the phosphorylated band by the loading control (ACTB or TUBA1A) or the total protein (phosphorylated plus nonphosphorylated form) corresponding to the amount of total protein lysate.
Reverse transcription real-time polymerase chain reaction
RNA was isolated from the cortex of flash frozen whole kidney using RNA STAT-60 (#CS-110; Tel Test) and Bullet Blender homogenizer with 0.5mm ZrO2 beads (Next Advance). DNase treatment and RNA cleanup were performed using the RNA Clean and Concentrate kit following the manufacturer's protocol (#R1016; Zymo Research). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription kit (#4368814; Life Technologies). Reverse transcription real-time polymerase chain reaction (RT-qPCR) was performed using iTaq Universal SYBR green Supermix (#172-5122; BioRad) and a StepOne Plus Real-Time PCR System (Applied Biosystems) or CFX384 (BioRad). All SYBR green primers and TaqMan primers are listed in Table 1. Results were calculated using the relative quantity equation for multiple housekeeping genes: relative gene expression = [(EGOI)ΔCt GOI]/{GeoMean[(EREF)ΔCt REF]} (2,17,39) and normalized to the geometric mean of glyceraldehyde 3-phsophate dehydrogenase (Gapdh) and gamma-actin (Actg1) based on a previous control gene analysis (11).
Table 1.
Primer Names, Sequences, and References
| Name | Full name | Strand | Sequence 5′-3′ | Reference |
|---|---|---|---|---|
| CVB4 | Coxsackievirus B4 | + | CCCACAGGACGCTCTAATA | (45) |
| − | CAGAGTTACCCGTTACGACA | |||
| Nfkb1 (p50) | Nuclear factor kappa B1 subunit p50 | + | CGGCAACTCACAGACAGAGA | (45) |
| − | ACGATTTTCAGGTTGGATGC | |||
| Rig1 | Retinoic acid inducible gene 1 | + | AAGAGCCAGAGTGTCAGAATCT | (45) |
| − | AGCTCCAGTTGGTAATTTCTTGG | |||
| Mda5 | Melanoma differentiation- associated protein 5 | + | AGACACAAGTTTGGCAGAAGG | (45) |
| − | GGCCACTTCCATTTGGTAAGG | |||
| Tlr3 | Toll-like receptor 3 | + | AATCCTTGCGTTGCGAAGTG | (45) |
| − | GGTTCAGTTGGGCGTTGTTC | |||
| Ifnb1 | Interferon-beta | + | ATAAGCAGCTCCAGCTCCAA | (45) |
| − | CTGTCTGCTGGTGGAGTTCA | |||
| Irf1 | Interferon regulatory factor 1 | + | ATGCCAATCACTCGAATGCG | (45) |
| − | TTGTATCGGCCTGTGTGAATG | |||
| Irf3 | Interferon regulatory factor 3 | + | GAGAGCCGAACGAGGTTCAG | (45) |
| − | CTTCCAGGTTGACACGTCCG | |||
| Irf5 | Interferon regulatory factor 5 | + | GGTCAACGGGGAAAAGAAACT | (45) |
| − | CATCCACCCCTTCAGTGTACT | |||
| Irf7 | Interferon regulatory factor 7 | + | CCTCTTGCTTCAGGTTCTGC | (45) |
| − | GCTGCATAGGGTTCCTCGTA | |||
| Tnfa | Tumor necrosis factor-alpha | + | CGGTCCCCAAAGGGATGAG | (45) |
| − | CCTTGAAGAGAACCTGGGAGTA | |||
| Actg1 | Gamma actin | + | ACCAACAGCAGACTTCCAGGAT | (24) |
| − | AGACTGGCAAGAAGGAGTGGTAA | |||
| Gapdh | Glyceraldehyde 3-phosphate dehydrogenase | + | TGTGTCCGTCGTGGATCTGA | (14) |
| − | CCTGCTTCACCACCTTCTTGA |
Urinary albumin and creatinine ratio and serum creatinine
Urine albumin and creatinine were measured according to the manufacturer's protocols using a mouse albumin ELISA (#E90-134; Bethyl Laboratories) and the creatinine companion (#1012; Exocell), respectively. Both assays use a standard curve to calculate μg/mL (albumin) and mg/mL (creatinine). The μg/mg urinary albumin creatinine ratio (UACR) was then calculated by dividing albumin by creatinine. Mouse serum collected at the time of euthanasia from 3, 7, 12, and 17 weeks post-CVB4 inoculation was sent to the University of Massachusetts Mouse Metabolic Phenotyping Core (Worchester, MA) for evaluation of serum creatinine (SrCr) concentration on a Cobas Clinical Chemistry Analyzer (Roche).
Statistical analyses
All statistical analyses were performed using R Statistical software (29). Shapiro–Wilk test and Levene's test were calculated to test for normality and equal variance, respectively. Data not falling within a normal distribution were transformed (log or square root) to best approach normality. Main effects were tested using two-way analysis of variance with CVB4 inoculation and time as factors. Tukey's post hoc test was performed when a main effect was found. Pearson's correlation coefficient was calculated to test for significant correlations. Student's t-test (two-tailed) was used for two-factor comparisons. Alpha was set at 0.05 for all statistical tests. Graphs were generated using GraphPad Prism 7.0. Dots depict individual samples, center line indicates the mean, and whiskers indicate standard error.
Results
CVB4 abundance and localization within the NOD mouse kidney
To study CVB4 inoculation in the kidneys of NOD mice, the presence and location of the virus in the kidney after a single intraperitoneal inoculation of CVB4 were assessed over time. Renal CVB4 abundance peaked 2 to 3 days postinoculation and was significantly reduced, but not completely cleared (did not return to baseline), by 14 days postinoculation (Fig. 1A). Moreover, renal CVB4 abundance was highly variable at each time point (between time points and within the same time point). For example, viral abundance 3 and 14 days postinoculation had a 171- and 5,000-fold difference between the kidneys with the most and least viral abundances at the indicated time point, respectively.
FIG. 1.

CVB4 abundance and location in the kidney over time. (A) RT-qPCR of CVB4 RNA normalized by the geometric mean of Gapdh and Actg1 (n = 4–8; *p < 0.05 compared with day 0, @p < 0.05 compared with day 2, &p < 0.05 compared with day 3). (B) Representative images from ISH analysis of +ssCVB4 RNA (genomic) 2 days (upper two panels) and 14 days (lower two panels) postinoculation. CVB4 RNA was detected in the glomerular (left) and tubulointerstitial (right) regions of the kidneys of inoculated mice 2 days postinoculation; however, 14 days postinoculation, CVB4 was only found in the glomerular region. Arrowheads point to labeled CVB4 RNA (representative images taken at 400 × magnification; n = 4). Actg1, gamma-actin; CVB4, coxsackievirus B4; Gapdh, glyceraldehyde 3-phsophate dehydrogenase; ISH, in situ hybridization; RT-qPCR, reverse transcription real-time polymerase chain reaction.
ISH detecting CVB4 (genomic) RNA was performed to identify the specific regions of the kidney where CVB4 was localized 2, 5, 9, and 14 days postinoculation. CVB4 RNA was found in glomeruli of all virus-inoculated NOD mouse kidneys examined (Fig. 1B and Table 2) and in the interstitial region of all virus-inoculated NOD mouse kidneys 2 days postinoculation (Fig. 1B and Table 2); however, after this time point, the presence of CVB4 RNA in the interstitial region varied (Table 2). At 5 days postinoculation, 25% (1/4) of the mice no longer had CVB4 RNA in the interstitial region, while at 9 days postinoculation, 50% of mice (2/4) no longer had CVB4 RNA in this region. It was not until 14 days postinoculation that CVB4 RNA was totally absent in the interstitial region of all kidney samples examined (Fig. 1B and Table 2).
Table 2.
Region and Number of Kidney Samples Containing CVB4 RNA per Total Number of Mice Analyzed
| Region | 2 Days | 5 Days | 9 Days | 14 Days |
|---|---|---|---|---|
| Glomerulus | 4/4 | 4/4 | 4/4 | 4/4 |
| Interstitium | 4/4 | 3/4 | 2/4 | 0/4 |
CVB4, coxsackievirus B4.
In accordance with our RT-qPCR results, ISH also revealed variability in the amount of CVB4 RNA present at all time points examined (Supplementary Fig. S1). No CVB4 RNA was detected by ISH in noninoculated mouse kidneys 2 days postinoculation (data not shown).
Renal morphological and functional changes following CVB4 inoculation
Histological evaluation of the kidneys of NOD mice by PAS staining was performed 3, 7, 10, and 14 days post-CVB4 inoculation to identify viral-induced alterations. No alterations, e.g., glomerulonephritis, glomerular tuft swelling, increased glomerular area, increased glomerular cellularity, tubular dilation, mesangial expansion, immune cell infiltration, proliferation, or apoptosis, were observed (Supplementary Fig. S2). Moreover, UACR, kidney weight, and body weight were measured; however, significant differences, but not meaningful differences, were observed at any time point following CVB4 inoculation (Supplementary Fig. S3).
Effects of CVB4 inoculation on viral pattern recognition receptors
Because CVB4 RNA was detected in the kidney, but no readily apparent, histological alterations were observed via PAS staining, CVB4-mediated effects on pattern recognition receptor (PRR) expression were assessed via RT-qPCR to identify any viral-induced molecular alterations that might underlie impending renal pathology.
Ifnb1 upregulation, indicating viral-mediated activation of the MyD88-independent branch of PRR signaling, was detected in the kidneys of seven of eight virus-inoculated mice at 3 days postinoculation and in the kidneys of one of four virus-inoculated mice at 5 days postinoculation (Fig. 2A). Moreover, there was a 130-fold variation in Ifnb1 expression 3 days postinoculation. Tnfa, a proinflammatory cytokine induced by the MyD88-dependent branch of PRR signaling (mediated via NF-κB), was upregulated in the kidneys of all CVB4-inoculated mice at all time points, peaking at day 3 postinoculation (Fig. 2B).
FIG. 2.

Viral PRR expression and downstream targets. RT-qPCR of (A) Ifnb, (B) Tnfa, (D) Irf3, (B) Irf1, (C) Irf5, and (D) Irf7 0, 2, 3, 5, 7, 9, 10, and 14 days post-CVB4 inoculation, normalized to the geometric mean of Gapdh and Actg1. Only seven of eight kidneys at 3 days and one of four kidneys at 5 days postinoculation had detectable levels of Ifnb (n = 4–8; *p < 0.05 compared with day 0, @p < 0.05 compared with day 2, &p < 0.05 compared with day 3). PRR, pattern recognition receptor.
To further demonstrate the presence of viral-induced PRR activation in the kidneys of CVB4-inoculated NOD mice, upstream PRR signaling that precedes Tnfa and Ifnb1 upregulation was also evaluated. The RNA expression of the transcription factors that upregulate type I interferon expression was evaluated by RT-qPCR.
Irf3 expression was unaffected by CVB4 inoculation at any time point (Fig. 2C); however, Irf1, Irf5, and Irf7 were significantly upregulated 2 and 3 days postinoculation (Fig. 2D–F). Irf5 and Irf7 returned to baseline by 5 days postinoculation (Fig. 2E, F, respectively), while Irf1 was still significantly upregulated at 14 days postinoculation (Fig. 2D).
IRF activation (measured by serine phosphorylation) at 3 days postinoculation was also evaluated by immunoprecipitation of phosphoserine (pSer) proteins from kidney lysates, followed by Western blot analysis (Fig. 3). IRF5 could not be reliably detected, even in the positive control pancreas of an NOD mouse inoculated with CVB4 for 3 days (Supplementary Fig. S4). IRF1 was not detectable in the kidney (Fig. 3A) and IRF3 was not significantly activated (Fig. 3B, D) compared with kidneys from noninoculated mice; however, phosphorylated IRF7 (pIRF7) was significantly increased in the kidneys of CVB4-inoculated mice compared with the kidneys of noninoculated mice (Fig. 3C, E).
FIG. 3.

IRF activation following CVB4 inoculation. Western blot analyses of 200 μg of total protein IP with pSer antibody and probed for (A) IRF1, (B) IRF3, and (C) IRF7. Separately, 20 μg of total protein was probed with total IRF1, IRF3, IRF7, and TUBA1A (Tubulin) or ACTB (Actin) as protein loading controls. V+ and V− indicate kidneys from CVB4-inoculated and noninoculated NOD mice, respectively. The final V+ lane contains 10 μg lysate from CVB4-inoculated NOD pancreas. Densitometry analysis of phospho/total, phospho/loading control, and total/loading control are depicted for (D) IRF3 and (E) IRF7 (n = 6; *p < 0.05). IP, immunoprecipitated; IRF, interferon regulatory factor; NOD, non-obese diabetic.
This increase in activated IRF7 appears to be driven by an increase in total IRF7 protein, with a proportional increase in phosphorylation (Fig. 3E). While levels of total and pIRF7 were quite variable, a Pearson's correlation coefficient comparing total IRF7 (IRF7/Actin) and pIRF7 (pIRF7/Actin) resulted in a significant correlation with an r value of 0.95 (data not shown). The level of pIRF7 and total IRF7 protein returned to baseline by 7 days postinoculation (Supplementary Fig. S4), which was similar to the Irf7 expression pattern (Fig. 2F).
We next assessed the DNA binding activity for the canonical NF-κB pathway, which results in the transcription of Tnfa. DNA binding of the canonical NF-κB pathway subunit containing a transcriptional activation domain, RELA, as well as its coactivating subunit, NFKB1, was not detectable by TransAM assay. NF-κB pathway activation, however, can participate in a positive feedback loop that induces the expression of NF-κB genes (36); therefore, expression of Nfkb1 was evaluated by RT-qPCR. Nfkb1 expression was upregulated 3 days post-CVB4 inoculation and returned to baseline by 10 days postinoculation (Fig. 4).
FIG. 4.
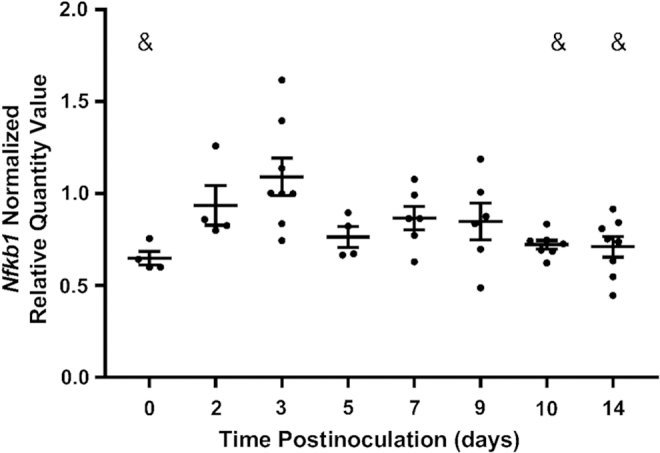
Gene expression of NF-κB subunits. RT-qPCR of Nfkb1 (p50) 0, 2, 3, 5, 7, 9, 10, and 14 days post-CVB4 inoculation, normalized to the geometric mean of Gapdh and Actg1 (n = 4–8; &p < 0.05 compared with day 3).
In an attempt to identify the PRR pathway responsible for the upregulation of Ifnb1 and Tnfa expression in CVB4-inoculated NOD mouse kidneys, the presence and/or expression of different PRRs (PKR, Tlr3, Rig1, and Mda5) were evaluated. There was no significant change in phosphorylated PKR (pPKR) or total PKR when normalized by TUBA1A in the kidneys of CVB4-inoculated compared with noninoculated NOD mice at 3 days postinoculation (Fig. 5A, B). There was a significant decrease in pPKR relative to total PKR in the kidneys of CVB4-inoculated compared with noninoculated NOD mice at 3 days postinoculation (Fig. 5A, B).
FIG. 5.

PRR activity and gene expression. (A) Western blot analysis of 200 μg of total protein IP with pThr antibody and 20 μg of input lysate probed with anti-PKR and anti-TUBA1A (Tubulin). Lysates were from noninoculated (V−) and CVB4-inoculated (V+) NOD mouse kidneys. V+ indicates 10 μg lysate from CVB4-inoculated NOD pancreas. (B) Densitometry analysis of pPKR/PKR, pPKR/αTubulin, and PKR/αTubulin signals (n = 6; *p < 0.05). Plots of RT-qPCR results of (C) Tlr3, (D) Rig1, and (E) Mda5 0, 2, 3, 5, 7, 9, 10, and 14 days post-CVB4 inoculation normalized to the geometric mean of Gapdh and Actg1 (n = 4–8; *p < 0.05 compared with day 0, @p < 0.05 compared with day 2, &p < 0.05 compared with day 3).
Expression of PRRs Tlr3, Rig1, and Mda5 was significantly upregulated 2 and 3 days postvirus inoculation and returned to baseline by 5 days postinoculation (Fig. 5C–E); however, we were unable to detect the proteins of each gene or positive control by Western blot analysis (all three tested) or immunofluorescence (only TLR3 tested) (Supplementary Fig. S5).
Correlation of PRR signaling pathways and CVB4 inoculation
Because CVB4 abundance was highly variable between mice, and to better understand the relationships between renal CVB4 abundance and PRR signaling pathways, a Pearson's correlation coefficient was calculated for many of the parameters evaluated in this study. Renal CVB4 abundance was positively correlated to Irf5, Irf7, Nfkb1, and Tnfa expression (Table 3, down) and to PRR expression of Tlr3, Rig1, and Mda5 (Table 3, across). Tlr3, Rig1, and Mda5 expression was also all positively correlated with Irf1, Irf5, Irf7, Nfkb1, and Tnfa expression. However, Tlr3 expression was the only PRR positively correlated with Ifnb1 expression and was also the only PRR to correlate with all PRR signaling pathway members. No correlation was found between renal CVB4 abundance and body weight, kidney weight, normalized kidney weight, or UACR.
Table 3.
Significant Pearson's Correlation Coefficients Between CVB4, Tlr3, Rig1, and Mda5 Gene Expression and Various Measured Parameters Following CVB4 Infection (p < 0.05)
| CVB4 | Tlr3 | Rig1 | Mda5 | |
|---|---|---|---|---|
| CVB4 | 0.52 | 0.50 | 0.46 | |
| Irf1 | 0.72 | 0.55 | 0.62 | |
| Irf5 | 0.33 | 0.74 | 0.67 | 0.66 |
| Irf7 | 0.56 | 0.89 | 0.95 | 0.93 |
| Ifnb1 | 0.72 | |||
| Nfkb1 | 0.35 | 0.60 | 0.50 | 0.50 |
| Tnfa | 0.51 | 0.75 | 0.53 | 0.61 |
The effect of TLR3 knockout on CVB4 abundance
To determine if TLR3 could be the PRR responsible for the upregulation of Tnfa, Ifnb1, Irf1, Irf5, Irf7, and Nfkb1 following CVB4 inoculation, a cohort of Tlr3−/− NOD mice were inoculated with CVB4 at 8 weeks of age, and renal CVB4 abundance was evaluated via RT-qPCR to detect CVB4 RNA. Renal CVB4 abundance (i.e., renal CVB4 RNA abundance) in Tlr3−/− NOD mice was highest at 2 and 3 days postinoculation (Fig. 6A, B, upper panels), in a trend similar to that observed in Tlr3+/+ NOD mice (Fig. 1A). Interestingly, renal CVB4 abundance in Tlr3−/− NOD mice returned to baseline levels by 9 days postinoculation (Fig. 6A), whereas renal CVB4 abundance in Tlr3+/+ NOD mice was still elevated (Fig. 1A).
FIG. 6.

CVB4 abundance and location over time in Tlr3−/− NOD mouse kidneys. (A) RT-qPCR of CVB4 RNA normalized by the geometric mean of Gapdh and Actg1 (n = 4–8; *p < 0.05 compared with day 0, @p < 0.05 compared with day 2, &p < 0.05 compared wiht day 3). (B) Representative images from ISH analysis of +ssCVB4 RNA 2 days (upper two panels) and 14 days (lower two panels) post-CVB4 inoculation. CVB4 was detected in the glomerular (left) and tubulointerstitial (right) regions of CVB4-inoculated mouse kidneys 2 and 14 days post-CVB4 inoculation. Arrowheads point to labeled CVB4 RNA (images taken at 400 × magnification; n = 4).
As was seen in the kidneys of Tlr3+/+ NOD mice, renal CVB4 abundance in Tlr3−/− NOD mice was highly variable, with a 640- and 139-fold difference between the highest and lowest CVB4 abundance at 3 and 14 days postinoculation, respectively. ISH also revealed that at 2 days postinoculation, CVB4 RNA is located in the glomerulus and interstitial regions (Fig. 6B, top row); however, unlike Tlr3+/+ NOD mouse kidneys, CVB4 was still present in both locations in kidneys of Tlr3−/− NOD mice, although barely detectable in the interstitial region, at 14 days postinoculation (Fig. 6B, bottom row).
When CVB4 abundance in Tlr3+/+ and Tlr3−/− NOD mouse kidneys was directly compared within the same experiment, renal CVB4 abundance was not significantly different between Tlr3+/+ and TLR3−/− NOD mouse kidneys at day 0; at all other time points, there was significantly more CVB4 in Tlr3−/− NOD mouse kidneys compared with Tlr3+/+ NOD mouse kidneys (Fig. 7).
FIG. 7.

The absence of TLR3 increases CVB4 abundance in the kidney. Plot of RT-qPCR of CVB4 RNA normalized by the geometric mean of Gapdh and Actg1 0, 2, 3, and 14 days post-CVB4 inoculation (n = 4–8; *p < 0.05 compared with Tlr3−/− at the same time point).
Chronic effect of a one-time CVB4 inoculation
To investigate longer term renal consequences of a one-time CVB4 inoculation, kidneys of Tlr3+/+ NOD mice 3, 7, 12, and 14 weeks after a single CVB4 inoculation were evaluated. While many of the visual signs of CVB4 infection (diarrhea, dehydration, decreased mobility) were no longer apparent, CVB4-inoculated NOD mouse body weight and kidney weight were significantly reduced by 20% 3 weeks postinoculation in comparison with age-matched, noninoculated controls, and returned to normal by 7 weeks postinoculation (Supplementary Fig. S6). However, these changes were proportional; kidney weight normalized to body weight did not differ between inoculated and noninoculated mice at any time point post-CVB4 inoculation. Correspondingly, CVB4 inoculation had no effect on UACR or SrCr at any of these later time points (Supplementary Fig. S6).
Long-term renal CVB4 abundance
CVB4 abundance was measured by RT-qPCR 0 and 3 days and 3, 7, 12, and 17 weeks post-CVB4 inoculation to determine how long after inoculation the CVB4 RNA was detectable in Tlr3+/+ NOD mouse kidneys. CVB4 abundance returned to baseline 3 weeks postinoculation (Fig. 8A). While CVB4 abundance by week 3 and onward was not significantly different from week 0, kidneys of 12 of 13 mice at 3 weeks, 5 of 9 mice at 7 weeks, 1 of 6 mice at 12 weeks, and 0 of 11 mice at 17 weeks postinoculation had higher CVB4 abundance than any kidney of week 0 mice (Fig. 8A).
FIG. 8.

Chronic effect of a one-time CVB4 inoculation. (A) Plots of RT-qPCR analysis of CVB4 RNA measured 0 and 3 days and 3, 7, 12, and 17 weeks post-CVB4 inoculation and normalized to the geometric mean of Gapdh and Actg1 (n = 4–13; %p < 0.05 compared with day 3 and week 17, &p < 0.05 compared with day 3). (B) Representative images of PAS staining of noninoculated (left panels) and CVB4-inoculated (right panels) NOD mouse kidneys 12 weeks (upper panels) and 17 weeks (lower panels) postinoculation. Plots of (C) PAS-positive glomerular area and (D) total glomerular area were plotted per individual glomerulus. PAS-positive area was normalized by dividing dark pink (PAS) glomerular staining by total glomerular area and multiplying by 100 for each glomerulus. Arrowheads point to dark pink PAS-positive areas. Images taken at 400 × magnification (n = 30–65 glomeruli per group; *p < 0.05 compared with age-matched noninoculated controls). PAS, periodic acid-Schiff.
Histopathological changes after CVB4 inoculation
PAS staining was performed on kidneys from CVB4-inoculated and noninoculated NOD mice 12 and 17 weeks post-CVB4 inoculation to assess chronic histological alterations, such as mesangial matrix expansion, following a one-time CVB4 inoculation. PAS-positive areas were observed at varying intensities in some, but not all, glomeruli and in only some portions of an individual glomerulus, indicating possible initiation of focal segmental glomerulosclerosis following CVB4 inoculation (Fig. 8B). To determine if the observed PAS-positive area was a result of CVB4 inoculation and not aging, since positive staining was observed in both virus-inoculated and noninoculated mouse kidneys, PAS-positive area relative to total glomerular area was quantified in five randomly selected glomeruli from each kidney section and expressed as a percentage (Fig. 8C).
Percentages were calculated for each individual glomerulus, not averaged per mouse, and all 5 glomeruli were represented as individuals in the graph, making the sample size for each time point rise from 6–13 mice per group to 30–65 glomeruli per group (e.g., 6 mice times 5 glomeruli equals 30, 13 mice times 5 glomeruli equals 65). There was no significant difference in PAS-positive area between noninoculated and CVB4-inoculated NOD mouse kidneys at 12 weeks postinoculation; however, at 17 weeks postinoculation, CVB4-inoculated NOD mouse kidneys had significantly more PAS-positive area per glomerulus than noninoculated, age-matched controls.
Glomerular area was also evaluated to ensure that the increase in PAS-positive area was not an artificial finding due to a reduction in glomerular size (Fig. 8D). There was no significant difference in glomerular area at 12 or 17 weeks postinoculation between noninoculated or CVB4-inoculated NOD mouse kidneys (Fig. 8D).
Discussion
CV exposure in humans is rarely reported to result in significant renal damage; however, when it is reported, the impact on the kidney is considerable (8,9,27). Currently, it is unknown why the majority of patients who become infected with CV do not have obvious negative renal outcomes, while others respond with renal injury and failure that can lead to death. Moreover, there is a dearth of knowledge regarding the short-term or long-term effects of CV exposure on the kidney. Thus, this study is the first to evaluate the short-term (i.e., acute) effects of a one-time CVB4 inoculation on the kidneys of NOD mice and how the presence of CVB4 in the kidney may set the stage for the development and/or acceleration of CKD.
The finding that CVB4 RNA is still present in the glomerulus 14 days postinoculation provides the first piece of evidence that CVB4 may predispose the kidney to future CKD. While the ISH studies could not delineate the glomerular cell types with persistent CVB4 RNA at 14 days postinoculation, Conaldi et al. previously demonstrated in human cell culture studies that mesangial cells, but not podocytes or proximal tubule cells, are able to maintain the presence of CVB4 greater than 50 days in culture (10).
These data suggest that CVB4 found in the glomeruli of NOD mouse kidneys 14 days postinoculation is most likely maintained by mesangial cells. Furthermore, at 17 weeks postinoculation, we observed more PAS-positive area in glomeruli of CVB4-inoculated mouse kidneys than noninoculated mouse kidneys, suggesting that a one-time inoculation initiated an increase in mesangial area. While this increase was subtle, it may indicate the initiation of CKD or an increased susceptibility to a future insult that might lead to CKD.
Perhaps some of the most clinically important findings of this study were the large variations of CVB4 abundance and clearance in NOD mouse kidneys. All mice were inoculated with the same amount of CVB4; however, at various time points after inoculation, the amount of detectable virus in the kidney was highly variable, ranging between 5,000-fold (14 days) and 2-fold (5 days) differences in abundance (most and least variable time points postinoculation, respectively). Moreover, while all kidneys 2 days postinoculation had observable CVB4 RNA in the glomerular and tubular regions, CVB4 location and timing of clearance from the kidney were highly variable at all subsequent time points.
This variability suggests the following: (i) CVB4 can travel to and remain in the kidney more readily in some animals than others, (ii) some animals are able to clear CVB4 more rapidly and efficiently than others, and/or (iii) CVB4 is able to evade clearance and persist in some animals better than others. Consideration of each of these possibilities will be important in determining individual patient susceptibility to CVB4-induced kidney injury.
After establishing that CVB4 RNA was present in the kidney in the absence of any readily apparent acute histological alterations, functional decline, or immune cell infiltration, we next wanted to determine if the kidney was responding locally to the presence of CVB4 via a PRR response. Tlr3, Rig1, and Mda5 were upregulated following CVB4 inoculation and evidence of their downstream signaling was also observed via an elevation in total and phosphorylated IRF7, as well as an elevation of Tnfa and Ifnb1 expression.
Since Tlr3 expression correlated with CVB4 abundance along with expression of Irf1, Irf5, Irf7, Nfkb1, Tnfa, and Ifnb1, while Rig1 and Mda5 expression correlated with expression of those same genes except Ifnb1, but did correlate with CVB4 abundance, it is tempting to hypothesize that TLR3 might be the primary PRR activated by CVB4 in the kidneys. This is of note since Ifnb1 expression was difficult to detect at any time point other than 3 days postinoculation and Tlr3 was the only PRR gene whose expression was correlated with Ifnb1 expression.
If TLR3 is one of the primary PRRs activated in response to CVB4 and responsible for its renal clearance, we would expect to see a higher abundance of CVB4 in the kidneys of Tlr3−/− NOD mice compared with Tlr3+/+ NOD mice, because this critical PRR response would be absent and unable to elicit a sufficient immune response to clear the virus from the kidney. Indeed, CVB4 abundance was significantly higher in the kidneys of Tlr3−/− NOD mice 2, 3, and 14 days post-CVB4 inoculation compared with that in the kidneys of age-matched Tlr3+/+ NOD mice (Fig. 7), suggesting that TLR3 does play an important role in renal CVB4 clearance. This finding is in agreement with a prior report that CVB4 viral titers are significantly higher in the pancreas of Tlr3−/− NOD mice at 3 days postinoculation than in Tlr3+/+ NOD mice inoculated with the same amount of virus (26).
Future studies will aim to evaluate other acute kidney parameters in Tlr3−/− mice, which may provide more insight into the precise role(s) TLR3 signaling plays in this process. For example, since Tlr3−/− mice have a higher abundance of CVB4 at day 3 postinoculation, does this result in any key histological or functional alterations? Moreover, are the same IRFs activated in the absence of TLR3 and are type I interferons transcribed? Answering these questions may help identify a more distinct role of TLR3 in the kidney following CVB4 inoculation.
We must note that our findings are not without limitation. While we supplied evidence to suggest mesangial cells are the primary renal cell type affected by CVB4 inoculation [presence of CVB4 in the glomeruli and increased mesangial area 17 weeks postinoculation, Conaldi's persistent infection data (10)], further cell-type-specific analysis is required to confirm this. Moreover, we were unable to determine why some animals had a strong innate immune response and others did not or why no immune cells were found in the kidney following PRR activity.
In addition, we did not purify CVB4 before using it to inoculate NOD mice; therefore, it is possible that the immune response we observed may have been influenced by cell lysate components from the virus preparation. Finally, while we can speculate that an increase in mesangial area could result in a CVB4-induced susceptibility to future kidney injury, further studies to confirm these findings are warranted.
While many questions remain, the key findings presented here include the following: (i) CVB4 does elicit a response in the kidneys of NOD mice, (ii) CVB4 and the subsequent response are highly variable between animals, (iii) there is a PRR response to CVB4 within the kidney, (iv) the presence of CVB4 and some CVB4-stimulated gene expression persists in the kidney at 14 days postinoculation, and (v) a one-time CVB4 inoculation results in a subtle increase in PAS-positive glomerular area 17 weeks postinoculation.
Together, these data suggest that CVB4 may be predisposing the kidney for susceptibility to future development of CKD and that TLR3 may play a pivotal role in this process. Studies in NOD mice will help us to better understand the reasons why some patients who are predisposed to develop T1D are protected from CVB4-induced kidney damage while others are not. Our findings do, however, indicate that CVB4 may play a larger role in the kidney than previously thought, and studying the effect of otherwise “innocent” viruses may provide insight for overall kidney health in individuals susceptible to T1D.
Supplementary Material
Acknowledgments
The authors acknowledge Roger Loria, Virginia Commonwealth University, Richmond, VA, for the gift of the CVB4 Edwards strain. In addition, they acknowledge Li Wen, Yale School of Medicine, New Haven, CT, for the Tlr3−/− NOD mice. They also thank the University of Massachusetts Mouse Metabolic Phenotyping Core (NIH 5U2C-DK093000) for evaluation of SrCr and the Ohio University histology core facility for PAS staining.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported, in part, by the JO Watson Endowment for Diabetes Research; the Ohio University Baker Fund, Student Enhancement Award, and Honors Tutorial College Student Support Fund; the John J. Kopchick Molecular & Cellular Biology/Translational Biomedical Sciences Student Support Fund; and the Ohio University Heritage College of Osteopathic Medicine McCall Research Support Fund.
Supplementary Material
References
- 1. Alexopoulou L, Holt AC, Medzhitov R, et al. . Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001;413:732–738 [DOI] [PubMed] [Google Scholar]
- 2. Andersen CL, Jensen JL, and Orntoft F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004;64:5245–5250 [DOI] [PubMed] [Google Scholar]
- 3. Andreoletti L, Hober D, Hober-Vandenberghe C, et al. . Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type 1 diabetes mellitus. Med Virol 1998;52:121–127 [DOI] [PubMed] [Google Scholar]
- 4. Aronson MD, and Phillips CA. Coxsackievirus B5 infections in acute oliguric renal failure. J Infect Dis 1975;132:303–306 [DOI] [PubMed] [Google Scholar]
- 5. Atkinson MA, and Gianani R. The pancreas in human type 1 diabetes: providing new answers to age-old questions. Curr Opin Endorcrinol Diabetes Obes 2009;16:279–285 [DOI] [PubMed] [Google Scholar]
- 6. Banatvala JE, Bryant J, Schernthaner G, et al. . Coxsackie B, mumps, rubella, and cytomegalovirus specific IgM responses in patients with juvenile-onset insulin-dependent diabetes mellitus in Britain, Austria, and Australia. Lancet Lond Engl 1985;1409–1412 [DOI] [PubMed] [Google Scholar]
- 7. Baron S, Fons M, and Albrecht T. Viral pathogenesis. In Baron S, ed. Medical Microbiology. Galveston, TX: University of Texas Medical Branch at Galveston, ch. 45, 1996;3–4 [PubMed] [Google Scholar]
- 8. Bayatpour M, Zbitnew A, Dempster G, et al. . Role of coxsackievirus B4 in the pathogenesis of acute glomerulonephritis. Can Med Assoc J 1973;109:873–875 [PMC free article] [PubMed] [Google Scholar]
- 9. Burch GE. Progressive coxsackie viral pancarditis and nephritis. Ann Intern Med 1969;71:963. [DOI] [PubMed] [Google Scholar]
- 10. Conaldi PG, Biancone L, Bottelli A, et al. . Distinct pathogenic effects of group B coxsackieviruses on human glomerular and tubular kidney cells. J Virol 1997;71:9180–9187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coschigano KT, Wetzel AN, Obichere N, et al. . Identification of differentially expressed genes in the kidneys of growth hormone transgenic mice. Growth Horm IGF Res 2010;20:345–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Drescher KM, Kono K, Bopegamage S, et al. . Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology 2004;329:381–394 [DOI] [PubMed] [Google Scholar]
- 13. Faulhaber JR, and Nelson PJ. Virus-induced cellular immune mechanisms of injury to the kidney. Clin J Am Soc Nephrol 2007;2:S2–S5 [DOI] [PubMed] [Google Scholar]
- 14. Fujimoto M, Maezawa Y, Yokote K, et al. . Mice lacking smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun 2003;305:1002–1007 [DOI] [PubMed] [Google Scholar]
- 15. Gale EAM. The rise of childhood type 1 diabetes in the 20th century. Diabetes 2002;51:3353–3361 [DOI] [PubMed] [Google Scholar]
- 16. Glassock RJ. Immune complex-induced glomerular injury in viral diseases: an overview. Kidney Int Suppl 1991;35:S5–S7 [PubMed] [Google Scholar]
- 17. Hellemans J, Mortier G, De Paepe A, et al. . qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 2007;8:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horwitz MS, Bradley LM, Harbertson J, et al. . Diabetes induced by coxsackie virus: initation by bystander damage and not molecular mimicry. Nat Med 1998;4:781–785 [DOI] [PubMed] [Google Scholar]
- 19. Hyoty H, Hiltunen M, Knip M, et al. . A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood diabetes in finland (DiMe) study group. Diabetes 1995;44:652–657 [DOI] [PubMed] [Google Scholar]
- 20. Isome M, Yoshida K, Suzuki S, et al. . Experimental glomerulonephritis following successive inoculation of five different serotypes of group B coxsackieviruses in mice. Nephron 1997;77:93–99 [DOI] [PubMed] [Google Scholar]
- 21. Jensen MM. Viruses and kidney disease. Am J Med 1967;43:897–911 [DOI] [PubMed] [Google Scholar]
- 22. Kamiyama S. Experimental glomerulonephritis induced by Coxsackie B4 virus in mice-glomerular changes associated with intermittent viral inoculations. Nihon Jinzo Gakkai Shi 1990;32:939–948 [PubMed] [Google Scholar]
- 23. Kaprio J, Tuomilehto J, Koskenvuo M, et al. . Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992;35:1060–1067 [DOI] [PubMed] [Google Scholar]
- 24. Lefever S, Vandesompele J, Speleman F, et al. . RTPrimerDB: the portal for real-time PCR primers and probes. Nucleic Acids Res 2009;37:D942–D945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCall KD, Schmerr MJ, Thuma JR, et al. . Phenylmethimazole suppresses dsRNA-induced cytotoxicity and inflammatory cytokines in murine pancreatic beta cells and blocks viral acceleration of type 1 diabetes in NOD mice. Endocrinology 2013;18:3841–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCall KD, Thuma JR, Courreges MC, et al. . Toll-like receptor 3 is critical for coxsackievirus B4-induced type 1 diabetes in female NOD mice. Endocrinology 2015;156:453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pasch A, and Frey FJ. Coxsackie B viruses and the kidney—a neglected topic. Nephrol Dial Transplant 2006;21:1184–1187 [DOI] [PubMed] [Google Scholar]
- 28. Prasad N, and Patel MR. Infection-induced kidney diseases. Front Med (Lausanne) 2018;5:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2014 [Google Scholar]
- 30. Rewers M, and Zimmet P. The rising tide of childhood type 1 diabetes—what is the elusive environmental trigger? Lancet 2004:364:17368-6. [DOI] [PubMed] [Google Scholar]
- 31. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serreze DV, Chapman HD, Varnum DS, et al. . Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol 1997;158:3978–3986 [PubMed] [Google Scholar]
- 33. Serreze DV, Ottendorfer EW, Ellis TM, et al. . Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 2000;49:708–711 [DOI] [PubMed] [Google Scholar]
- 34. Serreze DV, Wasserfall C, Ottendorfer EW, et al. . Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and gamma interferon. J Virol 2005;79:1045–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh HK, and Nickeleit V. Kidney disease caused by viral infections. Curr Diagn Pathol 2004;10:11–21 [Google Scholar]
- 36. Sun SC. The noncanonical NF-κB pathway. Immunol Rev 2012;246:125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tracy S, Drescher KM, Chapman NM, et al. . Toward testing the hypothesis that group B coxsackievirus (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB marketdly lowers diabetes incidence. J Virol 2002;76:12097–12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. USRDS. 2017. USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2017 [Google Scholar]
- 39. Vandesompele J, De Preter K, Pattyn F, et al. . Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Viruses and renal disease. JAMA 1968;204:539. [PubMed] [Google Scholar]
- 41. vonHerrath MG, Fujinami RS, and Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Nat Rev Microbiol 2003;1:151–157 [DOI] [PubMed] [Google Scholar]
- 42. Webb SR, Loria RM, Madge GE, et al. . Susceptibility of mice to group B coxsackie virus is influenced by the diabetic gene. J Exp Med 1976;143:1239–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wong FS, Hu C, Zhang L, et al. . The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci 2008;1150:146–148 [DOI] [PubMed] [Google Scholar]
- 44. Xiao X, Ma B, Dong B, et al. . Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J Autoimmun 2009;32:85–93 [DOI] [PubMed] [Google Scholar]
- 45. Ye J, Coulouris G, Zaretskaya I, et al. . Primer-BLASt: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012;13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoon JW, and Jun HS. Viruses cause type 1 diabetes in animals. Ann. N. Y. Acad. Sci 2006;1097:138–146 [DOI] [PubMed] [Google Scholar]
- 47. Yoshida K, Suzuki J, Suzuki S, et al. . Experimental IgA nephropathy induced by coxsackie B4 virus in mice. Am J Nephrol 1997;17:81–88 [DOI] [PubMed] [Google Scholar]
- 48. Zhou HT, Wang B, and Che XY. Nephrotic syndrome in hand, foot and mouth disease caused by coxsackievirus A16: a case report. Int J Infect Dis 2014;28:1–2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.