

Abstract
Airway epithelial barrier dysfunction is frequently observed in asthma and may have important implications. The physical barrier function of the airway epithelium is tightly interwoven with its immunomodulatory actions, while abnormal epithelial repair responses may contribute to remodelling of the airway wall. We propose that abnormalities in the airway epithelial barrier play a crucial role in the sensitization to allergens and pathogenesis of asthma. Many of the identified susceptibility genes for asthma are expressed in the airway epithelium, supporting the notion that events at the airway epithelial surface are critical for the development of the disease. However, the exact mechanisms by which the expression of epithelial susceptibility genes translates into a functionally altered response to environmental risk factors of asthma are still unknown. Interactions between genetic factors and epigenetic regulatory mechanisms may be crucial for asthma susceptibility. Understanding these mechanisms may lead to identification of novel targets for asthma intervention by targeting the airway epithelium. Moreover, exciting new insights have come from recent studies using single‐cell RNA sequencing (scRNA‐Seq) to study the airway epithelium in asthma. This review focuses on the role of airway epithelial barrier function in the susceptibility to develop asthma and novel insights in the modulation of epithelial cell dysfunction in asthma.
Keywords: airway remodelling, asthma, (epi)genetics, epithelial barrier, type 2 responses
Bullet points outlining future research perspective.
Future research unravelling the molecular mechanisms and regulatory networks underlying abnormal epithelial repair responses after exposure to environmental insults hold promise for the identification of novel intervention strategies in asthma.
Single‐cell RNA‐sequencing studies may lead to elucidating the cellular changes and causal gene regulatory networks underlying the different asthma endotypes.
Analysis of matched single‐cell RNA‐Sequencing data sets from airway wall biopsies, bronchial brushes and nasal brushes will allow identification of novel biomarkers for disease activity or treatment response using less invasive methodologies.
Better understanding of (epi)genetic regulatory mechanisms of airway epithelial abnormalities in asthma likely contributes to identification of novel targets for asthma intervention.
Box outlining the major milestone discoveries.
Loss of epithelial junctions not only results in increased susceptibility towards pathogens and allergens, but also propagates pro‐inflammatory responses and may contribute to airway remodelling.
E‐cadherin loss and activation of β‐catenin per se induce epithelial features reminiscent of the airway epithelium in asthma in in vitro and in vivo models.
Loss of airway epithelial barrier function in asthma is a consequence of interaction between environmental and genetic factors and epigenetic regulatory mechanisms.
Expression quantitative trait loci (eQTL) studies in human bronchial epithelial cells and bronchial alveolar lavage identified risk alleles that regulate expression of genes involved in epithelial function, including IL1RL1, IL33, TSLP, CDHR3, MUC5AC, KIF3A, EFHC1 and GSDMB, support the role of the airway epithelium as driver of asthma pathogenesis.
1. INTRODUCTION
Asthma is a chronic inflammatory airway disease characterized by coughing, wheezing, chest tightness, variable airflow limitation and airway hyper‐responsiveness (AHR) 1 to environmental specific (allergens such as house dust mite (HDM), pollen and animal dander) and nonspecific (eg tobacco smoke, air pollution) stimuli. Asthma is a heterogeneous disease with a complex aetiology. Allergen‐induced asthma is the most common form, with atopy and allergic sensitization being identified as major risk factors. 2 Other risk factors include increased viral infections during early childhood, exposure to tobacco smoke and air pollution. 3 In addition to elevated serum IgE, features of atopic asthma include chronic eosinophilic airway inflammation and airway remodelling with increased smooth muscle mass, subepithelial fibrosis, epithelial desquamation and goblet cell hyperplasia. Type‐2T‐helper (Th2) lymphocytes are key players in the eosinophilic airway inflammatory response of allergen‐sensitized individuals, giving rise to the pathological changes and clinical symptoms of asthma. 4 Other asthma endotypes include nonallergic eosinophilic asthma, which may be driven by type‐2 innate lymphocytes, mixed‐granulocytic asthma, type‐1 and type‐17‐mediated neutrophilic asthma, and paucigranulocytic asthma, without apparent neutrophilia and eosinophilia. 5
Susceptibility to asthma has a strong genetic component. Many asthma susceptibility genes are expressed in the airway epithelium (eg IL1RL1, IL33, TSLP, CDHR3, PCDH1, MUC5AC, KIF3A, EFHC1 and GSDMB, as outlined below), highlighting the importance of the airway epithelium in the development of asthma. Allergens, viruses and other inhaled environmental insults are in first contact with the airway epithelial barrier, which forms a continuous lining of the respiratory system from the nose to the trachea, bronchi, bronchioles and finally the alveoli. The upper airway epithelium has a different developmental origin than the epithelia of the lower airway and alveolar epithelium. The nature of the epithelium changes in the specific regions, being a pseudostratified columnar epithelium in the nose, trachea and bronchi, transitioning into cuboidal cells in the bronchioles and forming a single‐cell thick alveolar epithelium. The alveolar epithelium is highly vascularized and responsible for gas exchange. The alveoli receive air from the conducting airways, starting in the trachea, bifurcating into the bronchi and bronchioles and ending in the terminal bronchioles, which divide into the alveolar ducts from which the alveoli arise. The transitional region between terminal bronchioles and alveoli is referred to as the bronchioalveolar duct junction. Alveolar cells can be subdivided into alveolar type 1 (AT1) epithelial cells, flat‐shaped epithelial cells that accommodate the transfer of oxygen into the blood stream and cuboidal‐shaped AT2 cells that serve as progenitor cells for AT1 cells, contribute to alveolar tissue regeneration upon injury and produce surfactants to reduce the surface tension. The pseudostratified epithelial layer of the conducting airways is separated from the underlying mesenchyme by the basement membrane and consists of different epithelial cell types: basal, club, goblet and ciliated cells being the major ones. Basal cells serve as progenitors, being able to differentiate into secretory club cells, which can further differentiate into mucus producing goblet cells or mucus clearing ciliated cells. 6 Club cells are able to self‐renew and generate ciliated cells after injury, repopulating damaged airway tissue. Secretory cells also have the capacity to dedifferentiate into basal cells when these cells are ablated by diphteria toxin, underscoring the remarkable plasticity of the airway epithelium. 7 While some studies have shown that ciliated cells are terminally differentiated, 8 others have shown that ciliated cells can undergo dynamic changes in cell shape and gene expression to re‐differentiate into columnar cells upon naphthalene induced injury. 9 In the presence of IL‐13, ciliated cells also undergo transdifferentiation into goblet cells. 10 In addition to the physical barrier function and mucociliary clearance of foreign particles, the airway epithelium acts as chemical barrier against environmental insults by secreting, for example antimicrobial peptides, anti‐proteases and antioxidants, and is part of the innate immune system. Airway epithelial cells express pattern recognition receptors (PRRs) like toll‐like receptors (TLRs), retinoic acid‐inducible gene (RIG)‐I‐like receptors (RLRs), nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs), C‐type lectin receptors (CLRs), protease activated receptor (PAR)‐2 and purinergic receptors. 11 These recognize pathogen‐associated molecular patterns (PAMPs) from inhaled microbes, parasites and allergens as well as alarmins/damage‐associated molecular patterns (DAMPs) released from dying or damaged cells. Upon recognition of PAMPs or DAMPs, PRRs activate downstream signalling that promotes the release of pro‐inflammatory cytokines/chemokines, including IL‐6, IL‐8, CCL20, CCL17, TSLP, IL‐25, IL‐33 and GM‐CSF. These can attract and/or activate cells from the innate and adaptive immune system. Upon sensing of allergens by various PRRs, including purinergic receptors, multiprotein complexes termed inflammasome can be activated, leading to caspase‐1 activity and subsequent cleavage of IL‐1β and IL‐18 into active forms. 12 In particular, HDM has been shown to activate the nucleotide‐binding domain and leucine‐rich repeat protein 3 (NLRP3) inflammasome through PI3K/Akt pathway leading to inflammation in asthma. 13 , 14 During these allergen‐driven inflammatory responses, dendritic cells (DCs) induce the differentiation Th2 cells, which secrete cytokines such as IL‐4, IL‐5, IL‐9 and IL‐13 to induce IgE production by B‐lymphocytes, eosinophilic infiltration into the airways and goblet cell hyperplasia with excessive mucus production. Epithelial alarmins can drive similar responses (independent of allergens) through activation of type‐2 innate lymphoid cells (ILC2). 15
Upon damage, for example by exposure to allergens, the epithelial barrier is disrupted, promoting epithelial release of growth factors such as epidermal growth factor (EGF) and TGF‐β, which activate fibroblasts and myofibroblasts. 16 This promotes excessive deposition of extracellular matrix (ECM) components, for example collagens, in the lamina reticularis just below the basement membrane, termed as subepithelial fibrosis, resulting in airway wall thickening and increased smooth muscle mass. 17 In addition, release of vascular endothelial growth factor (VEGF) by airway epithelial cells increases the size of airway wall vessels and promotes angiogenesis. 18 These structural changes are characteristic of airway remodelling in asthma (Figure 1). Thus, the airway epithelium may be crucial in the pathophysiology of asthma. In this review, we will focus on airway epithelial barrier dysfunction as driver of asthma.
FIGURE 1.
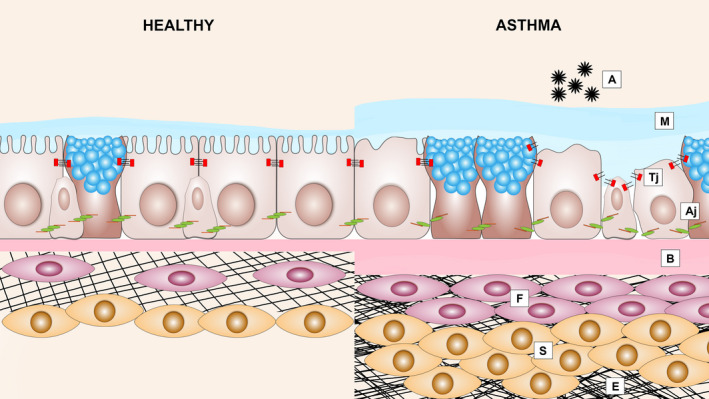
Structural changes in the airways of allergic asthma patients: Epithelial barrier dysfunction and airway remodelling. Asthmatic airway epithelium exposed to allergens (A) results in the disruption of adherens junctions (Aj) and tight junctions (Tj), which is accompanied by loss of ciliated cells, mucus hypersecretion (M), thickening of the basal membrane (B), subepithelial fibrosis (F), increased smooth muscle mass (S) and excessive deposition of ECM (E)
2. EPITHELIAL BARRIER DYSFUNCTION IN ASTHMA
The airway epithelial layer in asthma is disrupted, as indicated by detachment of ciliated cells, presence of epithelial cell aggregates (creola bodies) in sputum, increased permeability to allergens and reduced expression of cell‐cell adhesion molecule E‐cadherin. 19 , 20 Epithelial damage is a pathological feature observed in all phenotypes of asthma. 21 Structural changes have been observed in the airway epithelium of children with respiratory problems before the onset of airway inflammation and clinical diagnosis of asthma, suggesting that epithelial changes occur early in asthma pathogenesis. 2 This challenged the dogma that chronic airway inflammation induces airway remodelling. One of the key features of epithelial remodelling in asthma is the loss of cell‐cell contact proteins, which mechanically connect adjacent epithelial cells, thereby keeping the barrier intact. These intercellular junctions are mainly comprised of tight junctions (TJs), which are located most apically, adherens junctions (AJs) and (hemi)desmosomes, which are located basolaterally (Figure 2). Desmosomes form adhesive bonds with the filament cytoskeleton between adjacent cells or between cells and the lamina propria by nonclassical cadherins. 22 The major constituent of AJs is transmembrane protein E‐cadherin. Its extracellular domain binds homotypically to neighbouring cells, while the intracellular domain is linked to the actin cytoskeleton by a microtubule network of p120‐catenin, β‐catenin and α‐catenin proteins, providing mechanical support and intracellular signalling. E‐cadherin is thought to be crucial for formation of all other junctions, and its disruption results in delocalization of TJ proteins. 23 , 24 TJs are composed of the transmembrane proteins zona occludens‐1 (ZO‐1), occludin, claudins and junction adhesion molecules (JAMs) and are the main regulators of epithelial permeability. 25
FIGURE 2.
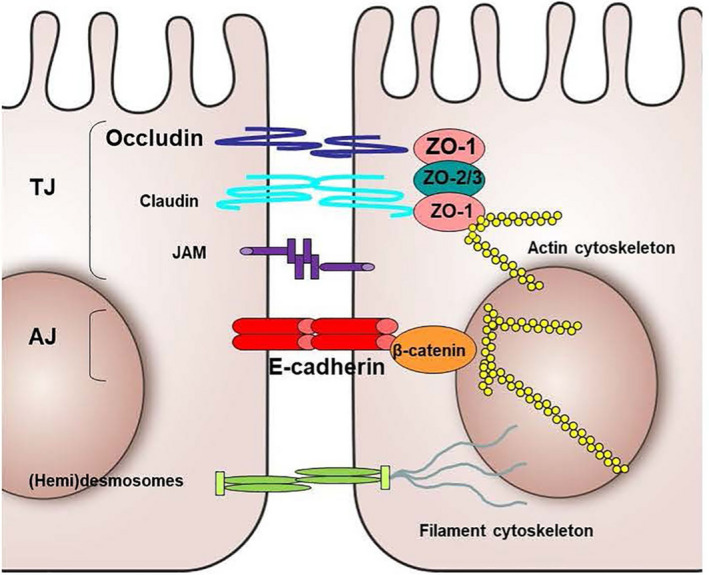
Schematic representation of the basic structural components of epithelial junctions. AJ, Adherens Junction; JAM, junctional adhesion molecule; TJ, Tight junction
Disrupted expression of E‐cadherin, β‐catenin, ZO‐1 and occludin has been observed in airway epithelium of asthma patients, 20 , 26 , 27 leading to impaired barrier function. 19 , 28 In murine studies, it has been demonstrated that the junctional proteins Zo‐1, Tjp2, Occludin and Claudins‐5,‐8,‐18 and ‐23 are decreased in all the three chronic HDM models of eosinophilic, neutrophilic and mixed granulocyte experimental asthma. 29 Animal models have also demonstrated that lung epithelial‐specific deficiency of E‐cadherin results in epithelial denudation with specific loss of ciliated cells 30 and that loss of E‐cadherin in club cells induces their proliferation while inhibiting differentiation, impairing epithelial repair upon injury. 31 Expression of E‐cadherin may not only be critical for the formation of a functionally intact epithelial layer, as downregulation of E‐cadherin is also crucial for epithelial plasticity, where cells lose their epithelial phenotype and gain mesenchymal characteristics, termed epithelial‐to‐mesenchymal transition (EMT). 32 Loss of E‐cadherin releases β‐catenin into the cytoplasm, where it is normally proteolytically degraded by a destruction complex including glycogen synthase kinase (GSK)‐3β. Inactivation of GSK‐3β, for example by active WNT signalling or TGF‐β, prevents the degradation of β‐catenin, resulting in nuclear translocation and transcriptional activation. Active β‐catenin, especially when bound to co‐activator CREB‐binding protein (CBP), promotes the expression of E‐cadherin repressors such as Snail and Slug as well as various mesenchymal genes, including fibronectin, EGF receptor (EGFR) and VEGF, which may contribute to airway wall remodelling. 22 The initial induction of a mesenchymal phenotype enables epithelial repair, promoting cell migration and proliferation. After this, cells differentiate into a pseudostratified epithelial layer. In asthma, this repair process may be disturbed, which is supported by the observed increase in basal cell markers (eg cytokeratin 5 and p63) 22 and repair markers (eg TGF‐β and EGFR) in the airway epithelium, representing a more proliferative, less differentiated phenotype. 22 HDM facilitates TGF‐β‐induced EMT in airway epithelial cells in vitro 33 and induces EMT‐like features in the airway epithelium of mice. 34 In asthma, epithelial cells are more susceptible to undergo TGF‐β‐induced EMT. 35 The Notch signalling pathway also plays a crucial role in controlling the fate of airway epithelial cells upon injury. Although the mechanisms by which Notch signalling modulates epithelial homeostasis and responses to environmental insults are incompletely understood, various Notch (target) genes are differently expressed in healthy and asthmatic airway epithelium. 36 , 37
The inability to reconstitute epithelial barrier function may have important pathophysiological consequences, not only resulting in increased permeability to allergens, but also propagating pro‐inflammatory and abnormal repair responses in the airways, leading to airway hyper‐responsiveness and airway remodelling 16 (Figure 3). Accordingly, airway epithelial damage has been shown to correlate with the severity of AHR. 38 Furthermore, the knock‐down of E‐cadherin in vitro resulted in EGFR activation and pro‐inflammatory responses. 32 Upon loss of E‐cadherin in vivo, the loss of ciliated cells was accompanied by spontaneous goblet cell metaplasia and infiltration of eosinophils and dendritic cells. 22 These features may at least in part be mediated by activation of β‐catenin, as inhibition of β‐catenin downstream activity attenuated airway inflammation, smooth muscle thickness, supepithelial fibrosis, hyper‐responsiveness and goblet cell metaplasia in mouse models of asthma. 39 Moreover, we recently demonstrated that inhibition of β‐catenin/CBP signalling not only improves epithelial barrier function, but also attenuates HDM‐induced airway epithelial pro‐inflammatory responses in vitro. 40
FIGURE 3.
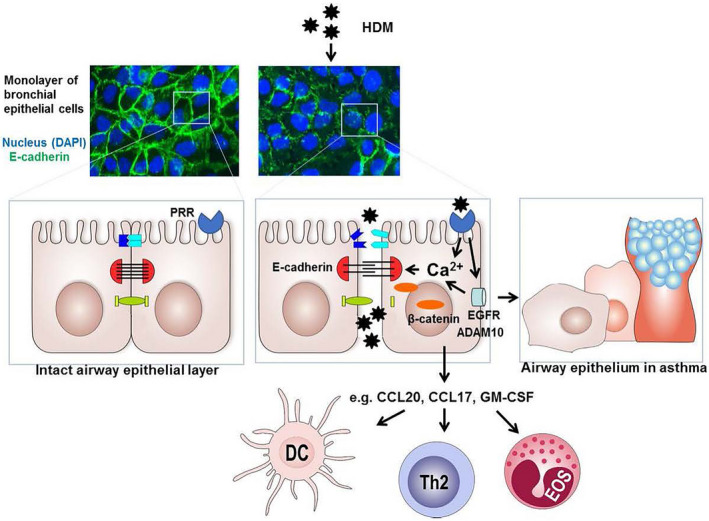
Proposed model of house dust mite (HDM)‐induced airway epithelium barrier dysfunction. Allergens including HDM can directly cleave epithelial junctions proteolytically or act on various pattern recognition receptors (PRRs), including PAR‐2, C‐type lectins (CLR) and purinergic receptors. Their activation can induce degradation and/or delocalization of junctional proteins, including E‐cadherin, in which intracellular Ca2+ signalling and subsequent activation of calpain may be involved and epidermal growth factor receptor (EGFR) activation. 161 EGFR can activate ADAM10, a sheddase of E‐cadherin as well as CCL20. 40 In addition, EGFR signalling can induce secretion of pro‐inflammatory mediators, such as CCL20, CCL17 and GM‐CSF that attract and/or activate dendritic cells (DCs), Th2 cells and eosinophils (EOS). 22 When epithelial repair and re‐differentiation is impaired, persistent loss of E‐cadherin can result in activation of β‐catenin‐mediated programs that cause further loss of epithelial characteristics, induction of a more basal/mesenchymal phenotype as well as goblet cell hyperplasia, with loss of ciliated cells, as is also characteristic of the epithelial phenotype in asthma
3. ENVIRONMENTAL RISK FACTORS AND EPITHELIAL BARRIER DYSFUNCTION IN ASTHMA
As described above, the development of asthma results from the interaction between genetic and environmental factors. Various in vitro studies have shown that allergens can disrupt the airway epithelial barrier. 41 Exposure of cultured airway epithelial cells to proteolytically active allergens from house dust mites (eg Der p1), ragweed, white birch, grass and pollen can lead to the cleavage of the junctional proteins. 22 Furthermore, house dust mite (HDM), cockroach, fungi and mould extracts have been shown to disrupt epithelial junctions via activation of PAR‐2 and downstream signalling. 42 Accordingly, exposure of human airway epithelial cells to HDM induces rapid, transient reduction in epithelial barrier function, 33 concomitant with delocalization of junctional proteins (Figure 3). Submerged cultures of airway epithelial cells from mild/moderate asthma patients were more susceptible to HDM‐induced barrier dysfunction than healthy subject‐derived cultures. Surprisingly, this was independent of serine and cysteine proteases. 43 , 44 Yet to be identified PRRs coupled to Ca2+/calpain‐dependent disruption of epithelial junctions may be involved. 43 In addition to direct effects of allergens, allergic sensitization may lead to epithelial barrier dysfunction as consequence of type‐2 mediated airway inflammation associated with atopic asthma. Both Th2 cells and ILC2 may contribute to the compromised epithelial barrier function through IL‐13 secretion, which induces many features of the airway epithelium in asthma, including mucus production, and has been reported to disrupt airway epithelial barrier function in vitro. 45 , 46 In fact, Th2‐derived IL‐13 and IL‐4 and type‐2 driving cytokine TSLP 47 have been shown to decrease barrier integrity in air‐liquid interface (ALI) cultured primary airway epithelial cells from healthy subjects, with delocalization of TJ proteins. 48 This was not observed in cultures derived from asthma subjects, in which barrier function was already compromised at baseline. 48 This may reflect cell‐intrinsic loss of airway epithelial cells in asthma to re‐differentiate and form an effective barrier upon ALI culture in vitro, as proposed previously. 2
In addition to allergens, early‐life sensitization to lower respiratory viral infections is an important environmental risk factor for developing asthma in childhood, with the highest risk for progression to persistent asthma when these environmental exposures coincide. 49 Two major respiratory viruses, rhinovirus (RV) and respiratory syncytial virus (RSV), bind to specific receptors on the airway epithelium, for example cadherin‐related family member 3 (CDHR3) and ICAM‐1 for RV 50 and CX3CR1 for RSV. 51 Upon internalization, uncoating and replication, the virus is recognized by TLR3 and RIG‐I like helicase, inducing production of anti‐viral type‐I interferons (IFNs), which eradicate pathogens and promote pro‐inflammatory cytokine release. Impaired epithelial barrier function is accompanied by compromised IFN responses in asthma, resulting in increased viral replication upon rhinovirus infection compared to nonasthma‐derived epithelial cultures. 52 Exposure of airway epithelial cells to double‐stranded RNA or infection with RV or RSV in vitro induces upregulation of TSLP 53 , 54 and may thus support type‐2 mediated inflammation. This may further impair epithelial barrier function in a vicious circle, viral exposure causing disruption of epithelial cell‐cell contacts. 55 , 56 RV has been shown to disrupt TJ integrity in human bronchial epithelial cell lines and ALI‐differentiated primary cultures via loss of ZO‐1 from TJs and airway epithelial cells cultures from healthy and asthmatic children, with more pronounced and sustained effects in asthmatic‐derived cultures. 57
Other environmental factors that may impact on epithelial integrity are those associated with nonatopic forms of asthma, for example noneosinophilic, neutrophilic asthma. Besides viral infections, these include smoking 58 and bacterial colonization. 21 Smoke exposure is well known to cause airway epithelial barrier dysfunction by disruption epithelial junctions. 59 Indirectly, smoking‐induced Th17‐mediated inflammation can reduce epithelial barrier function through Th17 cytokine IL‐17. 58 As colonization of the respiratory tract with bacteria, for example Streptococcus pneumoniae, Haemophilus influenzae or Moraxella catarrhalis, may increase the risk of asthma, it is of relevance that also bacteria can cause epithelial barrier dysfunction, as demonstrated for infection with S pneumonia in a bronchial epithelial cell line. 60
Finally, environmental pollutants such as particulate matter and ozone as well as household cleaning products may contribute to the development and/or worsening of asthma 61 and can disrupt the epithelial barrier. Particulate matter has been shown to attenuate ciliary beat frequency in bronchial epithelial cells and degrade TJ proteins in lung epithelial cells. 62 Diesel exhaust particles decreased the expression of TJ proteins and epithelial resistance in primary nasal epithelial cells. 63 Ozone was reported to cause rapid disruption of the epithelial barrier with increased permeability and diminished expression of TJ and AJ proteins in the absence of IL‐33. 64 Of interest, also laundry detergents were recently shown to compromise human bronchial epithelial integrity by disruption of tight junctions and may thus contribute to the development of asthma. 65
4. GENETIC FACTORS AND THE EPITHELIAL BARRIER IN ASTHMA
As mentioned above, in addition to environmental factors, a heredity component contributes to disease risk, with 35%‐95% of susceptibility thought to involve genetic factors. Positional cloning and more recently genome‐wide association studies (GWAS) have been highly successful in identifying risk alleles and loci for asthma and related phenotypes. 66
Expression quantitative trait loci (eQTL) studies in human bronchial epithelial cells and bronchial alveolar lavage identified that risk alleles regulate highly relevant genes involved in epithelial function, for example IL1RL1, IL33, TSLP, HLA‐DQB1, CDHR3, ZTB10, Corf30, DEX1 and GSDMB levels. 67 Similarly, Luo and colleagues combined asthma GWAS results and small and large airway epithelial eQTL data to demonstrate enrichment of airway epithelial eQTLs. 68 This supports the barrier hypothesis, where genetic alterations influence the ability of the skin and epithelial tissues to form a protective barrier from, for example pathogens and allergens. The finding that the majority of genetic variants associated with risk of developing asthma is shared risk factors for the development of atopic dermatitis and allergic rhinitis 69 further underlines this. Selected genes identified through asthma genetic studies and implicated in epithelial cell function are outlined in Table 1. Genetic changes in the epithelium may thus be important in mediating several aspects of relevance to asthma, including the inflammatory environment, for example IL33, TSLP, IL1RL1, responses to pathogens, for example CDHR3, mucociliary clearance, for example MUC5AC, KIF3A, EFHC1 and cell homeostasis and epithelial integrity, including proliferation, migration, cell‐cell adhesion, apoptosis and repair, for example PCDH1, SMAD3, GSDMB, ORMDL3 and PLAUR.
TABLE 1.
Selected genes identified through genetic studies of asthma implicated in airway epithelial cell homeostasis which may impact barrier properties and inflammation
| Chrs |
Gene Reported variants |
Main Asthma Phenotype(s) | Suggested role in HBEC homeostasis/epithelial gene expression | References |
|---|---|---|---|---|
| 2q12.1 |
a IL1RL1 rs3771166 |
Asthma, Asthma + Exacerbation, moderate‐severe asthma | IL33 receptor, regulates inflammation. Important in innate immune responses including responses to viruses and Type 2 inflammation. Expressed in HBEC | 74, 76, 138, 139 |
| 5q22.1 |
a TSLP rs1043828 |
Asthma, Asthma + Hay fever, moderate‐severe asthma | Can drive induction of allergic responses by effects on several cell types including dendritic cells. Regulates an IL‐13–dependent increase in bronchial epithelial cell proliferation | 138, 140, 141, 142, 143 |
| 5q31.1 |
KIF3A rs17690965 |
Atopic Dermatitis followed by Asthma | Molecular motor that transports molecules along microtubules, role in ciliary function. Role in epithelial apoptosis and inflammation | 70, 71, 144 |
| 5q31.3 |
PCDH1 rs3797054 rs3822357 |
Airway hyper‐responsiveness | Epithelial adhesion, differentiation, barrier formation | 78, 79, 145 |
| 6p12.2 |
EFHC1 rs9357733 |
Atopic Dermatitis followed by Asthma | Contains an EF‐hand motif which is able to bind Ca2+ ions. Involved in ciliary function | 70, 72, 73 |
| 7q22.3 |
a CDHR3 rs6967330 |
Asthma + Exacerbation | Epithelial polarity and cell‐cell interactions. Receptor for Rhinovirus C, the most common respiratory virus associated with exacerbations in asthma. Cys529Tyr regulates viral entry | 76, 77, 146 |
| 9p24.1 |
a IL33 rs1342326 |
Asthma, Asthma + Exacerbation, moderate‐severe asthma | Epithelium‐derived cytokine alarmin, regulates inflammation via interactions with ST2/IL1RL1 on several inflammatory cells. Type 2 inflammation, viral exacerbation. Also activates HBEC via ST2/IL1RL1 | 74, 76, 138, 147, 148 |
| 11p15.5 |
a MUC5AC rs11603634 |
Moderate‐Severe asthma | Oligomeric mucus/gel‐forming, a pathogenic mucin linked to allergic airway hyper‐reactivity. Elevated in bronchial epithelial cell brushing from severe asthma patients | 74, 75 |
| 15q22.33 |
SMAD3 rs744910 |
Asthma, Asthma + Hay fever | Signalling intermediate in the TGF‐β1 induced epithelial–mesenchymal transition | 69, 74, 75, 139, 149, 150 |
| 17q21.1 |
a GSDMB rs7216389 |
Asthma, childhood asthma + exacerbations, Asthma + Hay fever, childhood asthma, moderate–severe asthma | Member of gasdermin‐domain containing protein family, elevated in the airway epithelium in asthma and in mice increased expression led to spontaneous, remodelling and airway hyper‐responsiveness. Epithelial cell pyroptosis | 76, 82, 83, 140, 151 |
| 17q21.1 |
ORMDL3 rs7216389 |
Asthma, childhood asthma + exacerbations, Asthma + Hay fever, childhood asthma, moderate–severe asthma | Orosomucoid‑like protein isoform 3, regulates endoplasmic reticulum (ER) stress. Implicated in epithelial barrier formation, pro‐remodelling phenotype in vivo and in vitro. Sphingolipid regulation | 69, 76, 138, 151, 152, 153, 154, 155, 156 |
| 19q23 |
PLAUR rs4493171 rs2356338 rs2239372 |
Asthma, decline in lung function | Regulates activation of urokinase plasminogen activator (uPA), triggering the plasminogen/plasmin activation cycle. Epithelial repair, proliferation, pro‐remodeling phenotype | 157, 158, 159, 160 |
For a comprehensive review of asthma related phenotypes, these loci have been associated with see recent reviews. 54 , 142
Abbreviations: CDHR3 cadherin‐related family member 3; EFHC1, EF‐hand domain containing protein 1; IL1RL1, Interleukin 1 Receptor Like 1; IL33, Interleukin 33; KIF3A, Kinesin Family Member 3A; MUC5AC, Mucin 5AC, Oligomeric Mucus/Gel‐Forming; ORMDL3, ORMDL sphingolipid biosynthesis regulator 3; PCDH1, Protocadherin 1; PLAUR, plasminogen activator, urokinase receptor; SMAD3, GSDMB, gasdermin B; TSLP, Thymic stromal lymphopoietin.
Identified in eQTL studies using asthma risk alleles in airway epithelium.
While a discussion of all these genes is beyond this review, it is important to highlight selected genes particularly implicated in barrier function. In the GWAS of atopic dermatitis followed by asthma, two genes thought to be involved in ciliary function were implicated, that is KIF3A and EFHC1. 70 These genes encode for Kinesin Family Member 3A and EF‐hand domain containing protein 1, respectively. KIF3A is thought to function as a molecular motor transporting molecules along microtubules and has also been implicated in ciliary function in epithelial cells. Interestingly, mice deficient in KIF3A in the epithelium is more susceptible to allergen‐induced inflammation and epithelial cell apoptosis in an allergic airway model. 71 Mutations within EFHC1 have been associated with juvenile myoclonic epilepsy via a role in motile cilia and in regulating calcium channels. 72 , 73 Importantly, EFHC1 may be of relevance in cilia function in the airways, being expressed in the tracheal epithelium in mice. Therefore, KIF3A and EFHC1 may in part contribute to poor allergen and mucus clearance from the airways. Recently, in a GWAS of moderate‐severe asthma, a signal on chromosome 11 was identified that regulates expression of MUC5AC, 74 the main mucin found in the airways and linked to severe asthma, 75 emphasizing abnormal mucociliary clearance. In a GWAS of asthma with exacerbation, polymorphisms spanning CDHR3 were identified, including coding change Cys529Tyr. 76 CDHR3 is involved in epithelial polarity and cell‐cell interactions. As described above, recent data suggest that CDHR3 is the receptor for RV‐C and the Cys529Tyr mediates this interaction providing a putative mechanism. Interestingly, CDHR3 knock‐down also influences transepithelial resistance. 77 The PCDH1 gene also encodes an adhesion molecule localizes to cell‐cell junctions especially in differentiated airway epithelial. 78 , 79 PCDH1 has a dual function, supporting epithelial barrier function 79 and regulating TGF‐β/SMAD3 signalling. 80 Hence, PCDH1 may serve as cellular switch between TGF‐β driven EMT and epithelial repair vs epithelial differentiation and barrier formation. The gene ORMDL3 regulates cytosolic Ca2+ entry by the sarco‐endoplasmic reticulum (ER) Ca2+ ATPase (SERCA) pump, which we previously showed to be involved in HDM‐induced epithelial barrier dysfunction. 43 Moreover, the ORMDL3 gene product was recently shown to support RV replication in epithelia cells. 81 Finally, GSDMB encodes gasdermin B, which is a member of the gasdermin‐domain containing protein family linked to epithelial apoptosis. Recently, it has been shown that GSDMB is elevated in the airway epithelium in asthma. In mice, increased expression led to spontaneous airway hyper‐responsiveness, 82 and the GSDMB protein induces pyroptotic cell death in airway epithelium. 83 Although several asthma genes have been shown to act on airway epithelial function, a clear endotype of asthma driven by the loss of epithelial barrier specifically due to these asthma‐associated polymorphisms has not been identified. However, it is important to note that the asthma phenotypes associated with these selected genetic signals include bronchial hyper‐responsiveness (PCDH1, PLAUR, ORMDL3/GSDMB) and asthma exacerbation (IL33, IL1RL1, CDHR3, ORMDL3/GSDMB), potentially directly by effects on bronchial epithelial function. Similarly, genetic signals associate with blood eosinophil counts (IL33, IL1RL1, TSLP), time to asthma onset (IL33, IL1RL1, ORMDL3/GSDMB), atopic march (KIF3A, EFHC1) and self‐reported allergy (IL33, ORMDL3/GSDMB, IL1RL1), potentially via an indirect mechanism by the production of cytokines from bronchial epithelial cells leading to type‐2 inflammation. 84 , 85 The gene signature of the type‐2 high endotype of asthma, characterized by increased blood and BAL eosinophils and basal membrane thickness, lower PC20 threshold and a better lung function improvement after inhaled corticosteroids, identifies this asthma subphenotype as a steroid responsive signature of epithelial cells in asthma, 86 indicating the relevance of the airway epithelial phenotype in the disease. Two of these genes (CLCA1 and SERPINB2) are predominantly expressed in goblet cells, indicating that a true asthma endotype reflecting loss of epithelial barrier function is yet to be identified.
5. EPIGENETIC FACTORS AND THE EPITHELIAL BARRIER IN ASTHMA
As outlined, asthma‐associated polymorphisms can directly alter a gene's coding sequences, thereby altering protein function and, consequently, the biology of the airway epithelium. More frequently, however, asthma‐associated SNPs have a regulatory effect on gene expression, acting as eQTLs. A recent study shows that almost 59% of the asthma‐associated SNPs identified by the Trans‐National Asthma Genetic Consortium (TAGC) study is an eQTL in nasal epithelium and that in almost 90% of these cases, this effect is mediated by CpG methylation. 87 Clearly, epigenetic regulation of gene expression is highly relevant to the translation of disease susceptibility into altered biology of the airway epithelium. Epigenetic marks are highly responsive to environmental exposures relevant to asthma inception or exacerbations, further underscoring the relevance of epigenetics for understanding asthma pathophysiology. 88 , 89 , 90 , 91 Three main types of epigenetic marks can be distinguished: CpG methylation, histone modifications and small, noncoding RNAs.
Differences in DNA methylation patterns between asthma patients and healthy controls have been studied in (epi)genome‐wide analyses (EWAS). As CpG methylation patterns are also highly cell‐type dependent, cell‐type composition of the biological sample is an important cofounder of EWAS analyses. 92 Therefore, we here focus on the studies in upper (nasal) airway brushes, that mainly consist of epithelial cells, 93 and which were shown to have the best correlation to the DNA methylation patterns in bronchial epithelial cells. 94 In four studies reported to date, 95 , 96 , 97 , 98 methylation of the GJA4 gene, encoding Connexin37, a protein capable of forming heterotypic gap junctions, was consistently found to be reduced, although an association with altered gene expression levels was not detected. 97 Other genes relevant to epithelial barrier function (CDH26, CDHR3) were also found differentially methylated. 95 , 97 In addition to CDHR3, another genes selectively expressed in ciliated epithelial cells, ZMYND10 91 was found to be differentially methylated, which is consistent with an altered airway epithelial composition in asthma. Only one study to date analysed CpG methylation in bronchial biopsies from asthma patients and healthy controls, but this analysis was focussed on methylation patterns associated with remission of asthma. 99
Several studies have looked specifically into DNA methylation changes induced by relevant environmental factors, which affect epigenetic regulation of asthma genes. 100 , 101 , 102 RV infection‐induced DNA methylation patterns differed between nasal epithelial cells from asthmatic children and healthy controls, with enrichment for loci carrying genes involved in cell‐cell and cell‐matrix interactions. 100 Similarly, RV infection‐induced DNA methylation patterns differed between nasal epithelial cells from asthmatic adults and healthy controls. 102 In children who had early‐life rhinovirus‐induced wheezing, specific DNA methylation patterns associated with asthma later in life were identified, including increased methylation at the SMAD3 locus. 101 Finally, one elegant study analysed the effects of diesel exhaust particle exposure and (segmental) allergen challenge on DNA methylation patterns in airway epithelial cells obtained by bronchial brushing both 48 hours after exposure and after 4 weeks. 103 While both allergen challenge and diesel exhaust particle exposure induced DNA methylation changes in airway epithelial cells, the most pronounced effects were observed in individuals who received an allergen challenge 4 weeks prior to exhaust particles exposure, with genes annotated to cell adhesion being most enriched in the differentially methylated regions. 103 These data clearly indicate the relevance of environmental exposures for epigenetic regulation of gene expression in the airway epithelium and therefore for asthma. As the epigenetic signature of the airway epithelium integrates genetic susceptibility with the life history of relevant environmental exposures, it can be expected to be a strong biomarker for asthma development or even treatment response. 98
In addition to DNA methylation, epithelial gene expression can be modulated by miRNAs, which are small noncoding RNAs of about 21‐25 nucleotides that can bind to target mRNAs, leading to mRNA degradation or translational repression. Altered miRNA profiles have been observed in airway epithelium of asthma patients compared to healthy controls. 104 , 105 Several of the differentially expressed miRNAs modulate the expression of genes implicated in epithelial barrier function, repair, proliferation or apoptosis. For example, miR‐744, miR‐19a, miR‐221, miR‐27a, miR‐128 and miR‐34/449 are differentially expressed in bronchial epithelial cells from asthma patients compared to controls and have been described to modulate cell proliferation, apoptosis and ciliogenesis by targeting TGF‐β1, TGF‐βR2, SIRT1, SMAD2 (target of both miR‐27a and miR‐128) and Cp110, respectively (Figure 4). 106 , 107 , 108 , 109 , 110 , 111 Of interest, the discussed miRNAs were not all identified in patients with the same disease severity. The differential expression of miR‐744, miR‐221 and miR‐19 was shown in HBEC from severe asthma patients with an atopic and eosinophilic phenotype, 106 , 107 , 108 whereas miR‐34/449 was identified in mild atopic asthma. 105 While miR‐19 was higher in severe atopic eosinophilic asthma, its expression in mild asthma was similar as in healthy controls. 108 Moreover, a miR‐19 mimic induced more proliferation in HBEC from severe asthma patients than in control‐derived HBEC. The expression of miR‐744 was reduced in HBEC from severe asthma, but tended to increase in mild asthma compared to healthy controls. 106 These observations suggest that the impact of the miRNAs on the epithelium can differ with disease severity; however, this requires further investigation. It is also unknown whether miRNAs that affect epithelial barrier in asthma modify the treatment response, but for miR‐34/449, miR‐19 and miR‐223, there were no correlations between miRNA expression and treatment with inhaled corticosteroids. 105 , 108 , 112 Furthermore, miR‐155 and miR‐223 have been implicated in EMT by altering mesenchymal markers, 112 , 113 although their exact role in asthma airway epithelial cells is unknown. Also, differential expression of miR‐3162, miR‐125b, miR‐223 and miR‐330 in blood or sputum from asthma patients, possibly transported in extracellular vesicles (EVs), can affect the epithelial barrier function by influencing the expression of, for example β‐catenin, vimentin and mucins 112 , 114 , 115 , 116 , 117 (Figure 4). Moreover, airway epithelium itself can communicate by secreting EVs. Epithelial‐derived EVs play a role in airway homeostasis and airway epithelial remodelling by inducing amongst others mucin hypersecretion. 117 The miRNA signature in epithelial‐derived EVs is altered upon stimulation with IL‐13 compared to EVs obtained from untreated bronchial epithelial cells. 118 However, it is unclear whether similar changes can be observed in the miRNA profile of epithelial‐derived EVs from asthma patients and whether those changes in miRNA expression affect the epithelial barrier. In asthma murine models, lower miR‐448‐5p and higher miR‐106a levels were expressed in lung tissue compared to control mice. 111 , 119 In vitro up‐ or downregulation of these miRNAs resulted in altered protein levels of E‐cadherin, fibronectin, collagen IV and vimentin in bronchial epithelial cells after TGF‐β1 stimulation. 111 , 119
FIGURE 4.
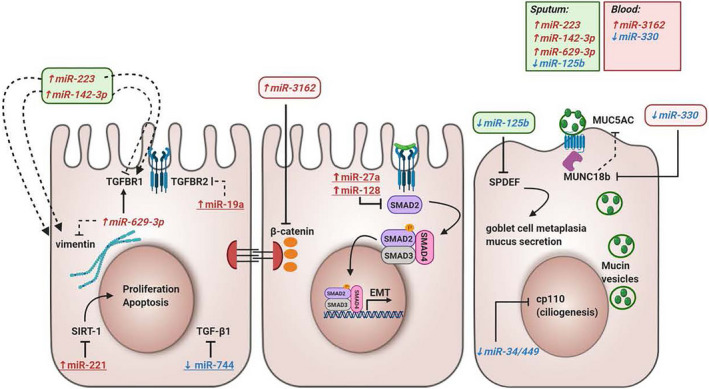
The influence of microRNAs in epithelial barrier function. This overview illustrates miRNAs that are differentially expressed in asthma and could contribute to epithelial barrier dysfunction in asthma. miRNAs coloured in red with upward arrow are upregulated in asthma, and miRNAs coloured in blue with downward arrow are downregulated in asthma. miRNAs with an underscore were measured in bronchial epithelial cells, and miRNAs in italic were measured in sputum or blood from asthma patients and controls. Black lines ending with a perpendicular line indicate inhibitory effects, and black lines ending with an arrow indicate a stimulatory effect. Full lines indicate direct effects, and half‐full lines indicate indirect effects. EMT, epithelial‐mesenchymal transition; LPS, lipopolysaccharide; SIRT‐1, Sirtuin 1; SPDEF, SAM Pointed Domain Containing ETS Transcription Factor; TGF‐β1, Transforming Growth Factor Beta 1; TGFBR1, Transforming Growth Factor Beta Receptor 1
Together, the interaction between genetic factors and epigenetic regulatory mechanisms may contribute to abnormalities in the airway epithelium and the development of asthma. Understanding these mechanisms may lead to identification of novel targets in airway epithelium for asthma intervention.
6. NEW INSIGHTS FROM SINGLE‐CELL SEQUENCING DATA
Further insight into the mechanisms of asthma and the role of the airway epithelium may come from technological advances. These include recent progress in single‐cell RNA sequencing (scRNA‐Seq), greatly enhancing the granularity at which the cellular composition of tissues can be characterized. 120 In addition, scRNA‐Seq allows the description of molecular cell phenotypes (or cellular “states”), predict cell‐cell interactions and cell state transitions at unprecedented detail. Using these technologies to study lung tissue, the ionocyte has recently been discovered as novel airway epithelial cell. 6 , 121 The pulmonary ionocyte is a relatively rare cell type, characterized by expression of ion transporters including V‐ATPase and the cystic fibrosis CFTR gene, indicating a role in regulation of ion and fluid transport across the airway epithelium as well as pH of the mucosal surface. While the application of these technologies to identify all cell types of the healthy human body, including lung, as pursued by the Human Cell Atlas consortium 122 , 123 are exciting, these novel techniques also hold great potential to increase our understanding of disease pathogenesis. A first description of the cellular landscape of healthy airway wall and the changes thereof in patients with childhood‐onset allergic asthma identified a unique disease‐associated airway epithelial cell state, as well as a remarkable shift in cell‐cell communication. 93 Various known changes in the asthmatic airway wall were recapitulated by scRNA‐Seq analysis, such as increased numbers of airway smooth muscle cells, goblet cells and mast cells, underscoring the validity of the approach (Figure 5). The study identified a subset of ciliated epithelial cells in asthma that was characterized by expression of MUC5AC and other goblet‐cell genes, a molecular phenotype of ciliated cells that was not observed in healthy airway walls. 93 This so‐called mucous ciliated cell type was mapped to the ciliated differentiation trajectory. Interestingly, these mucous ciliated cells as well as the goblet cells in asthma lacked expression of Notch target genes, but instead expressed a signature of IL4/IL13‐induced genes, which was in contrast to the (few) goblet cells present in airway wall from healthy donors. Therefore, mucous ciliated cells were proposed to represent a transitional cell state in the ciliated lineage—induced by IL‐4/IL‐13 signalling—leading to a mucous cell phenotype that contributes to mucous cell metaplasia in asthma. 93 As these pathogenic Th2 effector cells were exclusively observed in asthmatic airway walls, and the mucous ciliated cells showed evidence of IL‐13‐induced gene transcription, it seems likely that Th2 cytokines are responsible for these cell state changes in the asthmatic airway epithelium. Indeed, Th2 effector cells were found to dominate the predicted airway wall cell‐cell interactome in asthma. 93 We previously reported that Th2 cytokine production was suppressed by primary bronchial epithelial cells, a regulatory mechanism that seems to be attenuated in asthma. 124 The airway wall cellular interactome analysis also identified cell‐cell communication between epithelial cells and other structural or tissue‐resident cells, characterized by growth factor signalling. This interaction was present in healthy airway wall, but lost in asthma. 93 Therefore, it will be of great interest to study which cell‐cell interactions observed in healthy airway wall maintain the barrier function of airway epithelium, and how these can be restored in the asthmatic condition. Future studies in larger cohorts of patients and controls, as well as in a larger variety of asthma subphenotypes also hold the promise of charting the cellular changes and causal gene regulatory networks underlying a wider variety of asthma endotypes. Moreover, analysis of matched scRNA‐Seq data sets from airway wall biopsies, bronchial brushes and nasal brushes will allow design of novel biomarkers for disease activity or treatment response using less invasive methodologies.
FIGURE 5.
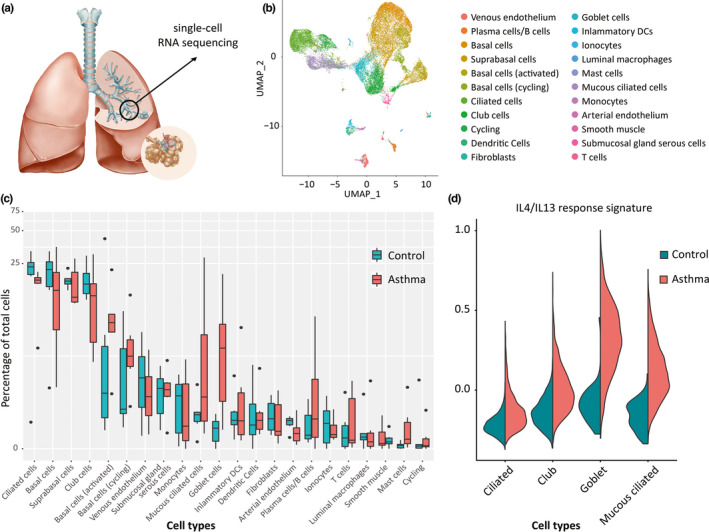
Analysis of airway epithelial cells in asthma using single‐cell RNA sequencing. (A) Airway wall biopsies are obtained from 5th‐7th generation airway through bronchoschopy, followed by tissue digestion and scRNA‐Seq analysis. (B) Unsupervised clustering identifies a large number of epithelial and nonepithelial cell types from airway wall. (C) Comparison of relative frequencies of cell types identified increased number of goblet cells and mucous ciliated cells, a novel, disease‐associated ciliated epithelial cell phenotype and increased numbers of mast cells and B cells in asthma compared to healthy. (D) Analysis of epithelial cell subset‐specific transcriptomes reveals presence of IL4/IL13‐induced gene transcription in goblet cells and mucous ciliated cells, specifically in asthma
7. THERAPEUTIC STRATEGIES TO IMPROVE BARRIER FUNCTION
Targeting the airway epithelial barrier may constitute a promising novel therapeutic strategy for asthma and related allergic diseases. Intrinsic abnormalities in the airway epithelium of asthmatics culminate in inappropriate immune and inflammatory responses as well as defective repair. Genetically supported targets could double the success rate in clinical development.
A number of pathways involved in maintaining or restoring epithelial barrier function are targetable; these include those (a) enhancing mucosal innate immunity, (b) decreasing epithelial permeability through effective assembly of TJ and AJ proteins and (c) restoring epithelial cell integrity by improving regeneration and regulating mucus production. Modulation of several developmental transcription factors has been shown to improve epithelial differentiation and as a consequence, barrier function. We recently demonstrated that inhibition of β‐catenin/CBP signalling inhibits EMT and promotes recovery of epithelial barrier function through restoration of E‐cadherin expression. 40 , 80 , 125 , 126 Notch signalling appears to be intimately involved in regulating mucus cell fate and mucus release. 127 Recent studies from our laboratory and others have shown that modulating Notch signalling has a dramatic effect on mucus secretion. 37 In addition, Smad3 inhibitors may reverse airway epithelial abnormalities as observed in asthma, as reviewed previously. 2 Because of the described effects of type‐2 cytokines on epithelial barrier function, we anticipate that new biologics may have beneficial effects on airway epithelial barrier function specifically in type‐2 driven asthma; however, to the best of our knowledge, there are no studies yet that assessed this.
The majority of patients respond well to a combination of inhaled corticosteroids (ICS) and bronchodilators. Whether or not ICS have direct beneficial effects on epithelial health or barrier function is unclear. Although corticosteroids failed to prevent the TGF‐β‐induced downregulation of E‐cadherin in a bronchial epithelial cell line, 128 findings in primary bronchial epithelial cells indicate that ICS protect against oxidative stress‐induced epithelial barrier dysfunction. 129 However, asthma epithelium was found less responsive to ICS. 129 Oxidative stress as well as IL‐17 may lead to ICS unresponsiveness by PI3K‐dependent post‐translational histone deacetylase (HDAC)2 modifications and proteasomal HDAC2 degradation. 130 , 131 Strategies to restore ICS sensitivity could be beneficial in improving epithelial barrier function in asthma in combination with ICS, including the use of antioxidants or α‐IL‐17 antibodies. 132 Endotype‐specific therapies that have been recently developed to mitigate symptoms in patients refractory to conventional ICS‐based therapy may largely have their impact through effects on immune/inflammatory components though.
8. CONCLUDING REMARKS
The airway epithelial phenotype induced by the interaction of genotype and environment plays a central role in the pathogenesis of asthma. Accumulating evidence indicates that multiple genetic variants associated with the risk of developing asthma in response to environmental factors regulate proteins of relevance to airway epithelial function, including roles in barrier function, inflammation, mucociliary clearance and homeostasis. In addition, alterations in epigenetic regulation contribute to abnormalities in the biology of the airway epithelium in asthma. Further insight into these regulatory mechanisms, for example by the use of scRNA‐seq, holds promise for identifying patients likely to benefit from epithelial‐focused therapies and the identification of targets for novel therapies strategies aimed at correcting dysfunctional epithelial barrier.
CONFLICT OF INTEREST
Dr Maes reports grants from Ghent University, Fund for Scientific Research Flanders (FWO; G053516N, G041819N, FWO‐EOS project G0G2318N), during the conduct of the study; personal fees from GlaxoSmithKline, outside the submitted work, and is shareholder of Oryzon Genomics and of Mendelion Lifesciences SL; Prof. Nawijn reports grants from the Netherlands Lung Foundation (LF 14.020 and LF 18.226), during the conduct of the study. Outside of the submitted work, Prof. Sayers laboratory reports grants from Asthma UK, British Lung Foundation Nottingham University Hospitals, National Institute for Health Research, Medical Research Council, GlaxoSmithKline and Boehringer Ingelheim; Prof. Nawijn reports grants from GSK; Prof. Heijink reports grants from the Netherlands Lung Foundation (LF 15.017) and Boehringer Ingelheim.
ACKNOWLEDGMENTS
We thank J. Eliasova (scientific illustrator) for support with the design of figures and M. Berg for support with creating the figures.
Heijink IH, Kuchibhotla VNS, Roffel MP, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. 2020;75:1898–1913. 10.1111/all.14421
Footnotes
The shared genetic origin of asthma, rhinitis and eczema was recently analysed in detail. 133 This approach revealed a striking overlap in risk SNPs between these three allergic disorders, with limited disease‐specific polymorphisms. The study identified a total of 132 plausible target genes, which were enriched for expression in blood and lung tissue. 133 These results clearly indicate that susceptibility to allergic diseases is mediated by at least in part shared biological mechanisms. Loss of epithelial barrier function has indeed been postulated as a central mechanism in allergic rhinitis 134 and eczema 135 as well, with loss of function variants in epidermal protein filaggrin being identified as major predisposing factor of atopic dermatitis. 136 In addition, GWAS studies have identified epithelial junction protein Desmoglein 1 as susceptibility gene for eosinophilic esophagitis. 137
REFERENCES
- 1. The Global Asthma Report 2018; 2018. http://www.globalasthmareport.org/. Accessed January 14, 2020.
- 2. Heijink IH, Nawijn MC, Hackett TL. Airway epithelial barrier function regulates the pathogenesis of allergic asthma. Clin Exp Allergy. 2014;44(5):620‐630. [DOI] [PubMed] [Google Scholar]
- 3. Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355(21):2226‐2235. [DOI] [PubMed] [Google Scholar]
- 4. Dunican EM, Fahy JV. The role of type 2 inflammation in the pathogenesis of asthma exacerbations. Ann Am Thorac Soc. 2015;12(Suppl 2):S144‐S149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Svenningsen S, Nair P. Asthma endotypes and an overview of targeted therapy for asthma. Front Med (Lausanne). 2017;4:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Montoro DT, Haber AL, Biton M, et al. A revised airway epithelial hierarchy includes CFTR‐expressing ionocytes. Nature. 2018;560(7718):319‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tata PR, Mou H, Pardo‐Saganta A, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503(7475):218‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rawlins EL, Hogan BL. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L231‐L234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park KS, Wells JM, Zorn AM, et al. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol. 2006;34(2):151‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turner J, Roger J, Fitau J, et al. Goblet cells are derived from a FOXJ1‐expressing progenitor in a human airway epithelium. Am J Respir Cell Mol Biol. 2011;44(3):276‐284. [DOI] [PubMed] [Google Scholar]
- 11. Wills‐Karp M. Allergen‐specific pattern recognition receptor pathways. Curr Opin Immunol. 2010;22(6):777‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee TH, Song HJ, Park CS. Role of inflammasome activation in development and exacerbation of asthma. Asia Pac Allergy. 2014;4(4):187‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim SR, Park HJ, Lee KB, et al. Epithelial PI3K‐delta promotes house dust mite‐induced allergic asthma in NLRP3 inflammasome‐dependent and ‐independent manners. Allergy Asthma Immunol Res. 2020;12(2):338‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu T, Zhou YT, Wang LQ, et al. NOD‐like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J Allergy Clin Immunol. 2019;144(3):777‐787.e9. [DOI] [PubMed] [Google Scholar]
- 15. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18(5):684‐692. [DOI] [PubMed] [Google Scholar]
- 16. Boxall C, Holgate ST, Davies DE. The contribution of transforming growth factor‐{beta} and epidermal growth factor signalling to airway remodelling in chronic asthma. Eur Respir J. 2006;27(1):208‐229. [DOI] [PubMed] [Google Scholar]
- 17. Fehrenbach H, Wagner C, Wegmann M. Airway remodeling in asthma: what really matters. Cell Tissue Res. 2017;367(3):551‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee CG, Ma B, Takyar S, et al. Studies of vascular endothelial growth factor in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2011;8(6):512‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hackett TL, Singhera GK, Shaheen F, et al. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol. 2011;45(5):1090‐1100. [DOI] [PubMed] [Google Scholar]
- 20. Xiao C, Puddicombe SM, Field S, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128(3):549‐556.e12 [DOI] [PubMed] [Google Scholar]
- 21. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. The Lancet. 2018;391(10122):783‐800. [DOI] [PubMed] [Google Scholar]
- 22. Nawijn MC, Hackett TL, Postma DS, van Oosterhout AJ, Heijink IH. E‐cadherin: gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 2011;32(6):248‐255. [DOI] [PubMed] [Google Scholar]
- 23. Heijink IH, Kies PM, Kauffman HF, Postma DS, van Oosterhout AJ, Vellenga E. Down‐regulation of E‐cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor‐dependent Th2 cell‐promoting activity. J Immunol. 2007;178(12):7678‐7685. [DOI] [PubMed] [Google Scholar]
- 24. Tunggal JA, Helfrich I, Schmitz A, et al. E‐cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005;24(6):1146‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol 2008;86(3):105‐112. [DOI] [PubMed] [Google Scholar]
- 27. Hackett TL, de Bruin HG, Shaheen F, et al. Caveolin‐1 controls airway epithelial barrier function. Implications for asthma. Am J Respir Cell Mol Biol. 2013;49(4):662‐671. [DOI] [PubMed] [Google Scholar]
- 28. Heijink IH, Brandenburg SM, Noordhoek JA, Postma DS, Slebos DJ, van Oosterhout AJM. Characterisation of cell adhesion in airway epithelial cell types using electric cell‐substrate impedance sensing. Eur Respir J. 2010;35(4):894‐903. [DOI] [PubMed] [Google Scholar]
- 29. Tan HT, Hagner S, Ruchti F, et al. Tight junction, mucin, and inflammasome‐related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy. 2019;74(2):294‐307. [DOI] [PubMed] [Google Scholar]
- 30. Post S, Heijink IH, Hesse L, et al. Characterization of a lung epithelium specific E‐cadherin knock‐out model: implications for obstructive lung pathology. Sci Rep. 2018;8(1):13275‐14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ceteci F, Ceteci S, Zanucco E, et al. E‐cadherin controls bronchiolar progenitor cells and onset of preneoplastic lesions in mice. Neoplasia. 2012;14(12):1164‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hackett TL. Epithelial–mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12(1):53‐59. [DOI] [PubMed] [Google Scholar]
- 33. Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite‐induced epithelial barrier dysfunction. Eur Respir J. 2010;36(5):1016‐1026. [DOI] [PubMed] [Google Scholar]
- 34. Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial‐mesenchymal transition in the large airways. PLoS One. 2011;6(1):e16175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hackett TL, Warner SM, Stefanowicz D, et al. Induction of epithelial‐mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor‐beta1. Am J Respir Crit Care Med. 2009;180(2):122‐133. [DOI] [PubMed] [Google Scholar]
- 36. Kuchibhotla VN, Heijink IH. Join or leave the club: jagged1 and Notch2 dictate the fate of airway epithelial cells. Am J Respir Cell Mol Biol. 2020. 10.1165/rcmb.2020-0104ED. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reid AT, Nichol KS, Chander Veerati P, et al. Blocking Notch3 signaling abolishes MUC5AC production in airway epithelial cells from individuals with asthma. Am J Respir Cell Mol Biol. 2020;62(4):513‐523. [DOI] [PubMed] [Google Scholar]
- 38. Laitinen LA, Heino M, Laitinen A, Kava T, Haahtela T. Damage of the airway epithelium and bronchial reactivity in patients with asthma. Am Rev Respir Dis. 1985;131(4):599‐606. [DOI] [PubMed] [Google Scholar]
- 39. Kumawat K, Koopmans T, Gosens R. Beta‐catenin as a regulator and therapeutic target for asthmatic airway remodeling. Expert Opin Ther Targets. 2014;18(9):1023‐1034. [DOI] [PubMed] [Google Scholar]
- 40. Kuchibhotla VNS, Jonker MR, de Bruin HG, Noordhoek JA, Knight DA, Nawijn MC, Heijink IH. Inhibition of β‐catenin/CBP signalling improves airway epithelial barrier function and suppresses CCL20 release. Allergy. 2020;75:1786‐1788. 10.1111/all.14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heijink IH, Noordhoek JA, Timens W, van Oosterhout AJ, Postma DS. Abnormalities in airway epithelial junction formation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(11):1439‐1442. [DOI] [PubMed] [Google Scholar]
- 42. Winter MC, Shasby SS, Ries DR, Shasby DM. PAR2 activation interrupts E‐cadherin adhesion and compromises the airway epithelial barrier: protective effect of beta‐agonists. Am J Physiol Lung Cell Mol Physiol. 2006;291(4):L628‐L635. [DOI] [PubMed] [Google Scholar]
- 43. Post S, Nawijn MC, Jonker MR, et al. House dust mite‐induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. Allergy. 2013;68(9):1117‐1125. [DOI] [PubMed] [Google Scholar]
- 44. Post S, Nawijn MC, Hackett TL, et al. The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax. 2012;67(6):488‐495. [DOI] [PubMed] [Google Scholar]
- 45. Schmidt H, Braubach P, Schilpp C, et al. IL‐13 impairs tight junctions in airway epithelia. Int J Mol Sci. 2019;20(13):3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sugita K, Steer CA, Martinez‐Gonzalez I, et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL‐13 in asthmatic patients. J Allergy Clin Immunol. 2018;141(1):300‐310.e11. [DOI] [PubMed] [Google Scholar]
- 47. Dong H, Hu Y, Liu L, et al. Distinct roles of short and long thymic stromal lymphopoietin isoforms in house dust mite‐induced asthmatic airway epithelial barrier disruption. Sci Rep. 2016;6:39559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wawrzyniak P, Wawrzyniak M, Wanke K, et al. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. J Allergy Clin Immunol. 2017;139(1):93‐103. [DOI] [PubMed] [Google Scholar]
- 49. Kusel MM, de Klerk NH, Kebadze T, et al. Early‐life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119(5):1105‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Basnet S, Palmenberg AC, Gern JE. Rhinoviruses and their receptors. Chest. 2019;155(5):1018‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Johnson SM, McNally BA, Ioannidis I, et al. Respiratory syncytial virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog. 2015;11(12):e1005318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wark PA, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mahmutovic‐Persson I, Akbarshahi H, Bartlett NW, et al. Inhaled dsRNA and rhinovirus evoke neutrophilic exacerbation and lung expression of thymic stromal lymphopoietin in allergic mice with established experimental asthma. Allergy. 2014;69(3):348‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee HC, Headley MB, Loo YM, et al. Thymic stromal lymphopoietin is induced by respiratory syncytial virus‐infected airway epithelial cells and promotes a type 2 response to infection. J Allergy Clin Immunol. 2012;130(5):1187‐1196.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Comstock AT, Ganesan S, Chattoraj A, et al. Rhinovirus‐induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol. 2011;85(13):6795‐6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178(12):1271‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Looi K, Buckley AG, Rigby PJ, et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin Exp Allergy. 2018;48(5):513‐524. [DOI] [PubMed] [Google Scholar]
- 58. Potaczek DP, Miethe S, Schindler V, Alhamdan F, Garn H. Role of airway epithelial cells in the development of different asthma phenotypes. Cell Signal. 2020;69:109523. [DOI] [PubMed] [Google Scholar]
- 59. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. 2018;58(2):157‐169. [DOI] [PubMed] [Google Scholar]
- 60. van den Berge M, Jonker MR, Miller‐Larsson A, Postma DS, Heijink IH. Effects of fluticasone propionate and budesonide on the expression of immune defense genes in bronchial epithelial cells. Pulm Pharmacol Ther. 2018;50:47‐56. [DOI] [PubMed] [Google Scholar]
- 61. Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. 2019;56(2):219‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. De Grove KC, Provoost S, Brusselle GG, Joos GF, Maes T. Insights in particulate matter‐induced allergic airway inflammation: focus on the epithelium. Clin Exp Allergy. 2018;48(7):773‐786. [DOI] [PubMed] [Google Scholar]
- 63. Kim N, Han DH, Suh MW, Lee JH, Oh SH, Park MK. Effect of lipopolysaccharide on diesel exhaust particle‐induced junctional dysfunction in primary human nasal epithelial cells. Environ Pollut. 2019;248:736‐742. [DOI] [PubMed] [Google Scholar]
- 64. Michaudel C, Mackowiak C, Maillet I, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL‐33. J Allergy Clin Immunol. 2018;142(3):942‐958. [DOI] [PubMed] [Google Scholar]
- 65. Wang M, Tan G, Eljaszewicz A, et al. Laundry detergents and detergent residue after rinsing directly disrupt tight junction barrier integrity in human bronchial epithelial cells. J Allergy Clin Immunol. 2019;143(5):1892‐1903. [DOI] [PubMed] [Google Scholar]
- 66. Hall R, Hall IP, Sayers I. Genetic risk factors for the development of pulmonary disease identified by genome‐wide association. Respirology. 2019;24(3):204‐214. [DOI] [PubMed] [Google Scholar]
- 67. Li X, Hastie AT, Hawkins GA, et al. eQTL of bronchial epithelial cells and bronchial alveolar lavage deciphers GWAS‐identified asthma genes. Allergy. 2015;70(10):1309‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Luo W, Obeidat M, Di Narzo AF, et al. Airway epithelial expression quantitative trait loci reveal genes underlying asthma and other airway diseases. Am J Respir Cell Mol Biol. 2016;54(2):177‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ferreira MA, McRae AF, Medland SE, et al. Association between ORMDL3, IL1RL1 and a deletion on chromosome 17q21 with asthma risk in Australia. Eur J Hum Genet. 2011;19(4):458‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marenholz I, Esparza‐Gordillo J, Ruschendorf F, et al. Meta‐analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. 2015;6:8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Geng G, Du Y, Dai J, Tian D, Xia Y, Fu Z. KIF3A knockdown sensitizes bronchial epithelia to apoptosis and aggravates airway inflammation in asthma. Biomed Pharmacother. 2018;97:1349‐1355. [DOI] [PubMed] [Google Scholar]
- 72. Annesi F, Gambardella A, Michelucci R, et al. Mutational analysis of EFHC1 gene in italian families with juvenile myoclonic epilepsy. Epilepsia. 2007;48(9):1686‐1690. [DOI] [PubMed] [Google Scholar]
- 73. Ikeda T, Ikeda K, Enomoto M, Park MK, Hirono M, Kamiya R. The mouse ortholog of EFHC1 implicated in juvenile myoclonic epilepsy is an axonemal protein widely conserved among organisms with motile cilia and flagella. FEBS Lett. 2005;579(3):819‐822. [DOI] [PubMed] [Google Scholar]
- 74. Shrine N, Portelli MA, John C, et al. Moderate‐to‐severe asthma in individuals of European ancestry: a genome‐wide association study. Lancet Respir Med. 2019;7(1):20‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Singhania A, Rupani H, Jayasekera N, et al. Altered epithelial gene expression in peripheral airways of severe asthma. PLoS One. 2017;12(1):e0168680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bonnelykke K, Sleiman P, Nielsen K, et al. A genome‐wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46(1):51‐55. [DOI] [PubMed] [Google Scholar]
- 77. Everman JL, Sajuthi S, Saef B, et al. Functional genomics of CDHR3 confirms its role in HRV‐C infection and childhood asthma exacerbations. J Allergy Clin Immunol. 2019;144(4):962‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Koning H, Sayers I, Stewart CE, et al. Characterization of protocadherin‐1 expression in primary bronchial epithelial cells: association with epithelial cell differentiation. FASEB J. 2012;26(1):439‐448. [DOI] [PubMed] [Google Scholar]
- 79. Faura Tellez G, Willemse BW, Brouwer U, et al. Protocadherin‐1 localization and cell‐adhesion function in airway epithelial cells in asthma. PLoS One. 2016;11(10):e0163967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Faura Tellez G, Vandepoele K, Brouwer U, et al. Protocadherin‐1 binds to SMAD3 and suppresses TGF‐beta1‐induced gene transcription. Am J Physiol Lung Cell Mol Physiol. 2015;309(7):L725‐L735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Y, Bochkov YA, Eickhoff JC, et al. Orosomucoid like 3 (ORMDL3) supports rhinovirus replication in human epithelial cells. Am J Respir Cell Mol Biol. 2020;62(6):783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Das S, Miller M, Beppu AK, et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc Natl Acad Sci U S A. 2016;113(46):13132‐13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Panganiban RA, Sun M, Dahlin A, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immunol. 2018;142(5):1469‐1478.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koppelman GH, Sayers I. Evidence of a genetic contribution to lung function decline in asthma. J Allergy Clin Immunol. 2011;128(3):479‐484. [DOI] [PubMed] [Google Scholar]
- 85. Schoettler N, Rodriguez E, Weidinger S, Ober C. Advances in asthma and allergic disease genetics: is bigger always better? J Allergy Clin Immunol. 2019;144(6):1495‐1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Woodruff PG, Boushey HA, Dolganov GM, et al. Genome‐wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104(40):15858‐15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim S, Forno E, Yan Q, et al. SNPs identified by GWAS affect asthma risk through DNA methylation and expression of cis‐genes in airway epithelium. Eur Respir J. 2019;55(4):1902079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Brook PO, Perry MM, Adcock IM, Durham AL. Epigenome‐modifying tools in asthma. Epigenomics. 2015;7(6):1017‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yang IV, Lozupone CA, Schwartz DA. The environment, epigenome, and asthma. J Allergy Clin Immunol. 2017;140(1):14‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hudon Thibeault AA, Laprise C. Cell‐specific DNA methylation signatures in asthma. Genes (Basel). 2019;10(11):932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Qi C, Jiang Y, Yang IV, et al. Nasal DNA methylation profiling of asthma and rhinitis. J Allergy Clin Immunol. 2020. 10.1016/j.jaci.2019.12.911. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Teschendorff AE, Relton CL. Statistical and integrative system‐level analysis of DNA methylation data. Nat Rev Genet. 2018;19(3):129‐147. [DOI] [PubMed] [Google Scholar]
- 93. Vieira Braga FA, Kar G, Berg M, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. 2019;25(7):1153‐1163. [DOI] [PubMed] [Google Scholar]
- 94. Brugha R, Lowe R, Henderson AJ, et al. DNA methylation profiles between airway epithelium and proxy tissues in children. Acta Paediatr. 2017;106(12):2011‐2016. [DOI] [PubMed] [Google Scholar]
- 95. Forno E, Wang T, Qi C, et al. DNA methylation in nasal epithelium, atopy, and atopic asthma in children: a genome‐wide study. Lancet Respir Med. 2019;7(4):336‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cardenas A, Sordillo JE, Rifas‐Shiman SL, et al. The nasal methylome as a biomarker of asthma and airway inflammation in children. Nat Commun. 2019;10(1):3095‐4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yang IV, Pedersen BS, Liu AH, et al. The nasal methylome and childhood atopic asthma. J Allergy Clin Immunol. 2017;139(5):1478‐1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Qi C, Xu CJ, Koppelman GH. The role of epigenetics in the development of childhood asthma. Expert Rev Clin Immunol. 2019;15(12):1287‐1302. [DOI] [PubMed] [Google Scholar]
- 99. Vermeulen CJ, Xu CJ, Vonk JM, et al. Differential DNA methylation in bronchial biopsies between persistent asthma and asthma in remission. Eur Respir J. 2020;55(2):1901280. [DOI] [PubMed] [Google Scholar]
- 100. Pech M, Weckmann M, Konig IR, et al. Rhinovirus infections change DNA methylation and mRNA expression in children with asthma. PLoS One. 2018;13(11):e0205275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lund RJ, Osmala M, Malonzo M, et al. Atopic asthma after rhinovirus‐induced wheezing is associated with DNA methylation change in the SMAD3 gene promoter. Allergy. 2018;73(8):1735‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. McErlean P, Favoreto S, Costa FF, et al. Human rhinovirus infection causes different DNA methylation changes in nasal epithelial cells from healthy and asthmatic subjects. BMC Med Genomics. 2014;7:37‐8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Clifford RL, Jones MJ, MacIsaac JL, et al. Inhalation of diesel exhaust and allergen alters human bronchial epithelium DNA methylation. J Allergy Clin Immunol. 2017;139(1):112‐121. [DOI] [PubMed] [Google Scholar]
- 104. Moheimani F, Hsu AC, Reid AT, et al. The genetic and epigenetic landscapes of the epithelium in asthma. Respir Res. 2016;17(1):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Solberg OD, Ostrin EJ, Love MI, et al. Airway epithelial miRNA expression is altered in asthma. Am J Respir Crit Care Med. 2012;186(10):965‐974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Huang H, Lu H, Liang L, et al. MicroRNA‐744 inhibits proliferation of bronchial epithelial cells by regulating Smad3 pathway via targeting transforming growth factor‐beta1 (TGF‐beta1) in severe asthma. Med Sci Monit. 2019;25:2159‐2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhang H, Sun Y, Rong W, et al. miR‐221 participates in the airway epithelial cells injury in asthma via targeting SIRT1. Exp Lung Res. 2018;44(6):272‐279. [DOI] [PubMed] [Google Scholar]
- 108. Haj‐Salem I, Fakhfakh R, Berube JC, et al. MicroRNA‐19a enhances proliferation of bronchial epithelial cells by targeting TGFbetaR2 gene in severe asthma. Allergy. 2015;70(2):212‐219. [DOI] [PubMed] [Google Scholar]
- 109. Martinez‐Nunez RT, Bondanese VP, Louafi F, et al. A microRNA network dysregulated in asthma controls IL‐6 production in bronchial epithelial cells. PLoS One. 2014;9(10):e111659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Song R, Walentek P, Sponer N, et al. miR‐34/449 miRNAs are required for motile ciliogenesis by repressing cp110. Nature. 2014;510(7503):115‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Song L, Sen S, Sun Y, Zhou J, Mo L, He Y. Ketamine inhalation ameliorates ovalbumin‐induced murine asthma by suppressing the epithelial‐mesenchymal transition. Med Sci Monit. 2016;22:2471‐2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Maes T, Cobos FA, Schleich F, et al. Asthma inflammatory phenotypes show differential microRNA expression in sputum. J Allergy Clin Immunol. 2016;137(5):1433‐1446. [DOI] [PubMed] [Google Scholar]
- 113. Cui W, Meng W, Zhao L, Cao H, Chi W, Wang B. TGF‐beta‐induced long non‐coding RNA MIR155HG promotes the progression and EMT of laryngeal squamous cell carcinoma by regulating the miR‐155‐5p/SOX10 axis. Int J Oncol. 2019;54(6):2005‐2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fang C, Lu W, Li C, et al. MiR‐3162‐3p is a novel MicroRNA that exacerbates asthma by regulating beta‐catenin. PLoS One. 2016;11(3):e0149257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu Z, Chen X, Wu Q, Song J, Wang L, Li G. miR‐125b inhibits goblet cell differentiation in allergic airway inflammation by targeting SPDEF. Eur J Pharmacol. 2016;782:14‐20. [DOI] [PubMed] [Google Scholar]
- 116. Su Y, Wang J, Zou J, Han W, Li S. miR‐330 regulates interleukin‐13‐induced MUC5AC secretion by targeting Munc18b in human bronchial epithelial cells. Int J Clin Exp Pathol. 2018;11(7):3463‐3470. [PMC free article] [PubMed] [Google Scholar]
- 117. Gupta R, Radicioni G, Abdelwahab S, et al. Intercellular communication between airway epithelial cells is mediated by exosome‐like vesicles. Am J Respir Cell Mol Biol. 2019;60(2):209‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bartel S, La Grutta S, Cilluffo G, et al. Human airway epithelial extracellular vesicle miRNA signature is altered upon asthma development. Allergy. 2020;75(2):346‐356. [DOI] [PubMed] [Google Scholar]
- 119. Yang ZC, Qu ZH, Yi MJ, et al. MiR‐448‐5p inhibits TGF‐beta1‐induced epithelial‐mesenchymal transition and pulmonary fibrosis by targeting Six1 in asthma. J Cell Physiol. 2019;234(6):8804‐8814. [DOI] [PubMed] [Google Scholar]
- 120. Vento‐Tormo R, Efremova M, Botting RA, et al. Single‐cell reconstruction of the early maternal‐fetal interface in humans. Nature. 2018;563(7731):347‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Plasschaert LW, Zilionis R, Choo‐Wing R, et al. A single‐cell atlas of the airway epithelium reveals the CFTR‐rich pulmonary ionocyte. Nature. 2018;560(7718):377‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Schiller HB, Montoro DT, Simon LM, et al. The human lung cell atlas: a high‐resolution reference map of the human lung in health and disease. Am J Respir Cell Mol Biol. 2019;61(1):31‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Regev A, Teichmann SA, Lander ES, et al. The human cell atlas. Elife. 2017;6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Schwarze J, Fitch PM, Heimweg J, et al. Viral mimic poly‐(I:C) attenuates airway epithelial T‐cell suppressive capacity: implications for asthma. Eur Respir J. 2016;48(6):1785‐1788. [DOI] [PubMed] [Google Scholar]
- 125. Hua K, Li Y, Zhou H, et al. Haemophilus parasuis infection disrupts adherens junctions and initializes EMT dependent on canonical wnt/beta‐catenin signaling pathway. Front Cell Infect Microbiol. 2018;8:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Moheimani F, Roth HM, Cross J, et al. Disruption of beta‐catenin/CBP signaling inhibits human airway epithelial‐mesenchymal transition and repair. Int J Biochem Cell Biol. 2015;68:59‐69. [DOI] [PubMed] [Google Scholar]
- 127. Lafkas D, Shelton A, Chiu C, et al. Therapeutic antibodies reveal notch control of transdifferentiation in the adult lung. Nature. 2015;528(7580):127‐131. [DOI] [PubMed] [Google Scholar]
- 128. Doerner AM, Zuraw BL. TGF‐beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL‐1beta but not abrogated by corticosteroids. Respir Res. 2009;10:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Heijink I, van Oosterhout A, Kliphuis N, et al. Oxidant‐induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax. 2014;69(1):5‐13. [DOI] [PubMed] [Google Scholar]
- 130. Marwick JA, Caramori G, Casolari P, et al. A role for phosphoinositol 3‐kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2010;125(5):1146‐1153. [DOI] [PubMed] [Google Scholar]
- 131. Ito K, Yamamura S, Essilfie‐Quaye S, et al. Histone deacetylase 2‐mediated deacetylation of the glucocorticoid receptor enables NF‐kappaB suppression. J Exp Med. 2006;203(1):7‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. To Y, Ito K, Kizawa Y, et al. Targeting phosphoinositide‐3‐kinase‐delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(7):897‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ferreira MA, Vonk JM, Baurecht H, et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet. 2017;49(12):1752‐1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol. 2017;12:331‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pigors M, Common JEA, Wong XFCC, et al. Exome sequencing and rare variant analysis reveals multiple filaggrin mutations in bangladeshi families with atopic eczema and additional risk genes. J Invest Dermatol. 2018;138(12):2674‐2677. [DOI] [PubMed] [Google Scholar]
- 136. Lopes C, Rocha L, Sokhatska O, et al. Filaggrin polymorphism Pro478Ser is associated with the severity of atopic dermatitis and colonization by staphylococcal aureus. J Investig Allergol Clin Immunol. 2016;26(1):70‐72. [PubMed] [Google Scholar]
- 137. Sherrill JD, Kc K, Wu D, et al. Desmoglein‐1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol. 2014;7(3):718‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Moffatt MF, Gut IG, Demenais F, et al. A large‐scale, consortium‐based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gordon ED, Palandra J, Wesolowska‐Andersen A, et al. IL1RL1 asthma risk variants regulate airway type 2 inflammation. JCI Insight. 2016;1(14):e87871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ferreira MA, Matheson MC, Tang CS, et al. Genome‐wide association analysis identifies 11 risk variants associated with the asthma with hay fever phenotype. J Allergy Clin Immunol. 2014;133(6):1564‐1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Soumelis V, Reche PA, Kanzler H, et al. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673‐680. [DOI] [PubMed] [Google Scholar]
- 142. Gras D, Martinez‐Anton A, Bourdin A, et al. Human bronchial epithelium orchestrates dendritic cell activation in severe asthma. Eur Respir J. 2017;49(3):1602399. [DOI] [PubMed] [Google Scholar]
- 143. Hui CC, Yu A, Heroux D, et al. Thymic stromal lymphopoietin (TSLP) secretion from human nasal epithelium is a function of TSLP genotype. Mucosal Immunol. 2015;8(5):993‐999. [DOI] [PubMed] [Google Scholar]
- 144. Kovacic MB, Myers JM, Wang N, et al. Identification of KIF3A as a novel candidate gene for childhood asthma using RNA expression and population allelic frequencies differences. PLoS One. 2011;6(8):e23714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Koppelman GH, Meyers DA, Howard TD, et al. Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am J Respir Crit Care Med. 2009;180(10):929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Basnet S, Bochkov YA, Brockman‐Schneider RA, et al. CDHR3 asthma‐risk genotype affects susceptibility of airway epithelium to rhinovirus C infections. Am J Respir Cell Mol Biol. 2019;61(4):450‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Altman MC, Lai Y, Nolin JD, et al. Airway epithelium‐shifted mast cell infiltration regulates asthmatic inflammation via IL‐33 signaling. J Clin Invest. 2019;129(11):4979‐4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Jackson DJ, Makrinioti H, Rana BM, et al. IL‐33‐dependent type 2 inflammation during rhinovirus‐induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Stefanowicz D, Hackett TL, Garmaroudi FS, et al. DNA methylation profiles of airway epithelial cells and PBMCs from healthy, atopic and asthmatic children. PLoS One. 2012;7(9):e44213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Tan QY, Cheng ZS. TGFbeta1‐smad signaling pathway participates in interleukin‐33 induced epithelial‐to‐mesenchymal transition of A549 cells. Cell Physiol Biochem. 2018;50(2):757‐767. [DOI] [PubMed] [Google Scholar]
- 151. Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448(7152):470‐473. [DOI] [PubMed] [Google Scholar]
- 152. Miller M, Tam AB, Mueller JL, et al. Cutting edge: targeting epithelial ORMDL3 increases, rather than reduces, airway responsiveness and is associated with increased sphingosine‐1‐phosphate. J Immunol. 2017;198(8):3017‐3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Miller M, Rosenthal P, Beppu A, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol. 2014;192(8):3475‐3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ono JG, Kim BI, Zhao Y, et al. Decreased sphingolipid synthesis in children with 17q21 asthma‐risk genotypes. J Clin Invest. 2020;130(2):921‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wang H, Liu Y, Shi J, Cheng Z. ORMDL3 knockdown in the lungs alleviates airway inflammation and airway remodeling in asthmatic mice via JNK1/2‐MMP‐9 pathway. Biochem Biophys Res Commun. 2019;516(3):739‐746. [DOI] [PubMed] [Google Scholar]
- 156. Yang R, Tan M, Xu J, Zhao X. Investigating the regulatory role of ORMDL3 in airway barrier dysfunction using in vivo and in vitro models. Int J Mol Med. 2019;44(2):535‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Barton SJ, Koppelman GH, Vonk JM, et al. PLAUR polymorphisms are associated with asthma, PLAUR levels, and lung function decline. J Allergy Clin Immunol. 2009;123(6):1391‐400.e17. [DOI] [PubMed] [Google Scholar]
- 158. Portelli MA, Hodge E, Sayers I. Genetic risk factors for the development of allergic disease identified by genome‐wide association. Clin Exp Allergy. 2015;45(1):21‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ierodiakonou D, Postma DS, Koppelman GH, et al. E‐cadherin gene polymorphisms in asthma patients using inhaled corticosteroids. Eur Respir J. 2011;38(5):1044‐1052. [DOI] [PubMed] [Google Scholar]
- 160. Stewart CE, Nijmeh HS, Brightling CE, Sayers I. uPAR regulates bronchial epithelial repair in vitro and is elevated in asthmatic epithelium. Thorax. 2012;67(6):477‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Post S, Rozeveld D, Jonker MR, Bischoff R, van Oosterhout AJ, Heijink IH. ADAM10 mediates the house dust mite‐induced release of chemokine ligand CCL20 by airway epithelium. Allergy. 2015;70(12):1545‐1552. [DOI] [PubMed] [Google Scholar]