

Abstract
Disruption of the circadian cycle is strongly associated with metabolic imbalance and reduced longevity in humans. Also, rodent models of circadian arrhythmia, such as the constitutive knockout of the clock gene Bmal1, leads to metabolic disturbances and early death. Although astrocyte clock regulates molecular and behavioral circadian rhythms, its involvement in the regulation of energy balance and lifespan is unknown. Here, we show that astrocyte‐specific deletion of Bmal1 is sufficient to alter energy balance, glucose homeostasis, and reduce lifespan. Mutant animals displayed impaired hypothalamic molecular clock, age‐dependent astrogliosis, apoptosis of hypothalamic astrocytes, and increased glutamate and GABA levels. Importantly, modulation of GABAA‐receptor signaling completely restored glutamate levels, delayed the reactive gliosis as well as the metabolic phenotypes and expanded the lifespan of the mutants. Our results demonstrate that the astrocytic clock can influence many aspects of brain function and neurological disease and suggest astrocytes and GABAA receptor as pharmacological targets to prevent the metabolic dysfunctions and shortened lifespan associated with alterations of circadian rhythms.
Keywords: astrocytes, circadian clock, GABA signaling, glutamate, lifespan, metabolism
Astrocyte clock controls energy balance, glucose homeostasis and longevity in mice.
Modulation of GABAA‐receptor signaling delays the metabolic phenotype and expanded the life span of mice with astrocytic deletion of BMAL1.
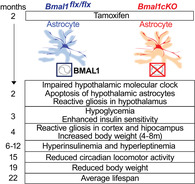
1. INTRODUCTION
The circadian clock is an endogenous, self‐sustaining oscillator that operates with a periodicity of 24 hr to maintain proper rhythms in gene expression, physiology, and behavior (reviewed in Hastings, Maywood, & Reddy, 2008). As evidenced by individuals working in the night or rotating shifts and in rodent models of circadian arrhythmia, disruption of the circadian cycle is strongly associated with metabolic imbalance and reduced longevity (reviewed in Chaudhari, Gupta, Makwana, & Kondratov, 2017; Karlsson, Knutsson, Lindahl, & Alfredsson, 2003; Parkes, 2002; van Amelsvoort, Schouten, & Kokn, 1999).
The timekeeping system includes cellular autonomous clocks that are entrained by hormonal and neuronal signals from a central pacemaker: the suprachiasmatic nuclei (SCN), located in the hypothalamus (Moore & Eichler, 1972; Stephan & Zucker, 1972). The molecular mechanisms driving circadian oscillations involve rhythmic and self‐sustained transcriptional–translational feedback loops of clock genes/proteins. The E‐box specific transcription factors BMAL1 (Brain and muscle Arnt‐like protein‐1) and CLOCK (Circadian locomotor output cycles kaput) are the positive limb of this oscillatory mechanism, which heterodimerize to activate transcription of the repressors Period (Per1/2/3) and Cryptochrome genes (Cry1/2; van der Horst et al., 1999; Zheng et al., 2001). The negative loop of the molecular clock is constituted by PER/CRY heterocomplex that, upon accumulation, lead to degradation of BMAL1/CLOCK dimers thus inhibiting their own transcription (Kume et al., 1999). CLOCK/BMAL1 drives rhythmic expression of clock‐controlled genes, which regulate many physiological processes including major components of energy homeostasis such as feeding behavior, locomotor activity, sleep–wake cycle and glucose metabolism (reviewed in Marcheva, Ramsey, Affinati, & Bass, 2009; Richards & Gumz, 2013). Indeed, the absence of BMAL1 in mice results in a loss of circadian rhythms, alteration in energy balance, acceleration of aging and shortened lifespan (Bunger et al., 2005; Kondratov, Kondratova, Gorbacheva, Vykhovanets, & Antoch, 2006; Lamia, Storch, & Weitz, 2008; Lee, Donehower, Herron, Moore, & Fu, 2006; Marcheva et al., 2010; Rudic et al., 2004; Shi, Ansari, McGuinness, Wasserman, & Johnson, 2006).
In mammals, neurons of the SCN have been classically considered as master pacemaker cells, controlling daily rhythms of physiology and behavior and coordinating the circadian programs of peripheral tissues (Yoo et al., 2004). However, several evidence have recently challenged this neurocentric view of the timekeeping system by uncovering that astrocytes autonomously function as a central circadian clock regulating molecular and behavioral circadian rhythms (Barca‐Mayo et al., 2017; Brancaccio et al., 2019; Brancaccio, Patton, Chesham, Maywood, & Hastings, 2017; Tso et al., 2017). In light of this new scenario, we hypothesize that astrocytic clock might have a crucial contribution to the circadian control of metabolism and lifespan.
Here, we provide evidence that adult disruption of the astrocytic clock, via genetic deletion of BMAL1, is sufficient to lead to the metabolic and age‐associated dysfunctions of constitutive Bmal1 knockout animals. Specifically, the deletion of BMAL1 in astrocytes impairs the hypothalamic circadian function and leads to increased GABA and glutamate levels, age‐dependent astrogliosis and apoptosis of hypothalamic astrocytes, altered glucose homeostasis, altered growth curves, and reduced lifespan. Remarkably, these phenotypes were delayed upon inhibition of GABAA receptor signaling. Our results demonstrate that the astrocytic molecular clock can influence many aspects of brain function and neurological disease and suggest astrocytes and GABAA receptor as targets for chronotherapies to prevent dysfunctions associated with chronic or acute alterations of circadian rhythms.
2. MATERIALS AND METHODS
2.1. Animals
All experiments and procedures were approved by the Italian Ministry of Health (Permit No. 214/2015‐PR) and the local Animal Use Committee, and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the European Community Council Directives and of Italian Ministry of Health. Bmal1flox/flox mice (Jackson Laboratory Stock 007668, B6.129S4 (Cg) Arntltm1Weit/J, RRID: IMSR_JAX:007668) were crossed with Glast (Glutamate aspartate transporter) creERT2+/− mouse line (RRID: MGI: 3830092). Bmal1 conditional knockout mice were generated by treating 6‐ to 8‐weeks‐old male Glast‐CreERT2 +/−:Bmal1 flox/flox with tamoxifen (TM; Bmal1cKO), as previously reported (Barca‐Mayo et al., 2017). TM‐treated Bmal1 flox/flox male littermates served as controls. Note that the functional glutamate uptake, live span, energy balance, and glucose homeostasis is unaffected in Glast‐CreERT2+/− mice (García‐Cáceres et al., 2016; Saab et al., 2012). Mice were housed with ad libitum access to food and water, and kept on a 12 hr (8 a.m. to 8 p.m.) light–dark cycle, in a room, maintained at 21°C at the animal facility of the Istituto Italiano di Tecnologia (IIT), Genoa, Italy. Pentylenetetrazole (PTZ) treatment of control and Bmal1cKO mice was performed as previously reported (Barca‐Mayo et al., 2017). Feeding behaviors were examined in mice housed in individual cages by measuring the daily, the daytime (immediately after the onset of the light period (8:00 a.m., Zeitgeber [ZT0]), and nighttime (immediately before the onset of the dark period [8:00 p.m., ZT12]), food intake. Specifically, food intake was assessed by subtracting the amount of food remaining in the cages from the amount provided to the animals the previous day or daytime (ZT0 and ZT12). Food spillage was minimal, and was assessed by visual inspection and accounted for when necessary. Averages represent the intake over 2 consecutive days.
2.2. Locomotor activity
Two to three or 15 months after TM treatment, male Bmal1cKO and control mice were single‐housed in cages equipped with running wheels (ENV‐044; Med Associates, Inc). Mice were adapted to the wheel for 3 days in standard light–dark cycles (12:12 hr, lights on at 8 a.m., ZT0). Running wheel activity monitoring started under these conditions during the next 3–5 days. Running wheel activity was recorded in 5 min (min) bins by Wheel Manager software (SOF‐860; Med Associates, Inc) as previously reported (Barca‐Mayo et al., 2017).
2.3. Operant conditioning
Two months after TM treatment, male control and Bmal1cKO mice were subjected to a restricted feeding schedule where the access to standard lab chow (Special Diet Services, UK) was restricted from 16:00 to 18:00 (i.e., ZT8 and ZT10, respectively). Behavioral experiments started 3 days after the restricted feeding schedule and were conducted between 14:00 and 16:00. The behavioral training took place in operant chambers (17.8 cm × 15.2 cm × 18.4 cm) in which two holes were placed on either side of a food magazine (Med Associates, St. Albans, VT). Sucrose pellets (SP, TestDiet, Indianapolis, IN) were delivered into the food magazine when the mouse nose poked into the “active” hole (ANP), whereas a poke into the “inactive” hole had no consequence. Initial nose poke training consisted of three daily sessions of fixed ratio (FR) schedule (FR‐1), in which every active nose poke was rewarded. After the FR1 sessions, mice were trained on a FR‐5 and FR‐25 schedules, in which every fifth or 25th active nose poke led to SP delivery, respectively. After three daily FR‐5 sessions and three daily FR‐25 schedules, mice were trained under a progressive ratio (PR) schedule for 3 days. In the PR‐schedule, the number of ANP required to obtain SP is increased with each completed trial (ANP = 5 × e0.2SP) so each successive SP required more ANP and the amount of ANP reflected the effort that was invested in the task.
2.4. Glucose tolerance test
Control and Bmal1cKO mice (at the age and time point specified) were fasted for 16 hr before the glucose tolerance test. Blood was obtained from a tail cut and was assessed for fasting glucose levels using an OneTouch Ultra 2 (LifeScan, Johnson & Johnson) glucometer. Mice then received a glucose solution (2 g/kg body weight) delivered by intraperitoneal injection. At 15, 30, 60, 90, and 120 min after the administration, dried blood was quickly removed from the tail wound and fresh blood was collected again to measure the glucose concentration.
2.5. Determination of serum leptin, insulin, corticosterone, and glucagon
Leptin, insulin, corticosterone, and glucagon serum levels were determined by ELISA using reagents kits and methods provided by Merck‐Millipore (insulin, EZRMI‐13K; leptin, EZML‐82K), Enzo Life Sciences (corticosterone, ADI‐900‐097) and Sigma (Glucagon, RAB0202).
2.6. Determination of GABA/glutamate levels in cerebrospinal fluid
GABA was quantified in mouse cerebrospinal fluid (CSF) samples, collected from the cisterna magna, by UPLC‐MS/MS (Ultra Performance Liquid Chromatography–Tandem mass spectrometry) as we previously described (Barca‐Mayo et al., 2017). Glutamate was extracted from CSF by precipitation with acetonitrile spiked with deuterated Glutamate (D5) as an internal standard (Sigma Aldrich). The analytes were then separated by HILIC chromatography (Hydrophilic Interaction Liquid Chromatography) using a BEH HILIC 2.1X100 mm. column and a short gradient of water in acetonitrile (5–40% in 2 min), with the eluent added with formic acid to a final 0.1% v/v, flow rate was kept at 0.45 ml/min. Glutamate was quantified on a Xevo TQ‐MS instrument operating in electrospray, positive ion mode and following the MRM (Multiple Reaction Monitoring) transitions. Both the column and the UPLC‐MS/MS systems were purchased from Waters Inc. (Milford). Glutamate quantification was performed using a standard calibration curve prepared by serial dilution in artificial CSF and extracted along with the samples. The investigators were blinded to group allocation during experiments.
2.7. Immunofluorescence
Mice were administered ketamine/xylazine (150 mg/kg, 10 mg/kg, respectively) and transcardially perfused with ice‐cold PBS followed by ice‐cold 4% PFA in PBS. Brains were post‐fixed overnight in 4% PFA in PBS and 30 μm slices were prepared Cryostat (Leica). Slices were permeabilized with 0.3% Triton X‐100 in PBS, blocked with 10% goat serum in PBS, and incubated at 4°C overnight with the primary antibody mouse anti‐Glial Fibrillary Acidic Protein (GFAP) 1:1,000 dilution (Sigma, G3893); rabbit anti‐BMAL1, dilution 1:200 (Abcam, ab93806); mouse anti‐S100 calcium‐binding protein β (S100β), 1:1,000 dilution (Sigma, AMAB91038); rabbit anti‐active CASPASE 3, 1:1,000 dilution (Cell Signaling Technology, 9579); mouse anti‐RNA Binding Fox‐1 Homolog 2 (FOX2) 1:500 dilution (Abcam, ab57154); rabbit anti‐KI67 1:200 dilution (Thermo Scientific MA5‐14520). The following day, sections were extensively washed, and incubated for 2 hr with goat anti‐rabbit or anti‐mouse Alexa‐488 or Alexa‐546 secondary antibodies (1:1,000 dilution). Slices were then washed, mounted with Prolong Gold and imaged with an inverted laser scanning confocal microscope (TCS SP5 microscope using a 20× or 40× objective, Leica Microsystems). Quantification and analysis were performed with ImageJ software (Wayne Rasband, NIH), by outlining the hypothalamus from the 4′,6‐diamidino‐2‐phenylindole (DAPI)‐stained image and using this template to measure the relative intensity of the immunostaining. When more than one section was analyzed from each animal, the mean of the measures from consecutive sections was used for that individual.
2.8. RNA isolation and quantitative real‐time RT‐PCR
Total RNA was extracted from hypothalamus using TRIzol reagent following the manufacturer's instructions. RNA was further cleaned using an RNeasy Mini Kit. cDNA was obtained by reverse transcription of 0.5 μg of total mRNA using the ImProm‐II™ Reverse Transcription System following the manufacturer's instructions. Real‐time RT‐PCR was done using the ABI PRISM.7900 (Applied Biosystems). For a 15 μl reaction, 9 ng of cDNA template was mixed with the primers to a final concentration of 200 nM and mixed with 7.5 μl of 2× QuantiFast SYBR Green PCR Master Mix. The reactions were done in duplicates using the following conditions: 5 min at 95°C followed by 40 cycles of 10 s at 95°C, 30 s at 60°C, and 1 min at 70°C. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) or beta‐actin transcripts were used as reference controls.
2.9. Statistical analysis
Statistical parameters including the exact value of n, and precision measures (mean ± SEM) and statistical significance are reported in the figures and figure legends. All statistical tests were two‐sided. A log‐rank test was used for survival curve analysis. Other statistical comparisons were done by Student's paired t test, or two‐way ANOVA with a post hoc Bonferroni. Data were checked for normality and equal variances between groups. Statistical significance of the rhythmic expression was determined by Cosinor analysis as previously reported (Barca‐Mayo et al., 2017). The cutoff for significance was *p < .05, **p < .01, ***p < .001, and ****p < .0001. Statistical analysis was performed with GraphPad PRISM 6 software.
3. RESULTS
3.1. Deletion of BMAL1 in astrocytes leads to early death, altered body weight, and glucose homeostasis
To investigate the contribution of astrocyte clock in the regulation of lifespan and energy balance, we genetically deleted Bmal1 in astrocytes expressing GLAST by crossing Bmal1 flox/flox mice with a tamoxifen (TM) inducible knock‐in Glast‐CreERT2 +/− deletor mouse line (Mori et al., 2006), here referred to as Bmal1cKO. We administered TM to 6‐ to 8‐weeks‐old Bmal1cKO male mice and controls (Bmal1 flox/flox), an approach that we have previously shown to achieve astrocyte‐ and time‐specific deletion of Bmal1, while avoiding functional abnormalities or compensations that might occur during development (Barca‐Mayo et al., 2017; Mori et al., 2006). Following TM administration, we kept mice in 12‐hr: 12‐hr light–dark cycles and allowed them to feed ad libitum with regular diet. We found that the lifespan of Bmal1cKO animals was significantly reduced compared to controls (Figure 1a). Specifically, most mutants died between 20 and 25 months of age (average of lifespan, 22 months after TM treatment; Figure 1a). Bmal1cKO mice were significantly heavier than control animals from 4 until 8 months after TM treatment, with increased adipose tissue mass, and their body weight progressively decreased after 19 months of TM treatment (Figure 1b). Similarly, higher body weight and greater adipose tissue mass than control animals were previously found in constitutive Bmal1 −/− mice at 1–2 months of age, followed by a steady decrease (Table S1), a phenotype that was regarded as hallmark of premature aging (Bunger et al., 2005; Kondratov et al., 2006; Lamia et al., 2008; Lee et al., 2006). Therefore, we concluded that the deletion of BMAL1 in astrocytes is sufficient to shorten the lifespan and alter the growth curves as previously observed in Bmal1 −/− mice (Table S1).
Figure 1.
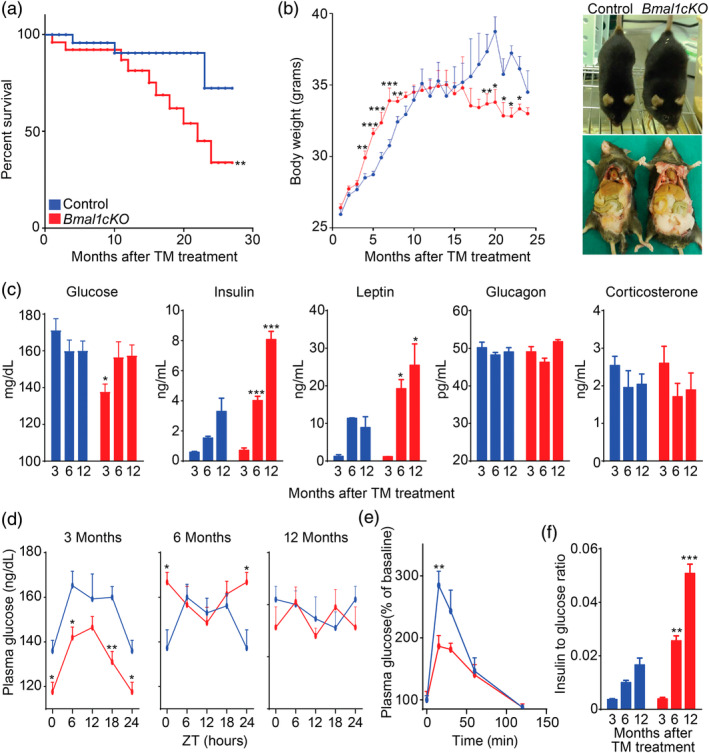
Loss of Bmal1 in astrocytes leads to early death, altered body weight and glucose homeostasis. (a) Kaplan‐Meyer survival curve of control and Bmal1cKO mice (n = 24 and n = 16, respectively, Log rank test, **p < .01). (b) Left panel, age‐dependent changes in body weight of control and Bmal1cKO males. Data are represented as mean ± SEM (n = 18, paired t‐test, *p < .05, **p < .01, and ***p < .001 vs. control animals). Right panel, gross appearance of control and Bmal1cKO mice 4 months after TM treatment, showing increased fat mass in the mutants. (c) Blood glucose, insulin, leptin, glucagon, and corticosterone levels in control and Bmal1cKO mice at 3, 6, and 12 months after TM treatment (ZT6). Data are represented as mean ± SEM (n = 8, paired t‐test, *p < .05 and ***p < .001 vs. control animals). (d) Blood glucose in control and Bmal1cKO animals after 3, 6, and 12 months of TM treatment. Data are represented as mean ± SEM (n = 8, paired t‐test, *p < .05 and **p < .01 vs. control animals). (e) Glucose tolerance test in control and Bmal1cKO mice at 3 months after TM treatment performed at ZT10. Data are represented as mean ± SEM (n = 5–6, paired t‐test, **p < .01 vs. control animals). (f) Insulin to glucose ratio of control and Bmal1cKO animals at 3, 6, and 12 months of TM treatment (ZT6). Data are represented as mean ± SEM (n = 5–6, paired t‐test, **p < .01 and ***p < .001 vs. control animals) [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether the altered growth curves of Bmal1cKO animals were associated with changes in metabolic markers, we analyzed blood levels of glucose, insulin, glucagon, leptin, and corticosterone in the mutants and controls at 3, 6, and 12 months after TM treatment, corresponding to the onset of increased weight gain (i.e., pre‐obese stage), transient obesity and to the period in which bodyweight of mutant mice was indistinguishable from control animals, respectively. We found that Bmal1cKO animals had hypoglycemia at 3 months after TM treatment and hyperinsulinemia and hyperleptinemia at 6 and 12 months after TM treatment (Figure 1c). These alterations were not due to changes in glucocorticoid production, because levels of corticosterone were indistinguishable between Bmal1cKO and controls (Figure 1c). Similarly, no differences were found in glucagon levels between both groups of mice (Figure 1c). Remarkably, at 3 months after TM treatment, Bmal1cKOs had lower levels of glucose throughout the day (Figure 1d). However, at later stages no differences were found in blood glucose between mutants and control at different times of the day, with the exception of significantly higher levels in the mutants at ZT0, 6 months after TM treatment (Figure 1d).
To gain insights into the hypoglycemia of Bmal1cKOs at the pre‐obese stage (i.e., 3 months after TM treatment), we performed a glucose tolerance test. Interestingly, mutant mice showed a blunted elevation of blood glucose compared to controls (Figure 1e). The mean peak increase in blood glucose level (normalized as a percentage of the initial level) of Bmal1cKOs was 34.47% lower than that of controls (186.43% ± 17.25 vs. 284.49% ± 22.89%, paired t test, p = .01). These results indicated enhanced insulin sensitivity in Bmal1cKOs and were consistent with previous findings in constitutive Bmal1−/− and ClockΔ19 animals (Table S1; Lamia et al., 2008; Marcheva et al., 2010; Rudic et al., 2004). Defective glucose regulation typically worsens with age (reviewed in Neubauer & Kulkarni, 2006). Indeed, Bmal1cKOs showed significantly higher levels of insulin at 6 and 12 months after TM treatment, leading to increased insulin to glucose ratio (Figure 1f). This indicates that Bmal1cKOs developed age‐dependent insulin resistance, likely associated with their increased body weight.
Altogether, our results indicate that adult deletion of Bmal1 in a subpopulation of astrocytes is sufficient reduce lifespan, alter body weight and glucose homeostasis as previously reported in constitutive Bmal1−/− animals (Table S1; Bunger et al., 2005; Kondratov et al., 2006; Lamia et al., 2008; Lee et al., 2006; Marcheva et al., 2010; Rudic et al., 2004; Shi et al., 2006).
3.2. Bmal1cKO mice showed increased food intake with no alterations in the brain reward systems
Bodyweight results from the homeostatic regulation to balance energy intake and energy expenditure. We monitored, at different times after TM treatment, the daily food intake of control and mutant animals that were kept in normal light–dark cycles. We observed that 1 month after TM treatment, Bmal1cKO animals had comparable food intake than control mice (Figure 2a). However, from 2 to 6 months after TM treatment, Bmal1cKOs had significantly increased food intake than controls (Figure 2a), therefore preceding their increase in the body weight (observed 4 months after TM treatment, Figure 1b) and their insulin resistance (detected 6 months after TM treatment, Figure 1f). Twelve months after TM treatment, when the bodyweight of mutant mice was indistinguishable from control animals (Figure 1b), the mutants showed no differences in the food intake compared to controls (Figure 2a). On the other hand, we found no differences in the daily activity between control and Bmal1cKO mice 2 months after TM treatment (Figure 2b), consistent with our previous observations (Barca‐Mayo et al., 2017). However, at this pre‐obese stage, Bmal1cKO animals had abnormal feeding behavior as shown by their significantly increased food intake at both day and night times (Figure 2c). It was shown that the metabolic dysfunctions and obesity of mouse models with genetic disruptions in core clock genes such as Clock, are directly linked to disturbed feeding rhythms and/or excessive daytime feeding (Turek et al., 2005). Therefore, we postulate that the altered feeding pattern of Bmal1cKO mice at the pre‐obese stage results in obesity and insulin resistance at later stages.
Figure 2.

Bmal1cKO mice showed increased food intake with no alterations in the brain reward systems. (a) Daily food intake was determined in control and Bmal1cKO mice after 1, 2, 3, 6, and 12 months of TM treatment. Animals were maintained in 12 hr:12 hr light–dark cycles and fed ad libitum with a standard mouse chow. Data are represented as mean ± SEM (n = 8, paired t‐test, **p < .01 vs. control animals). (b) Activity waveforms for control (n = 8) and Bmal1cKO (n = 7) mice, 2 months after TM treatment, in 12 hr: 12 hr light–dark cycles. Activity counts are expressed as the average amount of activity in 5 min bins. Data plotted is given in ZT, such that ZT0 = lights on. The value expresses the means + SEM. (c) Food intake was determined in control and Bmal1cKO mice after 2 months of TM treatment. Animals were maintained in 12 hr:12 hr light–dark cycles and fed ad libitum with a standard mouse chow. Data are represented as mean ± SEM (n = 8, paired t‐test, *p < .05 vs. control animals). (d) Number of nose spokes performed by control and Bmal1cKO mice, 2 months after TM treatment, during fixed ratio (FR), and progressive ratio (PR) sessions in the operant conditioning test. Data are represented as mean ± SEM (n = 4 for controls and n = 11 for Bmal1cKOs). (e) Mean ± SEM breakpoints in control (n = 4) and Bmal1cKO (n = 11) mice, 2 months after TM treatment, in the operant conditioning test [Color figure can be viewed at wileyonlinelibrary.com]
The main brain region regulating homeostatic food intake is the arcuate nucleus (ARC) of the hypothalamus through its connections with other hypothalamic nuclei and extra‐hypothalamic brain areas (reviewed in Coll, Farooqi, & O'Rahilly, 2007 and in Blouet & Schwartz, 2010; Dietrich & Horvath, 2009; van Vliet‐Ostaptchouk, Hofker, van der Schouw, Wijmenga, & Onl‐Moret, 2009). However, other brain circuits involved in the rewarding effects of food such as several limbic (nucleus accumbens, amygdala and hippocampus) and cortical brain regions (orbitofrontal cortex, cingulate gyrus, and insula) are also implicated in hedonic‐driven food consumption and obesity (reviewed in Coll et al., 2007 and in Belgardt, Okamura, & Brüning, 2009; Goldstone, 2006; Rolls, 2008). To address whether the increased food intake of the mutant was due to an alteration in hedonic system, we subjected to control and Bmal1cKO mice (after 2 months of TM treatment) to a progressive‐ratio schedule of food‐pellet reinforcement, a commonly used measure of reward strength (Hodos, 1961, 1963). Animals were trained to nose‐poke in an active hole to get a food reward under FR1, FR5, and FR25 schedules. No significant differences were detected between Bmal1cKO and control mice in the acquisition of the task (Figure 2d). Moreover, in the PR session, in which mice had to nose‐poke an increasing number of times to get the same reward (breakpoint), Bmal1cKO mice poked as control animals and earned the same rewards (Figure 2e). This result suggests that increased food intake in Bmal1cKO mice is due to an alteration in the hypothalamic homeostatic system and less likely to hedonic search for food.
Astrocytes have been recently postulated as key contributors to energy balance regulation and obesity due to the discovery of hypothalamic inflammation and gliosis, particularly in the ARC nucleus, in obese rodents and humans (reviewed in McNay, Briançon, Kokoeva, Maratos‐Flier, & Flier, 2012; Thaler et al., 2012; Valdearcos, Xu, & Koliwad, 2015). Remarkably, it was reported that BMAL1 is a potent regulator of astrocyte activation or gliosis (Lananna et al., 2018; Musiek et al., 2013). Therefore, we hypothesize that BMAL1 may connect dysregulation of circadian function to hypothalamic astrogliosis leading to the increased body weight and metabolic alteration of Bmal1cKO mice.
3.3. BMAL1 deletion in astrocytes globally impairs the hypothalamic molecular clock
To test our hypothesis, we crossed Bmal1cKO mice with a Cre‐inducible Red fluorescent reporter mouse line (Td‐Tomato), as we previously reported (Barca‐Mayo et al., 2017). We first ascertained the specificity and efficiency of GLAST‐Cre‐mediated BMAL1 deletion in hypothalamic astrocytes, by quantifying the co‐immunolocalization of TOMATO with the astrocyte markers Glial fibrillary acidic protein (GFAP) or S100 calcium‐binding protein β (S100β) and BMAL1 in Glast‐Cre‐Td‐Tomato (control) or Bmal1cKO‐Td‐Tomato animals. As expected, 2 months after TM, virtually all of the Cre‐recombined cells in the hypothalamus, as revealed by TOMATO, exhibited the stellated morphology characteristic of astrocytes, as well as immunoreactivity for GFAP and S100β (Table S2), consistently with previous observations (García‐Cáceres et al., 2016). BMAL1 was expressed in 48.5 ± 4.78% of TOMATO positive astrocytes in the ARC nucleus of control animals. In contrast, this proportion was reduced by approximately 60% in Bmal1cKO‐Td‐Tomato mice (paired t test, p = 8.39 × 10−6; Figure 3a,b). Similarly, the percentage of GFAP or S100β positive astrocytes expressing BMAL1 was significantly reduced by 60.1 and 62.3%, respectively, in the ARC of Bmal1cKO‐Td‐Tomato mice (paired t test, p = 6.9 × 10−8 for GFAP and p = 8.88 × 10−5 for S100β), compared to controls (Figure 3a).
Figure 3.
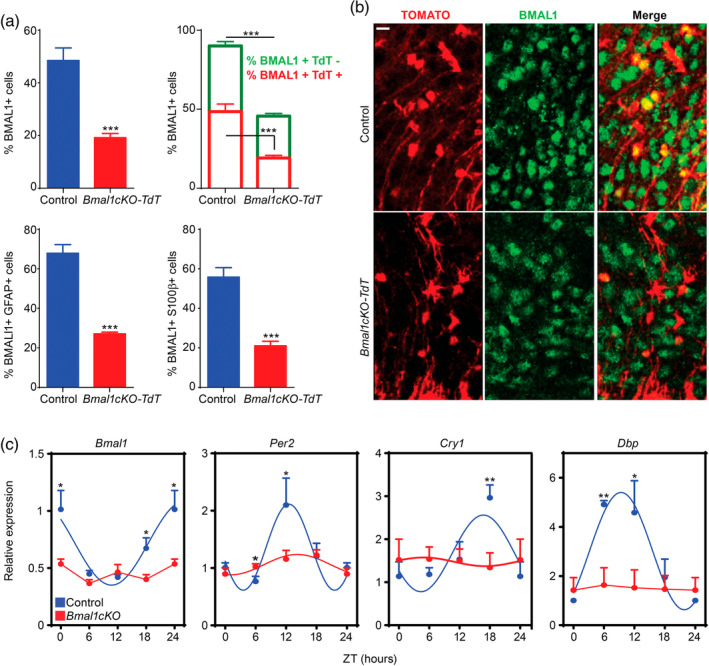
BMAL1 deletion in astrocytes globally impairs the hypothalamic molecular clock. (a) Upper left panel, a reduction of BMAL1 positive cells was observed in the ARC nucleus of Bmal1cKO mice compared with control animals 2 months after TM treatment (Y‐axis represents the percentage of total BMAL1‐positive cells in the ARC nucleus). Data are represented as mean ± SEM (n = 4, paired t‐test, ***p < .001 vs. control animals). Upper right panel, a 60% reduction of BMAL1‐positive cells was observed in the population of Td‐TOMATO‐positive cells of Bmal1cKO compared with control animals (red, paired t‐test, ***p < .001 vs. control animals). A 36% reduction of BMAL1‐positive cells in the population of Td‐TOMATO‐negative cells was found in Bmal1cKO compared with control animals (green, paired t‐test ***p < .001 vs. control animals). Data are represented as mean ± SEM (n = 4). Percent of BMAL1‐positive cells was significantly reduced in GFAP (lower left panel) or S100β (lower right panel) positive astrocytes in the ARC nucleus of Bmal1cKO‐Td‐Tomato mice compared with control animals. Data are represented as mean ± SEM (n = 4, paired t‐test, ***p < .001 vs. control animals). (b) Representative micrographs of BMAL1 immunostaining in the ARC nucleus of control or Bmal1cKO‐Td‐Tomato animals. Scale bar, 25 μm. (c) Analysis of clock transcripts (Bmal1, Cry1, Per2 and BMAL1 target, Dbp) in the hypothalamus of control and Bmal1cKO mice after 2 months of TM treatment. Experimental data were cosine fitted. The ZT24 time point is the ZT0 time point, shown again. Data are represented as mean ± SEM (n = 5–6, paired t‐test, *p < .05 and **p < .01) [Color figure can be viewed at wileyonlinelibrary.com]
Remarkably, in the mutants, the percentage of BMAL1 positive cells was also reduced by 36% in TOMATO negative cells (paired t test, p = 9.27 × 10−5; Figure 3a,b), suggesting that deletion of BMAL1 in GLAST positive astrocytes might also impact the clock in other cell populations of the hypothalamus, as we previously reported in cortex and hippocampus (Barca‐Mayo et al., 2017). To evaluate the impact of BMAL11 depletion on global oscillations in the hypothalamus, we quantified rhythmic expression of clock genes in controls and Bmal1cKOs, 2 months after TM treatment, at different ZTs. In control mice, we found rhythmic expression of Bmal1, Cry1, Per2, and BMAL1‐target Dbp, as previously reported (Barca‐Mayo et al., 2017) (Figure 3c). However, these oscillations were attenuated in Bmal1cKOs (Figure 3c), indicating that the deletion of BMAL1 in astrocytes globally impairs the molecular clock and, therefore, the circadian function of the hypothalamus.
3.4. Bmal1cKO mice showed age‐dependent astrogliosis and apoptosis of hypothalamic astrocytes
Next, we investigated whether the deregulation of the molecular clock in the brains of our mutants lead to reactive gliosis, as previously reported in cortex and hippocampus of constitutive Bmal1−/−, Nestin‐Cre‐Bmal1 or Aldh1l1‐CreERT2‐Bmal1 mice (Lananna et al., 2018; Musiek et al., 2013; Nakazato et al., 2017). Increased GFAP levels are generally regarded as hallmark of reactive glia, therefore we quantified Gfap transcripts or protein by qPCR or immunostaining, respectively, in different brain areas (cortical, hippocampal, and hypothalamic regions) of control and Bmal1cKO mice at 2 and 4 months after TM treatment. In agreement with previous reports (Lananna et al., 2018; Musiek et al., 2013; Nakazato et al., 2017), we did not detect the increased expression of Gfap in cortex or hippocampus 2 months after TM treatment (Figure 4a, Table S1). However, 4 months after TM treatment, we found increased GFAP immunoreactivity in different cortical regions and hippocampus of the mutants (Figure 4b and S1), confirming that Bmal1cKOs developed age‐dependent gliosis. Surprisingly, already at 2 months after TM treatment, we found elevated expression of Gfap in the hypothalamus of Bmal1cKO compared to control mice (Figure 4a). This result suggests that BMAL1 deletion in astrocytes leads to a temporal activation of this glial cell type which differs among different brain areas and, importantly that the hypothalamus is, among the different brain regions analyzed, the first in showing reactive gliosis.
Figure 4.
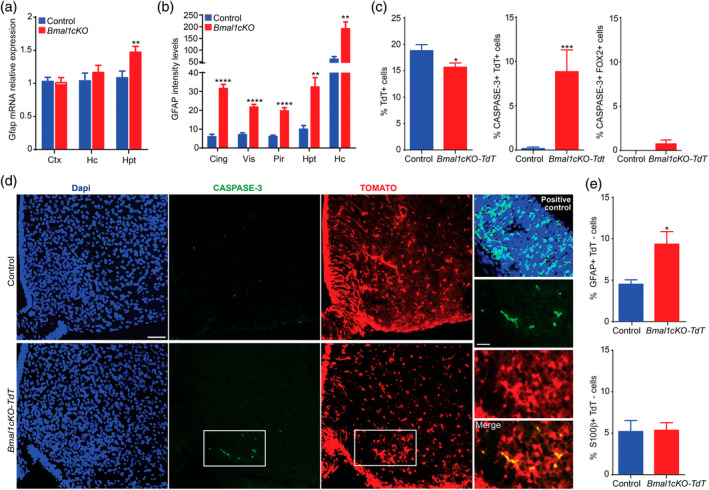
Bmal1cKO mice showed age‐dependent astrogliosis and apoptosis of hypothalamic astrocytes. (a) Gfap expression in cortex (Ctx), hippocampus (Hc) and hypothalamus (Hpt) of control and Bmal1cKO mice, 2 months after TM treatment. Data are represented as mean ± SEM (n = 5, paired t‐test, **p < .01). (b) Quantification of GFAP fluorescence intensity in cingulate (Cing), visual (Vis), piriform (Pir) cortex, Hpt and Hc of Bmal1cKOs, and controls, 4 months after TM treatment. Data are represented as mean ± SEM (n = 5, paired t‐test, **p < .01 and ****p < .0001 vs. controls). (c) Left panel, percentage of TOMATO‐positive cells in the ARC nucleus of control (Glast‐Cre‐Td‐Tomato) or Bmal1cKO‐Td‐Tomato animals, 2 months after TM treatment. Middle and right panel, percentage of active‐CASPASE 3 cells that co‐localized with TOMATO or FOX2 positive cells, respectively. Data are represented as mean ± SEM (n = 5, paired t‐test, *p < .05 and ***p < .001 vs. controls). (d) Representative micrographs of TOMATO and active CASPASE‐3 immunostaining in the ARC nucleus of control and Bmal1cKO‐Td‐Tomato mice 2 months after TM treatment. Scale bars, 50 and 25 μm in the higher magnification images. Cortex of Dgrc8 (DiGeorge Syndrome Critical Region Gene) knockout mice at embryonic day 13.5 was used as a positive control (right upper panel). (e) Percentage of GFAP (upper panel) or S100β (lower panel) positive cells that are TOMATO‐negative in the ARC nucleus of control and Bmal1cKO‐Td‐Tomato animals. Data are represented as mean ± SEM (n = 5, paired t‐test, *p < .05 vs. controls) [Color figure can be viewed at wileyonlinelibrary.com]
To gain further insights in this observation, we investigated whether reactivity was restricted to the subpopulation of GLAST positive astrocytes, in two different areas of the hypothalamus: The ARC nucleus, involved in the control of food intake and where inflammation and gliosis was linked to obesity in rodents and humans (reviewed in Valdearcos et al., 2015; McNay et al., 2012; Thaler et al., 2012) and the SCN, the main regulator of circadian locomotor activity in mammals (Ralph, Foster, Davis, & Menaker, 1990; Stephan & Zucker, 1972). Surprisingly, we found a 17% reduction of GLAST positive astrocytes (as revealed by TOMATO) in the ARC of Bmal1cKO‐Td‐Tomato mice compared to control animals, 2 months after TM treatment (Figure 4c). Moreover, we detected a significant decrease in the total number of cells per area, as determined by DAPI staining, in the ARC of the mutants 2 months after TM treatment (469.44 ± 39.99 for Bmal1cKOs vs. 585.14 ± 38.05 for controls, paired t test, p = .046). This decrease in the total cell numbers in the ARC of Bmal1cKO mice was not due to a reduction in the number of neurons, as shown by quantification of the neuronal marker RNA Binding Fox‐1 Homolog 2 (FOX2), a homolog of NeuN (Kim, Adelstein, & Kawamoto, 2009; Underwood, Boutz, Dougherty, Stoilov, & Black, 2005) (50.11% + 1.76 for controls and 53.49% + 2.64 for Bmal1cKOs, paired t‐test, p = .29). Indeed, in the ARC nucleus of Bmal1cKO‐Td‐Tomato mice we found a significant increase in the proportion of TOMATO‐positive astrocytes, but not in FOX2 positive neurons, that were also positive for the apoptosis marker active‐CASPASE‐3 (Figure 4c,d). Additionally, we observed a significant increase in the proportion of TOMATO negative cells that were positive for GFAP, while no differences were found in the expression of S100β among TOMATO positive or negative cells (Figure 4e and S2). Altogether, these results indicate a selective loss of GLAST positive astrocytes upon deletion of BMAL1 (i.e., TOMATO positive cells), and reactivity in the astrocytes that retained BMAL1 expression (i.e., TOMATO‐negative cells).
As we found reduced astrocyte numbers and increased the reactivity of astrocytes in the ARC after 2 months of TM treatment, we evaluated whether Bmal1 deletion was maintained at later time, specifically at 15 months post tamoxifen treatment. We found that the percent of GFAP positive cells was increased in the ARC nucleus of both control and Bmal1cKO mice with age (Figure S3b) as previously reported (reviewed in Palmer & Ousman, 2018). However, at this stage, Bmal1cKOs showed a significant increase in the percent of GFAP positive cells compared to control animals (Figure S3a,b). On the other hand, the percentage of BMAL1 positive cells in the ARC of control animals was reduced at 15 months after TM treatment compared to 2 months posttreatment (Figure S3c). This observation is consistent with previous reports showing a decline in the clock gene expression with age (reviewed in Hood & Amir, 2017). Despite, at this stage, we observed a reduction in the percentage of total BMAL1 positive cells in the ARC of the mutants compared to control animals, it did not reach significance (two‐way ANOVA, p = .19; Figure S3a,c). However, co‐immunostaining of GFAP and BMAL1 indicated a significant reduction among the BMAL1 positive that were GFAP positive or negative in the mutants (Figure S3d). Therefore, these results suggest that BMAL1 deletion is maintained at later time points after TM treatment. Indeed, despite that we detected a significant increase in the percentage of GFAP‐positive cells that were mitotically active (by coimmunostaining with the proliferative marker, KI67) after 15 months of TM treatment in the ARC nucleus of the mutants as compared to control animals (0.5% vs. 0.17%, respectively), this percentages were very small (Figure S4) and likely biologically irrelevant. Consistent with a previous report in Bmal1nestin−/− mice, these data suggest that the deletion of Bmal1 does not increase the number of astrocytes but rather increases the activation of pre‐existing ones (Nakazato et al., 2017).
Remarkably, in the SCN, no differences between control and Bmal1cKO animals was observed in the percentage of TOMATO (17.84% ± 1.51 vs. 17.23% ± 1.12, respectively, paired t test, p = .74), GFAP (16.47% ± 0.92 vs. 19.15% ± 0.82, respectively, paired t test, p = .58) or FOX2 positive cells (8.38% ± 0.46 vs. 10.5% ± 2, respectively, paired t test, p = .63), 2 months after TM treatment. Consistently, no differences were found in the proportion of TOMATO negative cells that were positive for GFAP between control and Bmal1cKO animals (7.11% ± 0.94 vs. 7.10% ± 0.72, respectively, paired t‐test, p = .99) indicating that BMAL1 deletion in SCN astrocytes do not lead to reactive gliosis 2 months after TM. As wheel‐running activity is widely used as an index of SCN circadian function (Ralph et al., 1990; Stephan & Zucker, 1972), this observation is consistent with the comparable pattern of daily activity in control and mutant animals in 12 hr–12 hr light–dark cycles (Figure 2b) indicating that the SCN circadian function was not altered in Bmal1cKO mice 2 months after TM treatment. It was reported that the circadian output measured at the level of circadian locomotor behavior is dampened with age (Nakamura et al., 2011). Consistent with this report, we found that the daily activity of control animals, at 15 months after TM treatment, were reduced as compared to younger mice (2 months after TM treatment; Figures 2b and S5). Remarkably, the circadian locomotor activity of the mutants, at this stage, was reduced as compared to controls (Figure S5).
Our results indicate that BMAL1 deletion in astrocytes lead to a temporal‐ and regional‐specific gliosis within the hypothalamus and, importantly that ARC nucleus is, among the different brain regions analyzed, the first showing astrocyte reactivity and apoptosis. This cascade of events likely accounts for the altered hypothalamic function of Bmal1cKO mice, which might lead to the increased body weight and metabolic alterations of the mutants. Together, these results (i.e., shorter lifespan, glucose imbalance, altered growth curves, age‐dependent gliosis and decrease of age‐related circadian locomotor activity) support our hypothesis that BMAL1 deletion in astrocytes may lead to premature aging.
3.5. GABAA receptor antagonist delayed the aging and metabolic phenotype of Bmal1cKO mice
Hypothalamic neurons expressing the orexigenic (appetite‐increasing) Neuropeptide Y/Agouti‐related peptide (NPY/AGRP) or the anorexigenic (appetite‐suppressing) Pro‐opiomelanocortin (POMC) receive inhibitory and excitatory inputs from GABA and glutamate. These two neurotransmitters account for most of the synaptic activity in the hypothalamus and therefore, are directly involved in appetite and energy balance regulation (reviewed in Delgado, 2013). We recently reported increased GABA levels in the CSF of Bmal1cKO mice in the light phase (day time, i.e., ZT6), 2 months after TM treatment (Barca‐Mayo et al., 2017). We now expand on these results by showing significantly higher levels of GABA also in the hypothalamus of Bmal1cKO mice in the light phase, compared to control animals (Figure 5a). Therefore, we hypothesized that the increased levels of GABA in the CSF and hypothalamus of Bmal1cKOs might be associated to their metabolic and age‐associated dysfunctions. Consistently, it was reported a stimulatory role for GABA in the regulation of hypothalamus‐dependent feeding behavior. Specifically, intracerebroventricular (ICV) administration of the GABAA receptor agonist muscimol stimulates feeding in satiated pigs, a response that is prevented by the specific GABAA receptor antagonist bicuculline (Baldwin, Ebenezer, & De La Riva, 1990). Also, systemic and ICV administration of the GABAB receptor agonist baclofen increases food intake in satiated pigs that can be abolished by pretreatment with the GABAB receptor antagonist phaclofen (Ebenezer & Baldwin, 1990).
Figure 5.
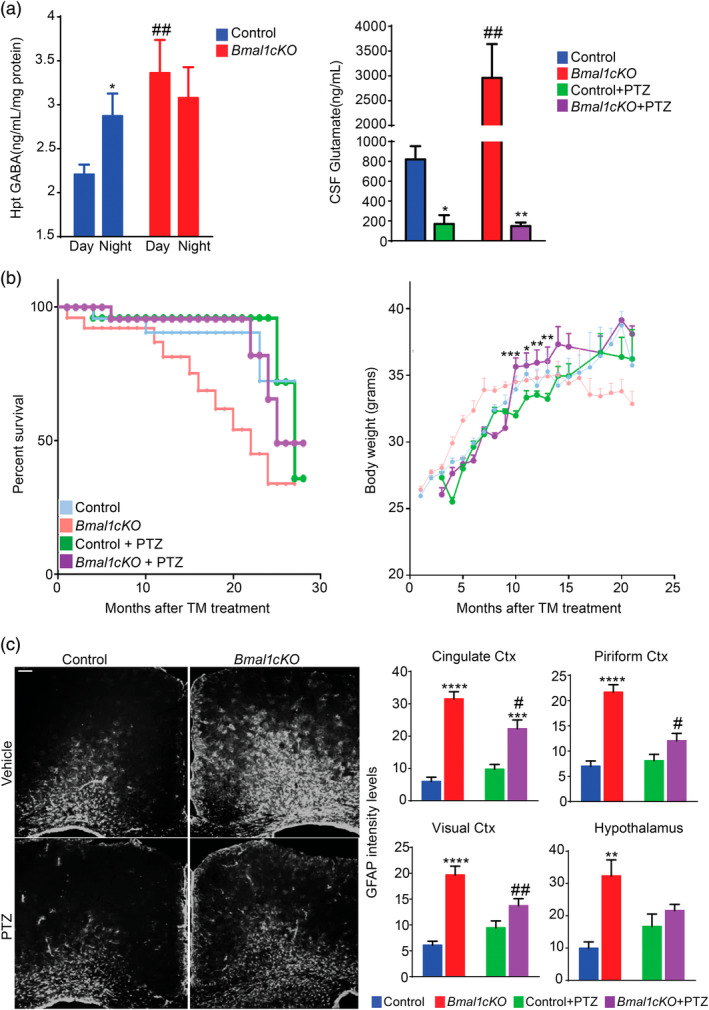
GABAA receptor antagonists delayed the aging and metabolic phenotype of Bmal1cKO mice. (a) Left panel, GABA levels in the hypothalamus of Bmal1cKOs and controls at day and night time, 2 months after TM treatment. Data are represented as mean ± SEM (n = 5, two‐way ANOVA, *p < .05 vs. daytime and ##p < .01 vs. controls). Right panel, CSF glutamate levels of naive or PTZ‐treated Bmal1cKO and control animals at ZT6. Data are represented as mean ± SEM (n = 5, two‐way ANOVA, *p < .05 and **p < .01 vs. naive animals; ##p < .01 vs. controls). (b) Left panel, Kaplan–Meyer survival curve of naive or PTZ‐treated control and Bmal1cKO mice. Right panel, age‐dependent changes in body weight of control and Bmal1cKO mice treated with PTZ. Data are represented as mean ± SEM (paired t‐test, *p < .05, **p < .01, and ***p < .001 vs. PTZ‐treated control animals). n = 10– 12 for PTZ‐treated control and Bmal1cKO animals. (c) Left panel, representative micrographs of GFAP immunostaining in the cingulate cortex of naive or PTZ‐treated control and Bmal1cKO mice, 4 months after TM treatment. Scale bar, 100 μm. Right panel, quantification of fluorescence intensity of GFAP levels in cingulate, piriform, and visual cortex (Ctx) as well as in the hypothalamus of naive or PTZ‐treated Bmal1cKO and control animals. Data are represented as mean ± SEM (n = 5, two‐way ANOVA, **p < .01, ***p < .001 and ****p < .0001 vs. control animals and #p < .05 and ##p < .01 vs. naive mice) [Color figure can be viewed at wileyonlinelibrary.com]
On the other hand, glutamate stimulates feeding upon the administration of its receptor agonists. ICV or lateral hypothalamic injection of glutamate, or its excitatory amino acid agonists (kainic acid, d,l‐alpha‐amino‐3‐hydroxy‐5‐methyl‐isoxazole propionic acid, and N‐methyl‐d‐aspartic acid) rapidly elicits intense food intake in rats (Stanley, Ha, Spears, & Dee, 1993; Stricker‐Krongrad, Beck, Nicolas, & Burlet, 1992). Likewise, ICV administration of metabotropic glutamate receptor 5 (mGluR5) agonists stimulate feeding whereas the mGluR5 receptor antagonist (R,S)‐2‐chloro‐5‐hydroxyphenylglycine, inhibits food intake (Ploj et al., 2010). Thereby, we also quantified glutamate levels in the CSF of control and Bmal1cKO animals 2 months after TM treatment at ZT6, following a previously described liquid chromatography–tandem mass spectrometry protocol (Buck, Voehringer, & Ferger, 2009). Control animals showed CSF glutamate levels in line with those previously reported (~7 μg ml−1; Espey, Kustova, Sei, & Basile, 1998). Remarkably, glutamate levels were significantly higher in the CSF of Bmal1cKOs (Figure 5a).
Previously, we showed that the increased GABA levels in the CSF of Bmal1cKO leads to the over‐inhibition of the circuits involved in learning and memory and to the uncoupling the SCN oscillators (Barca‐Mayo et al., 2017). Consistently, administration of GABAA receptor antagonists, following previously reported protocols (Colas et al., 2013; Fernandez et al., 2014; Ruby et al., 2013), restored the circadian locomotor activity and the cognitive functions of the mutants (Barca‐Mayo et al., 2017). To our surprise, we now found that glutamate levels in the CSF of both control and Bmal1cKO mice were strongly downregulated below normal levels 1 week after administration of the GABAA receptor antagonist pentylenetetrazole (PTZ, administered 2 months after TM treatment; Figure 5a). Importantly, the daily administration of PTZ at ZT6 (for 10 consecutive days during the pre‐obese stage) was sufficient to delay the transient obesity of Bmal1cKO mice for 6 months, while no differences were found in control animals (Figure 5b). Strikingly, PTZ treatment normalized the lifespan of Bmal1cKO animals (average lifespan 22 vs. 25 months after TM treatment, respectively, Log‐rank test, p = .049) while no differences were found in controls (Figure 5b). The long‐term effect of this protocol of PTZ treatment, at nonepileptic doses, is not surprising at it was previously shown to produce long‐lasting cognitive improvements after drug withdrawal in rodents (Colas et al., 2013; Contestabile, Magara, & Cancedda, 2017; Fernandez et al., 2014; Ruby et al., 2013). Interestingly, we also found that PTZ‐treated Bmal1cKO mice displayed significantly reduced levels of reactive gliosis in different areas of the cortex as well as in the hypothalamus in comparison to untreated animals at 4 months after TM treatment (i.e., 2 months after the PTZ administration; Figure 5c).
In sum, as the administration of GABAA receptor antagonist normalized glutamate levels, reduced astrocytes reactivity, delayed obesity and increased the lifespan of Bmal1cKO mice, we postulate that GABA and/or glutamate‐signaling are likely involved in astrocyte‐dependent control of lifespan and energy balance.
4. DISCUSSION
This study is the first demonstration that adult deletion of BMAL1 in a subpopulation of astrocytes is sufficient to shorten lifespan and alter energy balance, thus partly recapitulating phenotypes described in constitutive Bmal1−/− (Bunger et al., 2005; Kondratov et al., 2006; Lamia et al., 2008; Lee et al., 2006; Marcheva et al., 2010; Rudic et al., 2004; Shi et al., 2006). Importantly, as modulation of GABAA‐receptor signaling delayed the metabolic dysfunctions and the early death of Bmal1cKO mice, we demonstrate a crucial contribution of astrocytic clock in linking GABA signaling with the circadian regulation of metabolism and lifespan.
Astrocytes are widely distributed throughout the nervous system and express brain‐region‐specific genes. Thereby the selection of regulatory elements to target all astrocytes in vivo is almost impossible with today's tools. However, the general practice when using genetic approaches to perform recombination in all (or most) target cells might not be needed for astrocytes‐mediated phenotypes due to their anatomical properties. First, astrocytes are organized in structurally nonoverlapping domains in vivo where one astrocyte interacts with four neuron cell bodies, between 300 and 600 dendrites and more than 100,000 synapses (Bushong, Martone, Jones, & Ellisman, 2002; Halassa, Fellin, & Haydon, 2007). Second, astrocytes are interconnected through gap junction channels which allow metabolic or biochemical coupling with propagation distances ranging from four to up to 30 astrocytes (reviewed in Tian et al., 2006; Sul, Orosz, Givens, & Haydon, 2004). Therefore, despite not all astrocytes are directly targeted in our Bmal1cKO mice, the finding that they partially recapitulate the metabolic phenotype and early death of constitutive Bmal1−/− mice is not surprising and supported by the recent report that few GFAP positive astrocytes of the SCN are sufficient to instruct neurons to initiate and indefinitely sustain circadian patterns of activity and behavior (Brancaccio et al., 2019).
Our findings that the administration of a GABAA receptor antagonist normalized the glutamate levels, delayed the reactive gliosis and the metabolic phenotypes and increased the lifespan of Bmal1cKO mice, suggest that the altered GABA and/or glutamate astrocyte–neuron coupling underlie the phenotypes of our mutants. This hypothesis is supported by previous reports showing that astrocytes critically modulate hypothalamic neural circuits controlling energy homeostasis (Chen et al., 2016; Zhang, Reichel, Han, Zuniga‐Hertz, & Cai, 2017) by modulating extracellular GABA bioavailability (Zhang et al., 2017). Additionally, astrocytes can impact the molecular clock in cortical, hippocampal, SCN (Barca‐Mayo et al., 2017; Barca‐Mayo, Berdondini, & De Pietri Tonelli, 2019; Tso et al., 2017; Duhart et al., 2013) and hypothalamic neurons (present study). Indeed, we and others previously reported that GABA uptake and glutamate release coupled to astrocyte rhythms (Barca‐Mayo et al., 2017; Brancaccio et al., 2017, 2019; Tso et al., 2017) play a key role for rhythmic astrocyte‐neuron intercellular communication. In line with these observations, our study opens the possibility that astrocyte rhythmic regulation of GABA and/or glutamate might transmit timing information between this glial cell type and hypothalamic neuronal networks to optimize energy balance.
An important implication of our study is that while the deletion of BMAL1 is local, the impact on circulating leptin, glucose, and insulin provides a new link among circadian timing in astrocytes and systemic metabolic states or peripheral clocks. In the first case, by expressing the receptors for leptin and insulin (Cheunsuang & Morris, 2005; García‐Cáceres et al., 2016; Hsuchou et al., 2010; Hsuchou, Pan, Barnes, & Kastin, 2009; Jayaram et al., 2013; Kim et al., 2014; Pan et al., 2008), astrocytes are endowed with metabolic signal‐sensing properties, suggesting an alternative role for the regulation of energy homeostasis. Indeed, astrocytes have been reported to sense insulin and leptin, as well as to co‐regulate behavioral responses and metabolic processes via the control of brain glucose uptake and the glial ensheathment of POMC neurons in the ARC, respectively (García‐Cáceres et al., 2016; Kim et al., 2014), However, it is also conceivable that astrocyte clock control of leptin and glucose homeostasis may occur through interactions with peripheral clocks, leading to altered leptin rhythms or to inadequate response of peripheral tissues to circulating insulin respectively in the mutants. For example, astrocytes, as well‐known targets of glucocorticoids, might be sensitive to the negative feedback loop of the hypothalamo‐pituitary‐adrenal axis, thus linking peripheral and central oscillators. Importantly, it is widely accepted that while glucocorticoid signaling can reset peripheral clocks, it does not impact the central clock because SCN neurons do not express the glucocorticoid receptor Nuclear Receptor Subfamily 3 Group C Member 1 (NR3C1; Rosenfeld, Van Eekelen, Levine, & De Kloet, 1988). Thus, astrocytic feedback loops, via glucocorticoid signaling, could explain the so far puzzling results showing that the Per1‐Luc phases of SCN were affected significantly when adrenalectomized animals were treated with hydrocortisone administered in their drinking water (Pezük, Mohawk, Wang, & Menaker, 2012). It will be an enticing challenge to identify astrocyte rhythmic outputs or inputs from systemic cues, which transmits timing information between this glial cell type and peripheral tissues and/or clocks to optimize energy balance.
The effect of astrocytic BMAL1 deletion on the metabolic dysfunctions and early death cannot be completely disentangled from the effects on SCN‐mediated rhythms. However, several evidences suggest that these phenotypes of Bmal1cKO mice rely in extra‐SCN clocks. First, the circadian locomotor activity, commonly used as an index of SCN circadian function (Ralph et al., 1990; Stephan & Zucker, 1972), is not lost in mice with Bmal1 deletion in SCN astrocytes (Barca‐Mayo et al., 2017; Brancaccio et al., 2017; Tso et al., 2017). Second, BMAL1 deletion in SCN does not affect either lifespan or body weight despite complete loss of rhythmic behavior (Izumo et al., 2014). Consistently the circadian locomotor activity but not the ageing and metabolic disturbances of Bmal1−/− mice was rescued by restoring BMAL1 expression in the SCN (McDearmon et al., 2006). Third, BMAL1 specific ablation within steroidogenic factor 1 (SF1)‐neurons of the ventromedial hypothalamus is sufficient to alter energy expenditure (Orozco‐Solis et al., 2016). This study is in contrast with a report showing no alterations body weight and glucose homeostasis upon adult BMAL1 deletion, driven by tamoxifen‐regulated activation of the estrogen receptor 1 (Esr1)‐Cre system (Yang et al., 2016) selectively targeting SF1‐positive neurons, which express ESR1 (Musatov et al., 2007). A possible explanation for this apparent contradiction is that the timing of BMAL1 deletion, postnatally or from development, respectively, influences its effects on aging and survival (Yang et al., 2016). Interestingly, at present, expression of ESR1 has not been reported in hypothalamic astrocytes in vivo (Liu & Shi, 2015) thus providing a possible explanation for the phenotypic differences between Glast‐cre and Esr1‐cre‐driven Bma1cKO mice. Altogether, this suggests that a functional molecular clock in SCN astrocytes is neither sufficient nor required for controlling lifespan and energy balance.
Transcriptional activity induced by BMAL1 may have effects independent of the circadian clock that could impinge on aging‐associated dysfunctions and metabolism. Indeed, distinguishing the specific importance of circadian oscillation versus the “static” function of clock genes is a major challenge for all studies involving genetic manipulations of core clock genes. However, the BMAL1 DNA binding is highly rhythmic and regulated by the clock (Koike et al., 2012), making it difficult to separate entirely from circadian rhythms. For example, the reactive gliosis and astrocyte dysfunction observed in a neuronal‐specific Bmal1KO (Izumo et al., 2014) or in mice upon circadian disruption by exposure to 10 hr:10 hr light–dark conditions, which blunts BMAL1 oscillations in the brain (Lananna et al., 2018), suggest that it might be due to the loss of rhythms. On the other hand, it was also reported that arrhythmicity in the setting of increased BMAL1 expression, as in Per1/2 mutant mice, does not induce astrocyte activation (Lananna et al., 2018). Therefore, it is difficult to discern whether the reactive gliosis is dependent on suppression of BMAL1‐mediated transcription or to the loss of rhythms.
Reactivity in astrocytes typically increases their inflammatory phenotype and cause loss of their neuro‐supportive functions, thus rendering neurons vulnerable to hypo‐metabolic states, excitotoxicity and oxidative stress. Thus, gliosis is considered general hallmark of brain aging (Musiek et al., 2013) and age associated neurodegenerative disorders (reviewed in Camandola & Mattson, 2017). Importantly, reactive astrocytes abnormally produce and release GABA (Jo et al., 2014) and decrease glutamate uptake (Beurrier et al., 2010; Escartin et al., 2006). Indeed, suppressing GABA production or release from reactive astrocytes fully restores the synaptic plasticity and memory in a mouse model with Alzheimer's disease (Jo et al., 2014). On the other hand, aging is associated with increased levels of reactive oxygen species (ROS) and oxidized products in different tissues (reviewed in Balaban, Nemoto, & Finkel, 2005). Indeed, the free radical theory of aging postulates that the production of intracellular ROS is the major determinant of lifespan (reviewed in Balaban et al. (2005)). Remarkably, the cellular redox state is dependent on BMAL1 expression (Khapre, Kondratova, Susova, & Kondratov, 2011; Wang et al., 2012) and the acetylation of multiple critical mitochondrial proteins shows circadian oscillation, indicating clock‐mediated control of the redox state (Masri et al., 2013). ROS are important regulators of cellular metabolism, gene expression, and other molecular responses, playing key roles in the control of various physiological processes. The levels of external (food‐generated) and internal (metabolism/activity‐generated) oxidants change during the day as a result of fluctuations in food intake and behavior. Therefore, control of ROS homeostasis by the circadian system, which is intrinsically connected to an organism's daily activity, would provide the most effective protection from the damaging effects of oxidants at any given time of day. Indeed, the early aging phenotype in Bmal1−/− animals correlates with increased levels of ROS in some tissues, including brain, and treatment with the glutathione precursor N‐acetyl cysteine, extends lifespan in these mice (Kondratov et al., 2006; Kondratov, Vykhovanets, Kondratova, & Antoch, 2009; Kondratova, Dubrovsky, Antoch, & Kondratov, 2010). Similarly, targeted deletion of BMAL1 in neurons and glia promoted neuronal death in primary cultures and in mice treated with a chemical inducer of oxidative injury and neurodegeneration (Musiek et al., 2013). Consistent with our results, it was previously shown that BMAL1 deletion in astrocytes induces gliosis and inflammatory gene expression in vitro and in vivo, mediated in part by suppression of glutathione‐S‐transferase signaling (Lananna et al., 2018). Remarkably, supplementing BMAL1‐deficient astrocyte cultures or mice with NAC prevents astrogliosis (Lananna et al., 2018). Our results showing a lost in hypothalamic astrocytes upon BMAL1 deletion as well as the finding that the hypothalamus is the first region of the brain showing the reactive gliosis, suggest that arrhythmic astrocytes might lead to an inflammatory and hypometabolic state, rendering neurons to be more susceptible for neurodegeneration, excitotoxicity, and oxidative stress. Indeed, loss of BMAL1 in astrocytes promotes neuronal death in vitro to influence many aspects of brain function and neurological disease (Musiek et al., 2013). Failure of neurons to respond adaptively to a decline in basal metabolic rate and in energy‐driven tasks is a risk factor for age associated neurodegenerative disorders. At the same time, an imbalance between the circadian and ROS generating/metabolizing systems might increase damage due to oxidative stress, thus contributing to and/or complicating pathogenesis, aging, and lifespan.
Our finding that BMAL1 loss in astrocytes leads to increased GABA and glutamate levels and to age‐dependent gliosis has many potential implications for age‐related neurodegenerative diseases and suggests that further study of the regulation and function of astrocyte core clock genes in health and disease is warranted. Therefore, here we propose that circadian manipulation of GABA signaling, GABA uptake by astrocytes and/or bolstering astrocyte clock might have neuroprotective effects in noninvasive therapies for metabolic disorders and ageing.
CONFLICT OF INTEREST
The Authors declare that there is no conflict of interest.
Supporting information
Appendix S1: Supplementary material
ACKNOWLEDGMENTS
We thank Dr M. Götz (Physiological Genomics, Biomedical Center, Ludwig‐Maximilians‐University Munich, Germany) for kindly providing the Glast‐CreERT2 mouse line. We thank R. Pelizzoli and IIT‐NBT technical staff for their excellent support. We also thank M. Morini and staff of IIT Animal Facility Central Research labs for assistance in animal experiments. This work was supported by the European Research Executive Agency (REA) through the FP7‐PEOPLE‐2014‐IEF “ASTROCLOCK” (629867) and Fondazione CARIPLO research grant (2015‐0590). O.B.M. has a “Ramon y Cajal” contract (RYC2018‐026293‐I) from the Ministerio de Ciencia, Innovación y Universidades of Spain.
Barca‐Mayo O, Boender AJ, Armirotti A, De Pietri Tonelli D. Deletion of astrocytic BMAL1 results in metabolic imbalance and shorter lifespan in mice. Glia. 2020;68:1131–1147. 10.1002/glia.23764
Funding information European Research Executive Agency, Grant/Award Number: 629867; Fondazione CARIPLO Research, Grant/Award Number: 2015‐0590; Ministerio de Ciencia, Innovación y Universidades, Grant/Award Number: RYC2018‐026293‐I
The copyright line for this article was changed on 30 August 2020 after original online publication.
DATA AVAILABILITY STATEMENT
All raw/original relevant data are available upon request.
REFERENCES
- Balaban, R. S. , Nemoto, S. , & Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell, 120(4), 483–495. 10.1016/j.cell.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Baldwin, B. A. , Ebenezer, I. S. , & De La Riva, C. (1990). Effects of intracerebroventricular injection of muscimol or GABA on operant feeding in pigs. Physiology & Behavior, 48l(3), 417–421. 10.1016/0031-9384(90)90337-4 [DOI] [PubMed] [Google Scholar]
- Barca‐Mayo, O. , Berdondini, L. , & De Pietri Tonelli, D. (2019). Astrocytes and circadian rhythms: An emerging astrocyte‐neuron synergy in the timekeeping system In Di Benedetto B. (Ed.), Methods in molecular biology 1938 (1st ed., pp. 131–154). New York: Humana Press; 10.1007/978-1-4939-9068-9_10 [DOI] [PubMed] [Google Scholar]
- Barca‐Mayo, O. , Pons‐Espinal, M. , Follert, P. , Armirotti, A. , Berdondini, L. , & De Pietri Tonelli, D. (2017). Astrocyte deletion of Bmal1 alters daily locomotor activity and cognitive functions via GABA signaling. Nature Communications, 8, 14336 10.1038/ncomms14336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgardt, B. , Okamura, T. , & Brüning, J. C. (2009). Hormone and glucose signaling in POMC and AgRP neurons. The Journal of Physiology, 587(Pt 22), 5305–5314. 10.1113/jphysiol.2009.179192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurrier, C. , Faideau, M. , Bennouar, K. E. , Escartin, C. , Kerkerian‐Le Goff, L. , Bonvento, G. , & Gubellini, P. (2010). Ciliary neurotrophic factor protects striatal neurons against excitotoxicity by enhancing glial glutamate uptake. PLoS One, 5(1), e8550 10.1371/journal.pone.0008550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouet, C. , & Schwartz, G. J. (2010). Hypothalamic nutrient sensing in the control of energy homeostasis. Behavioral Brain Research, 209(1), 1–12. 10.1016/j.bbr.2009.12.024 [DOI] [PubMed] [Google Scholar]
- Brancaccio, M. , Edwards, M. D. , Patton, A. P. , Smyllie, N. J. , Chesham, J. E. , Maywood, E. S. , & Hastings, M. H. (2019). Cell‐autonomous clock of astrocytes drives circadian behavior in mammals. Science, 363(6423), 187–192. 10.1126/science.aat4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio, M. , Patton, A. P. , Chesham, J. E. , Maywood, E. S. , & Hastings, M. H. (2017). Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron, 93(6), 1420–1435. 10.1016/j.neuron.2017.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck, K. , Voehringer, P. , & Ferger, B. (2009). Rapid analysis of GABA and glutamate in microdialysis samples using high performance liquid chromatography and tandem mass spectrometry. Journal of Neuroscience Methods, 182(1), 78–84. 10.1016/j.jneumeth.2009.05.018 [DOI] [PubMed] [Google Scholar]
- Bunger, M. K. , Walisser, J. A. , Sullivan, R. , Manley, P. A. , Moran, S. M. , Kalscheur, V. L. , … Bradfield, C. A. (2005). Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal‐1 locus. Genesis, 41(3), 122–132. 10.1002/gene.20102 [DOI] [PubMed] [Google Scholar]
- Bushong, E. A. , Martone, M. E. , Jones, Y. Z. , & Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. Journal of Neuroscience, 22(1), 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola, S. , & Mattson, M. P. (2017). Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal, 36(11), 1474–1492. 10.15252/embj.201695810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari, A. , Gupta, R. , Makwana, K. , & Kondratov, R. (2017). Circadian clocks, diets and aging. Nutrition and Healthy Aging, 31(2), 101–112. 10.3233/NHA-160006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, N. , Sugihara, H. , Kim, J. , Fu, Z. , Barak, B. , Sur, M. , … Han, W. (2016). Direct modulation of GFAP‐expressing glia in the arcuate nucleus bi‐directionally regulates feeding. eLife, 5, e18716 10.7554/eLife.18716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheunsuang, O. , & Morris, R. (2005). Astrocytes in the arcuate nucleus and median eminence that take up a fluorescent dye from the circulation express leptin receptors and neuropeptide YY1 receptors. Glia, 52(3), 228–233. 10.1002/glia.20239 [DOI] [PubMed] [Google Scholar]
- Colas, D. , Chuluun, B. , Warrier, D. , Blank, M. , Wetmore, D. Z. , Buckmaster, P. , … Heller, H. C. (2013). Short‐term treatment with the GABAA receptor antagonist pentylenetetrazole produces a sustained pro‐cognitive benefit in a mouse model of Down's syndrome. British Journal of Pharmacology, 169(5), 963–973. 10.1111/bph.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, A. P. , Farooqi, I. S. , & O'Rahilly, S. (2007). The hormonal control of food intake. Cell, 129(2), 251–262. 10.1016/j.cell.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile, A. , Magara, S. , & Cancedda, L. (2017). The GABAergic hypothesis for cognitive disabilities in down syndrome. Frontiers in Cellular Neuroscience, 11, 54 10.3389/fncel.2017.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado, T. C. (2013). Glutamate and GABA in appetite regulation. Frontiers in Endocrinology, 4, 103 10.3389/fendo.2013.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, M. , & Horvath, T. (2009). Feeding signals and brain circuitry. European Journal of Neuroscience, 30(9), 1688–1696. 10.1111/j.1460-9568.2009.06963.x [DOI] [PubMed] [Google Scholar]
- Duhart, J. M. , Leone, M. J. , Paladino, N. , Evans, J. A. , Castanon‐Cervantes, O. , Davidson, A. J. , & Golombek, D. A. (2013). Suprachiasmatic astrocytes modulate the circadian clock in response to TNF‐α. The Journal of Immunology, 19(9), 4656–4664. 10.4049/jimmunol.1300450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenezer, I. S. , & Baldwin, B. A. (1990). Effect of intracerebroventricular administration of the GABAB‐receptor agonist baclofen on operant feeding in satiated pigs. British Journal of Pharmacology, 101(3), 559–562. 10.1111/j.1476-5381.1990.tb14120.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin, C. , Brouillet, E. , Gubellini, P. , Trioulier, Y. , Jacquard, C. , Smadja, C. , … Bonvento, G. (2006). Ciliary neurotrophic factor activates astrocytes, redistributes their glutamate transporters GLAST and GLT‐1 to raft microdomains, and improves glutamate handling in vivo. Journal of Neuroscience, 26(22), 5978–5989. 10.1523/JNEUROSCI.0302-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey, M. G. , Kustova, Y. , Sei, Y. , & Basile, A. S. (1998). Extracellular glutamate levels are chronically elevated in the brains of LP‐BM5‐infected mice: A mechanism of retrovirus‐induced encephalopathy. Journal of Neurochemistry, 71(5), 2079–2087. 10.1046/j.1471-4159.1998.71052079.x [DOI] [PubMed] [Google Scholar]
- Fernandez, F. , Lu, D. , Ha, P. , Costacurta, P. , Chavez, R. , Heller, H. C. , & Ruby, N. F. (2014). Dysrhythmia in the suprachiasmatic nucleus inhibits memory processing. Science, 346(6211), 854–857. 10.1126/science.1259652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Cáceres, C. , Quarta, C. , Varela, L. , Gao, Y. , Gruber, T. , Legutko, B. , … Tschöp, M. H. (2016). Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell, 166(4), 867–880. 10.1016/j.cell.2016.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstone, A. P. (2006). The hypothalamus, hormones, and hunger: Alterations in human obesity and illness. Progress in Brain Research, 153, 57–73. 10.1016/S0079-6123(06)53003-1 [DOI] [PubMed] [Google Scholar]
- Halassa, M. M. , Fellin, T. , & Haydon, P. G. (2007). The tripartite synapse: Roles for gliotransmission in health and disease. Trends in Molecular Medicine, 13(2), 54–63. 10.1016/j.molmed.2006.12.005 [DOI] [PubMed] [Google Scholar]
- Hastings, M. H. , Maywood, E. S. , & Reddy, A. B. (2008). Two decades of circadian time. Journal of Neuroendocrinology, 20(6), 812–819. 10.1111/j.1365-2826.2008.01715.x [DOI] [PubMed] [Google Scholar]
- Hodos, W. (1961). Progressive ratio as a measure of reward strength. Science, 134(3483), 943–944. 10.3791/3754 [DOI] [PubMed] [Google Scholar]
- Hodos, W. (1963). A simple method for description of interresponse time distributions. Journal of the Experimental Analysis of Behavior, 6, 90 10.1901/jeab.1963.6-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood, S. , & Amir, S. (2017). The aging clock: Circadian rhythms and later life. Journal of Clinical Investigation, 127(2), 437–446. 10.1172/JCI90328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsuchou, H. , Kastin, A. J. , Tu, H. , Joan Abbott, N. , Couraud, P. O. , & Pan, W. (2010). Role of astrocytic leptin receptor subtypes on leptin permeation across hCMEC/D3 human brain endothelial cells. Journal of Neurochemistry, 115(5), 1288–1298. 10.1111/j.1471-4159.2010.07028.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsuchou, H. , Pan, W. , Barnes, M. J. , & Kastin, A. J. (2009). Leptin receptor mRNA in rat brain astrocytes. Peptides, 30(12), 2275–2280. 10.1016/j.peptides.2009.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumo, M. , Pejchal, M. , Schook, A. C. , Lange, R. P. , Walisser, J. A. , Sato, T. R. , … Takahashi, J. S. (2014). Differential effects of light and feeding on circadian organization of peripheral clocks in a forebrain Bmal1 mutant. eLife, 3, e04617 10.7554/eLife.04617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram, B. , Pan, W. , Wang, Y. , Hsuchou, H. , Mace, A. , Cornelissen‐Guillaume, G. G. , … Kastin, A. J. (2013). Astrocytic leptin‐receptor knockout mice show partial rescue of leptin resistance in diet‐induced obesity. Journal of Applied Physiology, 114(6), 734–741. 10.1152/japplphysiol.01499.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, S. , Yarishkin, O. , Hwang, Y. J. , Chun, Y. E. , Park, M. , Woo, D. H. , … Lee, C. J. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nature Medicine, 20(8), 886–896. 10.1038/nm.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson, B. H. , Knutsson, A. K. , Lindahl, B. O. , & Alfredsson, L. S. (2003). Metabolic disturbances in male workers with rotating three‐shift work. Results of the WOLF study. International Archives of Occupational and Environmental Health, 76(6), 424–430. 10.1007/s00420-003-0440-y [DOI] [PubMed] [Google Scholar]
- Khapre, R. V. , Kondratova, A. A. , Susova, O. , & Kondratov, R. V. (2011). Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle, 10(23), 4162–4169. 10.4161/cc.10.23.18381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. G. , Suyama, S. , Koch, M. , Jin, S. , Argente‐Arizon, P. , Argente, J. , … Horvath, T. L. (2014). Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nature Neuroscience, 17(7), 908–910. 10.1038/nn.3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. K. , Adelstein, R. S. , & Kawamoto, S. (2009). Identification of neuronal nuclei (NeuN) as Fox‐3, a new member of the Fox‐1 gene family of splicing factors. The Journal of Biological Chemistry, 284(45), 31052–31061. 10.1074/jbc.M109.052969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike, N. , Yoo, S. H. , Huang, H. C. , Kumar, V. , Lee, C. , Kim, T. K. , & Takahashi, J. S. (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science, 338(6105), 349–354. 10.1126/science.1226339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov, R. V. , Kondratova, A. A. , Gorbacheva, V. Y. , Vykhovanets, O. V. , & Antoch, M. P. (2006). Early aging and age‐related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes & Development, 15(14), 1868–1873. 10.1101/gad.1432206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov, R. V. , Vykhovanets, O. , Kondratova, A. A. , & Antoch, M. P. (2009). Antioxidant N‐acetyl‐L‐cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging (Albany NY), 1(12), 979–987. 10.18632/aging.100113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratova, A. A. , Dubrovsky, Y. V. , Antoch, M. P. , & Kondratov, R. V. (2010). Circadian clock proteins control adaptation to novel environment and memory formation. Aging, 2(5), 285–297. 10.18632/aging.100142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume, K. , Zylka, M. J. , Sriram, S. , Shearman, L. P. , Weaver, D. R. , Jin, X. , … Reppert, S. M. (1999). mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell, 98(2), 193–205. 10.1016/s0092-8674(00)81014-4 [DOI] [PubMed] [Google Scholar]
- Lamia, K. A. , Storch, K. F. , & Weitz, C. J. (2008). Physiological significance of a peripheral tissue circadian clock. Proceedings of the National Academy of Sciences of the United States of America, 105(39), 15172–15177. 10.1073/pnas.0806717105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lananna, B. V. , Nadarajah, C. J. , Izumo, M. , Cedeño, M. R. , Xiong, D. D. , Dimitry, J. , … Musiek, E. S. (2018). Cell‐autonomous regulation of astrocyte activation by the circadian clock protein BMAL1. Cell Reports, 25(1), 1–9.e5. 10.1016/j.celrep.2018.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Donehower, L. A. , Herron, A. J. , Moore, D. D. , & Fu, L. (2006). Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One, 5, 6, e10995 10.1371/journal.pone.0010995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , & Shi, H. (2015). Regulation of estrogen receptor α expression in the hypothalamus by sex steroids: Implication in the regulation of energy homeostasis. International Journal of Endocrinology, 2015, 949085 10.1155/2015/949085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva, B. , Ramsey, K. , Affinati, A. , & Bass, J. (2009). Clock genes and metabolic disease. Journal of Applied Physiology, 107(5), 1638–1646. 10.1152/japplphysiol.00698.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva, B. , Ramsey, K. M. , Buhr, E. D. , Kobayashi, Y. , Su, H. , Ko, C. H. , … Bass, J. (2010). Disruption of the CLOCK components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature, 466(7306), 627–631. 10.1038/nature09253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri, S. , Patel, V. R. , Eckel‐Mahan, K. L. , Peleg, S. , Forne, I. , Ladurner, A. G. , … Sassone‐Corsi, P. (2013). Circadian acetylome reveals regulation of mitochondrial metabolic pathways. Proceedings of the National Academy of Sciences of the United States of America, 110(9), 3339–3344. 10.1073/pnas.1217632110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDearmon, E. L. , Patel, K. N. , Ko, C. H. , Walisser, J. A. , Schook, A. C. , Chong, J. L. , … Takahashi, J. S. (2006). Dissecting the functions of the mammalian clock protein BMAL1 by tissue‐specific rescue in mice. Science, 314(5803), 1304–1308. 10.1126/science.1132430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay, D. E. G. , Briançon, N. , Kokoeva, M. V. , Maratos‐Flier, E. , & Flier, J. S. (2012). Remodeling of the arcuate nucleus energy‐balance circuit is inhibited in obese mice. Journal of Clinical Investigation, 122(1), 142–152. 10.1172/JCI43134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, R. Y. , & Eichler, V. B. (1972). Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Research, 42(1), 201–206. 10.1016/0006-8993(72)90054-6 [DOI] [PubMed] [Google Scholar]
- Mori, T. , Tanaka, K. , Buffo, A. , Wurst, W. , Kühn, R. , & Götz, M. (2006). Inducible gene deletion in astroglia and radial glia‐a valuable tool for functional lineage analysis. Glia, 54(1), 21–34. 10.1002/glia.20350 [DOI] [PubMed] [Google Scholar]
- Musatov, S. , Chen, W. , Pfaff, D. W. , Mobbs, C. V. , Yang, X. J. , Clegg, D. J. , … Ogawa, S. (2007). Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America, 104(7), 2501–2506. 10.1073/pnas.0610787104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek, E. S. , Lim, M. M. , Yang, G. , Bauer, A. Q. , Qi, L. , Lee, Y. , … Fitzgerald, G. A. (2013). Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. Journal of Clinical Investigation, 123(12), 5389–5400. 10.1172/JCI70317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T. J. , Nakamura, W. , Yamazaki, S. , Kudo, T. , Cutler, T. , Colwell, C. S. , & Block, G. D. (2011). Age‐related decline in circadian output. Journal of Neuroscience, 31(28), 10201–10205. 10.1523/JNEUROSCI.0451-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato, R. , Kawabe, K. , Yamada, D. , Ikeno, S. , Mieda, M. , Shimba, S. , … Takarada, T. (2017). Disruption of Bmal1 impairs blood–brain barrier integrity via pericyte dysfunction. Journal of Neuroscience, 37(42), 10052–10062. 10.1523/JNEUROSCI.3639-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer, N. , & Kulkarni, R. N. (2006). Molecular approaches to study control of glucose homeostasis. ILAR Journal, 47(3), 199–211. 10.1093/ilar.47.3.199 [DOI] [PubMed] [Google Scholar]
- Orozco‐Solis, R. , Aguilar‐Arnal, L. , Murakami, M. , Peruquetti, R. , Ramadori, G. , Coppari, R. , & Sassone‐Corsi, P. (2016). The circadian clock in the ventromedial hypothalamus controls cyclic energy expenditure. Cell Metabolism, 23(3), 467–478. 10.1016/j.cmet.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, A. L. , & Ousman, S. S. (2018). Astrocytes and aging. Frontiers in Aging Neuroscience, 10, 337 10.3389/fnagi.2018.00337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, W. , Hsuchou, H. , He, Y. , Sakharkar, A. , Cain, C. , Yu, C. , & Kastin, A. J. (2008). Astrocyte leptin receptor (ObR) and leptin transport in adult‐onset obese mice. Endocrinology, 149(6), 2798–2806. 10.1210/en.2007-1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes, K. R. (2002). Shift work and age as interactive predictors of body mass index among offshore workers. Scandinavian Journal of Work, Environment & Health, 28(1), 64–71. 10.5271/sjweh.648 [DOI] [PubMed] [Google Scholar]
- Pezük, P. , Mohawk, J. A. , Wang, L. A. , & Menaker, M. (2012). Glucocorticoids as entraining signals for peripheral circadian oscillators. Endocrinology, 153(10), 4775–4783. 10.1210/en.2012-1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploj, K. , Albery‐Larsdotter, S. , Arlbrandt, S. , Kjaer, M. B. , Skantze, P. M. , & Storlien, L. H. (2010). The metabotropic glutamate mGluR5 receptor agonist CHPG stimulates food intake. NeuroReport, 21(10), 704–708. 10.1097/WNR.0b013e32833b4fe7 [DOI] [PubMed] [Google Scholar]
- Ralph, M. R. , Foster, R. G. , Davis, F. C. , & Menaker, M. (1990). Transplanted suprachiasmatic nucleus determines circadian period. Science, 247(4945), 975–978. 10.1126/science.2305266 [DOI] [PubMed] [Google Scholar]
- Richards, J. , & Gumz, M. L. (2013). Mechanism of the circadian clock in physiology. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 304(12), R1053–R1064. 10.1152/ajpregu.00066.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls, E. (2008). Taste, olfactory, and food texture reward processing in the brain and obesity. International Journal of Obesity, 85(4), 45–56. 10.1038/ijo.2010.155 [DOI] [PubMed] [Google Scholar]
- Rosenfeld, P. , Van Eekelen, J. A. , Levine, S. , & De Kloet, E. R. (1988). Ontogeny of the type 2 glucocorticoid receptor in discrete rat brain regions: An immunocytochemical study. Developmental Brain Research, 42(1), 119–127. [DOI] [PubMed] [Google Scholar]
- Ruby, N. F. , Fernandez, F. , Garrett, A. , Klima, J. , Zhang, P. , Sapolsky, R. , & Heller, H. C. (2013). Spatial memory and long‐term object recognition are impaired by circadian arrhythmia and restored by the GABAA antagonist pentylenetetrazole. PLoS One, 8(8), e72433 10.1371/journal.pone.0072433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudic, R. D. , McNamara, P. , Curtis, A. M. , Boston, R. C. , Pa, S. , Hogenesch, J. B. , & Fitzgerald, G. A. (2004). BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biology, 2(11), e377 10.1371/journal.pbio.0020377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab, A. S. , Neumeyer, A. , Jahn, H. M. , Cupido, A. , Šimek, A. A. , Boele, H. J. , … Kirchhoff, F. (2012). Bergmann glial AMPA receptors are required for fine motor coordination. Science, 337(6095), 749–753. 10.1126/science.1221140 [DOI] [PubMed] [Google Scholar]
- Shi, S. Q. , Ansari, T. S. , McGuinness, O. P. , Wasserman, D. H. , & Johnson, C. H. (2006). Circadian disruption leads to insulin resistance and obesity. Current Biology, 23(5), 372–381. 10.1016/j.cub.2013.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley, B. G. , Ha, L. H. , Spears, L. C. , & Dee, M. G. (1993). Lateral hypothalamic injections of glutamate, kainic acid, D,L‐alpha‐amino‐3‐hydroxy‐5‐methyl‐isoxazole propionic acid or N‐methyl‐D‐aspartic acid rapidly elicit intense transient eating in rats. Brain Research, 613(1), 88–95. 10.1016/0006-8993(93)90458-y [DOI] [PubMed] [Google Scholar]
- Stephan, F. K. , & Zucker, I. (1972). Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proceedings of the National Academy of Sciences of the United States of America, 69(6), 1583–1586. 10.1073/pnas.69.6.1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker‐Krongrad, A. , Beck, B. , Nicolas, J. P. , & Burlet, C. (1992). Central effects of monosodium glutamate on feeding behavior in adult long‐Evans rats. Pharmacology Biochemistry and Behavior, 43(3), 881–886. 10.1016/0091-3057(92)90421-b [DOI] [PubMed] [Google Scholar]
- Sul, J. Y. , Orosz, G. , Givens, R. S. , & Haydon, P. G. (2004). Astrocytic connectivity in the hippocampus. Neuron Glia Biology, 1(1), 3–11. 10.1017/s1740925x04000031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler, J. P. , Yi, C. X. , Schur, E. A. , Guyenet, S. J. , Hwang, B. H. , Dietrich, M. O. , … Schwartz, M. W. (2012). Obesity is associated with hypothalamic injury in rodents and humans. Journal of Clinical Investigation, 122(1), 153–162. 10.1172/JCI59660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, G. F. , Takano, T. , Lin, J. H. , Wang, X. , Bekar, L. , & Nedergaard, M. (2006). Imaging of cortical astrocytes using 2‐photon laser scanning microscopy in the intact mouse brain. Advanced Drug Delivery Reviews, 58(7), 773–787. 10.1016/j.addr.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Tso, C. F. , Simon, T. , Greenlaw, A. C. , Puri, T. , Mieda, M. , & Herzog, E. D. (2017). Astrocytes regulate daily rhythms in the suprachiasmatic nucleus and behavior. Current Biology, 27(7), 1055–1061. 10.1016/j.cub.2017.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek, F. W. , Joshu, C. , Kohsaka, A. , Lin, E. , Ivanova, G. , McDearmon, E. , … Bass, J. (2005). Obesity and metabolic syndrome in circadian clock mutant mice. Science, 308(5724), 1043–1045. 10.1126/science.1108750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood, J. G. , Boutz, P. L. , Dougherty, J. D. , Stoilov, P. , & Black, D. L. (2005). Homologues of the Caenorhabditis elegans fox‐1 protein are neuronal splicing regulators in mammals. Molecular and Cellular Biology, 25(22), 10005–10016. 10.1128/MCB.25.22.10005-10016.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdearcos, M. , Xu, A. W. , & Koliwad, S. K. (2015). Hypothalamic inflammation in the control of metabolic function. Annual Review of Physiology, 77, 131–160. 10.1146/annurev-physiol-021014-071656 [DOI] [PubMed] [Google Scholar]
- van Amelsvoort, L. G. , Schouten, E. G. , & Kokn, F. J. (1999). Duration of shiftwork related to body mass index and waist to hip ratio. International Journal of Obesity and Related Metabolic Disorders, 23(9), 973–978. 10.1038/sj.ijo.0801028 [DOI] [PubMed] [Google Scholar]
- van der Horst, G. T. , Muijtjens, M. , Kobayashi, K. , Takano, R. , Kanno, S. , Takao, M. , … Yasui, A. (1999). Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature, 398(6728), 627–630. 10.1038/19323 [DOI] [PubMed] [Google Scholar]
- van Vliet‐Ostaptchouk, J. V. , Hofker, M. H. , van der Schouw, Y. T. , Wijmenga, C. , & Onl‐Moret, N. C. (2009). Genetic variation in the hypothalamic pathways and its role on obesity. Obesity Reviews, 10(6), 593–609. 10.1111/j.1467-789X.2009.00597.x [DOI] [PubMed] [Google Scholar]
- Wang, T. A. , Yu, Y. V. , Govindaiah, G. , Ye, X. , Artinian, L. , Coleman, T. P. , … Gillette, M. U. (2012). Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science, 337(6096), 839–842. 10.1126/science.1222826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, G. , Chen, L. , Grant, G. R. , Paschos, G. , Song, W. L. , Musiek, E. S. , … FitzGerald, G. A. (2016). Timing of expression of the core clock gene Bmal1 influences its effects on aging and survival. Science Translational Medicine, 8(324), 324ra16 10.1126/scitranslmed.aad3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, S. H. , Yamazaki, S. , Lowrey, P. L. , Shimomura, K. , Ko, C. H. , Buhr, E. D. , … Takahashi, J. S. (2004). PERIOD2::LUCIFERASE real‐time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proceedings of the National Academy of Sciences of the United States of America, 111(15), 5339–5346. 10.1073/pnas.0308709101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Reichel, J. M. , Han, C. , Zuniga‐Hertz, J. P. , & Cai, D. (2017). Astrocytic process plasticity and IKKβ/NF‐κB in central control of blood glucose, blood pressure, and body weight. Cell Metabolism, 25(5), 1091–1102.e4. 10.1016/j.cmet.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, B. , Albrecht, U. , Kaasik, K. , Sage, M. , Lu, W. , Vaishnav, S. , … Lee, C. C. (2001). Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell, 105(5), 683–694. 10.1016/s0092-8674(01)00380-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary material
Data Availability Statement
All raw/original relevant data are available upon request.