

Abstract
Background
Colon cancer (CC) is a heterogeneous disease. Novel prognostic factors beyond pathological staging are required to accurately identify patients at higher risk of relapse. Integrating these new biological factors, such as plasma circulating tumour DNA (ctDNA), CDX2 staining, inflammation-associated cytokines and transcriptomic consensus molecular subtypes (CMS) classification, into a multimodal approach may improve our accuracy in determining risk of recurrence.
Methods
One hundred and fifty patients consecutively diagnosed with localised CC were prospectively enrolled in our study. ctDNA was tracked to detect minimal residual disease by droplet digital PCR. CDX2 expression was analysed by immunostaining. Plasma levels of cytokines potentially involved in disease progression were measured using ELISAs. A 96 custom gene panel for nCounter assay was used to classify CC into colorectal cancer assigner and CMS.
Results
Most patients were classified into CMS4 (37%) and CMS2 (28%), followed by CMS1 (20%) and CMS3 (15%) groups. CDX2-negative tumours were enriched in CMS1 and CMS4 subtypes. In univariable analysis, prognosis was influenced by primary tumour location, stage, vascular and perineural invasion together with high interleukin-6 plasma levels at baseline, tumours belonging to CMS 1 vs CMS2 +CMS3, ctDNA presence in plasma and CDX2 loss. However, only positive ctDNA in plasma samples (HR 13.64; p=0.002) and lack of CDX2 expression (HR 23.12; p=0.001) were found to be independent prognostic factors for disease-free survival in the multivariable model.
Conclusions
ctDNA detection after surgery and lack of CDX2 expression identified patients at very high risk of recurrence in localised CC.
Keywords: consensus molecular subtypes, CDX2 homeoprotein, colon cancer, interleukin-6, plasma circulating-tumor DNA
Key questions.
What is already known about this subject?
The TNM staging system continues to be the most important prognostic factor in patients with localised colon cancer.
However, up to 50% of them will relapse in spite of an optimal primary treatment.
Proof-of-concept studies have already demonstrated that the detection of circulating tumour DNA (ctDNA) in plasma provides evidence of minimal residual disease and identify patients at higher risk of relapse.
What does this study add?
ctDNA after surgery and the lack of CDX2 expression identify patients at very high risk of recurrence in localised colon cancer.
Consensus molecular subtype (CMS) 1 is associated with poorer disease-free survival.
Neither interleukin-6 levels nor CMSs are independent prognostic factors.
CDX2 loss and postoperative plasma ctDNA are stronger prognostic factors than other conventional clinicopathological features.
How might this impact on clinical practice?
Both ctDNA and CDX2 may be useful for identifying patients at a higher risk of relapse and for a better selection of patients in future adjuvant clinical trials.
Introduction
Colon cancer (CC) is a leading cause of cancer mortality, representing a group of molecularly heterogeneous diseases.1 Several factors seem to influence survival in patients with localised CC, among which recurrence is possibly the most important. Despite optimal surgery‚ the likelihood of recurrence is high. Accurate prognostic and predictive biomarkers for patients undergoing curative surgery are still needed.2 Identifying novel factors, together with better treatment strategies, may help in better selecting patients with higher risk of relapse and avoiding overtreatment for those harbouring lower risk. The last few years have seen many endeavours to molecularly classify CC with the aim of personalising treatment. A consensus molecular subtypes (CMS) combining the major published classifications and the TCGA database has been developed to improve transcriptomic characterisation of patients with CC3; however, this classification is not currently used to predict treatment in the clinical setting.
The CDX2 homeoprotein is an intestine-specific transcription factor regulating homeostasis and also involved in CC oncogenesis. CDX2 proteins interact with SMAD3, a major element of the TGFβ signalling pathway.4 Dalerba et al showed that lack of CDX2 expression is characteristic in a small subgroup of CC and indicates an aggressive phenotype associated with worse disease-free survival (DFS) in stage II–III patients with CC.5 They also demonstrated that administration of adjuvant chemotherapy resulted in improved DFS in CDX2-negative tumours. CDX2 expression, therefore, has potential as a prognostic biomarker to define high-risk localised CC and drive adjuvant treatment in this setting.
New evidence has highlighted the role of the tumour microenvironment as a key player in CC tumourigenesis and in development of metastases. For example, secretion of interleukin-11 (IL-11) and IL-6 by TGF-β-stimulated cancer-associated fibroblasts and macrophages, respectively, triggers GP130/STAT3 signalling in tumour cells.6 This crosstalk confers a survival advantage to metastatic cells. As the TGF-β stromal programme has a role in metastasis initiation, the tumour microenvironment potentially influences prognosis in localised colon cancerCC.7
Moreover, plasma circulating tumour DNA (ctDNA) has recently been shown to detect minimal residual disease (MRD) after curative treatment and can help identify a subset of patients at higher risk of recurrence.8–10
Our study aims at integrating novel biological factors into a multimodal approach to improve on our ability to pinpoint the risk of relapse. These include molecular alterations at multiple omic levels, such as plasma ctDNA, CDX2 staining, inflammation-associated cytokines and the Colorectal Cancer Assigner (CRCA) classification according to a novel validated assay,11 as well as CMS classification using a new assay method.12
Patients and methods
Study design
Study design and clinicopathological characteristics were previously reported13 also included as online supplemental table 1. In summary, a cohort of 150 localised CC patients was consecutively recruited at the Hospital Clínico Universitario de Valencia, Spain from October 2015 to October 2017. Primary tumours were collected at surgery in all cases. Plasma was taken at baseline and 6–8 weeks after surgery, and tracked every 4 months thereafter for ctDNA and inflammation-related cytokines. A consort diagram is provided in online supplemental figure 1.
esmoopen-2020-000847supp001.pdf (46.5KB, pdf)
esmoopen-2020-000847supp002.pdf (151.4KB, pdf)
Pathology
Pathology reports were reviewed for tumour site, lymph node yield, tumour differentiation, T stage and lymphovascular invasion. Mismatch repair (MMR) status was assessed by immunohistochemistry for MLH1, MSH2, MSH6 and PMS2 proteins using standard protocols. CDX2 immunostaining was carried out with a primary mouse anti-human CDX2 monoclonal antibody (clone DAK-CDX2, prediluted, Dako). CDX2 staining was graded semiquantitatively according to recommendations by a dedicated pathologist.5
CRCA and CMS
CRCA classification14 was performed using NanoCRCA assay. As previously described,12 100 ng of total RNA from FFPE tissues was used to perform the assay, using our custom 38-gene NanoCRCA assay using nCounter platform (NanoString Technologies). Data quality control was performed using nSolver Analysis Software (NanoString Technologies). CRCA subtypes were identified using our published CRCA-38 centroids and Pearson correlation. Samples with distance between first and second highest correlation coefficient ≤0.06 within a single sample were classed as undetermined. CMS subtypes3 were identified using a 33-gene (derived from the original CMS study custom nCounter platform-based biomarker assay12 and a centroid-based and gen and rank-based classifier method. Here, centroids refer to a summary statistics defining expression of each gene in each subtype.
Cytokines analysis
A commercially available ELISA (Abcam, Cambridge) kit was used to evaluate IL-6 in plasma at baseline, postsurgery and at time of relapse. All procedures were performed according to the respective manufacturer’s protocols. For each assay, samples were measured in duplicate also including in-house quality control. The interassay and intra-assay coefficients of variation were less than 10%.
ctDNA analysis
DNA was extracted from 4 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) according to the manufacturer’s instructions. Identification of pathological somatic variants to track MRD and ddPCR analysis of longitudinal plasma ctDNA samples were as previously reported.13
Statistical analysis
The cut-off for statistical significance was set as α=0.05, all tests are two sided. The primary outcome measure was DFS according to standard radiologic criteria. Comparison of IL-6 baseline ersus follow-up and follow-up versus relapse were carried out using Wilcoxon signed-rank test. Cytokine levels were categorised into high (≥3.8 pg/mL) and low (<3.8 pg/mL) according to median value. Univariable logistic regressions were performed to evaluate T stage, nodal involvement and stage effects on cytokines levels. Median DFS was estimated using the Kaplan-Meier method to evaluate the impact on survival of CMS, CDX2 and IL-6. CMS analysis was performed as previously described. Pairwise comparisons were performed within each CMS group using the Bonferroni p value correction. Kaplan-Meier curves and pairwise comparisons were performed with R package ‘survminer’.15 Univariable and multivariable analyses were performed using Cox regression with R package ‘survival’.16 All statistical analyses were carried out with R V.3.6.1.17
Results
CRCA and CMS subgroups to predict DFS
CC samples were classified into CRCA subtypes14 using our NanoCRCA assay based on a custom CRCA-38 gene using 117 formalin-fixed paraffin-embedded (FFPE) samples.12 Eighty-eight per cent of the samples were successfully classified into the five CRCA subtypes, with only 12% undetermined samples found, possibly owing to poor quality or a mixture of multiple subtypes. Among the five subtypes, the enterocyte and goblet-like subtypes were most prevalent with 24% and 22%, respectively, followed by transitamplifying in 16% and inflammatory and stem-like with 13% each (figure 1A).
Figure 1.

Distribution of CC samples according to CRCA and CMS subtyping (n=117). Proportion of each CRCA subtype (A) and CMS group (B–C) in our population. CC, colon cancer; CMS, consensus molecular subtype; CRCA, colorectal cancer assigner; TA, transit amplifying.
Furthermore, these 117 CC samples were classified into CMS subtypes3 using a new 38-gene CMS assay and a rank-based classifier based on NanoString Technology. The proportion of different subtypes include 22% CMS4 (generally associated with stem-like), 20% CMS2 (enterocyte and transit amplifying [TA]), 12% CMS3 (goblet-like) and 14% CMS1 (inflammatory). However, a substantial 32% of samples represented undetermined subtypes, which were similar to the ‘mixed’ subtype samples reported in the original publication.3 We further stratified the samples into all of the CMS subtypes using the nearest classifier: 37.0% were CMS4, 28% of CMS2, 20% of CMS1 and 15% of CMS3 (figure 1B, C).
The ‘nearestCMS’ and ‘predictedCMS’ classifications were compared with evaluate if DFS varied between these two methodologies. No statistical difference was found using partial likelihood ratio test for non-nested model (p=0.533). The ‘nearest CMS’ was selected to be included in our multivariable model.
After a median follow-up of 24.7 months, DFS was significantly different according to CMS subtyping (log-rank p=0.0068.) (figure 2). Pairwise comparison showed that CMS1 had significant lower DFS than CMS2 +3 (log-rank p=0.0068). No significant differences were found between CMS4 and CMS2 +3 (log-rank p=0.6492) or between CMS1 and CMS4 (log-rank p=0.2194).
Figure 2.
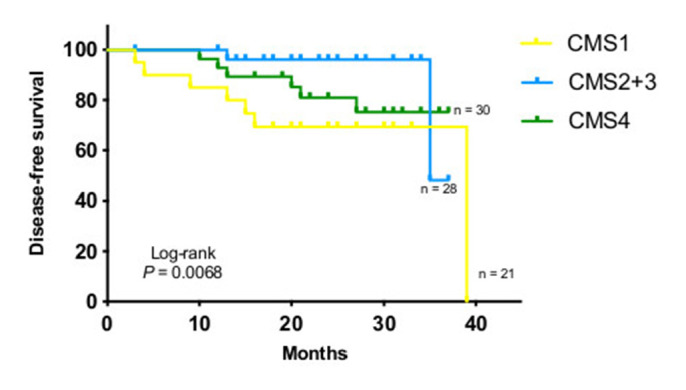
Disease-free survival (DFS) according to consensus molecular subtypes (CMS). Kaplan-Meier estimate of DFS by CMS subtypes. P value determined by log-rank test.
CDX2 expression
Loss of CDX2 expression was observed in only 5.3% of our patients (8/150), in line with other series.5 All patients with CDX2 loss had stage IIIB or IIIC disease at diagnosis. Six of eight CDX2 negative tumours were located in the right-side of the colon and three of them were MMR-deficient. CDX2 negative samples were enriched in RAS/RAF mutated tumours (6/8) highlighting the importance of the MAPK pathway as potential mediator of lack of CDX2 expression, vascular and perineural invasion (6/8) and CMS1 (5/6) y CMS4 subgroups (1/6). Seven patients received adjuvant chemotherapy and six experienced relapses. Lack of CDX2 expression was associated with poorer DFS (log-rank p<0.0001, figure 3).
Figure 3.
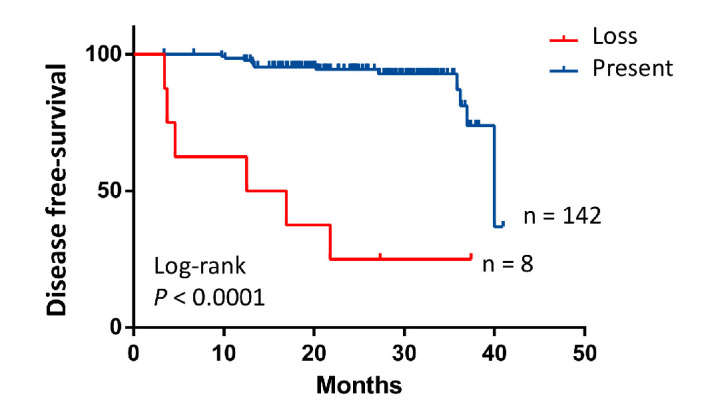
Disease-free survival (DFS) according to CDX2 expression. Kaplan-Meier estimate of DFS by CDX2 expression. P value determined by log-rank test.
Plasma inflammation-associated cytokines levels
We focused on IL-6 as a cytokine potentially involved in CC development and progression. IL-6 concentrations were detectable in all plasma samples with some interindividual variability (0–73 pg/mL). At baseline, IL-6 plasma concentrations were related to pathological stage, tumour bowel wall invasion and nodal involvement, being higher in T3 or T4 (T1 vs T3 p=0.002; T1 vs T4 p=0.009 online supplemental figure 2A). IL-6 was also more elevated in stage II/III relative to stage I (stage I vs II p=0.008; stage I vs III p=<0.0001) or n+tumours (online supplemental figure 2B and C). Baseline IL-6 concentrations were higher than follow-up levels in relapsing patients. IL-6 plasma levels were found to be more elevated than those without a relapse (baseline level vs follow-up p=0.0093; follow-up vs relapse level p=0.047 (online supplemental figure 2D), suggesting a relation with tumour recurrence. IL-6 levels were compared within CMS classification by means of Tukey’s post hoc test. Levels of IL-6 were found significantly higher in CMS1 versus CMS2 + CMS3 (p=0.036), but no difference was found between CMS2 + CMS3 and CMS4 (p=0.201) nor CMS1 vs CMS4 (p=0.607). In our cohort, higher levels of IL-6 at baseline were related to poorer DFS (p=0.018, figure 4).
Figure 4.
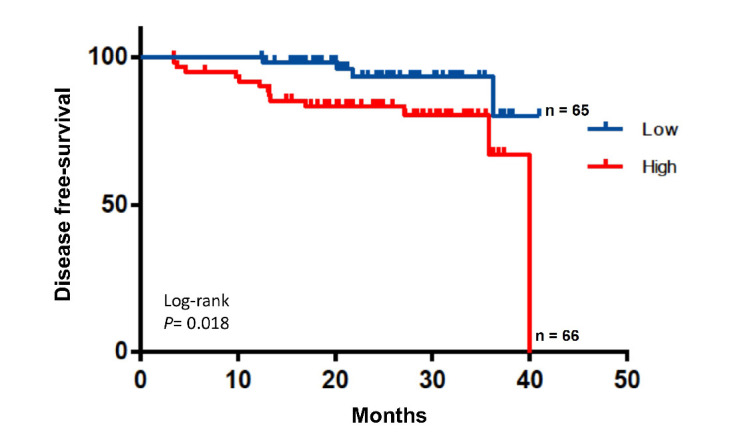
Disease-free survival (DFS) according to plasma interleukin-6 (IL-6) baseline levels. Kaplan-Meier estimate of DFS by IL-6 concentration. P value determined by log-rank test.
esmoopen-2020-000847supp003.pdf (496.7KB, pdf)
Integrating clinicopathological features and novel biological factors into a univariable and multivariable model
We integrated a series of conventional clinicopathological features, together with some novel biological factors studied in this cohort, to assess their prognostic impact on CC recurrence. Table 1 shows a Cox regression model to evaluate DFS analysed according to univariable and multivariable analyses. Right-sided tumours, advanced T stage, lymph node involvement and vascular and perineural invasion were significantly associated with poor prognosis in the univariable analysis. Among the biological factors tested, postoperative ctDNA, higher IL-6 plasma levels, CMS1 subtype and loss of CDX2 were also significantly related to a more unfavourable prognosis. In the multivariable analysis, none of the conventional clinicopathological features retained their independent status as a prognostic factor. IL-6 plasma levels and CMS subtyping also lost their independent contribution to the model. Only presence of postoperative plasma ctDNA and loss of CDX2 expression in the primary tumour remained independently related to DFS, after adjusting for tumour site, stage, nodal involvement, lymphovascular invasion, IL-6 levels and CMS groups. Most CDX2 negative patients belonged to CMS1 and CMS4 subtypes. We, thus, examined the prognostic impact of CDX2 separately in CMS1 and CMS4 subtypes and found that CDX2 loss was highly prognostic for DFS in CMS1 (log rank p<0.0001) and in CMS4 subtypes (log rank p=0.02) (online supplemental figure 3).
Table 1.
Cox regression model to evaluate recurrence-free survival based on patients’ characteristics including ctDNA status
| Variable | Univariable analysis (n=149, events=18) |
Multivariable analysis (n=61, events=15) |
||||
| HR | 95% CI | P value | HR | 95% CI | P value | |
| Age,<70 vs ≥70 | 2.08 | 0.78 to 5.55 | 0.143 | 1.11 | 0.23 to 5.1 | 0.895 |
| Sex, male versus female | 0.67 | 0.25 to 1.80 | 0.430 | 1.20 | 0.29 to 4.96 | 0.801 |
| Tumour site right versus left | 0.30 | 0.11 to 0.85 | 0.023 | 0.42 | 0.10 to 1.84 | 0.250 |
| Tumour differentiation poor versus well moderated | 0.54 | 0.12 to 2.25 | 0.376 | |||
| T stage (T1-T2-T3 vs T4) | 3.36 | 1.27 to 8.88 | 0.015 | |||
| Lymph-node yield (<12 vs≥12) | 2.84 | 0.37 to 21.45 | 0.313 | |||
| Nodal involvement (N0 vs N1 +N2) | 4.65 | 1.50 to 14.45 | 0.008 | |||
| Stage (II vs III) | 3.24 | 1.04 to 10.09 | 0.043 | 1.62 | 0.30 to 8.80 | 0.578 |
| Vascular invasion (yes vs no) | 0.14 | 0.05 to 0.38 | <0.001 | |||
| Perineural invasion (yes vs no) | 0.38 | 0.15 to 0.98 | 0.045 | |||
| MMR status (proficient vs deficient) | 2.80 | 0.78 to 10.08 | 0.115 | 1.43 | 0.18 to 11.44 | 0.735 |
| Stage II risk (low vs high) | 2.92 | 0.26 to 32.27 | 0.381 | |||
| Adjuvant chemotherapy (yes vs no) | 0.41 | 0.15 to 1.12 | 0.081 | |||
| Postoperative CEA (normal vs elevated) | 0.53 | 0.12 to 2.40 | 0.413 | |||
| Postoperative ctDNA status (negative vs positive) | 6.96 | 2.57 to 18.91 | <0.001 | 13.64 | 2.64 to 70.49 | 0.002 |
| IL-6 (≤3.45 vs>3.45) | 3.55 | 1.16 to 10.90 | 0.027 | |||
| CMS (CMS1 vs CMS2 +CMS3) | 0.12 | 0.03 to 0.59 | 0.009 | 2.19 | 0.19 to 25.31 | 0.529 |
| CMS (CMS1 vs CMS4) | 0.36 | 0.12 to 1.09 | 0.071 | 3.16 | 0.40 to 25.36 | 0.279 |
| CDX2 (present vs loss) | 12.68 | 4.63 to 34.69 | <0.001 | 23.12 | 3.59 to 149.05 | 0.001 |
Analysed according to a univariable and multivariable analyses.
CMS, consensus molecular subtype; ctDNA, circulating tumour DNA; IL-6, interleukin-6; MMR, mismatch repair.
esmoopen-2020-000847supp004.pdf (81KB, pdf)
Discussion
CC is a molecularly heterogeneous disease. At the most basic level, prognosis is estimated according to pathological staging; however, this often fails to accurately predict recurrence. A better risk stratification of patients with CC is needed to refine our selection of patients who may need adjuvant chemotherapy. We explored the value of adding novel biological variables to the conventional ones in estimating prognosis. In our previous study, we demonstrated that detection of plasma ctDNA after curative-intent surgery was related to MRD and therefore with higher risk of recurrence.13 In the current article, we underline the fact that postoperative ctDNA detection and lack of CDX2 expression are independent prognostic indicators for DFS, unlike stage, IL-6 levels in plasma or CMS subtypes.
On CMS classification, 14%, 20%, 12% and 22% of patients in our cohort were classified, respectively, into CMS1, CMS2, CMS3 and CMS4, in line with previous reports.11 However, subtyping was undetermined in 32% of our samples using the ‘predicted CMS’ classification, probably due to limited quality of samples or intratumoural heterogeneity.18 Moreover, we have previously reported intratumoural heterogeneity associated with transcriptional subtype of CRCA classification representing the presence of more than one subtype within a sample.19 Similarly, Marisa et al reported up to 5% of tumours harbouring three distinct subtypes within a sample.20
We identified CDX2 loss as a relevant biomarker for DFS despite its low prevalence (5.4%), in line with previous studies.5 We then demonstrated that CDX2-negative tumours were associated with shorter DFS. CDX2 expression loss was particularly prevalent in two CMS subtypes: CMS1 and CMS4. In contrast to reported data,21 loss of CDX2 expression was associated with poorer DFS in both CMS1 and CMS4 groups in our cohort. These discordant results could be justified by the use of different platforms, gene sets, assays and algorithms to analyse mRNA expression profile by each group.
In the era of precision medicine and immunotherapy, as commented above, microenvironment is also of great interest, to which end we integrate molecular and immune features of CC. We showed that higher IL-6 levels in patients at diagnosis was correlated with worse prognosis in the univariable analysis. The highest IL-6 levels were found in the CMS1 and CMS 4 subtypes (p=0.036). This finding may be due to the fact that CMS1 CCs are more closely associated with microsatellite instable phenotype and are therefore more immunogenic.22 Moreover, the CMS4 subtype is also enriched for certain immune cell types along with mesenchymal and stromal gene signatures, which may be derived from surrounding cells rather than being cancer specific.23 Higher plasma IL-6 levels and absent CDX2 expression in CMS1 and CMS4 subtypes might contribute to more aggressive phenotypes. However, neither IL-6 levels nor CMS subtypes reached independent significance in the multivariable analysis for DFS. Nevertheless, recent data support that tumour microenvironment markers could better reflect the risk of recurrence in localised CC than transcriptomic markers.7 Subtyping based on genes expressed by stromal cells other than epithelial tumour cells may provide a more accurate molecular characterisation of CC.24
Intratumoural heterogeneity leads to underestimation of the molecular landscape from single tumour-biopsy samples,25 leading to inaccuracy in molecular portrayal. On the other hand, ctDNA is released into the bloodstream by tumour cells and may capture a more faithful representation of tumour heterogeneity. Moreover, it is able to detect MRD after curative-intent surgery, overcoming the drawbacks of tumour tissue analysis.8–10 As we previously reported, detection of ctDNA after surgery implies MRD and patients at risk of relapse are identified earlier. Likewise, serial ctDNA levels during the follow-up precede clinical or radiological recurrence.13 Despite this, given the exploratory design and the small sample size of our study, these results will need to be further validated.
In conclusion, postoperative ctDNA status and CDX2 expression identified high-risk patients with localised CC with poor prognosis. CDX2 loss and high levels of IL-6 were enriched in CMS1 and CMS4 subgroups, suggesting involvement in similar pathways. However, CDX2 was reinforced as an independent prognostic factor for DFS in multivariable analysis. Taken together with postoperative plasma ctDNA, they could represent the pivotal stratification factor for future adjuvant trials.
Acknowledgments
We gratefully acknowledge the support of the INCLIVA BioBank (PT17/0015/0049; B.0000768 ISCIII) as part of the Valencian Biobanking Network and the Spanish National Biobanks Network, and especially wish to thank all patients and families for their participation in this study.
Footnotes
Twitter: @pirega83
DR and AC contributed equally.
Contributors: DR and AC contributed equally to this manuscript as corresponding authors.
Funding: This study was supported by grants from the Instituto de Salud Carlos III (PI15/02180 and PI18/01909 to AC; PI18/01508 to TF). VG was supported by the ESMO 2014 fellowship program, and by Rio Hortega contract CM18/00241 from the Carlos III Health Institute; TF is supported by Joan Rodes contract 17/00026 from the Carlos III Health Institute. NT was supported by Rio Hortega contract CM15/00246 from the Instituto de Salud Carlos III and by the ESMO 2013 fellowship program; DR was supported by Joan Rodes contract 16/00040 from the Instituto de Salud Carlos III. SZ has a CA18/00042 contract for bioinformaticians from the Instituto de Salud Carlos III. Part of the equipment used in this study has been funded by Generalitat Valenciana and cofinanced with ERDF funds (OP ERDF of Comunitat Valenciana 2014-2020).
Competing interests: AC declares the following disclosures: Consultant or Advisory Role: Merck Serono, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda, Astelas. Research Funding: Genentech, Merck Serono, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda, Astelas, Pierre Fabre, Fibrogen, Amcure, Sierra Oncology, Astra Zeneca, Medimmune, BMS, MSD. Speaking: Merck Serono, Roche, Angem, Bayer, Servier, Foundation Medicine. Grant support: Merck Serono, Roche. Others: Executive Board member of ESMO, Chair of Education ESMO, ESMO Education Director, INCLIVA General and Scientific Director, Associate Editor: Annals of Oncology and ESMO Open, Editor in chief: Cancer Treatment Reviews. AC declares institutional research funding from Genentech, Merck Serono, BMS, MSD, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda, Astellas and Fibrogen and advisory board or speaker fees from Merck Serono, Roche, Servier, Takeda and Astellas in the last five years. All remaining authors have declared no conflict of interest. AS has ownership interest as an inventor for a patent entitled ‘Colorectal cancer classification with differential prognosis and personalised therapeutic responses’ (patent number PCT/IB2013/060416). AS-Research Funding-Bristol-Myers Squibb, Merck KGaA and Pierre Fabre.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as online supplemental information.
References
- 1.Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA A Cancer J Clin 2020;70:145–64. 10.3322/caac.21601 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177–93. 10.3322/caac.21395 [DOI] [PubMed] [Google Scholar]
- 3.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calon A, Gross I, Lhermitte B, et al. Different effects of the Cdx1 and Cdx2 homeobox genes in a murine model of intestinal inflammation. Gut 2007;56:1688–95. 10.1136/gut.2007.125542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalerba P, Sahoo D, Paik S, et al. Cdx2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374:211–22. 10.1056/NEJMoa1506597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calon A, Espinet E, Palomo-Ponce S, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012;22:571–84. 10.1016/j.ccr.2012.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dienstmann R, Villacampa G, Sveen A, et al. Relative contribution of clinicopathological variables, genomic markers, transcriptomic subtyping and microenvironment features for outcome prediction in stage II/III colorectal cancer. Ann Oncol 2019;30:1622–9. 10.1093/annonc/mdz287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 2016;8:346ra92. 10.1126/scitranslmed.aaf6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Li L, Cohen JD, et al. Prognostic potential of circulating tumor DNA measurement in postoperative surveillance of nonmetastatic colorectal cancer. JAMA Oncol 2019;5:1118–23. 10.1001/jamaoncol.2019.0512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reinert T, Henriksen TV, Christensen E, et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol 2019;5:1124–31. 10.1001/jamaoncol.2019.0528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ragulan C, Eason K, Fontana E, et al. Analytical validation of multiplex biomarker assay to stratify colorectal cancer into molecular subtypes. Sci Rep 2019;9:7665. 10.1038/s41598-019-43492-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontana E, Ragulan C, Eason K, et al. Validated nCounter platform to stratify colorectal cancer (CRC) into consensus molecular subtypes (CMS) and CRCassigner subtypes in Asian population. Annals of Oncology 2017;28:mdx659.003:x43 10.1093/annonc/mdx659.003 [DOI] [Google Scholar]
- 13.Tarazona N, Gimeno-Valiente F, Gambardella V, et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann Oncol 2019;30:1804–12. 10.1093/annonc/mdz390 [DOI] [PubMed] [Google Scholar]
- 14.Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 2013;19:619–25. 10.1038/nm.3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassambara A, Kosinski M, Biecek P. survminer: Drawing Survival Curves using 'ggplot2'. R package version 0.4.5, 2019. Available: https://CRAN.R-project.org/package=survminer
- 16.Therneau T. A package for survival analysis in S_. version 2.38. Available: https://CRAN.R-project.org/package=survival.2015
- 17.R Core Team R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2018URL https://www.R-project.org/ [Google Scholar]
- 18.Fontana E, Eason K, Cervantes A, et al. Context matters-consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann Oncol 2019;30:520–7. 10.1093/annonc/mdz052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fontana E, Nyamundanda G, Cunningham D, et al. Molecular subtype assay to reveal anti-EGFR response sub-clones in colorectal cancer (CRC). JCO 2018;36:658 10.1200/JCO.2018.36.4_suppl.658 [DOI] [Google Scholar]
- 20.Marisa L, Ayadi M, Balogoun R, et al. Clinical utility of colon cancer molecular subtypes: validation of two main colorectal molecular classifications on the PETACC-8 phase III trial cohort. JCO 2017;35:3509 10.1200/JCO.2017.35.15_suppl.3509 [DOI] [Google Scholar]
- 21.Pilati C, Taieb J, Balogoun R, et al. Cdx2 prognostic value in stage II/III resected colon cancer is related to CMS classification. Ann Oncol 2017;28:1032–5. 10.1093/annonc/mdx066 [DOI] [PubMed] [Google Scholar]
- 22.Putoczki TL, Thiem S, Loving A, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013;24:257–71. 10.1016/j.ccr.2013.06.017 [DOI] [PubMed] [Google Scholar]
- 23.Dienstmann R, Vermeulen L, Guinney J, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer 2017;17:79–92. 10.1038/nrc.2016.126 [DOI] [PubMed] [Google Scholar]
- 24.Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet 2015;47:320–9. 10.1038/ng.3225 [DOI] [PubMed] [Google Scholar]
- 25.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
esmoopen-2020-000847supp001.pdf (46.5KB, pdf)
esmoopen-2020-000847supp002.pdf (151.4KB, pdf)
esmoopen-2020-000847supp003.pdf (496.7KB, pdf)
esmoopen-2020-000847supp004.pdf (81KB, pdf)