


Abstract
Class-II AP-endonuclease (XthA) and NAD+-dependent DNA ligase (LigA) are involved in initial and terminal stages of bacterial DNA base excision repair (BER), respectively. XthA acts on abasic sites of damaged DNA to create nicks with 3′OH and 5′-deoxyribose phosphate (5′-dRP) moieties. Co-immunoprecipitation using mycobacterial cell-lysate, identified MtbLigA-MtbXthA complex formation. Pull-down experiments using purified wild-type, and domain-deleted MtbLigA mutants show that LigA-XthA interactions are mediated by the BRCT-domain of LigA. Small-Angle-X-ray scattering, 15N/1H-HSQC chemical shift perturbation experiments and mutational analysis identified the BRCT-domain region that interacts with a novel 104DGQPSWSGKP113 motif on XthA for complex-formation. Isothermal-titration calorimetry experiments show that a synthetic peptide with this sequence interacts with MtbLigA and disrupts XthA–LigA interactions. In vitro assays involving DNA substrate and product analogs show that LigA can efficiently reseal 3′OH and 5′dRP DNA termini created by XthA at abasic sites. Assays and SAXS experiments performed in the presence and absence of DNA, show that XthA inhibits LigA by specifically engaging with the latter's BRCT-domain to prevent it from encircling substrate DNA. Overall, the study suggests a coordinating function for XthA whereby it engages initially with LigA to prevent the undesirable consequences of futile cleavage and ligation cycles that might derail bacterial BER.
INTRODUCTION
Mycobacterium tuberculosis is a deadly human pathogen that can survive for years in the human host. This is partly attributed to its ability to repair DNA damage caused by exposure to the hostile oxidative environment present within the host cell (1–3). In the absence of a mismatch repair (MMR) pathway in the mycobacterium, the non-canonical MMR by EndoMS/NucS (4,5) or DNA base excision repair (BER) pathway assumes importance for countering host-inflicted oxidative DNA damaging responses (6,7). BER is a multi-step process initiated by the recognition and removal of damaged DNA bases by DNA glycosylases (8–10), to generate non-coding, abasic sites which can block DNA replication and transcription if left unprocessed (11,12). Subsequently, Class II AP-endonucleases cleave the phosphodiester backbone of DNA at the abasic sites (13,14). The repair process is then completed by the concerted action of downstream enzymes, including DNA polymerase that fills in the nucleotide(s) and DNA ligase that seals the nick (15). It has been reported in higher eukaryotes that proteins are recruited and exchanged at the damaged site following a strict hierarchy which is controlled by specific protein-protein interactions (16).
Class II AP-endonucleases are multi-functional proteins that act on duplex DNA as a 3′-5′ exonuclease, 3′-phosphodiesterase and 3′-phosphatase (17). These activities are important for maintaining numerous cellular functions (18). Generally, deletion/knock-down of AP-endonuclease family proteins leads to increased sensitivity to DNA damaging agents in both prokaryotes and eukaryotes (19–22). Further, mutations in mycobacterial AP endonuclease (XthA) compromises the survival of mycobacterium within its host (23). AP-endonucleases hydrolyses the phosphodiester bond on the 5′ end of the abasic site, generating a 3′ hydroxyl (3′OH) terminus for extension by a DNA polymerase and a 5′ terminus with a 2′deoxyribose 5′ phosphate residue (5′dRP) to be removed by an AP lyase. AP-endonucleases functionally interact with several BER components to regulate the pathway as shown in eukaryotes (24). APE1 is the counterpart of XthA, and its knockout has been shown to be lethal in mice (25). Human APE1 stimulates the activities of DNA glycosylase (26–29) and FLAP-endonucleases (FEN1) (30) for more efficient BER initiation. APE1 also modulates AP lyase and polymerase activities of DNA polymerase β (31–33). Further, AP-endonuclease interacts with proliferating cell nuclear antigen (PCNA), suggesting that it has a coordinating role in BER (30,34,35). These activities are drawing attention to its potential as an emerging target for various human diseases.
BER terminates with the action of a DNA ligase, which catalyses the formation of phosphodiester bonds between 5′-phosphoryl and 3′-OH termini within a DNA strand (36–39). NAD+-dependent DNA ligase (LigA) is essential in bacteria and has a modular architecture with a nucleotide binding domain (AdD-Ia and Ib), an oligomer binding fold (OB), a zinc finger and helix–hairpin–helix domain (Zn-HhH) involved in nicked-substrate binding and a C-terminal BRCT domain (40,41). On the other hand, the human counterpart viz. DNA ligase I has low sequence similarity with LigA, and the organization of the domains is quite different (42) (Supplementary Figure S1 A). The BRCT domain of LigA plays an important role in making direct interactions with substrate DNA thereby allowing LigA to encircle its substrate for catalysis (43–47). In line with these reports, our previous study showed that the BRCT-deleted MtbLigA exhibited reduced ligase activity compared to the full-length protein, reaffirming that the BRCT domain is important for efficient DNA binding and ligation (48). In eukaryotes, the BRCT domain, via specific protein-protein interactions, acts as a DNA damage signal transducer element that conveys signals from the damage sensors to other components of DNA repair machinery (49,50).
Other important protein interactions governing BER in eukaryotes include those between human DNA glycosylase and FLAP-endonuclease (FEN-1) (51), DNA polymerase and DNA ligase (52) and other BER components with XRCC1 (53,54) or PCNA (34,55–57). This suggests that various steps of BER are regulated by multiple protein–protein interactions whereby components of the BER machinery receive and relay DNA substrates or products sequentially to the next appropriate enzyme. This could be essential for preventing exposure of DNA intermediates to the detrimental action of nucleases, recombination events, etc. While several crucial participants of mycobacterial BER, like DNA glycosylase MtbNei (58–61), MtbXthA (62–64), DNA polymerase (65,66), the DNA sliding β-clamp (67,68) and MtbLigA (48,69,70), have been studied in isolation, the protein-interaction network in mycobacterium, and bacteria in general, is relatively unexplored.
We have earlier reported that MtbXthA–Mtbβ–clamp interactions exist and also that these are mediated by DNA (63). Here, we have identified interactions between MtbXthA and MtbLigA that are involved in the initial and terminal stages of bacterial BER respectively. The complex formation between the two proteins is mediated specifically by a newly identified motif in MtbXthA that interacts with the BRCT domain in MtbLigA. The interaction of XthA with LigA inhibits the latter's activity. We have also shown that LigA can efficiently act on the product of XthA activity to ligate it. Our results suggest that XthA engages with the C-terminal BRCT domain of LigA to prevent ‘futile’ ligation of the former's product till an AP-Lyase is recruited to remove the 5′-dRP moiety.
MATERIALS AND METHODS
Cloning, expression and purification of MtbLigA, MtbLigA variants and MtbXthA
Full-length His-tagged MtbLigA and its deletion mutants MtbLigA1, MtbLigA2, MtbLigA3 and BRCT domain alone, were cloned, expressed and purified to homogeneity as previously described by our laboratory (48,69,70). MtbLigA mutants with hexa-histidine tag, viz. MtbLigAG614I, MtbLigAG621I and MtbLigAG639V, were generated using appropriate primers (Supplementary Table S1) and full-length pET21d-MtbLigA construct as the template (48), as per earlier reported protocols (71). Mutant proteins were also expressed and purified as described above. The clone constructs were expressed in Escherichia coli BL21 (DE3) cells (Novagen) and grown in LB medium containing 0.1 mg ml−1 ampicillin to A600 0.5, followed by 0.2–0.5 mM isopropyl β‐d‐thiogalactopyranoside (IPTG) induction for 16 h at 20°C. The cells were harvested by centrifugation at 6000 rpm and pellets were resuspended in Buffer A (50 mM Tris–HCl pH 8.0, 200 mM NaCl and 2 mM imidazole, 1 mM PMSF and 0.2 mg ml−1 lysozyme, DNase I) and sonicated. The homogenate was centrifuged at 12 000 rpm for 30 min and the clarified cell lysates were applied to pre-equilibrated nickel NTA column (Amersham Biosciences). The proteins were eluted using 300 mM imidazole in Buffer A. The eluted fractions containing the protein of interest were pooled and concentrated up to 4–6 mg/ml and subjected to pre-equilibrated (20 mM Tris–HCl pH 8.0, 100 mM NaCl) 1 ml MonoQ 5/5 columns on AKTA prime (GE Biosciences). Following four column volumes washes with equilibration buffer, a NaCl gradient in 20 mM Tris–HCl pH 8.0 was used to elute the proteins. The resultant fractions were pooled, concentrated and applied over a Superdex 200 size exclusion chromatography (SEC) column (GE Biosciences) in a buffer containing 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5% (v/v) glycerol. Fractions were pooled, concentrated and analysed on 12% SDS-PAGE.
6XHis-tagged and GST-tagged MtbXthA and its catalytic dead mutant E57A/D251A have been cloned, expressed and characterized in our lab as reported earlier (62,63). A GST-tagged triple-mutant, MtbXthAID (S110A, K111L, G112A), was generated using specific primers (Supplementary Table S1) and pGEX-MtbXthA construct as the template (63) using standard protocols (71). All expression constructs were sequenced to verify their integrity. Overexpression and purification of recombinant MtbXthA/variant proteins was carried out as explained above with minor modifications in cell lysis buffer (40 mM Tris–HCl pH 8.0, 500 mM NaCl, 0.1% N‐lauryl sarcosine, 10 mM imidazole, 1 mM PMSF and 0.2 mg ml−1 lysozyme, DNase I).
Anti-sera generation and co-immunoprecipitation (Co-IP)
Antibodies against purified MtbLigA and MtbXthA were generated by subcutaneous immunization of BALB/c mice and female NZ rabbit, respectively (experiments approved by the Animal Ethics Committee, CSIR-Central Drug Research Institute, Lucknow, India). Protein (200 μg) in Freund's complete adjuvant (1:1 v/v) was given as primary immunization followed by two booster doses of 100 μg each in incomplete adjuvant. Ten days after the second booster, the animals were bled to obtain polyclonal antiserum. Specificities of the antisera were checked by immunoblotting of M. tuberculosis cell lysate.
In vivo interaction between MtbLigA and MtbXthA was analysed by co-IP according to previously published procedures with some modifications (63). Briefly, exponentially growing cells of M. tuberculosis (H37Rv) were harvested, resuspended and lysed in 1.5 ml of buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.5% Sodium deoxycholate and 0.01% SDS). To remove any DNA mediated mode of interaction between the proteins, the mycobacterial cell extract was pre-treated with DNase I (15 units) at 25°C for 45 min. 50 μl slurry of protein A sepharose (GE Healthcare Biosciences) was washed twice with PBS (50 mM sodium phosphate, pH 7.5, 150 mM NaCl) and incubated on a rotary shaker with 25 μl of anti MtbLigA antibody. In control experiments MtbLigA antiserum was not added to the beads. Each reaction was washed twice with PBS followed by two washes using 0.2 M tri-ethanolamine, pH 8.0. The antibody was then cross-linked to protein A with dimethyl pimelimidate (Pierce) according to the manufacturer's protocol. After equilibration of the antibody-conjugated protein A sepharose with TEN buffer (50 mM Tris–HCl pH 8.0, 5 mM EDTA, 150 mM NaCl), 1 ml aliquot of mycobacterial cell extract (60 mg/ml) was added and incubated for 4 h at 4°C on a rotary shaker. The beads were washed thrice with TEN buffer and the complexes were eluted from beads using 0.1 M glycine, pH 2.5. Samples were neutralized with Tris–HCl pH 8.0 and run on 12% SDS-PAGE for western blotting using anti-MtbLigA and anti-MtbXthA antisera. Goat anti-mice/goat anti-Rabbit HRP conjugated were used as secondary antibodies and signals were detected using an enhanced chemiluminescent substrate (Thermo Fischer) and captured by X-ray film (GE Healthcare).
In vitro GST pull down assays
5 μM purified His-tagged MtbLigA, MtbLigA mutants (MtbLigA1, MtbLigA2, MtbLigA3 or BRCT) respectively, were incubated with GST-tagged MtbXthA in 500 μl of assay buffer (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 2 mM DTT, 10% glycerol) at 4°C for 3 h. The proteins were then mixed with 50 μl of glutathione sepharose beads (GE Healthcare) with constant rotation at 4°C for 1 h. The beads were then washed thrice with assay buffer containing 0.1% Triton X-100. Subsequently beads were boiled in 50 μl of 1× Laemmli buffer and the pulled-down protein complexes were run on 12% SDS-PAGE followed by immune blotting. The blot was probed using anti-His and anti-GST antibodies (Santa Cruz Biotech), respectively.
Size exclusion chromatography of complexes
MtbXthA–MtbLigA complex was prepared by mixing equimolar ratio of MtbXthA and MtbLigA in a buffer containing 50 mM Tris pH 8.0, 200 mM NaCl and 2 mM beta-mercaptoethanol, in absence or presence of nicked DNA substrate (S1). Complexes were incubated for at least 3–4 h on ice prior to SEC analysis. For MtbLigA-MtbXthA-nicked DNA complex, equimolar ratio of both the proteins were incubated on ice for 2 h and then S1 was added followed by further incubation for 30 min on ice such that the final molar ratio becomes 1:1:1. SEC was performed using Superdex 200 increase GL 10/30 column (GE Healthcare) in the same buffer in which complexes were made. The column was calibrated using blue dextran (to determine void volume) and standards Aprotinin (6.51 kDa, 1.3 nm), Ribonuclease (13.7 kDa, 2.4 nm), Ovalbumin (44 kDa, 4.4 nm), Aldolase (158 kDa, 6.62 nm), and Ferritin (440 kDa, 7.99 nm) of known molecular weights and Stoke's radii (Table 1, Supplementary Figure S2D, E).
Table 1.
Size exclusion chromatography of proteins and complexes
| Protein/protein complex | Theoretical molecular weight (kDa) | Average molecular weight (kDa) by SEC | Stoke's radii (nm) | 260/280 ratio |
|---|---|---|---|---|
| MtbXthA | 35 | 39.81 | 2.67 | 0.51 |
| MtbLigA | 77 | 121.04 | 4.02 | 0.57 |
| BRCT domain | 11 | 22.30 | 2.14 | 0.53 |
| MtbLigA1 | 66 | 115.80 | 3.91 | 0.56 |
| MtbXthA+BRCT domain | 46 | 45.68 | 2.79 | 0.54 |
| MtbXthA+MtbLigA | 112 | 106.45 | 3.83 | 0.59 |
| MtbLigA+Nicked DNA | 101 | 112.27 | 3.19 | 0.82 |
| MtbXthA+MtbLigA+Nicked DNA | 136 | 103.20 | 3.79 | 1.03 |
The 260/280 ratios were measured by nanodrop for eluted fractions.
Small angle X-ray scattering
SAXS experiments were performed at BM29, ESRF (Grenoble, France). SAXS measurements were carried out for MtbXthA, MtbLigA and MtbXthA–MtbLigA. Studies were also carried out on MtbLigA and MtbXthA–MtbLigA in complex with 27-mer nicked DNA (S1) at equimolar protein/DNA concentrations. The complex was formed by mixing the equimolar concentration of protein-protein/protein–DNA and then centrifuged for 5 min at 10,000 rpm to exclude any aggregated participants. Data were acquired at protein concentrations: 5.3 mg/ml for MtbXthA, 1.3 mg/ml for MtbLigA, 2.5 mg/ml for MtbLigA-nicked DNA complex, 2.5 mg/ml for MtbXthA–MtbLigA complex and 2.0 mg/ml for MtbXthA–MtbLigA-nicked DNA complex. All proteins were in buffer containing 50 mM Tris pH 8.0, 200 mM NaCl and 2 mM βME. Data collection was performed at 10°C and 10 frames of 1 s for samples were collected. (Table 2). Buffer scattering was measured to generate buffer subtracted intensity profiles. Data was processed using Primusqt implemented in ATSAS 2.8.1. The forward scattering intensity I(0) and radius of gyration R(g) were estimated using AUTOGNOM which was also used to evaluate the molecular size by plotting pair-distance distribution functions (PDDF). P(r) of scattering data is the representation in real space and reflects the particular particle's shape. P(r) approaches zero at its maximum dimension Dmax. AUTOGNOM results were used to generate 10 independent ab initio models through DAMMIF.
Table 2.
Small Angle X-ray scattering (SAXS) data collection and statistics
| MtbXthA | MtbLigA | MtbLigA+Nicked ds-DNA | MtbXthA+LigA | MtbXthA+LigA+Nicked ds-DNA | |
|---|---|---|---|---|---|
| SASBDB ID | SASDDD8 | SASDDN9 | SASDDV9 | SASDEW3 | SASDEX3 |
| Data collection parameters | |||||
| X-ray source | BM29,ESRF | BM29,ESRF | BM29,ESRF | BM29,ESRF | BM29,ESRF |
| Detector | Pilatus 1D | Pilatus 1D | Pilatus 1D | Pilatus 1D | Pilatus 1D |
| Wavelength (nm) | 0.081 | 0.081 | 0.081 | 0.081 | 0.081 |
| Exposure time per frame | 1 s | 1 s | 1 s | 1 s | 1 s |
| No. of frames collected | 10 | 10 | 10 | 10 | 10 |
| Concentration (mg/ml) | 5.3 | 1.3 | 2.5 | 2.5 | 2.0 |
| Measurement temperature (°C) | 10 | 10 | 10 | 10 | 10 |
| Structural parameters | |||||
| I(0) [from Guinier] | 13.63±0.012 | 10.86±0.11 | 14.5±0.13 | 197.66±0.46 | 8.15±0.073 |
| Rg (nm) [from Guinier] | 2.37±0.00 | 5.22±0.08 | 4.49±0.07 | 6.24±0.02 | 4.52±0.45 |
| I(0) [from P(r)] | 13.54±0.012 | 10.82 | 14.50 | 201.6±0.479 | 8.317±0.099 |
| Rg (nm) [from P(r)] | 2.35±0.003 | 5.20±0.06 | 4.49±0.06 | 6.66±0.0213 | 4.788±0.089 |
| Dmax (nm) | 7.3 | 16.7 | 14.79 | 23.86 | 17.06 |
| Modeling parameters | |||||
| Symmetry | P1 | P1 | P1 | P1 | P1 |
| Anisotropy | Unknown | Unknown | Unknown | Unknown | Unknown |
| Modelling iterations | 10 | 10 | 10 | 10 | 10 |
| Chi2 | 0.5765 | 0.79 | 0.64 | 1.02 | 0.8048 |
To generate the full-length model of MtbLigA, we modelled OB domain and HhH domain using E. coli. LigA (PDB: 2OWO) and BRCT domain was modelled by using Phyre2, using the structure of the BRCT domain from Human replication factor C large subunit 1 (PDB ID: 2EBU) as a template, which has 99.9% confidence and 99% coverage. Then all the domains were subjected to ensemble optimization modelling in EOM online software against SAXS data (72) to generate full length extended and closed forms of MtbLigA. For MtbXthA and MtbLigA complex, we docked the proteins using Cluspro (73). For MtbLigA + nicked DNA and MtbXthA + LigA + nicked DNA complexes, we built models on the basis of SAXS generated beads models in COOT (74) and their scattering was calculated and superimposed on SAXS scattering by using Supcomb (75) and Crysol (76). The model validation statistics is summarized in Supplementary Table S2. The models were subsequently superimposed using SUPCOMB. Figures were generated using PYMOL v0.99rc6/ Chimera 1.10.1.
Isothermal titration calorimetric analysis
Isothermal titration calorimetry (ITC) was performed on a MicroCal VP-ITC calorimeter (GE healthcare) and data analysis was done using Origin 7.0 software with Microcal add-on package. Proteins were purified as described above, concentrated and extensively dialyzed in 50 mM HEPES–Na pH 8.0, 200 mM NaCl and 2 mM βME. For peptide titrations, 0.4 mM of test peptides (i) DGQ (DGQPSWSGKP) (ii) YDV (YDVAHVGFD) were titrated in to 0.05 mM of protein (BRCT domain). Injections of 10 μl of substrate solution were added from a computer-controlled micro syringe at 3 min intervals into the protein solution (cell volume = 1.43 ml) with stirring at 394 rpm. Experimental data were fitted to a theoretical titration curve using software supplied by Microcal, with ΔH (binding enthalpy kcal mol−1), Ka (association constant) and n (number of binding sites per monomer), as adjustable parameters. All experiments were performed with c values 1 < c < 200, the quantity c = KaMt where Mt is the initial macromolecule concentration. Control experiments were performed for all the peptides and appropriate values subtracted from the final data sets. The calibration kit (supplied by manufacturer) was used to calibrate the instrument. Thermodynamic parameters were calculated from the Gibbs free energy equation.
15N/1H-HSQC NMR chemical shift perturbation (CSP) studies
Isotopically enriched 15N-labeled BRCT domain was expressed in minimal media consisting of vitamins and various salts and was purified as described above. Titration studies were carried out in uniform 15N labelled BRCT protein samples (∼0.2 mM) and unlabelled XthA (∼2 mM). The titration study was performed by adding aliquots of purified XthA to 15N labelled BRCT protein from BRCT:XthA ratio of 1:0, 1:0.2, 1:0.4, 1:0.6, 1:0.8 up to saturation level of 1:1 ratio. The 15N/1H-HSQC spectra were acquired at each titration point keeping all other experimental parameters constant. The NMR construct had an inadvertently introduced V667A conservative mutation. The pH of sample was monitored during titration and there were no significant changes in the pH till the end of the experiment. Chemical shift perturbations of the BRCT:XthA complex were determined from the deviation between peak position of the free and the complexed peak in the 15N/1H-HSQC spectra of the BRCT-domain.
Surface plasmon resonance (SPR)
SPR experiments were carried out using Biacore 3000, GE Healthcare. The surface of CM5 sensor chip was activated with N-hydroxy succinimide (NHS, 0.05 M)/N-ethyl-N-(diethylaminopropyl) and carbodiimide (EDC, 0.2 M). The GST antibody (sc-80998, Santa Cruz Biotech) was immobilized by amine coupling to the activated surface at concentration of 200 μg/ml in 10 mM sodium acetate buffer at pH 5.0 to a density of 8000 response units. After antibody immobilization the surface was blocked using ethanolamine at pH 7.4 and 50 mM NaOH was used as the regeneration solution. The GST-tagged MtbXthA and MtbXthAID were used as a ligand and diluted in running buffer (0.01 M HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% v/v Surfactant P20) at concentration of about 80 μg/ml (∼2.28 μM) and injected over one flow cell of the anti-GST Ab-coated sensor chip at 5 μl/min until 6000 response units of ligand was immobilized. The His-tagged MtbLigA and BRCT domain, MtbLigAG614I, MtbLigAG621I, MtbLigAG639V were injected respectively as the analyte at concentrations ranging from 0, 0.164, 0.329, 0.658, 1.32 and 2.63 μM. The 0.164 μM concentration was replicated twice in each experiment. A second flow cell served as a reference to account for bulk-shift responses and minor, nonspecific interactions with the anti-GST that was subtracted from the binding response. The kinetic experiment was run using application wizard of binding through the capturing molecule. Binding for 120 secs after end of each injection was measured at different analyte concentrations which were injected for 3 min at flow rate of 20 μl/min. Complete regeneration of surface was achieved with single 30 s injection of 50 mM NaOH. Sensograms were processed using BIA evaluation software version 4.1.1 (GE Healthcare). The steady state equilibrium constant (KD) was determined by fitting the data to a 1:1 Langmuir isotherm.
Fluorescence measurements
Complex disruption assay
MtbXthA was labelled with Oregon green 488 maleimide (MtbXthAOG) while BRCT domain of MtbLigA was labelled with Alexa flour 555 C2-maleimide (BRCTAF) using a protein labelling kit (Thermo Scientific). The degree of labelling was found to be three fluorophores per MtbXthA and two fluorophores per BRCT domain, respectively as determined by following the manufacturer's instructions. Interaction between the two labelled proteins was monitored by fluorescence resonance energy transfer (FRET). The donor MtbXthAOG (0.2 μM) and acceptor BRCTAF (2 μM) were incubated in 500 μl FRET buffer (50 mM HEPES–Na pH 8.0, 50 mM NaCl, 2 mM DTT, 8 mM MgCl2 and 0.5 mM EDTA) for 30 min on ice followed by measurement at room temperature using Cary Eclipse Fluorescence spectrophotometer (Agilent Technologies). Samples were excited at 488 nm and emission spectra were recorded from 495 to 600 nm. The complex disruption was tested by adding respective peptides and monitoring any change in the FRET efficiency. FRET efficiency (E) was calculated by; E = 1 – Fd’/Fd where Fd’ is the emission intensity of the donor in the presence of acceptor and Fd is the emission intensity of the donor alone. The peptides used in the study include (i) DGQ (DGQPSWSGKP) (ii) YDV (YDVAHVGFD) and (iii) Control peptide (IGQFDLFGV)
MtbLigA enzyme assays
The standard reaction mixture for in vitro ligation assay (20 μl) contained 10 nM DNA substrate (L1/L2, Supplementary Table S3) and 5 nM MtbLigA/MtbLigA1/LigA-K123A in 50 mM Tris–HCl (pH 7.8), 10 mM MgCl2, 5 mM DTT and 50 μM NAD+. Where indicated, the mentioned concentration of MtbXthA/ mutant E57A/D251A MtbXthA, was included in the reaction mixture. Reactions were assembled on ice, incubated at 37°C for 3 min, and quenched by adding 10 μl stop solution (98% formamide, 10 mM EDTA, 0.15% bromophenol blue). Samples were heated for 5 min at 100°C and chilled on ice before loading. Reaction products were resolved on 12% polyacrylamide gels containing 8 M urea. Gels were scanned using Image Quant LAS 4000 and band intensities were measured and quantified using ImageQuantTL 8.1 software. For monitoring the effect of MtbLigA on true incised abasic site containing nicked DNA, the DNA substrate L3 (1 nM) was first incubated with 1 unit of uracil DNA glycosylase (Thermo Fisher) for 30 min at 37°C following manufacturer protocol.
MtbXthA enzyme assays
A standard reaction mixture for AP-endonuclease activities (20 μl) contained 40 mM Tris–HCl (pH 7.8), 50 mM NaCl, 10 mM MgCl2, 2 mM DTT, 27 μM NAD+, 100 μg ml−1 BSA, 10 nM DNA substrate (N1, Supplementary Table S3) and 10 nM MtbXthA protein. Where indicated mentioned concentrations of MtbLigA/MtbLigA1/LigA-K123A and BSA, were added together with MtbXthA. Reactions were assembled on ice, incubated at 37°C for 10 min and quenched by adding 10 μl stop solution (98% formamide, 10 mM EDTA, 0.15% xylene cyanol and 0.15% bromophenol blue). Samples were heated for 5 mins at 100°C and chilled on ice before loading. Reaction products were resolved on 12% polyacrylamide gels containing 8 M urea. Gels were scanned using Image Quant LAS 4000 and band intensities were measured and quantified using ImageQuantTL 8.1 software.
Protein-DNA binding assay by electrophoretic mobility shift assay
Duplex DNA substrate L1/N1 (10 nM) was incubated with different concentration of MtbLigA/MtbXthA (0, 0.01, 0.03, 0.1, 0.3, 1, 3, 10 μM) in binding buffer (10 mM Tris pH 7.8, 10 mM NaCl, 2 mM MgCl2, 1 mM DTT, 20 mM EDTA, 2% glycerol and 0.1 mg ml−1 BSA) at room temperature for 1 h in a total reaction volume of 20 μl. For accessing the binding of MtbXthA to DNA substrate, EDTA was included in the buffer to prevent metal-ion dependent incision activity. After incubation, 8 μl of loading buffer (15% glycerol, 0.1% bromophenol blue) was added and the complexes were analysed by 6% native PAGE in 0.5× TBE buffer. Gels were scanned using Image Quant LAS 4000, bound DNA and free DNA were quantified by densitometry using the ImageQuantTL 8.1 software (GE Healthcare). The bound fraction was calculated as: intensity of protein-bound probe over the sum of the protein-bound and the free probe intensities, respectively. The fraction of DNA bound in each reaction was plotted versus the concentration of protein. The data were fitted using Prism software and non-linear regression to obtain the KD. The fraction of DNA bound in each reaction was plotted versus the concentration of protein:
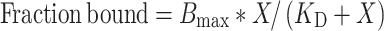 |
where X is the protein concentration, Bmax is the maximum binding and KD is the dissociation constant.
RESULTS
Co-immunoprecipitation of MtbXthA and MtbLigA from mycobacterial cell lysate suggests that they form a complex in vivo
We hypothesized that LigA might interact with XthA in mycobacteria/ bacteria based on reports involving human DNA ligase I and AP-endonuclease (77). In order to assess this, antibodies were raised against purified MtbLigA, which detected a specific band in M. tuberculosis lysate (Figure 1A, a). Protein A beads conjugated with anti-MtbLigA antibody were used to immunoprecipitate LigA along with any in vivo interacting proteins from DNAse I treated mycobacterial cell lysate. Co-immunoprecipitation of MtbXthA was detected by immunoblotting with anti-MtbXthA antibody in the fraction precipitated with anti-MtbLigA (Figure 1A, b, lane 2), while no signal was detected in the precipitates from the negative control where anti-MtbLigA antibody was excluded from the reaction (Figure 1A, b, lane 1). The same blot was stripped and re-probed with anti-MtbLigA antibody to confirm the immuno-precipitation (Figure 1A, c, lane 4). The specific signals for MtbLigA were absent in the negative control where anti-LigA antibody was not added to the reaction (Figure 1A, c, lane 3).
Figure 1.

Interactions between MtbLigA and MtbXthA and identification of regions involved in the interaction. (A) Antisera generation and physiological interactions between MtbLigA and MtbXthA. (A) To check specificity of raised antisera, immunoblots of Mtb cell lysate were probed with anti-MtbLigA antibody. (B, C) Co-IP was performed by precipitating Mycobacterium tuberculosis (H37Rv) cell lysate with anti MtbLigA Ab immobilized on protein A beads. No anti MtbLigA Ab was immobilized on protein A beads in control (b, c: lanes 1, 3). The co-precipitates were detected by western blot analysis using MtbXthA Abs (b: lane 2). The blot was also probed with anti MtbLigA Ab (c: lane 4) to detect immuno-precipitated MtbLigA. (B) Diagrammatic representation of domain architecture of MtbLigA and schematics showing various domain deletion mutants of MtbLigA used in this study. Sub-domains of Ia and Ib of adenylation domain are shown in purple and teal. Oligonucleotide binding (OB) domain is shown in pink, Zinc finger domain (Zn-HhH) is shown in beige and BRCT domain is shown in (Yellow), respectively. (C) GST pull down assay to probe MtbLigA-MtbXthA interactions. (A) GST-tagged WT MtbXthA was incubated with His-tagged wild type WT MtbLigA (lane 1) or LigA domain deletion mutants MtbLigA1 (lane 2), MtbLigA2 (lane 3), MtbLigA3 (lane 4), BRCT domain (lane 5) or (b) GST alone and mixed with glutathione sepharose beads. The immunoblot analysis of the pulled down complexes was carried out using appropriate antibodies as detailed in Materials and Methods. (D) Interaction analysis of proteins by surface plasmon resonance (SPR). MtbXthA-GST (∼2.28 μM) was immobilized as ligand on the chip coated with GST antibody and (I) His-tagged MtbLigA or (II) BRCT domain were passed as the analyte with concentration ranging from 0, 1.64e−7, 1.64e−7, 3.29e−7, 6.58e−7, 1.32e−6 and 2.36e−6 M. The KD values were determined by fitting the data to 1:1 Langmuir isotherm using Biaevaluation software 4.1.1.
C-terminal BRCT domain of MtbLigA mediates complex formation with MtbXthA
LigA has a multi-domain architecture with the adenylation domain at the N-terminal end and the BRCT domain at the C-terminal end (Figure 1B). To determine the site of the physical interactions between MtbXthA and MtbLigA, we created deletion mutants of MtbLigA as follows: BRCT alone, BRCT deleted (MtbLigA1), BRCT and HhH domains deleted (MtbLigA2), and BRCT, HhH and OB-fold domains deleted (MtbLigA3). Recombinant His-tagged full-length (FL) or domain deletion mutants of MtbLigA and GST-tagged MtbXthA were purified (Supplementary Figure S2A). As MtbXthA and MtbLigA are DNA binding proteins, steps (detailed in the Methods section) were taken to prevent co-purification of any bound DNA which was verified by UV-vis spectrophotometry (Supplementary Figure S2, B). We performed in vitro GST pull-down assays, where GST-MtbXthA (Figure 1C, a) or GST alone (Figure 1C, b) were respectively incubated with His-tagged FL MtbLigA or the domain deletion mutants before being precipitated by GST-binding glutathione beads. Immunoblotting of the pulled-down complexes with anti-His antibody showed a specific co-precipitation of the FL MtbLigA with GST-MtbXthA (Figure 1C, a, lane 1), while none of the domain deletion mutants were co-precipitated (Figure 1C, a, lanes 2–4). Interestingly, the LigA BRCT domain alone was also pulled-down by GST-MtbXthA (Figure 1C, a, lane 5). No co-precipitation of any complexes was observed with GST alone (Figure 1C, b, lanes 1–5). These results show that the BRCT domain of MtbLigA mediates interactions with MtbXthA.
Next, the respective interactions of MtbLigA and BRCT domain with MtbXthA were quantitatively analysed by surface plasmon resonance (SPR). GST-MtbXthA was immobilised on a GST-coated CM5 chip onto which His-tagged MtbLigA and BRCT-domain were respectively passed as analytes. MtbXthA binds to MtbLigA and the BRCT-domain respectively with similar equilibrium dissociation constant, KD, of 6.77e−8 M and 7.14e−8 M, respectively (Figure 1D I, II, Supplementary Table S4). The latter results suggest that the interaction between MtbXthA and MtbLigA is primarily mediated by the BRCT-domain. The result is additionally supported by the fact that the BRCT-deleted LigA mutant does not interact with MtbXthA (Figure 1C a, lane 2).
We also performed size exclusion chromatography (SEC) of the individual proteins and their complexes (Figure 2A and Supplementary Figure S2 D, E and F). The molecular mass and Stoke's radii were calculated using standard proteins (Supplementary Figure S2 D, E; Table 1). Fractions corresponding to the peaks were collected and analysed by SDS-PAGE (Figure 2A). MtbXthA, elutes with an apparent molecular weight 39.8 kDa (Figure 2A, green peak), while MtbLigA elutes at ∼121.04 kDa (Figure 2A, orange peak). When the two proteins were mixed together, MtbLigA and MtbXthA co-eluted in the molecular mass range ∼106.5 kDa (Figure 2A, purple peak). The UV–Vis spectra of the eluted peaks show the absence of any contaminating DNA (Supplementary Figure S2B). The BRCT deleted MtbLigA1 mutant failed to interact with MtbXthA as both proteins eluted separately (Figure 2A, blue peaks) with no shift evident in their individual elution volumes. Interestingly, purified BRCT domain (Supplementary Figure S2F) co-eluted with MtbXthA (Figure 2A, red peak). These results indicate that, a DNA independent binary complex forms between MtbXthA and MtbLigA, and that this interaction is mediated by the BRCT domain of the latter.
Figure 2.

Structure and dynamics of proteins/complexes. (A) Size exclusion chromatographic analysis. SEC profile of proteins/complexes as indicated. The elution volume (EV) in ml for proteins/complexes is shown on the side of respective eluted peaks. The fraction corresponding to the eluted peaks of proteins/complexes was analysed by 12% SDS-PAGE (shown as inset on right). The molecular weight marker (kDa) is indicated on each gel image. The stoke's radii calculated is depicted as superscript to indicated labelling in the graphs. The SEC profile for protein in complex with nicked DNA complex (S1) is show in dotted lines. The elution of standard proteins is marked as red arrows at the bottom of the chromatogram as 1: Ferritin (440 kDa), 2: Aldolase (158 kDa), 3: Ovalbumin (44 kDa), 4: Ribonuclease A (13.7 kDa) and 5: Aprotinin (6.5 kDa) for superdex 200 10/30 increase column. Small angle X-Ray scattering (SAXS) envelope of protein and complexes fitted with homology models. (B) MtbXthA alone (C) (i) Apo MtbLigA: SAXS reveals MtbLigA adopts extended conformation in solution while in the presence of 27 mer nicked DNA substrate (S1, top) a compact MtbLigA+DNA complex (ii) forms. The central cavity is seen lost and Ia, HhH and BRCT domains are in close vicinity surrounding DNA compared to apo MtbLigA structure. (D) MtbXthA+MtbLigA binary complex: The complex has ‘extended’ conformation where MtbLigA BRCT domain interacts with MtbXthA. The interaction interface is marked with dotted circle. (E) MtbXthA–MtbLigA-DNA ternary complex. SAXS shows MtbLigA only ‘partially closed’ around DNA as compared to MtbLigA-DNA complex. The BRCT domain is seen interacting with MtbXthA with interaction interface marked by dotted circle. (F) Normalized Pairwise interatomic distance distribution P[r] function of MtbXthA, MtbLigA and MtbXthA+MtbLigA complex demonstrates an increase in Dmax value for the complex. (G) Normalized Pairwise interatomic distance distribution P[r] function for MtbLigA+DNA and MtbXthA+MtbLigA+DNA. (H) Pictorial representation of MtbLigA ‘extended’ and ‘compact’ conformations around nicked DNA substrate in absence and presence of MtbXthA as revealed by SEC and SAXS analysis of individual components or protein complexes.
Structural characterization of MtbXthA–MtbLigA complex
To get an insight into the structure of the MtbXthA–MtbLigA complex, small-angle X-ray scattering (SAXS) was used to first characterize the solution behaviour of individual proteins MtbXthA and MtbLigA. While MtbXthA behaved as a globular protein with a radius of gyration (Rg) 2.37±0.00 nm and Dmax 7.3 nm (Figure 2B, Supplementary Figure S3A and Table 2), MtbLigA adopted an extended conformation with large Rg 5.22±0.08 nm and Dmax 16.7 nm (Figure 2C (i), Supplementary Figure S3B). All the domains of MtbLigA (AdD, OB, HhH and BRCT domain) were found to be present in the extended conformation in the bead model generated using DAMMIF (Supplementary Figure S3B and F(i)). The ab initio 3D bead model generated for MtbLigA using the SAXS data showed that the extended structure of MtbLigA fits the SAXS envelope derived from the average bead model with a normalized spatial discrepancy (NSD) of 2.93 by SUPCOMB (Supplementary Figure S3B, Table 2 and Supplementary Table S2).
The SEC analysis estimated a stoichiometric ratio of 1:1 interaction between MtbXthA–MtbLigA, therefore we studied the behaviour of MtbXthA–MtbLigA complex in solution at equimolar ratios. MtbXthA–MtbLigA complex appeared multi-domain and flexible in nature with 6.66 ± 0.02 nm Rg values and large Dmax 23.86 nm (Figure 2D, F, Supplementary Figure S3C and Table 2). The Kratky plot for the MtbXthA–MtbLigA complex was parabolic (Supplementary Figure S3C, iii purple) and the linearity of Guinier plot at lower q values suggests that the protein complex was devoid of any aggregation (Supplementary Figure S3C, ii). The χ2 values obtained for the experimental data and the SAXS profile calculated from the modeled structure supports the quality of the complex model (Table 2). Importantly, the interaction interface in the SAXS model shows that the extended MtbLigA engages with MtbXthA through its BRCT domain (Figure 2D).
MtbXthA, MtbLigA forms a ternary complex with nicked DNA
The incision by MtbXthA at an abasic site creates a nicked DNA intermediate on which downstream proteins including MtbLigA act to repair the DNA damage. We therefore wanted to study the interaction between the two proteins in the presence of nicked DNA. We first performed SEC analysis of MtbLigA incubated with a nicked DNA substrate analog (S1). The apparent molecular weight of the DNA–LigA complex was ∼112.27 kDa with a reduced Stoke's radii of 3.19 nm compared to MtbLigA alone (4.02 nm) (Figure 2A, yellow dotted peak and Table 1). The UV–Vis spectral analysis of the eluted peak shows the presence of DNA in the complex (Supplementary Figure S2B, red dotted). The result suggests that MtbLigA likely adopts a compact conformation in the presence of DNA. However, in the presence of MtbXthA, the Stoke's radius of the tri-complex increases to 3.79 nm indicating that inclusion of MtbXthA results in the formation of a larger oligomeric complex. (Figure 2A, red dotted peak). Both MtbXthA and MtbLigA were detected in SDS-PAGE analysis of the fraction from eluted peaks, indicating the complex formation between the two proteins (Figure 2A). The UV–Vis spectra of the eluted peak also confirmed the presence of DNA in the fraction volume (Supplementary Figure S2B, magenta). The SEC results overall show that MtbXthA–MtbLigA–DNA forms a ternary complex.
To get an insight into the structure of this ternary complex by SAXS we first determined the structural dynamics of MtbLigA binding to nicked DNA.
SAXS analysis of apo and DNA-bound MtbLigA identifies extended and compact conformations of MtbLigA respectively
We carried out SAXS analysis of MtbLigA in the presence and absence of nicked DNA substrate (S1) (Figure 2C, I & II). The full-length model of MtbLigA was built for the analysis using crystal structures of homologs and structures solved by our group as templates (details in Methods). The pairwise distance distribution function P(r) demonstrated a reduction in the Dmax from 16.7 nm (open, extended conformation of apo MtbLigA, Figure 2F, orange) to 14.79 nm indicating a closed/ compact conformation, (Figure 2G, salmon). This was accompanied by a decrease in the radius of gyration (Rg) from 5.20 to 4.49 nm in the presence of nicked DNA substrate, suggesting a DNA induced conformational change in LigA (Table 2). The comparative Kratky analysis at lower q values of scattering profiles of both data sets reveal the globularity attained by LigA in the presence of the nicked S1 (Supplementary Figure S3D, iii) as was also suggested by SEC data above. All samples were monodispersed and free from aggregation as determined by Guinier analysis (Supplementary Figure S3B, ii and D, ii).
The bead model generated using DAMMIF shows the presence of various domains of apo MtbLigA (AdD, OB, HhH and BRCT domain) in the extended conformation (Supplementary Figure S3F) where the distance between the AdD domain and HhH is prominent (2.5 nm) and central cavity is about 4.3 nm wide. Interestingly, SAXS data for MtbLigA-nicked DNA complex supports a ‘toroid’ shaped model. Conformational changes in the local spatial disposition of domains around the nicked DNA resulted in reducing the distance between Ia and HhH domains and a loss of the central cavity (Figure 2C, i and ii, Supplementary Figure S3F i and ii). The BRCT domain also rearranges and gets in close vicinity of Ia sub-domain compared to apo MtbLigA, contributing to the compact architecture. Comparison of SAXS model of MtbLigA–DNA complex (fitted with homology structure) with template crystal structures (2OWO, 1DGS), suggests a rotation of nearly 170° from Y324 as the centre of rotation (Supplementary Figure S3F, iii). The adoption of a compact conformation by MtbLigA may be a requisite for stabilizing its interaction with substrate DNA as suggested in earlier reports (43,78).
Structure and dynamics of MtbXthA–MtbLigA–nicked DNA ternary complex
We next analysed the MtbXthA–MtbLigA complex in the presence of nicked DNA substrate (S1) using SAXS (Figure 2D & E). The addition of DNA into the MtbXthA–MtbLigA complex introduced disorder at near qmin values as reflected by Kratky plot of the complex (Rg 4.52±0.45 nm) (Supplementary Figure S3E, iii, teal). Ternary complex exhibits a reduced Dmax (17.06 nm) compared to the binary MtbXthA–MtbLigA complex (23.86 nm) indicating adoption of more compact structure (Figure 2E and Table 2). While the Kratky plot for MtbXthA–MtbLigA complex was parabolic (Supplementary Figure S3C, iii), inclusion of DNA in the complex resulted in an upward trend in the plot, towards higher angles reflecting increased disorder (Supplementary Figure S3E, iii). The SAXS model of ternary complex showed an interaction between MtbXthA and BRCT domain of MtbLigA. In contrast to the compact conformation adopted by MtbLigA in its complex with nicked-DNA, it adopts a relatively extended conformation while interacting with XthA, both in the presence and in the absence of nicked DNA. The linearity of Guinier plot at lower q values suggests that the proteins and complexes were devoid of any aggregation (Supplementary Figure S3E, ii). The χ2 values obtained for the experimental data and the SAXS profile calculated from the modelled structure supports the quality of the modelled complex (Table 2). A schematic representation of the SEC and SAXS results is shown in Figure 2H.
Identification of a conserved MtbXthA motif and MtbLigA residues that mediate MtbXthA–MtbLigA interactions
It is clear from the above results that the C-terminal BRCT domain of MtbLigA interacts with MtbXthA. Correspondingly, we examined the SAXS derived MtbXthA–MtbLigA model to identify the potential region on MtbXthA involved in mediating the interactions of the complex. An examination of the interaction interface showed that the BRCT domain of MtbLigA engages with an amino acid stretch 104DGQPSWSGKP113 in MtbXthA (Figure 3A). We then designed a peptide corresponding to the interaction interface region in MtbXthA and performed Isothermal Titration Calorimetry (ITC) to determine its binding with the BRCT domain of MtbLigA. The test peptides used were: DGQ (104DGQPSWSGKP113) located on the interaction interface and a control YDV (75YDVAHVGFD83), present on an adjoining loop in proximity but away from the interaction interface. The DGQ peptide bound to BRCT domain with apparent Kd 2.47 μM, while the YDV peptide showed minimal interactions (Kd 88.49 μM) (Figure 3B and C). Peptides titrated against only the buffer did not show any non-specific binding of peptides (Supplementary Figure S4A and B). The thermodynamic analysis of DGQ peptide binding suggests the occurrence of favourable hydrogen and hydrophobic interactions (Supplementary Table S5).
Figure 3.

Validation of interaction interface between MtbXthA and MtbLigA. (A) SAXS envelope of MtbXthA–MtbLigA complex with fitted homology model showing interaction interface between MtbXthA and BRCT domain of MtbLigA. The BRCT domain and MtbXthA are represented in yellow and green, respectively. Selected residues (marked) forming contacts at the interface are shown in sticks (B) Isothermal calorimetry (ITC): The binding isotherms for DGQ peptide: located on interaction interface and (C) control YDV peptide, with the BRCT domain of MtbLigA are shown. Both raw ITC isotherms (top panels) and data after integration and normalization (bottom panels) are depicted. The solid lines (red) in the bottom panels show the fits with a one-site binding model. Thermodynamic parameters for ITC experiments are summarized in Supplementary Table S5. (D) Schematic representation of the Fluorescence resonance energy transfer (FRET) experiment to probe disruption of the MtbXthA‐BRCT domain complex by the respective peptides. (E) FRET analysis showed that the MtbXthA derived peptides disrupt the interaction between Oregon green 488 maleimide‐labelled MtbXthA (MtbXthAOG) and Alexa fluor 488‐labelled BRCT domain (BRCTAF). A decrease in FRET occurred in the presence of DGQ peptide (orange). The YDV (green) moderately disrupted FRET efficiency while control peptide (grey) could not elicit disruptive effects. (F) Bar graphs showing FRET efficiency of respective peptides depicted as: YDV (green), DGQ (orange) and control peptide (grey). The purple bar represents the control without peptides. Error bars correspond to standard deviation of two independent experiments.
We next performed peptide mediated disruption assays against the MtbXthA–BRCT complex. Fluorescently labelled MtbXthAOG and BRCTAF (Alexa fluor labelled) were used to monitor complex formation using FRET (Figure 3D). FRET between MtbXthAOG (donor) and BRCTAF (acceptor) was measured in the absence (Figure 3E-F; purple) and presence of peptides (DGQ-orange and YDV-green). Inclusion of DGQ peptide in the reaction mixture resulted in a marked decrease in FRET indicative of inhibition of MtbXthA–BRCT complex formation (orange), while the peptide YDV resulted in a poor decrease in FRET efficiency (green). No reduction in FRET was observed on inclusion of a negative control peptide (IGQFDLFGV) (grey). The above data suggests that the sequence motif, 104DGQPSWSGKP113 in MtbXthA is important for forming a complex with MtbLigA. To further probe the role of this motif in mediating complex formation, we mutated the 104DGQPSWSGKP113 motif in XthA to 104DGQPSWALAP113 (this interaction-defective mutant will be referred to as MtbXthAID). We found that MtbXthAID exhibited ten-fold lower binding affinity with LigA (KD 6.13e−7 M, Figure S5A) compared to the wild-type XthA with MtbLigA (KD 6.77e−8 M, Figure 1D). We also mutated three conserved glycine residues, viz. G614, G621 and G639, of the BRCT-domain of MtbLigA (Supplementary Figure S6D). These glycine residues are part of the interaction interface with MtbXthA as suggested by SAXS (Figure 3A). A previous study involving E. coli LigA (47) had suggested that the corresponding glycines in the E. coli enzyme are located at transitions from ordered secondary structures to loops, leading to significant local changes in the BRCT-domain. The analogous LigA BRCT-domain mutants, MtbLigAG614I, MtbLigAG621I and MtbLigAG639V were accordingly generated to probe the effects of disrupting the BRCT-domain interaction interface. We then used SPR to check the binding affinity of wild-type XthA with LigA mutants. Relatively poor binding was observed between MtbXthA and MtbLigAG639V (KD 2.77e−6 M), while moderate binding was observed with MtbLigAG614I and MtbLigAG621I (KD 7.81e−7 M and 1.54e−7 M, respectively) (Supplementary Figure S5B–D, Supplementary Table S4). Importantly, MtbXthAID and MtbLigAG639V exhibited very weak interactions (KD 9.01e−6 M) (Supplementary Figure S5E). The mutational results support the SAXS-derived model of the MtbXthA–MtbLigA complex and the protein-protein interaction interface.
We then carried out 15N/1H-HSQC chemical shift perturbation (CSP) studies using purified MtbXthA and 15N labelled BRCT-domain to further characterise the MtbXthA–BRCT interaction interface. Changes to the chemical shifts, either for 1H or 15N or both, were observed for several residues of the BRCT domain (Supplementary Figure S6A). The sequence-specific resonance assignment identified residues in the BRCT domain interface that undergo flexible/ allosteric changes upon binding with MtbXthA (Supplementary Figure S6C). Significant changes in the average chemical shift (Dδavg) were observed for BRCT-domain residues V619, T620, S622, L623, D629, K632, E633, T650, N651, V653, A655, G656, V671, E676 and L683 (Supplementary Figure S6A, B and C), many of which are also conserved (Supplementary Figure S6D). The above residues are part of the interaction interface as suggested by the SAXS-derived model (Figure 3A, close-up view) and agree well with the mutational analysis.
Overall, the above results support that MtbXthA–MtbLigA interactions are mediated by the 104DGQPSWSGKP113 motif of MtbXthA with a specific interaction interface of the BRCT-domain of LigA.
MtbXthA inhibits the ligase activity of MtbLigA
Human APE1 stimulates the activity of human DNA ligase I on a nicked substrate while inhibits the latter's activity on nick-THF substrates (product mimics of APE1) (77). Despite differences in domain organization, primary sequences and cofactor specificity between LigA (NAD+-dependent) and DNA ligase I (ATP-dependent) and also between XthA and APE1 (Supplementary Figure S1), we investigated if analogous functional interaction occurs between bacterial counterparts. We therefore investigated the functional significance of MtbXthA–MtbLigA interactions. Ligation activity of MtbLigA was monitored using a 5′-FAM labelled duplex DNA substrate (L1) containing a single stranded nick (Supplementary Figure S7A). The effect of MtbXthA on the in vitro ligation activity of MtbLigA was tested. Incubation of the DNA substrate in the presence of limiting concentration of MtbLigA alone resulted in 75% DNA ligation (Figure 4A, lane 3). However, the titration of MtbXthA into the reaction resulted in a progressive decrease in the ligation activity (Figure 4A, lanes 4–10). Notably, MtbXthA on its own had no effect on the nicked substrate (Figure 4A, lane 2).
Figure 4.

Regulation of MtbLigA ligation activity by MtbXthA. (Top) Diagram to show the nicked duplex DNA substrate (L1) used in the ligation assays. Asterisks denote the 6-FAM labelled termini. (A) MtbXthA dependent inhibition of MtbLigA ligation activity. (B) Effect of catalytic dead mutant of MtbXthA (E57A/D251A) on MtbLigA ligase activity. (C) Gel shift assay to monitor the affinity of MtbLigA and MtbXthA for nicked DNA. 10 nM Nicked DNA substrate (L1) was incubated with increasing concentration (0, 0.01, 0.03, 0.1, 0.3, 1, 3, 10 μM) of MtbXthA (lanes 1–8) and MtbLigA (lanes 9–16) respectively, to determine the affinity constant (shown in the side graph). (D) Effect of interaction defective (ID) XthA mutant (MtbXthA ID) on MtbLigA activity. In A, B and D, DNA substrate (10 nM) was incubated with MtbLigA (5 nM) in absence (lanes 3) or presence of increasing concentration (0.5, 1, 2, 5, 10, 20, 30 nM) of MtbXthA/mutants (lanes 4 onwards) under standard conditions. Lanes 1 and 2 respectively, contain no proteins and 5 nM MtbXthA, E57A/D251A, respectively, as controls. Reaction products were analysed on 8 M urea–12% polyacrylamide gels for A and B while 6% native polyacrylamide gel was used for product analysis in C, gel shift assay. Intensity of the fluorescent bands corresponding to the products of respective activities were scanned and quantified by using ImageQuant LAS 4000 and ImageQuantTL 8.1 software (GE Healthcare). The images are single representative image of experiments carried out in duplicate. A standard deviation of ±2.5 was obtained for the % DNA ligated. The position of the ligated product at 40 mer is indicated.
Nicked-DNA is known to be a weak substrate of XthA for its 3′-exonuclease activity (62). We therefore probed whether MtbXthA activity could oppose that of MtbLigA. However, we found that titration of a catalytically inactive mutant of MtbXthA, E57A/D251A (62), into the ligation assay also led to a similar decrease in ligation as seen by wt-MtbXthA (Figure 4B).
We then analysed whether there was mutual competition between MtbXthA and MtbLigA to bind to the nicked DNA substrate using gel shift assays (Figure 4C). We found that MtbLigA binds nicked DNA with KD ∼60 nM, while surprisingly MtbXthA did not form any stable detectable complex with nicked DNA, indicating that it may require additional factors to facilitate binding.
As shown by the SAXS experiments described earlier, when MtbXthA engages with MtbLigA, it prevents the latter from encircling the DNA substrate and keeps it in a relatively extended conformation (Figure 2E). It has been reported that complete encircling of DNA substrate by LigA is required for effective ligation (43). We hypothesise that this is prevented by XthA which directly engages with the BRCT-domain of LigA, thus leading to the observed inhibition of ligation activity. This is further supported by the fact that the LigA-interaction defective XthA mutant, MtbXthAID, did not affect LigA-activity on nicked DNA (Figure 4D).
MtbXthA prevents the ‘futile’ ligation activity of MtbLigA at incised abasic sites
Next, we were interested to see if mutual regulatory interactions exist between the two proteins. We evaluated the effect of MtbLigA on the activity of MtbXthA. We have shown earlier that MtbXthA is a multifunctional enzyme, possessing a major AP-endonuclease activity and also several other activities like, 3′-5′ exonuclease, 3′ phosphatase activities etc. (62). We analysed the effect of MtbLigA on the AP-endonuclease activity of MtbXthA using a 5′-FAM labelled apurinic/ apyrimidinic DNA (AP-DNA) substrate (N1) (Figure 5A, top) that has an abasic site analog, tetrahydrofuran, incorporated into it. While, the incubation of MtbXthA alone resulted in 70% DNA cleavage (Figure 5A, lane 3), the titration of MtbLigA into the reaction resulted in a progressive decrease in product formation, (lanes 4–10). In the absence of MtbXthA, MtbLigA on its own had no effect on the AP-DNA substrate (lane 2).
Figure 5.

Effect of MtbLigA on AP-endonuclease activity of MtbXthA. (Top) Diagram to show the abasic site (THF) containing DNA substrate (N1) used to assess the AP-endonuclease activity of MtbXthA. (A) Effect of MtbLigA on AP-endonuclease activity of MtbXthA. (B) Effect of MtbLigA1 on AP-endonuclease activity of MtbXthA. (C) Effect of catalytic dead LigA-K123A on AP-endonuclease activity of MtbXthA. DNA substrate (20 nM) were incubated with MtbXthA (10 nM) in absence (lanes 3) or presence of increasing concentration (5,10, 20, 40, 60, 80, 100 nM) of MtbLigA/LigA variants (lanes 4–10) under standard conditions. Lanes 1 and 2 respectively contain no proteins and 5 nM MtbLigA alone as controls. Reaction products were analyzed on 8 M urea-12% polyacrylamide gels. The position of the incision product at 32mer is indicated. Intensity of the fluorescent bands were scanned and quantified by using ImageQuant LAS 4000 and ImageQuantTL 8.1 software (GE Healthcare). % DNA hydrolysed is indicated below each gel picture. Asterisks denote the 6-FAM labelled termini. The position of the incised product at 75mer is indicated. BRCT domain in illustrated in red circle while dotted white circles represents mutated domains. The images are single representative image of experiments carried out in triplicate. A standard deviation of ±3 was obtained for the % DNA cleavage.
Earlier reports suggest that macromolecular crowding may affect an enzymatic reaction. To rule out that this inhibition is due to a non-specific effect of macromolecular crowding, we monitored the effect of BSA, at concentrations similar to LigA, on the AP-endonuclease activity of MtbXthA, and found that it exerted no effect (Supplementary Figure S7B). To rule out that the inhibition may be due to direct binding of MtbLigA to the AP-DNA, substrate of MtbXthA, we performed gel shift assay. While we had previously shown that MtbXthA possesses high affinity for AP-DNA (62,63), MtbLigA did not form a complex with THF containing AP-DNA (N1) (Supplementary Figure S7C). We next tested the effect of the BRCT-deleted mutant of MtbLigA (MtbLigA1) on the AP-endonuclease activity of MtbXthA. BRCT lacking MtbLigA1 itself has severely compromised DNA binding (48) and ligation activity (Supplementary Figure S7D) compared to full length MtbLigA. The deletion of the BRCT domain possibly renders MtbLigA unable to encircle and bind its DNA substrate effectively to carry out ligation (43,46). We found that increasing presence of MtbLigA1 had no effect on the AP-endonuclease activity of MtbXthA (Figure 5B). The inhibition of endonuclease activity only by full-length MtbLigA and not by the MtbLigA1 mutant indicates that the BRCT domain is crucial for the regulation of MtbXthA activity by MtbLigA. We thought that this may be facilitated by MtbXthA’s interaction with BRCT domain of LigA. Thus, we checked endonuclease activity of MtbXthAID mutant in the presence of LigA. Unexpectedly, we also observed reduction in incision activity of MtbXthAID mutant in the presence of full-length MtbLigA (Supplementary Figure S8A), suggesting that the apparent reduction in activity of MtbXthA in the presence of LigA is independent of interactions between the proteins and might be attributed to the action of MtbLigA on the reaction product of XthA activity.
To analyse the role of the ligase activity of MtbLigA on the inhibition of MtbXthA’s AP-endonuclease activity, we tested the effect of the catalytically inactive mutant of MtbLigA (LigA-K123A) (Supplementary Figure S8B). Interestingly, the mutant LigA-K123A did not show any inhibition of the AP-endonuclease activity of MtbXthA (Figure 5C, lanes 4–10). This suggests that the intrinsic ligase activity of MtbLigA is responsible for the observed inhibition of MtbXthA AP endonuclease activity. This result was in contrast with our earlier observation that MtbLigA does not bind to THF containing AP-DNA.
The above results led us to conjecture that instead of the intact AP-DNA, MtbLigA may be acting as a ligase on the incised, nick-containing AP-DNA that is obtained as a product of MtbXthA AP-endonuclease activity. This ligation would be a direct reversal of the MtbXthA endonuclease activity and would essentially be a futile step during AP-site DNA repair process. Indeed, such a step, if allowed by the cell, would be futile and potentially derail BER itself. To probe this conjecture, MtbLigA activity on an incised abasic DNA was tested. We measured the ligation activity of MtbLigA using a nicked duplex DNA substrate (L2) containing a 5′p-THF instead of a 5′phosphate moiety at the nick (Figure 6A, top). We found that MtbLigA could ligate the nicked AP-DNA (Figure 6A, lanes 2–10). We further evaluated the ability of MtbLigA to ligate a ‘true product’ of abasic site incision by using a mimic DNA substrate (L3), generated by treating a nicked DNA containing 5′-uracil at the nick with E. coli uracil DNA glycosylase, which removes the uracil base and creates a 5′-dRP residue (Figure 6B, top). Incubation of L3 with increasing concentrations of MtbLigA resulted in a progressive increase in the ligated product (Figure 6B, lanes 2–10). This indicates that MtbLigA can effectively reverse the action of the AP-endonuclease activity of MtbXthA at the abasic site in AP-DNA by resealing the nick the latter creates (Figure 6C). Interestingly, this ligation activity of MtbLigA on nicked AP-DNA is also inhibited by the presence of MtbXthA (Figure 6D) indicating that MtbXthA coordinates and checks futile ligation activity of MtbLigA.
Figure 6.

MtbLigA can reseal a nick with 5′-phosphorylated abasic site. (A) Top; Diagrammatic representation to show DNA substrate (L2) with abasic site (pTHF) at the nick. MtbLigA can affectively ligate incised abasic site containing substrate. (B) Top; Diagrammatic representation to show the generation of true incised abasic site containing DNA substrate that contains dRP moiety at the nick. DNA substrates (10 nM) were incubated with increasing concentration (0.01, 0.05, 0.1, 0.5, 1, 2, 3, 5,10, 20 nM) of MtbLigA (lanes 2–10) under standard conditions. Lane 1 contains no protein as control. (C) Schematics to show the action of MtbLigA at the abasic sites post MtbXthA mediated AP-site incision. MtbLigA can reseal the nick created after AP-site incision during base excision repair. This would lead to futile ligation. (D) 10 nM of DNA substrate (L2) was incubated with MtbLigA (5 nM) in the absence (lane 3) or presence of increasing concentration of MtbXthA (0.5, 1, 2, 5, 10, 20, 30 nM). Reaction products were analyzed on 8 M urea-12% polyacrylamide gels. Intensity of the fluorescent bands were scanned and quantified by using ImageQuant LAS 4000 and ImageQuantTL 8.1 software (GE Healthcare). % DNA ligated is indicated below each gel picture. Asterisks denote the 6-FAM labelled termini. The position of the ligated product at 40mer is indicated. The images are single representative image of experiments carried out in duplicate. A standard deviation of ±3 was obtained for the % DNA ligated.
DISCUSSION
Protein-protein interactions are critical for cellular processes. Often, the behaviour of individual proteins is widely modulated/ regulated when they act as part of complexes. We are characterizing prokaryotic BER complexes and their roles in the overall DNA repair process. Recently, we had identified DNA-mediated interactions between the Mtbβ-clamp and MtbXthA (63). An earlier report (77) regarding interactions between human APE1 and DNA Ligase I spurred our search for similar interactions, if they exist, in prokaryotes. It intrigued us because of the large differences in sequence and domain organization between human DNA Ligase I and bacterial LigA (Supplementary Figure S1A). On the other hand, the domain organization of XthA and APE1 are quite similar, except for ∼60-residue N-terminal extension in APE1 (Supplementary Figure S1B).
The pull-down assays involving Mtb lysate and also the assays involving recombinant purified proteins establish the existence of MtbXthA–MtbLigA interactions. Given the conserved nature of the proteins, we believe that these interactions are conserved in bacteria in general. The mutational analysis involving deletion of domains of LigA and checking for their roles in mediating interactions with XthA establishes that BRCT-domain of LigA is important for interacting with XthA. In higher eukaryotes, BRCT domains mediate various protein interactions as also in BER (79–82). However, to our knowledge, this is the first report to show that the BRCT domain of LigA mediates a protein–protein interaction in bacteria.
An important global difference between APE1 and XthA lies in an ∼60-residue N-terminal extension in APE1 compared to XthA. APE1 is known to interact with other proteins like the XRCC1-DNA Ligase III complex, via residues lying in its N-terminal disordered segment (81). Sequence alignment and in silico homology modelling showed that such an N-terminal extension is absent in MtbXthA (Supplementary Figure S1, B) (63). We had earlier identified a protein interacting peptide (PIP) motif on XthA that is involved in interactions with the Mtbβ-clamp (63). On the other hand, the SAXS derived structure of the MtbLigA–MtbXthA complex ruled out the PIP motif (Protein Interaction Peptide in clamp) in interactions and instead revealed an interaction interface with significant sequence conservation in MtbXthA (Supplementary Figure S9A). The ‘DGQ’ peptide derived from the interacting region, was able to specifically disrupt the MtbLigA-MtbXthA complex and also exhibited binding to the BRCT-domain of LigA as shown by ITC experiments. Disruption of 104DGQPSWSGKP113 motif in MtbXthA resulted in reduced ability to form a complex with MtbLigA as enunciated by the mutational analysis. Correspondingly, NMR-CSP and mutational analysis involving LigA BRCT-domain identified important residues for mediating interactions with MtbXthA. The latter results further support the SAXS-identified interface that mediates the interactions between the two proteins. Therefore, XthA has at least 2 protein interacting regions suggesting a coordinating role for it in BER (Supplementary Figure S9B). This is analogous to the reported roles of APE1 in coordinating the activities of Flap endonuclease 1 and DNA ligase I, amongst other roles (77). A key differentiator is that APE1 reportedly interacts through the N-terminal unstructured extension with protein partners.
MtbXthA acts at the 5′-side of the AP-site and generates a nicked DNA with 3′-OH and 5′-dRP moieties at the incised termini. The 5′-dRP moiety is then removed by an AP-Lyase to generate suitable DNA ends for further processing (83). While, ATP ligases like T4 DNA ligase and DNA Ligase I, are active on DNA substrates with 5′ incised AP-site as an AP-Lyase (83), no such activity has been yet been reported for bacterial LigA. On observing that MtbLigA does not exhibit affinity for abasic DNA but could still inhibit MtbXthA in AP-endonuclease assays, we hypothesized that MtbLigA does so by interacting with the incised abasic DNA intermediate generated by MtbXthA. Interestingly, we found MtbLigA could efficiently ligate nicks adjacent to 3′-OH and 5′-THF or 5′-dRP DNA termini. Therefore, the detected inhibition of the MtbXthA AP endonuclease activity by MtbLigA can be attributed to re-ligation of the incised abasic DNA generated by MtbXthA. This represents direct reversal of the action of MtbXthA at the abasic site and can derail BER itself. The result is strengthened by our observation that both catalytically inactive LigA-K123A mutant (capable of binding DNA), and the inefficient BRCT-deleted MtbLigA, did not affect the respective AP-endonuclease activity of MtbXthA and MtbXthAID mutant, demonstrating that the observed decrease in AP-endonuclease activity of MtbXthA was due to the intrinsic ligation activity of MtbLigA.
The present work suggests a novel coordination in mycobacterial BER pathway orchestrated by MtbXthA to prevent futile ligation. As supported by SAXS, it does so by physically engaging with the BRCT-domain of LigA and thereby prevents it from encircling the DNA substrate. Our previous study showed that β-clamp increases the affinity of MtbXthA for its substrate (63). Since β-clamp acts as a scaffold protein that recruits protein participants to the site of damage, we speculate that the β-clamp stays at the damage site with MtbXthA till an AP-Lyase (as yet unidentified) is recruited to remove the 5′-dRP moiety. This would result in the release of MtbXthA from the damage site and facilitate the action of DNA polymerase and ligase to resume their normal functions. It is important to note that the MtbXthA–MtbLigA interaction abrogates ligation activities against both nicked DNA substrate, and the ‘product’ of XthA activity. In contrast, APE1 enhances the activity of DNA Ligase I against nicked substrates and inhibits the activity against substrates that mimic a reduced abasic site (77). The authors hypothesized that the binding of DNA ligase I to THF-containing substrates is blocked by APE1 while it permits access to nicked DNA substrates, thereby enhancing the efficiency of eukaryotic BER overall and at the same time prevents unwanted ‘futile’ ligation. However, the exact mechanisms by which human APE1 stimulates or restricts DNA ligase I activity needs to be investigated.
Overall, our results suggest that it is important to inhibit the activity of LigA during earlier steps of BER to prevent futile cycles of substrate cleavage and ligation. In mycobacteria (and possibly bacteria in general), this is coordinated by XthA which engages with the BRCT-domain of LigA to keep it in a relatively extended conformation. This prevents LigA from encircling the substrate DNA and consequently from acting detrimentally against any DNA substrate during the initial steps of BER (Supplementary Figure S9C). It is remarkable that the control of unwanted ligase activity in the initial steps of BER is apparently a necessity and is enforced by eukaryotes and prokaryotes despite scant sequence similarity and different domain organization of the respective enzymes.
DATA AVAILABILITY
Small-angle X-ray scattering data have been submitted to the SASBDB database at https://www.sasbdb.org/ with accession numbers as follows: MtbXthA (SASDDD8), MtbLigA (SASDDN9), MtbXthA–MtbLigA complex (SASDEW3), MtbLigA-DNA complex (SASDDV9), MtbXthA–MtbLigA-DNA complex (SASDEX3). 15N/1H HSQC NMR- chemical shift perturbation data have been submitted to the Biological Magnetic Resonance Bank (BMRB) at http://www.bmrb.wisc.edu/ with accession number 28077.
Supplementary Material
ACKNOWLEDGEMENTS
T.K., A.S., M.A. acknowledge junior and senior research fellowships from the Council of Scientific and Industrial Research. R.K. Srivastava is acknowledged for help with raising of the antisera.
Author contributions: T.K., M.A. and A.S.: Design of experiments, performing experiments, data analysis and writing manuscript, S.K.: Computational calculations, H.S. and M.P.: Peptide synthesis, K.D. and Ashish: Helped with SAXS experiments, K.K.S.: Mtb cell lysate and genomic DNA, F.A. and R.S.A.: 15N/1H-HSQC NMR chemical shift perturbation analysis, R.R.: Design of experiments, computational analysis, interpretation of data and writing manuscript.
Notes
Present address: Taran Khanam, MRC-Protein Phosphorylation and Ubiquitylation Unit, University of Dundee, Scotland DD1 5EH, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
CSIR-CDRI [66/2018/RR]; Council of Scientific and Industrial Research [MLP0120]; Department of Science and Technology through DST [GAP0292].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ehrt S., Schnappinger D.. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 2009; 11:1170–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stallings C.L., Glickman M.S.. Is Mycobacterium tuberculosis stressed out? A critical assessment of the genetic evidence. Microbes Infect. 2010; 12:1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gorna A.E., Bowater R.P., Dziadek J.. DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: varying activities at different stages of infection. Clin. Sci. (Lond.). 2010; 119:187–202. [DOI] [PubMed] [Google Scholar]
- 4. Ishino S., Skouloubris S., Kudo H., l’Hermitte-Stead C., Es-Sadik A., Lambry J.C., Ishino Y., Myllykallio H.. Activation of the mismatch-specific endonuclease EndoMS/NucS by the replication clamp is required for high fidelity DNA replication. Nucleic Acids Res. 2018; 46:6206–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takemoto N., Numata I., Su’etsugu M., Miyoshi-Akiyama T.. Bacterial EndoMS/NucS acts as a clamp-mediated mismatch endonuclease to prevent asymmetric accumulation of replication errors. Nucleic Acids Res. 2018; 46:6152–6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizrahi V., Andersen S.J.. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence. Mol. Microbiol. 1998; 29:1331–1339. [DOI] [PubMed] [Google Scholar]
- 7. Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E. 3rd et al.. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998; 393:537–544. [DOI] [PubMed] [Google Scholar]
- 8. Hazra T.K., Hill J.W., Izumi T., Mitra S.. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:193–205. [DOI] [PubMed] [Google Scholar]
- 9. Mullins E.A., Rodriguez A.A., Bradley N.P., Eichman B.F.. Emerging roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci. 2019; 44:765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dizdaroglu M., Coskun E., Jaruga P.. Repair of oxidatively induced DNA damage by DNA glycosylases: mechanisms of action, substrate specificities and excision kinetics. Mutat. Res. 2017; 771:99–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boiteux S., Guillet M.. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst.). 2004; 3:1–12. [DOI] [PubMed] [Google Scholar]
- 12. Yu S.L., Lee S.K., Johnson R.E., Prakash L., Prakash S.. The stalling of transcription at abasic sites is highly mutagenic. Mol. Cell Biol. 2003; 23:382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dyrkheeva N.S., Lebedeva N.A., Lavrik O.I.. AP endonuclease 1 as a key enzyme in repair of Apurinic/Apyrimidinic sites. Biochemistry (Mosc). 2016; 81:951–967. [DOI] [PubMed] [Google Scholar]
- 14. Whitaker A.M., Freudenthal B.D.. APE1: a skilled nucleic acid surgeon. DNA Repair (Amst.). 2018; 71:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krokan H.E., Bjoras M.. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wallace S.S. Base excision repair: a critical player in many games. DNA Repair (Amst.). 2014; 19:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mol C.D., Hosfield D.J., Tainer J.A.. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 2000; 460:211–229. [DOI] [PubMed] [Google Scholar]
- 18. Tell G., Fantini D., Quadrifoglio F.. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell. Mol. Life Sci.: CMLS. 2010; 67:3589–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demple B., Halbrook J., Linn S.. Escherichia coli xth mutants are hypersensitive to hydrogen peroxide. J. Bacteriol. 1983; 153:1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carpenter E.P., Corbett A., Thomson H., Adacha J., Jensen K., Bergeron J., Kasampalidis I., Exley R., Winterbotham M., Tang C. et al.. AP endonuclease paralogues with distinct activities in DNA repair and bacterial pathogenesis. EMBO J. 2007; 26:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson R.E., Torres-Ramos C.A., Izumi T., Mitra S., Prakash S., Prakash L.. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes Dev. 1998; 12:3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramotar D., Popoff S.C., Gralla E.B., Demple B.. Cellular role of yeast Apn1 apurinic endonuclease/3′-diesterase: repair of oxidative and alkylation DNA damage and control of spontaneous mutation. Mol. Cell Biol. 1991; 11:4537–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puri R.V., Reddy P.V., Tyagi A.K.. Apurinic/apyrimidinic endonucleases of Mycobacterium tuberculosis protect against DNA damage but are dispensable for the growth of the pathogen in guinea pigs. PLoS One. 2014; 9:e92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thakur S., Dhiman M., Tell G., Mantha A.K.. A review on protein-protein interaction network of APE1/Ref-1 and its associated biological functions. Cell Biochem. Funct. 2015; 33:101–112. [DOI] [PubMed] [Google Scholar]
- 25. Wilson D.M. 3rd, Takeshita M., Demple B.. Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res. 1997; 25:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill J.W., Hazra T.K., Izumi T., Mitra S.. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001; 29:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vidal A.E., Hickson I.D., Boiteux S., Radicella J.P.. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001; 29:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sidorenko V.S., Nevinsky G.A., Zharkov D.O.. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair (Amst.). 2007; 6:317–328. [DOI] [PubMed] [Google Scholar]
- 29. Luncsford P.J., Manvilla B.A., Patterson D.N., Malik S.S., Jin J., Hwang B.J., Gunther R., Kalvakolanu S., Lipinski L.J., Yuan W. et al.. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair (Amst.). 2013; 12:1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dianova II, Bohr V.A., Dianov G.L.. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001; 40:12639–12644. [DOI] [PubMed] [Google Scholar]
- 31. Liu Y., Prasad R., Beard W.A., Kedar P.S., Hou E.W., Shock D.D., Wilson S.H.. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007; 282:13532–13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bennett R.A., Wilson D.M. 3rd, Wong D., Demple B.. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sukhanova M.V., Khodyreva S.N., Lebedeva N.A., Prasad R., Wilson S.H., Lavrik O.I.. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005; 33:1222–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kiyonari S., Tahara S., Shirai T., Iwai S., Ishino S., Ishino Y.. Biochemical properties and base excision repair complex formation of apurinic/apyrimidinic endonuclease from Pyrococcus furiosus. Nucleic Acids Res. 2009; 37:6439–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kiyonari S., Tahara S., Uchimura M., Shirai T., Ishino S., Ishino Y.. Studies on the base excision repair (BER) complex in Pyrococcus furiosus. Biochem. Soc. Trans. 2009; 37:79–82. [DOI] [PubMed] [Google Scholar]
- 36. Shuman S. DNA ligases: progress and prospects. J. Biol. Chem. 2009; 284:17365–17369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tomkinson A.E., Vijayakumar S., Pascal J.M., Ellenberger T.. DNA ligases: structure, reaction mechanism, and function. Chem. Rev. 2006; 106:687–699. [DOI] [PubMed] [Google Scholar]
- 38. Williamson A., Hjerde E., Kahlke T.. Analysis of the distribution and evolution of the ATP-dependent DNA ligases of bacteria delineates a distinct phylogenetic group ‘Lig E’. Mol. Microbiol. 2016; 99:274–290. [DOI] [PubMed] [Google Scholar]
- 39. Pergolizzi G., Wagner G.K., Bowater R.P.. Biochemical and structural characterisation of DNA ligases from bacteria and archaea. Biosci. Rep. 2016; 36:00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doherty A.J., Suh S.W.. Structural and mechanistic conservation in DNA ligases. Nucleic Acids Res. 2000; 28:4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilkinson A., Day J., Bowater R.. Bacterial DNA ligases. Mol. Microbiol. 2001; 40:1241–1248. [DOI] [PubMed] [Google Scholar]
- 42. Ellenberger T., Tomkinson A.E.. Eukaryotic DNA ligases: structural and functional insights. Annu. Rev. Biochem. 2008; 77:313–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J.Y., Chang C., Song H.K., Moon J., Yang J.K., Kim H.K., Kwon S.T., Suh S.W.. Crystal structure of NAD(+)-dependent DNA ligase: modular architecture and functional implications. EMBO J. 2000; 19:1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Benarroch D., Shuman S.. Characterization of mimivirus NAD+-dependent DNA ligase. Virology. 2006; 353:133–143. [DOI] [PubMed] [Google Scholar]
- 45. Wilkinson A., Smith A., Bullard D., Lavesa-Curto M., Sayer H., Bonner A., Hemmings A., Bowater R.. Analysis of ligation and DNA binding by Escherichia coli DNA ligase (LigA). Biochim. Biophys. Acta. 2005; 1749:113–122. [DOI] [PubMed] [Google Scholar]
- 46. Feng H., Parker J.M., Lu J., Cao W.. Effects of deletion and site-directed mutations on ligation steps of NAD+-dependent DNA ligase: a biochemical analysis of BRCA1 C-terminal domain. Biochemistry. 2004; 43:12648–12659. [DOI] [PubMed] [Google Scholar]
- 47. Wang L.K., Nair P.A., Shuman S.. Structure-guided mutational analysis of the OB, HhH, and BRCT domains of Escherichia coli DNA ligase. J. Biol. Chem. 2008; 283:23343–23352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Srivastava S.K., Dube D., Kukshal V., Jha A.K., Hajela K., Ramachandran R.. NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis: novel structure-function relationship and identification of a specific inhibitor. Proteins. 2007; 69:97–111. [DOI] [PubMed] [Google Scholar]
- 49. Sheng Z.Z., Zhao Y.Q., Huang J.F.. Functional evolution of BRCT domains from binding DNA to protein. Evol. Bioinform. Online. 2011; 7:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bork P., Hofmann K., Bucher P., Neuwald A.F., Altschul S.F., Koonin E.V.. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997; 11:68–76. [PubMed] [Google Scholar]
- 51. Hegde M.L., Theriot C.A., Das A., Hegde P.M., Guo Z., Gary R.K., Hazra T.K., Shen B., Mitra S.. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008; 283:27028–27037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prasad R., Singhal R.K., Srivastava D.K., Molina J.T., Tomkinson A.E., Wilson S.H.. Specific interaction of DNA polymerase beta and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J. Biol. Chem. 1996; 271:16000–16007. [DOI] [PubMed] [Google Scholar]
- 53. Nazarkina ZhK., Khodyreva S.N., Marsin S., Radicella J.P., Lavrik O.I.. Study of interaction of XRCC1 with DNA and proteins of base excision repair by photoaffinity labeling technique. Biochemistry (Mosc). 2007; 72:878–886. [DOI] [PubMed] [Google Scholar]
- 54. Caldecott K.W., McKeown C.K., Tucker J.D., Ljungquist S., Thompson L.H.. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell Biol. 1994; 14:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xia L., Zheng L., Lee H.W., Bates S.E., Federico L., Shen B., O’Connor T.R.. Human 3-methyladenine-DNA glycosylase: effect of sequence context on excision, association with PCNA, and stimulation by AP endonuclease. J. Mol. Biol. 2005; 346:1259–1274. [DOI] [PubMed] [Google Scholar]
- 56. Gary R., Kim K., Cornelius H.L., Park M.S., Matsumoto Y.. Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J. Biol. Chem. 1999; 274:4354–4363. [DOI] [PubMed] [Google Scholar]
- 57. Matsumoto Y. Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:129–138. [DOI] [PubMed] [Google Scholar]
- 58. Lata K., Afsar M., Ramachandran R.. Biochemical characterization and novel inhibitor identification of Mycobacterium tuberculosis Endonuclease VIII 2 (Rv3297). Biochem. Biophys. Rep. 2017; 12:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sidorenko V.S., Rot M.A., Filipenko M.L., Nevinsky G.A., Zharkov D.O.. Novel DNA glycosylases from Mycobacterium tuberculosis. Biochemistry (Mosc.). 2008; 73:442–450. [DOI] [PubMed] [Google Scholar]
- 60. Srinath T., Bharti S.K., Varshney U.. Substrate specificities and functional characterization of a thermo-tolerant uracil DNA glycosylase (UdgB) from Mycobacterium tuberculosis. DNA Repair (Amst.). 2007; 6:1517–1528. [DOI] [PubMed] [Google Scholar]
- 61. Kumar P., Bharti S.K., Varshney U.. Uracil excision repair in Mycobacterium tuberculosis cell-free extracts. Tuberculosis (Edinb.). 2011; 91:212–218. [DOI] [PubMed] [Google Scholar]
- 62. Khanam T., Shukla A., Rai N., Ramachandran R.. Critical determinants for substrate recognition and catalysis in the M. tuberculosis class II AP-endonuclease/3′-5′ exonuclease III. Biochim. Biophys. Acta. 2015; 1854:505–516. [DOI] [PubMed] [Google Scholar]
- 63. Khanam T., Rai N., Ramachandran R.. Mycobacterium tuberculosis class II apurinic/apyrimidinic-endonuclease/3′-5′ exonuclease III exhibits DNA regulated modes of interaction with the sliding DNA beta-clamp. Mol. Microbiol. 2015; 98:46–68. [DOI] [PubMed] [Google Scholar]
- 64. Abeldenov S., Talhaoui I., Zharkov D.O., Ishchenko A.A., Ramanculov E., Saparbaev M., Khassenov B.. Characterization of DNA substrate specificities of apurinic/apyrimidinic endonucleases from Mycobacterium tuberculosis. DNA Repair (Amst.). 2015; 33:1–16. [DOI] [PubMed] [Google Scholar]
- 65. Gu S., Li W., Zhang H., Fleming J., Yang W., Wang S., Wei W., Zhou J., Zhu G., Deng J. et al.. The beta2 clamp in the Mycobacterium tuberculosis DNA polymerase III alphabeta2epsilon replicase promotes polymerization and reduces exonuclease activity. Sci. Rep. 2016; 6:18418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Arrigo C.J., Singh K., Modak M.J.. DNA polymerase I of Mycobacterium tuberculosis: functional role of a conserved aspartate in the hinge joining the M and N helices. J. Biol. Chem. 2002; 277:1653–1661. [DOI] [PubMed] [Google Scholar]
- 67. Kukshal V., Khanam T., Chopra D., Singh N., Sanyal S., Ramachandran R.. M. tuberculosis sliding beta-clamp does not interact directly with the NAD+-dependent DNA ligase. PLoS One. 2012; 7:e35702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gui W.J., Lin S.Q., Chen Y.Y., Zhang X.E., Bi L.J., Jiang T.. Crystal structure of DNA polymerase III beta sliding clamp from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 2011; 405:272–277. [DOI] [PubMed] [Google Scholar]
- 69. Srivastava S.K., Dube D., Tewari N., Dwivedi N., Tripathi R.P., Ramachandran R.. Mycobacterium tuberculosis NAD+-dependent DNA ligase is selectively inhibited by glycosylamines compared with human DNA ligase I. Nucleic Acids Res. 2005; 33:7090–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Srivastava S.K., Tripathi R.P., Ramachandran R.. NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis. Crystal structure of the adenylation domain and identification of novel inhibitors. J. Biol. Chem. 2005; 280:30273–30281. [DOI] [PubMed] [Google Scholar]
- 71. Fisher C.L., Pei G.K.. Modification of a PCR-based site-directed mutagenesis method. BioTechniques. 1997; 23:570–571, 574. [DOI] [PubMed] [Google Scholar]
- 72. Tria G., Mertens H.D., Kachala M., Svergun D.I.. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015; 2:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vajda S., Yueh C., Beglov D., Bohnuud T., Mottarella S.E., Xia B., Hall D.R., Kozakov D.. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017; 85:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 75. Konarev P.V., Petoukhov M.V., Svergun D.I.. Rapid automated superposition of shapes and macromolecular models using spherical harmonics. J. Appl. Crystallogr. 2016; 49:953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Svergun D., Barberato C., Koch M.H.J.. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 77. Ranalli T.A., Tom S., Bambara R.A.. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J. Biol. Chem. 2002; 277:41715–41724. [DOI] [PubMed] [Google Scholar]
- 78. Nandakumar J., Nair P.A., Shuman S.. Last stop on the road to repair: structure of E. coli DNA ligase bound to nicked DNA-adenylate. Mol. Cell. 2007; 26:257–271. [DOI] [PubMed] [Google Scholar]
- 79. Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G.. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 1998; 18:3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taylor R.M., Wickstead B., Cronin S., Caldecott K.W.. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr. Biol. 1998; 8:877–880. [DOI] [PubMed] [Google Scholar]
- 81. Vidal A.E., Boiteux S., Hickson I.D., Radicella J.P.. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001; 20:6530–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dulic A., Bates P.A., Zhang X., Martin S.R., Freemont P.S., Lindahl T., Barnes D.E.. BRCT domain interactions in the heterodimeric DNA repair protein XRCC1-DNA ligase III. Biochemistry. 2001; 40:5906–5913. [DOI] [PubMed] [Google Scholar]
- 83. Bogenhagen D.F., Pinz K.G.. The action of DNA ligase at abasic sites in DNA. J. Biol. Chem. 1998; 273:7888–7893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Small-angle X-ray scattering data have been submitted to the SASBDB database at https://www.sasbdb.org/ with accession numbers as follows: MtbXthA (SASDDD8), MtbLigA (SASDDN9), MtbXthA–MtbLigA complex (SASDEW3), MtbLigA-DNA complex (SASDDV9), MtbXthA–MtbLigA-DNA complex (SASDEX3). 15N/1H HSQC NMR- chemical shift perturbation data have been submitted to the Biological Magnetic Resonance Bank (BMRB) at http://www.bmrb.wisc.edu/ with accession number 28077.