

Abstract
Purpose:
Treatment approaches using Hsp90 inhibitors at their maximum tolerated doses have not produced selective tumor toxicity. Inhibition of Hsp90 activity causes degradation of client proteins including those involved in recognizing and repairing DNA lesions. We hypothesized that if DNA repair proteins were degraded by concentrations of an Hsp90 inhibitor below those required to cause non-specific cytotoxicity, significant tumor-selective radiosensitization might be achieved.
Experimental design:
Tandem Mass Tagged (TMT)-mass spectrometry (MS) was performed to determine the effect of a sub-cytotoxic concentration of the Hsp90 inhibitor, AT13387 (Onalespib) on global protein abundance. The effect of AT13387 on in vitro radio-sensitization was assessed using a clonogenic assay. Pharmacokinetic (PK) profiling was performed in mice bearing xenografts. Finally, the effect of low-dose AT13387 on the radiosensitization of three tumor models was assessed.
Results:
A sub-cytotoxic concentration of AT13387 reduced levels of DNA repair proteins, without affecting the majority of Hsp90 clients. The PK study using one-third of the maximum tolerated dose (MTD) showed 40-fold higher levels of AT13387 in tumors compared to plasma. This low dose enhanced Hsp70 expression in peripheral blood mononuclear cells (PBMC), which is a biomarker of Hsp90 inhibition. Low dose monotherapy was ineffective but, when combined with radiotherapy (RT), produced significant tumor growth inhibition.
Conclusions:
The current study shows that a significant therapeutic ratio can be achieved by a low dose of Hsp90 inhibitor in combination with radiotherapy. Hsp90 inhibition, even at a low dose, can be monitored by measuring Hsp70 expression in PBMC in human studies.
Keywords: AT13387, Hsp90 inhibition, HNSCC, Radiotherapy, Chemotherapy, Radiosensitization
Introduction
Heat shock protein Hsp90 serves as an ATP-dependent molecular chaperone that aids in cellular protein maturation in a highly regulated process and stabilizes its clients during various stressful conditions. Over 600 identified clients of Hsp90 (http://www.picard.ch/downloads/Hsp90facts.pdf) are involved in a multitude of cellular processes (e.g. cell cycle control, proliferative/anti-apoptotic signaling). Several clients are crucial for normal cell growth while many of them are oncogenic (1). Hsp90 often exerts cytoprotective functions by preventing the degradation of proteins involved in the recognition and removal of a diverse array of DNA lesions in normal tissues (2,3). It has been considered as a promising therapeutic target since the realization that several mutated or aberrantly expressed oncogenes depend on Hsp90 for their stabilization (4).
Hsp90 cycles with client proteins in different ways. The classic client proteins, such as proteins involved in DNA repair, undergo cycles of protein•Hsp90 heterocomplex assembly/disassembly to produce complexes that are stabilized such that they can be analyzed by normal biochemical separation techniques. Client proteins that undergo cycles of stable hetero-complex assembly are very unstable in the absence of cycling with Hsp90, and treatment with Hsp90 inhibitors is followed by rapid client protein degradation. Other client proteins are more inherently stable, undergo more dynamic cycles of Hsp90 assembly, and exhibit slower degradation when Hsp90 is inhibited. Thus, the consequences of Hsp90 inhibition may be more severe with classic clients than with dynamically cycling clients (5).
However, this putative chemotherapeutic benefit of Hsp90 inhibitors has not been fully realized for patients in clinical trials. The tolerability profile in human subjects remains unclear, despite Hsp90’s druggability, with data from 20 inhibitors and over 70 clinical trials (completed/ withdrawn/ ongoing) with 11 specific inhibitors. The main difficulty lies in finding a therapeutic window in which efficacy outweighs toxicity. This is likely due to the critical normal functions that Hsp90 regulates in normal cells. In clinical studies where these inhibitors have been used at the maximally tolerated or effective dose levels to inhibit the target, they also accentuate night blindness and severe hematologic, cardiac, and hepatotoxicities (6–9). So far, none of these inhibitors has achieved FDA approval.
Inhibition of Hsp90 activity promotes degradation of client proteins that include a spectrum of oncogenes involved in cell survival, such as EGFR, ErbB2, Src, and cMet as well as molecules involved in DNA repair, such as ATM, ATR, RAD51 and DNA-PK (4,10–13). Degradation of these proteins correlates with the induction of γH2Ax, inhibition of DNA repair, cell death, and profound radiosensitization (14). Several groups have conducted preclinical studies and shown that inhibition of Hsp90 activity would be an ideal approach to sensitize tumor cells to localized ionizing radiation therapy (12,15–21). We have listed 70 clinical trials (Supplementary Table I) and observed only two trials (results not posted) that combined an Hsp90 targeted agent (ganetespib and AT13387) with chemo (cisplatin, paclitaxel, and carboplatin) and radiotherapy. Based on our understanding from published results, we can postulate a possibility of dose-related severe toxicities in human subjects receiving a combination of systemic therapy (both molecularly targeted agents and chemotherapy) and radiotherapy.
Thus, the main objective of the current study was to determine a method of using Hsp90 inhibitors that limits the toxicities previously observed in clinical studies while still sensitizing cancer cells to radiation therapy. As DNA repair proteins are among the Hsp90 clients which would be involved in repair after irradiation, we hypothesized that low concentrations/doses of Hsp90 inhibitors which are not toxic on their own could selectively radiosensitize by causing degradation of some DNA repair proteins. Therefore, we designed a study to determine if DNA repair proteins are affected by low concentrations/doses of Hsp90 targeting agents. Radiosensitization was assessed after we found selective degradation of DNA repair proteins. We found that non-cytotoxic concentrations of AT13387 radiosensitized a panel of HNSCC cell lines. Thus, we combined systemic administration of lower doses of an Hsp90 inhibitor prior to localized radiation therapy in xenograft models and found substantial radiosensitization of tumors, with minimal toxicity. We also examined potential biomarkers for validation of Hsp90 inhibition in an individual patient in future clinical studies.
Our data suggest that while Hsp90 activity inhibition monotherapy may not be effective in patients because of dose-related toxicities, lower, nontoxic doses of these inhibitors may be effective in combination with radiotherapy. This radiosensitization may relate to inhibition of DNA repair in tumors receiving targeted radiation.
Materials and Methods
Materials
AT13387 (Astex Pharmaceuticals, Cambridge, UK) was kindly provided by the CTEP, NIH, USA. EGFR (sc-03), Rad51 (sc-398587), Chk1 (sc-8408), and CyclinB1 (sc-245) antibodies were procured from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for Hsp70 (cat# 4872), ATR (cat# 13934), ATM (cat# 2873), DNA-PK (cat# 12311), Caspase3 (cat# 9662), cMet (cat# 3127), and GAPDH (cat# 2118) were purchased from Cell Signaling Technology (Danvers, MA). γH2Ax (cat# 05–636) antibody was purchased from Millipore. Rad51 (Ab133534), pT1989-ATR (ab227851), PT68-Chk2 (ab85743), and pS345-Chk1(ab47318) were bought from Abcam. pS1981-ATM (NB100–306) was purchased from Novus.
Cell Culture
Late and early passages of several human head and neck squamous cell carcinoma cell lines; UMSCC1, UMSCC10B, UMSCC11B, UMSCC29, UMSCC47, UMSCC74B, UMSCC92 were kindly provided by Dr. Thomas Carey (University of Michigan, Ann Arbor, MI). The SCCVII, MiaPaCa-2, Panc 1, and BxPC3 cell lines were purchased from the American Type Culture Collection (Manassas, VA). All cell lines were grown in RPMI 1640 medium, except for UMSCC92, MiaPaCa-2, and Panc 1 which were grown in DMEM, supplemented with 10% cosmic calf serum. Cell lines were regularly tested for mycoplasma. Though the donating laboratory and ATCC confirmed the genotypes, the genotypes of selected human cell lines were validated by the UM genotyping core using AMPFLSTR Identifier Plus Assay and Gene Mapper Software, Version 5 (Applied Biosystems). A keratinocyte cell line (BS4 mutant) was kindly provided by Dr. Stefan Stoll (University of Michigan, Ann Arbor, MI) and grown in EpiLife™ Medium from Gibco (Catalog # S0125).
Protein identification and relative quantitation by Tandem Mass Tag (TMT) labeling and Multinotch-MS3 Tandem Mass Spectrometry
UMSCC74B cells (1.5×106) were treated with either 100 nM or 3 μM AT13387, and cells were harvested at the beginning of the treatment (0 hours) and after 9 and 24 hours (n=2 at each time point for both concentrations). Protein identification and relative quantitation were performed by the TMT-MS approach as described by several groups (22,23). “The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8” and “The Protein Analysis Through Evolutionary Relationships-v.15.0 (PANTHER)” were used to understand the unbiased biological relevance of significantly (p ≤ 0.05) up or downregulated proteins upon 100 nM AT13387 treatment. The detailed method used to analyze the enriched/overrepresented pathways has also provided in the Supplementary Methods section.
Irradiation
Irradiation was carried out at a dose rate of either 2 Gy (for SCCVII and all HNSCC cell lines or xenografts) or 1.8 Gy (pancreatic cancer cell lines or xenograft)/minute using a Philips RT250 (Kimtron Medical) in the University of Michigan Comprehensive Cancer Center Experimental Irradiation Core. All animals were anesthetized with isoflurane before tumor irradiation and received irradiation only at flank tumors while the rest of the body was shielded from radiation. The dosimetry was carried out using an ion chamber that is directly traceable to a National Bureau of Standards. All irradiation was conducted at room temperature.
Immunoblotting
Cells were plated in 60-mm dishes at a density of 3×105 cells per dish and incubated overnight or to 70 % confluence. The cells were treated with the vehicle (DMSO) or AT13387 (100 nM). Media were changed immediately, and cells were harvested at various time points. To validate TMT-LC-MS/MS data, UMSCC74B cells were harvested at 0, 1, 3, 9, and 24 hours, and the rest of the lines were harvested at 0, 9, and 24 hours following AT13387 treatment. The pellets were washed twice with ice-cold PBS and re-suspended in lysis buffer for 30 min. After sonication, particulate material was removed by centrifugation at 13,000 rpm for 10 min at 4°C. The soluble protein fraction was heated to 95°C for 5 min and then applied to a 4–12% Bis-Tris precast gel (Invitrogen) and transferred onto a PVDF membrane. Membranes were incubated for 1 hour at room temperature in a blocking buffer consisting of 5% BSA and 1% normal goat serum in Tris-buffered saline (137 mM NaCl, 20 mM Tris-HCl (pH 7.6), 0.1% (v/v) Tween 20). Membranes were subsequently incubated overnight at 4°C with the primary antibody in blocking buffer, washed, and incubated for 1 hour with horseradish peroxidase-conjugated secondary antibody. After three additional washes in Tris-buffered saline, bound antibody was detected by enhanced chemiluminescence plus reagent. For quantification of relative protein levels, immunoblot films were scanned and analyzed using Image J 1.32j software. The relative protein levels shown represent a comparison with untreated controls.
Clonogenic cell survival assay
Clonogenic assays were performed using a standard technique described previously (24,25). Briefly, 250–10,000 cells were plated in 60-mm dishes in triplicate, and the next day, cells were treated with 100 nM of AT13387 or 17-AAG. After 24 hours of treatment, cells were irradiated at doses from 0 to 8 Gy. Immediately after IR, the media were changed, and the cells were maintained in normal culture conditions. Eight to fifteen days later, colonies were fixed with acetic acid/methanol (1:7, v/v), stained with crystal violet (0.5%, w/v), and counted using a stereomicroscope. Drug cytotoxicity (surviving drug-treated cells) was measured and normalized to the survival of the untreated control cells. Cell survival curves were fitted using the linear-quadratic equation, and the mean inactivation dose calculated according to the method of Fertil and colleagues (26). The cell survival enhancement ratio was calculated as the ratio of the mean inactivation dose under control conditions divided by the mean inactivation dose after drug exposure. A value significantly greater than 1 indicates radiosensitization.
DNA damage marker γH2Ax focus formation assay
Cells were plated on sterile glass coverslips in 100-mm dishes. The next day, cells were treated with either vehicle (DMSO) or 100 nM AT13387. Twenty-four hours after AT13387 treatment, cells were irradiated (2 Gy), and the culture medium was replaced immediately. The coverslips were removed after 30 mins, 1, 2, and 6 hours post-irradiation, and cells were fixed in 4 % paraformaldehyde in PBS for 15 minutes at room temperature, followed by a wash in PBS. The cells were then permeabilized in 0.5% Triton X-100 for 10 min and blocked in (5% goat serum, 1% BSA, in TBS) for an hour at room temperature. The cells were sequentially incubated with the γH2Ax antibody at 1:1500 dilutions overnight at 4 °C and Alexa Fluor 594-conjugated secondary antibody at 1:300 dilutions for 1 hour at 37 °C in PBS with 1% BSA. After three washes in 1xTBS to remove the non-specific antibody binding, the nuclei were counterstained with 4-diamidino-2-phenylindole dihydrochloride (DAPI). Fluorescence images were obtained using a DS-Fi1 (Nikon, Melville, NY) camera fitted on an Olympus Lx71 microscope. Fields were chosen at random based on DAPI staining. For quantitation of γH2Ax foci and γH2Ax stained cells, at least 100 cells from each of four independent experiments were visually scored for each condition. Cells with 10 or more γH2Ax foci were scored as positive.
The mean number of γH2Ax foci per cell each treatment group (AT13387 only, radiation only, and combination of AT13387 + radiation) were compared via a linear model that included main effect terms for the time elapsed from radiation (minutes) and the treatment group. Interaction terms were not included due to the small sample sizes of the SCCVII and UMSCC74B data sets (n=18). A quadratic time effect was incorporated into the model based on plots of the average γH2Ax foci per cell. Models were fit separately for each cell line. Differences between treatment groups were assessed using two-sided t-tests with a significance level of 0.05. All analysis was conducted in R version 3.6.2.
Cell cycle analysis
UMSCC74B, SCCVII and MiPaCa-2 cells (0.5×106) were grown overnight in 100 mm dishes and treated with 100 nM AT13387 for 24 hours. Cells were harvested with 1x ice-cold PBS and fixed in ice-cold 70 % ethanol for 30 min at 4 °C. Ethanol was removed and fixed cells were washed twice with 1xPBS by centrifuging at 850 g for 5 min. Each sample was incubated with 2 μg/mL RNase for 1 hour at 37°C, followed by staining with propidium iodide (final concentration 1.8 μg/mL) at 4°C in the dark for 2 hours. Cell acquisition and data analysis were done by ZE5 Cell Analyzer (Bio-Rad) and Everest Software respectively. In each case, 10,000 cells were analyzed in the Flow Cytometry Core of the University of Michigan Rogel Cancer Center.
Pharmacokinetic and pharmacodynamics (PK/PD) analysis
PK/PD parameters were determined after 40 mg/kg intraperitoneal (ip) administration of AT13387 to athymic BALB/c mice bearing subcutaneous UMSCC74B xenografts. In one group, blood and tumor samples were collected at 0.1, 2, 24, and 96 hours after the first administration. In another group, the second dose of AT13387 was administered 96 hours later, and blood and tumor samples were drawn at 98, 120, 192 hours from first ip injection. Blood was drawn from the heart into a heparinized syringe. Samples were stored at −80°C until analysis. Both tumor tissue homogenates and plasma were processed in the same manner for liquid chromatography-mass spectrometry (LC-MS/MS) at the core facility of the University of Michigan. A detailed protocol has been provided in the Supplementary methods section.
Isolation of peripheral blood mononuclear cells (PBMC)
Mouse PBMCs were isolated from whole blood by density gradient centrifugation using Ficoll as described elsewhere (27,28). Briefly, 2 ml of diluted blood (1:1, in phosphate buffer saline) (1xPBS) blood was carefully layered over 2 ml Histopaque®−1077 and centrifuged at 3,000 rpm for 30 min without brake and acceleration. After centrifugation, the interphase was collected and centrifuged again at 1,800 rpm for 10 min. Subsequently, the pellet was resuspended and centrifuged at 1,000 rpm for 10 min, two times with 1xPBS. Finally, the pellet was assessed for immunofluorescence as well as immunoblotting.
Tumor growth studies
To generate tumor xenografts, 1×106 UMSCC74B or MiaPaCa-2 cells were transplanted into the flanks of athymic nude Fox n1/nu mice (Harlan Laboratories, Indianapolis, IN). SCCVII cells were transplanted into the flanks of immune competent C3H/HeJ mice. When the tumors reached a volume of ~100 mm3 (approximately 5–10 days), the mice were randomized into four groups (one vehicle control and three experimental groups for AT13387, IR, and the combination of both AT13387 and IR), and treatment was initiated. Tumors (> 100 mm3) were separately harvested at various time points for pharmacodynamic studies. Based on the PK/PD data, we further reduced the dose of AT13387 to 30 mg/kg, and the mice were treated twice a week (Monday and Thursday). In the irradiation-only arm, UMSCC74B and SCCVII tumors were irradiated with 2 Gy fractions from Monday to Friday for two weeks (total 2 × 5 × 2 = 20 Gy). MiaPaCa-2 tumors were irradiated with 1.8 Gy fractions from Monday to Friday for two weeks due to their higher radiosensitivity (total 1.8 × 5 × 2 = 18 Gy). In the combination group, the mice were treated with AT13387, 2 hours prior to irradiation (only on Monday and Thursday). Body weight and other signs of toxicity were monitored daily, and the tumor volumes were recorded using calipers. Tumor volumes were calculated as follows: volume (mm3) = (LxW2) / 2. Animals from all four groups were euthanized when tumors reached 800 mm3 volume. Time to second tumor doubling (TD2) was calculated as the time in days from the start of treatment to the second instance of tumor doubling. TD2 was summarized using the Kaplan-Meier method, and survival curves were compared using the log-rank test (29,30). The effects of treatment on Hsp70 were analyzed by immunoblotting, immunohistochemistry, and immune fluorescence.
Tissue Microarray (TMA)
A TMA was used to study multiple human cancer/normal tissues in a single assay. Each tumor sample from all three xenograft models was collected at the indicated time point (6 hours following irradiation or 30 hours following AT13387 treatment in the combination arm). The TMA was developed, stained, and analyzed at the Research Histology and Immunohistochemistry Core, University of Michigan Rogel Cancer Center. To understand the in vivo mechanisms of radiosensitization, γH2Ax, Rad51, pChk1/2, pATM, and pATR were analyzed using these TMA samples. Statistical analysis (One-way ANOVA P<0.05, ** P<0.001, *** P<0.0001) was used to measure the level of significance.
Immunostaining
The Tissue and Histology Core of the University of Michigan Rogel Cancer Center and the Pathology Core for Animal Research at the University of Michigan assisted in preparing specimens for immunohistochemistry. After slides were deparaffinized in xylene and rehydrated using serial ethanol dilutions, antigen unmasking was performed by immersing slides in citrate buffer (pH 6.0) for 20 min at high pressure and temperature using a pressure cooker. Slides were then washed in PBS, blocked for 1 hour, and incubated in the primary antibody at 4°C overnight. Slides were then washed again in PBS, incubated in secondary antibody for 1 hour, rewashed, and prepared with a coverslip after a drop of Pro-Long Gold antifade reagent with 4′,6-diamidino-2-phenylindole (Molecular Probes) was added to each sample. Fluorescence images were acquired using DS-Fi1 (Nikon, Melville, NY) camera fitted on an Olympus IX-71 microscope.
Ethics Statement
Here we confirm that all the animal studies conducted here were approved by the University Committee on Use and Care of Animals (UCUCA) of the University of Michigan (Protocol # PRO00006797).
Results
Mechanisms of AT13387 mediated radiosensitization
We began by determining the effect of a cytotoxic concentration (3 μM) of AT13387 (treated cells failed to form colonies at this concentration) on the global protein abundance using high-resolution TMT-LC-MS/MS in UMSCC74B cells after a 24-hour exposure (9-hour data: Supplementary Table II). This concentration and time were selected based on the peak plasma concentration observed in a Phase I clinical study (26). A total of 5883 proteins with high confidence peptide-spectrum match (PSM) was detected. A standard volcano plot was developed using a total of 5086 proteins which show a low false discovery rate (FDR) (Figure 1). This analysis showed that the abundance, defined as log 2 of treated vs untreated condition when the limit was set to ≤ −0.5 or ≥ 0.5-fold, of over 20% of the proteins was significantly reduced. These proteins belong to a multitude of cellular processes including those involved in the regulation of cell survival (e.g. EGFR), proliferation (Transcription factor jun-B), signaling (Tyrosine-protein kinase JAK1), cell cycle (Cyclin-dependent kinases), transcription (CCAAT/enhancer-binding protein beta), genome/mitochondrial integrity (histone proteins), and DNA repair. This analysis also showed significant upregulation of over 15% of detected proteins involved in protein degradation machinery (Ubiquitin-conjugating enzyme E2), cell cycle progression (Aurora Kinase A), cell stress (e.g. Hsp co-chaperones) and cell death pathway (e.g. annexins, DnaJ proteins). The change in the global proteome at 3 μM AT13387 in UMSCC74 cells is listed in Supplementary Table II. The magnitude observed on protein levels (total 2145 significantly affected protein out of 5086) suggests that the 3 μM AT13387 concentration would not be selective. Therefore, when given at a dose producing such drug levels in patients, it seems unlikely that the cumulative effect of the drug would be selective for cancer treatment.
Figure 1. Effect of a cytotoxic concentration of AT13387 on the global proteome of an HNSCC cell line, UMSCC74B.
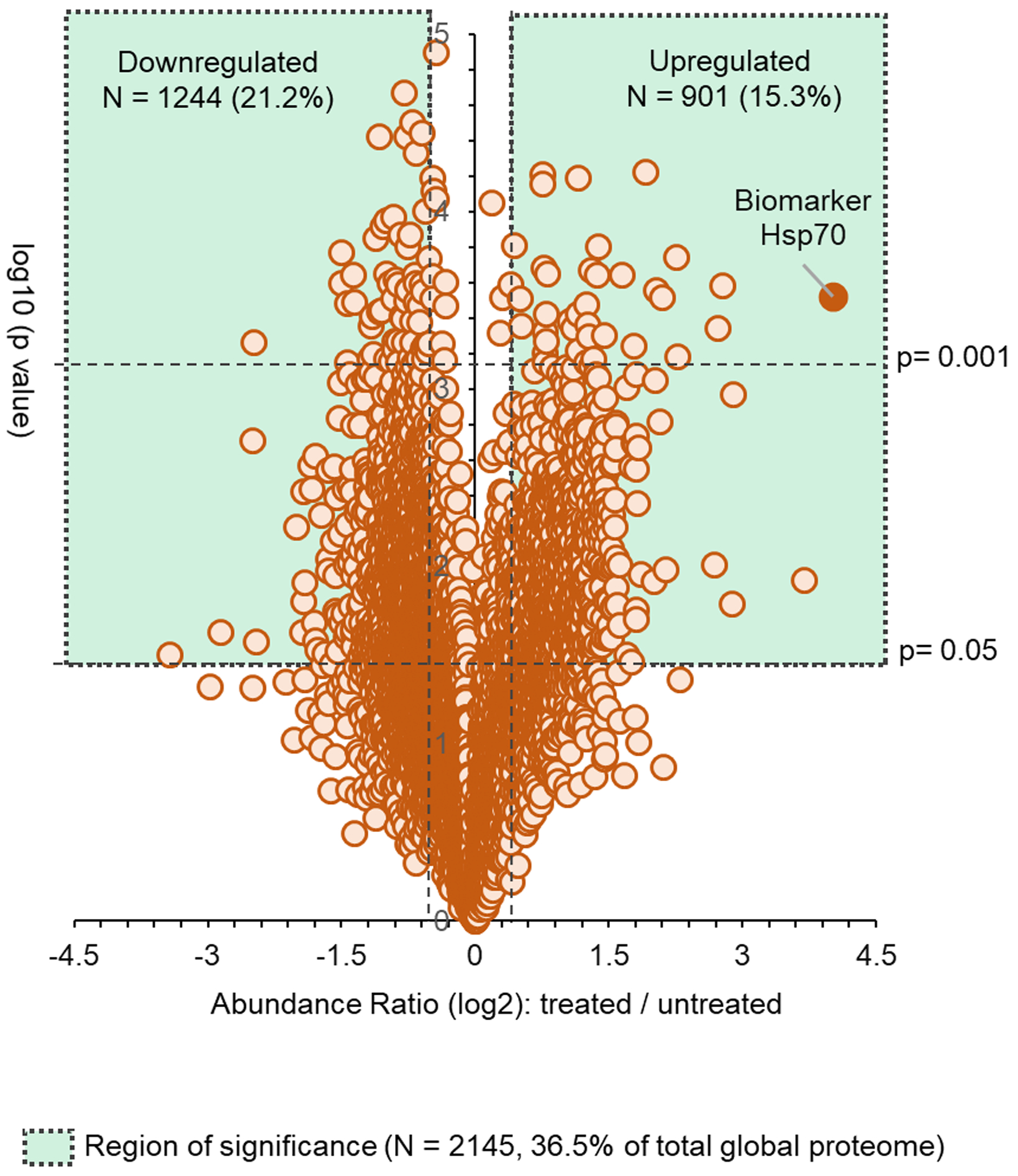
UMSCC74B cells were treated with a cytotoxic concentration (3 μM) of AT13387 for 24 hours. Whole-cell lysates were analyzed using TMT-LC-MS/MS method, and a standard volcano plot was developed to showcase the global proteome status. n=2 for each time point (0, 9, 24-hours). Only 24-hour data are shown.
Next, we set out to determine if a correlation exists between the inhibition of Hsp90 activity and the upregulation in Hsp70 protein expression. Hsp70 is a co-chaperone protein and its upregulation is known to serve as a biomarker of Hsp90 inhibition. UMSCC74B cells were treated for 2 hours with a range of AT13387 concentrations from 1 nM to 10 μM. Cell lysates were resolved on SDS-PAGE and change in the Hsp70 protein level was assessed by immunoblotting (SF1). We noted an approximately 2-fold increase in the Hsp70 protein levels at 100 nM AT13387 concentration. This effect was only minimally enhanced at the higher concentrations. This concentration is 30-fold less than the Cmax achieved in a Phase I clinical study (31), and is expected to be safe in patients. Also, a 24-hour exposure to this concentration resulted in greater than 70% survival of normal keratinocytes (SF9), demonstrating a relative lack of toxicity. Therefore, we repeated the proteomic experiment as described in Figure 1 using 100 nM AT13387. We hypothesized that this concentration would affect only the classic clients of Hsp90 that are primarily regulated by Hsp90 chaperone activity.
As shown in Figure 2, we found significant changes in only 3.6% of the global proteins (211 out of 5086 proteins, p< 0.05) at the 100 nM concentration at 24 hours post-treatment in UMSCC74B cells. Hsp70 was the most upregulated protein at this concentration as well (Log 2 of fold change: 1.86), confirming data shown in SF1, that this low concentration inhibits Hsp90 chaperone activity. (The entire list of identified proteins in 100 nM AT13387 treated UMSCC74 cell lysate is also provided in the Supplementary Table II.) The amplitude of the effects of 100 nM AT13387 on the global proteome was significantly lower than that of 3 μM AT13387. Interestingly, except for Hsp70, the abundance of any other protein did not reach beyond the 1.5-fold abundance (log scale) level. The majority of proteins either up- or down-regulated are known to be associated with several biological processes cell-cell adhesion, protein folding, DNA damage/repair, chromatin remodulation, transcription, RNA editing/splicing, mitochondrial function, oxidative stress, or immunomodulation pathways. We demonstrated enriched biological processes (genes-to-terms) associated with significantly affected (log2 fold change ≥ 0.5 and p ≤ 0.05) proteins in Supplementary Table II. A higher enrichment score (ES) for a group indicates that the group members are involved in more important (enriched) roles. Genes associated with the term ‘DNA damage or repair’ appeared later in the list (Cluster 8, 17, 19, etc.). However, several other proteins in this list are also indirectly involved in DNA damage or repair processes especially those enriched at the top. A functional annotation table for significantly downregulated proteins is included as Supplementary Table IV. In Figure 2B, selected up or downregulated protein (levels were changed either significantly or insignificantly in TMT-LC-MS/MS) are illustrated via a heat map. These include proteins associated with DNA repairs such as ATR, ATM, DNA-PK, and RAD50, cell cycle (e.g. cyclin B), survival/proliferation (e.g. EGFR, cMet) and death (caspase 3, LC3B I/II). The effect of the low concentration of AT13387 on these proteins in UMSCC74B, SCCVII, and MiaPaCa-2 cell lines was validated by immunoblotting (Figure 2C i, and SF2) and was compared with that of an immortal normal tissue cell line, Keratinocytes (Figure 2C ii).
Figure 2. A sub-cytotoxic concentration of AT13387 promotes selective inhibition of the DNA repair process.
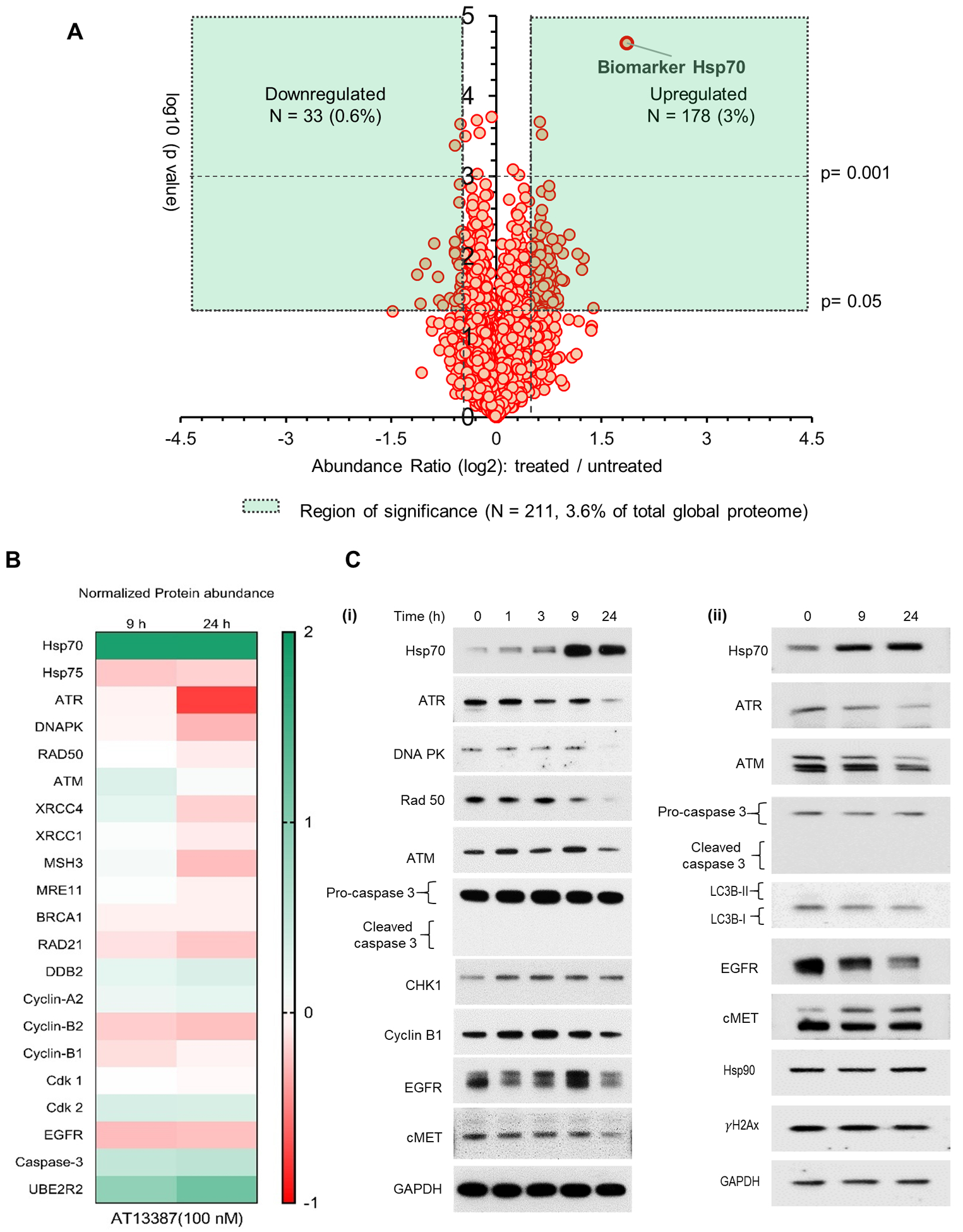
Whole-cell lysates from UMSCC74B cells treated with AT13378 were prepared and analyzed using the TMT-LC-MS/MS. (A) A volcano plot was developed using the protein abundance values obtained from the analysis. The demonstrated plot shows the cumulative effect of a low concentration of AT13387 (100 nM) on the global proteome of UMSCC74B cells following a 24-hour exposure. (B) A heat map was generated using log2 values of fold change (treated vs untreated) of DNA damage and repair associated proteins following 9 and 24 hours of AT13387 treatment. (C) Abundance of indicated proteins was validated by Immunoblotting in (i) UMSCC74B and (ii) Keratinocytes. n=2 for TMT-LC-MS/MS at each time point (0, 9, 24 hours) and n=3 for immunoblotting.
At this low concentration of AT13387, chaperone function of Hsp90 was affected. But the abundance of Hsp90 clients involved in the regulation of cell survival or cell cycle progression such as cyclin B, AKT, cyclin A2, cyclin B1, Cdk2, or Cdk7 was either minimally or not affected. Also, pro-apoptotic markers such as Caspase 3 or 7 both remained unchanged at this concentration suggesting that 100 nM AT13387 may not affect cell-cycle progression or cause apoptotic cell-death. We anticipated that this concentration would radiosensitize cells due to its significant effect on several proteins involved in promoting DNA damage and repair (32).
A sub-toxic concentration of AT13387 sensitizes HNSCC cells to IR
The effect of AT13387 and 17-AAG on the in vitro radiation sensitivity was assessed. The effect of AT13387 on the enhancement ratio in eight HNSCC cell lines and three pancreatic cell lines is tabulated in Figure 3A, and the radiosensitizing effect of 17-AAG in UMSCC74B, SCCVII, and MiaPaCa-2 is tabulated in SF3C. 100 nM AT13387 treatment caused radiosensitization in all 11 cell lines used in this study, irrespective of the presence of multiple driver mutations. The data shown in SF3A–B suggest that 100 nM AT13387 promotes better radiosensitization than 30 nM. A 24-hours exposure of 30 or 100 nM AT13387 resulted in over 80 and 70 % survival of immortalized keratinocytes, respectively, indicating the modest effect of the drug as a single agent (SF9), demonstrating a relative lack of toxicity. Though 100 nM concentration of both AT13387 and 17-AAG showed radiosensitization effect in all tested cancer cell lines, 17-AAG was cytotoxic in normal tissue Keratinocytes. Particularly, UMSCC74B, UMSCC10B, and UMSCC1 showed enhancement ratios of 2.87 ± 0.52, 2.3 ± 0.48, and 1.8 ± 0.24, respectively. A moderate sensitizing effect of AT13387 was observed in SCCVII, UMSCC29, UMSCC92, and UMSCC47 (enhancement ratios of 1.7 ± 0.3, 1.7 ± 0.42, 1.67 ± 0.37 and 1.4 ± 0.22, respectively) (Figure 3A). Line plots demonstrate the survival fraction of UMSCC74 cells (Figure 3B) and SCCVII cells (Figure 3C) following increasing doses of IR alone or in combination with 100 nM AT13387. Altogether, the data suggest that a sub-toxic concentration of AT13387 can cause radio-sensitization regardless of the specific oncogenic driver(s) present in the cell lines.
Figure 3. A subcytotoxic concentration of AT13387 sensitizes a panel of HNSCC and pancreatic cell lines to radiation.
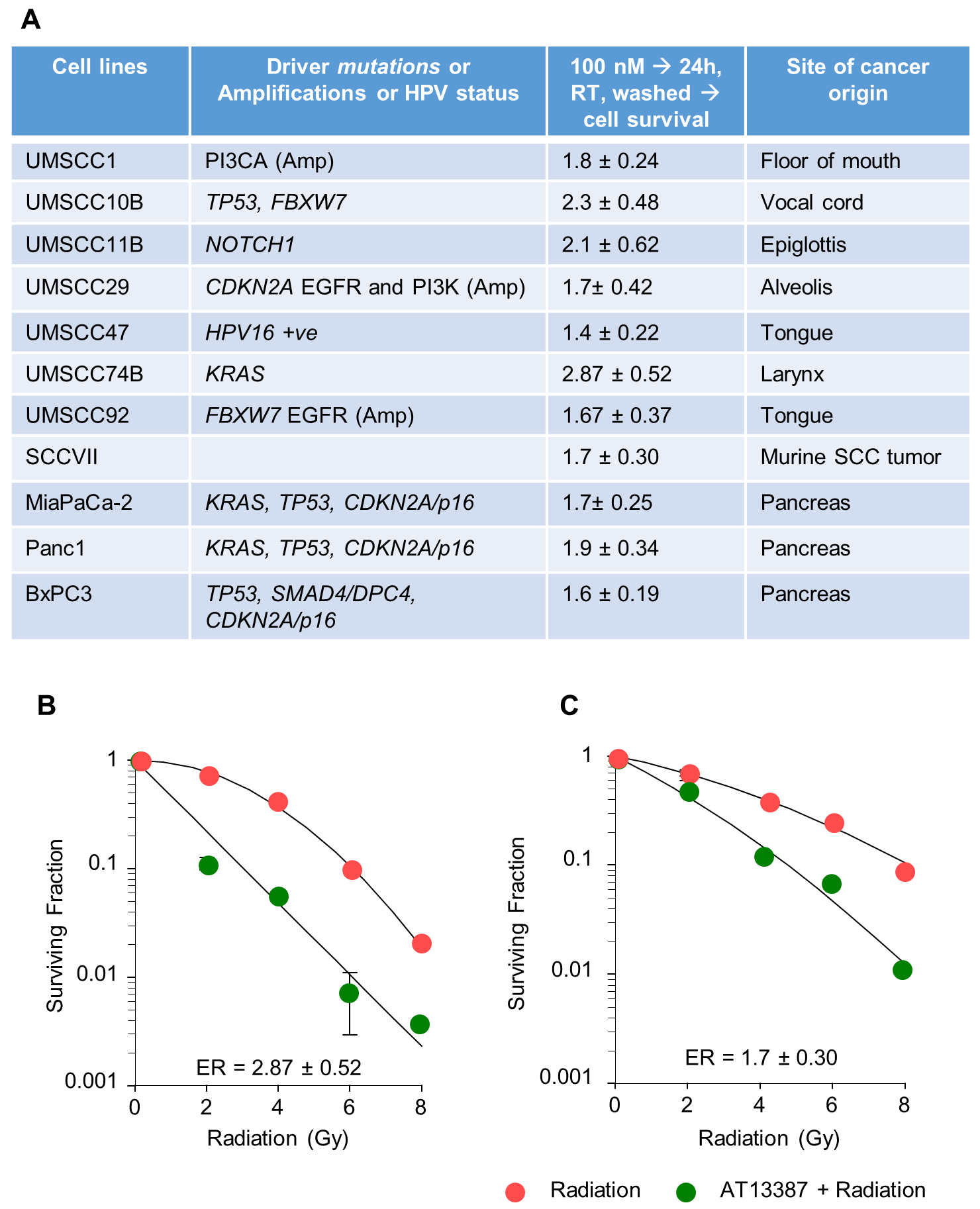
8 HNSCC and 3 pancreatic cell lines with different genetic backgrounds were treated with AT13387 (100nM) for 24 hours, Radiation (2, 4, 6, 8 Gy), or a combination of AT13387 and radiation. Cells were washed immediately after irradiation and incubated at 5% CO2 at 37 °C until detectable colonies (>50 cells) were observed in the untreated group. Colonies were fixed, stained using crystal violet solution, and counted. Enhancement ratios (ER) for the combination of radiation and AT13387 treatment relative to radiation alone were calculated. Genetic backgrounds and respective ER are tabulated (A). Representative survival curves are shown for UMSCC74B (B) and SCCVII (C) cells.
Combination of AT13387 and IR results in enhanced DNA damage (γH2Ax foci formation) and delays in repair (γH2Ax foci clearance)
Upon determination that a sub-toxic concentration of AT13387 radiosensitizes HNSCC and pancreatic cancer cell lines, we assessed whether radiation-induced DNA damage repair was affected by a sub-toxic concentration of AT13387. This was done by evaluating the γH2Ax foci formation, a biomarker for DNA damage, using an immunofluorescence assay. Representative images were captured at 6 hours following irradiation (or 30 hours following AT13387 treatment). In both the UMSCC74B and SCCVII cells, we observed a significant enhancement in γH2Ax foci formation in the cells treated with AT13387 before irradiation (Figure 4 A–B), which was maintained for up to 6 hours after radiation (Figure 4C) but not at 24 hours (SF10). The expected mean number of γH2Ax foci per cell (Figure 4D) was approximately 11.4 (p = 0.009; 95% CI 3.3–19.5) and 13.8 (p = 0.016; 95% CI 2.9–24.6) higher in the combination treatment group for SCCVII and UMSCC74B, respectively, when compared to the AT13387 only treatment group. The expected mean number of γH2Ax foci per cell was approximately 6.3 (p = 0.113; 95% CI −1.7–14.4) and 4.3 (p = 0.410; 95% CI −6.6, 15.1) higher in the combination treatment group for SCCVII and UMSCC74B, respectively, when compared to the radiation only treatment group. Immunoblotting data also confirmed a 2–3-fold increase in γH2Ax induction in the combination treatment group and a longer time to resolve the damage (SF4). These results suggested that exposure to sub-toxic concentration of AT13387 promoted greater DNA damage and delayed the irradiation-induced repair response.
Figure 4. Effect of AT13387 treatment on radiation-induced γH2Ax foci formation.
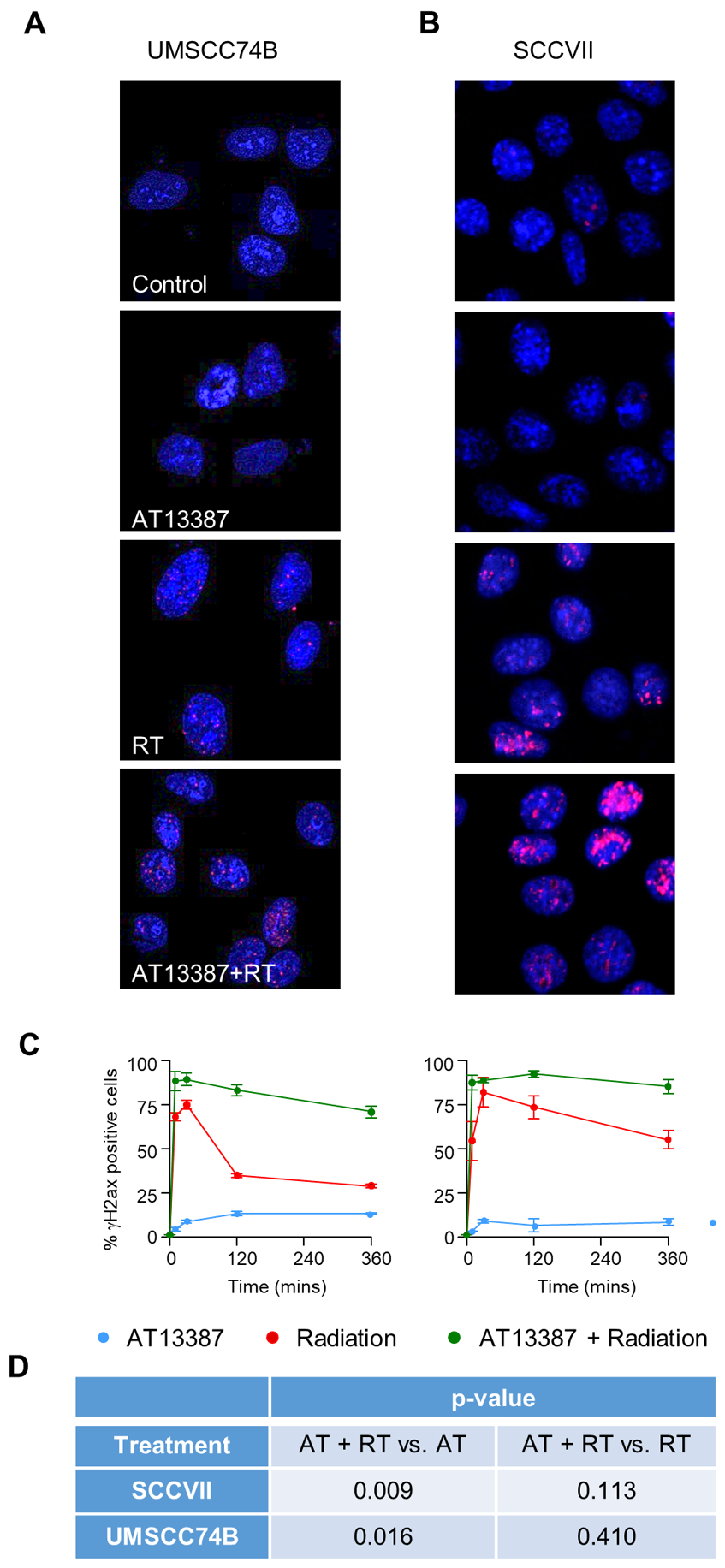
Cells were treated with AT13387 (100 nM), radiation (2 Gy) or a combination as described in Methods. For DNA repair analysis cells were fixed at different time points after irradiation. γH2Ax foci were visualized by immunofluorescence staining and the nucleus was stained with DAPI. Shown here are representative images of control, AT13387, and AT + RT group 6 hours post-radiation for UMSCC74B (A) and SCCVII (B) cells. Images were captured, cells with >10 γH2Ax foci were scored, and the percentage of positive cells is plotted as a function of time (C). Differences between treatment groups were assessed using two-sided t-tests with a significance level of 0.05. All analysis was conducted in R version 3.6.2 (D).
Another explanation for inhibition of the DNA repair process could be cell cycle redistribution as inhibition of Hsp90 is known to cause either G1 or G2/M cell cycle arrest. The mass spectrometry data suggested an insignificant or minimal effect on the proteins associated with cell-cycle regulation following a sub-toxic exposure to AT13387. Nevertheless, we investigated the effect of 100 nM AT13387 on the cell-cycle distribution by assessing DNA content using flow cytometry. UMSCC74B (WT TP53), SCCVII, MiaPaCa-2 (mutant TP53) cells were treated with 100 nM AT13387 or vehicle, and cell cycle analysis was performed at 24 hours using flow cytometry. The UMSCC74B cell line showed an increase in the G1 population (11%, p< 0.05), whereas a minimal change in the G1 phase, was observed in the SCC VII and MiaPaCa-2 cells (p> 0.05). This difference could be due to the difference in TP53 status. However, this treatment had minimal effect on the G2/M populations in all the cell lines, which is the most radiosensitive phase of the cell cycle, suggesting that the radiosensitization is largely independent of cell cycle redistribution by low concentration of AT13387 treatment (SF5).
Pharmacokinetic and biomarker studies to optimize dose for combination studies.
These in vitro data suggested that a sub-toxic concentration of AT13387 can cause significant radiosensitization of cancer cells. To translate these findings for animal studies, we wanted to determine a safe dose of AT13387 that can also be monitored using upregulation in the Hsp70 level, which is an established biomarker of Hsp90 inhibition (26). Therefore, we conducted pharmacokinetic and biomarker studies to evaluate if a lower than maximally tolerated (MTD) dose of AT13387 can reach xenograft tumors and if such a dose can inhibit Hsp90 activity as monitored by upregulation of Hsp70 protein levels in the tumor cells. We also evaluated if the effect of such a low concentration on Hsp90 activity can be monitored by Hsp70 upregulation in the peripheral blood mononuclear cells (PBMCs).
We began by assessing the pharmacokinetic effects of 40 mg/kg AT13387, given as either single or double i.p. injections administered at 0 and 96 hours, in plasma and UMSCC74B xenograft tumors. About half of the maximally tolerated dose (i.e. 90 mg/kg) was selected in this study to determine if the accumulation of AT13387 in the tumor would be enough for radiosensitization. The resulting concentrations of AT13387 in the plasma and tumor were determined at 0, 0.5, 2, 24, 96, 98, 120, 196 hours post-injection. A three-compartment model with first order rate of absorption from the peritoneum into the plasma with transfer rate constants from the plasma to tumor provided an excellent fit (R2= 0.998 [plasma], R2= 0.994 [tumor]) to the individual observed data (Supplementary Table III). The median transfer rate constant from plasma to the tumor was 2-fold higher than that from tumor to plasma implying tumor retention. The model predicted median [min, max] Cmax in plasma (Tmax= 0.4 hour) and tumor (Tmax=3.2 hour) were 299 [27.6, 354.5] ng/mL and 3003 [1426, 5301] ng/g, respectively. Similarly, the tumor median [min, max] AUC0-infinity was 90733 [24336, 168448] ng*hour/g, which was at least 40-fold more than the plasma median [min, max] AUC0-infinity: 2263 [168.8, 31787] ng*hour/mL) (Figure 5A and Supplementary Table III). The results show that the xenograft tumor exposures achieved even at less than half the MTD dose of AT13387 were higher than that required for radiosensitization and that AT13387 was cleared rapidly from the plasma and retained for a longer duration in the tumor as shown by other groups.
Figure 5. The pharmacokinetic and pharmacodynamic effects of AT13387.
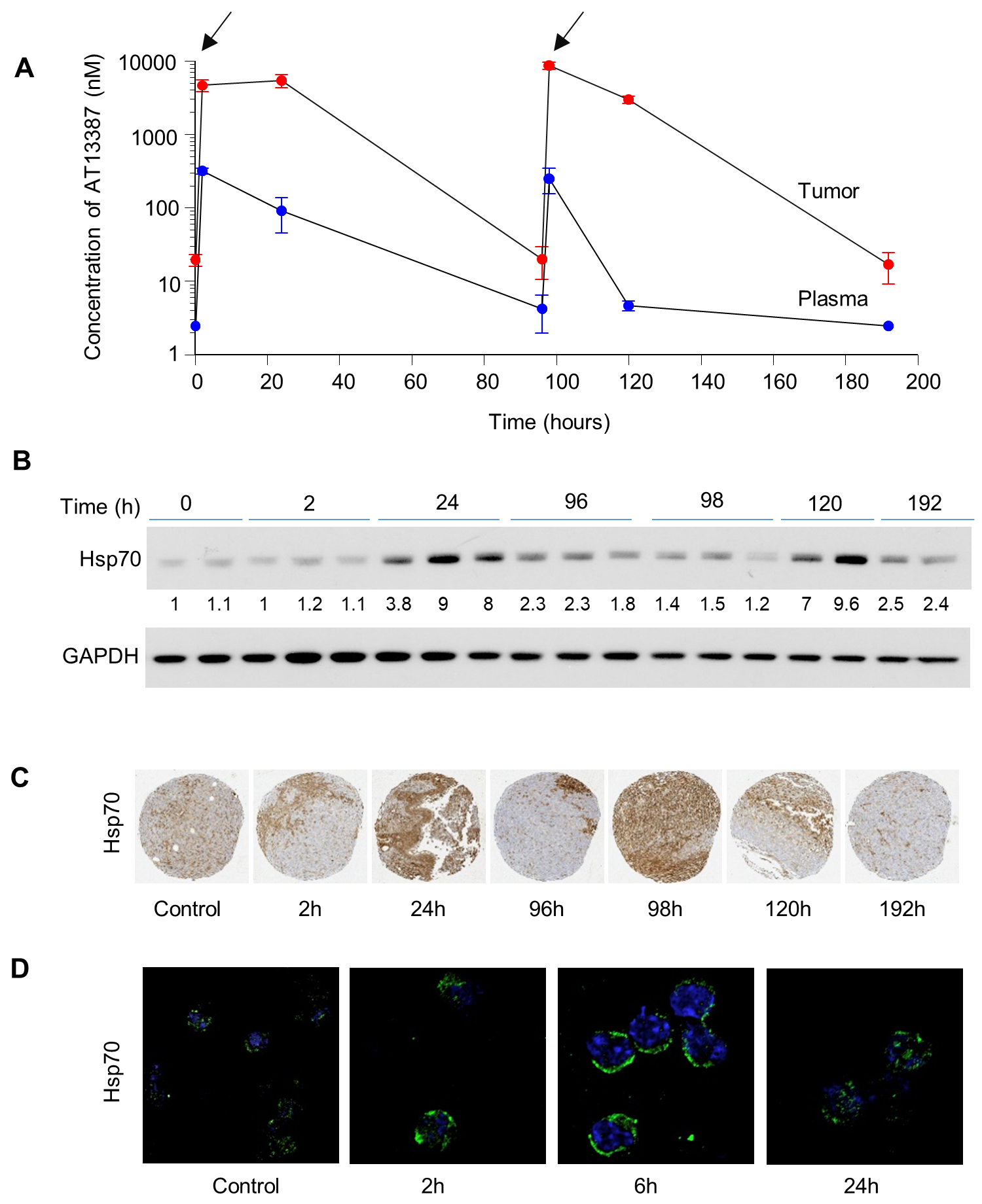
Mice bearing UMSCC74B xenografts were injected with 40 mg/kg dose of AT13387 at time 0 and again at 96 hours (marked by arrows). Mice were euthanized at 0, 2, 24, 96, 98, 120, and 196 hours after the first injection. The concentration of AT13387 was assessed in the plasma and xenografts at each time point (n=3) as described in the methods (A). Effect of AT13387 treatment on Hsp70 protein expression was determined using immunoblotting (B) and immunohistochemistry (C) in the xenograft tumors and by immunofluorescence in the PBMC cells (D).
Following the determination of the pharmacokinetic profile of AT13387, we sought to determine the duration for which AT13387 can exert its effects in vivo. To investigate these effects, a single 40 mg/kg dose of AT13387 was administered at 0 hours, and a second 40 mg/kg dose at 96 hours, to mice bearing UMSCC74B xenografts. The expression of Hsp70 was analyzed in the tumor at the various time points as previously described. Following administration, pharmacodynamics studies showed that the expression of chaperone protein Hsp70, our biomarker for Hsp90 activity, was upregulated after AT13387 treatment (Figure 5B). This result was confirmed by densitometry analysis, which showed an 8-fold upregulation of Hsp70 expression at 24 and 120 hours (Figure 5B). Nearly 2 to 5-fold increased Hsp70 expression was demonstrated using immunohistochemistry (Figure 5C). This upregulation suggests that AT13387 treatment showed the desired inhibitory effect of Hsp90 inhibition in vivo.
Determination of Hsp90 inhibition in PBMCs
In this study, we were also interested in establishing a biomarker to quantify Hsp90 inhibition by a low dose of inhibitor, which we expected to be non-toxic systemically. It has been observed that Hsp90 inhibition correlates with an increase in Hsp70. Using immunofluorescent staining (Figure 5D) and immunoblotting (SF6), we determined if a dose equaling one third of MTD could enhance Hsp70 expression in mouse PBMCs. At least 2-fold induction of Hsp70 in PBMC isolated from the blood of treated mice was observed. This induction in Hsp70 correlated well with the radiosensitizing concentration determined from in vitro experiments (SF1) and suggests that a less than half the MTD dose will be enough for inhibiting Hsp90 activity, followed by radiosensitization of HNSCC cell lines. It also suggests the potential usefulness of Hsp70 from blood cells as a clinical biomarker in future studies.
Pre-clinical assessment of radio-sensitization by a sub-toxic dose of AT13387 in a Pancreatic cancer, a Head & Neck human and syngeneic models
Pre-clinical experiments were conducted to determine the radiosensitizing effects of low dose AT13387 against several xenograft models. Based on the PK/PD data, we further reduced the dose of AT13387 to 30 mg/kg. Mice were randomized into four groups and treated as described in the Methods sections.
As expected, we observed a significant effect on both the tumor growth rate (Figure 6A, D, G) and the tumor doubling time (Figure 6B, E, H) in the combination treatment group. In the data displayed as a time to second tumor doubling (TD2), we observed a minimal effect on tumor doubling time with low dose AT13387 as monotherapy in all the three tumor models used in this study. Radiotherapy alone was minimally effective in UMSCC74B and SCCVII tumor models but had a substantial effect in MiaPaCa-2 tumors. The AT13387 treatment as monotherapy or in combination with radiation was well tolerated (Figure 6C, F, I).
Figure 6. Effect of low-dose AT13387 treatment on radiosensitivity in vivo.
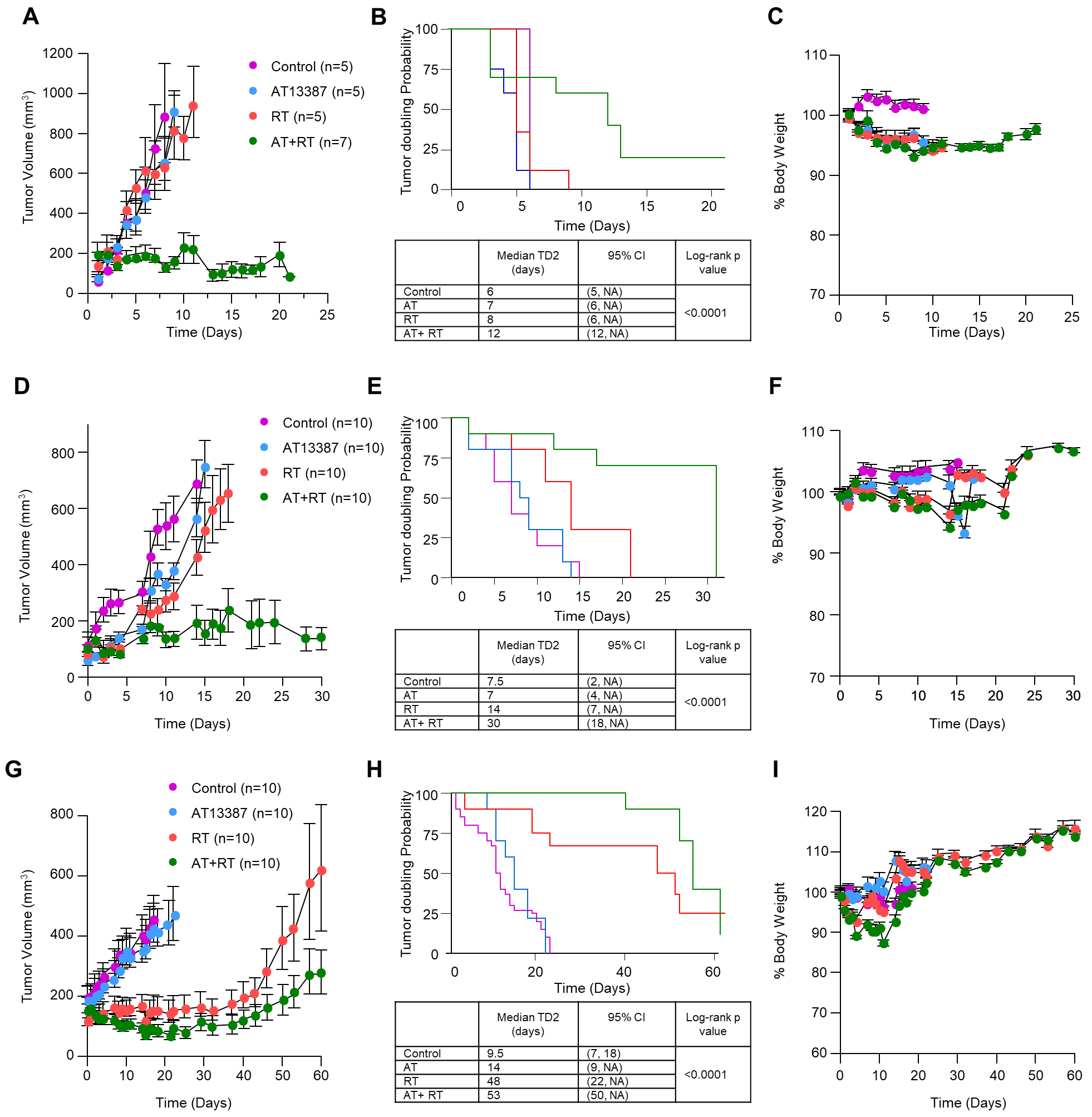
The effect of treatment on UMSCC74B (top panel), SCCVII (middle panel), and MiaPCa2 (bottom panel) tumors is shown. Mice bearing 100 mm3 or larger tumors were treated with AT13387, Radiation, or a combination as described in methods. Tumor volume was measured three times per week, and weight was recorded daily. Shown here are the tumor volumes as a function of time (A, D, and G), median times to second tumor doubling (TD2) along with log-rank p-values after initiation of treatment (B, E, and H) and the effect of treatment on mouse body weight (C, F, and I). Control: purple; AT13387/AT: Blue; Radiation/RT: Red; AT+RT: Green.
To explore the mechanism of AT13387 mediated radiosensitization tumors were harvested following either a single dose of 30 mg/kg AT13387 at 24 h, 2 Gy fraction of radiotherapy at 6h or a combination. A tissue microarray (TMA) was constructed and the change in DNA damage-repair markers such as γH2Ax, pChk1/2, Rad51, pATM, and pATR was assessed by counting the percent positive cells in the five random fields. The statistical analysis was performed using one-way ANOVA analysis. Representative images (SF7 and SF8) suggested that a single low dose of AT13387 and radiation-induced DNA damage at varying degrees across the xenografts. The increase in γH2Ax levels showed a trend in the combination treatment more than the single treatment condition across the tumor types. In the SCCVII syngeneic tumor model, this increase was statistically significant. A similar pattern (non-significant) was observed in the pChk1/2, pATM, and pATR levels in the combination treatment group (SF8 A–D).
Discussion
In this study, we evaluated a combination treatment regimen of AT13387 at a reduced dose to minimize potential off-target effects and yet preserve its radiosensitizing properties in preclinical models. We have found that an equivalent concentration (3 μM) to the plasma Cmax of AT13387 achieved upon an MTD dose to a human is non-selective in vitro in terms of its effect on Hsp90 client proteins. A substantial reduction in the in vitro concentration of AT13387 to 100 nM revealed a relatively selective effect on a sub-set of Hsp90 clients that are involved in DNA repair while not affecting most other Hsp90 client proteins.
Based on the effect of 100 nM AT13387 treatment on DNA repair proteins detected by the global proteome analysis, this concentration was used for each in vitro experiment. This concentration inhibited the Hsp90 chaperone function, causing nearly 2-fold upregulation of Hsp70 protein levels. Although a minimum percent inhibition of the Hsp90 activity requirement for radiosensitization remains unknown, we found that 100 nM AT13387 had minimal effect on the glucocorticoid receptor folding in vitro (SF11). Due to these small effects on clients, 100 nM AT13387 had minimal effect on the anti-proliferative or pro-apoptotic pathways, but reduced DNA repair proteins such as ATR, DNA-PK, and RAD51 levels. These data led to an assessment of the radiosensitizing potential of AT13387 against a panel of cancer cell lines with varying genetic backgrounds. This reduced AT13387 concentration remained a potent radiosensitizer across the 8 HNSCC and 3 pancreatic cancer cell lines used in this study. However, normal tissue keratinocytes remained unaffected at this low concentration. These in vitro studies also suggest the primary mechanism of radiosensitization appears to be independent of cell-cycle and specific genetic mutation. Observed radiosensitization was accompanied by degradation of several DNA repair client proteins of Hsp90, enhanced DNA damage, and delayed γH2Ax foci clearance in vitro.
This study also aimed to find a dose, schedule, and frequency of administration of AT13387 that reduces Hsp90 activity in tumors to a level that can cause radiosensitization without affecting normal tissue. To understand these parameters, detailed pharmacokinetics and pharmacodynamic studies were performed in mice bearing UMSCC74B xenografts. Hsp90 inhibitors are distributed throughout the body but are retained for an extended period in the tumors when compared to plasma or normal tissues, primarily due to the highly activated state of Hsp90 in tumors. To translate our in vitro findings into in vivo efficacy studies, we first assessed the effect of a dose of AT13387 that led to comparable compensatory up-regulation of Hsp70 levels in mouse PBMC and tumor cells as observed in vitro. We found that a single injection of 40 mg/kg AT13387, which is less than half of the MTD dose, induced Hsp70 expression by about 2-fold in PBMC within 2 hours and lasted up to 24 hours. Similarly, 2–4 fold induction of Hsp70 expression was also observed in the xenograft tumors. Results from the current study suggest that the upregulation of Hsp70 expression is a qualitative rather than quantitative biomarker of Hsp90 inhibition. In a Phase I study, an about 1.5-fold increase in Hsp70 expression in serum, PBMC’s or tumor was observed. This increase did not correlate with an increase in Hsp90 inhibitor from 100 mg to 800 mg dose level (33). This in vivo change in Hsp70 expression is similar to in vitro studies conducted at the low concentration (100 nM) of Hsp90 inhibitor suggesting that this concentration of AT13387 inhibits Hsp90 and is expected to be relatively safe to normal tissues. Here, we also determined the pharmacokinetic profile of AT13387 injected once or twice four days apart at the reduced dose level. The resulting concentration of AT13387 in the plasma was compared to the xenograft tumors to determine tumor-specific accumulation in addition to finding a potentially effective dose for radiosensitization. Further analysis of the PK parameters matched with each clinical observation even after using a low dose of AT13387. Despite using a low dose, the inhibitor can accumulate in the xenograft tumors at least 10-fold above plasma levels. Drug levels in the tumors even at the half MTD dose were well over radiosensitizing concentrations. To our knowledge, this is the first study evaluating Hsp70 as an immediate, detectable, and qualitative biomarker for mice treated with a low dose of Hsp90 inhibitor.
Though Canella et. al. demonstrated that long acting nature of AT13387 and its ability to cross the blood-brain barrier (BBB) makes it effective as a single agent, several clinical studies supported the view that the treatment failures were due in part to the use of MTD (34). Available results indicated that the combination therapy with a much lower dose of Hsp90 inhibitor may be the future of cancer therapy. Our data also revealed a plasma AUC of 2263 ng•hour/mL (5.52 μM•hour) based on a 40 mg/kg dose in mice that confirms the allometric translatability of information in the mouse model to that in humans. On the other hand, we observed a 40-fold difference in the AUC of 90733 ng•hour/mL (221.3 μM•hour) in the UMSCC74B xenograft tumor. This accumulation of AT13387 in tumor inhibited Hsp90 activity as reflected by the 6-fold upregulation of Hsp70 at 24 hours after injection. The observed concentration-time profile of AT13387 in this mouse model is consistent with the clinical trial experience with this agent. Previous dose-ranging studies in patients escalated the dose from 20 mg/m2 to 210 mg/m2 and included pharmacokinetic and safety assessments (35). These clinical data identified a linear plasma pharmacokinetic profile with an average AUC of 5.5–6 μM•h in patients receiving 120 mg/m2 which was associated with primarily hematologic Grade 1 adverse event. Based on in vivo modelling data, we estimated that a 30 mg/kg dose in mice would translate to doses of approximately 90 mg/m2 in patients, which is safe. This exploratory analysis supports the possibility of clinical benefits by combining low dose HSP90 inhibitors with RT.
In an attempt to reduce the potential off-target effects while maintaining the ability of AT13387 to enhance radiosensitivity, the efficacy of AT13387 at a further reduced level (30 mg/Kg) was determined against one pancreatic, two head and neck tumor models. As anticipated, treatment with a systemic lower dose of AT13387 was safe but it was not effective as monotherapy against any of these tumor models. However, significant inhibition of tumor growth with acceptable toxicity was observed in combination with radiotherapy. This effect correlated with an increase in γH2Ax foci formation after the first cycle of combination therapy suggesting that inhibition of Hsp90 activity compromised DNA repair resulting in radiosensitization. Similar tumor radiosensitizing results were obtained using the MiaPaCa-2 xenograft model even lower doses of radiation (1.8 Gy) with 30 mg/Kg AT13387 that were ineffective as a monotherapy. Overall, these findings demonstrate that treatment with sub-cytotoxic concentrations of an Hsp90 inhibitor can selectively radiosensitize cancer cells in vitro and in xenograft models. These results suggest that treatment with Hsp90 inhibitors can have potential clinical benefits in combination with radiotherapy.
Clinical studies of Hsp90 inhibitors used as dose-escalating monotherapies as well as in combination with chemotherapy have produced clinical responses, but, unfortunately, these responses have been accompanied by severe eye, liver, GI, cardiac or bone marrow toxicities. The toxicities of Hsp90 inhibitors may be made worse when chemotherapy was added to the treatment regime (and vice versa) (36). To enhance efficacy and reduce the toxicities observed in the combination of Hsp90 inhibitors with systemic chemotherapy, several pre-clinical or clinical studies have assessed the addition of other molecular targeted agents such as erlotinib, trastuzumab, and vemurafenib along with of Hsp90 inhibitors with or without chemotherapy. These efforts have also largely proven to be clinically ineffective, perhaps since Hsp90 inhibition targets the same clients (e.g. EGFR, Her2, and Braf) as that of the molecularly targeted agents. Therefore, clinical trials involving Hsp90 inhibitors have been either withdrawn before moving forward to phase III or failed to get FDA approval.
To the best of our knowledge, this is the first study that specifically demonstrates that sub-cytotoxic concentration of an Hsp90 inhibitor can enhance radiosensitization. Increased γH2Ax foci are often considered as a pharmacodynamic marker for radiation-induced DNA damage which correlates with radiosensitization (32). We also observed a trend of increased γH2Ax foci and other DNA repair markers across the tumor types in the combination group. After one cycle of treatment, the majority of γH2Ax foci were resolved but the enhanced radiosensitization suggests that either the repair was “error prone” or that repeated treatment had a cumulative effect on the treatment outcome.
In addition to the direct effect on DNA repair, Hsp90 and its co-chaperones such as Hsp70 also possess multiple immunomodulatory functions (37,38). Inhibition of Hsp90 activity is known to cause immunosuppression in patients. A recent study found that non-toxic concentration of Hsp90 inhibition can trigger an anti-tumor immune response (39) which was absent at cytotoxic concentration. We noted that inhibition of Hsp90 activity by AT13387 treatment greatly radiosensitized SCCVII xenografts, developed in syngeneic C3H/HeJ mice background. Whereas AT13387 mediated radiosensitizing effect against UMSCC74B and MiaPaCa-2 xenografts, developed in immunocompromised nude mice was less profound, indicating a potential role of the immune component in radiosensitization by AT13387. If these immunological findings are maintained or potentiated by radiation therapy, it would further support the revival of Hsp90 targeting agents as potential clinical radiosensitizers.
Supplementary Material
Translational Relevance:
At least twenty Hsp90 inhibitors have been developed by academic groups and pharmaceutical companies. However, tolerable monotherapy doses of all the Hsp90 inhibitors have shown only limited efficacy. This preclinical study demonstrates that a sub-cytotoxic concentration of an Hsp90 inhibitor can inhibit the DNA repair process and sensitize tumors to radiotherapy. These results show that while Hsp90 inhibition alone does not seem to be an effective approach in cancer therapy, significant improvement can be achieved without systemic toxicity if a lower dose of Hsp90 inhibitory therapy is combined with radiotherapy.
Acknowledgments
We thank the University of Michigan Pharmacokinetic, Proteomic, Flow cytometry, Histology, and DNA sequencing cores and animal facilities at the University of Michigan. AT13387 for this research was provided by the Cancer Therapy Evaluation Program (CTEP) at the NIH.
We would also like to thank Dr. Meredith A Morgan for providing valuable inputs on the discussion section, Mary A Davis for editing and Juan Cai for helping with the STR study.
Financial support: This work was supported by the National Institutes of Health [R01 CA131290 to M.K.N.], [ U01CA216449 to T.S.L.], [GM 077430 to Y.O.], and the Rogel Cancer Center support grant, P30CA46592. This project is a component of the Protein Folding Disease Research Initiative of the University of Michigan Medical School.
Abbreviations
- HNSCC
Head and neck squamous cell carcinoma
- Hsp90
Heat shock protein 90
- AT13387
(2,4-dihydroxy-5-isopropyl-phenyl)-[5-(4-methyl-piperazin-1-ylmethyl)-1,3-dihydro-isoindol-2-yl] thanone, l-lactic acid salt)
- nM
Nanomolar
- IHC
Immunohistochemistry
- SF
survival fraction
- DMF
Dose modifying factor
- IR
irradiation
- MED
minimum essential dose
- MTD
maximum tolerated dose
- PK
pharmacokinetics
- PD
pharmacodynamics
Footnotes
Disclosure of Potential Conflicts of Interest:
References
- 1.Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One 2011;6(10):e26044 doi 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sottile ML, Nadin SB. Heat shock proteins and DNA repair mechanisms: an updated overview. Cell Stress Chaperones 2018;23(3):303–15 doi 10.1007/s12192-017-0843-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, McAlpine SR. Regulating the cytoprotective response in cancer cells using simultaneous inhibition of Hsp90 and Hsp70. Org Biomol Chem 2015;13(7):2108–16 doi 10.1039/c4ob02531h. [DOI] [PubMed] [Google Scholar]
- 4.Neckers L Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends in Molecular Medicine 2002;8(4):S55–S61 doi 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 5.Pratt WB, Morishima Y, Peng HM, Osawa Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med (Maywood) 2010;235(3):278–89 doi 10.1258/ebm.2009.009250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancet JE, Gojo I, Burton M, Quinn M, Tighe SM, Kersey K, et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010;24(4):699–705 doi 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- 7.Walker AR, Klisovic R, Johnston JS, Jiang Y, Geyer S, Kefauver C, et al. Pharmacokinetics and dose escalation of the heat shock protein inhibitor 17-allyamino-17-demethoxygeldanamycin in combination with bortezomib in relapsed or refractory acute myeloid leukemia. Leuk Lymphoma 2013;54(9):1996–2002 doi 10.3109/10428194.2012.760733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Infante JR, Weiss GJ, Jones S, Tibes R, Bauer TM, Bendell JC, et al. Phase I dose-escalation studies of SNX-5422, an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumours. Eur J Cancer 2014;50(17):2897–904 doi 10.1016/j.ejca.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Johnson ML, Yu HA, Hart EM, Weitner BB, Rademaker AW, Patel JD, et al. Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer With Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J Clin Oncol 2015;33(15):1666–73 doi 10.1200/JCO.2014.59.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahsan A, Ray D, Ramanand SG, Hegde A, Whitehead C, Rehemtulla A, et al. Destabilization of the epidermal growth factor receptor (EGFR) by a peptide that inhibits EGFR binding to heat shock protein 90 and receptor dimerization. J Biol Chem 2013;288(37):26879–86 doi 10.1074/jbc.M113.492280 M113.492280 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elaimy AL, Ahsan A, Marsh K, Pratt WB, Ray D, Lawrence TS, et al. ATM is the primary kinase responsible for phosphorylation of Hsp90alpha after ionizing radiation. Oncotarget 2016;7(50):82450–7 doi 10.18632/oncotarget.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YF, Liu H, Diao LX, Potter A, Zhang JH, Qiao YW, et al. Hsp90 Inhibitor Ganetespib Sensitizes Non-Small Cell Lung Cancer to Radiation but Has Variable Effects with Chemoradiation. Clinical Cancer Research 2016;22(23):5876–86 doi 10.1158/1078-0432.ccr-15-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li T, Jiang HL, Tong YG, Lu JJ. Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery. J Hematol Oncol 2018;11(1):59 doi 10.1186/s13045-018-0602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehrlich ES, Wang T, Luo K, Xiao Z, Niewiadomska AM, Martinez T, et al. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci U S A 2009;106(48):20330–5 doi 10.1073/pnas.0810571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casal RG, Bhattacharya C, Epperly M, Socinski M, Levina V. The HSP90 inhibitor ganetespib potentiates effect of ionizing radiation in human non-small cell lung cancer. Cancer Research 2014;74(19):2 doi 10.1158/1538-7445.am2014-4894. [DOI] [Google Scholar]
- 16.Grabenbauer F, Katzer A, Fiedler V, Flentje M, Djuzenova CS. Radiation response of tumor cell lines after combined MEK and Hsp90 inhibition by PD184352 and NVP-AUY922: The role of drug-irradiation schedule. Strahlentherapie Und Onkologie 2015;191:S91–S. [Google Scholar]
- 17.Hashida S, Yamamoto H, Shien K, Ohtsuka T, Suzawa K, Maki Y, et al. Hsp90 inhibitor NVP-AUY922 enhances the radiation sensitivity of lung cancer cell lines with acquired resistance to EGFR-tyrosine kinase inhibitors. Oncology Reports 2015;33(3):1499–504 doi 10.3892/or.2015.3735. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y, Li HK, Masaoka A, Sunada S, Hirakawa H, Fujimori A, et al. The purine scaffold Hsp90 inhibitor PU-H71 sensitizes cancer cells to heavy ion radiation by inhibiting DNA repair by homologous recombination and non-homologous end joining. Radiotherapy and Oncology 2016;121(1):162–8 doi 10.1016/j.radonc.2016.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maki Y, Hashida S, Yamamoto H, Shien K, Ohtsuka T, Suzawa K, et al. Effect of Hsp90 inhibitor NVP-AUY922 with radiation on lung adenocarcinoma cell lines with acquired resistance to EGFR-tyrosine kinase inhibitors. Cancer Research 2015;75:2 doi 10.1158/1538-7445.am2015-742. [DOI] [PubMed] [Google Scholar]
- 20.Spiegelberg D, Dascalu A, Mortensen AC, Abramenkovs A, Kuku G, Nestor M, et al. The novel HSP90 inhibitor AT13387 potentiates radiation effects in squamous cell carcinoma and adenocarcinoma cells. Oncotarget 2015;6(34):35652–66 doi 10.18632/oncotarget.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang XT, Bao CH, Jia YB, Wang NN, Ma W, Liu F, et al. BIIB021, a novel Hsp90 inhibitor, sensitizes esophageal squamous cell carcinoma to radiation. Biochemical and Biophysical Research Communications 2014;452(4):945–50 doi 10.1016/j.bbrc.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun 2018;9(1):2845 doi 10.1038/s41467-018-05049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 2014;86(14):7150–8 doi 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan MA, Parsels LA, Maybaum J, Lawrence TS. Improving gemcitabine-mediated radiosensitization using molecularly targeted therapy: a review. Clin Cancer Res 2008;14(21):6744–50 doi 10.1158/1078-0432.CCR-08-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence TS. Reduction of doxorubicin cytotoxicity by ouabain: correlation with topoisomerase-induced DNA strand breakage in human and hamster cells. Cancer Res 1988;48(3):725–30. [PubMed] [Google Scholar]
- 26.Fertil B, Dertinger H, Courdi A, Malaise EP. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. Radiat Res 1984;99(1):73–84. [PubMed] [Google Scholar]
- 27.Boyum A Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 1968;97:77–89. [PubMed] [Google Scholar]
- 28.Mehta RK, Verma S, Pati R, Sengupta M, Khatua B, Jena RK, et al. Mutations in subunit interface and B-cell epitopes improve antileukemic activities of Escherichia coli asparaginase-II: evaluation of immunogenicity in mice. J Biol Chem 2014;289(6):3555–70 doi 10.1074/jbc.M113.486530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan EaM P Nonparametric estimation from incomplete observations J Amer Statist Assoc 1958;53 (282) doi 10.2307/2281868. [DOI] [Google Scholar]
- 30.Mantel N Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemotherapy Reports 1966;50(3):163–70. . [PubMed] [Google Scholar]
- 31.Shapiro GI, Kwak E, Dezube BJ, Yule M, Ayrton J, Lyons J, et al. First-in-human phase I dose escalation study of a second-generation non-ansamycin HSP90 inhibitor, AT13387, in patients with advanced solid tumors. Clin Cancer Res 2015;21(1):87–97 doi 10.1158/1078-0432.CCR-14-0979. [DOI] [PubMed] [Google Scholar]
- 32.Parsels LA, Parsels JD, Tanska DM, Maybaum J, Lawrence TS, Morgan MA. The contribution of DNA replication stress marked by high-intensity, pan-nuclear gammaH2AX staining to chemosensitization by CHK1 and WEE1 inhibitors. Cell Cycle 2018;17(9):1076–86 doi 10.1080/15384101.2018.1475827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saif MW, Takimoto C, Mita M, Banerji U, Lamanna N, Castro J, et al. A phase 1, dose-escalation, pharmacokinetic and pharmacodynamic study of BIIB021 administered orally in patients with advanced solid tumors. Clin Cancer Res 2014;20(2):445–55 doi 10.1158/1078-0432.CCR-13-1257. [DOI] [PubMed] [Google Scholar]
- 34.Canella A, Welker AM, Yoo JY, Xu J, Abas FS, Kesanakurti D, et al. Efficacy of Onalespib, a Long-Acting Second-Generation HSP90 Inhibitor, as a Single Agent and in Combination with Temozolomide against Malignant Gliomas. Clin Cancer Res 2017;23(20):6215–26 doi 10.1158/1078-0432.CCR-16-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Do K, Speranza G, Chang LC, Polley EC, Bishop R, Zhu W, et al. Phase I study of the heat shock protein 90 (Hsp90) inhibitor onalespib (AT13387) administered on a daily for 2 consecutive days per week dosing schedule in patients with advanced solid tumors. Invest New Drugs 2015;33(4):921–30 doi 10.1007/s10637-015-0255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slovin S, Hussain S, Saad F, Garcia J, Picus J, Ferraldeschi R, et al. Pharmacodynamic and Clinical Results from a Phase I/II Study of the HSP90 Inhibitor Onalespib in Combination with Abiraterone Acetate in Prostate Cancer. Clin Cancer Res 2019. doi 10.1158/1078-0432.CCR-18-3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graner MW. HSP90 and Immune Modulation in Cancer. Adv Cancer Res 2016;129:191–224 doi 10.1016/bs.acr.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Pockley AG. Heat shock proteins as regulators of the immune response. Lancet 2003;362(9382):469–76 doi 10.1016/S0140-6736(03)14075-5. [DOI] [PubMed] [Google Scholar]
- 39.Jaeger AM, Stopfer L, Lee S, Gaglia G, Sandel D, Santagata S, et al. Rebalancing Protein Homeostasis Enhances Tumor Antigen Presentation. Clin Cancer Res 2019. doi 10.1158/1078-0432.CCR-19-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.