

Summary
Prolactin (PRL) and its receptor (PRLr) play important roles in the pathogenesis of breast cancer. Cyclophilin A (CypA) is a cis-trans peptidyl-prolyl isomerase (PPI) that is constitutively associated with the PRLr and facilitates the activation of the tyrosine kinase Jak2. Treatment with the non-immunosuppressive prolyl isomerase inhibitor NIM811 or CypA short hairpin RNA inhibited PRL-stimulated signaling, breast cancer cell growth, and migration. Transcriptomic analysis revealed that NIM811 inhibited two-thirds of the top 50 PRL-induced genes and a reduction in gene pathways associated with cancer cell signaling. In vivo treatment of NIM811 in a TNBC xenograft lessened primary tumor growth and induced central tumor necrosis. Deletion of CypA in the MMTV-PyMT mouse model demonstrated inhibition of tumorigenesis with significant reduction in lung and lymph node metastasis. The regulation of PRLr/Jak2-mediated biology by NIM811 demonstrates that a non-immunosuppressive prolyl isomerase inhibitor can function as a potential breast cancer therapeutic.
Subject Areas: Medical Biochemistry, Molecular Biology, Cancer
Graphical Abstract
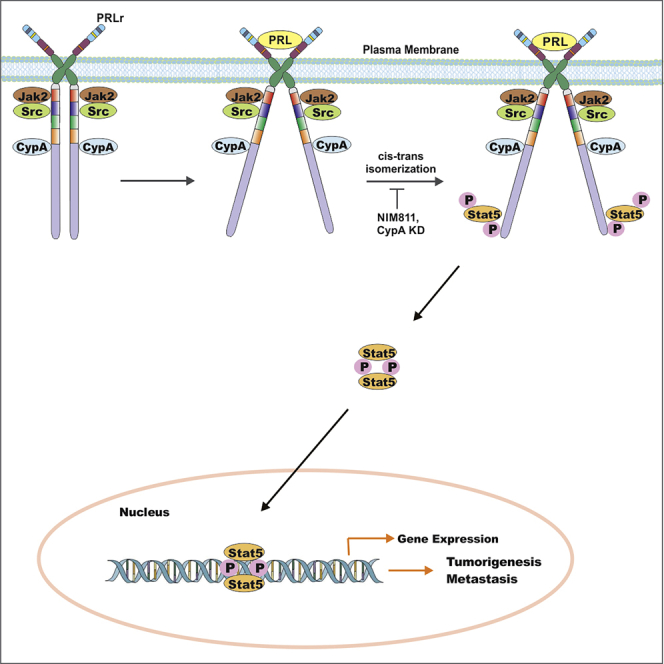
Highlights
-
•
CypA inhibition or knockdown blocks breast cancer cell signaling, growth, and migration
-
•
NIM811 inhibited PRL-induced genes and gene pathways relevant to cancer signaling
-
•
Deletion of CypA has shown reduction in tumorigenesis and metastasis in mice
Medical Biochemistry; Molecular Biology; Cancer
Introduction
Cyclophilins are highly conserved proteins throughout evolution (Wang and Heitman, 2005). Cyclophilin A (CypA) is involved in protein folding, trafficking, receptor assembly, cell cycle regulation, and signal transduction (Lammers et al., 2010). CypA is part of the peptidyl-prolyl isomerase (PPI) family of enzymes that catalyzes the cis-trans interconversion of imide bonds of proline residues inducing protein backbone conformational change (Gothel and Marahiel, 1999) and was initially identified as a binding partner of cyclosporine (CsA), an immunosuppressive drug (Handschumacher et al., 1984). The CypA-CsA complex allosterically inhibits calcineurin preventing IL2-mediated T cell activation (Roehrl et al., 2004). CypA is aberrantly expressed in cancer cells including the breast, lung, and pancreas (Lee and Kim, 2010). Female allograft recipients treated with CsA demonstrated a 50% reduction in incidence of breast cancer after 10 years (Stewart et al., 1995), suggesting a chemopreventive role for this agent within the breast. A potential target for CsA within the breast is the prolactin receptor (PRLr), as CypA constitutively interacts with this receptor (Clevenger et al., 2003; Zheng et al., 2008).
The polypeptide hormone prolactin has been implicated in the pathogenesis of breast cancer based on data at the cellular, epidemiologic, and genetic levels (Clevenger and Kline, 2001). PRL binds and signals through its cognate receptor at the cell surface of normal and breast tissue (Gertler et al., 1996). Breast cancer cells produce PRL, which contributes to increased proliferation, survival, motility, and invasion of breast cancer cells in vitro (Perks et al., 2004). In turn, its cognate receptor, PRLr is expressed in 98% of human breast cancers based on immunohistochemistry, in situ hybridization, and qRT-PCR analysis (Mertani et al., 1998). Mice that overexpressed PRL developed both ER+ and ER− tumors at both endocrine and autocrine/paracrine levels (Clevenger et al., 2003). In contrast, PRL knockout mice show significantly increased latency in developing oncogene-driven mammary tumors (Kelly et al., 2001). In vitro, PRLr knockdown impairs anchorage dependent and independent growth in breast cancer cells (Fiorillo et al., 2013).
Like growth hormone, PRL binding initiates a conformational change in the pre-dimerized PRL receptor (Gadd and Clevenger, 2006). The conformational change in the receptor leads to activation of tyrosine kinases like Janus kinase 2 (Jak2) and Src, which rapidly phosphorylate the C terminus of the PRLr (DaSilva et al., 1994), and these receptor proximal events enhance the activation of signaling pathways (Syed et al., 2003). Jak2 is constitutively associated with the PRLr, and PRL binding to the receptor leads to the auto-phosphorylation of Jak2 (Leonard and O'Shea, 1998). Auto-phosphorylated Jak2 in turn phosphorylates C-terminal PRLr residues, enabling Stat5 recruitment and tyrosine phosphorylation (Levy and Darnell, 2002). Tyrosine-phosphorylated Stat5 (Signal Transducer and Activator of Transcription 5) then dimerizes, translocates to the nucleus, and activates transcription of PRL-responsive gene like cyclin D1 and CISH (Brockman and Schuler, 2005; Schindler et al., 1992). Conditional loss of either Jak2 or Stat5 results in a lactation-deficient phenotype that closely mimics the PRLr knockout mouse model (Wagner et al., 2004). Thus the functions of the PRLr, Jak2, and Stat5 are closely associated with breast development and pathogenesis.
Previous studies from our laboratory found that the PPI activity of CypA contributes to PRLr and Jak2 function (Zheng et al., 2008). PRL-mediated signaling/gene expression is directly correlated with levels of CypA, and specifically its PPI activity. Furthermore, overexpression of a CypA interaction-defective PRLr mutant (P334A), was shown to significantly downregulate PRLr/Jak2-mediated signaling (Zheng et al., 2008). Pharmacologically, inhibition of CypA by the immunosuppressive prolyl isomerase inhibitor CsA blocks Jak2/Stat5 phosphorylation and growth/metastasis of ER−/+ breast cancer xenografts (Zheng et al., 2008). The use of immunosuppressive CsA in breast cancer is problematic, however, and its use can only be envisaged in the metastatic setting. Here, we sought to test the non-immunosuppressive CypA inhibitor, N-methyl-4-isoleucine-cyclosporine (NIM811), as well as the in vivo effect of CypA loss of function when introduced into a spontaneous mouse mammary cancer model. The introduction of a single side chain to the CsA backbone renders NIM811 non-immunosuppressive and virtually non-toxic, as was seen in a recent clinical trial (Lawitz et al., 2011). These studies found that non-immunosuppressive NIM811 was equipotent to CsA, resulting in significant inhibition of PRL-driven signaling, in vitro growth, and in vivo metastases in mouse models of breast cancer. Overall, these results reveal that this isomerase can be inhibited by a non-immunosuppressive PPI inhibitor and is a potential target for the therapeutic intervention of breast cancer metastasis.
Results
NIM811 Inhibits Phosphorylation of PRLr-Stat5 Signaling Intermediates
Based on recent studies, CypA has found to be a constitutively interacting protein with the PRLr (Leonard and O'Shea, 1998; Zheng et al., 2008). Inhibition of CypA activity with immunosuppressive PPI inhibitor CsA in breast cancer cells blocked PRLr signaling, as well as Jak2 and Stat5 phosphorylation (Zheng et al., 2008). CsA is a potent inhibitor of CypA and capable of modulating breast cancer functions but remains an immunosuppressive agent with toxic side effects. Therefore, the action of a non-immunosuppressive and non-toxic analog of CsA, termed NIM811, was explored.
Phospho-specific anti-PRLr, Jak2, and Src antibodies were utilized to test the effects of NIM811 on the PRL-induced phosphorylation given their significance to PRLr-associated signaling. To that end, ER/PR+ T47D breast cancer cells were pre-treated with NIM811 and then stimulated as a function of time with PRL. NIM811 significantly decreased tyrosine-phosphorylation of PRLr, Jak2, Src, and Stat5 (Figures 1A and 1B). There was a marked increase in pJak2-Y1007/1008 within 7.5–15 min following PRL stimulation; however, a 3- to 5-fold inhibition of p-YJak2 was observed with NIM811 treatment as a function of time (Figure 1), confirming the importance of CypA-PPI activity regulating Jak2 activation in breast cancer cells. Following PRL stimulation, there was also significant increase in levels of pPRLr-Y381 and pPRLr-Y587 within 7.5–15 min, whereas a concomitant 2- to 5-fold decrease in phosphorylation with NIM811 treatment was observed as a function of time (Figure 1). Src is an associated kinase of the PRLr/Jak2 complex, and Src-Y416 residue is involved in Src activation. In Figure 1, a marked increase in Src phosphorylation occurred within 7.5–15 min of PRL stimulation with a concomitant >3-fold decrease in phosphorylation observed when treated with NIM811. Stat5 phosphorylation is mediated by the Jak2 tyrosine kinase at tyrosine residue 694 (Clevenger, 2004). Following PRL stimulation, Stat5 was significantly tyrosine phosphorylated within 7.5–30 min while demonstrating an associated 5- to 7-fold decrease in level of p-Y694 with NIM811 treatment as a function of time (Figure 1). Furthermore, NIM811 significantly inhibited PRL-induced phosphorylation of downstream Akt and MAPK (Figure S1). Taken together, these data indicate that CypA inhibition by NIM811 significantly decreases phosphorylation/activation of PRLr-Stat5 signaling intermediates that have been implicated in regulating downstream gene expression and breast cancer cell functions.
Figure 1.

CypA Inhibition Modulates Phosphorylation of PRLr/Jak2 Signaling Intermediates
(A) T47D cells were serum starved for 16–24 h, pre-treated with NIM811 (10 μg/mL) or DMSO (vehicle control) for 4–6 h, and/or stimulated with human PRL (250 ng/mL) for the indicated times. Blots were probed with the indicated antibodies.
(B) Quantification of fold expression of phospho-proteins normalized to total proteins in (A) as indicated. Lines, mean of three independent experiments; error bars, ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001. Unpaired two-tailed t test was performed to determine significance.
Loss of Function or Rescue of CypA Modulates Phosphorylation of PRLr-Stat5 Signaling Intermediates
A loss-of-function approach was used to identify the CypA-binding site at proline 334 on the PRLr (Zheng et al., 2008). Previously, when cells expressed the CypA interaction-defective mutant PRLr-P334A, a significantly decreased CypA-PRLr association, Jak2/Stat5 phosphorylation, and PRL-induced gene expression were observed compared with CypA wild-type (WT) control (Zheng et al., 2008). Given the effects of NIM811 treatment on breast cancer cell signaling (Figures 1A and 1B) and PRLr mutant action on Jak2/Stat5 phosphorylation (Zheng et al., 2008), it was reasoned that such loss of function of CypA could alter phosphorylation/activations of PRLr-Stat5 signaling intermediates as well. To determine the consequences of molecular knockdown of CypA in breast cancer cell signaling, inducible short hairpin (shRNA)-mediated knockdown of CypA was performed in ER/PR+ T47D cells (Figure S2). Like the pharmacological inhibition of CypA PPI activity by NIM811, loss of its expression in breast cancer cells had significant effects on phosphorylation/activation of the PRLr, Jak2, Stat5, and Src in a PRL-dependent manner. Following PRL stimulation for 15 min, as expected, pYJak2 was significantly induced with prolactin stimulation in control cells, whereas >5-fold reduction in pYJak2 level was observed with the loss of CypA (Figures 2A and 2B). pPRLr-Y381 and pPRLr-Y587 were induced markedly in the shRNA control condition; however, in the CypA knockdown condition, a significant (>4-fold) decrease in phosphorylation was observed at these sites in T47D cells (Figures 2A and 2B). Similar to pSrc-Y416 levels in Figure 1, pSrc-Y416 was significantly induced within 7.5–15 min of PRL stimulation in control cells, whereas a >2-fold reduction in phosphorylation was observed in CypA knockdown cells (Figures 2A and 2B). Last, Jak2-mediated phosphorylation of Stat5 was significantly induced by PRL stimulation, whereas a >7-fold reduction in phosphorylation was observed in the absence of CypA (Figures 2A and 2B). To determine if the loss of phosphorylation of PRLr-Stat5 intermediates was directly due to CypA knockdown and not an off-target effect, a rescue overexpression was performed. T47D cells with CypA knockdown were transfected with CypA-WT plasmid (Zheng et al., 2008), and subsequently cell lysates were analyzed. The rescue overexpression of CypA in the CypA knockdown cells was adequate to restore p-YPRLr, pYJak2, pYStat5, and pYSrc levels to PRL-induced endogenous levels (Figures 2C and 2D). Taken together, these data indicate that loss of CypA had significant effects on tyrosine phosphorylation of PRLr-Stat5 intermediates, which was rescued by WT CypA expression.
Figure 2.

Silencing of CypA Modulates Phosphorylation of PRLr/Jak2 Signaling Intermediates and Can Be Rescued by CypA-WT
(A) Stable expression of non-silencing control or CypA shRNA in T47D cells. Cells were serum starved for 16–24 h and stimulated with PRL (250 ng/mL) for 15 min. Immunoblots were probed with the indicated antibodies.
(B) Quantification of fold expression of phospho-proteins normalized to total proteins in (A) as indicated. Columns, mean of at least three independent experiments: error bars, ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.005. Unpaired two-tailed t test was performed to determine significance.
(C) Control or CypA shRNA and/or CypA-WT (wild-type) were transfected and treated similarly as in (A).
(D) Quantification of fold expression of phospho-proteins normalized to total proteins in (C) as indicated. Columns, mean of three independent experiments: error bars, ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.005. Unpaired two-tailed t test was performed to determine significance.
Furthermore, NIM811 treatments of CypA-WT cell decreased levels of p-Stat5, whereas CypA knockdown cells demonstrated no significant further reduction in p-Stat5 levels following the addition of NIM811 (Figure S3). This demonstrates that off-target actions of NIM811 are not the principal basis of its action on the PRLr-Stat5 axis.
CypA Inhibition Decreases mRNA and Protein Expression Levels of CISH and Cyclin D1
PRL and its cognate receptor regulate downstream genes through the Jak2-Stat5 signaling pathway. PRL signaling in breast cancer cells induces several breast cancer-relevant genes including the estrogen receptor (ER), cyclin D1, and CISH (Fang et al., 2009). These genes have been implicated in the pathogenesis of breast cancer (Raccurt et al., 2003; Sherr, 1996). To that end, the alteration of PRL-responsive gene expression by the NIM811-mediated inhibition of CypA was examined. mRNA and protein expression of CISH and cyclinD1 were assessed in NIM811 pre-treated ER/PR+ T47D breast cancer cells utilizing qRT-PCR and immunoblot analysis, respectively. As demonstrated in Figure 3A, CISH and cyclin D1 mRNA levels were significantly down-regulated with NIM811 treatment in a PRL-dependent manner. Furthermore, NIM811 treatment also resulted in a highly significant decrease in PRL-induced expression of both CISH and cyclin D1 at the protein level (Figure 3B) based on quantification of their normalized expression (Figures 3C and 3D). Overall, these findings demonstrated that inhibition of CypA prolyl isomerase activity directly correlated with PRL-induced gene expression of CISH and cyclin D1.
Figure 3.

mRNA and Protein Expression of Prolactin-Responsive Genes, CISH and cyclin D1
(A) mRNA expression of CISH and cyclin D1. T47D cells pre-treated with NIM811 (10 μg/mL) for 4 h followed by PRL (250 ng/mL) stimulation for 2 h. Columns, mean of three independent experiments; error bars, SEM. ∗p < 0.05, ∗∗∗p < 0.005. Unpaired two-tailed t test was performed to determine significance. (B) Protein expression of CISH and cyclin D1 in T47D cells treated the same way as in (A). Quantification of % expression of CISH (C) and cyclin D1 (D) normalized to GAPDH in (B). Columns, mean of three independent experiments; error bars, SEM. ∗∗p < 0.01. Unpaired two-tailed t test was performed to determine significance.
CypA Inhibition Alters Global Gene Expression in Breast Cancer Cells
In a precedent study, microarray analysis revealed 120 PRL-induced genes up-regulated by WT, but not mutant (mutation in the trans-activation domain) PRLr in T47D cells (Fiorillo et al., 2013). We reasoned that inhibition of the PPI activity of CypA may alter PRL-induced global gene expression based on its role in structure/function relationships of the PRLr and associated proximal/downstream signaling. To determine the extent to which PRL and NIM811 altered global gene expression in human breast cancer cells, RNA derived from T47D cells treated with PRL and/or NIM811 was analyzed by expression microarrays. Analysis of the resultant data with a fold change cutoff of 1.2 revealed that PRL induced the expression of 535 genes and inhibited the expression of 372 genes, whereas NIM811 induced 1,725 genes and inhibited 1,737 genes. NIM811 inhibited expression of the PRLr target genes CCND1 and CEBPB and opposed the expression changes stimulated by PRL on a global scale (Figure 4). NIM811 significantly inhibited 57% of the top 100 prolactin-induced genes (Figure 4A) and significantly induced 62% of the top 100 PRL-inhibited genes (Figure 4B). Of the 535 and 372 genes induced and inhibited, respectively, by PRL, NIM811 significantly inhibited 50% (268/535) and induced 50% (185/372). The global effects of NIM811 on PRL-induced Stat5 gene expression were also examined. Indeed, utilizing a previously reported set of Stat5 target genes (Kang et al., 2014b), we found that of the 46 Stat5 target genes significantly induced by PRL, 52% (24/46) were also significantly inhibited by NIM811 (Figure 4C). Of the 26 Stat5 target genes significantly inhibited by PRL, 62% (16/26) were significantly induced by NIM811 (Figure 4D). Last, consistent with CypA's regulation of the PRLr with respect to Jak2 in the PRLr-Stat5 signaling cascade, gene set enrichment analysis comparing NIM811-inhibited genes with GEO (Gene Expression Omnibus) kinase perturbation experiments revealed highly significant similarity with Jak2 knockdown gene signatures (Figure 4E).
Figure 4.

NIM811 Opposes Prolactin-Induced Gene Expression Globally and at Stat5 Target Genes
(A and B) Heatmaps depict hierarchical clustering of the top 100 PRL-induced (A) and PRL-inhibited (B) genes (represented by rows) identified by microarray analysis. Samples are represented by columns and cluster with biological replicates according to the treatment conditions indicated above each dendrogram. Red and blue represent high and low gene expression values, respectively.
(C and D) Venn diagrams quantify the Stat5 target genes defined as those previously reported by Kang et al. (2014a) from the microarray analysis that are significantly induced (C) or inhibited (D) by PRL (fold change>1.2) and that are also significantly inhibited (C) or induced (D) by NIM811. Differential expression between treatment conditions was assessed by moderated t test adjusted for multiple hypotheses by the Benjamini and Hochberg method. The false discovery rate (FDR) was controlled so that only those probe sets with q < 0.01 were deemed significant.
(E) Bar graph depicts the highest scoring gene set enrichments when NIM811-inhibited genes (fold change>1.2) are compared with gene signatures from kinase perturbation experiments deposited in the Gene Expression Omnibus (GEO). The gene signatures with the highest concordance with NIM811-inhibited gene set are listed in descending order from top to bottom (# 1–8) based on p value ranking. (1) Jak2_knockdown_192_GSES4645, p < 3.18 × 10−12; (2) LRRK2_activemutant_159_GSE36321, p < 3.50 × 10−12; (3) LRRK2_mutant_GDS4401, p < 5.24 × 10−12; (4) GSK3β_knockdown_206_GDS4305, p < 2.27 × 10−9; (5) SYK_druginhibition_153_GSE34176, p < 5.22 × 10−9; (6) SYK_druginhibition_154_GSE34176, p < 1.23 × 10−8; (7) IGF1R_druginhibition_46_GSE14024, p < 1.62 × 10−7; and (8) JAK1_druginhibition_166_GSE38335, p < 3.79 × 10−7.
NIM811 Inhibits Breast Cancer Cell Viability, Anchorage-Independent Growth, Motility, and Migration
Breast cancer cell growth, particularly anchorage-independent growth, is a key hallmark of breast cancer development and progression (Paoli et al., 2013). Given that CypA function significantly impacts the PRLr and its associated signaling machinery that contributes to the pathogenesis of breast cancer (Rycyzyn et al., 2000), it was reasoned that utilization of non-immunosuppressive PPI inhibitor such as NIM811 might inhibit the malignant phenotype of breast cancer. Thus the effects of NIM811 treatment on breast cancer proliferation, viability, and anchorage-independent growth in vitro were examined. Proliferation of ER/PR+ (T47D) cells treated with NIM811 or CypA-knockdown was significantly inhibited in a PRL-dependent manner, as measured by CyQuant assay (Figure S4). When cultured in the presence of NIM811, both T47D and MDA231(ER/PR−) cells demonstrated a dose-dependent inhibition of cell viability (Figure 5A). Furthermore, there were similar effects in inhibition of cell viability of BT-474 (HER-2 positive) and T47D cells compared with MDA-231 cells (data not shown). Furthermore, there was a marked and dose-dependent increase in cleaved-caspase 3 and cleaved-PARP as a function of this treatment, indicating dose-related induction of apoptosis by NIM811 (Figure S5). To determine the effects of NIM811 on anchorage-independent growth, soft-agar colony formation assays were performed. As demonstrated in Figure 5B, NIM811 treatment markedly reduced anchorage-independent growth of T47D and MDA 231 breast cancer cells in a dose-dependent manner (Figure 5B). As a whole, these data indicate that as a non-immunosuppressive PPI inhibitor NIM811 can modulate in vitro malignant phenotypes of breast cancer cells in a dose-dependent manner.
Figure 5.

NIM811 Treatment of T47D and MDA231 Cells Markedly Decreased Cell Viability and Anchorage-Independent Growth
(A) T47D and MDA-MB-231 cells were incubated with various doses of NIM811. Viable cells were quantified every other day by trypan blue exclusion. Lines, mean of three independent experiments; error bars, ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.005. One-way ANOVA with Bonferroni's test to the control was used to determine significance.
(B) T47D and MDA-MB-231 cells incubated with or without various does of NIM811 were mixed with 0.45% agarose and overlaid over 0.8% bottom agar in 6-well plates. Cells were incubated for 14–21 days. Five randomly selected fields were counted for each well to assess anchorage-independent growth. Column, mean of three independent experiments; error bars, ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.005. One-way ANOVA with Bonferroni's test to the control was used to determine significance.
Prolactin also stimulates the cytoskeletal re-organization and motility of breast cancer cells by activating Nek3 and Vav2 pathways (Miller et al., 2005). Given this, the motility and migration of T47D and MDA231 cells was assessed utilizing wound-healing and trans-well migration assays. Highly metastatic MDA231 cells exhibited markedly reduced motility compared with control, while a more moderate but still significant reduction of T47D closure was noted (Figure 6A). To further explore this effect, trans-well migration assays were performed (Schmitt et al., 2016). As shown in Figure 6B, NIM811 significantly reduced the migration of both ER/PR+ and ER/PR− breast cancer cells (Figure 6B). These data demonstrated that NIM811 treatment significantly inhibits both the motility and migration of breast cancer cells.
Figure 6.

CypA Inhibition Significantly Reduced TNBC Cell Migration
(A) Wound-healing assay. Confluent MDA-MB-231 cells were wounded with a pipette tip and cultured in serum-free medium in the presence of various doses of NIM811. Representative images of wound closure assay were acquired with a phase-contrast microscope, and the percentage of the wound closed was quantified. Scale bar, 100 μm. Columns, mean of three independent experiments; error bars, ±SEM. ∗∗p < 0.01. One-way ANOVA with Dunnett's test was used to determine significance.
(B) Migration was measured by a modified Boyden chamber assay. PRL-stimulated cell migration in T47D and MDA-231 cells was inhibited by NIM811. SFM (serum-free media) was the negative control. Columns, mean of three independent experiments; error bars, ±SEM. ∗p < 0.05, ∗∗p < 0.01. One-way ANOVA with Dunnett's test was used to determine significance.
CypA Inhibition Induces Tumor Necrosis and Inhibits Lymph Node Metastasis in TNBC Xenografts
To test the in vivo effects of CypA inhibition by NIM811, various in vivo settings were examined. MDA-231 breast cancer cells were injected into the fourth lactiferous ducts of nude mice (Harrell et al., 2006), and tumors were allowed to establish for 2 weeks or tumor size at 100–200 mm3. Subsequently a twice daily gavage of NIM811 (or carrier control) was administered for 4 weeks. Although all NIM811-treated tumors revealed a trend toward a reduction in volume over the period, these parameters did not achieve statistical significance (Figure S6). However, two parameters were found to be markedly altered as functions of NIM811 treatment, namely, central tumor necrosis and lymph node metastasis. As demonstrated in Figure 7A, central necrosis of MDA-231 tumors treated with NIM811 was significantly increased compared with vehicle control. NIM811 effectively induced central tumor death at both 20 and 50 mg/kg/day doses based on quantification of necrotic area (Figure 7A). As shown in Figure 7B, NIM811 also markedly decreased lymph node metastasis of triple-negative breast cancer (TNBC) xenografts. Notably, 20 mg/kg/day dose of NIM811 was sufficient to significantly inhibit lymph node macro-metastasis (Figure 7B). Interestingly, although individual GFP-positive MDA-231 cells were observed in xenograft lymph nodes, the outgrowth of macro-metastasis was completely inhibited in these mice (Figure S7). Taken together, NIM811 therapy had significant effects on inhibition of the outgrowth of lymph node metastasis and extent of central tumor necrosis in TNBC xenografts.
Figure 7.

NIM811 Induces Tumor Necrosis and Inhibits Lymph Node Metastasis in TNBC Xenografts
(A) Quantification of the necrotic area of primary tumor based on histologic examination of H&E-stained sections. Percent of total necrotic area determined by a board-certified pathologist. Bars, mean ± SEM. ∗∗∗p < 0.005. One-way ANOVA with Dunnett's test was used to determine significance.
(B) Lymph node metastases were detected by histologic examination of H&E-stained section of lymph nodes harvested from mice treated for 12 weeks with either NIM811 or vehicle control. ∗∗p < 0.01, ∗∗∗p < 0.005. One-way ANOVA with Dunnett's test was used to determine significance.
Loss of CypA Reduces Tumor Burden and Lung and Lymph Node Metastasis in an Oncogene-Driven Breast Cancer Model
To demonstrate the loss of function of CypA (genetic knockout) in delaying or disrupting tumorigenesis and/or metastasis in a murine model with an intact immune system, the CypA−/- mouse was crossed into an MMTV-PyMT transgenic mammary mouse model (Ren et al., 2002; Sakamoto et al., 2010; Shen and Brown, 2005). This model demonstrates stochastic progression of tumorigenesis, with overlapping phases of hyperplasia, adenoma, invasion, and metastases. CypA+/- × FVB/NJ and PyMT × FVB/NJ mice were mated. The resultant female PyMT+ × CypA−/- progeny were followed for mammary tumor development and distal metastasis evaluated at 5–14 weeks of age. As presented in scatterplot in Figure S8A, a significant delay in tumor latency was noted in the PyMT+ × CypA−/- mice (PyMT+ × CypA+/+ mean latency = 36 days versus PyMT+ × CypA−/- = 51 days). Analysis of mammary tumor multiplicity in the mouse cohorts at the time of euthanasia/death showed that PyMT+ × CypA+/+ mice developed (average of 10 tumors/mouse) and demonstrated significantly more discrete carcinomatous foci than did PyMT+ × CypA−/- mice (average of 5 tumors/mouse) (Figure S8B). In parallel, survival of the PyMT+ × CypA−/- females was significantly enhanced (PyMT+ × CypA+/+ = 129 days versus PyMT+ × CypA−/- = 90 days) (Figure S8C). However, PyMT+ × CypA−/+ (heterozygous) females demonstrated no increase in tumor latency or survival, paralleling the curves of the PyMT+ × CypA+/+ mice. To assess mammary cancer progression in this model, morphology was examined in both the PyMT+ × CypA−/−and PyMT+ × CypA+/+. In regard to the classic PyMT+ FBV/NJ, age correlated to a particular cancer stage: 5 weeks to hyperplasia, 8 weeks to adenoma, 11 weeks to invasive carcinoma, and 13 weeks to metastasis (Figure 8A). PyMT+ × CypA+/+ and PyMT+ × CypA+/- mice demonstrated significantly more invasive carcinoma at 11 weeks and metastasis at 13 weeks compared with PyMT+ × CypA−/- mice (Figures 8A and S8D). The PyMT+ × CypA−/- cohort had a preponderance of tumors with hyperplasia but significantly less-invasive carcinoma compared with either CypA+/+ or CypA−/+ cohort at 13 weeks of age (Figure S8D). As the metastatic phenotype is related to primary tumor burden, tumorigenesis within the mammary glands of CypA+/+, CypA−/+, and CypA−/- mice was assessed. The primary tumor burden for CypA−/- mice was significantly decreased compared with CypA+/+ mice, whereas only moderate decrease was observed for CypA−/+ mice (Figure 8B). In addition to primary tumorigenesis, metastases to both lymph nodes and lung metastasis was evaluated. Quantification of percent positive lymph nodes of CypA+/+, CypA−/+, and CypA−/- mice demonstrated that CypA−/- mice had a marked reduction in lymph node metastasis compared with CypA+/+ (Figure 8C). To determine lung metastasis, both the metastases area and the number of metastases were measured. CypA−/- mice demonstrated a significant decrease in both the number of metastases (Figure 8D) and metastases area (Figure 8E). Even after correction for primary tumor burden (Figure 8F), CypA−/- mice demonstrated reduced levels of metastases compared with CypA+/+ mice. These results demonstrated that loss of CypA and its corresponding function has profound impacts on the reduction of primary tumor burden and metastasis of lymph nodes and lung in mammary cancer, even after correcting for tumor burden. These data demonstrate that CypA significantly contributes to multistage mammary cancer progression.
Figure 8.

Loss of CypA Reduces Tumor Burden and Lung and Lymph Node Metastases
(A) Representative images of H&E-stained mouse mammary gland containing tumor as a result of cross-breeding the WT (PyMT+) mouse to the Cyp A−/- mouse (n = 12). Different ages are representative of distinct cancer stages in the WT (PyMT+) model: 5 weeks, hyperplasia; 8 weeks, adenoma; 11 weeks, invasive carcinoma; 13 weeks, metastasis. Scale bar, 200 μm.
(B) Tumor burden was assessed as the sum of all primary mammary tumor volumes normalized to body weight in grams.
(C) Positivity of lymph nodes was determined as lymph nodes containing at least 1 macrometastasis from FFPE H&E-stained axillary and accessory axillary lymph nodes, assessed by C.V.C., a board-certified pathologist, under bright-field microscopy.
(D) Lung metastases from H&E-stained FFPE lung sections were counted.
(E) Borders were drawn of metastases identified in (D), and a scaled area was calculated for each metastasis as well as each lung lobe for each mouse. Total metastatic area was normalized to total lung area.
(F) Lung metastasis were counted and normalized per tumor burden. All experiments contained n = 12 mice per cohort. For (B and C), data reported as means with SEM. For primary tumor (B) and percent of total positive lymph nodes analysis (C), one-way ANOVA with Tukey's tests for multiple comparisons were used to determine significance.
For (D–F) medians with range were reported due to the lack of a normal distribution of the metastasis data. A non-parametric test for three groups was performed due to the lack of a normal distribution of the data. ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001.
Discussion
The activation of the Jak2-Stat5 pathway is thought to play an important role in mammary tumorigenesis (Wagner and Rui, 2008). However, inhibitors of Jak2/Stat5 or the receptors associated with this complex's activation have demonstrated limited success (Bousoik and Montazeri Aliabadi, 2018). In search for a better inhibitor of this pathway, our laboratory began to work with CypA, which through its prolyl isomerase activity is necessary for robust Jak2 activation. Use of CypA inhibitor, both in vitro and in vivo (Zheng et al., 2008), has significant effects on mammary tumorigenesis and progression. However, the immunosuppressive properties of CsA, through its engagement of calcineurin, preclude its use in patients with breast cancer with anything else other than end-stage disease. Thus, the development of non-immunosuppressive cyclosporines that remain fully functional as PPI inhibitors, such as NIM811, could have significant therapeutic impact. Herein, we demonstrate that NIM811 is fully capable at inhibiting the PRLr-triggered activation of the Jak2-Stat5 pathway and significantly alters breast cancer cell functions, mammary tumorigenesis, and metastasis.
The data presented here demonstrate that the PPI activity of CypA contributes to proximate PRLr receptor activation, enabling signal transduction through the PRLr/Jak2 complex. Inhibition of CypA PPI function by NIM811 blocked tyrosine phosphorylation of Jak2, Stat5, and Src at several sites that modulate the overall activity of these kinases. Similarly, NIM811 significantly reduced the phosphorylation of the PRLr itself, at residues 381 and 587, which are thought to play roles in the PRLr engagement of Src and Stat5, respectively (Brooks, 2012; Pezet et al., 1997). Like NIM811-mediated inhibition, CypA knockdown significantly suppressed phosphorylation of the PRLr, Jak2, and Src at those specific tyrosine residues (Figures 2A and 2B), confirming that CypA, and its prolyl isomerase activity, contributes to Jak2/Stat5 activity. Thus, the action of CypA may occur either through its direct inhibition of the Jak2 kinase or indirectly by inhibiting the larger interactions within the PRLr-Jak2 Complex.
NIM811 had significant effects on PRL-induced gene expression at both global and individual gene levels. Both CISH and cyclin D1 genes involved in the pathogenesis of breast cancer (Arnold and Papanikolaou, 2005; Borges et al., 2008) demonstrate significantly reduced expression at the RNA and protein levels (Figure 3), Transcriptomic analysis revealed the global contribution of CypA to PRLr-induced gene expression identifying several PRL-induced and NIM811-inhibited genes such as EGR3, KLF4, and LIN28A. Each of these genes has been found to contribute to the pathobiology of breast cancer, as follows: (1) ERG3 is a regulator of estrogen-mediated invasion and a potent prognostic factor in breast cancer (Suzuki et al., 2007); (2) KLF4 is required for maintenance of breast cancer stem cells, migration, and invasion (Yu et al., 2011); and (3) Lin28A facilitates breast cancer metastasis and promotes cell cycle by regulating cyclin D1 (Xiong et al., 2017). Functional analyses revealed that these data found cancer, cellular proliferation/growth, migration/invasion, and gene expression as the top molecular functions affected by NIM811 treatment. The fact that NIM811 down-regulated 57% of top 100 PRL-induced genes involved in a proliferative and migratory/invasive phenotype is strengthened by our observation that NIM811 also decreased the proliferation, anchorage-independent growth, and migration of breast cancer cells. Furthermore, when NIM811-inhibited genes were compared with gene signature from kinase perturbation experiments in the GEO database, significant similarities between Jak2 knockdown, LRRK2 (kinase) mutant, GSK3β knockdown, SYK and IGF1 inhibition, and NIM811-treated cohorts were observed. Not surprising, the Jak2 knockdown array generated the most similar gene signature as our CypA inhibition array, further confirming the relationship between these enzymes. It is interesting to note that leucine-rich repeat kinase 2 (LRRK2) mutant that induces progressive degeneration of human neural stem cells (Liu et al., 2012) is also associated with breast cancer (Waro and Aasly, 2018), but its mechanistic role is poorly understood. Glycogen synthase kinase 3β (GSKβ) interacts with the PRLr (Plotnikov et al., 2008) and is activated by the PRLr, hence the relation between it and the NIM811 gene set. Likewise, SYK kinase appears to be activated by the PRLr (Saha et al., 2009). Similarly, IGF signaling directly affects PRLr transduction and has a recognized role in breast cancer pathobiology (Carver et al., 2010). Taken together, these findings suggest that CypA is a key regulator of PRL-induced global genes that are implicated in inducing malignant phenotypes in breast cancer.
Treatment of murine xenograft model of TNBC breast cancer with NIM811 resulted in decreased lymph node metastasis and induction of necrosis, which are likely due to changes in receptor proximal signaling (Figures 1 and 2) and gene expression (Figures 3 and 4). Interestingly, whereas NIM811 therapy modestly affected tumor volume in vivo, treatment of TNBC xenografts with NIM811 induced central tumor cell death and the outgrowth of macro-metastasis. This phenomenon was also seen with the immunosuppressive, CsA (Zheng et al., 2008). There are a few of potential mechanisms through which these NIM811-mediated effects on xenograft progression may have been executed. The blockade of the PRLr-Jak2-Stat5 pathway, the activity of which has been linked to malignant phenotypes of human breast cancer (Holtkamp et al., 1984), is perhaps the most obvious. However, NIM811 treatment also significantly reduced PRL-responsive regulatory genes such as CISH and cyclinD1 as well as the global network of genes involved in promoting cancer malignancies (Figure 4).
The ability of NIM811 to block the malignant phenotype in vitro of breast cancer cells (Figures 5 and 6) revealed that NIM811 is equipotent to CsA at inhibiting proliferation, survival, motility, and anchorage-independent growth (Zheng et al., 2008). Similarly, NIM811 significantly inhibited lymph node and lung metastasis in vivo. As the sole functional difference between CsA and NIM811 is the ability of CsA to engage calcineurin (Ma et al., 2006), it is probable that the inhibition of calcineurin has little to do with the anti-cancer properties of the cyclosporine. Instead, these findings would argue that these properties reside in the ability of CsA and NIM811 to bind and block the prolyl isomerase activity of CypA, a finding further confirmed by the loss of signaling and anti-cancer of prolyl isomerase-defective form of CypA (Zheng et al., 2008).
This study used two complementary in vivo models of mammary carcinoma, namely, human xenografts and the PyMT mouse model. Each model has its advantages and disadvantages: whereas human xenograft models are performed in an immunocompromised model that lacks an immune system, the PyMT model is purely murine, but has an intact immune system. TNBC cells were introduced by teat injection (Harrell et al., 2006) into immunocompromised nude mice, and a significant reduction in lymph node metastasis (Figure 7B) and an increase in central tumor necrosis (Figure 7A) were observed. Although all NIM811-treated tumors revealed a trend toward a reduction in volume over the period, these parameters did not achieve statistical significance (Figure S6). Interestingly, although individual GFP-positive MDA231 cells were observed in xenograft lymph nodes, the outgrowth of macro-metastasis was completely inhibited in these mice (Figure S7). This would argue that NIM811 affects metastatic outgrowth, but does not alter micro-metastatic seeding, and further studies are planned to examine the mechanistic basis for this effect. Significant alterations in mammary cancer progression were also observed in the PyMT spontaneous model (Volker et al., 2018). CypA−/- × PyMT mice demonstrated a highly significant decrease in tumor burden in comparison with the CypA+/+ × PyMT cohorts (Figure 7). Loss of CypA in the PyMT mice demonstrated a significant difference in lymph node metastases compared with the WT control, even after correcting for tumor burden. Although the CypA−/- and Jak2−/− mice show parallels in their effects on tumor initiation (Sakamoto et al., 2009, 2010; Zheng et al., 2008), CypA−/- mice demonstrated a significant inhibition of metastasis, which was not observed in the Jak2−/− mice. This may be due to the additional signaling pathways inhibited by CypA, such as Src, Akt, and MAPK (Figure S1) (Garcia-Martinez et al., 2010; Watkin et al., 2008) (Zheng et al., 2008).
We have found that non-immunosuppressive NIM811 has profound effects on tumorigenesis, metastasis, and associated signaling in breast cancer cell and mammary cancer models. As a therapeutic agent, NIM811 and other non-immunosuppressive cyclosporines, such as Debio025 (alisporivir) SCY-635 and CRV431 have minimal toxicity and have successfully undergone phase I–III clinical trials for safety in patients (Lawitz et al., 2011; Sweeney et al., 2014). Additional pre-clinical studies with approved and experimental breast cancer therapeutics are warranted to identify additional synergies of these agents, given that their benign toxicity profile also hold promises as chemo-preventative agents.
Limitations of the Study
Investigation of mechanisms behind CypA regulation of the structure/functions relationships of the prolactin receptor would strengthen the conclusions of the study, which was beyond the scope of this article. Furthermore, NIM811 treatment of a PDX model would further demonstrate the efficacy of the drug in a relevant breast cancer model in addition to the mammary xenografts study included in this article.
Resources Availability
Lead Contact
Further information and requests should be directed to the Lead Contact, Charles V. Clevenger (Charles.clevenger@vcuhealth.org).
Materials Availability
Any unique reagents are available from the Lead Contact on request.
Data and Code Availability
The datasets supporting the current study are available from the corresponding author on request. The accession number for the microarray data reported in this paper is NCBI GEO: GSE151635.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Xianke Zeng for help with mouse xenograft studies and helpful discussions. We thank Senthil Radhakrishnan for critical review of this paper. Services and products in support of research project were generated by the Virginia Commonwealth Transgenic/knockout Mouse Core, Cancer Mouse Models Core Laboratory, and the VCU Microscopy Facility, supported in part with funding to the Massey Cancer Center from NIH-NCI Cancer Center Support Grant P30 CA016059 and by the Department of Pathology, Anatomic Pathology Research Services, at VCU Health. We thank Novartis for gifting NIM811. This work was supported in part by grants from the NIH, R01 CA173305.
Author Contributions
S.H. performed all the experiments; S.H., J.M.C., and J.E.K., performed all analysis, and all authors wrote and edited the paper.
Declaration of Interests
No potential conflicts of interest were disclosed by the authors.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101581.
Contributor Information
Shawn Hakim, Email: hakims@vcu.edu.
Charles V. Clevenger, Email: charles.clevenger@vcuhealth.org.
Supplemental Information
References
- Arnold A., Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. 2005;23:4215–4224. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]
- Borges S., Moudilou E., Vouyovitch C., Chiesa J., Lobie P., Mertani H., Raccurt M. Involvement of a JAK/STAT pathway inhibitor: cytokine inducible SH2 containing protein in breast cancer. Adv. Exp. Med. Biol. 2008;617:321–329. doi: 10.1007/978-0-387-69080-3_30. [DOI] [PubMed] [Google Scholar]
- Bousoik E., Montazeri Aliabadi H. "Do we know jack" about JAK? A closer look at JAK/STAT signaling pathway. Front. Oncol. 2018;8:287. doi: 10.3389/fonc.2018.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman J.L., Schuler L.A. Prolactin signals via Stat5 and Oct-1 to the proximal cyclin D1 promoter. Mol. Cell Endocrinol. 2005;239:45–53. doi: 10.1016/j.mce.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C.L. Molecular mechanisms of prolactin and its receptor. Endocr. Rev. 2012;33:504–525. doi: 10.1210/er.2011-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver K.C., Piazza T.M., Schuler L.A. Prolactin enhances insulin-like growth factor I receptor phosphorylation by decreasing its association with the tyrosine phosphatase SHP-2 in MCF-7 breast cancer cells. J. Biol. Chem. 2010;285:8003–8012. doi: 10.1074/jbc.M109.066480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger C.V. Roles and regulation of stat family transcription factors in human breast cancer. Am. J. Pathol. 2004;165:1449–1460. doi: 10.1016/S0002-9440(10)63403-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger C.V., Furth P.A., Hankinson S.E., Schuler L.A. The role of prolactin in mammary carcinoma. Endocr. Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger C.V., Kline J.B. Prolactin receptor signal transduction. Lupus. 2001;10:706–718. doi: 10.1191/096120301717164949. [DOI] [PubMed] [Google Scholar]
- DaSilva L., Howard O.M., Rui H., Kirken R.A., Farrar W.L. Growth signaling and JAK2 association mediated by membrane-proximal cytoplasmic regions of prolactin receptors. J. Biol. Chem. 1994;269:18267–18270. [PubMed] [Google Scholar]
- Fang F., Rycyzyn M.A., Clevenger C.V. Role of c-Myb during prolactin-induced signal transducer and activator of transcription 5a signaling in breast cancer cells. Endocrinology. 2009;150:1597–1606. doi: 10.1210/en.2008-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo A.A., Medler T.R., Feeney Y.B., Wetz S.M., Tommerdahl K.L., Clevenger C.V. The prolactin receptor transactivation domain is associated with steroid hormone receptor expression and malignant progression of breast cancer. Am. J. Pathol. 2013;182:217–233. doi: 10.1016/j.ajpath.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S.L., Clevenger C.V. Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol. Endocrinol. 2006;20:2734–2746. doi: 10.1210/me.2006-0114. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez J.M., Calcabrini A., Gonzalez L., Martin-Forero E., Agullo-Ortuno M.T., Simon V., Watkin H., Anderson S.M., Roche S., Martin-Perez J. A non-catalytic function of the Src family tyrosine kinases controls prolactin-induced Jak2 signaling. Cell Signal. 2010;22:415–426. doi: 10.1016/j.cellsig.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Gertler A., Grosclaude J., Strasburger C.J., Nir S., Djiane J. Real-time kinetic measurements of the interactions between lactogenic hormones and prolactin-receptor extracellular domains from several species support the model of hormone-induced transient receptor dimerization. J. Biol. Chem. 1996;271:24482–24491. doi: 10.1074/jbc.271.40.24482. [DOI] [PubMed] [Google Scholar]
- Gothel S.F., Marahiel M.A. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell. Mol. Life Sci. 1999;55:423–436. doi: 10.1007/s000180050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschumacher R.E., Harding M.W., Rice J., Drugge R.J., Speicher D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- Harrell J.C., Dye W.W., Allred D.C., Jedlicka P., Spoelstra N.S., Sartorius C.A., Horwitz K.B. Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res. 2006;66:9308–9315. doi: 10.1158/0008-5472.CAN-06-1769. [DOI] [PubMed] [Google Scholar]
- Holtkamp W., Nagel G.A., Wander H.E., Rauschecker H.F., von Heyden D. Hyperprolactinemia is an indicator of progressive disease and poor prognosis in advanced breast cancer. Int. J. Cancer. 1984;34:323–328. doi: 10.1002/ijc.2910340307. [DOI] [PubMed] [Google Scholar]
- Kang K., Yamaji D., Yoo K.H., Robinson G.W., Hennighausen L. Mammary-specific gene activation is defined by progressive recruitment of STAT5 during pregnancy and the establishment of H3K4me3 marks. Mol. Cell Biol. 2014;34:464–473. doi: 10.1128/MCB.00988-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K., Yamaji D., Yoo K.H., Robinson G.W., Hennighausen L. Mammary-specific gene activation is defined by progressive recruitment of STAT5 during pregnancy and the establishment of H3K4me3 marks. Mol. Cell Biol. 2014;34:464–473. doi: 10.1128/MCB.00988-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly P.A., Binart N., Freemark M., Lucas B., Goffin V., Bouchard B. Prolactin receptor signal transduction pathways and actions determined in prolactin receptor knockout mice. Biochem. Soc. Trans. 2001;29:48–52. doi: 10.1042/0300-5127:0290048. [DOI] [PubMed] [Google Scholar]
- Lammers M., Neumann H., Chin J.W., James L.C. Acetylation regulates cyclophilin A catalysis, immunosuppression and HIV isomerization. Nat. Chem. Biol. 2010;6:331–337. doi: 10.1038/nchembio.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawitz E., Godofsky E., Rouzier R., Marbury T., Nguyen T., Ke J., Huang M., Praestgaard J., Serra D., Evans T.G. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antivir. Res. 2011;89:238–245. doi: 10.1016/j.antiviral.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Lee J., Kim S.S. Current implications of cyclophilins in human cancers. J. Exp. Clin. Cancer Res. 2010;29:97. doi: 10.1186/1756-9966-29-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard W.J., O'Shea J.J. Jaks and STATs: biological implications. Annu. Rev. Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Levy D.E., Darnell J.E., Jr. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Liu G.H., Qu J., Suzuki K., Nivet E., Li M., Montserrat N., Yi F., Xu X., Ruiz S., Zhang W. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491:603–607. doi: 10.1038/nature11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Boerner J.E., TiongYip C., Weidmann B., Ryder N.S., Cooreman M.P., Lin K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 2006;50:2976–2982. doi: 10.1128/AAC.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertani H.C., Garcia-Caballero T., Lambert A., Gerard F., Palayer C., Boutin J.M., Vonderhaar B.K., Waters M.J., Lobie P.E., Morel G. Cellular expression of growth hormone and prolactin receptors in human breast disorders. Int. J. Cancer. 1998;79:202–211. doi: 10.1002/(sici)1097-0215(19980417)79:2<202::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Miller S.L., DeMaria J.E., Freier D.O., Riegel A.M., Clevenger C.V. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol. Endocrinol. 2005;19:939–949. doi: 10.1210/me.2004-0443. [DOI] [PubMed] [Google Scholar]
- Paoli P., Giannoni E., Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta. 2013;1833:3481–3498. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- Perks C.M., Keith A.J., Goodhew K.L., Savage P.B., Winters Z.E., Holly J.M. Prolactin acts as a potent survival factor for human breast cancer cell lines. Br. J. Cancer. 2004;91:305–311. doi: 10.1038/sj.bjc.6601947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet A., Ferrag F., Kelly P.A., Edery M. Tyrosine docking sites of the rat prolactin receptor required for association and activation of stat5. J. Biol. Chem. 1997;272:25043–25050. doi: 10.1074/jbc.272.40.25043. [DOI] [PubMed] [Google Scholar]
- Plotnikov A., Li Y., Tran T.H., Tang W., Palazzo J.P., Rui H., Fuchs S.Y. Oncogene-mediated inhibition of glycogen synthase kinase 3 beta impairs degradation of prolactin receptor. Cancer Res. 2008;68:1354–1361. doi: 10.1158/0008-5472.CAN-07-6094. [DOI] [PubMed] [Google Scholar]
- Raccurt M., Tam S.P., Lau P., Mertani H.C., Lambert A., Garcia-Caballero T., Li H., Brown R.J., McGuckin M.A., Morel G. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br. J. Cancer. 2003;89:524–532. doi: 10.1038/sj.bjc.6601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S., Cai H.R., Li M., Furth P.A. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4335–4339. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- Roehrl M.H., Kang S., Aramburu J., Wagner G., Rao A., Hogan P.G. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc. Natl. Acad. Sci. U S A. 2004;101:7554–7559. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycyzyn M.A., Reilly S.C., O'Malley K., Clevenger C.V. Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Mol. Endocrinol. 2000;14:1175–1186. doi: 10.1210/mend.14.8.0508. [DOI] [PubMed] [Google Scholar]
- Saha S., Gonzalez J., Rosenfeld G., Keiser H., Peeva E. Prolactin alters the mechanisms of B cell tolerance induction. Arthritis Rheum. 2009;60:1743–1752. doi: 10.1002/art.24500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K., Lin W.C., Triplett A.A., Wagner K.U. Targeting janus kinase 2 in Her2/neu-expressing mammary cancer: implications for cancer prevention and therapy. Cancer Res. 2009;69:6642–6650. doi: 10.1158/0008-5472.CAN-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K., Triplett A.A., Schuler L.A., Wagner K.U. Janus kinase 2 is required for the initiation but not maintenance of prolactin-induced mammary cancer. Oncogene. 2010;29:5359–5369. doi: 10.1038/onc.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C., Shuai K., Prezioso V.R., Darnell J.E., Jr. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992;257:809–813. doi: 10.1126/science.1496401. [DOI] [PubMed] [Google Scholar]
- Schmitt D., Andrews J., Tan M. Determination of breast cancer cell migratory ability. Methods Mol. Biol. 2016;1406:171–180. doi: 10.1007/978-1-4939-3444-7_14. [DOI] [PubMed] [Google Scholar]
- Shen Q., Brown P.H. Transgenic mouse models for the prevention of breast cancer. Mutat. Res. 2005;576:93–110. doi: 10.1016/j.mrfmmm.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Stewart T., Tsai S.C., Grayson H., Henderson R., Opelz G. Incidence of de-novo breast cancer in women chronically immunosuppressed after organ transplantation. Lancet. 1995;346:796–798. doi: 10.1016/s0140-6736(95)91618-0. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Inoue A., Miki Y., Moriya T., Akahira J., Ishida T., Hirakawa H., Yamaguchi Y., Hayashi S., Sasano H. Early growth responsive gene 3 in human breast carcinoma: a regulator of estrogen-meditated invasion and a potent prognostic factor. Endocr. Relat. Cancer. 2007;14:279–292. doi: 10.1677/ERC-06-0005. [DOI] [PubMed] [Google Scholar]
- Sweeney Z.K., Fu J., Wiedmann B. From chemical tools to clinical medicines: nonimmunosuppressive cyclophilin inhibitors derived from the cyclosporin and sanglifehrin scaffolds. J. Med. Chem. 2014;57:7145–7159. doi: 10.1021/jm500223x. [DOI] [PubMed] [Google Scholar]
- Syed F., Rycyzyn M.A., Westgate L., Clevenger C.V. A novel and functional interaction between cyclophilin A and prolactin receptor. Endocrine. 2003;20:83–90. doi: 10.1385/ENDO:20:1-2:83. [DOI] [PubMed] [Google Scholar]
- Volker S.E., Hedrick S.E., Feeney Y.B., Clevenger C.V. Cyclophilin A function in mammary epithelium impacts jak2/stat5 signaling, morphogenesis, differentiation, and tumorigenesis in the mammary gland. Cancer Res. 2018;78:3877–3887. doi: 10.1158/0008-5472.CAN-17-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.U., Krempler A., Triplett A.A., Qi Y., George N.M., Zhu J., Rui H. Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol. Cell Biol. 2004;24:5510–5520. doi: 10.1128/MCB.24.12.5510-5520.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.U., Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J. Mammary Gland Biol. Neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- Wang P., Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waro B.J., Aasly J.O. Exploring cancer in LRRK2 mutation carriers and idiopathic Parkinson's disease. Brain Behav. 2018;8:e00858. doi: 10.1002/brb3.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkin H., Richert M.M., Lewis A., Terrell K., McManaman J.P., Anderson S.M. Lactation failure in Src knockout mice is due to impaired secretory activation. BMC Dev. Biol. 2008;8:6. doi: 10.1186/1471-213X-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H., Zhao W., Wang J., Seifer B.J., Ye C., Chen Y., Jia Y., Chen C., Shen J., Wang L. Oncogenic mechanisms of Lin28 in breast cancer: new functions and therapeutic opportunities. Oncotarget. 2017;8:25721–25735. doi: 10.18632/oncotarget.14891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Li J., Chen H., Fu J., Ray S., Huang S., Zheng H., Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., Koblinski J.E., Dutson L.V., Feeney Y.B., Clevenger C.V. Prolyl isomerase cyclophilin A regulation of Janus-activated kinase 2 and the progression of human breast cancer. Cancer Res. 2008;68:7769–7778. doi: 10.1158/0008-5472.CAN-08-0639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study are available from the corresponding author on request. The accession number for the microarray data reported in this paper is NCBI GEO: GSE151635.