

Abstract
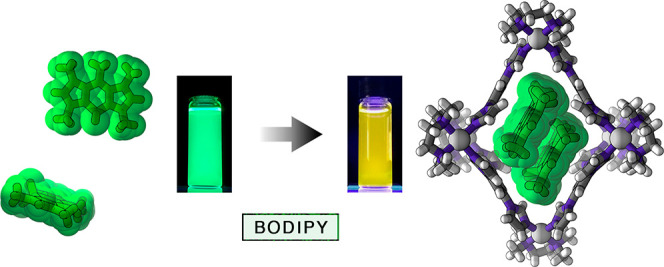
Aggregation of organic molecules can drastically affect their physicochemical properties. For instance, the optical properties of BODIPY dyes are inherently related to the degree of aggregation and the mutual orientation of BODIPY units within these aggregates. Whereas the noncovalent aggregation of various BODIPY dyes has been studied in diverse media, the ill-defined nature of these aggregates has made it difficult to elucidate the structure–property relationships. Here, we studied the encapsulation of three structurally simple BODIPY derivatives within the hydrophobic cavity of a water-soluble, flexible PdII6L4 coordination cage. The cavity size allowed for the selective encapsulation of two dye molecules, irrespective of the substitution pattern on the BODIPY core. Working with a model, a pentamethyl-substituted derivative, we found that the mutual orientation of two BODIPY units in the cage’s cavity was remarkably similar to that in the crystalline state of the free dye, allowing us to isolate and characterize the smallest possible noncovalent H-type BODIPY aggregate, namely, an H-dimer. Interestingly, a CF3-substituted BODIPY, known for forming J-type aggregates, was also encapsulated as an H-dimer. Taking advantage of the dynamic nature of encapsulation, we developed a system in which reversible switching between H- and J-aggregates can be induced for multiple cycles simply by addition and subsequent destruction of the cage. We expect that the ability to rapidly and reversibly manipulate the optical properties of supramolecular inclusion complexes in aqueous media will open up avenues for developing detection systems that operate within biological environments.
Introduction
Owing to their high chemical stability and photostability, sharp and intense absorption bands, and high fluorescence quantum yields, boron–dipyrromethene (BODIPY) derivatives are among the most widely studied fluorescent dyes.1−3 Furthermore, BODIPY chromophores can be easily and diversely functionalized; this has been used to modulate their optical properties,4−7 append them with solubilizing8−10 and analyte-binding11−15 groups, and attach them to larger entities, such as biomacromolecules.16−19 An additional advantage of BODIPY dyes is that their optical properties are strongly dependent on and can be modulated by the degree of dye aggregation as well as the mutual orientation of the dye molecules within the aggregates.20 The optical properties of such aggregates are typically rationalized by Kasha’s molecular exciton theory, which was described21 4 years before the first synthesis22 of BODIPY. According to this model, dipole–dipole interactions between two chromophore units split the energy of the excited state within the dimer into two energy levels. The HOMO/LUMO energy gap of the resulting dimer depends on the slip angle, θ, and the center-to-center distance between the coplanarly aligned transition dipoles of the two chromophore units. Despite certain limitations,23 the molecular exciton theory is a well-established model that explains how high slip angles of θ > 54.7° result in a blue shift of the main absorption band (due to the S1←S0 transition) in these so-called H-type aggregates (due to higher excitation energies; “H” after “hypsochromic”), whereas arrangements with θ < 54.7° give rise to decreased excitation energies (J-type aggregates; “J” after E. E. Jelley24).
One way to place two BODIPY units in close proximity is to connect them covalently through a short linker. Akkaya et al. installed two BODIPY groups on a xanthene scaffold;25 as expected, the cofacial arrangement of the two groups (i.e., θ ≈ 90°) resulted in a hypsochromic shift of the main absorption band from 492 to 478 nm. By systematically decreasing θ using various scaffolds, from xanthene to dibenzothiophene, Alamiry, Benniston, and co-workers tuned the spectral properties of their bis-BODIPYs and reported an efficient excimer emission for the latter scaffold.26 More recently, Bröring et al. coined the term “DYEmers” to describe the covalent oligomers of BODIPY units connected in different ways, including sulfur bridges27,28 and C=C bonds formed by the olefin cross-metathesis reaction.29
An alternative approach for inducing dye–dye communication is based on noncovalent aggregation of BODIPYs in nonsolvents and various other media.30 In an early study, Pagano et al. observed the formation of fluorescent J-type aggregates upon embedding a BODIPY derivatized with a long alkyl chain within lipid membranes.31 More recently, Fernández and co-workers designed a bolaamphiphilic BODIPY derivative that self-assembles into fibrillar H-aggregates,32 whereas Chen et al. observed the coexistence of H- and J-aggregates within vesicular aggregates formed by an amphiphilic BODIPY,33 among numerous other examples of controlled self-assembly in water.34−37 Other media, in which BODIPY aggregation has been investigated, include liquid crystals,38−40 micelles and vesicles,16,41 latex beads,42 and sodium silicate sol–gel glass.43 However, the above systems all suffer from a lack of control over the number of BODIPY units within the aggregates.
Formation of well-defined aggregates of BODIPY comprising a defined number of chromophore units arranged in a particular way might be possible using the host–guest approach. Over the past two decades, an increasing number of molecular and supramolecular capsules, cages, and boxes have been developed.44−46 Among them, metal–organic (coordination) cages are particularly interesting.47−52 These cages often carry multiple charges and have good water solubility; at the same time, their interiors are usually strongly nonpolar, facilitating the encapsulation of various organic “guest” molecules by means of the hydrophobic effect.53 When these interiors are sufficiently large, the simultaneous binding of two or more guests is possible.54 Notable examples include a PdII-based cylindrical cage capable of encapsulating three molecules of pyrene-4,5-dione,55 a cubic FeII-based cage stabilizing coronene trimers,56 and a PtII-based cage capable of coencapsulating a BODIPY derivative with planar aromatic guests, such as phenanthrene and anthracene.53a Recently, Yoshizawa and co-workers reported that two structurally related polyaromatic PtII2L4 cages could simultaneously bind two molecules of an asymmetrically substituted BODIPY derivative.57 However, a general methodology for encapsulating various BODIPY derivatives as dimers has remained unknown.
Here, we worked with a polycationic [Pd6L4]12+ cage, where L = 1,3,5-triimidazoylbenzene (TIm) (1 in Figure 1a).58 Owing to its high water solubility, hydrophobic interior, and high flexibility,59 cage 1 can encapsulate—and effectively solubilize in water—a wide range of structurally diverse guests.59−62 We show that cage 1 can undergo a structural distortion to accommodate two molecules of various BODIPY dyes. The mutual arrangement of the dye molecules within these BODIPY dimers is remarkably similar to that found in the crystal structures of respective BODIPYs, which feature an antiparallel arrangement of the neighboring molecules (H-aggregation). The unique stabilization of BODIPY dimers allowed us to study the spectral properties of the smallest possible noncovalent BODIPY H-aggregates, i.e., H-dimers. We also worked with a BODIPY derivative that has a tendency to form J-aggregates featuring parallel packing of the chromophore units with relatively small slip angles. We found that cage 1 drastically affects the aggregation behavior of this dye, forcing it to form the otherwise unstable H-dimers. Taking advantage of this finding, we developed a system allowing us to toggle the mutual orientation of the dye molecules and thus the optical properties reversibly for at least several cycles.
Figure 1.
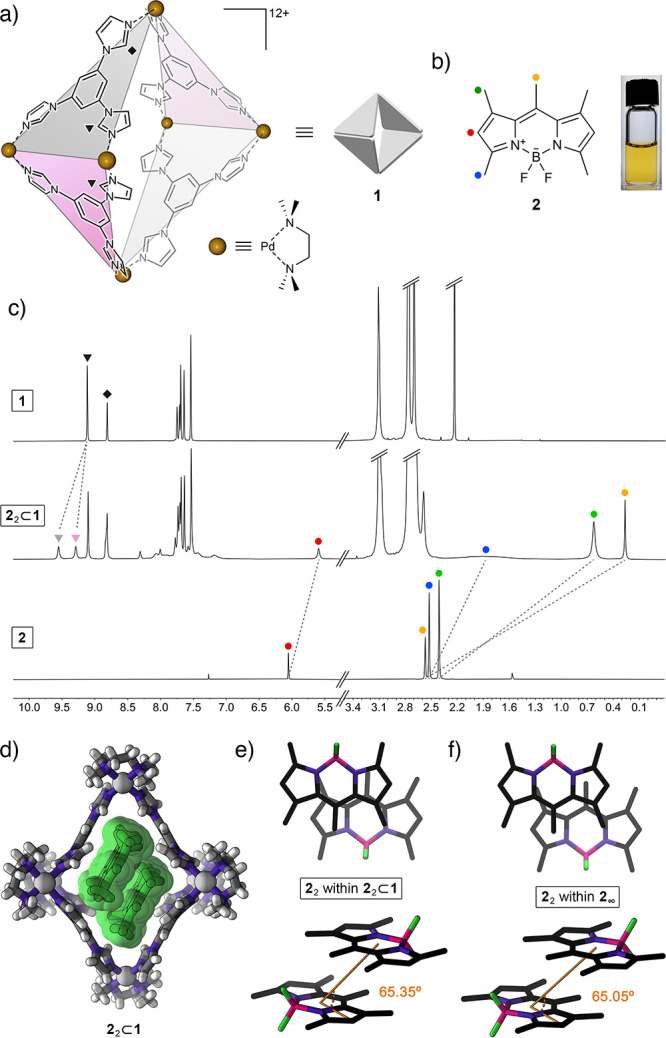
(a) Structural formula of coordination cage 1 based on six Pd2+ nodes and four triimidazole (TIm) ligands. Two kinds of acidic imidazole protons (equivalent in free TIm; nonequivalent within the cage) are denoted by triangles (equatorial acidic imidazole protons) and a diamond (axial acidic imidazole protons). Guest binding within the cage can further differentiate between equatorial protons within TIm ligands oriented parallel to the bound guest (pink panels) and perpendicular to it (gray panels). (b) Structural formula of pentamethyl-substituted BODIPY 2 (left), and photograph of 2 dissolved in an aqueous solution of cage 1. (c) Partial 1H NMR spectra of the empty cage 1 (top), 22⊂1 (middle) (500 MHz, D2O), and free 2 (bottom; 400 MHz, CDCl3). (d) Crystal structure of 22⊂1. (e) Top view (top) and side view (bottom) of the packing motif of 2 within the crystal of the 22⊂1 inclusion complex (cage 1 is not shown for clarity). (f) Top view (top) and side view (bottom) of the packing motif within the crystal of 2.66
Results and Discussion
We previously reported that cage 1 forms inclusion complexes with various azobenzene derivatives.60 Within these complexes, two azobenzene guest molecules occupy the nonpolar cavity of the host cage. On the basis of some structural similarity between azobenzene and common BODIPY dyes, we hypothesized that cage 1 would similarly form 1:2 complexes with various BODIPYs. We began our study with the widely investigated pentamethyl-substituted BODIPY 2 (Figure 1b).63−66 We were pleased to find that upon stirring with solid 2 (otherwise insoluble in water), the transparent aqueous solution of 1 turned intense yellow, indicative of the complexation of 2. The 1H NMR spectrum obtained upon stirring a solution of 1 in D2O over an excess of solid 2 at room temperature for 24 h is shown in Figure 1c (middle). The spectrum shows the presence of a complex comprising two molecules of guest inside the cage (i.e., 22⊂1; see below for the stoichiometry determination) and the residual free cage. To differentiate between the peaks due to empty and filled 1, we removed undissolved 2 and titrated the mixture of 22⊂1 and 1 with an excess of free 1 (see Figure S16).
The DOSY spectrum showed that all of the species in the solution had the same diffusion coefficient, confirming that 2 was quantitatively associated with cage 1 (Figure S19). We comprehensively characterized 22⊂1 with a suite of 2D NMR techniques, including COSY, NOESY, HSQC, HMBC, and HOESY (Figures S20–S28).
Integrating the signals in the 1H NMR spectrum (Figure 1c (middle) and Figure S15) showed that empty and filled 1 coexisted in an ∼1:1 ratio (we verified that stirring for a longer time, heating, and applying sonication did not increase the encapsulation yield of 2 and the other BODIPYs (see below)). The fact that only ∼50% of the cages become filled may seem surprising given the presence of an excess of the guest. However, we observed a similar effect previously and explained it59,60 by the very low aqueous solubility of free guest, [2], which, even for a high association constant (Kassoc = [22⊂1]/[1]·[2]2), will leave a substantial amount of the cage unfilled (i.e., [1] ≫ 0). The low solubility of free 2 in water makes the determination of Kassoc difficult. However, experiments in which we subjected cage 1 to competitive binding of 2 and azobenzene (a guest whose association constant with 1 was previously determined60) allowed us to conclude that Kassoc > 109 M–2 (Supporting Information, Section 11).
Further evidence for the 1:1 mixture of 22⊂1 and 1 (as opposed to a hypothetical 2⊂1 complex) was obtained by evaporating water from an aqueous solution of 1 saturated with 2. In this experiment, we observed the formation of a mixture of colorless crystals of the unfilled cage and red crystals, verified by a single-crystal X-ray diffraction structure solution as 22⊂1 (see below).
The 1H NMR spectrum of 22⊂1 shows that all four signals due to 2’s protons are upfield shifted, which can be attributed to the presence of 2 inside the hydrophobic, aromatic cavity of cage 1; indeed, the largest shift of ∼2.3 ppm was observed for 2’s central methyl group (yellow circle in Figure 1). Detailed analysis of 1D and 2D NMR spectra reveals some interesting insights into the structure and dynamics of the 22⊂1 complex. First, the signal due to protons adjacent to the BF2 moiety (blue in Figure 1) shows very significant broadening, which can be explained by through-space 1H–19F coupling (C–H···F distance = 2.50 Å in the crystal structure66 of free 2). This coupling is not observed in a chloroformic solution of free 2 (Figure 1c, bottom) due to efficient solvation and fast rotation of the methyl group. We hypothesize, however, that encapsulating 2 inside the cavity of 1 restricts the rotation, reinforcing the intramolecular hydrogen bonding. Second, we found that upon binding two molecules of guest 2 within the cage, many signals originating from 1’s TIm walls split into sets of two signals—a phenomenon that was not observed previously for complexes incorporating one guest molecule59,62 or two guests lacking extended aromatic systems.60,61 For 22⊂1, this splitting is best seen for 1’s equatorial acidic imidazole protons (denoted by triangles in Figure 1); it originates from interactions with the aromatic system of guest 2, resulting in the desymmetrization of two adjacent TIm panels (pink and gray in Figure 1a). Specifically, two molecules of BODIPY 2 within cage 1 are oriented parallel to each other and parallel to the “pink” panels and roughly perpendicular to the “gray” panels, which explains why the “pink” protons of 1 appear upfield compared with its “gray” protons (this differentiation is better illustrated for complex 32⊂1; see below). Third, the 1H–19F HOESY spectrum of 22⊂1 (Figure S28) shows a pronounced through-space correlation between 2’s fluorine and hydrogen on the opposite side of the molecule (the meso position; yellow in Figure 1b). This result suggests that the two molecules of 2 are arranged inside 1 in an antiparallel fashion. Furthermore, 1H–19F HOESY allowed us to observe strong correlations between the BF2 groups of the guests and 1’s axial acidic imidazole protons (diamond in Figure 1a), offering insights into the orientation of the antiparallel 2 dimer within 1. Taken together, these results provide a detailed picture of the structure of 22⊂1. Notably, this picture is in full agreement with the structure elucidated by X-ray crystallography (see below).
Single crystals of 22⊂1 were obtained by slow water evaporation from an aqueous solution of 1 saturated with 2. As mentioned above, this experiment afforded a mixture of colorless crystals of unfilled 1 (these crystals were discarded) and red crystals of 22⊂1, which were collected for X-ray diffraction. The crystal structure of 22⊂1 is shown in Figure 1d. Compared with the structure of empty 1, the cage containing two molecules of 2 is significantly extended in the vertical direction with a relatively large (18.44 Å) distance between the two axial Pd2+ centers (compare with 16.86 Å for the undeformed, empty 1(58)). Although this deformation is unfavorable,67 it is compensated by attractive interactions with the encapsulated guest molecules. Overall, the structure of 22⊂1 is similar to that of the previously reported 2:1 complexes of cage 1.60,61
Within the cavity of cage 1, two molecules of BODIPY 2 are arranged coplanar to each other and to two TIm panels of the cage, giving rise to an extended TIm···2···2···TIm π–π stack (Figure 1d). As expected, the two guests are arranged in an antiparallel fashion with (i) their BF2 moieties pointing toward the axial palladium metal centers and (ii) the methyl groups at the meso positions and their CH3 neighbors (yellow and green in Figure 1b, respectively) residing within the equatorial area of the cage. We found that the mutual orientation of the two guests within the cage (Figure 1e) is very similar to that of two neighboring molecules within the crystal of pure 2 (Figure 1f). Specifically, the plane-to-plane distances amount to 3.52 and 3.58 Å for free and encapsulated 2, whereas the slip angles, θ, are 65.05° and 65.35° for free and encapsulated 2, respectively (see Figure 1e and 1f and Supporting Information, Section 8). On the basis of this remarkable similarity, we postulate that cage 1 has the ability to isolate and stabilize the smallest repeating unit of crystalline 2. Importantly, 1’s ability to stabilize the noncovalent dimer of 2 provides us with the opportunity to investigate the optical properties of the minimal H-type aggregate of 2, i.e., the H-dimer.
Figure 2a (top) shows the absorption and emission spectra of an aqueous solution of the 22⊂1 inclusion complex. Compared with a solution of free 2 in acetonitrile (Figure 2a (bottom); λmax = 492 nm), the main absorption band is less intense, broadened, and blue-shifted (λmax = 480 nm), signifying an increased HOMO/LUMO gap for the caged dimer. On the other hand, the emission of 2 within 1 is largely quenched (with the fluorescence quantum yield decreasing from ΦF = 1.00 for free 2 in chloroform4 to ΦF = 0.13 for 22⊂1 in water), and it is broadened and red-shifted, appearing yellow (Figure 2a (top); λmax = 544 nm), in sharp contrast to the characteristic bright green emission of free 2 in an organic solution (Figure 2a (bottom); λmax = 502 nm).
Figure 2.
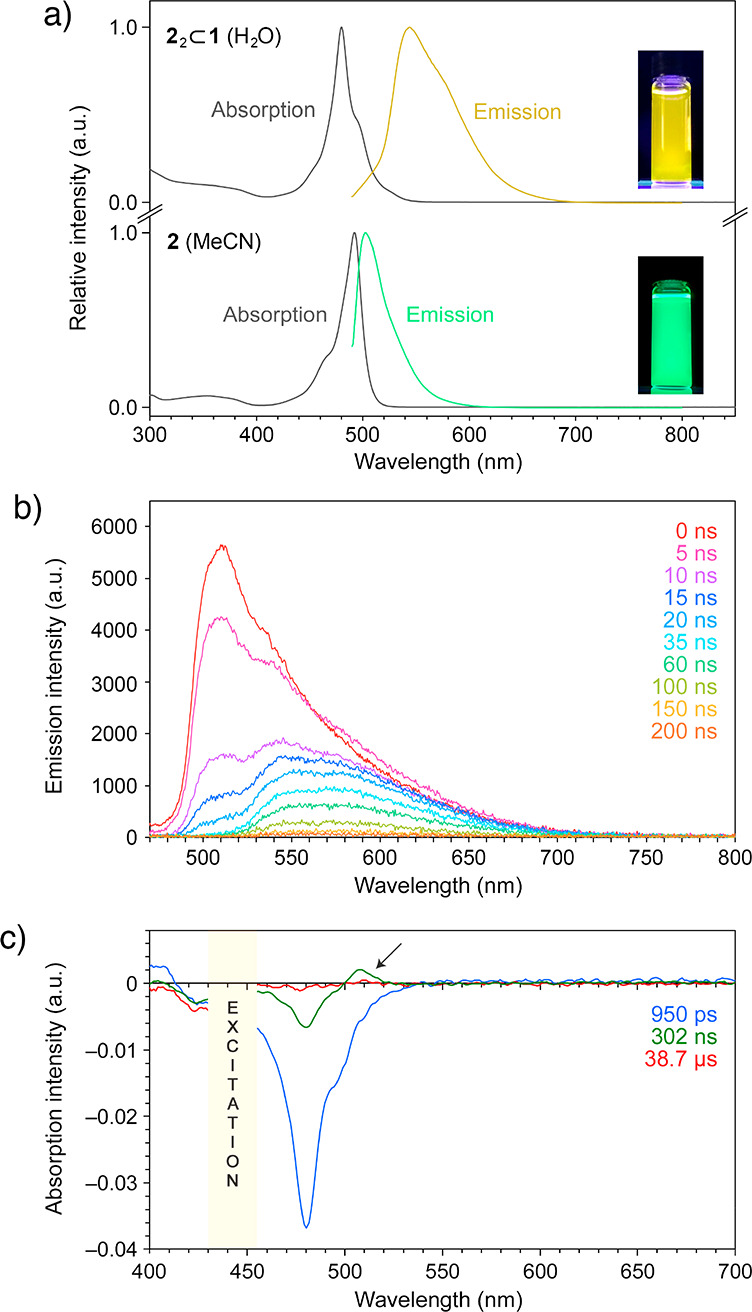
(a) Steady-state absorption and fluorescence (λexc = 460 nm) spectra of inclusion complex 22⊂1 in water (top) and free BODIPY 2 in acetonitrile (bottom). (b) Time-resolved fluorescence spectra of inclusion complex 22⊂1 in water. (c) Time-resolved absorption spectra of 22⊂1 in water. Arrow denotes transient absorption due to the triplet excited state.
To obtain further insight into the optical properties of 22⊂1, we performed transient fluorescence and absorption spectroscopy studies. The series of time-resolved emission spectra in Figure 2b show a major peak at ∼510 nm whose decay can be fitted to a single exponential with a time constant 8.70 (±0.43) ns. A similar behavior was found for a MeCN solution of free 2, which features a single peak at 508 nm, decaying with a similar time constant of 6.71 (±0.15) ns (Supporting Information, Figure S62a), in agreement with previous reports on free 2 and 2 encapsulated as a monomer.53a The inclusion complex 22⊂1 exhibited a more complex emission profile with two additional broad bands at ∼540 and ∼570 nm whose decay could not be fitted with single exponentials (instead, they were fitted with double exponentials with time constants of 11.4 (±1.6) and 89.5 (±43.9) ns for the ∼540 nm band and 4.58 (±0.63) and 56.4 (±1.9) ns for the ∼570 nm band).
Furthermore, we studied the optical properties of free and encapsulated 2 by transient absorption spectroscopy. Compound 2 in MeCN exhibited a pronounced bleach at 492 nm (Supporting Information, Figure S65a). The recovery of the bleach was fitted with a single exponential with a time constant of τ1/2 = 5.99 (±0.16) ns. For 22⊂1 in water, the main band due to bleach and stimulated emission was shifted to ∼480 nm, and it decayed significantly slower with τ1/2 = 57.5 (±2.0) ns (Figure 2c and Supporting Information, Figure S65b). In addition, we observed a transient absorption peak at ∼510 nm which could be attributed to the triplet state. The decay of this absorption was fitted with a single exponential with a time constant of τ1/2 = 14.5 (±5.1) μs. Overall, the optical properties of 22⊂1 are in agreement with Kasha’s model of exciton coupling in molecular dimers21 and with the antiparallel packing of 2 into H-dimers as elucidated by X-ray crystallography. At the same time, we note that whereas the optical properties of 22⊂1 are governed largely by 2···2 interactions, they can also be affected by the host cage.
The binding of 2 within the cavity of 1 in water is driven largely by hydrophobic interactions; therefore, the significant shift in the optical properties upon encapsulation of 2 could be reversed upon treating aqueous solutions of 22⊂1 with water-miscible organic solvents (Figures S52 and S53). We found, for example, that by titrating a solution of 22⊂1 with MeCN, both the absorption and the emission peaks increased in intensity until the values identical to those of free 2 in organic solvent were restored, which required addition of less than one volume of MeCN. Importantly, the quantitative release of 2 from the cage and the large difference in the optical properties of free and encapsulated 2 allowed us to confirm the percentage of cages filled with the guests. In this experiment, we first saturated a known amount of 1 dissolved in water with 2 and discarded excess solid 2. Then we released 2 with MeCN and compared the absorbance of the resulting solution with that of MeCN containing a known amount of 2 (the molar absorption coefficient of 2 in MeCN, ε ≈ 80 000 L mol–1 cm–1). We concluded that a solution of the cage contained 1.0 equiv of encapsulated 2, i.e., a 1:1 mixture of 1 and 22⊂1, in agreement with the encapsulation efficiency determined by 1H NMR spectroscopy.
To verify that the observed H-dimerization is not limited to compound 2, we worked with another BODIPY, 3 in Figure 3a. Similar to 2, compound 3 is insoluble in water but can readily be solubilized in an aqueous solution of cage 1. Owing to the presence of two additional methyl groups and a CHO group at the meso position (the mesomeric effect), the main absorption bands of 3 and 32⊂1 (in MeCN and water, respectively) are strongly red-shifted with respect to 2 and 22⊂1 (by 42 and 40 nm, respectively, giving rise to pink-colored solutions; see Figure 3b); in addition, the fluorescence of 3 is largely quenched. Similar to 2, the encapsulation of 3 resulted in a 12 nm blue shift of the main absorption band, suggesting the presence of H-dimers inside the cavities of 1. Interestingly, 1H NMR spectroscopy (Figure 3c) showed the presence of a single species, 32⊂1, demonstrating that 3, unlike BODIPY 2, did not leave any cages unfilled. This result can be explained by a higher aqueous solubility of 3 and/or tighter binding within the cage (Figure S46). Importantly, the lack of residual signals of empty 1 (Figure 3c, top) allowed us to characterize the inclusion complex more comprehensively (Figures S29–S41), including the unambiguous assignment of all of the signals in the 1H NMR spectrum (Figure S29). We found that the shifts of proton resonances accompanying the encapsulation of 3 were very similar to those of 2 (compare Figure 3c with Figure 1c) with significant broadening of the signal due to the CH3 adjacent to the BF2 moiety (blue in Figure 3a and 3c). Furthermore, we observed strong NOE correlations between the peripheral CH3 protons (red in Figure 3a) and the equatorial imidazole protons within the “pink” TIm panels (δ = 7.51 and 7.17 ppm), which, in the absence of similar correlations with the imidazole protons of the “gray” TIm panels (δ = 7.62 and 7.97 ppm), confirms that the guests are bound parallel to the “pink” walls.
Figure 3.
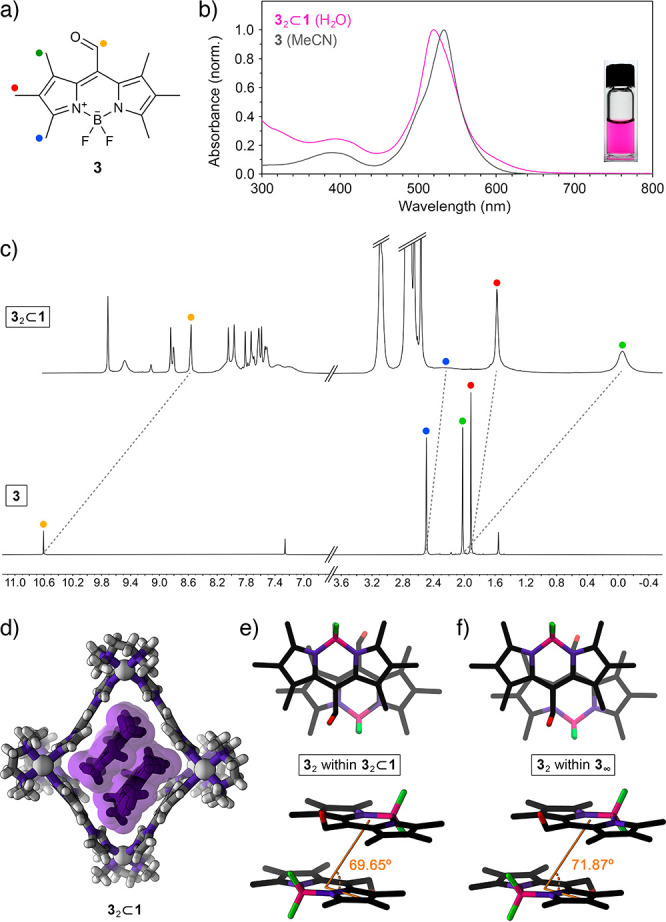
(a) Structural formula of CHO-substituted BODIPY 3. (b) UV–vis absorption spectra of 3 in MeCN (gray; λmax = 533 nm) and 32⊂1 in water (pink; λmax = 520 nm). (Inset) Photograph of 3 dissolved in an aqueous solution of cage 1. (c) Partial 1H NMR spectra of 32⊂1 (top; 500 MHz, D2O) and free 3 (bottom; 400 MHz, CDCl3). (d) Crystal structure of 32⊂1 (note that 32⊂1 crystallized as a 1:1 mixture of two conformers differing slightly in their structure; for the other conformer, see Figure S47). (e) Top view (top) and side view (bottom) of the packing motif of 3 within the crystal of the 32⊂1 inclusion complex (cage 1 is omitted for clarity; slip angle θ is denoted in orange; for the other conformer, θ = 73.45°). (f) Top view (top) and side view (bottom) of the packing motif within the crystal of 3.
Inclusion complex 32⊂1 crystallized as a 1:1 mixture of two conformers that differ slightly in the structure of the cage and the mutual orientation of the guests (see Supporting Information, Section 8). Most importantly, the slip angles, θ, amounted to 69.65° and 73.45° for the two conformers, within ∼2° of θ within the crystalline 3 (71.87°) and significantly different from θ for the encapsulated and crystalline 2 (65.35° and 65.05°, respectively). This result indicates that the structures of inclusion complexes 22⊂1 and 32⊂1 are governed primarily by BODIPY–BODIPY interactions (rather than BODIPY–cage interactions), allowing us to contend that cage 1 has the ability to isolate and stabilize BODIPY dimers in their near-native form (i.e., as they appear within extended H-aggregates).
The above results show that in the presence of BODIPYs 2 and 3, cage 1 undergoes structural deformation to accommodate in its cavity the minimal aggregates of these two chromophores. Next, we turned our attention to trifluoromethyl-substituted BODIPY derivative 4 (Figure 4a). Kim et al. reported that 4, despite its structural similarity to 2, has a tendency to form J-type (rather than H-type) aggregates, featuring a parallel orientation of the chromophores, with a large center-to-center distance and a small slip angle (θ = 37.86°).4,68 Typical UV–vis absorption spectra of nonaggregated 4 in MeCN and 4 aggregated in a water/MeCN mixture are shown in Figure 4b as gray and blue traces, respectively. Aggregated 4 forms a blue suspension with an intense absorption band centered above 600 nm, a sharp emission band at ∼626 nm (Figure S68), and a very small Stokes shift characteristic for J-aggregates. Interestingly, the aggregation of 4 increases ΦF by a factor of ∼40, transforming the virtually nonemissive free 4 into the fluorescent J-aggregates.4
Figure 4.
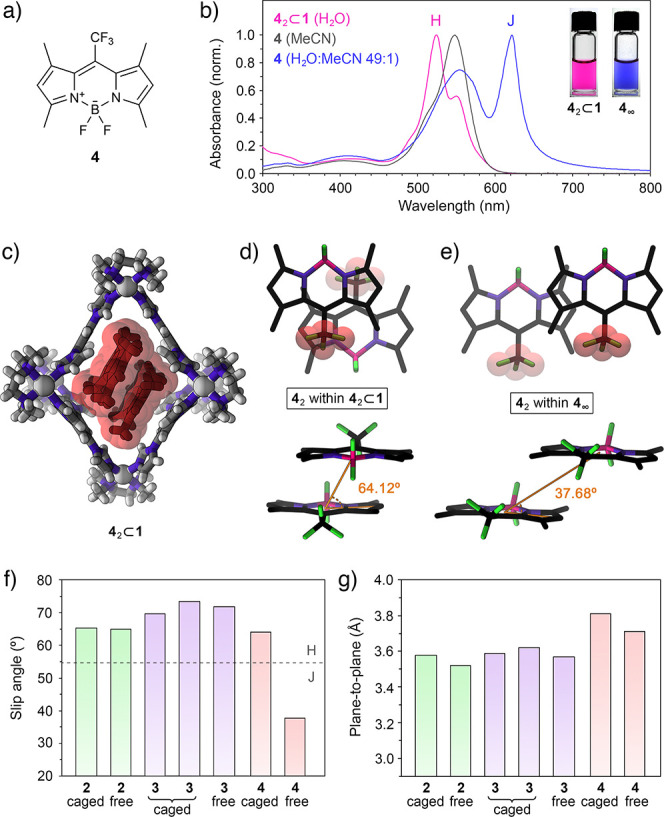
(a) Structural formula of CF3-substituted BODIPY 4. (b) UV–vis absorption spectra of free 4 in MeCN (gray), aggregated 4 in a 49:1 water/MeCN mixture (blue), and 42⊂1 in water (pink). (Inset) Photographs of aqueous solution of 42⊂1 (left) and aqueous suspension of 4 (right). (c) Crystal structure of 42⊂1. (d) Top view (top) and side view (bottom) of the packing motif of 4 within the crystal of the 42⊂1 inclusion complex (cage 1 is omitted for clarity). (e) Top view (top) and side view (bottom) of the packing motif within the crystal of 4.4 Van der Waals radii were included around the CF3 groups to emphasize the antiparallel (d) and parallel (e) arrangement of 4. (f) Summary of the slip angles for free and encapsulated BODIPYs 2–4. Dashed line denotes the transition between the H- and the J-aggregates at slip angle = 54.7°. (g) Summary of the plane-to-plane distances for free and encapsulated BODIPYs 2–4.
Encapsulation of the putative J-type dimer of 4 (see Figure 4e) would entail a prohibitively large structural deformation of 1. However, we found that an aqueous solution of 1 could readily dissolve BODIPY 4. The optical properties of the resulting solution (λmax = 524 nm; see the spectrum and photograph in Figure 4b) are remarkably similar to those of 32⊂1. Indeed, X-ray crystallography revealed that 4 was encapsulated as an H-type dimer (Figure 4c), indicating that the encapsulation of 4 as a J-dimer is too demanding for 1; instead, the cage forces 4 to form an otherwise unstable H-dimer, similar to 2 and 3.
Inclusion complex 42⊂1 formed with an ∼62% yield, as determined by release experiments (Figure S53d–f) (i.e., 62% of the cages became filled in the presence of an excess of solid 4). Unfortunately, this complex featured relatively broad signals in the 1H NMR spectra (Figure S42) and could not be comprehensively characterized by 2D NMR techniques. However, the crystal structure of 42⊂1 (Figure 4c) was remarkably similar to that of 22⊂1 and 32⊂1 (Figures 1d and 3d, respectively) except for the larger 4···4 distance (3.81 Å; compare with 3.58 Å for 2···2 and 3.59 Å/3.62 Å for 3···3) due to the relatively bulky CF3 groups.
Having found that the presence of cage 1 greatly affects the supramolecular organization of 4 in aqueous environments (i.e., J-aggregates vs H-dimers), we hypothesized that the repeated addition and removal of 1 could be used to reversibly modulate the optical properties of this BODIPY. To this end, we first titrated a solution of J-aggregates of 4 (obtained by diluting a methanolic solution of free 4 with 49 volumes of water) with cage 1 and found (see Supporting Information, Figure S67) that addition of 0.5 equiv of the cage with respect to 4 resulted in a spectrum identical to that of the 42⊂1 complex; no further changes were observed upon continued addition of 1 (except for sustained increase in absorption at <350 nm due to the accumulation of free cage). The disassembly of J-aggregates proceeded rapidly: upon adding 1.0 equiv of 1, formation of 42⊂1 was complete within 3 min (Figure 5a). The process could also be tracked by fluorescence spectroscopy (Figure S71).
Figure 5.
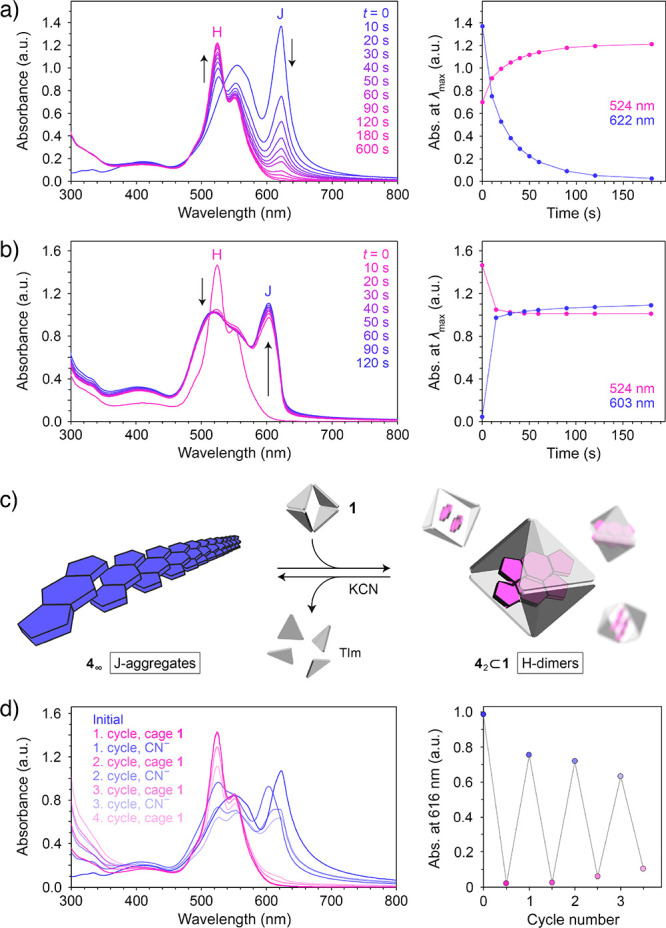
(a) Changes in the UV–vis absorption spectra of an aqueous suspension of J-aggregates of 4 induced by addition of 1.0 equiv of cage 1 (left), and changes in absorption at the wavelength of the maximum absorption of J-aggregates (here, 622 nm) and H-dimers (524 nm) (right). (b) Changes in the UV–vis absorption spectra of an aqueous solution of 42⊂1 induced by addition of 1.0 equiv of KCN (per Pd2+) (left), and changes in absorption at the wavelength of the maximum absorption of J-aggregates (here, 603 nm) and H-dimer (524 nm) (right). (c) Schematic representation of the reversible transformation between the J-aggregates and the H-dimers of 4. (d) Reversible changes in the UV–vis absorption spectra accompanying an alternating addition of cage 1 (0.7 equiv with respect to 4) and KCN (5.14 equiv per cage = 0.86 equiv with respect to Pd2+) (left), and changes in absorption due to J-aggregates over three cycles (note that the wavelength of the maximum absorption of the J-aggregates of 4 fluctuates in the 603–625 nm range depending on the degree of aggregation; thus, we followed the absorbance at a fixed wavelength within this range; 616 nm).
Next, we anticipated that the J-aggregates could be formed by releasing 4 encapsulated within 1. Such release could be accomplished by adding either a competitive guest with a high affinity to the cage cavity or a ligand with a high affinity to Pd2+, which would displace the TIm ligands from the Pd2+ centers, thus disintegrating the coordination cage.69 Here, we studied the controlled decomposition of the cage using CN–, which forms a strong complex with palladium(II), [Pd(CN)4]2–. Figure S69, Supporting Information, shows the evolution of the 1H NMR spectra of 1 upon gradual addition of KCN. Interestingly, addition of only 1.0 equiv of CN– (with respect to Pd2+) results in a spectrum in which the two characteristic peaks at 8.84 and 9.12 ppm (originating from the 1’s acidic imidazole protons) can no longer be seen, indicating that CN– is highly potent in disassembling cage 1. Addition of 4.0 equiv of CN– affords a spectrum equivalent to that of pure TMEDA in D2O, indicating that the reaction:
has taken place quantitatively (note that TIm is insoluble in water and precipitates from the solution). Similarly, substoichiometric amounts of KCN were sufficient for disintegrating the inclusion complex 42⊂1: we titrated 42⊂1 with KCN and found that formation of the J-aggregates of 4 was complete after adding <0.8 equiv of CN– (with respect to Pd2+; i.e., only 15 mol % of the amount required to form the [Pd(CN)4]2– complex; see Supporting Information, Figure S70). Moreover, the process was remarkably fast: formation of J-aggregates was largely complete within 1 min (Figure 5b; see also Supporting Information, Figure S71). In a control experiment, we verified that KCN added in excess to a solution of 4 had no effect on its optical properties.
Having optimized the conditions for the J-to-H and H-to-J transformations independently, we proceeded to study the reversible cycling between the J-aggregates and the H-dimers of 4 (Figure 5c). In the experiment shown in Figure 5d, we alternately added 0.7 equiv of 1 (with respect to 4) and disassembled it using 0.6 equiv of KCN (the number of equivalents with respect to Pd2+ centers). Three cycles of reversible switching between J- and H-dimers70 are shown in Figure 5d (right). Although further cycling is, in principle, possible, the accumulating waste byproducts ([Pd(CN)4]2–, TMEDA, and TIm) gradually interfere with formation of well-defined H-dimers and extended J-aggregates.
Conclusions
In summary, we studied the noncovalent encapsulation of three different BODIPY dyes within the cavity of a flexible metal–organic cage in aqueous media. For BODIPY 2 and 3, which tend to aggregate in an antiparallel fashion (H-aggregation), the cage underwent a structural distortion in order to encapsulate two molecules of each dye in an orientation (“H-dimer”) mimicking the mutual arrangement of neighboring chromophore units within the crystals of the free dyes. As the third dye, we thus selected a CF3-substituted BODIPY 4, which is known to aggregate into stacks featuring a parallel orientation of the neighboring molecules (J-aggregation). Owing to the large center-to-center distances within these J-aggregates, encapsulation of the putative J-dimers of 4 would entail a prohibitively large deformation of the cage. Instead, the cage was found to encapsulate and stabilize the H-dimer, which is otherwise unstable for this BODIPY. All three encapsulated BODIPYs exhibited optical properties that differ significantly from those of free dyes in organic solvents; most notably, we observed pronounced hypsochromic shifts of the S1←S0 transition. This change of optical properties was rationalized by Kasha’s model of exciton coupling, which predicts an increased HOMO/LUMO gap for dyes stacked in a coplanar and antiparallel fashion. In all cases, addition of an organic solvent triggered the release of the encapsulated dyes, restoring the behavior of the system to that of free dyes in organic solvents. When the H-dimers of 4 were released in water by disintegrating the cages using KCN, they quickly rearranged into J-aggregates and then equally rapidly regenerated H-dimers after a new batch of empty cage was added. Given that the conversion of 42 H-dimers into 4∞ J-aggregates could be accomplished by a substoichiometric amount of cyanide and that it is accompanied by a pronounced color change, possible use of 42⊂1 for detecting cyanide in aqueous solutions is suggested. Another promising direction is based on replacing one of the two encapsulated BODIPY molecules with another (structurally similar) guest X, resulting in 1:1:1 (BODIPY·X)⊂1 heterodimeric complexes. Here, we envision that the large windows of the cage will facilitate the rapid exchange dynamics of the guests between the cages. Overall, we expect that the ability to rapidly and reversibly manipulate the optical properties of supramolecular inclusion complexes in aqueous media will open up avenues for developing detection systems that are compatible with and can operate within biological environments.
Acknowledgments
We acknowledge funding from the European Union’s Horizon 2020 research and innovation program under the European Research Council grant agreement no. 820008 and the Marie Skłodowska-Curie grant agreement no. 812868. We also acknowledge support from the Minerva Foundation with funding from the Federal German Ministry for Education and Research. We gratefully acknowledge Prof. Dan Oron and Dr. Miri Kazes for sharing their expertise in fluorescence spectroscopy and for helpful discussions. We also thank Dr. Iddo Pinkas for recording time-resolved fluorescence and absorption spectra and Dr. Liat Avram for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c08589.
Materials and methods; synthesis of cage 1; synthesis and characterization of BODIPY dyes 2, 3, and 4; preparation, NMR characterization, and X-ray crystallography of inclusion complexes 22⊂1, 32⊂1, and 42⊂1; steady-state and time-resolved optical properties of 2–4 and their inclusion complexes; study of competitive binding of 2 vs azobenzene guests; study of controlled decomposition of cage 1 using KCN; study of reversible switching between H- and J-aggregates of BODIPY 4; X-ray data collection and structure refinement; supplementary references (PDF)
X-ray crystal structure of 3 (CIF)
X-ray crystal structure of 22⊂1 (CIF)
X-ray crystal structure of 32⊂1 (CIF)
X-ray crystal structure of 42⊂1 (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Loudet A.; Burgess K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- Banuelos J. BODIPY Dye, the Most Versatile Fluorophore Ever?. Chem. Rec. 2016, 16, 335–348. 10.1002/tcr.201500238. [DOI] [PubMed] [Google Scholar]
- Ulrich G.; Ziessel R.; Harriman A. The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed. Angew. Chem., Int. Ed. 2008, 47, 1184–1201. 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- Choi S.; Bouffard J.; Kim Y. Aggregation-induced emission enhancement of a meso-trifluoromethyl BODIPY via J-aggregation. Chem. Sci. 2014, 5, 751–755. 10.1039/C3SC52495G. [DOI] [Google Scholar]
- Zhang D.; Wen Y.; Xiao Y.; Yu G.; Liu Y.; Qian X. Bulky 4-tritylphenylethynyl substituted boradiazaindacene: pure red emission, relatively large Stokes shift and inhibition of self-quenching. Chem. Commun. 2008, 4777–4779. 10.1039/b808681h. [DOI] [PubMed] [Google Scholar]
- Tian D.; Qi F.; Ma H.; Wang X.; Pan Y.; Chen R.; Shen Z.; Liu Z.; Huang L.; Huang W. Domino-like multi-emissions across red and near infrared from solid-state 2-/2,6-aryl substituted BODIPY dyes. Nat. Commun. 2018, 9, 2688. 10.1038/s41467-018-05040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Mack J.; Yang Y.; Shen Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. 10.1039/C4CS00030G. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Dorh N.; Zhang J.; Vegesna G.; Li H.; Luo F.-T.; Tiwari A.; Liu H. Highly water-soluble neutral near-infrared emissive BODIPY polymeric dyes. J. Mater. Chem. 2012, 22, 2781–2790. 10.1039/C2JM14920F. [DOI] [Google Scholar]
- Kand D.; Liu P.; Navarro M. X.; Fischer L. J.; Rousso-Noori L.; Friedmann-Morvinski D.; Winter A. H.; Miller E. W.; Weinstain R. Water-Soluble BODIPY Photocages with Tunable Cellular Localization. J. Am. Chem. Soc. 2020, 142, 4970–4974. 10.1021/jacs.9b13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G.; Yang L.; Chen Z. Water-soluble BODIPY and aza-BODIPY dyes: synthetic progress and applications. Front. Chem. Sci. Eng. 2014, 8, 405–417. 10.1007/s11705-014-1445-7. [DOI] [Google Scholar]
- Que E. L.; Bleher R.; Duncan F. E.; Kong B. Y.; Gleber S. C.; Vogt S.; Chen S.; Garwin S. A.; Bayer A. R.; Dravid V. P.; Woodruff T. K.; O’Halloran T. V. Quantitative mapping of zinc fluxes in the mammalian egg reveals the origin of fertilization-induced zinc sparks. Nat. Chem. 2015, 7, 130–139. 10.1038/nchem.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashokkumar P.; Ashoka A. H.; Collot M.; Das A.; Klymchenko A. S. A fluorogenic BODIPY molecular rotor as an apoptosis marker. Chem. Commun. 2019, 55, 6902–6905. 10.1039/C9CC03242H. [DOI] [PubMed] [Google Scholar]
- Gotor R.; Costero A. M.; Gaviña P.; Gil S.; Parra M. Binding and Fluorescent Sensing of Dicarboxylates by a Bis(calix[4]pyrrole)-Substituted BODIPY Dye. Eur. J. Org. Chem. 2013, 2013, 1515–1520. 10.1002/ejoc.201201504. [DOI] [Google Scholar]
- Nierth A.; Kobitski A. Y.; Nienhaus G. U.; Jäschke A. Anthracene–BODIPY Dyads as Fluorescent Sensors for Biocatalytic Diels–Alder Reactions. J. Am. Chem. Soc. 2010, 132, 2646–2654. 10.1021/ja9084397. [DOI] [PubMed] [Google Scholar]
- Boens N.; Leen V.; Dehaen W. Fluorescent indicators based on BODIPY. Chem. Soc. Rev. 2012, 41, 1130–1172. 10.1039/C1CS15132K. [DOI] [PubMed] [Google Scholar]
- Karolin J.; Johansson L. B. Å.; Strandberg L.; Ny T. Fluorescence and Absorption Spectroscopic Properties of Dipyrrometheneboron Difluoride (BODIPY) Derivatives in Liquids, Lipid Membranes, and Proteins. J. Am. Chem. Soc. 1994, 116, 7801–7806. 10.1021/ja00096a042. [DOI] [Google Scholar]
- Bergström F.; Mikhalyov I.; Hägglöf P.; Wortmann R.; Ny T.; Johansson L. B. Å. Dimers of Dipyrrometheneboron Difluoride (BODIPY) with Light Spectroscopic Applications in Chemistry and Biology. J. Am. Chem. Soc. 2002, 124, 196–204. 10.1021/ja010983f. [DOI] [PubMed] [Google Scholar]
- Albrecht M.; Lippach A.; Exner M. P.; Jerbi J.; Springborg M.; Budisa N.; Wenz G. Site-specific conjugation of 8-ethynyl-BODIPY to a protein by [2 + 3] cycloaddition. Org. Biomol. Chem. 2015, 13, 6728–6736. 10.1039/C5OB00505A. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Tsao K.; Acton S. L.; Keillor J. W. A Green BODIPY-Based, Super-Fluorogenic, Protein-Specific Labelling Agent. Angew. Chem., Int. Ed. 2018, 57, 12390–12394. 10.1002/anie.201805482. [DOI] [PubMed] [Google Scholar]
- Descalzo A. B.; Ashokkumar P.; Shen Z.; Rurack K. On the Aggregation Behaviour and Spectroscopic Properties of Alkylated and Annelated Boron-Dipyrromethene (BODIPY) Dyes in Aqueous Solution. ChemPhotoChem. 2020, 4, 120–131. 10.1002/cptc.201900235. [DOI] [Google Scholar]
- Kasha M.; Rawls H. R.; Ashraf El-Bayoumi M. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. 10.1351/pac196511030371. [DOI] [Google Scholar]
- Treibs A.; Kreuzer F.-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Liebigs Ann. 1968, 718, 208–223. 10.1002/jlac.19687180119. [DOI] [Google Scholar]
- Würthner F.; Kaiser T. E.; Saha-Möller C. R. J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials. Angew. Chem., Int. Ed. 2011, 50, 3376–3410. 10.1002/anie.201002307. [DOI] [PubMed] [Google Scholar]
- Jelley E. E. Spectral Absorption and Fluorescence of Dyes in the Molecular State. Nature 1936, 138, 1009–1010. 10.1038/1381009a0. [DOI] [Google Scholar]
- Saki N.; Dinc T.; Akkaya E. U. Excimer emission and energy transfer in cofacial boradiazaindacene (BODIPY) dimers built on a xanthene scaffold. Tetrahedron 2006, 62, 2721–2725. 10.1016/j.tet.2005.12.021. [DOI] [Google Scholar]
- Alamiry M. A.; Benniston A. C.; Copley G.; Harriman A.; Howgego D. Intramolecular Excimer Formation for Covalently Linked Boron Dipyrromethene Dyes. J. Phys. Chem. A 2011, 115, 12111–12119. 10.1021/jp2070419. [DOI] [PubMed] [Google Scholar]
- Ahrens J.; Böker B.; Brandhorst K.; Funk M.; Bröring M. Sulfur-Bridged BODIPY DYEmers. Chem. - Eur. J. 2013, 19, 11382–11395. 10.1002/chem.201300893. [DOI] [PubMed] [Google Scholar]
- Ahrens J.; Scheja A.; Wicht R.; Bröring M. Excitonic Coupling in Acyclic and Cyclic Dithioaryl-Linked BODIPY DYEmers. Eur. J. Org. Chem. 2016, 2016, 2864–2870. 10.1002/ejoc.201600359. [DOI] [Google Scholar]
- Ahrens J.; Haberlag B.; Scheja A.; Tamm M.; Bröring M. Conjugated BODIPY DYEmers by Metathesis Reactions. Chem. - Eur. J. 2014, 20, 2901–2912. 10.1002/chem.201303468. [DOI] [PubMed] [Google Scholar]
- Solomonov A. V.; Marfin Y. S.; Rumyantsev E. V. Design and applications of dipyrrin-based fluorescent dyes and related organic luminophores: From individual compounds to supramolecular self-assembled systems. Dyes Pigm. 2019, 162, 517–542. 10.1016/j.dyepig.2018.10.042. [DOI] [Google Scholar]
- Chen C.-S.; Martin O. C.; Pagano R. E. Changes in the spectral properties of a plasma membrane lipid analog during the first seconds of endocytosis in living cells. Biophys. J. 1997, 72, 37–50. 10.1016/S0006-3495(97)78645-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allampally N. K.; Florian A.; Mayoral M. J.; Rest C.; Stepanenko V.; Fernández G. H-Aggregates of Oligophenyleneethynylene (OPE)-BODIPY Systems in Water: Guest Size-Dependent Encapsulation Mechanism and Co-aggregate Morphology. Chem. - Eur. J. 2014, 20, 10669–10678. 10.1002/chem.201402077. [DOI] [PubMed] [Google Scholar]
- Yang L.; Fan G.; Ren X.; Zhao L.; Wang J.; Chen Z. Aqueous self-assembly of a charged BODIPY amphiphile via nucleation–growth mechanism. Phys. Chem. Chem. Phys. 2015, 17, 9167–9172. 10.1039/C5CP00207A. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Liu Y.; Wagner W.; Stepanenko V.; Ren X.; Ogi S.; Würthner F. Near-IR Absorbing J-Aggregate of an Amphiphilic BF2-Azadipyrromethene Dye by Kinetic Cooperative Self-Assembly. Angew. Chem., Int. Ed. 2017, 56, 5729–5733. 10.1002/anie.201701788. [DOI] [PubMed] [Google Scholar]
- Tokoro Y.; Nagai A.; Chujo Y. Nanoparticles via H-aggregation of amphiphilic BODIPY dyes. Tetrahedron Lett. 2010, 51, 3451–3454. 10.1016/j.tetlet.2010.04.120. [DOI] [Google Scholar]
- Rödle A.; Lambov M.; Mück-Lichtenfeld C.; Stepanenko V.; Fernández G. Cooperative nanoparticle H-type self-assembly of a bolaamphiphilic BODIPY derivative in aqueous medium. Polymer 2017, 128, 317–324. 10.1016/j.polymer.2016.12.050. [DOI] [Google Scholar]
- Olivier J. H.; Widmaier J.; Ziessel R. Near-Infrared Fluorescent Nanoparticles Formed by Self-Assembly of Lipidic (Bodipy) Dyes. Chem. - Eur. J. 2011, 17, 11709–11714. 10.1002/chem.201101407. [DOI] [PubMed] [Google Scholar]
- Benstead M.; Rosser G. A.; Beeby A.; Mehl G. H.; Boyle R. W. Addressing fluorescence and liquid crystal behaviour in multi-mesogenic BODIPY materials. New J. Chem. 2011, 35, 1410–1417. 10.1039/c0nj00990c. [DOI] [Google Scholar]
- Frein S.; Camerel F.; Ziessel R.; Barberá J.; Deschenaux R. Highly Fluorescent Liquid-Crystalline Dendrimers Based on Borondipyrromethene Dyes. Chem. Mater. 2009, 21, 3950–3959. 10.1021/cm9008078. [DOI] [Google Scholar]
- Olivier J.-H.; Barberá J.; Bahaidarah E.; Harriman A.; Ziessel R. Self-Assembly of Charged Bodipy Dyes to Form Cassettes That Display Intracomplex Electronic Energy Transfer and Accrete into Liquid Crystals. J. Am. Chem. Soc. 2012, 134, 6100–6103. 10.1021/ja3007935. [DOI] [PubMed] [Google Scholar]
- Pagano R. E.; Chen C.-S. Use of BODIPY-labeled Sphingolipids to Study Membrane Traffic along the Endocytic Pathway. Ann. N. Y. Acad. Sci. 1998, 845, 152–160. 10.1111/j.1749-6632.1998.tb09668.x. [DOI] [PubMed] [Google Scholar]
- Descalzo A. B.; Xu H.-J.; Xue Z.-L.; Hoffmann K.; Shen Z.; Weller M. G.; You X.-Z.; Rurack K. Phenanthrene-Fused Boron–Dipyrromethenes as Bright Long-Wavelength Fluorophores. Org. Lett. 2008, 10, 1581–1584. 10.1021/ol800271e. [DOI] [PubMed] [Google Scholar]
- Tleugabulova D.; Zhang Z.; Brennan J. D. Characterization of Bodipy Dimers Formed in a Molecularly Confined Environment. J. Phys. Chem. B 2002, 106, 13133–13138. 10.1021/jp027126y. [DOI] [Google Scholar]
- Conn M. M.; Rebek J. Self-Assembling Capsules. Chem. Rev. 1997, 97, 1647–1668. 10.1021/cr9603800. [DOI] [PubMed] [Google Scholar]
- Murray J.; Kim K.; Ogoshi T.; Yao W.; Gibb B. C. The aqueous supramolecular chemistry of cucurbit[n]urils, pillar[n]arenes and deep-cavity cavitands. Chem. Soc. Rev. 2017, 46, 2479–2496. 10.1039/C7CS00095B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommet A. B.; Feller M.; Klajn R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 2020, 15, 256–271. 10.1038/s41565-020-0652-2. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Chen C.; Liu J.; Stang P. J. Recent developments in the construction and applications of platinum-based metallacycles and metallacages via coordination. Chem. Soc. Rev. 2020, 49, 3889–3919. 10.1039/D0CS00038H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Ronson T. K.; Nitschke J. R. Functional Capsules via Subcomponent Self-Assembly. Acc. Chem. Res. 2018, 51, 2423–2436. 10.1021/acs.accounts.8b00303. [DOI] [PubMed] [Google Scholar]
- Datta S.; Saha M. L.; Stang P. J. Hierarchical Assemblies of Supramolecular Coordination Complexes. Acc. Chem. Res. 2018, 51, 2047–2063. 10.1021/acs.accounts.8b00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen S.; Clever G. H. Mixed-Ligand Metal–Organic Frameworks and Heteroleptic Coordination Cages as Multifunctional Scaffolds—A Comparison. Acc. Chem. Res. 2018, 51, 3052–3064. 10.1021/acs.accounts.8b00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepehrpour H.; Fu W.; Sun Y.; Stang P. J. Biomedically Relevant Self-Assembled Metallacycles and Metallacages. J. Am. Chem. Soc. 2019, 141, 14005–14020. 10.1021/jacs.9b06222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of inclusion complexes encapsulating a single BODIPY guest within coordination cages, see:; a Yamashina M.; Sartin M. M.; Sei Y.; Akita M.; Takeuchi S.; Tahara T.; Yoshizawa M. Preparation of Highly Fluorescent Host–Guest Complexes with Tunable Color upon Encapsulation. J. Am. Chem. Soc. 2015, 137, 9266–9269. 10.1021/jacs.5b06195. [DOI] [PubMed] [Google Scholar]; b Gou X.-X.; Peng J.-X.; Das R.; Wang Y.-Y.; Han Y.-F. On/off fluorescence emission induced by encapsulation, exchange and reversible encapsulation of a BODIPY-guest in self-assembled organometallic cages. Dalton Trans. 2019, 48, 7236–7241. 10.1039/C8DT05103H. [DOI] [PubMed] [Google Scholar]; c Tsutsui T.; Catti L.; Yoza K.; Yoshizawa M. An atropisomeric M2L4 cage mixture displaying guest-induced convergence and strong guest emission in water. Chem. Sci. 2020, 11, 8145–8150. 10.1039/D0SC03223A. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example of an inclusion complex encapsulating a single BODIPY guest within a molecular capsule, see:; d Gupta M.; Parvathi K.; Mula S.; Maity D. K.; Ray A. K. Enhanced fluorescence of aqueous BODIPY by interaction with cavitand cucurbit[7]uril. Photochem. Photobiol. Sci. 2017, 16, 499–506. 10.1039/C6PP00325G. [DOI] [PubMed] [Google Scholar]
- Rizzuto F. J.; von Krbek L. K. S.; Nitschke J. R. Strategies for binding multiple guests in metal–organic cages. Nat. Rev. Chem. 2019, 3, 204–222. 10.1038/s41570-019-0085-3. [DOI] [Google Scholar]
- Yamauchi Y.; Yoshizawa M.; Akita M.; Fujita M. Discrete stack of an odd number of polarized aromatic compounds revealing the importance of net vs. local dipoles. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10435–10437. 10.1073/pnas.0810319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng W.; Breiner B.; Rissanen K.; Thoburn J. D.; Clegg J. K.; Nitschke J. R. A Self-Assembled M8L6 Cubic Cage that Selectively Encapsulates Large Aromatic Guests. Angew. Chem., Int. Ed. 2011, 50, 3479–3483. 10.1002/anie.201100193. [DOI] [PubMed] [Google Scholar]
- Dobashi H.; Catti L.; Tanaka Y.; Akita M.; Yoshizawa M. N-Doping of Polyaromatic Capsules: Small Cavity Modification Leads to Large Change in Host–Guest Interactions. Angew. Chem., Int. Ed. 2020, 59, 11881–11885. 10.1002/anie.202004168. [DOI] [PubMed] [Google Scholar]
- Samanta D.; Mukherjee S.; Patil Y. P.; Mukherjee P. S. Self-assembled Pd6 open cage with triimidazole walls and the use of its confined nanospace for catalytic Knoevenagel- and Diels–Alder reactions in aqueous medium. Chem. - Eur. J. 2012, 18, 12322–12329. 10.1002/chem.201201679. [DOI] [PubMed] [Google Scholar]
- Samanta D.; Galaktionova D.; Gemen J.; Shimon L. J. W.; Diskin-Posner Y.; Avram L.; Král P.; Klajn R. Reversible chromism of spiropyran in the cavity of a flexible coordination cage. Nat. Commun. 2018, 9, 641. 10.1038/s41467-017-02715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Gemen J.; Chu Z.; Diskin-Posner Y.; Shimon L. J. W.; Klajn R. Reversible photoswitching of encapsulated azobenzenes in water. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9379–9384. 10.1073/pnas.1712787115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanopolskyi A. I.; De S.; Białek M. J.; Diskin-Posner Y.; Avram L.; Feller M.; Klajn R. Reversible switching of arylazopyrazole within a metal–organic cage. Beilstein J. Org. Chem. 2019, 15, 2398–2407. 10.3762/bjoc.15.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton M.; Grommet A. B.; Pesce L.; Gemen J.; Li S.; Diskin-Posner Y.; Credi A.; Pavan G. M.; Andréasson J.; Klajn R. Improving Fatigue Resistance of Dihydropyrene by Encapsulating within a Coordination Cage. J. Am. Chem. Soc. 2020, 142, 14557–14565. 10.1021/jacs.0c06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B.; Peng X.; Cui A.; Wu Y.; Tian M.; Zhang L.; Chen X.; Gao Y. Synthesis and spectral properties of new boron dipyrromethene dyes. Dyes Pigm. 2007, 73, 206–210. 10.1016/j.dyepig.2005.11.007. [DOI] [Google Scholar]
- Nepomnyashchii A. B.; Bröring M.; Ahrens J.; Bard A. J. Synthesis, Photophysical, Electrochemical, and Electrogenerated Chemiluminescence Studies. Multiple Sequential Electron Transfers in BODIPY Monomers, Dimers, Trimers, and Polymer. J. Am. Chem. Soc. 2011, 133, 8633–8645. 10.1021/ja2010219. [DOI] [PubMed] [Google Scholar]
- Duan C.; Zhou Y.; Shan G.-G.; Chen Y.; Zhao W.; Yuan D.; Zeng L.; Huang X.; Niu G. Bright solid-state red-emissive BODIPYs: facile synthesis and their high-contrast mechanochromic properties. J. Mater. Chem. C 2019, 7, 3471–3478. 10.1039/C8TC06421K. [DOI] [Google Scholar]
- Wang H.; Vicente M. G. H.; Fronczek F. R.; Smith K. M. Synthesis and Transformations of 5-Chloro-2,2′-Dipyrrins and Their Boron Complexes, 8-Chloro-BODIPYs. Chem. - Eur. J. 2014, 20, 5064–5074. 10.1002/chem.201304310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce L.; Perego C.; Grommet A. B.; Klajn R.; Pavan G. M. Molecular Factors Controlling the Isomerization of Azobenzenes in the Cavity of a Flexible Coordination Cage. J. Am. Chem. Soc. 2020, 142, 9792–9802. 10.1021/jacs.0c03444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Bouffard J.; Kim Y. Tailoring the Solid-State Fluorescence Emission of BODIPY Dyes by meso Substitution. Chem. - Eur. J. 2015, 21, 17459–17465. 10.1002/chem.201503040. [DOI] [PubMed] [Google Scholar]
- Previous reports on controlled decomposition of coordination cages:; a Riddell I. A.; Smulders M. M. J.; Clegg J. K.; Nitschke J. R. Encapsulation, storage and controlled release of sulfur hexafluoride from a metal–organic capsule. Chem. Commun. 2011, 47, 457–459. 10.1039/C0CC02573A. [DOI] [PubMed] [Google Scholar]; b Mal P.; Schultz D.; Beyeh K.; Rissanen K.; Nitschke J. R. An Unlockable–Relockable Iron Cage by Subcomponent Self-Assembly. Angew. Chem., Int. Ed. 2008, 47, 8297–8301. 10.1002/anie.200803066. [DOI] [PubMed] [Google Scholar]; c Lewis J. E. M.; Gavey E. L.; Cameron S. A.; Crowley J. D. Stimuli-responsive Pd2L4 metallosupramolecular cages: towards targeted cisplatin drug delivery. Chem. Sci. 2012, 3, 778–784. 10.1039/C2SC00899H. [DOI] [Google Scholar]; d Kishi N.; Akita M.; Kamiya M.; Hayashi S.; Hsu H.-F.; Yoshizawa M. Facile Catch and Release of Fullerenes Using a Photoresponsive Molecular Tube. J. Am. Chem. Soc. 2013, 135, 12976–12979. 10.1021/ja406893y. [DOI] [PubMed] [Google Scholar]
- Gadde S.; Batchelor E. K.; Weiss J. P.; Ling Y.; Kaifer A. E. Control of H-and J-Aggregate Formation via Host–Guest Complexation using Cucurbituril Hosts. J. Am. Chem. Soc. 2008, 130, 17114–17119. 10.1021/ja807197c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.