

Abstract
IFN-γ and IL-17A can each elicit ocular autoimmunity independently of the other. Since absence of IFN-γ or IL-17A individually failed to abolish pathology of experimental autoimmune uveitis (EAU), we examined EAU development in the absence of both these cytokines. Ifng−/−Il17a−/− mice were fully susceptible to EAU with a characteristic eosinophilic ocular infiltrate, as opposed to a mononuclear infiltrate in WT mice. Retinal pathology in double-deficient mice was ameliorated when eosinophils were genetically absent or their migration was blocked, supporting a pathogenic role for eosinophils in EAU in the concurrent absence of IFN-γ and IL-17A. In EAU-challenged Ifng−/−Il17a−/− mice, ocular infiltrates contained increased GM-CSF-producing CD4+ T cells, and supernatants of retinal antigen-stimulated splenocytes contained enhanced levels of GM-CSF that contributed to activation and migration of eosinophils in vitro. Systemic or local blockade of GM-CSF ameliorated EAU in Ifng−/−Il17a−/− mice, reduced eosinophil peroxidase levels in the eye and in the serum and decreased eosinophil infiltration to the eye. These results support the interpretation that, in the concurrent absence of IFN-γ and IL-17A, GM-CSF takes on a major role as an inflammatory effector cytokine and drives an eosinophil-dominant pathology. Our findings may impact therapeutic strategies aiming to target IFN-γ and IL-17A in autoimmune uveitis.
Keywords: Eosinophils, GM-CSF, IFN-γ, IL-17A, Experimental autoimmune uveitis, Neuroinflammation
1. Introduction
Autoimmune uveitis and its animal model of experimental autoimmune uveitis (EAU) are characterized by the infiltration of retina-specific Th cells into the eyes, where they become reactivated and drive massive recruitment of inflammatory leukocytes [1]. EAU is part of a group of tissue specific autoimmune disease models elicited with a tissue-specific antigen, which also include experimental autoimmune encephalomyelitis (EAE) and autoimmune arthritis. These autoimmune disease models share essential effector mechanisms, including pathogenic IFN-γ-producing Th1 and IL-17A-producing Th17. Initially, Th1 cells were thought to be the main pathogenic effectors in these disease models, which was attributed at least in part to their production of IFN-γ [2]. This notion was supported by the finding that transgenic expression of IFN-γ in the tissue leads to inflammation and severe pathology [3,4]. Paradoxically, however, systemic neutralization or genetic deletion of IFN-γ exacerbated these tissue specific autoimmune disease models and was associated with an elevated Th17 response and Th17-polarized Ifng−/− effector T cells infused into Ifng−/− recipients, induced undiminished EAU [5–9]. In the wake of such findings, it was not surprising that IL-17A-producing Th17 cells, identified in 2005, promptly supplanted the Th1 cells as the T effector lineage universally blamed for inflammatory autoimmunity [10–14]. However, findings quickly emerged that were at odds with this notion. For example, although systemic deficiency or neutralization of IL-17A ameliorated disease, this was in many cases partial and variable [15], and experiments with IL-17A fate-reporter mice indicated that Th17 cells may actually have to convert to an IFN-γ-producing Th1-like phenotype within the target tissue in order to effect pathology [16].
The debate as to the relative importance of Th1 and Th17 lineages was in part reconciled by our finding in the EAU model that both lineages are standalone and independent effector phenotypes, and each is by itself sufficient to drive tissue pathology [15]. However, none of the studies cited above adequately addressed the roles of and requirement for their respective signature cytokines IFN-γ and IL-17A, or clarified what would happen if both these cytokines were absent.
In this study, we aimed to examine the pathogenesis of autoimmune uveitis in the concurrent deficiency of IFN-γ and IL-17A. We demonstrate that severe EAU pathology still develops in the IFN-γ/IL-17A double-deficient setting, but is driven by an alternative effector pathway. Our data reveal a compensatory production of other cytokines, primarily granulocyte-macrophage colony-stimulating factor (GM-CSF), that plays a non-redundant role in pathogenesis by activating and recruiting eosinophils to the eye.
2. Materials and methods
2.1. Mice
Wild type (WT) C57BL/6, B10.RIII, and Ifng−/− mice on the C57BL/6 background were purchased from The Jackson Laboratory. Il17a−/− [17], Il17f−/− and Il17a−/− Il17f−/− [18] mice on C57BL/6 background were obtained from Dr. Yoichiro Iwakura (Tokyo University of Science, Chiba, Japan), and ΔdblGATA (deletion of a palindromic GATA-binding site) [19,20] mice were from Dr. Avery August (Cornell University, NY). Ifng−/−Il17a−/−, Ifng−/−Il17a−/− Il17f−/−, and Ifng−/−Il17a−/− x ΔdblGATA mice were derived from the respective knockout (KO) mice and were subsequently bred homo-zygously. The mutations were also introgressed onto the highly EAU-susceptible B10. RIII background by backcrossing for at least 8 generations. Controls were the WT progeny of the original heterozygous colony that were maintained in the same animal room at the National Institutes of Health (NIH) in Bethesda, Maryland under specific pathogen-free (SPF) facility and given water and standard laboratory chow ad libitum. Care and use of the animals were in compliance with Institutional guidelines. The animal study protocol was approved by the Institutional Animal Care and Use Committee (ASP# NEI-581, renewed as NEI-688 in 2019).
2.2. EAU induction
WT and KO mice on C57BL/6 background were actively immunized subcutaneously (s.c.) with a mixture of 150 μg interphotoreceptor retinoid binding (IRBP) and 300 μg IRBP peptide 1–20 (p1–20; sequence GPTHLFQPSLVLDMAKVLLD) emulsified in an equal volume of complete Freund’s adjuvant (CFA) containing 2.5 mg/mL Mycobacterium tuberculosis H37RA (Difco, Detroit, MI). IRBP was isolated from bovine retinas, as described previously [21]. Pertussis toxin (0.2 μg, Sigma-Aldrich, St. Louis, MO) was injected intraperitoneally (i.p.) on day 0 and 2 after immunization. In some experiments, as indicated, 100 μg of IRBP peptide 651–670 (p651–670; LAQGAYRTAVDLESLASQLT) with simultaneous administration of 1 μg Pertussis toxin i.p. was used for immunization of EAU in mice on C57BL/6 background [22]. For induction of EAU in B10.RIII background mice, 10 μg of IRBP peptide 161–180 (p161–180; SGIPYIISYLHPGNTILHVD) was used. For induction of EAU by adoptive transfer, enriched T cells (CD3+ T cell enrichment columns, R&D) from pooled draining lymph nodes and spleens of immunized mice were isolated on day 7 after immunization and activated in vitro with 30 μg/mL IRBP1–20 and 30 μg/mL IRBP protein in the presence of irradiated (3000 rads) splenocytes as antigen-presenting cells. On the third day, cells were collected and adoptively transferred to naïve WT recipient mice by i.p. injection (10 millions/mouse).
2.3. In vivo treatment with antibodies or antagonists
For systemic antibody treatment, experimental animals were injected i.p. with the following antibodies: 300 μg of anti-GM-CSF antibody (purified clone MP1–22E9 from BioXcell, or clarified ascites produced by Harlan Bioproducts for Science Inc.), 300 μg of anti–IFN–γ antibody (clone R4–6A2 in the form of ascites produced by Harlan Bioproducts for Science Inc.), 600 μg of anti-IL-17A antibody (clone MM17F3 in the form of ascites from Harlan Bioproducts for Science Inc.) or their respective isotype Ig controls (Rat IgG2a, Rat IgG1, or mouse IgG1). Serum levels of rat IgG2a were determined by ELISA (Invitrogen) to check persisting levels of anti-GM-CSF antibody in circulation. For local antibody treatment, animals were given anti-GM-CSF antibody (100 μg/2 μl/eye), purified from ascites and concentrated, or isotype Ig (Rat IgG2a) on day 10 post-immunization as a single bilateral intravitreal injection. For blocking C–C chemokine receptor type 3 (CCR3) signaling, 200 μg SB328437 (CCR3 antagonist, Abcam), suspended in phosphate-buffered saline (PBS) containing 0.1% tween 80, was administered subcutaneously every day from 1 day before immunization [23].
2.4. EAU scoring
For clinical scoring of EAU, mice were anesthetized by i.p. injections of ketamine/xylazine and dilating drops (Phenylephrine hydrochloride and Tropicamide ophthalmic solution) were applied. Ocular disease was evaluated by fundus examination and was scored on a scale of 0–4, based on the severity of inflammation. The criteria for scoring and example photographs are described elsewhere [24]. For histology, eyes were enucleated and immediately fixed for 1 h in 4% Glutaraldehyde (Fisher Scientific) in PBS and then transferred into 10% phosphate-buffered formaldehyde (Sigma-Aldrich) until processing. Fixed and dehydrated tissue was embedded in methacrylate or paraffin. Sections (4–7 μm) were cut through the pupillary-optic nerve plane and were stained with hematoxylin and eosin (H&E). Severity of EAU was scored in a masked fashion on a scale of 0–4 using previously published criteria [25].
2.5. Isolation of eye-infiltrating cells
Eyes were enucleated and external tissues and lenses were removed. The remaining tissue was minced in HL-1 media (Lonza, Walkersville, MD) and incubated with 1 mg/mL collagenase D (Roche, Indianapolis, IN) for 45 min at 37 °C. Cells were then dispersed by trituration using a pipette, washed, filtered through 40 μm cell strainer, and resuspended in RPMI supplemented with 10% fetal bovine serum (FBS).
2.6. Intracellular cytokines staining and FACS analysis
Cells were incubated with Fc-block (clone 2.4G2, BioXcell) and stained with the following antibodies (clones) from BioLegend, eBioscience or BD Biosciences: anti-CD3 (clone 145–2C11), anti-CD4 (RM4.5), anti-CD8 (53–6.7), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD45 (30-F11), anti-Ly6C (HK1.4), anti-Ly6G (1A8), anti-SiglecF (1RNM44 N), and anti-CCR3 (J073E5). Fixable viability dyes, such as Ghost Dyes (Tonbo) and ViaKrome 808 (Beckman Coulter) were used to exclude dead cells from the analyses. For intracellular cytokine staining, cells were pulsed for 4 h with PMA (phorbol 12-miristate 13-acetate, 50 ng/mL), ionomycin (500 ng/mL), and Brefeldin A (GolgiPlug, BD), followed by 4% paraformaldehyde fixation and permeabilized (BD Biosciences) before intracellular cytokine staining for GM-CSF (MP1–22E9), IFN-γ (XMG1.2) and IL-17A (TC11–18H10.1). Stained cells were collected on MACSQuant analyzer (Miltenyi Biotec, Germany) or CytoFLEX (Beckman Coulter, Brea, CA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
2.7. Quantitation of cytokine levels and eosinophil peroxidase (EPO)
At the peak of EAU (day 14–17), splenocytes were harvested and stimulated with 10 μg/mL of IRBP651–670 or IRBP protein and 10 mg/mL of α-methyl-D-mannopyranoside (α-MMP; Sigma Aldrich). After 48 h, GM-CSF production in the culture supernatant was evaluated by ELISA (R&D system). Eosinophil peroxidase (EPO) in the serum and in the eye extracts was quantitated using customized LEGENDplex (Biolegend).
2.8. Eosinophil culture and chemotaxis assay
Bone marrow-derived eosinophils (bmEos) were differentiated as previously described [26]. Briefly, bone marrow cells were collected from the femurs and tibiae of WT C57BL/6 mice by flushing the bones with PBS. Cells were lysed by ACK buffer (Thermo Fisher Scientific). After centrifugation, cells were washed in PBS, and cultured at 1 × 106/mL in RPMI medium supplemented with 20% FBS, 100 ng/mL stem cell factor (SCF; PeproTech) and 100 ng/mL Fms-like tyrosine kinase 3 ligand (Flt3L; PeproTech). On day 4, the medium was replaced with medium supplemented with 10 ng/mL recombinant mouse IL-5 (Peprotech). Every second day from day 8, a half of the medium was replaced with fresh medium containing IL-5 and the cell concentration was maintained at 1 × 106 cells/mL. The purity of bmEos was determined using flow cytometry.
In some experiments, differentiated bmEos were cultured with supernatants of re-stimulated splenocytes from EAU-induced WT or Ifng−/−Il17a−/− mice in presence or absence of anti-GM-CSF antibody (10 μg/mL), or mouse GM-CSF (100 ng/mL). After 2 days, cells were harvested and stained for CD11b and SiglecF.
For the migration assay, a transwell plate (Corning) with 5.0-μm pore size polycarbonate membrane was used. The supernatant of re-stimulated splenocytes from EAU-induced WT or Ifng−/−Il17a−/− mice was placed in the lower chamber. BmEos (0.5 × 105) were placed in the upper chamber. Recombinant mouse eotaxin (Biolegend) or GMCSF (Biolegend) were used as positive control. After 3 h, cells in the lower chamber were counted and normalized to medium-only controls.
2.9. Statistical analysis
Statistical data analysis was performed using GraphPad Prism version 7.0 (GraphPad Software, La Jolla, CA). Student’s t-test (parametric data) or Mann-Whitney U test (non-parametric data) was used for two-group comparisons. One-way ANOVA with Bonferroni correction for multiple comparison test was used for multiple group analysis. EAU scores of mice intravitreally treated with anti-GM-CSF antibody were analyzed using Wilcoxon matched-pairs signed rank test. Correlation between disease score and cytokine production were calculated using Spearman correlation test. Data are displayed as mean ± SEM. Experiments were repeated at least twice, and usually three or more times.
3. Results
3.1. IFN-γ and IL-17A double deficient mice are fully susceptible to EAU and develop an eosinophilic inflammation in the eye
Our previous data show that IL-17A neutralization by systemic antibody treatment prevents and reverses EAU, and that the intensity of Th17 responses correlates with disease severity [15]. Furthermore, it has been proposed that IL-17A/IFN-γ double-producer T cells are the actual pathogenic effectors in the central nervous system (CNS) disease [16]. Paradoxically, however, Ifng−/− mice develop exacerbated autoimmunity compared to WT controls [5–7,27], and EAU could develop in Il17a−/− mice, albeit with lower scores than in WT mice [15,28], suggesting that different effector pathways can lead to comparable autoimmune pathology. To test whether concurrent lack of IFN-γ and IL-17A would be able to support EAU pathology, we immunized IFN-γ and IL-17A double deficient (Ifng−/−Il17a−/−) mice with the retinal autoantigen IRBP to induce EAU. Consistent with our previous findings, fundoscopy and histology scores were significantly lower in Il17a−/− mice, whereas these EAU scores were significantly higher in Ifng−/− mice, compared with their WT controls (Fig. 1A and B). Intriguingly, Ifng−/−Il17a−/− mice were fully susceptible to EAU and developed disease scores similar to WT controls (Fig. 1A and B). To test whether these patterns of disease severity in single or double KO mice cross antigenic specificity or differ between mouse strains, we immunized these single and double KO mice with a different IRBP peptide, IRBP651–670, which has higher uveitogenicity than IRBP1–20 [22]. We also immunized Ifng−/−Il17a−/− mice on the B10.RIII background with IRBP161–180, a major pathogenic epitope for this strain. The same pattern of response in terms of disease severity was maintained, irrespective of epitope specificity and background strain (Supplementary Fig. 1).
Fig. 1.
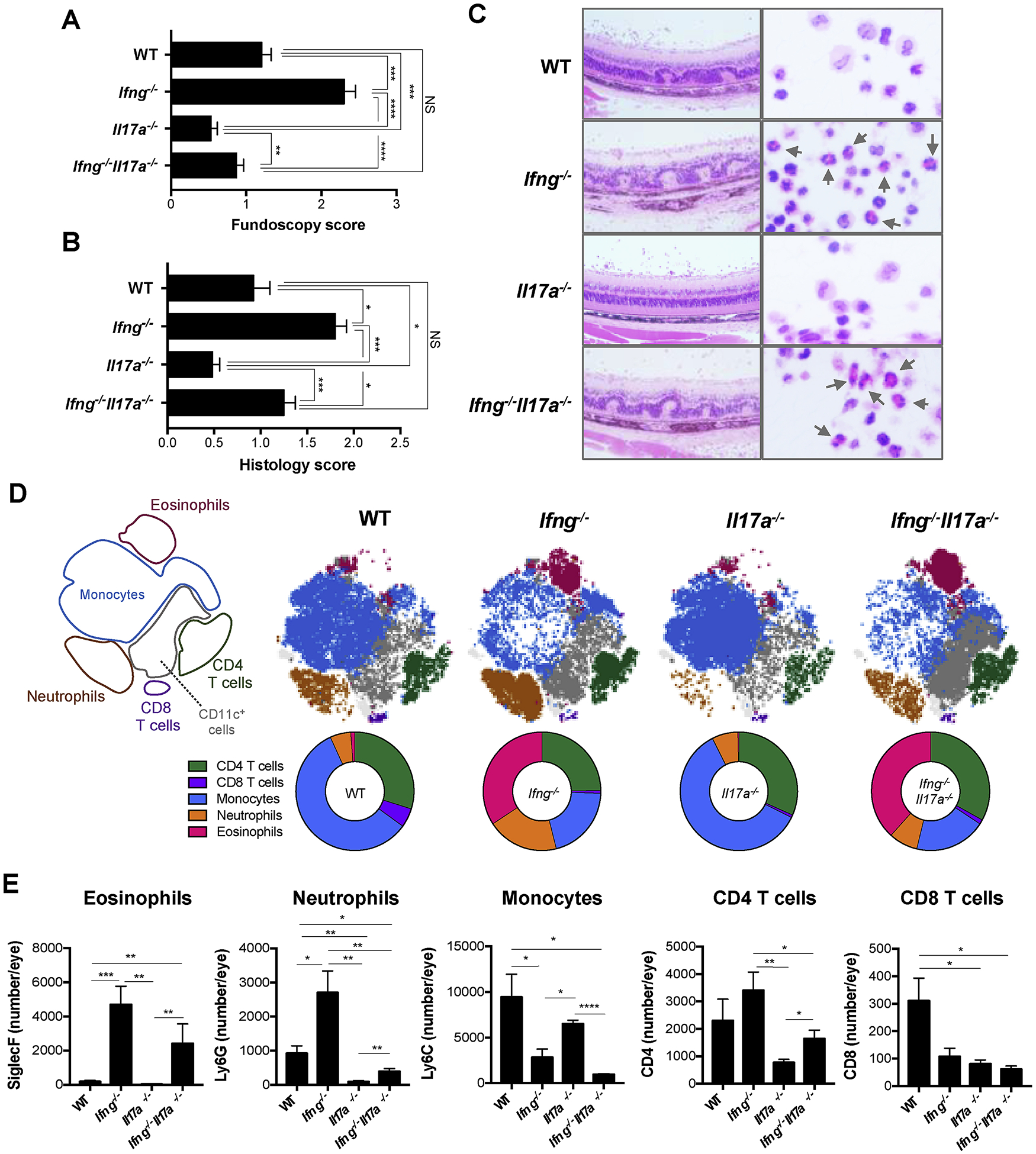
Ifng−/−Il17a−/− mice are susceptible to EAU and show eosinophil dominant infiltration in the eyes. WT, Ifng−/−, Il17a−/−, and Ifng−/−Il17a−/− mice on C57BL/6 background were immunized with 150 μg IRBP and 300 μg p1–20. (A, B) EAU scores were evaluated by fundoscopy (A) and histology (B) 21 days after immunization. (C) Representative histopathology of uveitic retina in each group with H&E stain. Original mag, 20x (left) and 150x (right). Arrows show eosinophils. (D) CD45+ live cells in the EAU eyes on day 16–18 were analyzed by algorithm-based t-SNE visualization and FlowSOM clustering based on their surface marker expression. Shown are the scheme for the cell-type cluster (left most), and a representative plot and parts of whole graph from each group. (E) The number of SiglecF+ (Eosinophils), Ly6C−Ly6G+ (Neutrophils) or Ly6C+Ly6G− (Monocytes) in CD11b+ gates, and CD4+ or CD8+ T cells in the eyes. (A) Data combined from 4 experiments (WT; n = 41, Ifng−/−; n = 26, Il17a−/−; n = 27, Ifng−/−Il17a−/−; n = 34). (B–E) Representative data from 4 independent experiments, and each group contains at least 3 mice. Data shown as mean ± SEM. Significance was determined using Mann-Whitney U test (A, B), ANOVA (E). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS; not significant.
Notably, however, the composition of ocular inflammatory infiltrate in Ifng−/−Il17a−/− and Ifng−/− mice differed from that of WT mice. Whereas the ocular infiltrate in WT mice was dominated by monocytes, confirming previous reports, the infiltrate in both Ifng−/−Il17a−/− and Ifng−/− mice was dominated by eosinophils, suggesting that it may be due to the lack of IFN-γ ([29], Fig. 1C–E). Flow cytometry confirmed that the proportion and number of eosinophils in inflamed eyes of EAU-induced Ifng−/−Il17a−/− and Ifng−/− mice were significantly higher compared to those in WT mice (Fig. 1D and E). Consistent with our previous observations [29], ocular infiltrates in Ifng−/− mice also contained increased neutrophils in addition to eosinophils. In Ifng−/−Il17a−/− mice, however, eosinophils were the only granulocyte type that was significantly increased compared to WT mice (Fig. 1D and E).
3.2. Eosinophils contribute to pathogenesis of EAU in the absence of both IFN-γ and IL-17A
To define the role of eosinophils in EAU pathogenesis in the absence of both IFN-γ and IL-17A, we crossed ΔdblGATA mice, which have a deletion in the high-affinity GATA-binding site of the promoter of the gene encoding the transcription factor GATA1, to double KO mice. This mutation in ΔdblGATA mice specifically blocks the development of mature eosinophils without affecting other cell types during hematopoiesis [20]. Eosinophil-deficient Ifng−/−Il17a−/− mice developed ameliorated EAU compared to eosinophil-sufficient Ifng−/−Il17a−/− mice (Fig. 2A and B). The ΔdblGATA mutation eliminated eosinophils from the eyes of Ifng−/−Il17a−/− mice (Fig. 2C), whereas the numbers of infiltrating neutrophils and monocytes were maintained (Fig. 2C). This result implicated eosinophils as a functionally important component of the ocular inflammatory infiltrate of Ifng−/−Il17a−/− mice during EAU.
Fig. 2.
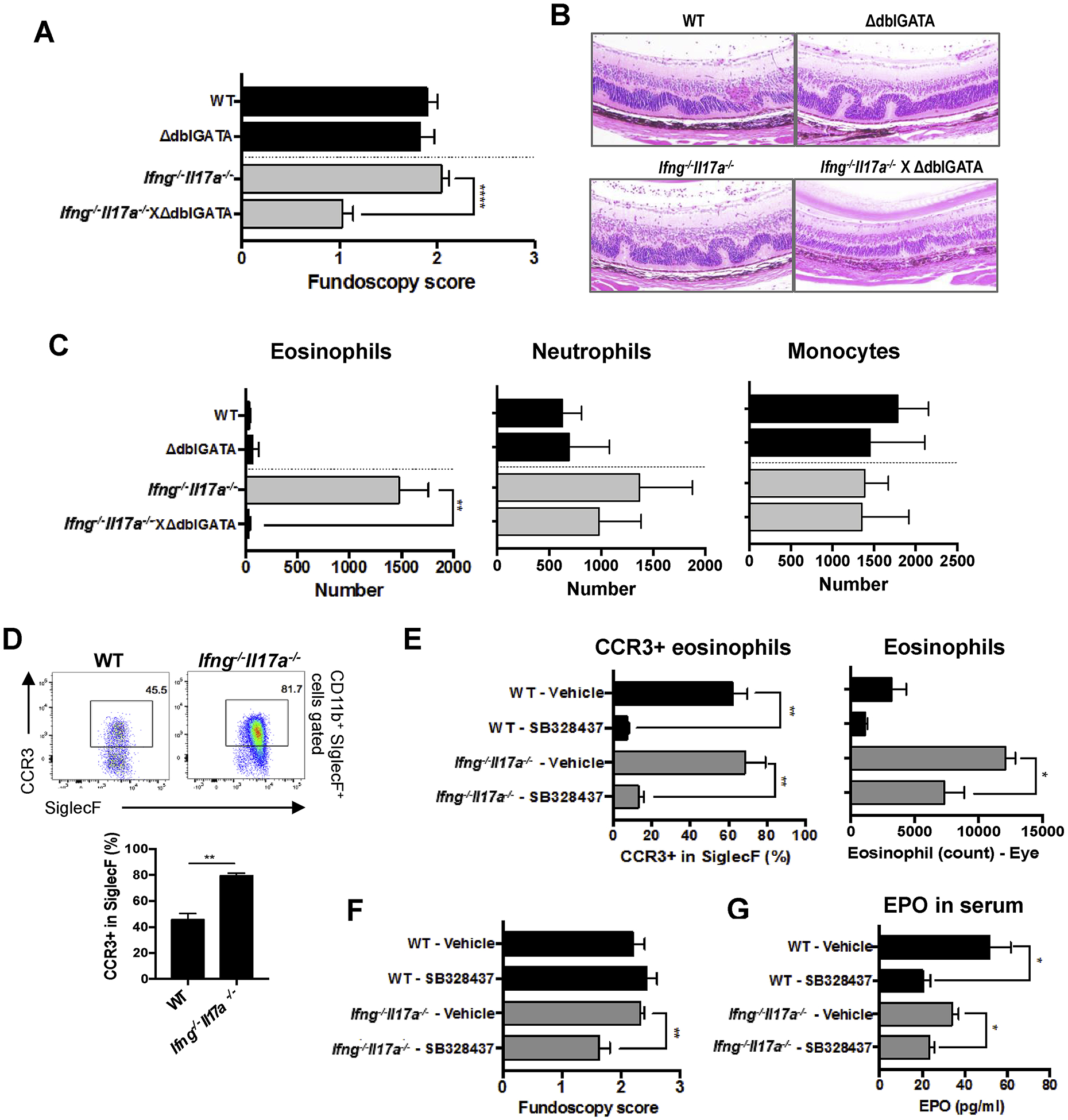
Eosinophils contribute to the pathogenesis of EAU in the absence of IFN-γ and IL-17A. (A–C) WT, ΔdblGATA, Ifng−/−Il17a−/−, Ifng−/−Il17a−/−ΔdblGATA mice on C57BL/6 background were immunized with 100 μg of p651–670. (A, B) EAU scores were evaluated by (A) fundoscopy, and (B) histology with H&E stain. Original mag, 20x. (C) The number of SiglecF+, Ly6C−Ly6G+, or Ly6C+Ly6G− cells in the CD45+CD11b+CD11c− gates from eyes on day 16. (D) The frequency of CCR3+ cells in eye-infiltrating eosinophils was determined by FACS. (E, F, G) WT and Ifng−/−Il17a−/− mice were s.c. treated with 200 μg of a CCR3 antagonist (SB328437) every day from one day before immunization. (E) The frequency of CCR3+ eosinophils and total eosinophils in eye-infiltrating cells were determined by FACS. (F) Fundoscopy score on day 15 days after immunization. (G) Levels of Eosinophil Peroxidase (EPO) in the serum were measured by LEGENDplex on day 16. (A) Data combined from 3 experiments (WT; n = 14, ΔdblGATA; n = 10, Ifng−/−Il17a−/−; n = 14, Ifng−/−Il17a−/− × ΔdblGATA; n = 5). (B–G) Representative data from 2 independent experiments, and each group contains at least 3 mice. Data shown as mean ± SEM. Significance was determined using Mann-Whitney U test (A, F), or unpaired t-test (C, D, E, G). *p < 0.05, **p < 0.01.
As another approach to reducing eosinophils in the eyes of EAU-challenged mice, we blocked CCR3, a receptor for eotaxin, which plays a critical role in trafficking of eosinophils [30]. We confirmed that eosinophils in the inflamed eyes of EAU-induced Ifng−/−Il17a−/− mice showed high expression of CCR3 (Fig. 2D). Mice were treated with a CCR3 antagonist, SB328437, during EAU to block migration of eosinophils into the eyes [23]. SB328437 effectively blocked expression of CCR3 on eosinophils and significantly reduced their migration into the eyes (Fig. 2E). This resulted in decreased EAU scores in Ifng−/−Il17a−/− mice, but not in WT mice (Fig. 2F), where EAU was apparently not dependent on eosinophils. Furthermore, SB328437 treatment reduced serum levels of EPO (Fig. 2G), which is produced exclusively by eosinophils and was shown to be toxic to tissues [31]. These data indicate that eosinophils are required to exacerbate pathology of EAU when both IFN-γ and IL-17A are absent.
3.3. Lack of both IFN-γ and IL-17A enhances the production of GM-CSF
Next, we focused on GM-CSF production in EAU immunized Ifng−/−Il17a−/− mice, or single KO mice. Eye-infiltrating cells were isolated when disease was fully developed on day 17–20, and immediately analyzed for GM-CSF–producing cells by intracellular cytokine staining after a PMA/Ionomycin pulse. A significantly higher frequency and total number of GM-CSF producing CD4+ T cells were detected in the eyes of Ifng−/−Il17a−/− mice than in WT mice (Fig. 3A). In addition, antigen-stimulated cultures of splenocytes from Ifng−/−Il17a−/− mice produced a significantly increased amount of GM-CSF in a recall response to the immunizing antigen IRBP (Fig. 3B). Although in splenocyte cultures activated T cells could produce GMCSF, all GM-CSF production was in response to autoantigen stimulation, therefore, we consider it relevant to disease.
Fig. 3.
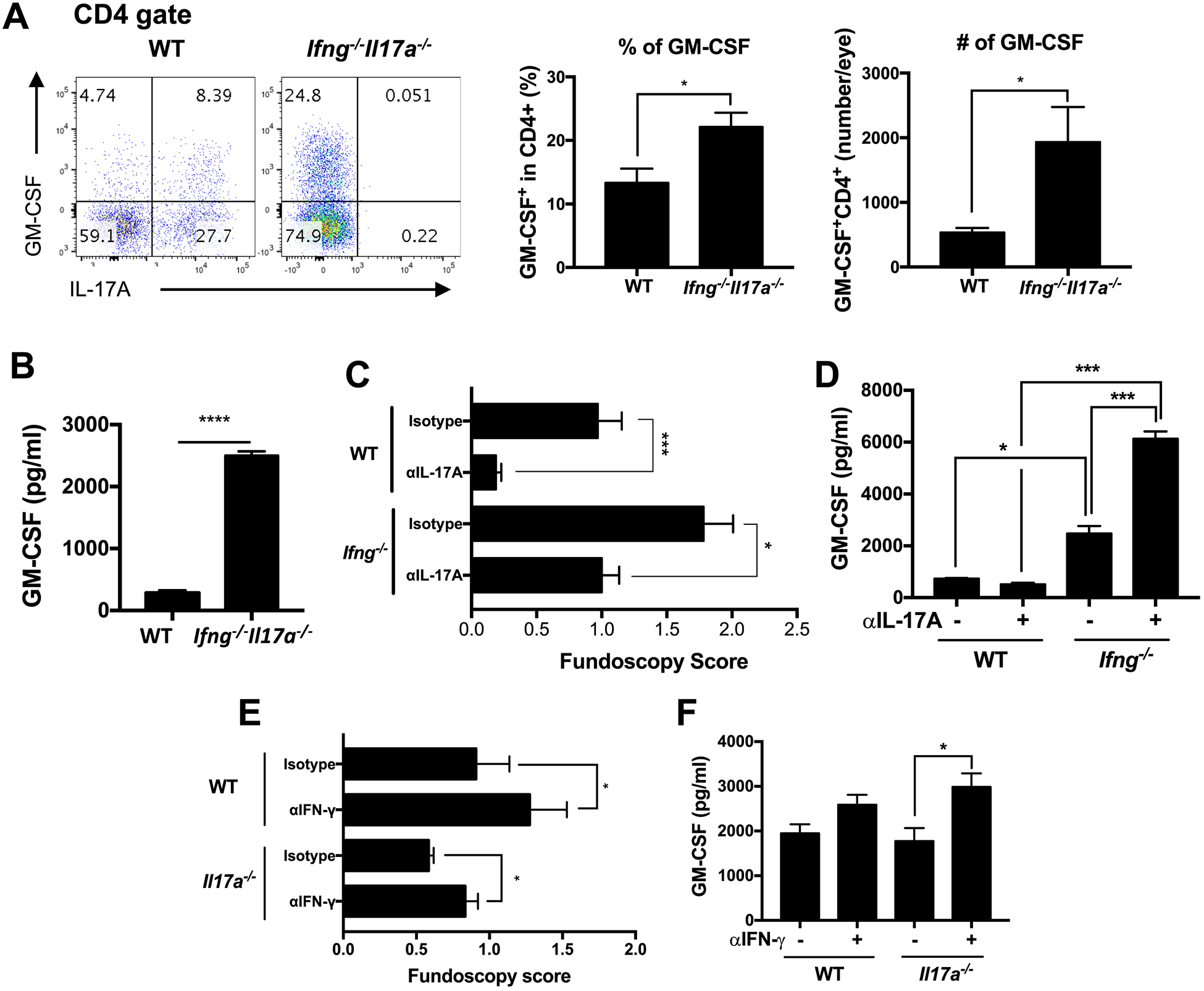
Production of GM-CSF is enhanced in cells that lack both IFN-γ and IL-17A. (A) On day 18 after EAU immunization of WT and Ifng−/−Il17a−/− mice on C57BL/6 background with IRBP and p1–20, eye-infiltrating cells were collected and intracellular cytokine staining was performed after a PMA/ionomycin pulse. Representative FACS plots (left), frequencies (middle) or numbers (right) of GM-CSF positive cells gated on CD4+ T cells. (B) GM-CSF secretion in supernatants of splenocytes was measured by ELISA after in vitro recall stimulation with IRBP. (C) WT and Ifng−/− mice were treated i.p. with 600 μg of anti-IL-17A Ab (MM17F3) or isotype control (mouse IgG1) every other day starting on day −1 relative to EAU immunization with IRBP and p1–20. (D, F) GM-CSF in splenocyte culture supernatant was measured by ELISA after in vitro recall stimulation with p651–670. (E) WT and Il17a−/− mice on C57BL/6 background were treated i.p. with 300 μg of anti–IFN-γ Ab (R4–6A2) or isotype control (Rat IgG1) every other day, starting on day −1 relative to EAU immunization with p651–670. Shown are representative data of 2 (C, D), 3 (E, F) or 4 (A, B) independent experiments, and each group contains at least 3 mice. Data shown as mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001, (A, B) Student’s t-test, (D, F) One way ANOVA with Bonferroni correction, (C, E) Mann-Whitney test.
In line with findings in other models, our earlier data showed that systemic depletion of IFN-γ by treatment with a neutralizing antibody exacerbates EAU in WT mice [8]. Furthermore the Th17 response, as judged by IRBP-stimulated IL-17A production, was higher in Ifng−/− mice with uveitis than in WT controls [15]. To investigate whether the increased disease severity due to systemic depletion of IFN-γ was due to a compensatory increase in IL-17A, we neutralized IL-17A in Ifng−/− mice starting from the afferent (priming) phase of EAU induction. As was previously observed [15], treatment with anti-IL-17A antibody significantly ameliorated EAU in WT mice. Interestingly, disease was also ameliorated in Ifng−/− mice which developed typically exacerbated EAU, but the severity of disease did not go below untreated WT controls (Fig. 3C). Notably, antigen-induced GM-CSF production from splenocytes in Ifng−/− mice was higher compared to WT mice, and was further upregulated when IL-17A was neutralized (Fig. 3D).
We also examined whether the GM-CSF response is enhanced in the opposite setting, in which IFN-γ was neutralized in Il17a−/− mice starting from the afferent (priming) phase of EAU induction. Exacerbated disease following anti–IFN-γ antibody treatment was observed in Il17a−/− mice, similarly to WT, suggesting that IFN-γ did not become indispensable for tissue pathology due to loss of IL-17A (Fig. 3E). In this setting as well, the antigen-stimulated immune cells from anti–IFN-γ antibody treated Il17a−/− mice produced increased amounts of GM-CSF in response to IRBP peptide than isotype control treated Il17a−/− mice (Fig. 3F). These data, with antibody neutralization of one cytokine in single KO mice lacking the other, support the finding in double-knockout mice that concurrent unavailability of both IFN-γ and IL-17A under inflammatory conditions precipitates GMCSF production.
3.4. Systemic or local neutralization of GM-CSF in Ifng−/− Il17a−/− mice ameliorates EAU severity
GM-CSF is known to promote eosinophil development and activation [32]. To examine the notion that GM-CSF plays a necessary and non-redundant role in EAU in the absence of IFN-γ and IL-17A, we employed neutralizing antibodies to deplete GM-CSF either systemically, or locally. First, we blocked GM-CSF by administering neutralizing antibody systemically during the afferent (priming) phase or the efferent (effector) phase of disease (Fig. 4A, C). Treatment with anti-GM-CSF antibody during the afferent phase prevented development of EAU in Ifng−/−Il17a−/− mice (Fig. 4B). The anti-GM-CSF antibody injected during the afferent phase of EAU were detected in the blood until day 21 after immunization (Supplementary Fig. 2), indicating that this antibody remains in circulation and may have continued to neutralize GM-CSF during the entire course of disease. Neutralization of GM-CSF during the efferent phase alone (starting on day 7 relative to immunization) also ameliorated the EAU severity in Ifng−/−Il17a−/− mice, albeit to a lesser extent (Fig. 4D). This suggested that GM-CSF is needed both during priming, and effector phase for EAU pathogenesis in the absence of IFN-γ and IL-17A.
Fig. 4.
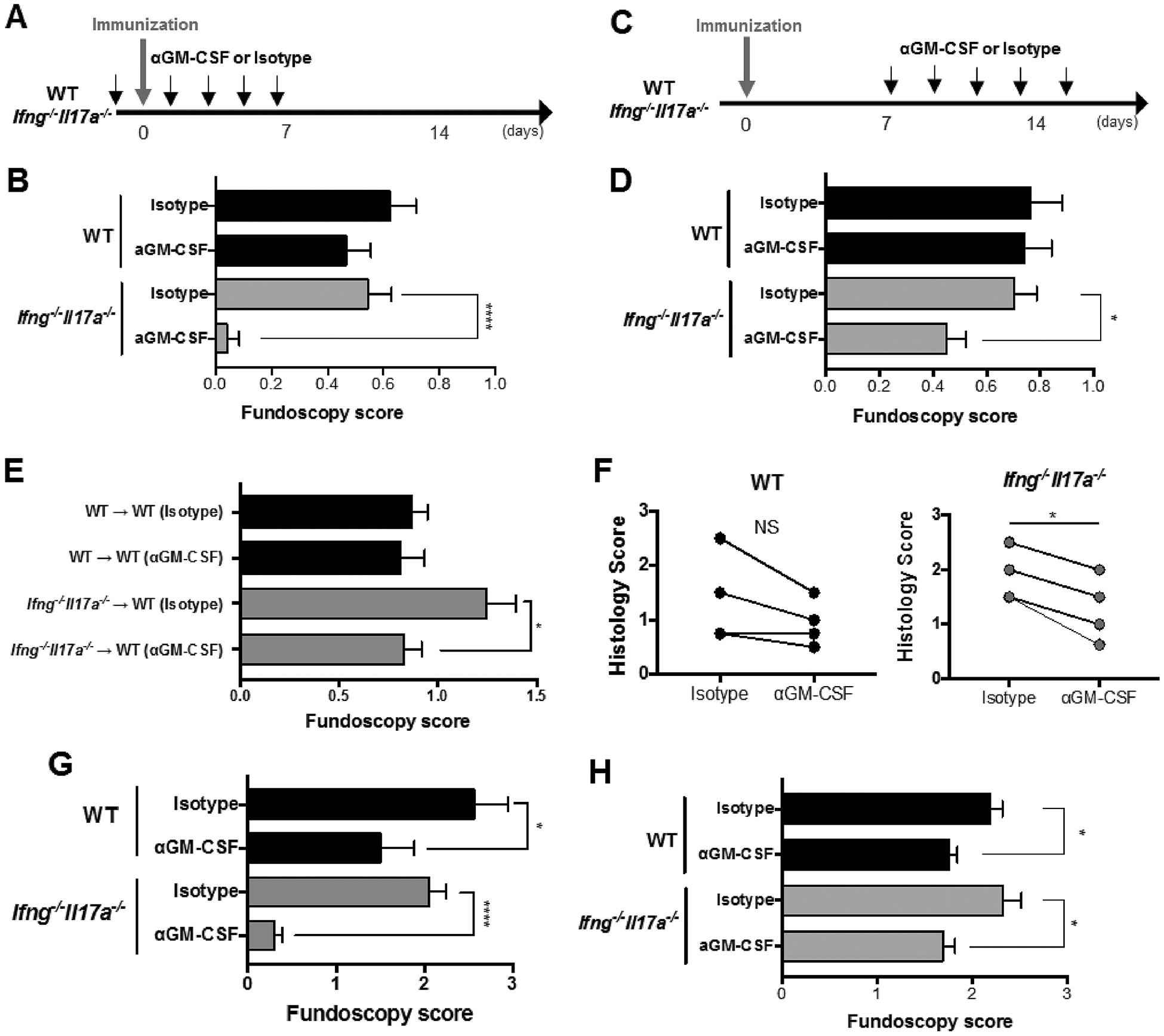
Treatment with anti-GM-CSF antibody at either induction or effector phase of EAU suppresses disease in Ifng−/−Il17a−/− mice. WT and Ifng−/−Il17a−/− mice either on C57BL/6 (immunized with IRBP and p1–20) (B, D, E) or on B10.RIII background (immunized with p161–180) (G, H) were i.p. treated with anti-GMCSF mAb (MP1–22E9.11) or isotype Ig (Rat IgG2a) from the induction phase (starting on day −1) (A, B, G) or from the effector phase (starting on day 7) (C, D, H) of EAU induction every other day. (E) T cells were enriched from LNs and spleens of EAU-induced WT and Ifng−/−Il17a−/− mice, were restimulated in vitro with IRBP for 3 days and were adoptively transferred to naïve recipient mice that received treatment of 300 μg of anti-GM-CSF mAb or isotype control every other day starting from the day of cell transfer. (F) WT and Ifng−/−Il17a−/− mice were immunized for EAU. On day 10, 100 μg of anti-GM-CSF mAb in 2 μl of PBS was intravitreally injected into the right eye and the isotype control was injected into the left eye of each mouse. EAU scores were assessed by histology on day 12. Representative data from 2 (E, F) or 3 (B, D, G, H) independent experiments, and each group contains at least 3 mice. Data shown as mean ± SEM. *p < 0.05, ****p < 0.0001, (B, D, E, G, H) Mann-Whitney U test, (F) Wilcoxon matched-pairs signed rank test. NS, not significant.
To more directly evaluate whether GM-CSF has a role in the effector phase of EAU, we adoptively transferred in vitro IRBP-reactivated T cells originating from EAU-challenged WT or Ifng−/−Il17a−/− mice into WT recipient mice, and treated the recipients with anti-GM-CSF antibody. Blockade of GM-CSF in recipients of WT T cells did not alter subsequent disease scores. In contrast, it significantly attenuated disease scores in recipients of Ifng−/−Il17a−/− cells (Fig. 4E). We further examined whether GM-CSF production in situ, within the inflamed eye, has a pathogenic role in EAU by injecting anti-GM-CSF antibody into one eye on the day of disease onset. In Ifng−/−Il17a−/− mice, the eye that received anti-GM-CSF antibody developed significantly less disease than the contralateral eye that received the isotype control (Fig. 4F). In the aggregate, these data implicate GM-CSF as a major uveitogenic cytokine in C57BL/6 mice lacking both IFN-γ and IL-17A.
Next, we tested whether this finding extends to the B10.RIII strain, which is the most susceptible strain to EAU. Unlike C57BL/6, neutralization of GM-CSF effectively ameliorated EAU irrespective of IFN-γ and IL-17A availability and there was a strong correlation between EAU scores and the proportion of GM-CSF producing CD4+ T cells in the inflamed eyes in WT B10.RIII mice (Supplementary Fig. 3). However, the dependence on GM-CSF appeared more pronounced in the absence of IFN-γ and IL-17A both during afferent (Fig. 4G) and efferent phase (Fig. 4H). These results suggest that the degree of dependence on GMCSF to effect ocular pathology is, at least to some extent, strain-dependent.
3.5. GM-CSF produced by autoantigen-stimulated Ifng−/−Il17a−/− cells promotes eosinophil activation and migration
Given the important roles of IL-5 and GM-CSF in eosinopoiesis and survival of eosinophils [33], we examined whether these factors play a role in eosinophil migration and activation during EAU in Ifng−/−Il17a−/− mice. To address this question, we cultured bone marrow-derived eosinophils (bmEos) with supernatants of IRBP-stimulated splenocytes from EAU-induced WT or Ifng−/−Il17a−/− mice, and measured the fluorescence intensity of the eosinophil activation markers CD11b and SiglecF [34,35]. Enhanced expression of both CD11b and SiglecF was observed in bmEos cultured with splenocyte supernatants from Ifng−/−Il17a−/− mice, compared to control supernatants produced from WT mice (Fig. 5A). Anti-GM-CSF antibody abrogated enhancement of both activation markers by supernatants of Ifng−/−Il17a−/− splenocytes, whereas addition of GM-CSF elevated their expression in presence of supernatants from WT splenocytes to Ifng−/−Il17a−/− levels (Fig. 5A). In contrast, addition of anti-IL-5 antibody had no effect on activation of eosinophils by the Ifng−/−Il17a−/− supernatant as judged by expression of CD11b and SiglecF (data not shown). These results indicate that activation of eosinophils in Ifng−/−Il17a−/− mice is likely due to increased production of GM-CSF, but not IL-5. In addition, in a Boyden chamber chemotaxis assay the supernatant of IRBP-stimulated splenocytes from EAU-induced Ifng−/−Il17a−/− mice promoted eosinophil migration better than that supernatant from parallel splenocytes WT mice, and this migration was inhibited by anti-GM-CSF antibody (Fig. 5B). These results suggest that GM-CSF could functionally contribute to activation and migration of eosinophils in the absence of IFN-γ and IL-17A.
Fig. 5.
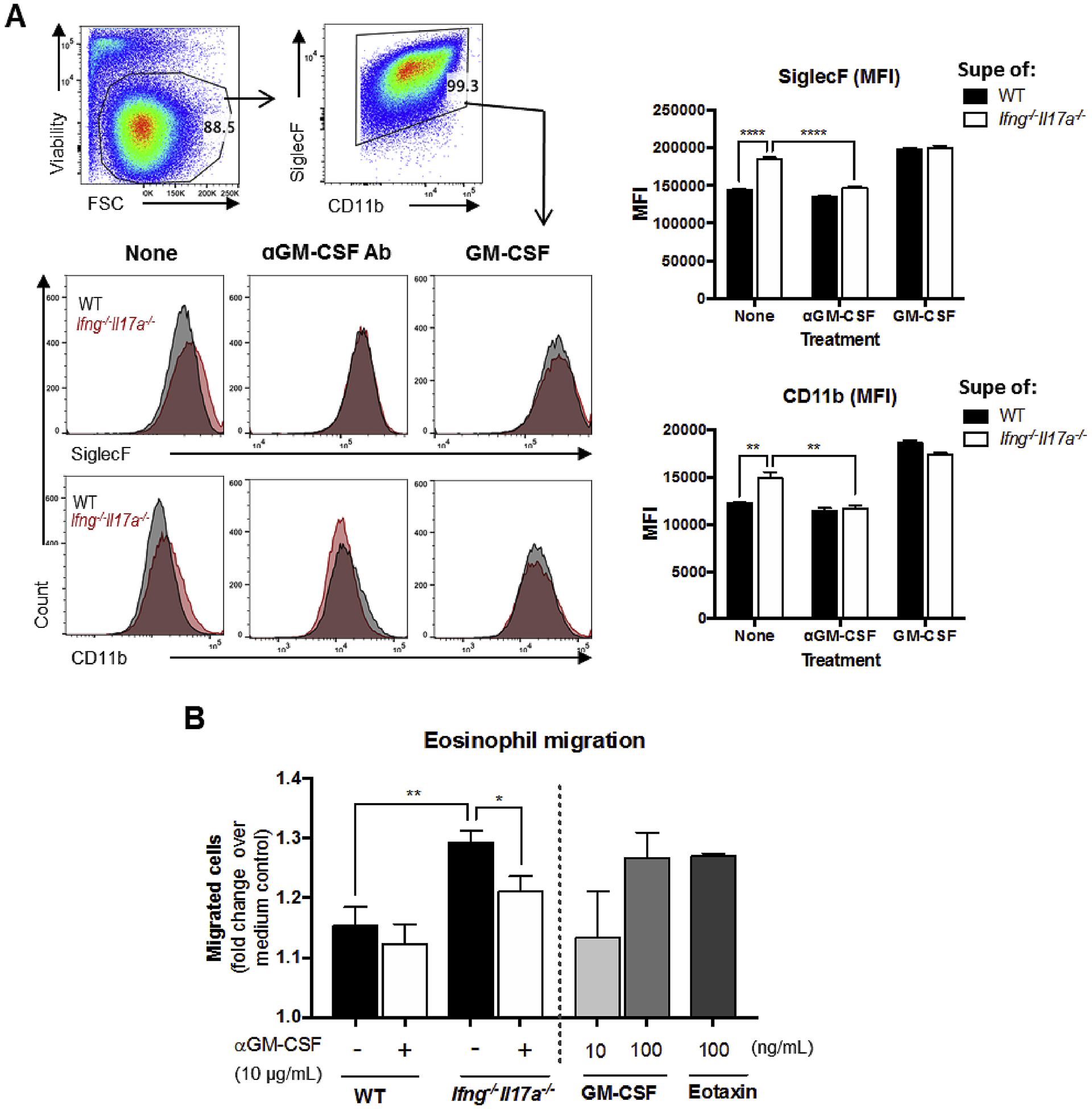
GM-CSF produced by autoantigen-stimulated Ifng−/−Il17a−/− cells promotes eosinophil activation and migration (A) Bone-marrow derived eosinophils (bmEos) from WT B10.RIII mice were cultured with the supernatant of antigen (IRBP) re-stimulated splenocytes from EAU-induced WT or Ifng−/−Il17a−/− mice in the presence or absence of anti-GM-CSF antibody (10 μg/mL) or GM-CSF (100 ng/mL). After 2 days, the mean fluorescence intensity (MFI) of SiglecF and CD11b on bmEos was determined by FACS. Representative FACS plots with gates and histograms are shown on the left. (B) BmEos were placed into the upper chamber of a 96-well transwell chemotaxis assay plate with the supernatant of re-stimulated splenocytes in the lower chamber and incubated for 3 h. GM-CSF or eotaxin were used as positive controls. The total number of live cells that had migrated into the bottom chamber with the supernatant was counted and divided by the number of live cells that had migrated to medium alone to give the fold change over the medium control. Representative data from 2 independent experiments, each group contains 2 individual samples, and each sample was duplicated. Data shown as mean ± SEM. Significance was determined using unpaired t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.6. The pathogenic effects of GM-CSF in Ifng−/−Il17a−/− EAU are connected to its effects on eosinophils
As the next question, we asked whether GM-CSF in fact contributes to pathogenicity at least in part through its effects on eosinophils. These experiments were performed in B10.RIII as well as in C57BL/6 background mice. In inflamed eyes of B10.RIII mice treated with anti-GMCSF antibody during the efferent phase, there was a drastic decrease in the number of eosinophils, which was particularly striking in the Ifng−/−Il17a−/− genotype (Fig. 6A and B). In addition, there was a significant reduction in EPO levels in the serum and eye homogenate supernatants of animals that received anti-GM-CSF antibody treatment (Fig. 6C). Similar inhibition in eosinophils recruitment to the eye by anti-GM-CSF antibody treatment during the efferent phase was observed in the C57BL/6 strain (Supplementary Fig. 4), and in both strains of mice treated with anti-GM-CSF antibody post immunization throughout the course of EAU (data not shown). These results demonstrate that the accumulation of eosinophils in the eyes of EAU-induced Ifng−/−Il17a−/− mice, which is causally related to disease (Fig. 2), is due at least in part to GM-CSF.
Fig. 6.
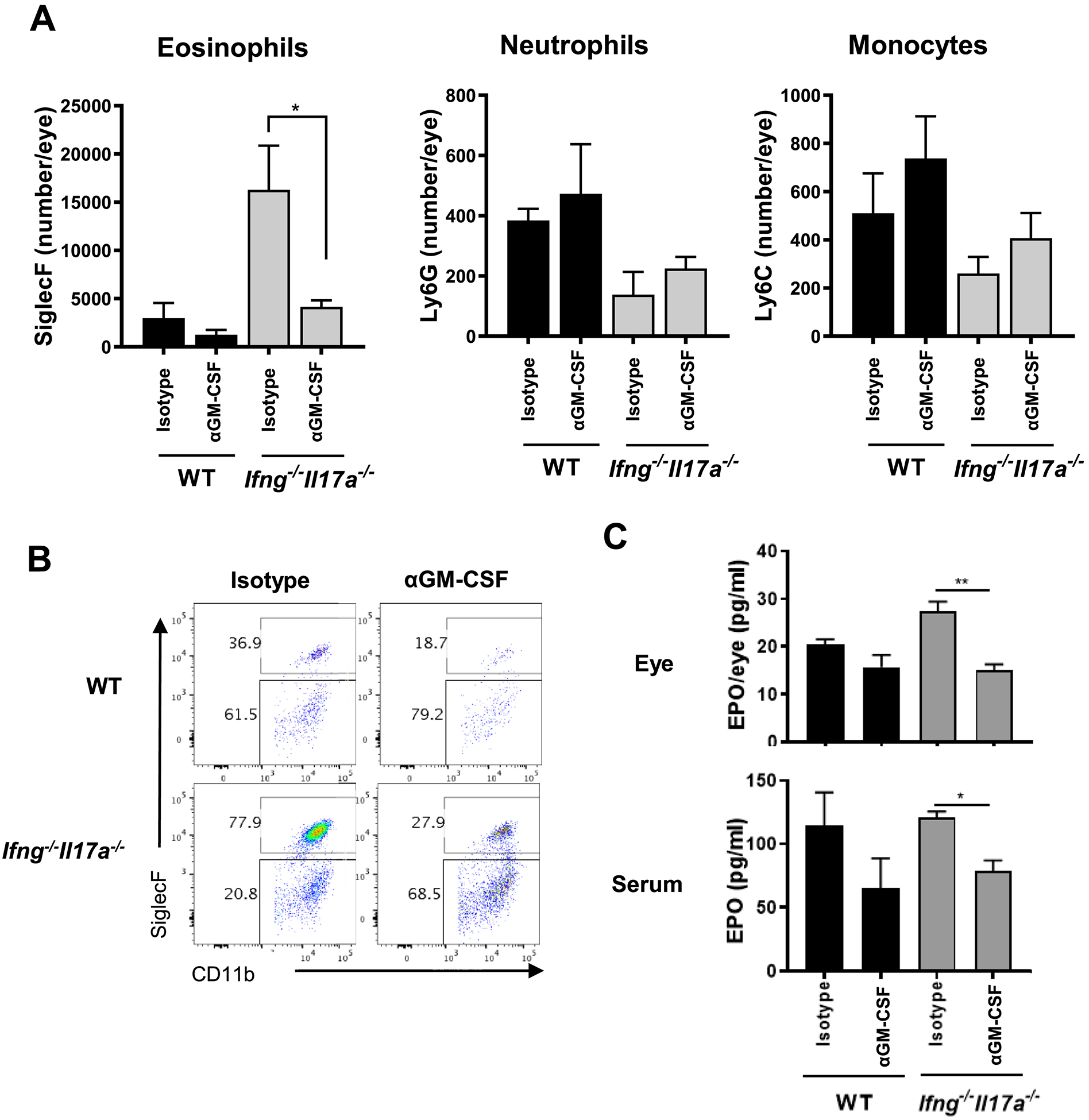
Blockade of GM-CSF decreases disease by suppressing migration and function of eosinophils. WT and Ifng−/−Il17a−/− mice on B10.RIII background were immunized with p161–180 and i.p. treated with anti-GM-CSF mAb (MP1–22E9.11) or isotype Ig (Rat IgG2a) from the effector phase (starting on day 7). (A) The number of eosinophils (SiglecF+CD11b+CD45+), neutrophils (Ly6G+Ly6C−CD11b+CD45+), monocytes (Ly6G−Ly6C+CD11b+CD45+) in eye-infiltrating cells was determined by FACS. (B) Representative FACS plots of eye-infiltrating eosinophils. (C) Eosinophil peroxidase (EPO) in the eye-supernatant and the serum were assessed by LEGENDPlex Representative data from 3 independent experiments, and each group contains at least 3 mice. Data shown as mean ± SEM. *p < 0.05, **p < 0.01, Student’s t-test.
3.7. IL-17F is also an important cytokine for EAU in Ifng−/−Il17a−/− mice, but does not contribute to the eosinophilic infiltration
IL-17F is a member of the IL-17 family and has the highest homology to IL-17A [36]. IL-17F is pathogenic in commensal micro-biota-mediated colitis, but unlike IL-17A, the role of IL-17F in autoimmune diseases has been elusive [37,38]. In fact, the severity of EAE was not affected by lack of IL-17F alone [10,18,39], and we detected enhanced IL-17F production in IRBP-stimulated Ifng−/−Il17a−/− spleen cells from EAU mice (data not shown). We, therefore, wished to examine whether IL-17F takes on a pathogenic role in EAU when other major pathogenic cytokines are absent. No significant differences in EAU scores were observed between WT and Il17f−/− mice (Fig. 7A). However, EAU in triple KO Ifng−/−Il17a−/−Il17f−/− mice was significantly ameliorated compared to double KO Ifng−/−Il17a−/− mice (Fig. 7B). In Ifng−/−Il17a−/− mice, IL-17F deficiency reduced the neutrophil component in inflamed eyes but did not affect the eosinophil or monocyte components. (Fig. 7C). These data suggest that (a) IL-17F plays a pathogenic role in EAU only when both IFN-γ and IL-17A are absent, by recruiting neutrophils, but it does not appear to contribute to the eosinophil-related pathology in Ifng−/−Il17a−/− mice; (b) not only eosinophils, but also neutrophils contribute to EAU pathology in Ifng−/−Il17a−/− double KO mice, as their loss in the absence of IL-17F makes a statistically significant (although modest) difference in EAU scores.
Fig. 7.
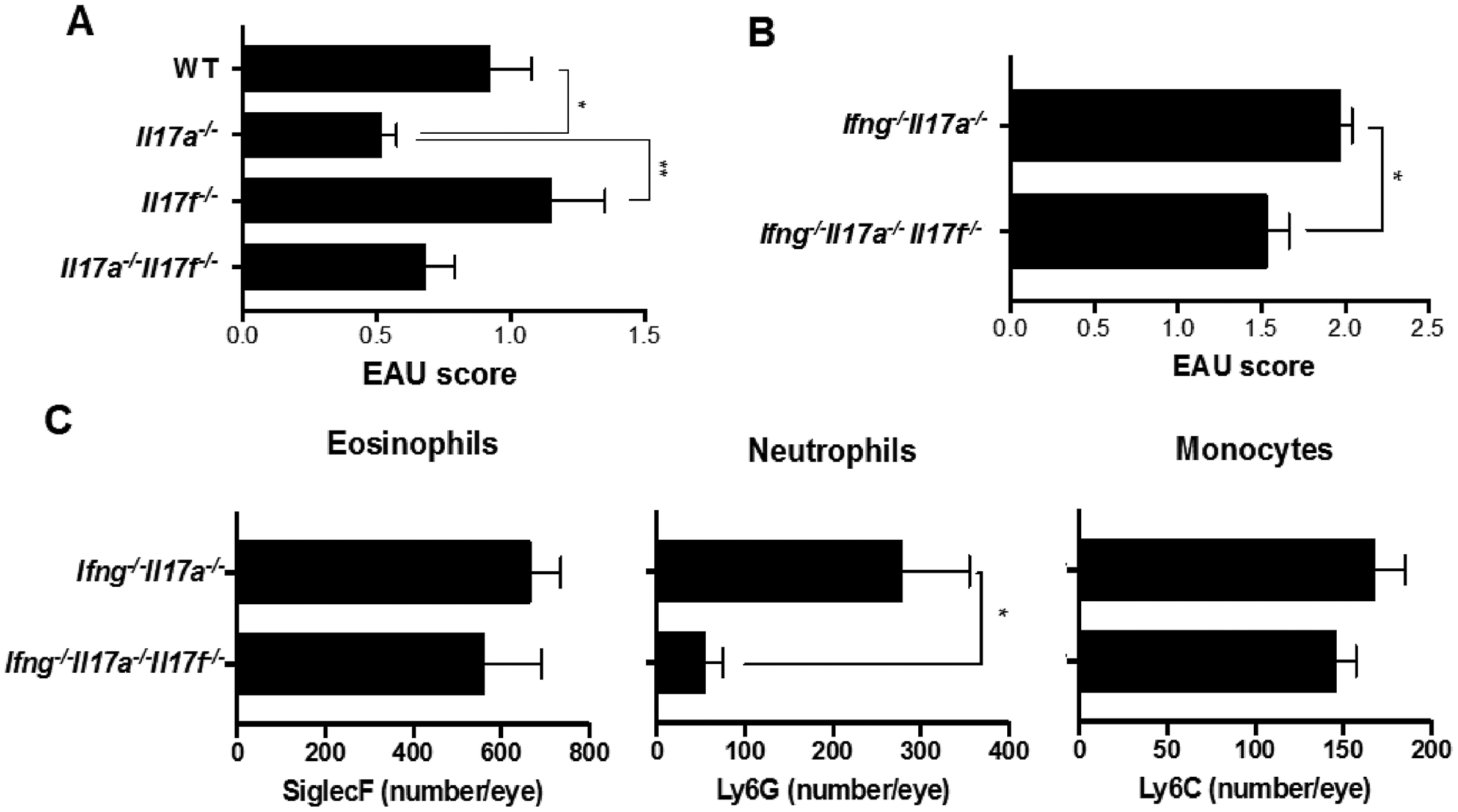
IL-17F plays a pathogenic role in EAU-challenged Ifng−/−Il17a−/− mice by recruiting neutrophils in the eyes. (A) EAU scores were evaluated by histology of eyes from C57BL/6 WT, Il17a−/−, Il17f−/−, and Il17a−/−Il17f−/− mice 21 days after immunization with IRBP and p1–20. (B, C) Ifng−/−Il17a−/−, Il17a−/−Il17f−/−Ifng−/− on C57BL/6 background were immunized with 150 μg p651–670. (B) EAU scores were evaluated by fundoscopy on day 16 after immunization. (C) The number of SiglecF+, Ly6C−Ly6G+, or Ly6C+Ly6G− cells in the CD45+CD11b+CD11c− gates from eyes. Representative data from 3 independent experiments. Data shown as mean ± SEM. Significance was determined using Mann-Whitney U test (A, B) or Student’s t-test (C). *p < 0.05, **p < 0.01.
4. Discussion
In this study, we wished to examine what would happen to expression of autoimmune disease affecting the neuroretina in the absence of the effector cytokines defining both major T cell lineages that have been implicated in its pathology, Th1 and Th17. We found that “all roads lead to Rome”, or in this case, to uveitis, but the pathology is caused by a distinct effector response. Severity of EAU in Ifng−/−Il17a−/− mice was similar to that in WT mice, but unlike in WT mice that develop a primarily mononuclear infiltrate in the eyes, and unlike Ifng−/− single KO mice that develop a mixed granulocytic infiltrate dominated by neutrophils [29], the inflammatory leukocyte infiltrate in the eyes of Ifng−/−Il17a−/− mice was strongly dominated by eosinophils (Fig. 1). Since IL-17 (A and F) recruit granulocytes to inflammatory sites, the granulocytic nature of the ocular infiltrate in single Ifng−/− mice can probably be explained by their increased IL-17 response [15]. It remains unclear, however, why IL-17F, which has many overlapping activities with IL-17A, was not sufficient to maintain neutrophil recruitment in Ifng−/−Il17a−/−, resulting in an eosinophil-dominant ocular inflammation.
The chemokine that plays a central role in the pathology of eosinophil-dominant inflammation is the CC chemokine CCL11/eotaxin [23] whose receptor is CCR3. Of note, CCR3 is expressed also by Th2 cells, basophils and mast cells [30,40] and is involved in eosinophil-associated as well as atopic diseases. The CCR3 antagonist used in this study, SB328437, inhibits cellular calcium flux induced by CCR3 agonists [23] and has beneficial effects in asthma and allergy [41]. Interestingly, CCR3 appears to play an important role also in a non-allergic setting. In a laser injury model used to simulate ‘wet’ age-related macular degeneration (AMD), SB328437 effectively diminished choroidal neovascularization, suggesting that CCR3 is also involved in angio-genesis [42]. Although we showed that SB328437 reduced EAU scores in Ifng−/−Il17a−/− mice by inhibiting eosinophilic infiltration, it is conceivable that additional mechanisms downstream of CCR3 blockade may have contributed to attenuation of EAU in Ifng−/−Il17a−/− mice.
Published studies have demonstrated that common β-chain cytokines, IL-3, IL-5 and GM-CSF are involved in eosinophil development and biology [43]. Our findings that neutralization of GM-CSF, but not of IL-5, reduced EAU scores in Ifng−/−Il17a−/− mice, coupled with the ability of GM-CSF produced by their immune cells to directly promote eosinophil activation and migration, support the interpretation that GM-CSF, rather than IL-5, drives the characteristic eosinophilic ocular inflammation that developed in these mice.
GM-CSF is produced by various types of hematopoietic cells, including activated T cells, and non-hematopoietic cells, such as endothelial cells and fibroblasts [44]. It has multifaceted effects on development, maturation, trafficking and function of immune cells, in particular, myeloid cells and their precursors [45]. Among its other activities, GM-CSF enhances the activity of monocyte/macrophages and neutrophils, which are the proximal effectors of tissue damage in autoimmune neuroinflammation. However, its precise role and effects on pathology are still controversial. While some investigators concluded that GM-CSF produced by antigen-specific effector T cells has a necessary and nonredundant role in EAE development [46,47], others demonstrate convincingly that GM-CSF modulates, rather than determines, EAE expression [48] and its role in that regard is fully redundant with that of IL-17A. Our data in the EAU model are more compatible with a modulatory, than an obligatory role of GM-CSF. The effect of GM-CSF neutralization on EAU pathology was pronounced only in the concurrent absence of IFN-γ and IL-17A, but was low to moderate in WT mice. This is also supported by our observation that IL-17A/IFN-γ-sufficient GM-CSF KO mice [49] immunized for EAU developed only moderately lower disease scores than their heterozygous littermate controls (Wloka et al., unpublished data). Additionally, while in EAE, the main cells targeted by GM-CSF are macrophage/monocytes and neutrophils [48,50,51], in EAU, GM-CSF promoted an eosinophilic inflammation, whereas IL-17 (A more than F) appeared to attract neutrophils. The importance and effects of GM-CSF in these different studies may in part be influenced also by the genetic background, C57BL/6 ([46,47,50,51], and our work) vs. C3HeB/FeJ [48] vs. B10.RIII (our work), adding another layer of complexity.
What remains to be settled, is the well recognized paradox that deficiency of IFN-γ, known to be a proinflammatory cytokine and a product of pathogenic Th1 cells, inhibits autoimmune neuroinflammation in EAE and EAU. Notably, IFN-γ strongly represses the development of GM-CSF producing effector T cells [47,48]. As a separate pathway, we showed that “innate” IFN-γ produced by cells such as NK and NKT during the priming phase of EAU dampens development of the adaptive Th1 and Th17 effector responses, at least in part through induction of NOS2 and elicitation of IL-27 [52–54].
Eosinophils are usually associated with a Th2 response, which can itself result in pathology. Although in cell-mediated tissue specific autoimmunity, such as EAE and EAU, the Th2 response has been considered regulatory, previous data demonstrated that under some circumstances an unopposed Th2 response could cause ocular pathology [55]. Relevant to this, we detected enhanced Th2-like responses (IL-4, IL-5, and IL-13 production) in IRBP-stimulated Ifng−/−Il17a−/− spleen cells from EAU mice (data not shown). Furthermore, eosinophils themselves can secrete Th2 cytokines, including IL-4 and IL-13 [56]. However, in the current scenario the eosinophils did not appear to represent an underlying Th2 response. In fact, blockade of the IL-4/IL-13 receptor heterodimer signaling by antibody to IL-4Rα exacerbated, rather than reduced, EAU in Ifng−/−Il17a−/− mice (data not shown), suggesting that, if anything, the Th2 response plays a protective role in this situation. Of note, eosinophils have been implicated in tissue pathology also in some other autoimmune conditions. While it is beyond our scope to provide a comprehensive discussion of the subject, the topic has recently been reviewed [57].
In conclusion, we have shown that increased GM-CSF production contributes to the pathogenesis of EAU in the concurrent absence of IFN-γ and IL-17A. Mechanistically, we propose that GM-CSF directly activates eosinophils in the periphery, enhances their migration to the eye and thereby promotes ocular inflammation. The finding that “all roads lead to uveitis”, where loss of one inflammatory cytokine is compensated for by another without reducing pathology, raises a note of caution when therapeutically targeting cytokines in a complex autoimmune disease, and may help explain why IL-17A-neutralizing therapy has been disappointing as treatment for uveitis [58]. GM-CSF or GM-CSF receptor targeting antibodies are currently being examined in clinical trials for rheumatoid arthritis and multiple sclerosis [59]. We propose that blockade of the GM–CSF–eosinophils pathway(s) might be a viable therapeutic approach for some patients with autoimmune uveitis, while taking into account potential consequences to host defense of targeting a major effector cytokine.
Supplementary Material
Acknowledgements
The authors thank Drs. Yoichiro Iwakura (Tokyo University of Science) and Avery August (Cornell University) for providing knockout mice, NEI Flow Cytometry Core and Histology Core for their assistance, and the members of Laboratory of Immunology, NEI, NIH for critical discussions. This work was supported by NEI/NIH Intramural funding, project # EY000184.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jaut.2020.102507.
References
- [1].Caspi RR, A look at autoimmunity and inflammation in the eye, J. Clin. Invest 120 (2010) 3073–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Damsker JM, Hansen AM, Caspi RR, Th1 and Th17 cells: adversaries and collaborators, Ann. N. Y. Acad. Sci 1183 (2010) 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Egwuagu CE, Mahdi RM, Chan CC, Sztein J, Li W, Smith JA, et al. , Expression of interferon-gamma in the lens exacerbates anterior uveitis and induces retinal degenerative changes in transgenic Lewis rats, Clin. Immunol 91 (1999) 196–205. [DOI] [PubMed] [Google Scholar]
- [4].Egwuagu CE, Sztein J, Mahdi RM, Li W, Chao-Chan C, Smith JA, et al. , IFN-gamma increases the severity and accelerates the onset of experimental autoimmune uveitis in transgenic rats, J. Immunol 162 (1999) 510–517. [PubMed] [Google Scholar]
- [5].Jones LS, Rizzo LV, Agarwal RK, Tarrant TK, Chan CC, Wiggert B, et al. , IFN-gamma-deficient mice develop experimental autoimmune uveitis in the context of a deviant effector response, J. Immunol 158 (1997) 5997–6005. [PubMed] [Google Scholar]
- [6].Krakowski M, Owens T, Interferon-γ confers resistance to experimental allergic encephalomyelitis, Eur. J. Immunol 26 (1996) 1641–1646. [DOI] [PubMed] [Google Scholar]
- [7].Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. , Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE), J. Immunol 156 (1996) 5–7. [PubMed] [Google Scholar]
- [8].Caspi RR, Chan CC, Grubbs BG, Silver PB, Wiggert B, Parsa CF, et al. , Endogenous systemic IFN-gamma has a protective role against ocular autoimmunity in mice, J. Immunol 152 (1994) 890–899. [PubMed] [Google Scholar]
- [9].Luger D, Caspi RR, New perspectives on effector mechanisms in uveitis, Semin. Immunopathol 30 (2008) 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. , IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis, J. Immunol 177 (2006) 566–573. [DOI] [PubMed] [Google Scholar]
- [11].Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, et al. , Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis, Cell. Immunol 237 (2005) 123–130. [DOI] [PubMed] [Google Scholar]
- [12].Nakae S, Nambu A, Sudo K, Iwakura Y, Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice, J. Immunol 171 (2003) 6173–6177. [DOI] [PubMed] [Google Scholar]
- [13].Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. , TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1, Nat. Med 13 (2007) 711–718. [DOI] [PubMed] [Google Scholar]
- [14].Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. , IL-23 drives a pathogenic T cell population that induces autoimmune inflammation, J. Exp. Med 201 (2005) 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. , Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category, J. Exp. Med 205 (2008) 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. , Fate mapping of IL-17-producing T cells in inflammatory responses, Nat. Immunol 12 (2011) 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, et al. , Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses, Immunity 17 (2002) 375–387. [DOI] [PubMed] [Google Scholar]
- [18].Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. , Differential roles of interleukin-17A and −17F in host defense against mucoepithelial bacterial infection and allergic responses, Immunity 30 (2009) 108–119. [DOI] [PubMed] [Google Scholar]
- [19].Walsh ER, Sahu N, Kearley J, Benjamin E, Kang BH, Humbles A, et al. , Strain-specific requirement for eosinophils in the recruitment of T cells to the lung during the development of allergic asthma, J. Exp. Med 205 (2008) 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y, et al. , Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo, J. Exp. Med 195 (2002) 1387–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pepperberg DR, Okajima T-IL, Ripps H, Chader GJ, Wiggert B, Functional properties OF interphotoreceptor retinoid-binding protein*, Photochem. Photobiol 54 (1991) 1057–1060. [DOI] [PubMed] [Google Scholar]
- [22].Mattapallil MJ, Silver PB, Cortes LM, St Leger AJ, Jittayasothorn Y,Kielczewski JL, et al. , Characterization of a new epitope of IRBP that induces moderate to severe uveoretinitis in mice with H-2b haplotype, Investig. Ophthalmol. Visual Sci 56 (2015) 5439–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].White JR, Lee JM, Dede K, Imburgia CS, Jurewicz AJ, Chan G, et al. , Identification of potent, selective non-peptide CC chemokine receptor-3 antagonist that inhibits eotaxin-, eotaxin-2-, and monocyte chemotactic protein-4-induced eosinophil migration, J. Biol. Chem 275 (2000) 36626–36631. [DOI] [PubMed] [Google Scholar]
- [24].Caspi RR, Experimental autoimmune uveoretinitis in the rat and mouse, Curr. Protoc. Im 53 (1) (2003) 15.6.1–15.6.20 (Chapter 15):Unit 15.6. [DOI] [PubMed] [Google Scholar]
- [25].Chen J, Qian H, Horai R, Chan CC, Caspi RR, Mouse models of experimental autoimmune uveitis: comparative analysis of adjuvant-induced vs spontaneous models of uveitis, Curr. Mol. Med 15 (2015) 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dyer KD, Moser JM, Czapiga M, Siegel SJ, Percopo CM, Rosenberg HF, Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow, J. Immunol 181 (2008) 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Avichezer D, Chan C-C, Silver PB, Wiggert B, Caspi RR, Residues 1–20 of IRBP and whole IRBP elicit different uveitogenic and immunological responses in interferon gamma deficient mice, Exp. Eye Res 71 (2000) 111–118. [DOI] [PubMed] [Google Scholar]
- [28].Yoshimura T, Sonoda KH, Miyazaki Y, Iwakura Y, Ishibashi T, Yoshimura A, et al. , Differential roles for IFN-gamma and IL-17 in experimental autoimmune uveoretinitis, Int. Immunol 20 (2008) 209–214. [DOI] [PubMed] [Google Scholar]
- [29].Su SB, Grajewski RS, Luger D, Agarwal RK, Silver PB, Tang J, et al. , Altered chemokine profile associated with exacerbated autoimmune pathology under conditions of genetic interferon-gamma deficiency, Investig. Ophthalmol. Visual Sci 48 (2007) 4616–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Humbles AA, Lu B, Friend DS, Okinaga S, Lora J, Al-Garawi A, et al. , The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness, Proc. Natl. Acad. Sci. U.S.A 99 (2002) 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fulkerson PC, Rothenberg ME, Targeting eosinophils in allergy, inflammation and beyond, Nat. Rev. Drug Discov 12 (2013) 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Esnault S, Malter JS, GM-CSF regulation in eosinophils, Arch. Immunol. Ther. Exp 50 (2002) 121–130. [PubMed] [Google Scholar]
- [33].Kouro T, Takatsu K, IL-5- and eosinophil-mediated inflammation: from discovery to therapy, Int. Immunol 21 (2009) 1303–1309. [DOI] [PubMed] [Google Scholar]
- [34].Voehringer D, van Rooijen N, Locksley RM, Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages, J. Leukoc. Biol 81 (2007) 1434–1444. [DOI] [PubMed] [Google Scholar]
- [35].Walker C, Rihs S, Braun RK, Betz S, Bruijnzeel PL, Increased expression of CD11b and functional changes in eosinophils after migration across endothelial cell monolayers, J. Immunol 150 (1993) 4061–4071. [PubMed] [Google Scholar]
- [36].Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, et al. , IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding, EMBO J 20 (2001) 5332–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. , Regulation of inflammatory responses by IL-17F, J. Exp. Med 205 (2008) 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. , IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice, J. Clin. Invest 119 (2009) 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Horai R, Caspi RR, Cytokines in autoimmune uveitis, J. Interferon Cytokine Res 31 (2011) 733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].James E, Pease. Targeting chemokine receptors in allergic disease, Biochem. J 434 (2011) 11–24. [DOI] [PubMed] [Google Scholar]
- [41].Pease JE, Horuk R, Recent progress in the development of antagonists to the chemokine receptors CCR3 and CCR4, Expet Opin. Drug Discov 9 (2014) 467–483. [DOI] [PubMed] [Google Scholar]
- [42].Takeda A, Baffi JZ, Kleinman ME, Cho WG, Nozaki M, Yamada K, et al. , CCR3 is a target for age-related macular degeneration diagnosis and therapy, Nature 460 (2009) 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Esnault S, Kelly EA, Essential mechanisms of differential activation of eosinophils by IL-3 compared to GM-CSF and IL-5, Crit. Rev. Immunol 36 (2016) 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. , Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know, Cell Res. 16 (2006) 126–133. [DOI] [PubMed] [Google Scholar]
- [45].Zhan Y, Lew AM, Chopin M, The pleiotropic effects of the GM-CSF rheostat on myeloid cell differentiation and function: more than a numbers game, Front. Immunol 10 (2019) 2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. , RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation, Nat. Immunol 12 (2011) 560–567. [DOI] [PubMed] [Google Scholar]
- [47].El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. , The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF, Nat. Immunol 12 (2011) 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pierson ER, Goverman JM, GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease, JCI Insight 2 (2017) e92362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. , Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis, Science 264 (1994) 713–716. [DOI] [PubMed] [Google Scholar]
- [50].Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P, et al. , Fate-Mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-1beta, Immunity 50 (2019) 1289–1304. [DOI] [PubMed] [Google Scholar]
- [51].Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, et al. , Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system, Immunity 46 (2017) 245–260. [DOI] [PubMed] [Google Scholar]
- [52].Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, et al. , Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis, J. Exp. Med 189 (1999) 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Grajewski RS, Hansen AM, Agarwal RK, Kronenberg M, Sidobre S, Su SB,et al. , Activation of invariant NKT cells ameliorates experimental ocular auto-immunity by a mechanism involving innate IFN-gamma production and dampening of the adaptive Th1 and Th17 responses, J. Immunol 181 (2008) 4791–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chong WP, van Panhuys N, Chen J, Silver PB, Jittayasothorn Y,Mattapallil MJ, et al. , NK-DC crosstalk controls the autopathogenic Th17 response through an innate IFN-gamma-IL-27 axis, J. Exp. Med 212 (2015) 1739–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim SJ, Zhang M, Vistica BP, Chan CC, Shen DF, Wawrousek EF, et al. , Induction of ocular inflammation by T-helper lymphocytes type 2, Investig. Ophthalmol. Visual Sci 43 (2002) 758–765. [PubMed] [Google Scholar]
- [56].Wen T, Rothenberg ME, The regulatory function of eosinophils, Microbiol. Spectr 4 (2016), 10.1128/microbiolspec.MCHD-0020-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Diny NL, Rose NR, Čiháková D, Eosinophils in autoimmune diseases, Front. Immunol 8 (2017) 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dick AD, Tugal-Tutkun I, Foster S, Zierhut M, Melissa Liew SH, Bezlyak V, et al. , Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials, Ophthalmology 120 (2013) 777–787. [DOI] [PubMed] [Google Scholar]
- [59].Shiomi A, Usui T, Mimori T, GM-CSF as a therapeutic target in autoimmune diseases, Inflamm. Regen. 36 (2016) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.