

The derepression of HERVs in tumors leads to the up-regulation of hundreds of KZFPs, which are potential tumor suppressors.
Abstract
Gene expression aberration is a hallmark of cancers, but the mechanisms underlying such aberrations remain unclear. Human endogenous retroviruses (HERVs) are genomic repetitive elements that potentially function as enhancers. Since numerous HERVs are epigenetically activated in tumors, their activation could cause global gene expression aberrations in tumors. Here, we show that HERV activation in tumors leads to the up-regulation of hundreds of transcriptional suppressors, namely, Krüppel-associated box domain–containing zinc-finger family proteins (KZFPs). KZFP genes are preferentially encoded nearby the activated HERVs in tumors and transcriptionally regulated by these adjacent HERVs. Increased HERV and KZFP expression in tumors was associated with better disease conditions. Increased KZFP expression in cancer cells altered the expression of genes related to the cell cycle and cell-matrix adhesion and suppressed cellular growth, migration, and invasion abilities. Our data suggest that HERV activation in tumors drives the synchronized elevation of KZFP expression, presumably leading to tumor suppression.
INTRODUCTION
Aberrant gene expression is a hallmark of cancers. The gene expression status in tumors is highly diverse among patients and is associated with the phenotypes of tumors, such as proliferation, invasion/metastasis capacity, and therapeutic response, as well as the clinical outcome of patients (1). In particular, many genes that are aberrantly expressed in tumors and associated with cancer progression have been identified (2); however, the abnormality of the gene regulatory network underlying the aberrant expression of these genes in tumors is poorly understood (3–5).
Decades of research have highlighted the significance of regulatory sequences derived from human endogenous retroviruses (HERVs) in the modulation of human gene expression (6). HERVs are a type of transposable element (TE) that originates from ancient retroviral infection in host germ cells (7). There are several hundred types of HERVs in the human genome, constituting 8% of the genome (8). Unlike other TEs, HERVs have long terminal repeat (LTR) sequences that particularly densely contain transcriptional regulatory elements (9, 10) and function as viral promoters (7). In addition, HERV LTRs have the potential to function as promoters or enhancers of adjacent genes (6). While most HERVs are epigenetically silenced in normal tissues by DNA methylation and repressive histone modifications, some HERVs function as a part of the host gene regulatory network and play crucial roles in diverse biological events (6, 11–16). For instance, HERVs harboring STAT1 (signal transducer and activator of transcription 1)– and IRF1 (interferon regulatory factor 1)–binding sites are essential for the interferon inducibility of genes related to the innate immune response (17).
The expression of HERVs in normal tissues is controlled by epigenetic mechanisms (18, 19); in contrast, HERV expression is highly elevated in various types of cancers (20–24). Since the elevation of HERV expression in tumors is presumably caused by epigenetic reactivation, the expressed HERVs could up-regulate the expression of adjacent genes. Therefore, it is possible that the derepression of numerous HERVs in tumors globally alters host gene expression and changes the characteristics of cancers (25, 26). To test this hypothesis, we investigated the multiomics dataset of tumors provided by The Cancer Genome Atlas (TCGA) (27) and assessed the effects of HERV activation on host gene expression. We found that genome-wide HERV activation in tumors is associated with the up-regulation of potent transcriptional suppressor genes, namely, Krüppel-associated box (KRAB) domain–containing zinc-finger family protein (KZFP) genes (28), which are preferentially located in the vicinity of activated HERVs. Although KZFPs are widely known as transcriptional silencers against TEs, including HERVs (28), our data highlight that the expression of KZFP genes is induced by adjacent HERVs in tumors, presumably leading to global gene expression alterations and phenotypic changes.
RESULTS
Characterization of expressed HERVs across 12 types of solid tumors
We investigated the tumor RNA sequencing (RNA-seq) data of 5470 patients provided by TCGA (table S1). Only RNA-seq reads that were uniquely mapped to the human genome were analyzed. A total of 11,011 loci of expressed HERVs were identified across 12 types of solid tumors (Fig. 1A and table S2). While some HERVs were detected in only specific types of cancers, most of the expressed HERVs were detected in multiple types of cancers, and the sets of the expressed HERV loci were highly similar among all cancer types (fig. S1, A and B). In 9 of the 12 types of cancers, the overall expression levels of HERVs in tumors were increased compared to the levels in the normal tissues adjacent to the tumors (Fig. 1B), consistent with the findings of previous reports (20–24). Dimension reduction analysis based on HERV expression profiles showed that each type of cancer displayed a distinguishable pattern of HERV expression (Fig. 1C and fig. S1C). The expressed HERVs preferentially overlapped with the open chromatin regions determined by assay for transposase-accessible chromatin sequencing (ATAC-seq) in tumors (Fig. 1D), suggesting that the HERVs expressed in tumors have high chromatin accessibility and the potential to modulate adjacent gene expression.
Fig. 1. Landscape of HERV expression in 12 types of solid cancers.
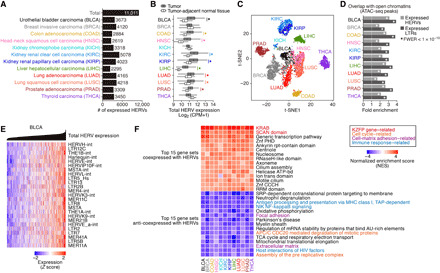
(A) Numbers of expressed HERV loci identified in the respective types of cancers. (B) Total expression levels of HERVs [log2 (counts per million (CPM) + 1)] in tumors and tumor-adjacent normal tissues. A significant increase is denoted as an asterisk [family-wise error rate (FWER) < 0.05 in two-sided Wilcoxon rank sum test]. (C) t-SNE plot representing the expression patterns of HERVs in tumor samples. The expression levels of the 1000 most highly expressed HERVs were used. (D) Fold enrichments of the overlaps between expressed HERV loci (entire HERVs or LTRs) and open chromatin regions (ATAC-seq peaks) in respective types of cancers. The enrichment value relative to random expectation is shown. Statistical significance was evaluated by a genome perturbation test. (E) Normalized expression levels of the respective HERV subfamilies in BLCA tumors. The 25 most highly expressed HERV subfamilies are shown. Tumors were ordered according to the total HERV expression. (F) Gene set enrichment analysis (GSEA) (29) representing the transcriptome signature associated with global HERV activity. Spearman’s correlation scores between the expression levels of respective genes and the total expression level of HERVs were calculated, and GSEA was subsequently performed on the basis of those scores. For the positive and negative correlations, the top 15 highest-scored gene sets (regarding the mean value among cancer types) are shown. Redundant gene sets were filtered.
Transcriptome signatures associated with the global derepression of HERVs in tumors
Although HERV expression levels tended to be elevated in tumors compared to the corresponding normal tissues (Fig. 1B), the genome-wide expression levels of HERVs in tumors were highly heterogeneous among patients, even within the same cancer type (Fig. 1E and fig. S1D). Such global HERV activation occurred regardless of the type of HERV (Fig. 1E and fig. S1, E and F), although the regulatory sequences of these HERVs were highly diverse (10). In many types of cancers, the global expression levels of HERVs were negatively correlated with the DNA methylation levels of CpG sites that are on or proximal (<1 kb) to the expressed HERVs (fig. S1, G and H), suggesting that derepression due to DNA demethylation is a cause of the elevation of HERV expression in tumors.
To elucidate the effects of the global derepression of HERVs on host gene expression in tumors, we investigated the genes whose expression was associated with HERV derepression in tumors. We assessed the correlation of the expression level of each gene with the total expression level of HERVs in tumors and subsequently performed gene set enrichment analysis (GSEA) (29) based on the correlation scores above. We found that the genes showing a correlation with HERVs were highly similar among distinct types of cancers (fig. S1I). KZFP genes (i.e., genes having the KRAB domain) were highly up-regulated upon the elevation of HERV expression in multiple types of tumors (Fig. 1F). Most KZFP genes were coexpressed with each other (fig. S1J) and with most HERV subfamilies in tumors (fig. S1K). Furthermore, genes related to the cell cycle, cell-matrix adhesion, and immune response were down-regulated upon the up-regulation of HERV and KZFP genes (Fig. 1F and fig. S2A). We investigated another RNA-seq dataset of cancer cell lines provided by the Cancer Cell Line Encyclopedia (CCLE) (30) and verified that the expression of HERVs was positively associated with that of KZFP genes and negatively associated with that of genes related to the cell cycle, cell-matrix adhesion, and immune response in cancer cell lines (fig. S2B). These results suggest that the gene expression fluctuation associated with HERVs observed in primary tumors originates from the gene expression alteration that occurs in cancer cells themselves rather than the change in the composition of noncancer cells (e.g., infiltrating lymphocytes).
Transcriptional up-regulation of KZFP genes by surrounding HERVs
We hypothesized that the derepressed HERVs near KZFP genes induce the expression of these genes, leading to the synchronized expression of HERVs and KZFP genes in tumors. It is known that KZFP genes form genomic clusters, particularly on chromosome 19 in the human genome (31). We found that the expressed HERVs in tumors were predominantly present in these clusters of KZFP genes (Fig. 2A). The expressed HERVs in tumors and those overlapping with open chromatin regions or enhancers defined by GeneHancer (32) were highly enriched in the vicinity of the transcriptional start sites (TSSs) of KZFP genes (Fig. 2B and fig. S3A). Several types of HERV LTRs, such as LTR70, LTR25, LTR5B, and LTR5_Hs, showed particularly strong enrichment around the TSSs of KZFP genes (Fig. 2C).
Fig. 2. Transcriptional upregulation of KZFP genes by adjacent HERVs.
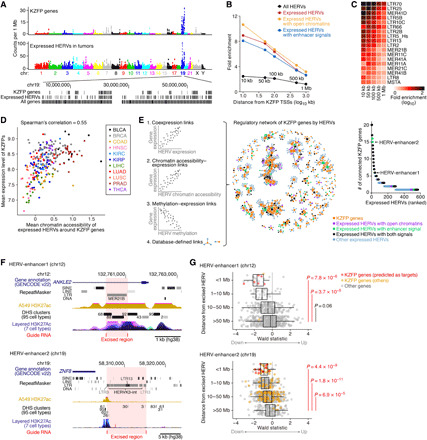
(A) The genomic densities (top) and locations (bottom) of KZFP genes and the expressed HERVs in tumors. (B) Enrichments of the expressed HERVs in tumors around the transcription start sites (TSSs) of KZFP genes. Fold enrichments of the respective categories of HERVs in the regions within the indicated distances from the KZFP TSSs are shown. (C) Enrichments of respective subfamilies of expressed HERVs (LTRs) around the KZFP TSSs. LTR subfamilies that were significantly [false discovery rate (FDR) < 0.05 in a genome permutation test] enriched within 50 kb from the TSSs are shown. (D) Association between the mean expression level of KZFPs and the mean chromatin accessibility of the expressed HERVs nearby (<50 kb) KZFP genes in tumors. (E) Prediction of the genes regulated by the expressed HERVs. (Left) Schematics of the regulatory relationship prediction. (Middle) Integrated network representing the predicted regulation of KZFP genes by HERVs. (Right) Numbers of KZFPs connected to the respective HERV nodes in the network. (F) UCSC genome browser view of the excised HERVs. (G) Effect of HERV excision on the expression of adjacent genes in LUAD (A549) cells. The x axis indicates the Wald statistic, in which the positive and negative values indicate the up- and down-regulation, respectively, of the gene expression compared to that in the nontarget control cells. Genes were stratified according to the distance from the excised HERV.
We next investigated the association between the transcriptional up-regulation of KZFP genes and the epigenetic derepression of adjacent HERVs in tumors. The mean expression level of KZFP genes was associated with the mean chromatin accessibility of the expressed HERVs around those genes in tumors (Fig. 2D). In addition, the mean expression level of KZFP genes in tumors was negatively correlated with the mean DNA methylation level of the CpG sites that are on or proximal (<1 kb) to the expressed HERVs around those genes (fig. S3B). These findings suggest that the expression of KZFP genes in tumors is up-regulated by the epigenetic derepression (i.e., decreasing DNA methylation and increasing chromatin accessibility) of adjacent HERVs.
Next, we searched for the genes that are likely to be regulated by respective HERV loci according to the coexpression, chromatin accessibility–expression, and DNA methylation–expression relationships, as well as the predefined enhancer–gene links (Fig. 2E, left) (table S3) (32). In these four types of predictions, KZFP genes were highly enriched in the set of genes that are likely to be regulated by HERVs (fig. S3C), supporting the significance of HERVs in the transcriptional regulation of KZFP genes. On the basis of these interactions, we constructed a network representing the regulation of KZFP genes by HERVs (Fig. 2E, middle). We identified several “hub” HERV loci, which are connected to many KZFP genes in the network and are likely to be involved in the transcriptional regulation of these genes (Fig. 2E, right).
To experimentally address the significance of HERVs in the transcriptional modulation of KZFP genes in cancer cells, we performed CRISPR-Cas9 excision of two hub HERV loci (HERV-enhancer1 and HERV-enhancer2; Fig. 2E, right) in a human lung adenocarcinoma (LUAD) cell line (A549 cells) (Fig. 2F and fig. S4). We selected these HERV loci because they displayed active histone marks in A549 cells (Fig. 2F). We demonstrated that the homozygous excision of these HERV loci decreased the expression of adjacent genes, including many KZFP genes (Fig. 2G). These results suggest that these HERVs work as parts of enhancers modulating adjacent genes, including KZFP genes, in LUAD cells.
Associations of the expression status of KZFPs and HERVs with the clinical outcomes of cancer patients
Since KZFPs are potent transcriptional suppressors (28), it is possible that the synchronized induction of many KZFPs in tumors alters gene expression globally and changes the characteristics of tumors. In addition, we found that somatic mutations accumulated particularly in the DNA-binding interfaces of KZFPs in tumors (fig. S5A), highlighting the aberrations of KZFPs in tumors. We therefore investigated the associations of the expression of KZFPs and HERVs with the clinical outcomes of cancer patients and found the following marked associations: in 3 [bladder carcinoma (BLCA), head and neck squamous cell carcinoma (HNSC), and LUAD] of 12 types of cancers, patients with high expression levels of KZFPs and HERVs in tumors tended to show a better prognosis than those with low expression levels [family-wise error rate (FWER) < 0.05] (Fig. 3A and fig. S5B). In addition, the investigation of the association of the expression levels of human genes and HERV loci with cancer prognosis using the Cox proportional hazards model revealed that KZFP genes and HERVs tended to show a stronger association with better prognosis than the other expressed genes in four cancer types [BLCA, HNSC, LUAD, and kidney renal papillary cell carcinoma (KIRP)] (Fig. 3B and fig. S5C). We performed GSEA on the basis of the results of the Cox proportional hazards analysis and found that KZFPs and HERVs were one of the gene sets exhibiting the strongest association with better prognosis in the four cancer types above (Fig. 3C and fig. S5D). Conversely, the gene sets related to the cell cycle and cell-matrix adhesion showed the strongest association with worse prognosis (fig. S5D). We further examined the association of KZFP expression levels and cancer stage, which reflects the degree of invasion and metastasis of tumors. The overall expression level of KZFPs decreased as the cancer stage progressed in multiple types of cancers (Fig. 3, D and E, and fig. S5, E and F). Conversely, the expression of genes related to the cell cycle and cell-matrix adhesion increased as the cancer stage progressed (fig. S5F). Together, these results suggest the possibility that the increased expression of KZFPs exerts suppressive effects on tumor progression.
Fig. 3. Associations of the expression status of KZFPs and HERVs in tumors with disease conditions.
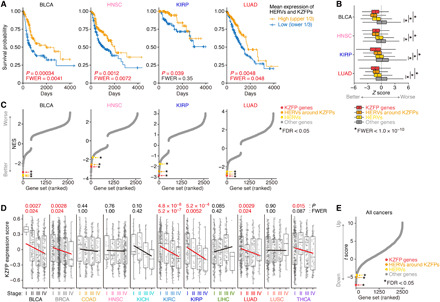
(A) Survival plots of cancer patients with high or low expression levels of HERVs and KZFPs (see also fig. S5B). The patients were stratified according to the mean value of the gene set–wise expression scores [GSVA scores (49)] between KZFPs and HERVs. Statistical significance was evaluated by the two-sided log-rank test. (B) Associations of respective genes and HERVs with the prognosis of cancer patients. Z score was calculated using the Cox proportional hazards model. Positive and negative Z scores indicate associations with worse or better prognoses, respectively. The Z score distributions were compared among KZFPs, HERVs, HERVs around KZFPs (within 50 kb), and the other genes expressed in respective types of cancers with a two-sided Wilcoxon rank sum test. (C) Results of GSEA based on the Z scores in the Cox proportional hazards model. Positive and negative NES values indicate associations with worse or better prognoses, respectively. The gene sets of interest are highlighted. See also fig. S5D. (D) Overall expression levels (GSVA scores) of KZFPs in respective cancer stages. Statistical significance was evaluated by single linear regression. (E) Multiple linear regression analysis evaluating pan-cancer associations of the expression levels of respective gene sets with cancer progression. Positive and negative t scores indicate the tendencies of increase and decrease, respectively, in the GSVA scores along with cancer progression. See also fig. S5F.
Gene expression and phenotypic changes induced by the overexpression of KZFP genes in LUAD cells
Analysis of the chromatin immunoprecipitation sequencing (ChIP-seq) dataset of KZFPs provided by Imbeault et al. (33) showed that many KZFPs preferentially bound to genes related to the cell cycle and cancer-associated signaling pathways, such as transforming growth factor (TGF)–related pathways (TGF-β, bone morphogenetic protein, SMAD2/3 pathways) and the Wnt pathway (fig. S6). These pathways are critical for the regulation of cell-matrix adhesion and are associated with cell migration/invasion and proliferation in cancers (34, 35). The expression levels of genes related to the cell cycle and cell-matrix adhesion were negatively correlated with those of KZFP genes in tumors (fig. S2A) and associated with worse disease conditions (fig. S5, D and F). Moreover, many KZFPs preferentially bound to genes that were anti-coexpressed with KZFPs in tumors (fig. S6). These data suggest the possibility that a variety of KZFPs suppress the expression of these genes in tumors and modulate cancer phenotypes.
To assess the effects of elevated KZFP expression on cancer cells, we established a panel of A549 LUAD cells overexpressing 30 types of KZFPs (referred to as A549/KZFP cells) (fig. S7). These KZFPs were coexpressed with HERVs in tumors and harbored expressed HERVs in the vicinity of their TSSs. A549 cells were selected as the parental cells since the expression levels of KZFPs (and HERVs) were relatively low in this cell line (fig. S7A). The expression levels of overexpressed KZFPs in A549/KZFP cells were the highest among LUAD tumors and lung cancer cells but did not abnormally deviate from those in naturally existing tumors and cancer cells (fig. S7C). In addition, the expression levels of overexpressed KZFPs in A549/KZFP cells were less than the upper quantile of those of all protein-coding genes expressed (fig. S7D). These results suggest that our A549/KZFP cell panel is a reasonable system that mimics cancer cells with higher expression levels of KZFPs. Using this A549/KZFP cell panel, we investigated the phenotypic and gene expression changes caused by the increased expression of these KZFPs. Twenty-nine of the 30 KZFPs tested induced apoptosis, while many KZFPs suppressed cell growth, migration, and invasion (Fig. 4A, bottom, and fig. S7E). Furthermore, we examined the tendencies of the phenotypic alterations caused by the increased expression of KZFPs: We tested whether the distribution of the log2 fold change values of the phenotype scores in A549/KZFP cells (compared to A549/empty vector cells) deviated from zero (Fig. 4B). In apoptosis, the mean value of A549/KZFP cells significantly and positively deviated from zero. In growth, migration, and invasion, the mean value significantly and negatively deviated from zero. These results suggest that KZFPs tend to induce apoptosis and suppress cellular growth and the migration and invasion abilities of cancer cells.
Fig. 4. Phenotypic and gene expression changes caused by the overexpression of KZFPs in LUAD cells.
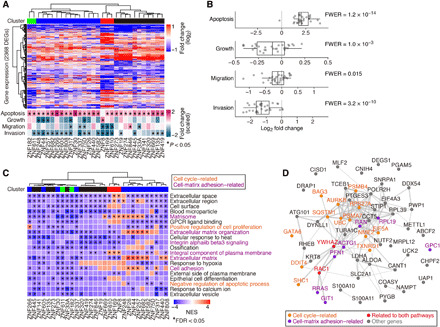
LUAD (A549) cells overexpressing 30 types of KZFPs were established (referred to as A549/KZFP cells), and the phenotypic and gene expression changes compared to empty vector–transduced cells were examined. (A) Phenotypic and gene expression changes in A549/KZFP cells. (Top) Heatmap showing the gene expression alterations of 2368 differentially expressed genes (DEGs) identified in any of the A549/KZFP cells. (Bottom) Heatmap summarizing the phenotype alterations observed in A549/KZFP cells (see also fig. S7E). An asterisk denotes a significant change (P < 0.05). Heatmap color shows the “scaled” log fold change (i.e., the standard deviation was adjusted at 1). (B) Distribution of the log2 fold change values of the phenotypes of A549/KZFP cells. Statistical significance was evaluated by the two-sided one-sample t test. (C) Results of GSEA summarizing genes with expression levels commonly down-regulated in A549/KZFP cells. An asterisk denotes a significant (FDR < 0.05) down-regulation of the gene set in certain A549/KZFP cells. The top 20 gene sets with respect to the number of cells exhibiting significant down-regulation are shown. Redundant gene sets were filtered. Gene expression–based clusters [shown in (A)] are displayed. (D) Genes that are likely to be targeted by KZFPs and critical for cancer progression. The details are described in fig. S8. Protein-protein interactions defined by STRING (version 11.0) (59) are shown.
RNA-seq analysis revealed that the expression of 2368 genes was significantly altered by the overexpression of any of the KZFPs tested [false discovery rate (FDR) < 0.05; absolute value of log2 fold change >1] (Fig. 4A). Genes related to the cell cycle and cell-matrix adhesion tended to be down-regulated by KZFP overexpression (Fig. 4C). Although the phenotypic and gene expression alterations caused by KZFPs were relatively similar among all types of A549/KZFP cells (Fig. 4, A and B, and fig. S7F), the alterations in cellular phenotype and gene expression were related to each other (fig. S7G and S7H). These data suggest that the phenotypic changes in A549/KZFP cells, which are associated with tumor suppression, could presumably be attributed to the alteration of gene expression by KZFPs.
To infer the mechanisms of KZFP-mediated tumor suppression, we identified the genes that are likely to be targeted by KZFPs and are critical for cancer progression. Of the genes that were bound by many (≥10) KZFPs, we extracted the genes showing the following: (i) a negative correlation with KZFPs in the TCGA tumor and CCLE cancer cell line datasets; (ii) an association with worse prognosis and stage progression in the TCGA tumor dataset; and (iii) decreased expression in A549/KZFP cells (fig. S8). Of the extracted genes, the genes related to the cell cycle and cell-matrix adhesion were highly enriched (Fig. 4D and fig. S8D). In particular, many genes related to cytoskeletal regulation (i.e., ACTG1, GIT1, PFN1, RAC1, and RRAS), which are critical for cell-matrix adhesion and modulate cell migration/invasion and proliferation (36), were identified as candidate KZFP targets. In addition, a serine-threonine kinase gene (AURKB) and ubiquitin-proteasome pathway genes (UBC, RPS27A, PSMB4, and PSMA7), which are critical for cell cycle regulation (37, 38), were identified.
To show further evidence supporting the tumor-suppressive effects of KZFPs, we analyzed a publicly available dataset of the CRISPR loss-of-function screening on cancer cell viability, provided by Cancer Dependency Map (DepMap) (39). We examined the genes in which knocking-out exerted the positive effects on cancer cell viability (i.e., suppressor genes of cancer cell viability). We extracted the top 100 of such suppressor genes in the data of A549 cells and found that eight KZFPs, as well as several well-characterized tumor suppressor genes (i.e., PTEN, NF2, TP53, and TSC1/2) (40), were included in the top 100 suppressor genes (fig. S9A). Moreover, KZFP genes were significantly enriched in the top 100 suppressor genes in a substantial fraction of cancer cell lines, including A549 cells (fig. S9, A and B). Together, these data suggest that the loss of function of several KZFPs up-regulate the viability in cancer cell lines, supporting the tumor-suppressive effects of KZFPs.
Furthermore, we examined whether the predicted target genes of KZFPs (Fig. 4D) are essential for cancer cell viability using the DepMap dataset. As shown in fig. S9C, we found that the knockout of the predicted KZFP target genes tended to be more critical for cancer cell viability than that of the other expressed genes, supporting the importance of those genes for cancer cell proliferation.
Transcriptional modulation of cancer phenotype-associated KZFP genes by adjacent HERVs in LUAD cells
Last, we validated whether the cancer phenotype-associated KZFP genes are transcriptionally modulated by adjacent HERVs. ZNF75D is a good candidate for this validation because the up-regulation of ZNF75D was capable of altering all four cancer phenotypes we investigated (Fig. 4A and fig. S7E). In the region approximately 5 kb upstream of a TSS of ZNF75D, two HERV integrants, LTR5_Hs and THE1D-int, were present (Fig. 5A), and the THE1D-int was coexpressed with ZNF75D in LUAD tumors (Fig. 5B). A luciferase reporter assay revealed that these two HERV elements exhibited enhancer activity in A549 cells (Fig. 5C and fig. S10) regardless of their orientation (fig. S10E). Furthermore, we excised these two HERVs in A549 cells using the CRISPR-Cas9 system (fig. S4) and demonstrated that the deletion of these HERVs decreased ZNF75D expression in an allelic number–dependent manner (Fig. 5D). These results suggest that these HERVs are involved in the transcriptional modulation of ZNF75D in LUAD cells.
Fig. 5. Transcriptional modulation of ZNF75D by adjacent HERVs in LUAD cells.
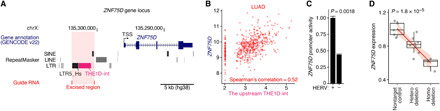
(A) Schematic view of the ZNF75D gene locus. (B) Expressional correlation between ZNF75D and an upstream HERV (THE1D-int) in tumors. (C) Effect of the HERV integrant on the promoter activity of ZNF75D. The effect was assessed by a luciferase reporter assay in A549 cells. A pair of reporter plasmids harboring the ZNF75D promoters with and without the HERV were constructed, and subsequently, the promoter activities were compared. Error bars indicate the SEM. (D) Effect of CRISPR-Cas9 excision of the HERV on the expression of ZNF75D in A549 cells. The mRNA expression level of ZNF75D in each clone of cells was measured by qRT-PCR. The P value was calculated using linear regression.
Moreover, for 12 of the 30 KZFP genes tested in A549 cells, we investigated the potential of transcriptional modulation of the adjacent HERVs by performing a luciferase reporter assay. HERVs in the vicinity of seven KZFP genes (ZNF141, ZNF248, ZNF30, ZNF320, ZNF44, ZNF611, and ZNF846) enhanced the promoter activities of these genes in A549 cells (fig. S10, F and G). Together, these results support the significance of HERVs in the transcriptional regulation of these KZFP genes in cancer cells.
DISCUSSION
In the present study, we found that the global activation of HERVs occurred in a substantial fraction of tumors (Fig. 1E and fig. S1D). Although the ultimate cause of HERV activation in tumors remains unclear, the attenuation of the epigenetic silencing (e.g., DNA demethylation) of HERVs would be a trigger of HERV activation (fig. S1, G and H). HERV activation was associated with the coordinated up-regulation of many KZFPs in tumors (Fig. 1F and fig. S1, J and K). Further analyses, including in vitro experiments, showed that HERVs in the vicinity of KZFP genes play a critical role in the transcriptional regulation of KZFPs (Figs. 2 and 5). We also provided evidence that the coordinated induction of KZFP expression is associated with better disease conditions in multiple types of cancers (Fig. 3). Moreover, we demonstrated that the increased expression of KZFPs in cancer cells tends to alter the expression of genes related to the cell cycle and cell-matrix adhesion and suppress some properties of cancers, such as cellular growth, migration, and invasion (Fig. 4, A and B). Furthermore, the analysis of the CRISPR loss-of-function screening dataset (39) suggested that several KZFPs are critical for suppressing the growth and viability of cancer cells (fig. S9, A and B). These results suggest that the increased expression of many KZFPs could exert suppressive effects on tumors. Since several notorious cancer-related genes [e.g., RAC1 (41) and AURKB (37)] and essential genes for cancer cell viability [e.g., RAN (fig. S9, A and C)] were identified as candidate KZFP targets in tumors (Fig. 4D), these genes may function in KZFP-mediated tumor suppression. Collectively, our data suggest that the activation of HERVs in tumors induces the coordinated expression of multiple KZFP genes, presumably leading to the suppression of the progressive characteristics of cancer cells by modulating gene expression.
Although our data highlight the significance of HERVs in the transcriptional activation of KZFP genes (Fig. 2), it is widely considered that one of the primary functions of KZFPs is to silence the disordered expression of TEs, including HERVs (28). Such seemingly paradoxical findings suggest the presence of a transcriptional negative feedback loop between HERVs and KZFPs—once HERVs are derepressed globally, the regulatory activity of the HERVs around KZFP genes is elevated simultaneously, resulting in the induction of KZFP expression. In other words, KZFP genes seem to use HERVs as their regulatory sequences to respond to the global derepression of HERVs. A previous report proposed the possibility that the HERV-KZFP negative feedback loop functions during embryogenesis to silence the activation of TEs, including HERVs (42). Overall, our data suggest that the HERV-KZFP regulatory axis, which works physiologically in embryogenesis, also functions in cancers and presumably contributes to tumor suppression.
We showed that many KZFPs are highly coexpressed in tumors (fig. S1J) and concordantly associated with better disease conditions (Fig. 3). The analysis of the public ChIP-seq dataset (33) showed that many KZFPs tended to bind similar sets of genes (fig. S6). Furthermore, the increased expression of individual KZFPs tended to cause similar phenotypic and gene expression alterations (Fig. 4, A and B, and fig. S7F). These results suggest that many KZFPs coordinately and further redundantly function in tumors. Moreover, it is known that some KZFPs target other KZFPs for transcriptional suppression and form a mutual suppressive regulatory network (33, 43). In addition, the present study and Pontis et al. (42) proposed that the HERV-KZFP negative feedback loop contributes to the formation of the complex network of KZFPs (Figs. 2 and 5). Together, these results suggest that each KZFP functions in tumors not individually but as a part of the complex network of KZFPs. Therefore, to further elucidate the roles of KZFPs as well as the complex HERV-KZFP network in tumors, experimental systems that can perturb the entire HERV-KZFP network in cancer cells are needed in the future.
In conclusion, we highlighted the presence of tumor heterogeneity driven by the gene regulatory network composed of HERVs and KZFPs. The present study provides new insights into the potential functions of KZFPs, the largest transcriptional repressor family in the human genome, as well as the complex regulatory networks of HERVs and KZFPs in tumors.
MATERIALS AND METHODS
Ethical approval
The utilization of the TCGA multiomics dataset was authorized by the National Cancer Institute (NCI) data access committee through the Database of Genotypes and Phenotypes (http://dbgap.ncbi.nlm.nih.gov) for the following projects: “Systematic identification of reactivated human endogenous retroviruses in cancers (#15126),” “Effects of the genome-wide activation of human endogenous retroviruses on gene expression and cancer phenotypes (#18470),” and “Screening of subclinical viral infections in healthy human tissues (#19481).”
Construction of the gene-HERV transcript model for RNA-seq analysis
For the gene transcript model, GENCODE version 22 (for GRCh38/hg38) obtained from the GENCODE website (http://www.gencodegenes.org/) was used. For the HERV transcript model, the RepeatMasker output file (15 January 2014; for GRCh38/hg38) obtained from the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/) was used. For the gene model, transcripts with the flag “retained intron” were excluded. For the HERV model, HERV loci with low reliability scores (i.e., Smith-Waterman score < 2500) were excluded. In addition, the regions of HERV loci overlapping with the gene transcripts were also excluded. A gene-HERV transcript model was generated by concatenating the gene and HERV models. This model includes 60,483 protein-coding/noncoding genes in addition to 138,124 HERV loci, which occupy 3.4% of the genome.
RNA-seq data analysis of the TCGA dataset
Poly A–enriched RNA-seq (mRNA-seq) data provided by TCGA were analyzed. Of the RNA-seq data, we analyzed only the data produced by paired-end sequencing with a read length of 48 to 50 bp. The BAM-formatted read alignment file (for GRCh38/hg38) of the RNA-seq data was downloaded from the Genomic Data Commons (GDC) data portal site (http://portal.gdc.cancer.gov/) using the GDC Data Transfer Tool (http://gdc.cancer.gov/access-data/gdc-data-transfer-tool/). Data for tumors and tumor-adjacent normal tissues were downloaded. To measure the expression levels of HERVs and genes, RNA-seq fragments mapped on HERVs and the exons of genes were counted using Subread featureCounts (44) with the BAM file and the gene-HERV transcript model. The option “fracOverlap” was set at 0.25. The RNA-seq fragments assigned to multiple features were not counted.
To control the quality of the RNA-seq data used in the present study, we checked the proportion of nonassigned RNA-seq fragments (i.e., the fragments that were uniquely mapped on the reference genome but not on HERVs or the exons of genes) in each sequence library. For this proportion of fragments, outlier libraries were detected recursively using the Smirnov-Grubbs test (the threshold was set at 0.05). These outlier libraries were excluded from the downstream analyses. The final RNA-Seq data (for both tumors and tumor-adjacent normal tissues) used in this study are summarized in table S4.
The expression count matrices of the RNA-seq data were separately prepared for the datasets of the respective types of cancers. In addition, an expression matrix including all tumor data was prepared, and an expression matrix including the data from the tumors and corresponding normal adjacent tissues was also prepared for each type of cancer. Genes and HERVs with low expression levels were removed from the expression matrices as follows. The counts per million (CPM) value of each gene and HERV locus were calculated in the respective RNA-seq libraries. Subsequently, genes and HERVs were discarded from the expression matrices if the 90th percentile of CPM values was less than 0.2.
In each type of cancer, the expressed HERVs in tumors, which are HERVs included in the expression matrix of the corresponding types of cancers, were determined.
The total expression level of the HERVs was normalized as CPM. The expression levels of genes and HERV loci were normalized using variance-stabilizing transformation (VST) implemented in DESeq2 (version 1.18.1) (45). This VST-normalized expression level was used unless otherwise noted.
RNA-seq data analysis of the CCLE dataset
The BAM-formatted read alignment file (for GRCh37/hg19) of the mRNA-seq data was downloaded from the GDC data portal site (http://portal.gdc.cancer.gov/) using the GDC Data Transfer Tool (http://gdc.cancer.gov/access-data/gdc-data-transfer-tool/). The RNA-seq data of CCLE used in this study are summarized in table S5. Since the gene-HERV transcript model prepared above is for GRCh38/hg38, the genomic coordinates of the gene-HERV transcript model were converted to those in GRCh37/hg19 using UCSC liftOver (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/liftOver). The option “minMatch” was set at 0.95. The generation of the expression count matrix, filtering of genes and HERVs with low expression levels, and normalization of the expression data were performed using the same procedures as those in the above section (“RNA-Seq data analysis of the TCGA dataset”).
RNA-seq analysis of A549/KZFP cells
The RNA-seq sample information is summarized in table S6. Low-quality sequences in RNA-seq fragments were trimmed using Trimmomatic (version 0.36) (46) with the option “SLIDINGWINDOW:4:20.” RNA-seq fragments were mapped to the human reference genome (GRCh38/hg38) using STAR (version 2.5.3a) (47) with the gene-HERV transcript model. STAR was run using the same options and parameters as those used in the GDC mRNA Analysis Pipeline (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline). The generation of the expression count matrix, filtering of genes and HERVs with low expression levels, and normalization of the expression data were performed using the same procedures as those in the above section (“RNA-seq data analysis of the TCGA dataset”).
Quantification of the expression levels of overexpressed KZFPs in A549/KZFP cells
The gene expression matrix was generated with the same procedure described in the “RNA-seq analysis of A549/KZFP cells” section except for the following points. In the RNA-seq fragment mapping step, the genome index including both the human reference genome (GRCh38/hg38) and the 3xHA (hemagglutinin) tag sequence used in the KZFP overexpression vector [Imbeault et al. (33)] was used. In the RNA-seq fragment counting step, the fragments mapped on the HA sequence were also counted, and the count of the HA sequence was added to that of the overexpressed KZFP gene.
Comparison of total HERV expression levels between tumors and normal tissues
The total HERV expression level was compared between tumors and tumor-adjacent normal tissues via data provided by TCGA. Since only a portion of patients (586 of 5470) had both tumor and tumor-adjacent normal tissue data in the TCGA dataset, an unpaired comparison was performed using a two-tailed Wilcoxon rank sum test.
Dimension reduction analysis of HERV expression profiles using t-SNE
The expression matrix including all tumor data was used in this analysis. The expression levels of the 1000 most highly expressed HERVs were used in the analysis. t-distributed stochastic neighbor embedding (t-SNE) analysis was performed using the “Rtsne” R package. For the analysis, the first 10 principal components of the HERV expression profiles were used, and the parameter “perplexity” was set at 70.
ATAC-seq data analysis
The ATAC-seq data of tumors and normal adjacent tissues provided by TCGA (TCGA-ATAC_PanCan_Log2Norm_Counts.rds) were downloaded from the GDC website (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG). This file contains the normalized read count matrix comprising all ATAC-seq samples (n = 796) and ATAC-seq peaks (open chromatin regions) (n = 562,709) analyzed in the previous study (4). In the respective types of cancers, the upper one-fourth of open chromatin regions with respect to the mean value were regarded as the open chromatin regions that are active in the corresponding cancer types.
To calculate the fold enrichment of the overlaps between the expressed HERVs in tumors and open chromatin regions, randomization-based enrichment analysis was performed as follows: genomic regions of open chromatin regions were randomized using BEDTools “shuffle” (48), and subsequently, the number of open chromatin regions on the expressed HERVs was counted. This process was repeated 1000 times, and the mean value of the counts in the randomized datasets was regarded as the random expectation value. The fold enrichment was calculated by dividing the observed count by the random expectation value. The P value was calculated according to the assumption of a normal distribution.
DNA methylation data analysis
The DNA methylation data [produced by the methylation microarray HumanMethylation450 (Illumina, San Diego, CA)] of tumors and normal tissue controls were downloaded from the GDC data portal (http://portal.gdc.cancer.gov/) using the GDC Data Transfer Tool (http://gdc.cancer.gov/access-data/gdc-data-transfer-tool/). These data describe the methylation level (beta value; proportion of methylated CpGs at a CpG site) of each probe in the array. Probes overlapping with single-nucleotide polymorphisms (SNPs) with >0.05 minor allele frequency were excluded from the analysis using the function “rmSNPandCH” implemented in the “DMRcate” library in R. The CpG sites that were on or proximal (<1 kb) to HERVs were extracted using the “slop” and “intersect” functions in BEDTools (48). The DNA methylation data used in this study are summarized in table S7.
Preparation of gene sets for enrichment analyses
As sources of gene sets, “GO biological process,” “GO cellular component,” “MSigDB canonical pathway,” and “InterPro” were used. The gene sets in these sources were concatenated and used. InterPro is a collection of gene sets according to protein families or domains and includes the gene set “KRAB,” representing the KZFP family genes. GO biological process and GO cellular component were obtained from the Gene Ontology (GO) consortium (http://geneontology.org/; GO validation date: 30 August 2017); “canonical pathway” was obtained from MSigDB (http://software.broadinstitute.org/gsea/msigdb; version 6.1); and InterPro was obtained from BioMart on the Ensembl website (www.ensembl.org; on 13 February 2018).
In addition, we defined the gene sets “HERVs” and “HERVs around KZFP genes.” The “HERV” gene set included all expressed HERVs in tumors, while “HERVs around KZFP genes” included the HERVs present in the genomic regions within 50 kb from the TSSs of KZFP genes expressed in tumors. These gene sets were used in Fig. 3 (C and E) and fig. S5 (D and F), in addition to the predefined gene sets.
Furthermore, we defined gene sets according to their negative expression correlation with HERVs or KZFP genes as follows. In the respective tumor datasets of TCGA, Spearman’s correlations between the expression levels of the respective genes and the total expression level of HERVs were calculated, and the genes were ranked according to their median value in the datasets. The top 100, 200, and 500 genes with respect to their negative expression correlation with HERVs were used as gene sets. Using the same procedures as above, the top 100, 200, and 500 genes with respect to their negative expression correlation with KZFP genes were extracted and used as gene sets. As the representative value of KZFP expression, the gene set–wise expression score [Gene Set Variation Analysis (GSVA) score (49)] of the KZFP genes was used. The GSVA score is described in the following section (“Calculation of the gene set–wise expression score using GSVA”). These gene sets were used in fig. S6 in addition to the predefined gene sets.
Calculation of the gene set–wise expression score using GSVA
The VST-normalized expression matrix was converted to a gene set–wise expression score matrix using GSVA (49) with the gene sets prepared above. The option “minimum size of gene set” was set at 20.
Gene set enrichment analysis
To perform GSEA (29), the R package “fgsea” (50), a fast implementation of GSEA, was used. The parameters of “number of permutations” and “minimum size of gene set” were set at 10,000 and 50, respectively. In the analyses of Fig. 1F and fig. S2B, Spearman’s correlations between the expression levels of the respective genes and the total expression level of HERVs were used as statistical scores. In the analysis of fig. S2A, Spearman’s correlations between the expression levels of the respective genes and the GSVA score of the KZFP genes were used. In the analyses of Fig. 3C and fig. S5D, the Z scores from the Cox proportional hazards regression were used (the Z score is described in the “Survival analysis of the cancer patients” section). In the analysis of Fig. 4C, the Wald statistics of the respective genes in the differential expression analysis were used (the Wald statistic is described in the “Differential expression analysis” section). Gene sets with FDR < 0.05 were regarded as significant.
Summarizing the results of GSEA and GO enrichment analysis by removing redundant gene sets
Since the gene members of some gene sets highly overlapped with each other, redundant gene sets were removed from the results of the enrichment analyses as follows. Gene sets were ranked according to the score of interest [e.g., the mean value of the normalized enrichment score (NES)]. If the gene members of a certain gene set highly overlapped with those of the upper-ranked gene sets, the gene set was removed from the result. As a statistic of the overlap, the Szymkiewicz-Simpson coefficient was used, and two gene sets were regarded as highly overlapped if the coefficient was greater than 0.7. This gene set filtering was applied to the analyses shown in Figs. 1F and 4C and figs. S5D, S6, and S7H, which show only the top-ranked gene sets.
GO enrichment analysis to identify gene sets that are preferentially present in the vicinity of the expressed HERVs
Randomization-based GO enrichment analysis was performed as follows. Only genes whose expression levels were detected in the TCGA tumor datasets were used. Regions of interest were defined as the regions within 50 kb from the TSSs of the gene members of a certain gene set. The genomic regions of HERVs were randomized using the “shuffle” function of BEDTools (48), and subsequently, the number of HERVs in the region of interest was counted. This process was repeated 1000 times, and the mean value of the counts in the randomized datasets was regarded as the random expectation value. The fold enrichment was calculated by dividing the observed count by the random expectation value.
In addition, we calculated the fold enrichments of the HERVs in the regions within 10, 100, and 500 kb and 1 Mb from the TSSs of the KZFP genes using the same procedures as above.
Prediction of genes regulated by HERVs
The regulatory interactions between HERV loci and genes were predicted according to the following information: coexpression between HERVs and genes, positive correlations between HERV chromatin accessibility and gene expression, negative correlations between HERV DNA methylation and gene expression, and predefined links between the regulatory sequences on HERVs and genes. The coexpression interaction was used only for pairs of HERVs and genes within 50 kb of each other, while the chromatin accessibility–expression, methylation-expression, and predefined interactions were used only for pairs of HERVs and genes within 500 kb of each other. A coexpression interaction was defined if the expression of a HERV and a gene were positively correlated (Spearman’s correlation > 0.4) in any type of cancer in TCGA. A methylation-expression interaction was defined if the DNA methylation level of the CpG site that is on or proximal (<1 kb) to a HERV and the expression of a gene were negatively correlated (Spearman’s correlation < −0.3) in any type of cancer or in the pan-cancer dataset in TCGA. As the source of chromatin accessibility–expression interactions, the interactions defined in a previous study (4) were used. As the source of predefined regulatory interactions, the interactions recorded in GeneHancer version 4.7 obtained from the GeneLoc database (https://genecards.weizmann.ac.il/geneloc/index.shtml) were used.
Mutation analysis
To define the DNA-binding amino acids of KZFP genes, we first determined the precise genomic positions of the KRAB and C2H2 zinc-finger domains as follows. For both the KRAB and C2H2 zinc-finger domains, hidden Markov model (HMM) profiles were generated using hmmbuild from HMMER2 (http://hmmer.org/). Multiple sequence alignments used to build the HMM profiles were generated from the seed sequences downloaded from Pfam (https://academic.oup.com/nar/article/44/D1/D279/2503120). Next, the human reference genome (GRCh37/hg19) was scanned using hmmpfam from HMMER2 with the built HMM profiles. Both strands of chromosomes translated in three reading frames were scanned. KZFP genes were collected if a KRAB domain had ≥2 downstream C2H2 zinc fingers found on the same strand within 40 kb, which corresponds to the maximum length from the first base of the KRAB domain to the last base of the zinc finger domain. Detected KZFP genes were then annotated according to the Ensembl annotation (version 92; for GRCh37/hg19). Last, the DNA-binding amino acid positions were inferred from the C2H2 zinc fingers annotated above, taking the 4th, 6th, 7th, and 10th positions (also called positions −1, +2, +3, and + 6) after the second cysteine of C2H2. Only zinc fingers with a canonical C2H2 structure and associated with a KRAB domain were taken into account.
Processed mutation data were obtained from the International Cancer Genome Consortium (release 27) (https://icgc.org/). Then, we measured the somatic missense mutation density (counts per megabase per patient) of KZFP genes in the DNA-binding amino acids and the whole coding regions of the canonical transcript.
Survival analysis of the cancer patients
The overall survival rate of the cancer patients was used for survival analyses with the R package “survival.” The survival curve of the patients was estimated by the Kaplan-Meier method, and statistical significance was evaluated by the two-sided log-rank test. With respect to the expression level of interest, the upper and lower third of patients were regarded as patients with higher and lower expression statuses, respectively. In Fig. 3A and fig. S5B, the patients were stratified according to the mean value of the GSVA scores of the HERVs and KZFPs in tumors.
To examine the association of the expression level of each gene and HERV locus with the prognosis of cancer patients, Cox proportional hazards regression analysis was performed with adjustment for the effects of patient sex and race. In addition to HERVs, genes that were included in any of the gene sets prepared above were used.
Association analysis of gene expression and cancer stage progression
Prostate adenocarcinoma (PRAD) tumors were excluded from the analysis since information on cancer stage for most PRAD patients was not available from TCGA. In the analysis, cancer stage was regarded as an interval scale. For each type of cancer, the association between the expression of each gene and the progression of the cancer stage was evaluated by single linear regression. Similarly, the association between the GSVA score of each gene set and the progression of cancer stage for each type of cancer was evaluated using the same procedure. To evaluate the pan-cancer association of the GSVA score of each gene set and the progression of cancer stage, multiple linear regression analysis with adjustment for the effects of cancer type was performed.
Analysis of a publicly available ChIP-seq dataset of KZFPs
This analysis was based on a publicly available ChIP-seq dataset of KZFPs in human embryonic kidney–293 (HEK293) T cells presented in a previous study [Imbeault et al. (33); Gene Expression Omnibus (GEO) accession no. GSE78099]. Information on predefined ChIP-seq peaks (GSE78099_RAW.tar) was downloaded from the GEO database (www.ncbi.nlm.nih.gov/geo/). Since these ChIP-seq peaks [referred to as transcription factor binding sites (TFBSs)] are for GRCh37/hg19, the genomic coordinates of these TFBSs were converted to those in GRCh38/hg38 using UCSC liftOver (https://genome-store.ucsc.edu/). The option “minMatch” was set at 0.95. If multiple technical replicates of ChIP-seq are available for one KZFP, the replicate files were merged using the BEDTools “merge” (48) function with the option “-c 5 -o mean.” KZFPs were removed from the downstream analyses if the total number of TFBSs was less than 500. If >10,000 TFBSs were available for one KZFP, only the top 10,000 highest scoring TFBSs were used for the analyses.
To identify sets of genes that are preferentially targeted by a certain KZFP, genomic region enrichment analysis (51) was performed as follows. Only genes whose expression was detected in the TCGA tumor datasets were used. Regions of interest were defined as the regions within 10 kb from the TSSs of the gene members of a certain gene set. Regions of background were defined as the regions within 10 kb from the TSSs of genes belonging to any of the gene sets. The lengths of the regions of interest and regions of background were calculated and referred to as Li and Lb, respectively. In the regions of interest and regions of background, the numbers of TFBSs were counted [referred to as counts of interest (Ci) and counts of background (Cb), respectively]. The fold enrichment value was calculated by dividing Ci/Cb by Li/Lb, and the statistical significance was evaluated using a binomial test.
Differential expression analysis
Differential expression analysis was performed using DESeq2 (version 1.18.1) (45) in R. Genes that were included in any of the gene sets prepared above were used. A549/KZFP cells and empty vector–transduced cells were compared (Fig. 4A). Statistical significance was evaluated by the Wald test with FDR correction. In addition, a comparison was conducted between A549 cells in which HERV-enhancer1 or HERV-enhancer2 were excised and the nontarget control cells (Fig. 2G).
Scoring system of genes for predicting the targets of KZFPs critical for cancer progression
The scheme is summarized in fig. S8A. For each gene, the following scores were defined. The TCGA expressional correlation score was defined as the Spearman’s correlation between the expression of each gene and the GSVA score of KZFPs in the TCGA dataset (the median value among all cancer types was used). The CCLE expressional correlation score was also defined using the same procedure but on the CCLE dataset. The prognosis score was defined as the Z score representing the association of each gene with the prognosis of cancer patients (the mean value among BLCA, HNSC, KIRP, and LUAD tumors was used). This Z score was described in the “Survival analysis of the cancer patients” section. The progression score was defined as the t score representing the association of each gene with cancer progression (the mean value among BLCA, BRCA, KIRC, KIRP, LUAD, and thyroid carcinoma tumors was used). This t score was described in the “Association analysis of gene expression and cancer stage progression” section. The suppression score was defined as the mean value of the Wald scores in the differential expression analysis of the A549/KZFP cells. This Wald score was described in the “RNA-seq analysis of A549/KZFP cells” section. Regarding the TCGA and CCLE correlation scores and suppression scores, the signs of the scores were inverted. All scores were standardized as Z scores and subsequently quantile-normalized. Genes were extracted if the minimum score was greater than 0.5 and the median score was greater than 1. Of the extracted genes, genes targeted by ≥10 KZFPs were further extracted and regarded as the target genes of KZFPs critical for cancer progression. A gene was regarded as the target of a certain KZFP if the KZFP bound to the regions within 10 kb from the TSSs of the gene. In this analysis, only TSSs of “principal transcripts” (principals 1 to 3) defined by APPRIS (52) were used. If >1000 genes were assigned to a certain KZFP as its targets, only the top 1000 genes with high-scored TFBSs were used.
Analysis of DepMap CRISPR loss-of-function screening dataset
The dataset of the loss-of-function screening using CRISPR library provided by the DepMap Achilles project (39) was analyzed. The preprocessed data of the estimated knockout effect scores [CERES scores (39)] of individual genes in respective cancer cell lines [“Achilles_gene_effect.csv” (version: Public 20Q2)] were downloaded from the DepMap portal (https://depmap.org/portal/depmap/). The preprocessed data of RNA-seq for DepMap cell lines [“CCLE_expression.csv” (version: Public 20Q2)] were also downloaded. Data of cancer cell lines in which both CRISPR screening and RNA-seq data are available were used in the downstream analysis. In each cancer cell line, the genes that are not expressed were excluded from the analysis, and the genes with positive high scores are regarded as the suppressor genes of cancer cell viability. Statistical enrichment of KZFP genes in the top 100 of the suppressor genes of cancer cell viability was evaluated using the two-sided Fisher exact test in each cancer cell line. To compare the knockout effects between the predicted KZFP target genes and other expressed genes, the mean value of knockout effect scores of each gene in respective cancer types was calculated. The statistical significance was evaluated by the two-sided Wilcoxon rank sum test.
Data visualization
All visualizations were performed in R. Graphs were plotted using the “ggplot2” package or the preimplemented function “plot” unless otherwise noted. Heatmaps were drawn using the “ComplexHeatmap” package. Networks were plotted using the “igraph” package. Kaplan-Meier plots were drawn using the “ggsurvplot” function in the “survminer” package.
Cell culture
HEK293T cells [CRL-11268; American Type Culture Collection (ATCC), Manassas, VA] were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, #D6046) with 10% fetal bovine serum (FBS; Sigma-Aldrich, #172012-500ML) and 1% penicillin-streptomycin (Sigma-Aldrich, #P4333-100ML). A549 cells (CCL-185; ATCC) were cultured in Ham’s F-12K (Kaighn’s) medium (Thermo Fisher Scientific, Waltham, MA, #21127022) with 10% FBS (guaranteed doxycycline free; Thermo Fisher Scientific, #2023-03) and 1% penicillin-streptomycin. A549/KZFP cells were cultured in F-12K medium with puromycin (1.0 μg/ml; Invivogen, San Diego, CA, #ant-pr-1). An A549 cell line stably expressing Cas9 (A549/Cas9 cells) was cultured in F-12K medium with 10% FBS (guaranteed doxycycline free; Thermo Fisher Scientific, #2023-03) and blasticidin (5.0 μg/ml; Invivogen, #ant-bl-1). All cells were cultured in 5% CO2 at 37°C.
Establishment of a panel of A549/KZFP cells
We selected 30 types of KZFP genes satisfying the following criteria: (i) showing a positive correlation (Spearman’s correlation > 0.3) between its expression and the total expression of HERVs in >2 types of cancers; (ii) having expressed HERVs within the vicinity (<20 kb) of its TSSs in tumors; (iii) showing a positive correlation (Spearman’s correlation > 0.3) between its expression and the expression of HERV loci in the vicinity (<20 kb) of its TSSs in >2 types of cancers; and (iv) having available ChIP-seq data presented by a previous study [Imbeault et al. (33)]. Information on the selected KZFP genes is summarized in table S8.
To prepare lentiviral vectors expressing 3xHA-tagged KZFPs, HEK293T cells were cotransfected with 12 μg of pCAG-HIVgp (RDB04394, kindly provided by H. Miyoshi), 10 μg of pCMV-VSV-G-RSV-Rev (RDB04393, kindly provided by H. Miyoshi), and 17 μg of pEXPpSIN-TRE-GW ZNF-3xHA (33) by the calcium phosphate method. The pEXPpSIN-TRE-GW ZNF-3xHA plasmids encoded the respective HA-tagged KZFP proteins. After 12 hours of transfection, the culture medium was changed to fresh F-12K medium. After 48 hours of transfection, the culture supernatant including lentivector particles was collected. A549 cells were infected with these particles at a multiplicity of infection (MOI) of 0.1. After 2 days of infection, the cells were selected with puromycin (1 μg/ml) for 7 days. Three days before the start of the experiments, doxycycline (1.0 μg/ml) was added to induce the expression of KZFP. The expression of KZFP was verified by Western blotting with an HA-specific antibody (Roche, Basel, Switzerland, #12013819001). Empty vector–transduced A549 cells [referred to as negative control (NC) cells] were established according to the procedures described above.
Apoptosis detection assay
A549/KZFP cells and NC cells were stained with Annexin V conjugated to Alexa Fluor 647 (Invitrogen Carlsbad, CA, #S32357). After staining, the number of Annexin V–positive cells was counted by a FACSCalibur system (BD Biosciences, San Jose, CA), and the rate of apoptotic cells was calculated. A single set of triplicate experiments was performed, and the mean and SEM values are shown in fig. S7E.
Cell growth assay
A549/KZFP cells and NC cells were seeded at 1.0 × 105 cells per well in six-well plates (Thermo Fisher Scientific). After 72 hours of seeding, the number of cells was counted manually under a microscope, and the growth rate of the cells was calculated. Single-replicate experiments were performed at least seven times independently, and the mean and SEM values are shown in fig. S7E.
Cell scratch assay [wound-healing assay (53)]
A549/KZFP cells and NC cells were seeded in 12-well plates (Thermo Fisher Scientific) and cultured until >90% confluence. A single straight wound was formed in each well by scratching with a sterile 1000-μl pipette tip. The cells were washed with phosphate-buffered saline (PBS), and 2 ml of F-12K medium was added. Images were taken under a microscope immediately after the scratch and again after 24 hours. Using ImageJ (54) software with in-house scripts, the area (pixels) in which cells migrated for 24 hours was calculated. Triplicate experiments were performed independently twice. Regarding the mean and SEM, the average values between the two sets of experiments are shown in fig. S7E. Two-sided Student’s t test with a threshold of 0.05 was performed for each set of experiments. Only if a significant difference was observed in both sets of experiments was the comparison considered significant.
Cell invasion assay
An invasion assay was performed using a 96-well Transwell plate (8.0-μm pore size) (Corning, Corning, NY, #3374) with Corning Matrigel Basement Membrane Matrix (Corning, #354234). The Matrigel matrix was diluted 50-fold with serum-free F-12K medium. To coat the Transwell insert plate, 30 μl of Matrigel matrix was dispensed into the insert plate. After 2 hours of incubation, 20 μl of the supernatant was removed from the coated Transwell plate. Subsequently, A549/KZFP cells and NC cells were seeded at 5.0 × 104 cells per well in the insert plate. The insert plate was filled with serum-free F-12K medium, while the reservoir plate was filled with F-12K medium with 10% FBS. After incubation at 37°C for 48 hours, the cells that had invaded the Matrigel and migrated to the opposite side of the insert plate were washed with PBS, stripped with trypsin-EDTA, and stained with calcein AM (Invitrogen, #C3100MP). To evaluate the degree of cell invasion, the fluorescence intensity of the cells was measured using a 2030 ARVO X multilabel counter (PerkinElmer, Waltham, MA). The relative fluorescence intensity was calculated as (FIi − FIb)/(FIc − FIb), where FIi denotes the fluorescence intensity of the A549/KZFP cells of interest, FIb denotes the intensity of the blank, and FIc denotes the intensity of the NC cells. Triplicate experiments were performed independently twice. Regarding the mean and SEM, the average values between the two sets of experiments are shown in fig. S7E. Two-sided Student’s t test with a threshold of 0.05 was performed for each set of experiments. Only if a significant difference was observed in both sets of experiments was the comparison considered significant.
Construction of plasmids for the luciferase reporter assay
Genomic DNA from the human peripheral blood lymphocytes of a healthy donor was used as the DNA source. A luciferase reporter vector, pGL3-basic (Promega, Madison, WI), was used. Using nested polymerase chain reaction (PCR), the genomic region indicated by the arrow in fig. S10 (A and B) was cloned into pGL3-basic.
Information on the plasmids and primers prepared in this section is summarized in tables S9 and S10, respectively.
Luciferase reporter assay to assess the promoter activity of genes
A549 cells were seeded at 1.0 × 105 cells per well in 12-well plates (Thermo Fisher Scientific). After 24 hours of seeding, the luciferase reporter plasmid was transfected using polyethylenimine transfection. To fairly compare the reporter activities of the two plasmids with different sequence lengths, 1 μg of the longer plasmid and the same molar of the shorter plasmid were used for the transfection. After 12 hours of transfection, the culture medium was changed to fresh F-12K medium. After 48 hours of transfection, the luminescence intensity of the transfected cells was measured using a 2030 ARVO X multilabel counter (PerkinElmer) or a GloMax Explorer Multimode Microplate Reader 3500 (Promega) with a BrillianStar-LT assay system (Toyo-B-Net, Tokyo, Japan, #307-15373 BLT100). A single set of triplicate experiments was performed, and the mean and SEM values are shown in Fig. 5C and fig. S10 (E to G).
Establishment of HERV-excised cells
First, an A549 cell line stably expressing Cas9 (referred to as A549/Cas9 cells) was established as follows. To prepare lentiviral vectors expressing Cas9, HEK293T cells were cotransfected with 12 μg of pCAG-HIVgp, 10 μg of pCMV-VSV-G-RSV-Rev, and 17 μg of plentiCas9-Blast (Addgene, Watertown, MA, #52962) by the calcium phosphate method. After 12 hours of transfection, the culture medium was changed to fresh F-12K medium. After 48 hours of transfection, the culture supernatant including lentivector particles was collected. A549 cells were infected with these particles at an MOI of 0.1. After 2 days of infection, the cells were selected with blasticidin (5 μg/ml) for 7 days. After selection, single cell clones were obtained through the limiting dilution method. By screening the expression level of Cas9 among the candidate clones, A549/Cas9 cells were established.
To excise the target HERV, a pair of guide RNAs (gRNAs) were designed in the upstream and downstream regions of the HERV using the web applications sgRNA designer (55) (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) or CRISPOR (56) (http://crispor.tefor.net). The gRNA information is summarized in table S11. The gRNA was cloned into a gRNA expression plasmid, lentiGuide-Puro (Addgene, #52963). A pair of gRNA expression plasmids was cotransfected into A549/Cas9 cells by electroporation using the NEON Transfection System (Thermo Fisher Scientific) (1200 V, 30 ms, 2 pulses, 1.0 × 105 cells, and 500 ng of each plasmid). After transfection, the cells were selected with puromycin (1 μg/ml) for 3 days. After selection, single cell clones were obtained through the limiting dilution method. Of these candidate clones, the clones in which homozygous or heterozygous excision of the target HERV occurred were screened using PCR (fig. S4). Regarding homozygous clones, the PCR fragments were checked through molecular cloning into a TOPO vector (Invitrogen, #450245) followed by Sanger sequencing.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from cells by the QIAamp RNA Blood Mini Kit (QIAGEN, Hilden, Germany, #52304) and subsequently treated with DNase I, Amplification Grade (Invitrogen, #18068015). cDNA was synthesized by reverse transcription of the total RNA using SuperScript III reverse transcriptase (Life Technologies, #18080044) with Oligo(dT)12-18 Primer (Invitrogen, #18418012). Quantitative reverse transcription (qRT)–PCR was performed on the cDNA using a CFX Connect Real-Time PCR Detection System (Bio-Rad, Richmond, CA, #1855201 J1) with a TaqMan Gene Expression Assay kit (Thermo Fisher Scientific). The primer and TaqMan probe information are listed in table S12. GAPDH was used as an internal control.
Preparation of RNA-seq samples and sequencing
Cells were seeded at 1.0 × 106 cells in 100-mm dishes (Thermo Fisher Scientific, EasYDish, #150466). After 48 hours of seeding, the cells were harvested and stored at −80°C. Total RNA was extracted from the cells by the QIAamp RNA Blood Mini Kit (QIAGEN, #52304) and subsequently treated with RNase-Free DNase Set (QIAGEN, #79254).
Quality checks, library construction, and sequencing were performed by Novogene (https://en.novogene.com). Paired-end 150-bp read length sequencing was performed on an Illumina NovaSeq 6000 system.
Statistical analysis
Statistical significance was evaluated by two-sided Student’s t test unless otherwise noted. To address multiple testing problems, the FWER and FDR were calculated by the Holm method (57) and Benjamini-Hochberg method (58), respectively.
Supplementary Material
Acknowledgments
We would like to thank N. Misawa (Institute for Frontier Life and Medical Sciences, Kyoto University, Japan) and K. Nomura (Yamaguchi University, Japan) for technical support and J. Pontis [School of Life Sciences, Ecole Polytechnique Federale de Lausanne (EPFL), Switzerland], S. Kojima and J. Kawasaki (Institute for Frontier Life and Medical Sciences, Kyoto University, Japan), and S. Yamada (Tokai University, Japan) for thoughtful comments. The super-computing resource, SHIROKANE, was provided by Human Genome Center, The Institute of Medical Science, The University of Tokyo, Japan. The results shown here are in part based on data generated by the TCGA Research Network (www.cancer.gov/tcga). Funding: This study was supported in part by AMED J-PRIDE JP19fm0208006 (to K.S.); AMED Research Program JP19fk0410014 (to Y.K. and K.S.) and JP19fk0410019 (to K.S.); JST CREST (to K.S.); JSPS KAKENHI Scientific Research B JP18H02662 (to K.S.); JSPS KAKENHI Scientific Research on Innovative Areas JP16H06429 (to K.S.), JP16K21723 (to K.S.), JP17H05813 (to K.S.), and JP19H04826 (to K.S.); JSPS KAKENHI Grant-in-Aid for Early-Career Scientists JP20K15767 (to J.I.); JSPS Research Fellow PD JP19J01713 (to J.I.) and DC1 JP19J20488 (to I.K.); JSPS Core-to-Core program (A. Advanced Research Networks) (to Y.K. and K.S.); Joint Usage/Research Center program of Institute for Frontier Life and Medical Sciences, Kyoto University (to K.S.); Takeda Science Foundation (to K.S.); ONO Medical Research Foundation (to K.S.); Ichiro Kanehara Foundation (to K.S.); Lotte Foundation (to K.S.); Mochida Memorial Foundation for Medical and Pharmaceutical Research (to K.S.); and the European Research Council (KRABnKAP, no. 268721; Transpos-X, no. 694658) and the Swiss National Science Foundation (310030_152879 and 310030B_173337) (to D.T.). Author contributions: J.I. conceived the study; J.I. and A.C. mainly performed bioinformatics analyses; I.K., H.N., I.I., P.T., and D.T. supported bioinformatics analyses; I.K. mainly performed experimental analyses; A.S. and Y.K. supported experimental analyses; Y.K., P.T., and D.T. provided reagents; J.I., I.K., and K.S. prepared the figures; J.I., I.K., and K.S. wrote the initial draft of the manuscript; all authors contributed to data interpretation, designed the research, revised the paper, and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The RNA-seq data reported in this paper are available in GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141803). The raw data related to the present study are available on the Mendeley Data repository (http://dx.doi.org/10.17632/c7r7dw9p42.1). The computer codes used in the present study are available on the GitHub repository (https://github.com/TheSatoLab/HERV_Pan-cancer_analysis).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabc3020/DC1
REFERENCES AND NOTES
- 1.Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F., Sanli K., von Feilitzen K., Oksvold P., Lundberg E., Hober S., Nilsson P., Mattsson J., Schwenk J. M., Brunnström H., Glimelius B., Sjöblom T., Edqvist P.-H., Djureinovic D., Micke P., Lindskog C., Mardinoglu A., Ponten F., A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Bradner J. E., Hnisz D., Young R. A., Transcriptional addiction in cancer. Cell 168, 629–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corces M. R., Granja J. M., Shams S., Louie B. H., Seoane J. A., Zhou W., Silva T. C., Groeneveld C., Wong C. K., Cho S. W., Satpathy A. T., Mumbach M. R., Hoadley K. A., Robertson A. G., Sheffield N. C., Felau I., Castro M. A. A., Berman B. P., Staudt L. M., Zenklusen J. C., Laird P. W., Curtis C.; Cancer Genome Atlas Analysis Network, Greenleaf W. J., Chang H. Y., The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H., Li C., Peng X., Zhou Z., Weinstein J. N.; Cancer Genome Atlas Research Network, Liang H., A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell 173, 386–399.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuong E. B., Elde N. C., Feschotte C., Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 18, 71–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.J. M. Coffin, Retroviruses (Cold Spring Harbor Laboratory Press, 2002). [PubMed] [Google Scholar]
- 8.Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J. P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann Y., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J. C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R. H., Wilson R. K., Hillier L. W., McPherson J. D., Marra M. A., Mardis E. R., Fulton L. A., Chinwalla A. T., Pepin K. H., Gish W. R., Chissoe S. L., Wendl M. C., Delehaunty K. D., Miner T. L., Delehaunty A., Kramer J. B., Cook L. L., Fulton R. S., Johnson D. L., Minx P. J., Clifton S. W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J. F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R. A., Muzny D. M., Scherer S. E., Bouck J. B., Sodergren E. J., Worley K. C., Rives C. M., Gorrell J. H., Metzker M. L., Naylor S. L., Kucherlapati R. S., Nelson D. L., Weinstock G. M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D. R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H. M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R. W., Federspiel N. A., Abola A. P., Proctor M. J., Myers R. M., Schmutz J., Dickson M., Grimwood J., Cox D. R., Olson M. V., Kaul R., Raymond C., Shimizu N., Kawasaki K., Minoshima S., Evans G. A., Athanasiou M., Schultz R., Roe B. A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W. R., de la Bastide M., Dedhia N., Blocker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J. A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D. G., Burge C. B., Cerutti L., Chen H. C., Church D., Clamp M., Copley R. R., Doerks T., Eddy S. R., Eichler E. E., Furey T. S., Galagan J., Gilbert J. G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L. S., Jones T. A., Kasif S., Kaspryzk A., Kennedy S., Kent W. J., Kitts P., Koonin E. V., Korf I., Kulp D., Lancet D., Lowe T. M., McLysaght A., Mikkelsen T., Moran J. V., Mulder N., Pollara V. J., Ponting C. P., Schuler G., Schultz J., Slater G., Smit A. F., Stupka E., Szustakowki J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y. I., Wolfe K. H., Yang S. P., Yeh R. F., Collins F., Guyer M. S., Peterson J., Felsenfeld A., Wetterstrand K. A., Patrinos A., Morgan M. J., de Jong P., Catanese J. J., Osoegawa K., Shizuya H., Choi S., Chen Y. J., Szustakowki J.; International Human Genome Sequencing Consortium , Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Sundaram V., Cheng Y., Ma Z., Li D., Xing X., Edge P., Snyder M. P., Wang T., Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 24, 1963–1976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito J., Sugimoto R., Nakaoka H., Yamada S., Kimura T., Hayano T., Inoue I., Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLOS Genet. 13, e1006883 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunarso G., Chia N.-Y., Jeyakani J., Hwang C., Lu X., Chan Y.-S., Ng H. H., Bourque G., Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Pi W., Zhu X., Wu M., Wang Y., Fulzele S., Eroglu A., Ling J., Tuan D., Long-range function of an intergenic retrotransposon. Proc. Natl. Acad. Sci. U.S.A. 107, 12992–12997 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emera D., Casola C., Lynch V. J., Wildman D. E., Agnew D., Wagner G. P., Convergent evolution of endometrial prolactin expression in primates, mice, and elephants through the independent recruitment of transposable elements. Mol. Biol. Evol. 29, 239–247 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Wang J., Xie G., Singh M., Ghanbarian A. T., Raskó T., Szvetnik A., Cai H., Besser D., Prigione A., Fuchs N. V., Schumann G. G., Chen W., Lorincz M. C., Ivics Z., Hurst L. D., Izsvak Z., Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516, 405–409 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Ferreira L. M., Meissner T. B., Mikkelsen T. S., Mallard W., O’Donnell C. W., Tilburgs T., Gomes H. A., Camahort R., Sherwood R. I., Gifford D. K., Rinn J. L., Cowan C. A., Strominger J. L., A distant trophoblast-specific enhancer controls HLA-G expression at the maternal–fetal interface. Proc. Natl. Acad. Sci. U.S.A. 113, 5364–5369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y., Li T., Preissl S., Amaral M. L., Grinstein J. D., Farah E. N., Destici E., Qiu Y., Hu R., Lee A. Y., Chee S., Ma K., Ye Z., Zhu Q., Huang H., Fang R., Yu L., Izpisua Belmonte J. C., Wu J., Evans S. M., Chi N. C., Ren B., Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 51, 1380–1388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuong E. B., Elde N. C., Feschotte C., Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351, 1083–1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slotkin R. K., Martienssen R., Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Deniz Ö., Frost J. M., Branco M. R., Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 20, 417–431 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Rooney M. S., Shukla S. A., Wu C. J., Getz G., Hacohen N., Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith C. C., Beckermann K. E., Bortone D. S., De Cubas A. A., Bixby L. M., Lee S. J., Panda A., Ganesan S., Bhanot G., Wallen E. M., Milowsky M. I., Kim W. Y., Rathmell W. K., Swanstrom R., Parker J. S., Serody J. S., Selitsky S. R., Vincent B. G., Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Invest. 128, 4804–4820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solovyov A., Vabret N., Arora K. S., Snyder A., Funt S. A., Bajorin D. F., Rosenberg J. E., Bhardwaj N., Ting D. T., Greenbaum B. D., Global cancer transcriptome quantifies repeat element polarization between immunotherapy responsive and T cell suppressive classes. Cell Rep. 23, 512–521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panda A., de Cubas A. A., Stein M., Riedlinger G., Kra J., Mayer T., Smith C. C., Vincent B. G., Serody J. S., Beckermann K. E., Ganesan S., Bhanot G., Rathmell W. K., Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 3, e121522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Attig J., Young G. R., Hosie L., Perkins D., Encheva-Yokoya V., Stoye J. P., Snijders A. P., Ternette N., Kassiotis G., LTR retroelement expansion of the human cancer transcriptome and immunopeptidome revealed by de novo transcript assembly. Genome Res. 29, 1578–1590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Babaian A., Mager D. L., Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 7, 24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang H. S., Shah N. M., Du A. Y., Dailey Z. Z., Pehrsson E. C., Godoy P. M., Zhang D., Li D., Xing X., Kim S., O’Donnell D., Gordon J. I., Wang T., Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 51, 611–617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutter C., Zenklusen J. C., The cancer genome atlas: Creating lasting value beyond its data. Cell 173, 283–285 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Ecco G., Imbeault M., Trono D., KRAB zinc finger proteins. Development 144, 2719–2729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghandi M., Huang F. W., Jané-Valbuena J., Kryukov G. V., Lo C. C., McDonald E. R. III, Barretina J., Gelfand E. T., Bielski C. M., Li H., Hu K., Andreev-Drakhlin A. Y., Kim J., Hess J. M., Haas B. J., Aguet F., Weir B. A., Rothberg M. V., Paolella B. R., Lawrence M. S., Akbani R., Lu Y., Tiv H. L., Gokhale P. C., de Weck A., Mansour A. A., Oh C., Shih J., Hadi K., Rosen Y., Bistline J., Venkatesan K., Reddy A., Sonkin D., Liu M., Lehar J., Korn J. M., Porter D. A., Jones M. D., Golji J., Caponigro G., Taylor J. E., Dunning C. M., Creech A. L., Warren A. C., McFarland J. M., Zamanighomi M., Kauffmann A., Stransky N., Imielinski M., Maruvka Y. E., Cherniack A. D., Tsherniak A., Vazquez F., Jaffe J. D., Lane A. A., Weinstock D. M., Johannessen C. M., Morrissey M. P., Stegmeier F., Schlegel R., Hahn W. C., Getz G., Mills G. B., Boehm J. S., Golub T. R., Garraway L. A., Sellers W. R., Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huntley S., Baggott D. M., Hamilton A. T., Tran-Gyamfi M., Yang S., Kim J., Gordon L., Branscomb E., Stubbs L., A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 16, 669–677 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M., Lancet D., Cohen D., GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imbeault M., Helleboid P.-Y., Trono D., KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Horbelt D., Denkis A., Knaus P., A portrait of transforming growth factor β superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 44, 469–474 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Wang Y., Wnt/Planar cell polarity signaling: A new paradigm for cancer therapy. Mol. Cancer Ther. 8, 2103–2109 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Hall A., The cytoskeleton and cancer. Cancer Metastasis Rev. 28, 5–14 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Dar A. A., Goff L. W., Majid S., Berlin J., El-Rifai W., Aurora kinase inhibitors—Rising stars in cancer therapeutics? Mol. Cancer Ther. 9, 268–278 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakayama K. I., Nakayama K., Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Meyers R. M., Bryan J. G., McFarland J. M., Weir B. A., Sizemore A. E., Xu H., Dharia N. V., Montgomery P. G., Cowley G. S., Pantel S., Goodale A., Lee Y., Ali L. D., Jiang G., Lubonja R., Harrington W. F., Strickland M., Wu T., Hawes D. C., Zhivich V. A., Wyatt M. R., Kalani Z., Chang J. J., Okamoto M., Stegmaier K., Golub T. R., Boehm J. S., Vazquez F., Root D. E., Hahn W. C., Tsherniak A., Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherr C. J., Principles of tumor suppression. Cell 116, 235–246 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Kazanietz M. G., Caloca M. J., The Rac GTPase in cancer: From old concepts to new paradigms. Cancer Res. 77, 5445–5451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pontis J., Planet E., Offner S., Turelli P., Duc J., Coudray A., Theunissen T. W., Jaenisch R., Trono D., Hominoid-specific transposable elements and KZFPs facilitate human embryonic genome activation and control transcription in naive human ESCs. Cell Stem Cell 24, 724–735.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frietze S., O’Geen H., Blahnik K. R., Jin V. X., Farnham P. J., ZNF274 recruits the histone methyltransferase SETDB1 to the 3’ ends of ZNF genes. PLOS ONE 5, e15082 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hänzelmann S., Castelo R., Guinney J., GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sergushichev A. A., Fast gene set enrichment analysis. bioRxiv 10.1101/060012 , (2016). [Google Scholar]
- 51.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez J. M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J.-J., Lopez G., Valencia A., Tress M. L., APPRIS: Annotation of principal and alternative splice isoforms. Nucleic Acids Res. 41, 110–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang C.-C., Park A. Y., Guan J.-L., In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanson K. R., Hanna R. E., Hegde M., Donovan K. F., Strand C., Sullender M. E., Vaimberg E. W., Goodale A., Root D. E., Piccioni F., Doench J. G., Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Concordet J.-P., Haeussler M., CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holm S., A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6, 65–70 (1979). [Google Scholar]
- 58.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 59.Szklarczyk D., Gable A. L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N. T., Morris J. H., Bork P., Jensen L. J., von Mering C., STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabc3020/DC1