

Abstract
Hydrogels have long been explored as attractive materials for biomedical applications given their outstanding biocompatibility, high water content, and versatile fabrication platforms into materials with different physiochemical properties and geometries. Nonetheless, conventional hydrogels suffer from weak mechanical properties, restricting their use in persistent load-bearing applications often required of materials used in medical settings. Thus, the fabrication of mechanically robust hydrogels that can prolong the lifetime of clinically suitable materials under uncompromising in vivo conditions is of great interest. This review focuses on design considerations and strategies to construct such tough hydrogels. Several promising advances in the proposed use of specialty tough hydrogels for soft actuators, drug delivery vehicles, adhesives, coatings, and in tissue engineering settings are highlighted. Whilst challenges remain before these specialty tough hydrogels will be deemed translationally acceptable for clinical applications, promising preliminary results undoubtedly spurs great hope in the potential impact this embryonic research field can have on the biomedical community.
Keywords: tough hydrogels, drug delivery, tissue engineering, tough adhesives, tough hydrogel coatings
1. Introduction
As one of the most popular biomaterials designed for clinical use, hydrogels – water-swollen, three-dimensional networks of crosslinked polymers – are fascinating materials whose application potential has tremendously expanded from traditional research areas in biomaterials and drug delivery systems to now include microfluidics, soft electronics, and nanotechnology.[1] In recent years, hydrogels with substantially improved and tunable physicochemical properties for applications in clinical settings have been enabled by interdisciplinary, collaborative efforts to rationalize design at molecular levels while controlling multiscale architecture.[2] Yet, despite several idyllic traits (e.g., biocompatibility, biodegradability, ease of fabrication, controllable permeability, etc.), conventional hydrogels suffer from being mechanically weak (stiffness ~10 kPa and toughness <10 J m−2), tremendously limiting their application in continuous load bearing scenarios such as tissue engineering given that tissues typically possess high toughness (1,000 J m−2), high tensile strength (30 MPa), and high stiffness (1 MPa).[3] In addition, the biodegradation profiles of many polymer networks are not suitable for long term in vivo applications because many hydrogels degenerate too quickly - especially in electrolyte solutions - to achieve desirable therapeutic effects, with degradation often being accompanied by the generation of acidic byproducts.[4, 5] Moreover, propagation and accumulation of damage within hydrogel networks can adversely cause a loss of structural integrity, potentially limiting the functional lifespan of the hydrogel. These limitations have drawn attention to designing materials with enhanced stretchable and toughness properties.[5–14] However, formulating mechanically robust hydrogels that are also morphologically and chemically stable for clinical use remains a challenge. For example, disruption of covalent bonds in tough interpenetrating network hydrogels can lead to irreparable network damage, employing hydrophobic associations is hampered by poor solubility of hydrophobes, competition from water for binding sites limits the association strength of hydrogen bonds, and ionic crosslinks are vulnerable to mobile ions typically encountered in physiological conditions.[9, 15, 16] All these issues could lead to undesirable swelling and degradation of mechanical properties. In fact, many tough hydrogels reported to date cannot maintain their mechanical properties during tissue culture or after implantation.[17–19] Thus, there remains a need to develop high-water-content, mechanically robust hydrogels that can prolong the lifetime of clinically suitable materials by reliably avoiding property degradation under aggressive in vivo conditions.
This review, as organized in Figure 1, presents current advances in the field of tough hydrogels while highlighting roadblocks in translating such materials for robust use in medical settings. We begin by briefly introducing underlying factors influencing material failure in conventional hydrogels, followed by commonly employed strategies to construct tough hydrogels given design considerations. Finally, we highlight some promising advances in biomedical applications of tough hydrogels, specifically focusing on tough hydrogels used as actuators, drug delivery vehicles, adhesives, hydrogel coatings, and in tissue engineering applications (i.e. cartilage, cardiovascular, and corneal tissue engineering). While many of these specialty tough hydrogels are not yet viable for clinical trials/applications, promising preliminary in vitro and in vivo data emphasize the exciting potential use of materials to tackle a wide array of challenging and pressing medical needs.
Figure 1. Overview of different strategies to make tough hydrogels and their biomedical applications.

Schematic representation of strategies used to formulate tough hydrogels, namely, Tetra-PEG, click chemistry, radiation crosslinked, slide ring, fiber/filler enforced networks, interpenetrating network hydrogels, double network, nanocomposite, and macromolecular microsphere composite hydrogels. Such specialty tough hydrogels have shown exciting promise as tough coatings and adhesives, and for use in soft actuators, drug delivery, and tissue engineering applications.
2. Design Considerations for Hydrogel Toughening
To design hydrogel devices and components with precisely tuned properties, evaluating the damage process at the crack tip becomes critical to determine what underlying factors lead to material failure and, consequently, material toughening.[20] When subjected to cyclic loading, hydrogels will undergo fatigue, meaning the progressive degradation of material properties or the nucleation and growth of cracks that may cause material fracture.[21] Consequently, a hydrogel’s strength is governed by its ability to withstand mechanical loads that lead to the formation and subsequent propagation of cracks: material failure occurs when the tip of a crack begins to propagate at its weakest point.[22] A static crack will lose stability and begin propagating when the change in potential energy released after an infinitesimal extension of the crack surpasses the fracture energy threshold (i.e. the energy needed to form two new surfaces).[23] Specifically, the energy dissipated during crack growth is equal to the energy where network strands are extended to the equivalent point of their dissociation energy.[24]
Unlike conventionally elastic materials, hydrogels exhibit large strain values at their fracture point and undergo deformation predominantly influenced by entropic rather than enthalpic phenomenon given that exerted forces drive changes in polymer chain configurations.[11] As such, during crack propagation, polymer chains undergo large deformation both along the crack path and in the surrounding area called the process zone (Figure 2).[11] In the process zone, all energetic dissipative processes that occur when the material is stretched beyond linear elastic deformation are assumed to take place, which are represented on stress-strain curves of materials as hysteresis loops.[22, 25] The size of the process zone varies significantly from material to material and can range anywhere from a few nanometers to a few millimeters. The added deformation in the process zone protects the crack tip from extending further from additional forces supplied by external loads and accounts for an added increase in the fracture energy of hydrogels. Subsequently, hydrogels with higher fracture energy would be better able to sustain greater stress and strain levels prior to rupture and could thereby be considered to have higher toughness.
Figure 2. Schematic representation of crack propagation in a hydrogel network.

When a crack forms, energy is released and transferred to the crack tip. The crack will continue to propagate if this energy is sufficient to rupture the polymer chains lying across the crack plane. The fracture energy of hydrogels is divided into two parts: fracture energy from polymer chains rupturing at the crack surface, and the mechanical energy dissipated by loading/unloading the hydrogel in the process zone. Adapted with permission.[26] Copyright 2013, The Royal Chemistry Society.
Generally, however, synthetic hydrogels are very brittle (fracture energy ~10 Jm−2 compared to 1000 Jm−2 for cartilage and above 10,000 Jm−2 for natural rubber) upon mechanical loading from a lack of significant energy dissipation in the process zone.[27] This problem is further intensified due to several other factors, including irregular distribution of crosslinking points, variations in polymer chain lengths at crosslinking junctions, and/or high water content.[12] Structural inhomogeneity inside polymer networks tends to arise as a result of differences in reactivity between monomers and crosslinkers: more densely cross-linked microgels initially form, which are then connected into a larger network, leading to an inhomogeneous network.[27] Along the same lines, it is difficult to ensure that crosslinking of polymer chains occurs at equal intervals when using crosslinking agents, thereby resulting in a wide range of chain lengths.[12] The resulting heterogeneity of the material, in turn, gives rise to defects that act as stress concentrators much greater than those experienced by the overall hydrogel. Regarding water uptake by the material, higher water contents result in a reduced crosslinking density, thereby reducing fracture energy given that fewer chains are broken for crack propagation. Moreover, crystallization or viscoelastic energy dissipation – additional energy dissipation mechanisms— are reduced by the greater separation distance of network chains in swollen hydrogel networks.[12, 24] While increasing the crosslinking density or polymer concentrations can slightly enhance a hydrogel’s mechanical strength, the mechanical properties of single network hydrogels remains insufficiently robust.
From a quantitative perspective, as reviewed by Zhao, the fracture energy of hydrogels is predominantly governed by two components: (i) the intrinsic fracture energy (i.e. the energy required to break the polymer chains lying across the crack path) and (ii), the fracture energy from mechanical dissipation in regions surrounding the crack path (i.e. process zone).[26] As mentioned above, the intrinsic fracture of polymers is highly dependent on the swelling ratio of hydrogels given that increased swelling leads to a reduction in the number of chains per cross-sectional unit area, thereby lowering intrinsic fracture energy.[24] Additionally, for polymers with fixed monomer units and crosslinking density, the value of the intrinsic fracture energy becomes relatively constant and very difficult to significantly increase. On the other hand, hydrogels that can sustain higher levels of stress and strain with more substantial stress–strain hysteresis and process zones lead to materials with higher fracture energy.[28] This was quantitatively modeled by Zhang et al., who used cohesive-zone and Mullins-effect models to show that toughening of a soft material relies on high intrinsic fracture energy of the material, high value of maximum hysteresis, and quick transition to the maximum hysteresis in the material under deformation.[25]
Considering these material properties and design constraints, there are broadly three overarching approaches to constructing endurant, tough hydrogels that maintain structural integrity following large deformation: (i) Reducing the presence of heterogeneities in the material by forming homogenous gels to evenly distribute load over a significant fraction of chains, thereby presenting fewer sites for micro-crack formation; (ii) Introducing one or several strategies to dissipate mechanical energy in order to limit macro-crack propagation; (iii) Incorporating both mechanisms by using multifunctional crosslinkers (e.g. micro and nano particles) to control inter-crosslinking distances, thereby forming homogenous networks while allowing for energy dissipation from rupture of bonds between the particles and polymer chains. In the following sections, different hydrogel fabrication methods and toughening mechanisms based on these three approaches are evaluated.
3. Strategies to synthesize and fabricate tough hydrogels
Several strategies have been proposed to fabricate which are broadly reviewed as three groups: (1) Homogeneous tough hydrogels, which encompasses tetra- polyethylene glycol (Tetra-PEG) hydrogels, click chemistry hydrogels, radiation crosslinked hydrogels, and slide ring hydrogels; (2) Energy-dissipating hydrogels, specifically interpenetrating network hydrogels, double network hydrogels, and fiber-reinforced hydrogels; and finally (3) Tough hydrogels with multifunctional crosslinkers (Table 1).
Table 1.
Schematic representation and chemical structure of different strategies (e.g. homogenizing polymer networks, introducing energy dissipation mechanisms, or combining both mechanisms) used to make tough hydrogels. Adapted with permission.[12] Copyright 2015, Elsevier.
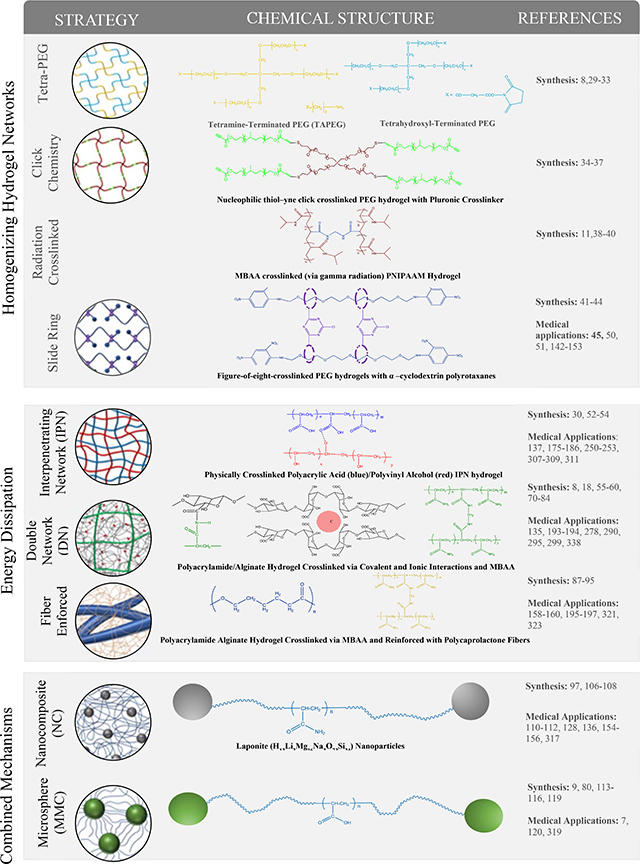 |
3.1. Homogenizing polymer networks to form tough hydrogels
Traditional hydrogels generally suffer from inhomogeneous networks due to poorly controllable crosslinking methods. Thus, if one can carefully synthesize hydrogels with increased network cooperativeness, resulting gels would be endowed with improved mechanical properties compared to inhomogeneous counterparts, given an even distribution of load over network chains. The following section discusses some example strategies to synthesize homogenous hydrogels with mechanically superior properties.
3.1.1. Tetra PEG Hydrogel networks
Asymmetrical combinations between multifunctional crosslinkers and telechelic polymers endow hydrogel networks with an increased degree of freedom that results in the formation of micro inhomogeneities such as loops and entanglements.[8] To limit the formation asymmetric combinations and form uniform hydrogel networks, one can decrease the degree of freedom of the reactants involved in the reaction.[11] To do so, Sakai et al. developed a tetra- polyethylene glycol (PEG) gel combining two symmetrical macromers (tetraamine-terminated PEG (TAPEG) and tetra-NHS-glutarate-terminated PEG (TNPEG)) with equal sizes and defined lengths.[8] The polycondensation reaction obeyed second-order kinetics regardless of gelation threshold, suggesting that the prepolymers mix homogeneously after initiation, with reactions occurring between adjacent prepolymers.[29] Given that the length of the PEG arms define the nano-structural organizations of the hydrogel, resulting networks were found to have extremely homogenous 3D tetrahedral structures; topological defects (e.g. entanglements and loops) were found to be negligible in the hydrogel network.[30] The resulting high mechanical strength of the gel was found to be comparable to that of human cartilage: at a one—to—one mole ratio of the two PEG macromers, the gel had a compressive strength of 2.5 MPa and compression modulus of 40 kPa.[31] When investigating gelation mechanisms of Tetra-Peg hydrogels, gelation was independent of pre-polymer molecular weight and polymer concentration polymer concentration, thereby indicating that a reaction-limited rather than diffusion-limited reaction was key to fabricate the homogeneous polymer networks.[32] Further functionalization with poly(ethylglycidylether) (PEGE) to form amphiphilic Tetra-PEG–PEGE gels endowed the tough hydrogel with rapid phase transition behavior similar to PNIPAM-based hydrogels.[33] This facile method to fabricate homogenous, tough hydrogels is limited in scope of monomers and polymers available since symmetrical macromers are necessary to successfully synthesize such materials.
3.1.2. Click Chemistry Hydrogels
A similar strategy to formulate hydrogels with improved mechanical performance through controlled architecture is employing “click chemistry.” “Click chemistry” is used to describe a reaction that is high yielding, wide in scope, stereospecific, and generates inoffensive byproducts that are removable via nonchromatographic techniques.[34] Through copper (I)-catalyzed cycloaddition chemistry, diacetylene-functionalized and tetraazide-functionalized PEG derivatives were used to form a tough-PEG based hydrogel with a well-defined network structure.[35] The drastically improved mechanical properties of the gel compared to photochemically crosslinked PEG hydrogels are attributed to an even dispersion of crosslinking points stemming from a more controlled crosslinking reaction. Moreover, material properties can be further fine-tuned via changes in azide/acetylene ratios, with unreacted azide and/or acetylene groups left available for subsequent functionalization, allowing for chemical tailoring to form a diverse repertoire of tough hydrogels. Other groups have similarly employed click chemistry to formulate tough hydrogels via controlled polymer network structures.[36] However, despite the benefit of spatiotemporal control and fast gelation times of thiol-ene and thiol-yne click chemistry reactions, potential toxicity from photo-initiators and radicals, along with cross-reactivity with thiols, remain concerning.[37] Likewise, copper (I) catalyzed reactions tend to use toxic copper catalysts, strain-promoted azide-alkyne cycloaddition is limited by difficult synthesis of cyclooctynes, and native chemical ligation suffers from cross-reactivity with amines.[37] Thus, complicated synthesis routes and potential side reactions between biomolecules and the hydrogels should be considered when choosing click-chemistry methods for developing tough hydrogels for medical applications.
3.1.3. Radiation Crosslinked Hydrogels
Norisuye et al. compared the structural profile of poly(N-isopropylacrylamide) (PNIPAM) gels prepared via conventional radical polymerization versus those prepared via γ-ray irradiation (γ-ray gels) using small-angle neutron scattering.[38] From their structural analysis, they found that γ-ray gels were more homogenous from a microscopic point of view than the chemical gels. This phenomenon is attributed the increased probability of achieving an even distribution of crosslinks via γ-ray irradiation mediated crosslinking because radicals can disperse evenly throughout the hydrogel network and crosslinking is independent of a monomer’s reactivity or concentration.[11] Using γ-ray irradiation mediated polymerization, Wang et al. synthesized polyacrylic acid (PAA) and polyacrylamide (PAAm) hydrogels.[39] The mechanical properties of the hydrogel were enhanced compared to classically polymerized PAA/PAAm hydrogels. More recently, Xu et al. fabricated a double crossed linked hydrogel from hyperbranched PEG-polymers that formed hydrogels in situ and could be further strengthened by γ-ray irradiation.[40] While controlling the degree of crosslinking is advantageous to form homogenous networks, a balance of risk to benefits must be considered, as extended exposure to γ-ray irradiation may result in un-intended toxicity, loss of biocompatibility, and potential loss of functionality of the hydrogel for medical settings where in situ polymerization is required.
3.1.4. Slide Ring Hydrogels
Okumura and Ito developed a novel polyrotaxane gel based on PEG chains and crosslinked cyclic α-cyclodextrin (α-CD) molecules.[41] In the so-called “slide-ring” hydrogel, polymer chains with bulky end groups are not chemically nor physically crosslinked but instead are interlocked by figure-of-eight crosslinks.[42] The freely movable crosslinks can slide along the threading polymer chain to equalize the tension i.e. the pulley effect.[42] This unique feature allows the hydrogel to stretch up to 24 times in length, and have a substantial volume change of up to 24,000 times in weight. [43] Furthermore, by modifying the hydroxyl groups of CDs with different functional groups, tough hydrogels with different stimuli-responsive properties have been formulated such as thermo-responsive, photo-responsive, and slide ring elastomer materials.[44] Although much less frequently reported, polyrotaxane gels can also be diversified with cyclic molecules other than CDs, such as pillararenes, cucurbiturils, and calixarenes.[44] Likewise, efforts to use polymer backbones other than PEG (e.g. poly (dimethyl siloxane), polybutadiene, polyester, a copolymer of poly(propylene glycol) and PEG or polyethyleneimine) have been explored, although efforts have been greatly limited due to the complexity of synthesis.[44] Ongoing research efforts to improve and simplify the synthesis of functionalized polyrotaxanes will undoubtedly expand the potential use of such materials for an array of medical applications.[45]
3.2. Incorporating Mechanical Energy-Dissipating Systems
Rather than homogenizing the polymer network, another approach to formulate tough hydrogels focuses on exploiting heterogeneity to produce an energy dissipation mechanism. As previously explained, when a crack forms in a hydrogel, energy released is transferred to the crack tip. The crack will continue to propagate only if this energy is sufficient to rupture polymer chains along the crack path. By incorporating an energy-dissipating system to diffuse this energy, the energy transferred to the crack tip will be insufficient to allow the crack to continue propagating in the hydrogel, thereby “toughening” the hydrogel. Such energy dissipation typically occurs from the rupture of bonds – physical and/or covalent—in the polymer network or from the fracture of embedded fibers.[11] Interpenetrating polymer networks and fiber/filler-reinforced composite hydrogels are two kinds of hydrogels fabricated with such energy-dissipating mechanisms.
3.2.1. Interpenetrating Polymer Networks
Interpenetrating polymer networks (IPNs) are amalgams of two or more crosslinked polymeric chains interconnected through noncovalent means that form unique multicomponent materials with distinct property profiles compared to their individual counterparts. According to their fabrication procedure, they can be classified as one of two kinds of IPNs: (1) Simultaneous IPNs in which precursors of the networks are mixed and synthesized simultaneously by independent, noninterfering routes (e.g. free radical and condensation polymerization) or; (2) Sequential IPNs, where after forming a hydrophilic polymer network, that network is then swollen in a second monomer solution, after which the latter is polymerized to form the resulting hydrogel.[46, 47] If one of the polymer chains is not crosslinked (i.e. linear), a semi IPN is formed, which can later be prepared into a full IPN by selective crosslinking of the linear polymer chain. IPNS are advantageous given that dense hydrogel matrices can be produced with stiffer and more widely controllable mechanical properties, allowing for more versatile combinations compared to single network counterparts. [47] Typically, long chains are interpenetrated with short chains that may be fractured or physically de-crosslinked under deformation, thereby dissipating energy while the long network chains maintain the elasticity of the network.[26] It must also be noted, however, that with IPNS, the polymers may become interpenetrated to such an extent that releasing encapsulated bioactive molecules becomes difficult, not to mention that the quality of the final polymer is highly susceptible to process parameters including reaction mechanism, reactor type, and reactor operating conditions.[48, 49] Likewise, limited fatigue resistance can restrict the clinical translation of many chemically crosslinked tough IPNS, thereby promoting more investigation into designing facile fabrication methods to produce IPNs that are both tough and self-healing with tunable application properties.[50]
Double Network Hydrogels
Pioneered by Gong et al., double network (DN) hydrogels are IPNs with tremendous mechanical properties that are typically composed of a stiff yet brittle first network and a ductile secondary network.[6] Under optimized conditions, DN hydrogels -with a water content as high as 90% - possess hardness (elastic modulus of 0.1–1.0 MPa), strength (failure tensile stress 1–10 MPa, strain 1000–2000%, failure compressive stress 20– 60 MPa, strain 90–95%) and toughness (tearing fracture energy of 100–1000 Jm2).[51–53] Such incredible properties are attributed to following essential features of DN: (1) A rigid and brittle polymer as the first network and a soft and ductile polymer as the second network; (2) A molar concentration of the second network 20–30 times that of the first network; (3) Tight crosslinking of the first network while the second loosely cross-linked, requiring a very high molecular weight of the second polymer.[6, 16]
Okumura, Brown, and Tanaka have proposed theories explaining the large fracture toughness of double network hydrogels.[54] The Brown-Tanaka model stipulates that crack propagation in DN gels occurs in two parts: (i) Failure of the first network -due to its brittle nature- causes the formation of multiple micro-cracks. As the external strain increases, these cracks are prevented from coalescing into macro-cracks by the second network through viscous dissipation, leading to the formation of damage zones around the crack tip.[55] The formation of these internal micro-cracks is a prominent form of energy dissipation, with the value of fracture energy increasing linearly with the size of the damage zone.[11, 56] Moreover, due to local necking and yielding around the crack tip, the gel becomes much softer, thus tremendously reducing stress concentration and increasing toughness.[53, 57, 58] Large hysteresis from the breakage of first network bonds, covalent interconnections, increased physical entanglements, and molecular associations are likewise stipulated to contribute to the superior mechanical strength of DN gels; and (ii) Second network failure leads to crack propagation that causes overall material failure.[52, 58, 59]
While DN hydrogels are traditionally formed by two-step free radical polymerization processes, molecular stent methods, one-pot synthesis methods, extrusion 3D printing, and free shapeable methods of production have also been explored.[6, 60] Void DN gels, biopolymer-based DNs, microgel enforced particle DN gels, liquid crystalline DN gels, bi-layered tough gels, and ultrathin DN gels are amongst the many novel techniques used to further enhance the mechanical properties of DN for practical use in biomedical applications.[61–63]
One consideration to keep in mind, however, is that while both classical and “molecular stent” strategies expanded the range of polymers available to fabricate DN hydrogels (e.g. neutral and polyelectrolyte polymers), both strategies necessitate multi-step synthesis. As such, the following limitations are encountered: (i) multistep synthesis is time-consuming as it generally involved swelling, diffusion, and two polymerization processes, resulting in a one to two day process to complete the DN hydrogel; (ii) only simple sheets and disc-like DN gels have been reported because it may be challenging to use the multi-step method to prepare complex shaped gels; and (iii) both “molecular stent” methods result in chemically cross-linked hydrogel networks and so the fracture of those networks—particularly the first network—causes irreversible damages to the DN hydrogels.[61]
To overcome these limitations, “one-pot” synthesis methods can prepare hybrid physically-chemically crosslinked DN gels in a faster and controllable way, while both extrusion 3D printing and free shapeable methods of production have facilitated the production of complex shaped DN hydrogels.[61] While these and other novel preparation methods of DN hydrogels have led to the formation of hybrid DN gels that are better able to withstand damage than traditionally chemically crosslinked hydrogels from reversible binding of physical crosslinks, poor fatigue resistance remains a key limitation of DN hydrogels.[61] Further exploration is warranted to design novel preparation to develop DN gels with robust mechanical properties, self-healing abilities, and multifunctional properties (e.g. magnetic, optical, electric properties, etc.) in an efficient manner.
3.2.2. Fiber Reinforced Composite Tough Hydrogels
Another method to design tough hydrogels involves embedding stretchy fibers or fillers into the hydrogel matrix with the idea that the fracture of fibers/fillers along the crack plane requires additional energy. This energy, coupled to the energy required to overcome cohesive forces of the matrix, increases the availability of dissipative energy, thereby increasing the overall toughness of the composite hydrogel network. For example, steel wool fibers, woven poly(ϵ-caprolactone) fiber scaffold, and polyacrylonitrile nanofibers were incorporated to improve the mechanical properties of alginate-PAAm gels.[64] Similarly, poly(ε-caprolactone), PLA, cellulose, and silk nanofiber composite tough hydrogels have been produced for intended medical applications, particularly for cell encapsulation purposes given that 3D woven fiber scaffolds can mimic layered structures innate to many tissues.[65, 66] Moreover, embedded fibers/fillers can enhance the compressive and wear properties of the gel scaffolds compared to single network hydrogels. Further biochemical modification of the hydrogel can likewise enhance the overall biocompatibility of the matrix. Additional investigation into architectural and spatial control of fiber distribution within the scaffolds will undoubtedly improve network cooperativeness of the composite to enhance the mechanical and biomimetic properties of such composites, as current hydrogels are yet to be able to suitably mimic the diverse microstructures and compositions of tissue ECMs.
3.3. Multifunctional Crosslinkers
Micro and nano-composite hydrogels are cross-linked polymer networks swollen with micro or nanostructures that endow the gel with higher elasticity and strength compared to their individual counterparts. The micro/nanoparticles can either crosslink the gel, be used to attach or absorb polymer chains, or add new properties (i.e. responsiveness to mechanical, optical, thermal, magnetic, electric stimulation, etc.) by physical entrapment within the network.[67] Multifunctional cross-linking via these micro/nano structures can allow for improved control over crosslinking densities and inter-crosslinking distances and thus facilitate better load redistribution within hydrogel networks to prevent macro-crack propagation. Similarly, reversible breaking of physical bonds between the particles and hydrogels serves to effectively dissipate energy to prevent crack propagation via an increase of fracture toughness. The rupture and reformation of bonds between the particles and polymer chains also advantageously endow such materials with superior self-healing properties compared to other tough hydrogels discussed prior.
3.3.1. Nanocomposite Tough Hydrogels
Haraguchi et al. developed transparent nanocomposite (NC) gels with excellent mechanical properties and structural homogeneity by using water-swellable silicate nanoplatelets (Laponite) as multifunctional crosslinkers for N-iso-propylacrylamide, N,N-dimethylacrylamide, or acrylamide polymers.[68] The inorganic clays are first exfoliated and uniformly dispersed in an aqueous media, after which radical polymerization is initiated thermally from the clay surface, thereby effectively crosslinking polymer chains by the clay sheets. Rather than being randomly crosslinked with haphazard and broad distributions of chain lengths as is common in traditional radical polymerization, the large distance between clay sheets allows the polymer chains to adopt long and flexible orientations with random conformations. Given that multiple polymer chains attach to individual clay NPs, detachment of single chains from NP has negligible effects on the overall structure of the hydrogel network.[11] Moreover, since the polymer chains can easily be reattached to the clay NPs via physical bonding interactions, the overall integrity and fatigue resistance of the hydrogel are enhanced. [11]
Regarding the mechanical properties of NC gels, the hydrogel demonstrate superior mechanical toughness, with the tensile modulus and tensile strength increasing almost proportionally with clay content, whereas the elongation at break tends to decrease slightly on increasing clay.[69, 70] However, at sufficiently high clay contents, the coil-to-globule transition of poly(N-isopropylacrylamide (PNIPAM) chains is inhibited due to steric hinderance. NC gels generally withstand 90% compression, are highly stretchable (elongation at break > 1000%), and under high loading conditions, can withstand sliding friction; yet frictional forces are affected by environmental conditions (wet or in-air), clay content, and drying of the gel surface.[69, 71] Interestingly, using other synthesis methods –simply mixing clay and polymer solutions or using other inorganic nanoparticles instead of clay—led to poorer mechanical performances of the resulting gels, thereby implying that the formation of the tough, organic/inorganic networks innate to this hydrogel are specifically realized via in situ polymerization in the presence of clay.[72]
The surface of the NC gels, while primarily hydrophobic in nature due to the spontaneous alignment of N-isopropyl groups at the hydrogel-air interface, exhibits reversible hydrophilic-hydrophobic changes based on exposure to different environmental conditions (e.g. in water versus air).[73, 74] The NC gels also demonstrated self-healing capacities and reversible swelling behavior from salt promoted coil-to-globule transitions of PNIPAM.[75] The fabrication process could be easily adapted to form platinum polymer clay nanocomposite hydrogels, copolymer nanocomposite hydrogels, and porous nanocomposites with characteristic layered morphologies.[76] Furthermore, NC gels were able to support the culture of HepG2 human hepatoma cells, human dermal fibroblasts, and human umbilical vein endothelial cells.[77] Other materials such as carbon-based nanomaterials (carbon nanotubes (CNTs), graphene, nano-diamonds), polymeric nanoparticles (polymer nanoparticles, dendrimers, hyperbranched polyesters), inorganic/ceramic nanoparticles (hydroxyapatite, silica, silicates, calcium phosphate), and metal/metal-oxide nanoparticles (gold, silver, iron-oxide) have since been combined with polymeric networks to obtain novel and tough nanocomposite hydrogels for medical applications.[78] One important consideration when using such materials for medical purposes is the in vivo degradation and subsequent the fate of the nanoparticles released from the composite materials.[13] Moreover, choosing the appropriate nanoparticles becomes critical, as different nanoparticles demonstrate different levels of biocompatibility, and the mechanical and self-recovery properties of the composite gels have been found to be affected by the choice of nanoparticle.[13]
3.3.2. Macromolecular Microsphere Composite Hydrogels
With a similar concept to nanocomposite tough hydrogels, the mechanical properties of hydrogels can likewise be reinforced by adding microspheres or microgels to form macromolecular microsphere composites. These composites allow for controlled crosslinking density and the inter-crosslinking distance through polymer chain attachment to microspheres, thereby enabling the network to sustain the stress cooperatively to enhance the overall mechanical integrity of the hydrogel.[12]
Huang et al. irradiated macromolecular microspheres (MMs)- composed of styrene, butyl acetate- with 60Co γ-rays in oxygen, forming peroxides on the surface of the evenly distributed MMs that then decompose under heat to form free radicals that act to initiate the grafting of acrylic acid monomers onto the MMs surface and initiate PAA homo-polymerization.[7] Transient inter and intramolecular hydrogen bonds between PAA chains act to dissipate energy, while the well-defined structure formed by long PAA chains crosslinked via MMs acts to sustain stress cooperatively, thereby forming a tough hydrogel whose crosslinking density and the inter-crosslinking distance can be tuned by adjusting the peroxide concentration and/or the concentration of MMs.[12] Notably, the composite was able to sustain a compressive stress of 10.2 MPa at a strain of 97.9% while being able to elastically recover to its original shape for strains greater than 90% despite having a high water content (~89%).[7] Hydrogels prepared by using peroxidized MMs and AAm exhibited moderate moduli (60− 270 kPa), high fracture tensile stresses (up to 0.54 MPa), high extensibilities (up to 2500%), and high fracture energies (270− 770 J m−2).[79]
Building on this concept, Xu et al. prepared multi-responsive composite hydrogels by treating core-shell microgels- composed of PNIPAA as the core and poly(vinyl amine) (PVAm) as the shell- with potassium persulfate to generate radicals on the amine nitrogens of PVAm that can be used to initiate graft polymerization of AAm onto the microgels.[80] The gels demonstrated high compressive strength (17–30 MPa) and rapid pH induced volume changes. He et al. developed an elegant synthesis method to formulate high mechanical strength microgels from micelles.[81] These micelles are formed by a nonionic surfactant in water, with subsequent nanoparticle formation via crosslinking under γ-ray irradiation; radiation-peroxidized micelles promote the grafting of polymer chains following the thermal decomposition of peroxides formed on the surface of the micelles. Composite networks of poly(2-acrylamido-2-methylpropanesulfonic sodium) (PNaAMPS) with sparsely crosslinked PAAm networks microgels have also been developed.[63, 82] The toughness and strength of the composites hydrogels depended on the PNaAMPS concentration in the microgel and the molar ratio of the PAAm to PNaAMPS.[83] Like DN hydrogels, the polyelectrolyte networks of the microgels serve as sacrificial bonds to increase the facture energy of the resulting network.[84] Remarkably, evaluation of hysteresis curves showed that DN microgels had a four-fold higher fracture efficiency from the rupture of polyelectrolyte networks than conventional DN gels at the same strain.[83]
To investigate the mechanical properties of temperature-sensitive microgel composites, Meid et al. embedded temperature-sensitive PNIPAAm microgels within PAAm hydrogel networks.[85] Results showed an increasing elastic modulus and storage modulus with increasing microgel content, with effects shown to be more pronounced for temperatures above the volume phase transition temperature.[85] Interestingly, swelling experiments showed a decrease in swelling capacity of composite materials – microgels promote chain entanglements, thereby reducing the ability of the material to absorb water – indicating that incorporating temperature-responsive microgel particles can endow composite hydrogels with temperature-sensitive mechanical behaviors.
Like nanocomposite tough hydrogels, MMCs overcome a classic limitation of organic crosslinkers (e.g. low number of reactive groups) given that the reactive surface of the microspheres presents several binding sites for polymer chains, forming strong yet reversible binding to aid in energy dissipation and self-healing processes. In contrast to NCs hydrogels that are typically made from highly specific polymers with water-swellable clay, MMC hydrogels only require organic components, with different compositions and properties easily formulated by changing microspheres and monomers. Many MMC tough hydrogels, however, are formulated with synthetic particles, which might present biocompatibility and biodegradability issues. While clinically ready materials have yet to come to fruition, several microgel organic/inorganic composites have been developed with promising applications for regenerative medicine applications.[5, 86] With further exploration into the incorporation of non-synthetic microparticles will undoubtedly facilitate the fabrication of desirable and translatable MMC hydrogels with ideal biocompatibility and mechanical properties for medical uses.
Overall, many of the strategies to enhance the mechanical properties of hydrogels discussed in this review have undoubtedly transformed and expanded the scope of hydrogel applications. However, only a small niche has shown feasible translatability towards medical applications. Complex fabrication methods with harsh solvents and extensive curing/irradiation time remain impractical, excessive swelling in aqueous solutions results in a tremendous reduction of mechanical properties, limited anti-fatigue properties leading to irreversible structural damage, and limited control over degradation profiles with non-toxic byproducts are a few unresolved challenges that still need to be addressed to design tough hydrogel devices and components that maintain their structural integrity for robust clinical use. In the following section, promising efforts to achieve the latter with tough hydrogels specifically for medical applications are reviewed.
4. Applications in Medicine
The intriguing properties of hydrogels that enable them to superficially resemble native soft tissues make them ideal material candidates for devices and components used in clinically relevant therapies. In the following section, proposed applications of tough hydrogels as soft actuators, vehicles for drug delivery, adhesives, coatings, and in tissue engineering are focused on and reviewed. While many materials have yet to be used in clinical settings/trials, promising preliminary in vitro and in vivo results using various animal models presage the use of such specialty, tough hydrogels for a wide array of medical applications with further optimization.
4.1. Soft Actuators
The use of soft matter in robotics has tremendously expanded the medical application of robots in the field, particularly in areas where soft patient interactions are preferred such as for replacement of limbs via prosthetics, artificial organs, body-part simulators, and drug delivery vehicles.[87] The principle behind using hydrogel-based soft actuators is that the hydrogel material is able to swell/shrink in response to external stimuli, which allows for the generation of various actuations and motions.[88] One application of interest of such actuator systems is developing artificial skin that is able to mimic both the flexibility and sensory sensitivity capabilities of the native skin, which has been explored using a wide range of materials from compliant conductors, semiconductors, to dielectrics.[89] Specifically, “electronic skin” is generally regarded as a stretchable sheet with area above 10 cm2 carrying sensors for various stimuli, including deformation, pressure, light, and temperature.[90] Using a hydrogel actuator, Sun et al. developed a transparent sensory sheet termed “ionic skin” from polyacrylamide (PAAm) hydrogels that could detect a wide range of stimuli (strains from 1% to 500%).[90] However, ionic skins made from chemically crosslinked PAAm generally suffer from poor self-healing properties propelling efforts towards the use of tough and/or autonomous self-healing hydrogels in the development of the next generation of ionic skins.[90, 91] For example, Lei et al. physically crosslinked calcium carbonate (ACC) nanoparticles in a PAA and alginate hydrogel to form an ionic skin with high pressure sensitivity (up to 1 kPa) and > 90% recovery even after ten drying–swelling cycles (Figure 3).[92] In the same spirit, Pu et al. developed a soft skin-like triboelectric nanogenerator TENG (STENG) using a PAAm-LiCl hydrogel- elastomer (PDMS and 3M VHB 9469) composite.[93] The sandwich structure of the STENG imparted the material with ultrahigh stretchability (uniaxial strain, 1160%) and transparency (average transmittance, 96.2% for visible light).
Figure 3. Soft actuator composed of a polyacrylic acid (PAA)/alginate hydrogel with calcium carbonate (AAC) nanoparticles to form an ionic skin.

a) Schematic of the hydrogel structure consisting of ACC, PAA, and alginate. b) SEM image of the composite ACC/PAA/alginate hydrogel following lyophilization. c) Storage modulus (G′) and loss modulus (G″) of the ACC/PAA/alginate and ACC/PAA hydrogels as a function of frequency. d) Images demonstrating facile manipulation of ACC/PAA/alginate hydrogel into different shapes. e) Images demonstrating dynamic adherence of the hydrogel to irregular surfaces and an ability to accommodate motion of the prosthetic finger. Reproduced with permission.[92] Copyright 2017, WILEY-VCH.
Stimuli-responsive polyelectrolyte hydrogels have also harnessed attention given their ability to transform chemical energy into mechanical motion without requiring external mechanical stimulation. A walking gel actuator made of cationic (acrylamide/sodium acrylate) and anionic (acrylamide/quaternized dimethylaminoethyl methacrylate) legs achieved unidirectional motion by alternate bending of each leg in response to changes in the direction of the applied electrical field.[94] A six-layered muscle-like actuator was developed that could undergo linear contraction (rather than bending) in response to an applied electric field.[95] Similarly, tough polyelectrolyte hydrogels with ionic nano-micelle macro-crosslinkers actuated by electric fields when immersed in salt solutions were recently reported.[96] Both positively and negatively charged hydrogels physically crosslinked via F127DA micelles were produced, yet cationic nano-micelle hydrogels interestingly decreased the toughness and strength of hydrogels compared to anionic analogs: anionic nano-micelle hydrogels demonstrated ionic strength dependent swelling behaviors, and exhibited significant, cyclic electric field sensitivity (e.g., bending angle up to 87° in 120s). Fatigue resistant, controllable, fast-acting actuation affords exciting opportunities to produce biosensors and artificial muscles, thereby broadening the potential for the development of “smart” prosthetics.
In terms of bi-layered tough hydrogel-based actuators, Liu et al. developed nanocomposite polyelectrolyte hydrogels composed of negatively charged (acrylamide and 2-acrylamido-2-methylpropanesulfonic acid) and positively charged (dimethylaminoethyl methacrylate methylchloride) hydrogel-exfoliated sodium montmorillonite nanosheets.[98] The actuator showed excellent fatigue resistance and high tensile strength toughness while demonstrating reversible actuation from the contrasting responsiveness of each gel component to the ionic strength of the immersion buffer. Zheng et al. developed a tough bi-layered actuator based on an interpenetrating network hybrid of physically cross-linked alginate and chemically cross-linked PNIPAM, which was used to make a four-arm robotic gripper that could sustain an estimated maximum load ∼1.21 g.[99] Similarly employing tough alginate/PNIPAM, Yuk et al. developed an optically transparent robust hydrogel actuator that could impressively catch, lift, and release a live ryukin goldfish via hydraulic pressure mediated actuation (Figure 4).[97] Further optimization of such materials can lead to the development of powerfully strong yet soft devices that improve the biomimetic stimulation of biological phenomena.
Figure 4. Example of an optically transparent, robust hydrogel actuator.

(a) Image of a leptocephalus in a marine environment. (b) Schematic of a hydrogel actuator inspired by the leptocephalus (c) Schematic of the hydrogel development process for hydraulic actuators. (d) Images of the hydraulic hydrogel actuator’s rapid actuation process. (e) Images demonstrating the implementation of a hydraulic hydrogel actuator to safely and rapidly capture and release a live ryukin goldfish. Adapted with permission.[97] Copyright 2017, Springer Nature.
Dalaney et al. constructed a valve within a microfluidic device using photo-responsive spiropyran functionalized onto thermoresponsive pNIPAAm hydrogels.[100] Initially, the hydrogel is in a swollen state, blocking flow through the valve. Once stimulated by LED light, the photo-responsive spiropyran is photoisomerized, contracting the hydrogel to allow fluid to move through the channel. Santiello et al. similarly developed a hydrogel carbon nanotube composite valve that could be used to control flow via bending actuation.[101] Such tough materials expand the potential of creating physical models to investigate human physiological and pathological conditions, such as controllable opening and closing of body passages and orifices. Several other biomedical, hydrogel-based sensors and actuators have been explored as reviewed by Banjernee et al., however, the design and implantation of biomimetic hydrogel actuators is very much still in early developmental stages given that many systems have yet to show in vivo functionality.[88] Moreover, despite the increase in mechanical stability provided by tough hydrogels, soft actuators that demonstrate excellent fatigue resistance and self-healing properties after continuous use while maintaining optical clarity, high water content, and strong adherence to robotic parts has yet to be achieved. Further collaborative, cross-disciplinary efforts to develop mechanically stable, high water content hydrogels for robotic applications will be critical to propel such technologies into clinically ready materials.
4.2. Drug Delivery
Hydrogel delivery systems should ideally- whilst optimizing patient compliance- maintain drug bioactivity through appropriate packing, transport, and storage, while the delivery should maximize the drug’s efficacy to achieve desirable therapeutic outcomes.[102] Though there are still challenges to overcome before these systems can be clinically applied, significant progress has been made in improving drug release kinetics from hydrogels, expanding the kind of drugs which can be delivered, and the efficacy of delivery.[103] Below are some examples of tough hydrogel-based drug delivery vehicles.
4.2.1. Polyrotaxanes for Drug and Gene Delivery
Polyrotaxanes consisting of α-CD molecules and PEG chains have garnered much attention for drug delivery applications given that CDs are FDA approved and PEG has been successfully employed to conjugate biologically active agents (e.g. proteins and drugs).[104] As such, several supramolecular polymers based on cyclodextrans have been developed specifically for drug and gene transfection activity (Figure 5).[105, 106] In one such example, Liu et al. immobilized hydrophobic cinnamic acid molecules onto the terminal groups of PEG (Cin-PEG) that were then threaded into α-CDs to form polyrotaxane nanoparticles.[107] Given that the polyrotaxanes are amphiphiles, they could self-assemble into vesicle-like nanoparticles used to encapsulate the hydrophobic antitumor drug doxorubicin, thereby increasing encapsulation efficiency compared to most polymer micelles. The polyrotaxane nanoparticles were non-toxic to NIH 3T3 fibroblasts and exhibited better in vivo tumor growth suppression than nanoparticles loaded with doxorubicin hydrochloride. By further functionalizing the hydroxyl groups of α-CDs with functional moieties, polyrotaxanes can be endowed active targeting properties.[108] For example, Ooya et al. found that saccharide modified polyrotaxanes had enhanced binding affinities to targeted lectins, while Yui et al. developed self-assembling micelles with triblock copolymers as a promising as a drug delivery carrier.[109] However, the clinical translation of these polyrotaxanes is limited by their poor degradability, unfavorable biocompatibility, and complicated synthesis routes of block copolymers. To remedy these limitations, Liu et al. recently developed α-CD and PEG polyrotaxane capped with cholic acid to load the anticancer drug doxorubicin, which interestingly exhibited selective recognition with cancer cells 4T1 via their high level of expression of glucose transporters.[110]
Figure 5. Schematic illustrations of synthesis routes for various polyrotaxanes used for drug and gene delivery.

Schematic illustration of the self-assembly (a) Fabrication of cationic polyrotaxanes with multiple β-CD rings featuring OEI grafting. (b) α-CD structure and polyrotaxane synthesis using α-CD and PEO-diamine. (c) α-CD–OEI star polymer fabrication process and structures. Adapted with permission.[106] Copyright 2008, Elsevier.
4.2.2. Composite Hydrogels for Drug Delivery
Considerable research efforts have promoted exciting progress in the use of hydrogel nanocomposite technologies for controlled drug delivery.[111] For instance, Li et al. developed an in vivo biocompatible alginate-clay nanoparticle composite for localized release of PEGylated insulin-like growth factor 1 (IGF1) mimetic protein.[112] At a neutral pH, the protein has a net positive charge and is therefore able to interact with the negatively charged surface of clay nanoparticles through electrostatic interactions. The nanoparticles enable significant loading of PEGylated IGF1 (8 mg/mL for 8% clay), while protein release – enabled by an ionic exchange—was upregulated by the low pH of injured tissues (Figure 6).[112] Choi et al. developed super tough hydrogel composites via an alginate/polyacrylamide DN embedded with mesoporous silica particles, and demonstrated the potential of such a network to exhibit on-demand BSA drug release via external mechanical stimulation.[113] Carboxymethyl guar gum (CMG)-chemically modified multiwalled carbon nanotube (MCNT) hybrid hydrogels were similarly developed for sustained trans-dermal release of diclofenac sodium an anti-inflammatory pain reliever and analgesic drug.[114]
Figure 6. Composite hydrogel with high loading and local release of IGF1.

(a) Clay composite microparticles containing Laponite nanoparticles (represented by gray discs) and adsorbed drug molecules (red spheres) localized within a biodegradable alginate network containing micro-sized basins (gray ovals) and free drug (not sequestered within the microparticle). (b) In vitro drug release profiles from composite hydrogels containing various percentages of clay in cPBS medium at a pH of 7.4. (c) In vitro drug release profiles from 6% clay composite hydrogels within release medium at different values of pH. (d) Use of drug-releasing scaffolds (green) to treat Achilles tendon injury in a rat model. (e) DAPI staining (blue) of tissue sections to examine the distribution of the fluorescein-labeled protein (red) as it changes over time in vivo. Adapted with permission.[112] Copyright 2018, WILEY-VCH.
When exposed to an external stimulus, embedded nano or micro particles can trigger material phase transitions propelling the hydrogel network to expel large amounts of water and absorbed therapeutics, subsequently delivering encapsulated drugs. For example, Servant et al. used electrosensitive poly(methylacrylic acid) (PMAA) containing multiwalled carbon nanotubes (pMWNT) for controllable pulsatile drug (i.e. sucrose) release-via reversible de-swelling of the gel matrix-upon applying an electrical field stimulation.[115] While sucrose release was sharper in response to electric fields both in vitro and in vivo, the quantity of sucrose release upon the second electrical stimulation was lower than control gels because of surface damage on the gels. Using graphene rather than pMWNT improved the antifatigue properties of the gels, allowing for large deformation and volumetric changes under applied electric fields without material degradation after repeated stimulation.[116] Similarly, a chip like device formed from a PVA matrix with graphene oxide (GO) nanosheets sustained repeated, pulsatile release of lidocaine hydrochloride, an anesthetic, with high precision under electrical field treatment.[117] Another electrically modulated chip-like device, this time composed of genipin-cross-linked carboxymethyl-hexanoyl chitosan (CHC)–silica hydrogels, supported the in vitro the anticonvulsant drug, ethosuximide.[118]
Drug delivery via optically induced stimuli from nanocomposite hydrogels has also been explored. GO sheets interconnected by hierarchical peptide- composed of pyrene at the N terminus, a glycine-alanine (GA) repeat sequence and tyrosine at the C-terminus- interactions were used to form an injectable composite for pulsatile triggered delivery: localized temperature increase from NIR irradiation results in partial unfolding of β-sheets and weakening of π–π interactions between GO and GA repeats, triggering the release of encapsulated drugs.[119] In vivo release of the anticancer drug doxorubicin (2 mg mL−1) successfully reduced tumor size in mice after a treatment course of 23 days. Similarly, poly(N-isopropylacrylamide-co-acrylamide) hydrogel with silica–gold nano-shells likewise employed NIR irradiation to deliver to doxorubicin co-delivered with dsDNA for potential applications in cancer therapies.[120] Delivery of water-insoluble paclitaxel and other hydrophobic drugs, as well as magnetically mediated composite drug delivery have also shown promise.[121]
4.2.3. Interpenetrating Network Hydrogels for Drug Delivery
Several IPN hydrogel systems have been developed towards anticancer, anti-asthmatic, antibiotic, anti-inflammatory, anti-tuberculosis and anti-hypertensive drug delivery applications, as has been extensively reviewed.[48, 122] In recent developments, Pacelli et al. developed a DN hydrogel by using a methacrylate derivative of gellan gum (GG-MA) with clay Laponite – incorporated to increase the stiffness, while reducing the swelling ability of the GG-MA hydrogel- as the brittle network and polyethylene glycol dimethacrylate (PEG-DMA) as the second soft and elastic network. [123] The DN IPN was non-cytotoxic when cultured with HUVECs. Moreover, incorporating Laponite improved the loading efficiency of the antibiotic, ofloxacin, while reducing its in vitro release rate, promoting the possibility for local, sustained release of antibiotics. Similarly, a DN composed of allylated chitosan and PNIPAAm was used to bind the anti-inflammatory drug diclofenac sodium (DCF) via electrostatic attractions between the carboxylic acid groups of DCF and amino groups on the chitosan chain.[124] The in vitro release of DCF was conducted in simulated gastric fluid (SGF) at pH 1.2, and simulated intestinal fluid (SIF) at pH 7.4, with higher dosages observed to be released at lower pH due to activation of Schiff base bonds increasing the pore size of the gel, thus promoting greater drug unloading. Another anti-inflammatory drug (i.e. Ibuprofen; (IBF)), was delivered from a poly(vinylalcohol) (PVA) poly(ethyleneglycol) (PEG) IPN crosslinked via glutaraldehyde.[125] Incorporation of solid inclusion complexes of IBF in β–cyclodextrin improved the aqueous solubility, bioavailability and dissolution properties of normally poorly soluble IBF and thus the gels were better able to support sustained drug release behavior. A different pH-responsive IPN based on cellulose and poly-dopamine was developed to deliver the hydrophobic drug, ciprofloxacin.[126] Dopamine’s active amino group can be exploited to combine with carboxyl or carbonyl groups typically found on hydrophobic drugs to form a pH-sensitive amide bond that can be used to self-release bound medication at desired sites. In vitro release experiments showed an almost three order higher drug release at pH=4 than at pH=7, demonstrating the strong acidic responsive property of the material. The drug-carried hydrogel sustained a five-day antibiotic effect on Escherichia coli, demonstrating a promising potential application of such a system to treat bacterial infections on wound surfaces. While some in vivo drug release studies have been performed with IPNs with promising results, further investigation is needed to prevent issues such a burst release/incorrect dosing or device failure, either from fibrotic engulfment or mechanical damage, while promoting long term release.[127]
4.2.4. Bioactive delivery via cellular encapsulation
Cell encapsulation involves immobilizing cells within a device that is generally surrounded by a polymeric membrane that permits the exchange of nutrients and oxygen and the egress of therapeutic protein products.[128] Significant effort has been devoted to create artificial 3D scaffolds for cell encapsulation as an alternative strategy to overcome current difficulties with whole organ graft rejection, limited availability of functional donor organs, and potentially harmful secondary side effects from the use of heavy immunosuppressive drugs.[129] When designing devices for cellular encapsulation, a careful balance must be found between biocompatibility, durability, and diffusional properties to guarantee long-term functionality of cells to, in turn, allow for long-term drug delivery for treatment of diseases. Moreover, the mechanical properties of the cell encapsulating material become critical given that hydrogels may lose their functionality and structural integrity following exposure to the host’s immune system.[130]
Truong et al. used click chemistry to prepared DN hydrogels under physiological conditions that displayed high compressive and tensile stresses without fracture or hysteresis and could be used to encapsulate human mesenchymal stem cells with excellent viability, although cultured for a short time period (48 hours).[131] Similarly employing a DN hydrogel, Zhang et al. designed and formed an alginate sericin IPN via calcium ion and glutaraldehyde crosslinking agents.[132] The IPN supported cell adhesion, mouse myoblast migration and proliferation, while showing more stable degradation kinetics compared to pure alginate hydrogels. Interestingly, the IPN hydrogels inherited sericin’s photoluminescence property with sufficient transdermal transmission that could be used for in vivo tracking. In vitro horseradish peroxidase (HRP) release from the IPN hydrogels showed promising results that such a gel could become a versatile platform for cellular and drug delivery with further optimization.
Forming a functional cellular delivery device, Zhang et al. devised resilient, high-water-content PEG-based hydrogels that could be fabricated into hydrogel tubes.[18] The covalently crosslinked, highly coiled water-swellable PEG chains endowed the material with elasticity, while the incorporated N-acryloyl glycinamide (NAGA) contained reversible, swelling resistant, dual hydrogen bonding that resisted deformation and dissipated energy during mechanical loading, thus forming a robust tough hydrogel. By optimizing the NAGA content, the devices exhibited low fibrotic response when implanted in vivo in mice, and supported in vitro MDA-MB231 cells and human islets (for five days and three days, respectively) with no loss of mechanical properties. Incorporating nanofibers to form robust hydrogel composites, An et al. developed nanofiber enabled encapsulation devices (NEEDs)- with tubular or planar geometries- for cell encapsulation.[134] Poly(caprolactam) (Nylon 6), polysulfone (PSU), polyacrylonitrile (PAN) and polycaprolactone (PCL), Polystyrene (PS) were used to form porous nanofibers that are then infiltrated by alginate, PEG-DA, collagen, or chitosan solutions via surface tension-driven wicking, forming a tough interlocked hydrogel nanofiber device. NEED was able to correct the diabetes of C57BL/6 mice with minimal fibrosis for two months when encapsulated with rat pancreatic islets. Similarly employing surface tension-driven wicking, a robust device produced from Ca2+-releasing poly(methyl methacrylate) (PMMA) coated nylon fibers was used to form a Thread-Reinforced-Alginate-Fiber-For-Islets-enCapsulation (TRAFFIC) device.[133] By coating the modified thread with a solution of islets suspended in alginate, diabetes correction was achieved in C57BL/6 mice using rat islets for three months and in SCID-Beige mice using human islets for four months, with scale-up potential and facile retrievability shown in a large animal model study (Figure 7). Mechanically robust, injectable scaffolds and tough composite hydrogels have likewise been explored for cellular-based delivery.[135]
Figure 7. TRAFFIC device for potential treatment of Type 1 Diabetes.

(a) Schematic of the design and fabrication process of the TRAFFIC device. (b) SEM images of the thread featuring nanoporous modifications uniform across the surface. (c) Fluorescent images of the thread, coated thread, and a cell-free TRAFFIC device. (d) Microscopic image of TRAFFIC device containing isolated rat islets. (e) Laparoscopic images demonstrating the device retrieval process from the trocar in a dog. Adapted with permission.[133] Copyright 2018, National Academy of Sciences.
Yet, tough hydrogel-based cell encapsulation devices are currently unable to sustain long term in vivo implantation given that incorporating cells and simply maintaining their viability in vitro often tremendously decreases the mechanical properties of the encapsulating hydrogel. This mechanical failure, in turn, reduces the functional lifetime of the device prior to achieving desired therapeutic results. Moreover, such devices are often susceptible to immune attacks from the host in response to the antigens presented by the encapsulated therapeutic cells, potentially leading to the formation of a thick fibrotic capsule around the implant that would likewise reduce its clinical lifetime. Thus, more development is warranted to formulate tough hydrogels with desirable mechanical toughness and immunomodulatory properties that can structurally protect transplanted cells for long-term clinical use.
4.2.5. Gastric Drug Release Devices
Liu et al. tested the in vivo use of triggerable tough hydrogels (TTH) as prolonged gastric resident drug depots in Yorkshire pig animal models.[136] The TTHs consist of intertwined and separately crosslinked alginate and polyacrylamide networks by stimuli-responsive bonds (ionic Ca2+ and disulfide bonds), which can be triggered to with biocompatible agents (i.e. ethylenediaminetetraacetic acid and glutathione) on demand. A single gastric administration of the TTH was able to maintain a constant blood drug concentration of lumefantrine, an antimalarial drug, for four days.[136] Insulin, rifampicin, and dimethyl sulfoxide (DMSO) were likewise deliverable with the TTH device, demonstrating that such a device could potentially be applicable for other drug delivery uses.
Inspired by the pufferfish, Liu et al. created a device composed of uperabsorbent polyacrylic acid particles encapsulated in a freeze-thawed PVA membrane.[137] The superabsorbent hydrogel facilitated rapid water uptake while the porous and antifatigue PVA membrane maintains the long-term robustness of the device (Figure 8). The hydrogel device (original size ~3 cm3) swelled to a size of ~50 cm3 after absorbing gastric fluids for 60 minutes. The device impressively retained its swollen shape for 9 to 29 days before evacuation through the pylorus. [137] Beyond applications as a gastric drug depot device, a temperature sensor was implanted in the device which could monitor and record the temperature of porcine stomachs for 29 days, demonstrating the feasibility of such a device to monitor in-situ physiological signals for an extended period of time.
Figure 8. Ingestible hydrogel device for long term gastric retention and physiological monitoring.

a) Schematic and images of the synthesis and functionalities of the hydrogel device. b) Proposed mechanism of action of the gastric-retentive hydrogel device as it enters the stomach through the esophagus as hydrogel pill, swells and is retained within the stomach for a prolonged period, and finally exits through the pylorus in the form of a shrunken capsule and small particles. c) Endoscopic images demonstrating rapid swelling of the hydrogel device as it resides within the porcine stomach. d) X-ray images demonstrating the residence of the hydrogel device within the porcine stomach before eventually being emptied into the GI tract (shown here for a period of 29 days within the stomach). Adapted with permission.[137] Copyright 2019, Springer Nature.
Wu et al. synthesized a pH-responsive, mechanically tough, biodegradable hydrogel via photoinitiated copolymerization of 2-vinyl-4,6-diamino-1,3,5-triazine (VDT) and GelMA.[138] The excellent mechanical properties of PVDT-GelMA hydrogel -compressive strength 7.24 MPa, tensile stress 1.87 MPa, and compressive strength 13.9 MPa at pH 7.4 -deteriorated when placed in simulated gastric fluid solution (pH 1.2) due to disruption of diaminotriazine (DAT)-DAT hydrogen bonding: protonation of DAT leads to an increased swelling degree from an increase in charged DAT moieties, thereby loosening the polymer network and decreasing mechanical properties. When implanted in a rabbit model experiment, the PVDT-GelMA hydrogel resided in the stomach for 48 hours before being completely degraded, with no in vivo toxicity report from the metabolites.
4.3. Tough Hydrogel Adhesives
Hydrogel adhesion is dependent on anchoring stretchy polymer networks onto substrates, with high interfacial strength and capacity for energy dissipation being key to enhance interfacial toughness.[139] In addition to forming strong interfacial bonds, the adhesive must maintain cohesive strength after application throughout its service lifespan (Figure 9). [140] In terms of applications, surgical adhesives have emerged as an appealing alternative to conventional closure techniques (e.g. sutures, tacks, or stables) due to advantageous features including ease of application, reduction in surgical time and complications (e.g. infections), hemostasis, and no removal requirement. [141] However, commercially available tissue adhesives (e.g. fibrin glues, collagen adhesives) are expensive, exhibit relatively poor mechanical and tissue-bonding properties, and risk being pro-inflammatory given that many are protein-based, thereby propelling ongoing efforts to develop robust adhesives with improved biocompatibility and mechanical properties than those currently commercially available.[142] Several excellent reviews have evaluated the biomechanical/chemical properties of adhesives for biomedical applications.[140, 143] In this section, protein-based, polysaccharide based, and synthetic tough hydrogel adhesives are briefly reviewed.
Figure 9. Interfacial bonds employed by bioadhesives.

(a-d) Various interfacial crosslinking chemistries using NHS-activated ester, isocyanate, aldehyde, and catechol with nucleophilic functional groups abundant on soft tissue surfaces in order to functionalize tissue with adhesives. (e) Adhesive functionalization utilizing catechol’s ability to form coordination bonds with metal oxide surfaces (potential application to surface functionalization of implants or devices). Reproduced with permission.[140] Copyright 2019, WILEY-VCH
4.3.1. Protein-Based Adhesives
Albumin, an abundant blood serum protein, has been widely explored as an adhesive. In recent efforts, an adhesive termed BCD composed of albumin, citrate acid, and dopamine was synthesized using a two-step EDC/NHS coupling reaction.[144] The biocompatible adhesive showed 10-fold greater wet adhesion than commercial fibrin glue, and in rat mastectomy model and rat hemorrhaging liver model it was shown that the BCD adhesive could successfully be used for seroma prevention and in vivo hemostatic usage. The adhesive also exhibited a controllable degradation rate (1–25 days), but the use of PEG resulted in excessive swelling of the hydrogel adhesive. Using hydrophobic 1,8-octanediol instead resulted in stronger adhesion with improved control over the water content and thus swelling properties of the adhesive.[145] BioGlue® and ProGel® are currently two FDA-approved albumin-based hydrogel adhesives. [146] BioGlue® -made of bovine albumin and glutaraldehyde- is used to repair acute thoracic aortic dissections and leak sealing in large blood vessels in conjunction to sutures and staples, while ProGel® -made with non-toxic poly(ethylene glycol) disuccinimidyl succinate instead of glutaraldehyde- is used to seal leaks in surgical lung resection.[147] Gelatin, a protein prepared from collagen, has also been explored as a hydrogel for use as a tough adhesive.[148, 149] However, a primary concern of using protein-based hydrogel adhesives remains that they may be vectors for allergic reactions and infectious diseases given that the proteins are typically derived from animal sources.[147] Moreover, primary crosslinkers used with these adhesives (e.g. formaldehyde and glutaraldehyde) have been shown to have cytotoxic side effects.[147]
4.3.2. Polysaccharide-Based Adhesives
Using polysaccharide-based hydrogel adhesives is of interest given that polysaccharides are biocompatible, biodegradable, and induce minimal immune responses.[147] Chitosan is a natural polysaccharide that has been greatly explored for wound closure, hemostatic applications, and as mucoadhesive materials when combined with catechol-containing moieties.[150] A dextran aldehyde-based tissue adhesive was applied to a 5 mm corneal incision on an enucleated rabbit eye and successfully sealed the corneal incisions to pressures of >10 psi (500 mmHg).[148] A dually crosslinked methacrylated gelatin dopamine (GMD) hydrogel has also shown promise as a polysaccharide based adhesive.[151] A concern with using polysaccharides, however, is that they are poorly soluble in aqueous solutions and thus need fuctionalization along the backbone to introduce water-soluble moieties.[147] Likewise, when cross-linked via imine groups, hydrolysis is rapid and thus adhesion strength is often reduced prior to proper wound healing, preventing their use in applications where long adhesion times are necessary.
4.3.3. Synthetic Polymer-Based Adhesives
Given the limitations of polysaccharide and protein-based adhesives, extensive efforts have been dedicated to the exploration of synthetic polymer-based adhesives. Tunable mechanical and adhesive properties have enabled investigators to design several novel hydrogels via selective tailoring of cross-linking agents and optimization of gelation time that are biocompatible, biodegradable, and mechanically robust during their service lifetime.
DOPA Chemistry-Based Adhesives
Mussels secrete specialized adhesive proteins containing a high content of the catecholic amino acid 3,4-dihydroxy-L-phenylalanine (DOPA), which can form extremely strong (dissociation force 800 pN) and reversible bonds with surfaces.[152] Moreover, the catechol group exhibits highly dynamic crosslinking given that it can form non-covalent metallo-catecholate complexes with multivalent metal ions (e.g. Fe3+, Cu2+, and Ti3+) and can be oxidized into o-quinone under alkaline or oxidation environment, forming irreversible covalent crosslinks with biological surfaces via amino groups on tissues.[144] As such, catechol-based adhesives provide an attractive option to synthesize tough hydrogels with strong wet adhesive properties.
Fan et al. developed a double-crosslinked tissue adhesive (DCTA) mimicking the formation of byssal cuticles in mussels.[153] By grafting dopamine onto a gelatin backbone via EDC/NHS chemistry, gluing is rapidly achieved through catechol-Fe3+ coordination complexation on tissues, while genipin crosslinking provides stable, long term covalent crosslinks through primary amino groups found in gelatin. The double crosslinking mechanism enhances the hydrogel’s toughness and adhesive properties, endowing it with higher wet tissue adhesion to porcine skin and cartilage compared to commercially available fibrin glue. Likewise, employing catechol-metal ion coordination, Gao et al. developed dopamine-functionalized hydrophobic association polyacrylamide hydrogels.[154] Reversible crosslinking points between catechol-Fe3+ complexes and disentanglement of hydrophobic segments from embedded micelles effectively dissipated large amounts of energy to endow the hydrogel with tough mechanical properties. The hydrogel also demonstrated temperature and pH-dependent self-repairing behavior and exhibited excellent adhesion to various mice tissues via catechol-matrix hydrogen bonding.
A pH-responsive adhesive was developed by copolymerizing dopamine methacrylamide (DMA) and 3-acrylamido phenylboronic acid (AAPBA).[155] To trigger catechol-boronate complexation via changes in pH, the oxidation state and thus adhesive strength of the catechol side chain of DMA was controlled by adding acrylic acid: incorporating acrylic acid shifted the catechol-boronate complexation to a more basic pH, allowing the adhesive to function at physiological relevant pH ranges.[156] Regarding injectable adhesives, a tough adhesive was developed by using citric acid to provide pendant reactive carboxyl groups to conjugate dopamine and prepare biodegradable polyesters with PEG.[157] Likewise, a series of tough, biocompatible, injectable, and self-healing hydrogels were designed via in situ crosslinking of hydrazide-modified poly (L-glutamic acid) (PLGA-ADH) and dually functionalized alginate (catechol- and aldehyde-modified alginate, ALG-CHO-Catechol).[158] The adhesive supported the culture of adipose stem cells and demonstrated hemostatic capacities in vivo.
Composite Adhesives
Feng et al. developed a mechanically robust, PEG-catechol adhesive containing collagen and embedded hydroxyapatite nanoparticles.[159] The ionic nanoparticles function as multivalent crosslinkers that bind with dopamine end groups in the PEG chain while the catechol units react with nucleophiles on collagen to form an overall tough, interconnected network. The material showed tunable adhesion and when testing the adhesive strength on wet/blood-covered porcine skin, the adhesive outperformed commercially available cyanoacrylate (six-fold) and fibrin (12-fold) based adhesives, as determined by adhesion strength calculated from the maximal of the stress vs. strain curves. An injectable, synthetic nano-silicate (i.e. Laponite) PEG-catechol-based adhesive likewise enhanced the mechanical strength and toughness of the hydrogel while promoting cellular infiltration at the device tissue interface.[160] However, the formation of the thick fibroblast may limit the applications of such an adhesive for tissue repair requiring rich vasculature.
Han et al. developed a tough polydopamine-clay polyacrylamide (PDA-clay-PAM) hydrogel adhesive where limited oxidation of DA between clay nanosheets is used to control the amount of free catechol groups used for strong interfacial binding.[161] A chitosan (CS)/GO and polydopamine (PDA) composite hydrogel with electroconductive properties enhanced the cell viability and proliferation of human embryonic stem cell-derived fibroblasts and cardiomyocytes (CMs) compared to controls, portending this adhesive’s potential application in electroactive tissue engineering applications. [162]
Li et al. developed an oyster inspired mineral crosslinked polyelectrolyte hydrogel adhesive.[163] The organic-inorganic hybrid hydrogel material is formed via crosslinking of polyacrylic acid via calcium carbonate nanoparticles, with enhanced adhesive strength achieved by incorporating secondary inorganic nanoparticle crosslinkers (i.e. gold, Fe3O4, Laponite, or Cu2O nanoparticles). The formulated hydrogel was optically clear, injectable, and showed comparable adhesive performance to dopamine-based adhesives for both wet and dry conditions. Aluminum hydroxide nanocomposite hydrogels (Al-NC gels), tannic acid-coated cellulose nanocrystals composites, and PEG hydrogels embedded with nanoparticle composites have also been explored to formulate composite hydrogels with strong adhesive properties towards clinical applications.[164]
Interpenetrating Network Adhesives
Mooney’s group developed a bilayered tough adhesive: (i) an interpenetrating, charged polymer adhesive and (ii) a dissipative matrix (e.g. Alg-PAAm).[165] The bridging polymer (e.g. chitosan, polyallylamine (PAA), polyethylenimine, collagen, and gelatin) can form bonds with tissues via electrostatic interactions, covalent bonds, and physical interpenetration with coupling agents (i.e. N-hydroxysulfosuccinimide and 1-ethyl-3-(3 dimethylaminopropyl)carbodiimide)) facilitating covalent bond formation, thereby forming strong adhesions with interfacial toughness over 1000 J m−2.[165] The adhesives strongly adhered to multiple porcine tissues paving the way for exciting potential applications of this adhesive for wound dressings and/or tissue repair (Figure 10).
Figure 10. Tough adhesive for adhesion onto wet and dynamic surfaces.

(a) Schematic of adhesive system consisting of an adhesive surface featuring a bridging polymer with primary amines (green lines), and a dissipative matrix constructed with hydrogel containing both ionically cross-linked (with calcium, as represented by red circles) and covalently cross-linked polymers (represented by black and blue lines). Upon crack formation and propagation, a process zone (represented by orange area) is able to dissipate significant amounts of energy as the ionic cross-links break. (b) Use of the TA as a cardiac tissue sealant to prevent leakage as the porcine heart undergoes inflation. (c) Adherence of TA to the liver as it remains bonded while being stretched out to 14 times its initial length (λ). Scale bars, 20 mm. (d) In vivo implementation of TA on blood-exposed surface of beating porcine heart. Adapted with permission.[165] Copyright 2017, Science.
An IPN of chondroitin sulfate–polyethylene glycol (CS–PEG) crosslinked by six arm PEG amine formed a robust adhesive that could covalently bind to proteins on tissues via amide bonds. [166] The adhesive induced a minor inflammatory response following subcutaneous implantation in rat models and demonstrated and ideal in vitro enzymatic degradation profile. Moreover, the material adhesive can be combined with intraoperative biologics (IOB) (e.g. bone marrow and platelet-rich plasma) to form hydrogels with promising applications for musculoskeletal tissue repair.[167] Similarly, a 4-azidobenzoic acid-modified chitosan and PEG semi- interpenetrating hydrogel precursor was applied to and directly photo-crosslinked onto nerve stumps, forming a strong in situ adhesive that can be used for anastomosing and stabilizing the injured nerves.[168] These IPN tough hydrogel adhesives show tremendous potential to be used as tissue adhesives and hemostatic dressings with further development and optimization.
Overall, several advances have been made in the use of bioinspired adhesives to form robust and durable bonds between the adhesive and substrate, which has enabled promising advances in the medical field. However, achieving similarly strong adhesions under “wet” conditions (e.g. aqueous and blood environments) remains quite challenging. In general, achieving robust wet adhesion is limited due to the presence of a hydrated water film that severely prevents chemical contact between the substrate and adhesive, thereby tremendously reducing the surface energy the adherent provides for adhesion.[169] This greatly reduces the wet adhesion strength of the adhesive and in some cases even eliminates it completely. To address these challenges, Cui et al. recently proposed a hyperbranched polymer adhesive, where when coming into contact with water, the hydrophobic chains self-aggregated to form coacervates, quickly displacing water molecules on the surface, thereby allowing exposed catechol groups to form strong adhesions in both wet and dry conditions.[170] The adhesive was biodegradable, biocompatible, and functioned as an injectable sealant for hemostasis of deep wound bleeding in pig and rat in vivo models. Yuk et al. likewise developed an adhesive capable of displacing water from the binding surface via a dry, double-sided tape (DST) made from gelatin or chitosan and crosslinked PAA grafted with N-hydrosuccinimide ester (Figure 11).[171] The DST achieved strong adhesion to wet, dynamic tissues in ex vivo porcine, in vivo rat, and in vitro mouse models. While these works are quite promising, further efforts are required to develop adhesives that can usefully serve as tissue adhesive and sealant or in adhering implantable and wearable devices to wet tissues.
Figure 11. Tissue adhesive fabricated via diffusion-bonding and dry-crosslinking.

(a) Schematic of application of liquid tissue adhesive to skin surface via existing diffusion-based process. (b) Schematic of existing adhesive fabrication process relying primarily on passive transport of monomers/polymers towards tissue. (c) Schematic of proposed tissue adhesive applied as a two-sided “tape.” (d) Schematic of DST fabrication via dry-crosslinking, which combines interfacial water drying and dry DST swelling, temporary crosslinking, and covalent bond formation between amine groups available on the tissue surface and the DST. Adapted with permission.[171] Copyright 2019, Springer Nature.
4.4. Tough Hydrogel Coatings
Medical devices play an essential part in the treatment of infirmities given that they are often used to replace and sometimes restore biological function. Yet, synthetic materials used for orthopedics, catheters, infusion lines, vascular stents and grafts, and sutures often trigger a foreign body response (FBR) that results in biofouling, thereby limiting the clinical lifetime of the device.[172] Given the costly complications of having to replace fouled medical devices, designing devices with surfaces that can prevent fouling while supporting the device’s integration with the surrounding environment is of great interest. Using both passive materials such as hydrophilic surfaces that form a physical hydration layer barrier, hydrophobic surfaces to repel biomolecule attachment, zwitterionic coatings that maintain a charge-neutral surface while forming a tight hydration layer, and/or peptide-functionalized polymers to impart antimicrobial activity is a common technique to modify surfaces of medical devices to minimize biofouling.[173] Using active therapeutic materials (e.g. drug-eluting surfaces and nitric oxide release materials) have also been reported for surface modification purposes.[174] In another effort, a biomimetic adhesive was formulated by coating gecko-mimetic PDMS pillars with a mussel mimetic poly-(dopamine methacrylamideco-methoxyethyl acrylate) p(DMA-co-MEA) polymer film.[152] However, grafted polymers are damage prone and even may even peel off due to shearing or abrasions, coatings usually suffer from poor hydrogel–substrate adhesion, and coatings have yet to provide tissue matching properties including high water content or mechanical compliance.[175]
To improve the robustness of tough hydrogel-surface anchoring, Kurokawa et al. employed a DN formation technique to anchor PAMPS/PAAm DN gels onto surfaces (glass, polyethylene, and sponge), however, such a technique is limited to porous substrates for binding to occur.[177] To overcome this limitation, adhering a particle gel-based double network (P-DN) hydrogel to nonporous solid surfaces was recently developed.[178] To bind the hydrogel to the desired surface (e.g., plastics, rubbers, ceramics, and metals), a primer layer containing the radical initiators poly(vinyl acetate) (PVAc) and benzophenone is first used to coat the surface to form a strong bonding interface.[178] After that, a pre-gel solution can be applied to the treated surface, followed by photo-induced polymerization, thereby forming a thin and robust tough hydrogel coating with a bonding strength of 1000 J m−2. This technique was adopted from Zhao’s group, who treated elastomer surfaces with benzophenone to form robust elastomer/hydrogel hybrid interfaces with strong interfacial toughness (over 1000 J m−2) (Figure 12).[176] They likewise were able to anchor long chains of PAAm and polyethylene glycol diacrylate (PEGDA) to various modified surfaces including glass, silicon, titanium, aluminum, and mica ceramics.[179] Robust hydrogel–elastomer–hydrogel laminates, using adhesive dispersion techniques, and acryloylated surfaces are a few other approaches used to form tough, hydrogel coated surfaces, helping to solve previous issues of poor hydrogel substrate adhesions.[180]
Figure 12. Elastomer/hydrogel hybrids with strong interfacial toughness.

(a) Schematic of the fabrication and synthesis of a microstructures hydrogel-elastomer hybrid. Following their individual production, the hydrogel and elastomer are combined and subsequently treated with ultraviolet irradiation in order to initiate further polymerization. Due to the covalently anchored polymer network in the hydrogel following ultraviolet irradiation, the resultant hydrogel–elastomer hybrid creates a robust and resilient interface on the elastomer surface. Further, microstructure patterns in the elastomers and hydrogels are also maintained and interfacial failure of the hybrid is avoided. (b) PAAm-alginate hydrogel bonded to the Ecoflex elastomer surface is capable of resisting large deformations (stretch ∼7) without undergoing debonding from the surface. Further, the bonding between the elastomer and hydrogel remains intact following fracture of the hybrid. Reproduced with permission.[176] Copyright 2016, Springer Nature.
Expanding on these techniques, both Yu et al. and Yong et al. developed methods to coat thin, conformal layers of tough hydrogels onto commercially available catheters. [175, 181] Yong et al. used a three-step process (e.g. shape-forming, gradient cross-linking, and swell-peeling) to coat a thin layer of tough, biocidal, and antifouling AAm−Agar−SBMA−HA gel that increased the lubricity and significantly reduced biofilm formation (90%) of the coated catheter compared to uncoated commercial catheters.[181] Yu et al. formed multifunctional “hydrogel skins” on various substrates by employing benzophenone to robustly attach a variety of hydrogels (e.g., AAm, AA, DMAA, VP, and HEMA) that likewise led to low-friction, antifouling, and ionically conductive surfaces without compromising original mechanical properties and geometries (Figure 13).[175] While there still remains no technique to bind materials (e.g. elastomers) onto hydrogels, these advances show promise to formulate robust coatings with tough hydrogels that can potentially be used in clinical settings to improve the lifetime of medical devices.
Figure 13. Strategies used to coat thin, robust hydrogel layers onto commercially available catheters.


(a) Schematic of hydrogel skin fabrication steps. Polymer-based substrates are first exposed to an organic hydrophobic initiator solution before undergoing immersion into an aqueous solution containing hydrogel monomers and hydrophilic initiators. Following curing and washing steps, thin and uniform hydrogel skins are developed on the polymer substrate via the surface-bound formation of hydrogel-polymer interpenetrating networks. (b) Images demonstrating the application of hydrogel skins onto medical devices. (c) Schematic of fabrication procedure implemented to produce conformal and lubricating hydrogel coatings on a catheter surface. (d) Images of a catheter and inflated balloon tip coated with hydrogel via fabrication using SGS. (a-b) Adapted with permission.[175] Copyright 2018 WILEY-VCH. (c-d) Adapted with permission.[181] Copyright 2019, American Chemical Society.
4.5. Tissue Engineering
The extracellular matrix (ECM) is a hydrophilic 3D micro-matrix of natural tissues that provides structural and biochemical support to cells and is thus is a substantial guide in the design of scaffolds for tissue engineering.[182] Given that hydrogels are both soft and rubbery in their swollen state and demonstrate idyllic biocompatibility, their inherent similarity to the ECM makes them ideal candidates for tissue engineering applications. Moreover, facile control over the shape, porosity, surface morphology, and size of the hydrogel provides the opportunity to fine-tune bio-active scaffolds that mimic native tissue architecture while maintaining providing an ideal environment to support cell survival. Tough hydrogels offer an additional advantage given that their superior mechanical properties (tensile strength of 0.1–1 MPa and fracture energy of 102-103 Jm−2) better match those of native tissues, such as cartilage and tendons, that possess high toughness (1,000 J) and strength (30 MPa).[3, 14] In the following section, tough hydrogels for potential applications in cartilage, cardiovascular, and corneal tissue engineering applications are reviewed.
4.5.1. Cartilage Tissue Engineering
Articular cartilage is a thin, multilayered, hypocellular, avascular tissue whose primary function is to provide a lubricated surface for low friction articulation and facilitate load transmission to the subchondral bone.[183–185] The tissue’s critical wear resistance properties stems from a highly synchronized relationship between proteoglycan aggregates, the collagen matrix, and the surrounding interstitial fluid; entrapment of negatively charged glycosaminoglycans increases the tissue’s osmolarity, thereby attracting water which leads to an increased swelling pressure that is countered by the tensile strength provided by the dense collagen matrix. [65, 183] Yet, given that cartilage tissue is devoid of blood, nerve, and lymphatic supply, has a sparse distribution of chondrocytes, and is continuously subjected to harsh mechanical loading, damaged articular cartilage has incredibly limited regenerative capacities.[184, 186] Moreover, the success of current therapeutic options, which overarchingly fall into either palliative, reparative, and restorative therapies, have often been hindered due to complex surgical procedures with high risks of post-operative infections, donor site morbidity, limited availability of donors, poor integration with surrounding native tissue, and formation of fibrocartilage rather than hyaline cartilage, etc.[186] Thus, the need to develop biocompatible materials with mechanical properties comparable to those of cartilage tissue that demonstrate tunable degradability, facilitate new ECM deposition by maturing cells, and provide scaffolding support without loss of mechanical strength are of great interest.
Acellular Cartilage Scaffolds
Poly(2-acrylamide-2-methyl-propane sulfonic acid) (PAMPS)/poly(N,N’-dimethyl acrylamide) (PDMMAm) DN tough hydrogels with comparable wear resistance to clinically available ultra-high molecular weight polyethylene were developed as a promising material for an acellular articular cartilage scaffold.[187] The excellent mechanical properties of the PAMPS/PDMMAm hydrogels were maintained and even showed a significant increase in ultimate stress and tangent modulus after six-week subcutaneous implantation in a rabbit model, all while retaining a water content above 90%.[188] The hydrogels induced a mild inflammatory response after one week, but showed similar levels of inflammation as negative controls at four and six weeks post-implantation in both para-vertebral muscle and subcutaneous tissue rabbit models [189]. An ex vivo evaluation indicated that the friction coefficient between the PAMPS/PDMMAm hydrogel and normal cartilage was lower than normal-to-normal cartilage articulation (Figure 14).[190] When implanted for four weeks in osteochondral defects, the PAMPS/PDMMAm DN gel generated a matrix rich in type 2 collagen, Aggrecan, and SOX9 expression, all indicative of hyaline-cartilage tissue generation, although expression was lower than in normal cartilage.[187] Further analysis of the gene expression profiles of the regenerated tissue showed that it was genetically similar however not identical to hyaline articular cartilage.[191]
Figure 14. A DN tough hydrogel developed as a promising material for an acellular articular cartilage repair.

(a) Image of hydrogel plug consisting of PAMPS/PDMAAm DN. (b) Schematic of proposed implantation of DN plug into an osteochondral defect within the patellofemoral joint at the femoral groove. (c) Schematic of the cross-section of the plug within the defect at the femoral groove (d) Histology revealing fibrous and bone tissues flanking the DN-gel implanted defect (left) and Safranin-O-stained areas at the gel-bone interface (right) (e) The sham operated patella (left) and the DN-gel implanted patella (right) reveal a nearly regular and natural appearance at 4 weeks. Adapted with permission.[190] Copyright 2009, WILEY-VCH.
Using a molecular stent method, Zhao et al. developed a series of tough, bioactive DN hydrogels from polyelectrolyte biopolymers (i.e. chondroitin sulfate proteoglycans, chondroitin sulfate, and sodium hyaluronate) in tandem with PDMAAm.[192] The biopolymers cause the PDMAAm to swell from an increase in ionic osmotic pressure, thereby allowing the PDMAAm precursor solution to diffuse into the primary network, forming a tough DN gel. The mechanical strength of the DN gels were similar and even superior to native cartilage, however when cultured with rabbit and human articular chondrocytes, the DN gels only showed weak adhesion.
Cellular Cartilage Scaffolds
Given that acellular scaffolds require a patient’s progenitor cells to infiltrate, propagate, and differentiate within the scaffold for appropriate tissue regeneration, incorporating stem cells or chondrocytes in the scaffold may improve the generation of functional tissue. Mesenchymal stem cells (MSCs) are often utilized in such scaffolds given their multipotent capacity to differentiate into cartilage, bone, muscle, fat, and marrow stroma with a lack of significant immunogenicity.[193] For example, autologous MSC laden polyglycolic acid-hyaluronan tough hydrogel scaffolds promoted chondrogenic lineage development in vitro and hyaline-like cartilage repair in vivo.[194] Similarly, a mechanically robust, biodegradable, fibrin/hyaluronic acid methacrylate hydrogel promoted in vitro bone marrow-derived MSCs proliferation with phenotypes ideally suited for chondrogenesis.[195] Huang et al. used a chitosan thermogel-demineralized bone matrix tough hydrogel that promoted robust MSC chondrogenic differentiation in vitro without osteogenesis or hypertrophy.[196]
To encapsulate chondrocytes, Fan et al. developed a tough, double network hydrogel by combining oligo (2,2- dimethyltrimethylene carbonate)-poly(ethylene glycol)-oligo (2,2 dimethyltrimethylene carbonate)-diacrylate chains and methacrylated hyaluronic acid.[197] The tough hydrogel showed excellent biocompatibility, comparable mechanical strength to natural cartilage, and facilitated in vitro chondrocyte proliferation and synthesis of cartilage-specific extracellular matrix. While these advances are incredibly promising, further work is needed to truly develop robust cellular-based scaffolds that can be used in practical settings given that incorporating cells and maintaining their viability in culture medium often decreases the mechanical properties of the encapsulating hydrogel.
Injectable Cartilage Scaffolds
Besides implanting preformed hydrogels, injectable hydrogels for articular cartilage are of significant interest due to their minimally invasive delivery, capacity to fill geometrically irregular defects, and ability to undergo gelation at physiological conditions.[186, 198] Various biomaterials, both synthetic and natural, have been investigated for injectable hydrogels for cartilage engineering applications, however very few are characterized as tough hydrogels.[199] Zhao et al. crosslinked poly(vinyl alcohol) via 4-carboxyphenylboronic acid in the presence of calcium ions to form mechanically tough, injectable hydrogels with high fracture energy.[200] This dual dynamic crosslinked hydrogel scaffold degraded after three months in vivo implantation in osteochondral defects of rabbit models and was replaced by calcified cartilage. Shen et al. developed porous chitosan–gelatin scaffolds with comparable compressive strength and modulus to human cartilage via an in situ precipitation method.[142] The injectable and tough hydrogel showed a degradation rate matching that of the regeneration rate of cartilage and supported in vitro human thyroid cartilage cell adhesion and growth. Boyer et al. mixed laponites with silated hydroxypropylmethyl cellulose, forming an injectable interpenetrating network that led to the formation of cartilage-like tissue when injected with chondrogenic cells after six weeks in vivo implantation in mice.[201] Other nanocomposite and IPN systems have also been explored as injectable hydrogel-based scaffolds for cartilage regeneration.[139, 202]
4.5.2. Cardiovascular Tissue Engineering
The layered and hierarchical structure of the cardiovascular system, coupled to persistent cyclic loading, innately requires its component structures to have an enormous amount of strength, flexibility and durability, while simultaneously having the adaptive capacity to accommodate changes in physical activity, growth, and pathological condition.[203] Munoz-Pinto et al. developed a collagen-PEGDA IPN that improved the stiffness, strength, physical stability, and blood compatibility of pure collagen gels.[204] The hydrogel supported the initial stages of smooth muscle cell lineage progression and demonstrated thromboresistant properties, as clot formation was qualitatively reduced and the degree of platelet adhesion was 40% lower than pure collagen controls. Peng et al. similarly reinforced collagen matrices with chitosan and showed that subcutaneously implanted collagen–chitosan matrices stimulated greater vascular growth and recruited more endothelial and angiogenic cells than the collagen-only matrix.[205] To improve the mechanical properties of cellulose constructs, a loosely chemically cross-linked gel-fabricated by synthesizing cellulose with epichlorihydrin in a LiOH/urea solution- was pre-stretched and aligned in the stretching direction to form a temporarily oriented structure.[206] Subjecting the gel to sulfuric acid to rapidly fixed the alignment and shape of the hydrogel: acid treatment destroys the alkali/urea solvent shell on the cellulose chains, thereby removing the solvent molecules from the network to lock the designed shape of the hydrogels via intermolecular hydrogen bonds between cellulose.[206] Micropattern scaffolds from these gels promoted the adhesion and orientation of neonatal rat ventricular myocyte, resulting in the ability of the cardiomyocytes to connect with each other and contract together. A bi-layered fibrinogen construct- one with smooth muscle cells to promote matrix remodeling and contractility and a cell-free layer of high fibrinogen concentration to provide mechanical strength -for tissue-engineered vascular grafts increased the mechanical properties of gels without compromising smooth muscle contractile function.[207]
In terms of injectable scaffolds, Clarkin et al. developed a composite scaffold by using gallium-based glass particles to control the gelation of alginate hydrogels and thus, enhance alginate’s mechanical properties.[208] The composite exhibited strengths over four times the strength needed to withstand hypertensive blood pressure (19 kPa), with the mechanical strength and elastic modulus increasing significantly up to 7 days due to an increase in crosslinking density caused by the continued release of ions from the glass phase. Dong et al. developed self-healing injectable hydrogels by mixing chitosan-graft-aniline tetramers and benzaldehyde capped PEG: introduction of the aniline tetramers endowed the material with antimicrobial properties, while aldehyde groups endowed the hydrogel with tissue adhesive properties.[209] The gel had a conductivity (~2 X 10−3cm s−1) comparable to that of the native myocardium, could be used to viably co-encapsulate C2C12 myoblasts and ADMSCs cardiac cells, showed a tunable release rate of H9c2 and C2C12 cells, and showed stable in vivo degradation over a period of 45 days, suggesting a promising cell therapy vehicle for cardiac tissue repair. Steele et al. developed a polymer-nanoparticle hydrogel with notable shear thinning and self-healing behavior by crosslinking hydroxyapatite (HA) by core shell PEG-PLA NPs.[210] The HA was hydrophobically functionalized with tetradecylamine, dodecylamine, or octylamine, and by further tuning the molecular weight of HA and varying the NP loading content, an over 13-fold range of hydrogel strengths could be fabricated. The reversible polymer–NP crosslinks allow the hydrogel to be easily injected through a 31-G needle without loss of mechanical integrity, demonstrating its capacity for minimally invasive delivery (Figure 15). Dual release- from the hydrogel and NP phase- of stromal cell-derived factor 1α (SDF-1α) for angiogenic therapy showed a 12-fold increase in cellular viability of HUVECs compared to the untreated groups.
Figure 15. A polymer-nanoparticle hydrogel for cardiovascular tissue engineering applications.

(a) HA–NP hydrogel development using hyaluronic acid and nanoparticles with biodegradable capabilities. (b) Images demonstrating the HA–NP hydrogel injectability as performed with a high gauge needle. Adapted with permission.[210] Copyright 2019, WILEY-VCH.
Regarding composite scaffolds, Smith et al. assembled PEG microspheres around HL-1 cardiomyocytes to produce highly porous modular scaffolds with improved mechanical strength.[211] RGD peptides were incorporated in the microsphere to promote cell adhesion.[211] When cultured in the microspheres scaffold, HL-1 cells expressed high levels proteins involved in the excitation and contraction of myocytes. [211] Such results show promising results that such a scaffold can help cells retain their cardiomyocyte phenotype and potentially be used to implant functional cardiac tissues. Kharaziha et al. electrospun biodegradable poly(glycerol sebacate) (PGS)-gelatin nanofibrous scaffolds with adjustable chemical and mechanical compositions that supported the attachment, proliferation, differentiation and alignment of neonatal rat cardiac fibroblast cells.[212] The scaffolds with 33 wt% of PGS led to optimized synchronous contractions and alignment of cardiomyoctes seeded on the scaffold. Khan et al. fabricated tubular scaffolds by coating electrospun poly(1,4 cyclohexane dimethylene isosorbide terephthalate) nanofiber-based tubes with PVA hydrogels with adequate tensile strength and biocompatibility for artificial blood vessels.[213]
Camci-Unal et al. formulated hyaluronic methacrylate-gelatin methacrylate (GelMA) hydrogels whose compressive modulus could be tuned from 5.0 ± 2.5 to 73.0 ± 11.1 kPa by varying prepolymer compositions before UV crosslinking.[214] GelMA’s cell-interactive functional groups induced HUVEC cellular spreading in the hybrid scaffolds, demonstrating a potentially promising scaffold with tunable mechanical properties for tissue engineering applications. Similarly employing the use of GelMA, Shin et al. seeded neonatal rat cardiomyocytes onto carbon nanotube (CNTs)-GelMA hydrogels, with the idea that the CNTs can form strengthen the gelatin scaffold while reducing the electrical impedance.[215] Indeed, incorporating CNTs enhanced the strength of the network (from 10 to 32 kPa) while promoting cardiomyocyte maturation and local alignment. Optimal electrophysiological function was found at a concentration of 3 mg/mL of CNT, while 5 mg/mL of CNTs demonstrated maximum protective capabilities against a cardio toxic and a cardio-inhibitor. Kageyama et al. elegantly used gold-coated cylindrical needles modified with a monolayer of oligopeptides to attach GFP-HUVECs which were then electrochemically transferred to an in situ cross-linkable gelatin-HA hydrogel, forming perfusable microchannels enveloped with endothelial cells.[216] Microfabricated electroconductive hydrogels have also been explored to develop scaffolds with advanced architectures and functionalities to be used in cardio tissue engineering applications.[217] Although significant progress has been made in the use of biomaterials to engineer substrates that mechanically support hemodynamic function while responding to various biochemical and mechanical cues, considerable work remains to optimize clinically viable cardiovascular tissue engineering therapies.
4.5.3. Corneal Tissue Engineering
The optically transparent cornea is a multilayered, avascular connective tissue that is highly innervated and serves a two-fold purpose; protecting the inner portion of the eye and providing the majority of the eye’s refractive power.[218] As such, the cornea must be mechanically robust while maintaining a fixed shape to precisely focus incoming visible light, both of which are properties dictated by the hierarchical arrangement of the corneal stroma’s constituents.[219] Currently, there are two clinically available artificial corneas, the Boston type keratoprosthesis (Boston KPro) and the osteo-odonto-KPro (OOKP).[220] The Boston KPro is a three-component implant machined from PMMA that is sutured to the eye; during assembly, donated corneal tissue is sandwiched in between a front plate containing an optical stem and a back plate punctured with 16 holes for improved nutrient exchange that are locked in place with a titanium c-ring.[220] The OOKP, on the other hand, requires a two-stage operation and uses an osteo-odonto-lamina from a donor tooth — typically canine or premolar— to fabricate a bolt-shaped structure that is fitted into a PMMA optical cylinder.[221] A buccal mucous membrane graft is used to cover the ocular surface where the implant will be inserted to allow the formation of a new blood supply and in the meantime, the device is implanted into a sub-muscular pouch. After a period of two to five months, the OOKP is removed from the submuscular pouch and inserted into the eye, with the buccal mucous membrane repositioned and sutured in place to allow the anterior part of the optical cylinder to protrude.[221] Due to the persistent need for immune suppression and antibiotics following surgery, coupled with the severe risks of complications from the procedure, such implants are only available to those who are unviable candidates for corneal grafts.[222] Thus, designing keratoprosthetics that sufficiently mimic the mechanical, optical, and diffusive properties of the cornea while maintaining structural integrity and function to replace the diseased tissue are of key interest. Given that type I collagen is the main structural component of the cornea, collagen-based hydrogels are of great interest for matrix replacement scaffolds for corneal tissue engineering. However, conventional collagen hydrogel scaffolds are mechanically weak, displaying low stiffness and strength - a factor further worsened by cell adhesion and proliferation within the scaffold- and thus require stabilization e.g. by chemical crosslinking or plastic compression.[223] Several studies have explored the use of different techniques to improve the mechanical stiffness, strength, and degradation to cell-mediated contraction of collagen-based scaffolds for corneal tissue engineering applications.[223–227]
For example, Rafat et al. developed a hybrid collagen–chitosan composite hydrogel via bifunctional crosslinkers.[225] While mechanically inferior to native corneas, the biomimetic porcine collagen corneal substitute promoted regeneration of corneal epithelium stroma and nerves, tear film, and touch sensitivity. To minimize potential transmission of infectious agents from animal-derived materials, the crosslinking strategy was adapted to incorporate human recombinant collagen (HRC) type I and III.[228] The implants demonstrated ideal suturability, stable host graft integration, and facilitated regeneration of corneal cells and nerves similar to that in allograft tissues when evaluated in mini pig models. When comparing HRC I versus HRC III, no significant difference was found in terms of mechanical properties, inflammatory responses, or mass transfer properties, however HRC III showed better optical properties.[229] In an early clinical study where the biosynthetic corneas were implanted in ten human patients through anterior partial keratosplay surgery, all implants were well integrated with observable regeneration of host corneal epithelium after 6–7 months.[142] In a two year follow up, the biosynthetic corneas remained well integrated and avascular without requiring long term steroid immunosuppression beyond prophylaxis, with vision improving in six out of ten patients and corneal sensitivity returning within the first 12 months of surgery, albeit at lower levels than those with intact innervation.[230] The regenerated epithelium was morphologically normal in all patients, however, initial delays in epithelial closure from retained sutures caused some implant thinning and fibrosis. In a four year follow up, the recombinant human collagen implants achieved stable corneal regeneration, with future efforts focused on improving visual acuity through materials with better shape retention.[231]
In a separate study with high-risk patients, a second network of 2 -methacryloxylethyl phosphocoline (MPC) was incorporated into the carbodiimide-crosslinked recombinant HRC III corneal implant.[218, 229] Patients with ulcers/scarring from infection showed the most improvement, followed by those with burns, while those with immune and degenerative disorders performed most poorly. While only three out of six patients showed significant vision improvement, touch sensitivity was restored to near normal levels for all patients.[229]
Myung et al. developed an artificial cornea composed of a PEG/PAA double network hydrogel core interpenetrated around the periphery with a microperforated PHEA layer using photolithography patterning: the two-part design was fabricated to promote epithelialization on the surface and fibroblast ingrowth in the periphery to anchor the implant to the surrounding stroma (Figure 16).[227] The implant, which was surface modified to tether a thin layer of collagen type I, demonstrated high mechanical strength, optical clarity, and nutrient permeability, fell short of the refractive strength and tensile strength of the natural cornea. Nevertheless, primary rabbit corneal epithelial cells were successfully cultured on both the core and periphery surface, which was contingent upon the presence of collagen. While the in vivo performance of this implant was not tested, the biocompatibility and optical clarity of PEG/PAA double networks when used as corneal inlay in a rabbit deep corneal stromal pocket implantation model was evaluated: a normal amount of epithelial cell layers devoid of immune cell infiltration, inflammation or neovascularization was observed after 6 months of the operation.[226] Despite these promising results, there is still a need to develop a biomaterials approach that mimics the mechanical properties of the cornea while maintaining proper optical clarity.
Figure 16. Schematic and image of a tough hydrogel used to form an artificial cornea.

(a) Image of artificial cornea consisting of a central optic region and peripheral perimeter, as patterned with photolithographically. (b) Schematic of the proposed use of a bifunctional crosslinker in order to create covalent bonds between collagen type I and the PEG/PAA and PHEA hydrogel surfaces. (c) Schematic of proposed surface epithelialization and integration of tissue within peripheral pores following implantation of the cornea. Adapted with permission.[227] Copyright 2007, Springer.
5. Conclusion
Specialty tough hydrogels have undoubtedly opened new frontiers in medical applications of biomaterials by broadening the development of novel therapies and facilitating innovative, interdisciplinary research that has been previously limited by the traditionally weak mechanical properties of conventional hydrogels. Quite simply, tough hydrogels should ideally be able to maintain their structural integrity following large deformation. To do so, one can design hydrogels with homogenous network structures to distribute evenly loads and prevent micro-crack formation, or incorporate energy-dissipating mechanisms to diffuse energy away from the crack tip to prevent crack propagation, or incorporate both mechanisms via multi-functional crosslinkers. These various strategies have produced a wide array of tough hydrogels from Tetra-PEG to DN to nano/micro composite hydrogels. Several of these tough hydrogels have shown promising preliminary applications in various medical settings, including as soft actuators, for drug delivery purposes, as tough adhesives and coatings on medical devices, and for tissue engineering applications.
Despite the tremendous progress in the field, using these tough hydrogels as clinically ready materials is still very much in its embryonic stage. This is because promising tough hydrogels often become unstable in tissue culture medium or fail after long term in vivo use. Additionally, tough hydrogel swelling in aqueous solutions often results in a reduction of mechanical stability, long term exposure to implanted devices might induce undesirable immune responses, and tunable degradation profiles- with non-cytotoxic byproducts- remains elusive, yet is critical to achieving desirable therapeutic results. Likewise, harsh fabrication conditions remain impractical, not to mention that both cell encapsulation and injectable tough hydrogels suffer from weaker mechanical properties.
As the field of tough hydrogel progresses, an interesting and promising direction the field is considering is to incorporate self-healing properties in designing hydrogels for medical applications, particularly for persistent load-bearing applications where self-healing properties are ideal. The ability to self-heal, an attractive characteristic innate to many tissues and living organisms, refers to a material’s ability to automatically repair and recuperate its original function in response to damage.[232] As reviewed by Amaral and Pasparakis, there are two main strategies to synthesize self-healing materials: (1) incorporating catalysts that re-form polymer networks when activated at the fracture point and (2) incorporating dynamic, reversible bonds in the polymer matrix such that the dissociation of such bonds increases the mobility of polymer chains in the damaged area, thereby allowing for the formation of new bonds that repair the network.[50]
Unfortunately, there have been very limited practical applications of self-healable materials given the following problems: (1) the mechanical properties of self-healing hydrogels is poor as a consequence of the reversible and weaker nature of dynamic interactions; (2) chemistries employed are often cytotoxic or require very high temperatures; (3) self-healing times are long, ranging from several hours to even a few days; (4) healing capacity deteriorates with time; (5) involved chemistries lose functionality in complex environments (e.g., biological conditions or open air) and can be incompatible with other device components.[50, 233] Moreover, many tough hydrogels suffer from fatigue failure, as resistance to fatigue crack propagation comes from intrinsic fracture energy and is thus unaffected by additional energy dissipation mechanisms introduced in tough hydrogels.[234] As a result, most self-healable materials are based on relatively weak polymeric systems.[235]
In recent efforts to address these challenges, Zhao’s group hypothesized that the anti-fatigue properties of biological tissues arise from the partial crystallinity of collagen fibers.[234] Inspired by this notion, the group introduced highly crystalline regions in PVA hydrogels via computer-aided design of electrical circuits to induce localized heat treatments in pristine hydrogels. Both mesh patterned and ring patterned highly crystalline regions on PVA hydrogels outperformed conventional synthetic hydrogels in terms of fatigue thresholds, all while maintaining a water content above 80% and low Young’s moduli. Thus, this work demonstrates that the fatigue threshold of tough hydrogels can be enhanced by the introduction and design of crystalline domains. Other groups have likewise explored the use of catechol-based tough hydrogels, nanocomposite sheets (graphene oxide, boron-nitride, etc.), and dipole-dipole/hydrogen bond reinforced DN hydrogels to produce tough hydrogels with rapid self-healing properties.[17, 232, 235, 236] Further investigation will undoubtedly lead to the development of novel strategies to produce mechanically strong hydrogels with self-healing behaviors under mild conditions.
Along the same lines, improving other aspects such as biocompatibility and functionalization with cell-binding motifs, growth factors, immunomodulatory entities, etc. is equally important and requires further research efforts. The functionalization of hydrogels can facilitate the development of artificial matrices with instructive biochemical and biophysical microenvironments that can pave the way for functional tissues and improve cell/matrix interactions. Such interdisciplinary work will undoubtedly lead to the development of specialty tough hydrogels with enhanced mechanical properties, bioactivity, and refined micro/nano-architecture for robust use in various biomedical applications.
Acknowledgments
We would like to thank Dr. Daniel T. Bowers and Alexander U. Ernst for their critical feedback on this manuscript. This work was partially supported by the National Science Foundation (SNM-1530522), Juvenile Diabetes Research Foundation (JDRF), the Hartwell Foundation, the National Institutes of Health (NIH, 1R01DK105967-01A1) and the Novo Nordisk Company.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
6 References
- [1].Kopecek J, Journal of Polymer Science Part a-Polymer Chemistry 2009, 47, 5929; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hoffman A, Advanced Drug Delivery Reviews 2012, 64, 18. [DOI] [PubMed] [Google Scholar]
- [2].Zhang Y, Khademhosseini A, Science 2017, 356, eaaf3627; [DOI] [PMC free article] [PubMed] [Google Scholar]; Peak C, Wilker J, Schmidt G, Colloid and Polymer Science 2013, 291, 2031. [Google Scholar]
- [3].Taylor D, Panhuis M, Advanced Materials 2016, 28, 9060. [DOI] [PubMed] [Google Scholar]
- [4].Kamata H, Akagi Y, Kayasuga-Kariya Y, Chung U, Sakai T, Science 2014, 343, 873. [DOI] [PubMed] [Google Scholar]
- [5].Utech S, Boccaccini A, Journal of Materials Science 2016, 51, 271. [Google Scholar]
- [6].Gong J, Katsuyama Y, Kurokawa T, Osada Y, Advanced Materials 2003, 15, 1155. [Google Scholar]
- [7].Huang T, Xu H, Jiao K, Zhu L, Brown H, Wang H, Advanced Materials 2007, 19, 1622. [Google Scholar]
- [8].Sakai T, Matsunaga T, Yamamoto Y, Ito C, Yoshida R, Suzuki S, Sasaki N, Shibayama M, Chung U, Macromolecules 2008, 41, 5379. [Google Scholar]
- [9].Tuncaboylu D, Sari M, Oppermann W, Okay O, Macromolecules 2011, 44, 4997. [Google Scholar]
- [10].Sun J, Zhao X, Illeperuma W, Chaudhuri O, Oh K, Mooney D, Vlassak J, Suo Z, Nature 2012, 489, 133; [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu Y, He W, Zhang Z, Lee BP, Gels 2018, 4, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chung H, Charaya H, Liu L, Li X, in Hybrid Organic-Inorganic Interfaces: Towards Advanced Functional Materials, Vol. 2 (Eds: Delville M-H, Taubert A), Wiley; 2017, Ch. 12, p. 535. [Google Scholar]
- [12].Costa A, Mano J, European Polymer Journal 2015, 72, 344. [Google Scholar]
- [13].Grijalvo S, Eritja R, Díaz Díaz D, Gels 2019, 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang W, Narain R, Zeng H, Frontiers in Chemistry 2018, 6, doi: 10.3389/fchem.2018.00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li J, Suo Z, Vlassak J, Journal of Materials Chemistry B 2014, 2, 6708; [DOI] [PubMed] [Google Scholar]; Appel E, del Barrio J, Loh X, Scherman O, Chemical Society Reviews 2012, 41, 6195; [DOI] [PubMed] [Google Scholar]; Huey D, Hu J, Athanasiou K, Science 2012, 338, 917; [DOI] [PMC free article] [PubMed] [Google Scholar]; Keplinger C, Sun J, Foo C, Rothemund P, Whitesides G, Suo Z, Science 2013, 341, 984. [DOI] [PubMed] [Google Scholar]
- [16].Gong J, Soft Matter 2010, 6, 2583. [Google Scholar]
- [17].Li L, Yan B, Yang J, Chen L, Zeng H, Advanced Materials 2015, 27, 1294. [DOI] [PubMed] [Google Scholar]
- [18].Zhang Y, An D, Pardo Y, Chiu A, Song W, Liu Q, Zhou F, McDonough S, Ma M, Acta Biomaterialia 2017, 53, 100. [DOI] [PubMed] [Google Scholar]
- [19].Shibayama M, Li X, Sakai T, Industrial & Engineering Chemistry Research 2018, 57, 1121. [Google Scholar]
- [20].Fu J, Panhuis M, Journal of Materials Chemistry B 2019, 7, 1523. [DOI] [PubMed] [Google Scholar]
- [21].Bai R, Yang J, Suo Z, European Journal of Mechanics a-Solids 2019, 74, 337. [Google Scholar]
- [22].Bouchbinder E, Goldman T, Fineberg J, Reports on Progress in Physics 2014, 77, 046501. [DOI] [PubMed] [Google Scholar]
- [23].Lake G, Thomas A, Proceedings of the Royal Society of London Series a-Mathematical and Physical Sciences 1967, 300, 108. [DOI] [PubMed] [Google Scholar]
- [24].Naficy S, Brown H, Razal J, Spinks G, Whitten P, Australian Journal of Chemistry 2011, 64, 1007. [Google Scholar]
- [25].Zhang T, Lin S, Yuk H, Zhao X, Extreme Mechanics Letters 2015, 4, 1. [Google Scholar]
- [26].Zhao X, Soft Matter 2014, 10, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Furukawa H, Horie K, Nozaki R, Okada M, Phys Rev E Stat Nonlin Soft Matter Phys 2003, 68, 031406. [DOI] [PubMed] [Google Scholar]
- [28].Zhao X, Proceedings of the National Academy of Sciences of the United States of America 2017, 114, 8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kamata H, Chung U, Shibayama M, Sakai T, Soft Matter 2012, 8, 6876. [Google Scholar]
- [30].Akagi Y, Matsunaga T, Shibayama M, Chung U, Sakai T, Macromolecules 2010, 43, 488. [Google Scholar]
- [31].Sakai T, Akagi Y, Matsunaga T, Kurakazu M, Chung U, Shibayama M, Macromolecular Rapid Communications 2010, 31, 1954. [DOI] [PubMed] [Google Scholar]
- [32].Nishi K, Fujii K, Katsumoto Y, Sakai T, Shibayama M, Macromolecules 2014, 47, 3274. [Google Scholar]
- [33].Sakai T, Reactive & Functional Polymers 2013, 73, 898. [Google Scholar]
- [34].Kolb H, Finn M, Sharpless K, Angewandte Chemie-International Edition 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- [35].Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason A, Hedrick J, Liao Q, Frank C, Kingsbury K, Hawker C, Chemical Communications 2006, 2774 DOI: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- [36].Xu X, Chen C, Wang Z, Wang G, Cheng S, Zhang X, Zhuo R, Journal of Polymer Science Part a-Polymer Chemistry 2008, 46, 5263; [Google Scholar]; Truong V, Tsang K, Forsythe J, Biomacromolecules 2017, 18, 757; [DOI] [PubMed] [Google Scholar]; Crescenzi V, Cornelio L, Di Meo C, Nardecchia S, Lamanna R, Biomacromolecules 2007, 8, 1844; [DOI] [PubMed] [Google Scholar]; Ossipov D, Hilborn J, Macromolecules 2006, 39, 1709; [Google Scholar]; van Dijk M, van Nostrum C, Hennink W, Rijkers D, Liskamp R, Biomacromolecules 2010, 11, 1608. [DOI] [PubMed] [Google Scholar]
- [37].Buwalda S, Vermonden T, Hennink W, Biomacromolecules 2017, 18, 316. [DOI] [PubMed] [Google Scholar]
- [38].Norisuye T, Masui N, Kida Y, Ikuta D, Kokufuta E, Ito S, Panyukov S, Shibayama M, Polymer 2002, 43, 5289. [Google Scholar]
- [39].Wang X, Wang H, Brown H, Soft Matter 2011, 7, 211. [Google Scholar]
- [40].Xu Q, Sigen A, McMichael P, Creagh-Flynn J, Zhou D, Gao Y, Li X, Wang X, Wang W, Acs Macro Letters 2018, 7, 509. [DOI] [PubMed] [Google Scholar]
- [41].Okumura Y, Ito K, Advanced Materials 2001, 13, 485. [Google Scholar]
- [42].Creton C, Macromolecules 2017, 50, 8297. [Google Scholar]
- [43].Ito K, Polymer Journal 2007, 39, 489. [Google Scholar]
- [44].Noda Y, Hayashi Y, Ito K, Journal of Applied Polymer Science 2014, 131, 40509. [Google Scholar]
- [45].WENZ G, Angewandte Chemie-International Edition 1994, 33, 803; [Google Scholar]; Loethen S, Kim J, Thompson D, Polymer Reviews 2007, 47, 383. [Google Scholar]
- [46].SPERLING L, Klempner D, Sperling L, Utracki L, Interpenetrating Polymer Networks 1994, 239, 3; [Google Scholar]; Myung D, Waters D, Wiseman M, Duhamel P, Noolandi J, Ta C, Frank C, Polymers For Advanced Technologies 2008, 19, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dragan E, Chemical Engineering Journal 2014, 243, 572. [Google Scholar]
- [48].Lohani A, Singh G, Bhattacharya S, Verma A, Journal of Drug Delivery 2014, 2014, 583612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Somya G, Nayvar P, Akanksha B, Pramod-Kumar S, Egyptian Pharmaceutical Journal 2015, 14, 75. [Google Scholar]
- [50].Amaral A, Pasparakis G, Polymer Chemistry 2017, 8, 6464. [Google Scholar]
- [51].Huang M, Furukawa H, Tanaka Y, Nakajima T, Osada Y, Gong J, Macromolecules 2007, 40, 6658; [Google Scholar]; Na Y, Kurokawa T, Katsuyama Y, Tsukeshiba H, Gong J, Osada Y, Okabe S, Karino T, Shibayama M, Macromolecules 2004, 37, 5370; [Google Scholar]; Tanaka Y, Kuwabara R, Na Y, Kurokawa T, Gong J, Osada Y, Journal of Physical Chemistry B 2005, 109, 11559. [DOI] [PubMed] [Google Scholar]
- [52].Nakajima T, Furukawa H, Tanaka Y, Kurokawa T, Osada Y, Gong J, Macromolecules 2009, 42, 2184; [Google Scholar]; Tsukeshiba H, Huang M, Na Y, Kurokawa T, Kuwabara R, Tanaka Y, Furukawa H, Osada Y, Gong J, Journal of Physical Chemistry B 2005, 109, 16304. [DOI] [PubMed] [Google Scholar]
- [53].Na Y, Tanaka Y, Kawauchi Y, Furukawa H, Sumiyoshi T, Gong J, Osada Y, Macromolecules 2006, 39, 4641. [Google Scholar]
- [54].Okumura K, Europhysics Letters 2004, 67, 470; [Google Scholar]; Brown H, Macromolecules 2007, 40, 3815; [Google Scholar]; Tanaka Y, Epl 2007, 78, 56005. [Google Scholar]
- [55].Xin H, Saricilar S, Brown H, Whitten P, Spinks G, Macromolecules 2013, 46, 6613. [Google Scholar]
- [56].Yu Q, Tanaka Y, Furukawa H, Kurokawa T, Gong J, Macromolecules 2009, 42, 3852. [Google Scholar]
- [57].Tanaka Y, Kawauchi Y, Kurokawa T, Furukawa H, Okajima T, Gong J, Macromolecular Rapid Communications 2008, 29, 1514. [Google Scholar]
- [58].Webber R, Creton C, Brown H, Gong J, Macromolecules 2007, 40, 2919. [Google Scholar]
- [59].Tominaga T, Tirumala V, Lee S, Lin E, Gong J, Wu W, Journal of Physical Chemistry B 2008, 112, 3903; [DOI] [PubMed] [Google Scholar]; Tominaga T, Tirumala V, Lin E, Gong J, Furukawa H, Osada Y, Wu W, Polymer 2007, 48, 7449. [Google Scholar]
- [60].Saito J, Furukawa H, Kurokawa T, Kuwabara R, Kuroda S, Hu J, Tanaka Y, Gong J, Kitamura N, Yasuda K, Polymer Chemistry 2011, 2, 575; [Google Scholar]; Nakajima T, Sato H, Zhao Y, Kawahara S, Kurokawa T, Sugahara K, Gong J, Advanced Functional Materials 2012, 22, 4426; [Google Scholar]; Bakarich S, Balding P, Gorkin R, Spinks G, Panhuis M, Rsc Advances 2014, 4, 38088; [Google Scholar]; Bakarich S, Panhuis M, Beirne S, Wallace G, Spinks G, Journal of Materials Chemistry B 2013, 1, 4939; [DOI] [PubMed] [Google Scholar]; Nakajima T, Takedomi N, Kurokawa T, Furukawa H, Gong J, Polymer Chemistry 2010, 1, 693; [Google Scholar]; Tang T, Takasu A, Rsc Advances 2015, 5, 819; [Google Scholar]; Chen Q, Zhu L, Zhao C, Wang Q, Zheng J, Advanced Materials 2013, 25, 4171. [DOI] [PubMed] [Google Scholar]
- [61].Chen Q, Chen H, Zhu L, Zheng J, Journal of Materials Chemistry B 2015, 3, 3654. [DOI] [PubMed] [Google Scholar]
- [62].Nakajima T, Furukawa H, Tanaka Y, Kurokawa T, Gong J, Journal of Polymer Science Part B-Polymer Physics 2011, 49, 1246; [Google Scholar]; Nakayama A, Kakugo A, Gong J, Osada Y, Takai M, Erata T, Kawano S, Advanced Functional Materials 2004, 14, 1124; [Google Scholar]; Hagiwara Y, Putra A, Kakugo A, Furukawa H, Gong J, Cellulose 2010, 17, 93; [Google Scholar]; Yang W, Furukawa H, Gong J, Advanced Materials 2008, 20, 4499; [Google Scholar]; Liang S, Yu Q, Yin H, Wu Z, Kurokawa T, Gong J, Chemical Communications 2009, 7518 DOI: 10.1039/b916581a; [DOI] [PubMed] [Google Scholar]; Haque M, Kurokawa T, Kamita G, Gong J, Macromolecules 2011, 44, 8916; [Google Scholar]; Haque M, Kamita G, Kurokawa T, Tsujii K, Gong J, Advanced Materials 2010, 22, 5110; [DOI] [PubMed] [Google Scholar]; Haque M, Kurokawa T, Gong J, Polymer 2012, 53, 1805. [Google Scholar]
- [63].Hu J, Hiwatashi K, Kurokawa T, Liang S, Wu Z, Gong J, Macromolecules 2011, 44, 7775. [Google Scholar]
- [64].He Q, Wang Z, Yan Y, Zheng J, Cai S, Extreme Mechanics Letters 2016, 9, 165; [Google Scholar]; Illeperuma W, Sun J-Y, Suo Z, Vlassak J, Extreme Mechanics Letters 2014, 1, 90; [Google Scholar]; Liao IC, Moutos FT, Estes BT, Zhao X, Guilak F, Adv Funct Mater 2013, 23, 5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yodmuang S, McNamara S, Nover A, Mandal B, Aganwal M, Kelly T, Chao P, Hung C, Kaplan D, Vunjak-Novakovic G, Acta Biomaterialia 2015, 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yang X, Abe K, Biswas S, Yano H, Cellulose 2018, 25, 6571; [Google Scholar]; Moutos F, Freed L, Guilak F, Nature Materials 2007, 6, 162; [DOI] [PubMed] [Google Scholar]; Kosik-Koziol A, Constantini M, Bolek T, Szöke K, Barbetta A, Brinchmann J, Święszkowski W, Biofabrication 2017, 9, 044105; [DOI] [PubMed] [Google Scholar]; Jordan A, Kim S, Van de Voorde K, Pokorski J, Korley L, Acs Biomaterials Science & Engineering 2017, 3, 1869; [DOI] [PubMed] [Google Scholar]; Coburn J, Gibson M, Bandalini P, Laird C, Mao H, Moroni L, Seliktar D, Elisseeff J, Smart Structures and Systems 2011, 7, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Schexnailder P, Schmidt G, Colloid and Polymer Science 2009, 287, 1. [Google Scholar]
- [68].Haraguchi K, Takehisa T, Advanced Materials 2002, 14, 1120. [Google Scholar]
- [69].Haraguchi K, Farnworth R, Ohbayashi A, Takehisa T, Macromolecules 2003, 36, 5732. [Google Scholar]
- [70].Haraguchi K, Takehisa T, Fan S, Macromolecules 2002, 35, 10162. [Google Scholar]
- [71].Haraguchi K, Takada T, Macromolecular Chemistry and Physics 2005, 206, 1530. [Google Scholar]
- [72].Haraguchi K, Li H, Matsuda K, Takehisa T, Elliott E, Macromolecules 2005, 38, 3482. [Google Scholar]
- [73].Haraguchi K, Li H, Okumura N, Macromolecules 2007, 40, 2299. [Google Scholar]
- [74].Haraguchi K, Current Opinion in Solid State & Materials Science 2007, 11, 47. [Google Scholar]
- [75].Haraguchi K, Kimura Y, Shimizu S, Soft Matter 2018, 14, 927; [DOI] [PubMed] [Google Scholar]; Haraguchi K, Uyama K, Tanimoto H, Macromolecular Rapid Communications 2011, 32, 1253. [DOI] [PubMed] [Google Scholar]
- [76].Haraguchi K, Matsuda K, Chemistry of Materials 2005, 17, 931; [Google Scholar]; Haraguchi K, Li H, Xu Y, Li G, Polymer 2016, 96, 94; [Google Scholar]; Haraguchi K, Varade D, Polymer 2014, 55, 2496. [Google Scholar]
- [77].Haraguchi K, Takehisa T, Ebato M, Biomacromolecules 2006, 7, 3267. [DOI] [PubMed] [Google Scholar]
- [78].Song F, Li X, Wang Q, Liao L, Zhang C, Journal of Biomedical Nanotechnology 2015, 11, 40; [DOI] [PubMed] [Google Scholar]; Gaharwar A, Peppas N, Khademhosseini A, Biotechnology and Bioengineering 2014, 111, 441; [DOI] [PMC free article] [PubMed] [Google Scholar]; Carrow J, Gaharwar A, Macromolecular Chemistry and Physics 2015, 216, 248. [Google Scholar]
- [79].Jiang F, Huang T, He C, Brown H, Wang H, Journal of Physical Chemistry B 2013, 117, 13679. [DOI] [PubMed] [Google Scholar]
- [80].Xu K, Tan Y, Chen Q, An H, Li W, Dong L, Wang P, Journal of Colloid and Interface Science 2010, 345, 360. [DOI] [PubMed] [Google Scholar]
- [81].He C, Jiao K, Zhang X, Xiang M, Li Z, Wang H, Soft Matter 2011, 7, 2943. [Google Scholar]
- [82].Hu J, Kurokawa T, Hiwatashi K, Nakajima T, Wu Z, Liang S, Gong J, Macromolecules 2012, 45, 5218. [Google Scholar]
- [83].Hu J, Kurokawa T, Nakajima T, Sun T, Suekama T, Wu Z, Liang S, Gong J, Macromolecules 2012, 45, 9445. [Google Scholar]
- [84].Hu J, Kurokawa T, Nakajima T, Wu Z, Liang S, Gong J, Macromolecules 2014, 47, 3587. [Google Scholar]
- [85].Meid J, Dierkes F, Cui J, Messing R, Crosby A, Schmidt A, Richtering W, Soft Matter 2012, 8, 4254. [Google Scholar]
- [86].Pich A, Adler H, Polymer International 2007, 56, 291. [Google Scholar]
- [87].Cianchetti M, Laschi C, Menciassi A, Dario P, Nature Reviews Materials 2018, 3, 143. [Google Scholar]
- [88].Banerjee H, Suhail M, Ren H, Biomimetics (Basel) 2018, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Chen D, Pei Q, Chemical Reviews 2017, 117, 11239. [DOI] [PubMed] [Google Scholar]
- [90].Sun J, Keplinger C, Whitesides G, Suo Z, Advanced Materials 2014, 26, 7608. [DOI] [PubMed] [Google Scholar]
- [91].Kim C, Lee H, Oh K, Sun J, Science 2016, 353, 682; [DOI] [PubMed] [Google Scholar]; Chen B, Lu J, Yang C, Yang J, Zhou J, Chen Y, Suo Z, Acs Applied Materials & Interfaces 2014, 6, 7840; [DOI] [PubMed] [Google Scholar]; Larson C, Peele B, Li S, Robinson S, Totaro M, Beccai L, Mazzolai B, Shepherd R, Science 2016, 351, 1071; [DOI] [PubMed] [Google Scholar]; Yang C, Chen B, Zhou J, Chen Y, Suo Z, Advanced Materials 2016, 28, 4480. [DOI] [PubMed] [Google Scholar]
- [92].Lei Z, Wang Q, Sun S, Zhu W, Wu P, Advanced Materials 2017, 29, DOI: 10.1039/b916581a. [DOI] [PubMed] [Google Scholar]
- [93].Pu X, Liu M, Chen X, Sun J, Du C, Zhang Y, Zhai J, Hu W, Wang Z, Science Advances 2017, 3, e1700015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Morales D, Palleau E, Dickey M, Velev O, Soft Matter 2014, 10, 1337. [DOI] [PubMed] [Google Scholar]
- [95].Liu Z, Calvert P, Advanced Materials 2000, 12, 288. [Google Scholar]
- [96].Li Y, Sun Y, Xiao Y, Gao G, Liu S, Zhang J, Fu J, Acs Applied Materials & Interfaces 2016, 8, 26326; [DOI] [PubMed] [Google Scholar]; Sun Y, Liu S, Du G, Gao G, Fu J, Chemical Communications 2015, 51, 8512; [DOI] [PubMed] [Google Scholar]; Sun Y, Gao G, Du G, Cheng Y, Fu J, Acs Macro Letters 2014, 3, 496. [DOI] [PubMed] [Google Scholar]
- [97].Yuk H, Lin S, Ma C, Takaffoli M, Fang N, Zhao X, Nature Communications 2017, 8, 14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Liu S, Gao G, Xiao Y, Fu J, Journal of Materials Chemistry B 2016, 4, 3239. [DOI] [PubMed] [Google Scholar]
- [99].Zheng W, An N, Yang J, Zhou J, Chen Y, Acs Applied Materials & Interfaces 2015, 7, 1758. [DOI] [PubMed] [Google Scholar]
- [100].Delaney C, McCluskey P, Coleman S, Whyte J, Kent N, Diamond D, Lab on a Chip 2017, 17, 2013. [DOI] [PubMed] [Google Scholar]
- [101].Santaniello T, Migliorini L, Locatelli E, Monaco I, Yan Y, Lenardi C, Franchini M, Milani P, Smart Materials and Structures 2017, 26, 085030. [Google Scholar]
- [102].Li J, Mooney D, Nature Reviews Materials 2016, 1, DOI: 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Hoare T, Kohane D, Polymer 2008, 49, 1993. [Google Scholar]
- [104].Yui N, Katoono R, Yamashita A, in Inclusion Polymers, Vol. 222 (Ed: Wenz G), Springer-Verlag Berlin Heiderlberg, Advanced Polymer Science 2009, Ch. 2, p. 55. [Google Scholar]
- [105].Higashi T, Chemical & Pharmaceutical Bulletin 2019, 67, 289; [DOI] [PubMed] [Google Scholar]; Badwaik V, Mondjinou Y, Kulkarni A, Liu L, Demoret A, Thompson D, Macromolecular Bioscience 2016, 16, 63; [DOI] [PMC free article] [PubMed] [Google Scholar]; Moon C, Kwon Y, Lee W, Park Y, Chang L, Yang V, Journal of Biomedical Materials Research Part a 2008, 84A, 238; [DOI] [PubMed] [Google Scholar]; Bai S, Hou M, Shi X, Chen J, Ma X, Gao Y, Wang Y, Xue P, Kang Y, Xu Z, Carbohydrate Polymers 2018, 193, 153. [DOI] [PubMed] [Google Scholar]
- [106].Li J, Loh X, Advanced Drug Delivery Reviews 2008, 60, 1000. [DOI] [PubMed] [Google Scholar]
- [107].Liu R, Lai Y, He B, Li Y, Wang G, Chang S, Gu Z, International Journal of Nanomedicine 2012, 7, 5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Hyun H, Yui N, Macromolecular Rapid Communications 2011, 32, 326; [DOI] [PubMed] [Google Scholar]; Hu X, Gao J, Luo Y, Wei T, Dong Y, Chen G, Chen H, Macromolecular Rapid Communications 2017, 38, 1700434. [DOI] [PubMed] [Google Scholar]
- [109].Ooya T, Eguchi M, Yui N, Journal of the American Chemical Society 2003, 125, 13016; [DOI] [PubMed] [Google Scholar]; Tamura A, Tanaka H, Yui N, Polymer Chemistry 2014, 5, 4511. [Google Scholar]
- [110].Liu S, Jin J, Jia Y, Wang J, Mo L, Chen X, Qi D, Chen Y, Ren L, Macromolecular Bioscience 2019, 19, 1800478. [DOI] [PubMed] [Google Scholar]
- [111].Merino S, Martin C, Kostarelos K, Prato M, Vazquez E, Acs Nano 2015, 9, 4686; [DOI] [PubMed] [Google Scholar]; Sharma G, Thakur B, Naushad M, Kumar A, Stadler F, Alfadul S, Mola G, Environmental Chemistry Letters 2018, 16, 113. [Google Scholar]
- [112].Li J, Weber E, Guth-Gundel S, Schuleit M, Kuttler A, Halleux C, Accart N, Doelemeyer A, Basler A, Tigani B, Wuersch K, Fornaro M, Kneissel M, Stafford A, Freedman B, Mooney D, Advanced Healthcare Materials 2018, 7, e1701393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Choi S, Choi Y, Jang M, Lee J, Jeong J, Kim J, Advanced Functional Materials 2017, 27, 1703826. [Google Scholar]
- [114].Giri A, Bhowmick M, Pal S, Bandyopadhyaya A, International Journal of Biological Macromolecules 2011, 49, 885. [DOI] [PubMed] [Google Scholar]
- [115].Servant A, Methven L, Williams RP, Kostarelos K, Adv Healthc Mater 2013, 2, 806. [DOI] [PubMed] [Google Scholar]
- [116].Servant A, Leon V, Jasim D, Methven L, Limousin P, Fernandez-Pacheco E, Prato M, Kostarelos K, Advanced Healthcare Materials 2014, 3, 1334. [DOI] [PubMed] [Google Scholar]
- [117].Liu H, Hu S, Chen Y, Chen S, Journal of Materials Chemistry 2012, 22, 17311. [Google Scholar]
- [118].Huang W, Lee T, Hsiao C, Chen S, Liu D, Journal of Materials Chemistry 2011, 21, 16077. [Google Scholar]
- [119].Wu J, Chen A, Qin M, Huang R, Zhang G, Xue B, Wei J, Li Y, Cao Y, Wang W, Nanoscale 2015, 7, 1655. [DOI] [PubMed] [Google Scholar]
- [120].Strong L, Dahotre S, West J, Journal of Controlled Release 2014, 178, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Bajpai A, Gupta R, Journal of Materials Science-Materials in Medicine 2011, 22, 357; [DOI] [PubMed] [Google Scholar]; Chen Y, Kang S, Yu J, Wang Y, Zhu J, Hu Z, Journal of the Mechanical Behavior of Biomedical Materials 2019, 92, 179; [DOI] [PubMed] [Google Scholar]; Abandansari H, Nabid M, Rezaei S, Niknejad H, Polymer 2014, 55, 3579; [Google Scholar]; Liu T, Wu T, Liu H, Ke B, Huang H, Jiang Z, Xie M, Journal of Applied Polymer Science 2014, 131, DOI: 10.1002/app.40438; [DOI] [Google Scholar]; Zhong D, Liu Z, Xie S, Zhang W, Zhang Y, Xue W, Journal of Applied Polymer Science 2013, 129, 767; [Google Scholar]; Gwak G, Paek S, Oh J, European Journal of Inorganic Chemistry 2012, 2012, 5269; [Google Scholar]; Watanabe K, Nishio Y, Makiura R, Nakahira A, Kojima C, International Journal of Pharmaceutics 2013, 446, 81; [DOI] [PubMed] [Google Scholar]; Ju C, Sun J, Zi P, Jin X, Zhang C, Journal of Pharmaceutical Sciences 2013, 102, 2707; [DOI] [PubMed] [Google Scholar]; Satarkar N, Hilt J, Journal of Controlled Release 2008, 130, 246; [DOI] [PubMed] [Google Scholar]; Hawkins A, Bottom C, Liang Z, Puleo D, Hilt J, Advanced Healthcare Materials 2012, 1, 96. [DOI] [PubMed] [Google Scholar]
- [122].Matricardi P, Di Meo C, Coviello T, Hennink W, Alhaique F, Advanced Drug Delivery Reviews 2013, 65, 1172; [DOI] [PubMed] [Google Scholar]; Aminabhavi T, Nadagouda M, More U, Joshi S, Kulkarni V, Noolvi M, Kulkarni P, Expert Opinion on Drug Delivery 2015, 12, 669. [DOI] [PubMed] [Google Scholar]
- [123].Pacelli S, Paolicelli P, Avitabile M, Varani G, Di Muzio L, Cesa S, Tirillo J, Bartuli C, Nardoni M, Petralito S, Adrover A, Casadei M, European Polymer Journal 2018, 104, 184. [DOI] [PubMed] [Google Scholar]
- [124].Xu S, Li H, Ding H, Fan Z, Pi P, Cheng J, Wen X, Carbohydrate Polymers 2019, 214, 8. [DOI] [PubMed] [Google Scholar]
- [125].Das S, Subuddhi U, Journal of Pharmaceutical Analysis 2019, 9, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Yan Q, Liu L, Wang T, Wang H, Colloid and Polymer Science 2019, 297, 705. [Google Scholar]
- [127].Kulkarni R, Boppana R, Mohan G, Mutalik S, Kalyane N, Journal of Colloid and Interface Science 2012, 367, 509; [DOI] [PubMed] [Google Scholar]; Kulkarni R, Mutalik S, Mangond B, Nayak U, Journal of Pharmacy and Pharmacology 2012, 64, 530; [DOI] [PubMed] [Google Scholar]; Changez M, Koul V, Krishna B, Dinda A, Choudhary V, Biomaterials 2004, 25, 139; [DOI] [PubMed] [Google Scholar]; Changez M, Koul V, Dinda A, Biomaterials 2005, 26, 2095; [DOI] [PubMed] [Google Scholar]; Jaiswal M, Naz F, Dinda A, Koul V, Biomedical Materials 2013, 8, 045004. [DOI] [PubMed] [Google Scholar]
- [128].Orive G, Santos E, Pedraz J, Hernandez R, Advanced Drug Delivery Reviews 2014, 67–68, 3. [DOI] [PubMed] [Google Scholar]
- [129].Orive G, Hernandez R, Gascon A, Calafiore R, Chang T, De Vos P, Hortelano G, Hunkeler D, Lacik I, Shapiro A, Pedraz J, Nature Medicine 2003, 9, 104; [DOI] [PubMed] [Google Scholar]; Orive G, Hernandez R, Gascon A, Calafiore R, Chang T, de Vos P, Hortelano G, Hunkeler D, Lacik I, Pedraz J, Trends in Biotechnology 2004, 22, 87; [DOI] [PubMed] [Google Scholar]; de Vos P, Bucko M, Gemeiner P, Navratil M, Svitel J, Faas M, Strand B, Skjak-Braek G, Morch Y, Vikartovska A, Lacik I, Kollarikova G, Orive G, Poncelet D, Pedraz J, Ansorge-Schumacher M, Biomaterials 2009, 30, 2559; [DOI] [PubMed] [Google Scholar]; Nicodemus G, Bryant S, Tissue Engineering Part B-Reviews 2008, 14, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Schmidt J, Rowley J, Kong H, Journal of Biomedical Materials Research Part a 2008, 87A, 1113. [DOI] [PubMed] [Google Scholar]
- [131].Truong V, Ablett M, Richardson S, Hoyland J, Dove A, Journal of the American Chemical Society 2015, 137, 1618. [DOI] [PubMed] [Google Scholar]
- [132].Zhang Y, Liu J, Huang L, Wang Z, Wang L, Scientific Reports 2015, 5, 12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].An D, Chiu A, Flanders J, Song W, Shou D, Lu Y, Grunnet L, Winkel L, Ingvorsen C, Christophersen N, Fels J, Sand F, Ji Y, Qi L, Pardo Y, Luo D, Silberstein M, Fan J, Ma M, Proceedings of the National Academy of Sciences of the United States of America 2018, 115, E263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].An D, Ji Y, Chiu A, Lu Y, Song W, Zhai L, Qi L, Luo D, Ma M, Biomaterials 2015, 37, 40. [DOI] [PubMed] [Google Scholar]
- [135].Ciriza J, del Burgo L, Virumbrales-Munoz M, Ochoa I, Fernandez L, Orive G, Hernandez R, Pedraz J, International Journal of Pharmaceutics 2015, 493, 260; [DOI] [PubMed] [Google Scholar]; Young S, Sherman S, Cooper T, Brown C, Anjum F, Hess D, Flynn L, Amsden B, Biomaterials 2018, 159, 146; [DOI] [PubMed] [Google Scholar]; Yan Y, Li M, Yang D, Wang Q, Liang F, Qu X, Qiu D, Yang Z, Biomacromolecules 2017, 18, 2128; [DOI] [PubMed] [Google Scholar]; Weaver J, Stabler C, Acta Biomaterialia 2015, 16, 136; [DOI] [PMC free article] [PubMed] [Google Scholar]; Cai L, Heilshorn S, in Abstracts of Papers of the American Chemical Society, Vol. 248, San Francisco, CA, USA: 2014. [Google Scholar]
- [136].Liu J, Pang Y, Zhang S, Cleveland C, Yin X, Booth L, Lin J, Lee Y, Mazdiyasni H, Saxton S, Kirtane A, von Erlach T, Rogner J, Langer R, Traverso G, Nature Communications 2017, 8, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Liu X, Steiger C, Lin S, Parada G, Liu J, Chan H, Yuk H, Phan N, Collins J, Tamang S, Traverso G, Zhao X, Nature Communications 2019, 10, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wu T, Xu Z, Zhang Y, Wang H, Cui C, Chang B, Feng X, Liu W, Macromolecular Materials and Engineering 2018, 303, 1800290. [Google Scholar]
- [139].Zhang T, Yuk H, Lin S, Parada G, Zhao X, Acta Mechanica Sinica 2017, 33, 543. [Google Scholar]
- [140].Pinnaratip R, Akram Bhuiyan M, Meyers K, Rajachar R, Lee B, Advanced Healthcare Materials 2019, 8, 1801568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Reece T, Maxey T, Kron I, American Journal of Surgery 2001, 182, 40S. [DOI] [PubMed] [Google Scholar]
- [142].Shen Z, Cui X, Hou R, Li Q, Deng H, Fu J, Rsc Advances 2015, 5, 55640. [Google Scholar]
- [143].Lee B, Messersmith P, Israelachvili J, Waite J, Clarke D, Fratzl P, Annual Review of Materials Research, Vol 41 2011, 41, 99; [DOI] [PMC free article] [PubMed] [Google Scholar]; Forooshani P, Lee B, Journal of Polymer Science Part a-Polymer Chemistry 2017, 55, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; Bouten P, Zonjee M, Bender J, Yauw S, van Goor H, van Hest J, Hoogenboom R, Progress in Polymer Science 2014, 39, 1375; [Google Scholar]; Duarte A, Coelho J, Bordado J, Cidade M, Gil M, Progress in Polymer Science 2012, 37, 1031. [Google Scholar]
- [144].Zhu W, Peck Y, Iqbal J, Wang D, Biomaterials 2017, 147, 99. [DOI] [PubMed] [Google Scholar]
- [145].Ji Y, Ji T, Liang K, Zhu L, Journal of Materials Science-Materials in Medicine 2016, 27, 30. [DOI] [PubMed] [Google Scholar]
- [146].SUMMARY OF SAFETY AND EFFECTIVENESS DATA ProGelTM Pleural Air Leak Sealant, https://www.accessdata.fda.gov/cdrh_docs/pdf/P010047b.pdf, accessed: June 24, 2019;; Summary of Safety and Effectiveness CryoLife, Inc., BioGlue® Surgical Adhesive, https://www.accessdata.fda.gov/cdrh_docs/pdf/P010003b.pdf, accessed: June 24, 2019.
- [147].Ghobril C, Grinstaff M, Chemical Society Reviews 2015, 44, 1820. [DOI] [PubMed] [Google Scholar]
- [148].Bhatia S, Arthur S, Chenault H, Figuly G, Kodokian G, Current Eye Research 2007, 32, 1045. [DOI] [PubMed] [Google Scholar]
- [149].BONCHEK L, BRAUNWALD N, Annals of Surgery 1967, 165, 420; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tatooles CJ, Braunwald NS, Surgery 1966, 60, 857; [PubMed] [Google Scholar]; Nomori H, Horio H, Suemasu K, Surg Today 2000, 30, 244; [DOI] [PubMed] [Google Scholar]; Chang WH, Chang Y, Lai PH, Sung HW, J Biomater Sci Polym Ed 2003, 14, 481. [DOI] [PubMed] [Google Scholar]
- [150].Ryu J, Lee Y, Kong W, Kim T, Park T, Lee H, Biomacromolecules 2011, 12, 2653; [DOI] [PubMed] [Google Scholar]; Azad A, Sermsintham N, Chandrkrachang S, Stevens W, Journal of Biomedical Materials Research Part B-Applied Biomaterials 2004, 69B, 216; [DOI] [PubMed] [Google Scholar]; Rao S, Sharma C, Journal of Biomedical Materials Research 1997, 34, 21; [DOI] [PubMed] [Google Scholar]; Kim K, Ryu J, Lee H, Biomaterials 2015, 52, 161; [DOI] [PubMed] [Google Scholar]; Xu J, Soliman G, Barralet J, Cerruti M, Langmuir 2012, 28, 14010. [DOI] [PubMed] [Google Scholar]
- [151].Kim J, Ryu S, Park K, Journal of Industrial and Engineering Chemistry 2018, 58, 105. [Google Scholar]
- [152].Lee H, Lee B, Messersmith P, Nature 2007, 448, 338. [DOI] [PubMed] [Google Scholar]
- [153].Fan C, Fu J, Zhu W, Wang DA, Acta Biomater 2016, 33, 51. [DOI] [PubMed] [Google Scholar]
- [154].Gao Z, Duan L, Yang Y, Hu W, Gao G, Applied Surface Science 2018, 427, 74. [Google Scholar]
- [155].Narkar A, Barker B, Clisch M, Jiang J, Lee B, Chemistry of Materials 2016, 28, 5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Narkar A, Lee B, Langmuir 2018, 34, 9410. [DOI] [PubMed] [Google Scholar]
- [157].Mehdizadeh M, Weng H, Gyawali D, Tang L, Yang J, Biomaterials 2012, 33, 7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Yan S, Wang W, Li X, Ren J, Yun W, Zhang K, Li G, Yin J, Journal of Materials Chemistry B 2018, 6, 6377. [DOI] [PubMed] [Google Scholar]
- [159].Feng J, Ton XA, Zhao S, Paez JI, Del Campo A, Biomimetics (Basel) 2017, 2, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Liu Y, Meng H, Konst S, Sarmiento R, Rajachar R, Lee BP, ACS Appl Mater Interfaces 2014, 6, 16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Han L, Lu X, Liu K, Wang K, Fang L, Weng L, Zhang H, Tang Y, Ren F, Zhao C, Sun G, Liang R, Li Z, Acs Nano 2017, 11, 2561. [DOI] [PubMed] [Google Scholar]
- [162].Jing X, Mi H, Napiwocki B, Peng X, Turng L, Carbon 2017, 125, 557. [Google Scholar]
- [163].Li A, Jia Y, Sun S, Xu Y, Minsky B, Stuart M, Colfen H, von Klitzing R, Guo X, Acs Applied Materials & Interfaces 2018, 10, 10471. [DOI] [PubMed] [Google Scholar]
- [164].Joseph C, McCarthy C, Tyo A, Hubbard K, Fisher H, Altscheffel J, He W, Pinnaratip R, Liu Y, Lee B, Rajachar R, Acs Biomaterials Science & Engineering 2019, 5, 959; [DOI] [PMC free article] [PubMed] [Google Scholar]; Li F, Zhang G, Wang Z, Jiang H, Yan S, Zhang L, Li H, Acs Applied Materials & Interfaces 2019, 11, 15071; [DOI] [PubMed] [Google Scholar]; Shao C, Wang M, Meng L, Chang H, Wang B, Xu F, Wang J, Wan P, Chemistry of Materials 2018, 30, 3110; [Google Scholar]; Pinnaratip R, Meng H, Rajachar RM, Lee BP, Biomed Mater 2018, 13, 025003; [DOI] [PMC free article] [PubMed] [Google Scholar]; Gaharwar A, Rivera C, Wu C, Schmidt G, Acta Biomaterialia 2011, 7, 4139; [DOI] [PubMed] [Google Scholar]; Wu C, Wilker J, Schmidt G, Macromolecular Bioscience 2013, 13, 59. [DOI] [PubMed] [Google Scholar]
- [165].Li J, Celiz A, Yang J, Yang Q, Wamala I, Whyte W, Seo B, Vasilyev N, Vlassak J, Suo Z, Mooney D, Science 2017, 357, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Strehin I, Nahas Z, Arora K, Nguyen T, Elisseeff J, Biomaterials 2010, 31, 2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Simson J, Crist J, Strehin I, Lu Q, Elisseeff J, Journal of Orthopaedic Research 2013, 31, 392. [DOI] [PubMed] [Google Scholar]
- [168].Amoozgar Z, Rickett T, Park J, Tuchek C, Shi R, Yeo Y, Acta Biomaterialia 2012, 8, 1849. [DOI] [PubMed] [Google Scholar]
- [169].Rapp MV, Maier GP, Dobbs HA, Higdon NJ, Waite JH, Butler A, Israelachvili JN, J Am Chem Soc 2016, 138, 9013; [DOI] [PubMed] [Google Scholar]; Kamino K, Nakano M, Kanai S, FEBS J 2012, 279, 1750; [DOI] [PubMed] [Google Scholar]; Ahn Y, Jang Y, Selvapalam N, Yun G, Kim K, Angew Chem Int Ed Engl 2013, 52, 3140. [DOI] [PubMed] [Google Scholar]
- [170].Cui C, Fan C, Wu Y, Xiao M, Wu T, Zhang D, Chen X, Liu B, Xu Z, Qu B, Liu W, Adv Mater 2019, e1905761 DOI: 10.1002/adma.201905761. [DOI] [PubMed] [Google Scholar]
- [171].Yuk H, Varela CE, Nabzdyk CS, Mao X, Padera RF, Roche ET, Zhao X, Nature 2019, 575, 169. [DOI] [PubMed] [Google Scholar]
- [172].Harding J, Reynolds M, Trends in Biotechnology 2014, 32, 140. [DOI] [PubMed] [Google Scholar]
- [173].Costa F, Carvalho I, Montelaro R, Gomes P, Martins M, Acta Biomaterialia 2011, 7, 1431; [DOI] [PubMed] [Google Scholar]; Francolini I, Crisante F, Martinelli A, D’Ilario L, Piozzi A, Acta Biomaterialia 2012, 8, 549; [DOI] [PubMed] [Google Scholar]; S. Chen, L. Li, C. Zhao, J. Zheng, Polymer 2010, 51, 5283; [Google Scholar]; Li J, Kao W, Biomacromolecules 2003, 4, 1055; [DOI] [PubMed] [Google Scholar]; Li J, Kleintschek T, Rieder A, Cheng Y, Baumbach T, Obst U, Schwartz T, Levkin P, Acs Applied Materials & Interfaces 2013, 5, 6704; [DOI] [PubMed] [Google Scholar]; Epstein A, Wong T, Belisle R, Boggs E, Aizenberg J, Proceedings of the National Academy of Sciences of the United States of America 2012, 109, 13182; [DOI] [PMC free article] [PubMed] [Google Scholar]; Privett B, Youn J, Hong S, Lee J, Han J, Shin J, Schoenfisch M, Langmuir 2011, 27, 9597; [DOI] [PMC free article] [PubMed] [Google Scholar]; Timofeeva L, Kleshcheva N, Applied Microbiology and Biotechnology 2011, 89, 475; [DOI] [PubMed] [Google Scholar]; Siedenbiedel F, Tiller J, Polymers 2012, 4, 46; [Google Scholar]; Jiang S, Cao Z, Advanced Materials 2010, 22, 920; [DOI] [PubMed] [Google Scholar]; Blanco C, Ortner A, Dimitrov R, Navarro A, Mendoza E, Tzanov T, Acs Applied Materials & Interfaces 2014, 6, 11385; [DOI] [PubMed] [Google Scholar]; Forbes S, McBain A, Felton-Smith S, Jowitt T, Birchenough H, Dobson C, Biomaterials 2013, 34, 5453; [DOI] [PubMed] [Google Scholar]; Waterhouse A, Wise S, Yin Y, Wu B, James B, Zreiqat H, McKenzie D, Bao S, Weiss A, Ng M, Bilek M, Biomaterials 2012, 33, 7984; [DOI] [PubMed] [Google Scholar]; Bilek M, Bax D, Kondyurin A, Yin Y, Nosworthy N, Fisher K, Waterhouse A, Weiss A, dos Remedios C, McKenzie D, Proceedings of the National Academy of Sciences of the United States of America 2011, 108, 14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Stefanini G, Holmes D, New England Journal of Medicine 2013, 368, 254; [DOI] [PubMed] [Google Scholar]; Mowery K, Schoenfisch M, Saavedra J, Keefer L, Meyerhoff M, Biomaterials 2000, 21, 9; [DOI] [PubMed] [Google Scholar]; Charville G, Hetrick E, Geer C, Schoenfisch M, Biomaterials 2008, 29, 4039; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lu Y, Slomberg D, Sun B, Schoenfisch M, Small 2013, 9, 2189; [DOI] [PubMed] [Google Scholar]; Vasilev K, Poulter N, Martinek P, Griesser H, Acs Applied Materials & Interfaces 2011, 3, 4831; [DOI] [PubMed] [Google Scholar]; Ruckh T, Oldinski R, Carroll D, Mikhova K, Bryers J, Popat K, Journal of Materials Science-Materials in Medicine 2012, 23, 1411; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kakade S, Mani G, Drug Design Development and Therapy 2013, 7, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Yu Y, Yuk H, Parada G, Wu Y, Liu X, Nabzdyk C, Youcef-Toumi K, Zang J, Zhao X, Advanced Materials 2019, 31, 1807101. [DOI] [PubMed] [Google Scholar]
- [176].Yuk H, Zhang T, Parada G, Liu X, Zhao X, Nature Communications 2016, 7, 12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Kurokawa T, Furukawa H, Wang W, Tanaka Y, Gong J, Acta Biomaterialia 2010, 6, 1353. [DOI] [PubMed] [Google Scholar]
- [178].Takahashi R, Shimano K, Okazaki H, Kurokawa T, Nakajima T, Nonoyama T, King D, Gong J, Advanced Materials Interfaces 2018, 5, DOI: 10.1002/admi.201801018. [DOI] [Google Scholar]
- [179].Yuk H, Zhang T, Lin S, Parada G, Zhao X, Nature Materials 2016, 15, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Liu X, Yin C, Yang J, Liang M, Wei J, Zhang Z, Wang H, Wang Q, Journal of Materials Chemistry a 2016, 4, 17933; [Google Scholar]; Parada G, Yuk H, Liu X, Hsieh A, Zhao X, Advanced Healthcare Materials 2017, 6, 1700520; [DOI] [PubMed] [Google Scholar]; Wirthl D, Pichler R, Drack M, Kettlguber G, Moser R, Gerstmayr R, Hartmann F, Bradt E, Kaltseis R, Siket C, Schausberger S, Hild S, Bauer S, Kaltenbrunner M, Science Advances 2017, 3, e1700053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Yong Y, Qiao M, Chiu A, Fuchs S, Liu Q, Pardo Y, Worobo R, Liu Z, Ma M, Langmuir 2019, 35, 1927. [DOI] [PubMed] [Google Scholar]
- [182].El-Sherbiny IM, Yacoub MH, Glob Cardiol Sci Pract 2013, 2013, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Ulrich-Vinther M, Maloney MD, Schwarz EM, Rosier R, O’Keefe RJ, J Am Acad Orthop Surg 2003, 11, 421. [DOI] [PubMed] [Google Scholar]
- [184].Sophia Fox AJ, Bedi A, Rodeo SA, Sports Health 2009, 1, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yang P, Temenoff J, Tissue Engineering Part B-Reviews 2009, 15, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Singh Y, Moses J, Bhardwaj N, Mandal B, Journal of Materials Chemistry B 2018, 6, 5499. [DOI] [PubMed] [Google Scholar]
- [187].Yasuda K, Gong J, Katsuyama Y, Nakayama A, Tanabe Y, Kondo E, Ueno M, Osada Y, Biomaterials 2005, 26, 4468. [DOI] [PubMed] [Google Scholar]
- [188].Azuma C, Yasuda K, Tanabe Y, Taniguro H, Kanaya F, Nakayama A, Chen Y, Gong J, Osada Y, Journal of Biomedical Materials Research Part a 2007, 81A, 373. [DOI] [PubMed] [Google Scholar]
- [189].Tanabe Y, Yasuda K, Azuma C, Taniguro H, Onodera S, Suzuki A, Chen Y, Gong J, Osada Y, Journal of Materials Science-Materials in Medicine 2008, 19, 1379. [DOI] [PubMed] [Google Scholar]
- [190].Arakaki K, Kitamura N, Fujiki H, Kurokawa T, Iwamoto M, Ueno M, Kanaya F, Osada Y, Gong J, Yasuda K, Journal of Biomedical Materials Research Part a 2010, 93A, 1160. [DOI] [PubMed] [Google Scholar]
- [191].Imabuchi R, Ohmiya Y, Kwon H, Onodera S, Kitamura N, Kurokawa T, Gong J, Yasuda K, Bmc Musculoskeletal Disorders 2011, 12, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Zhao Y, Nakajima T, Yang J, Kurokawa T, Liu J, Lu J, Mizumoto S, Sugahara K, Kitamura N, Yasuda K, Daniels A, Gong J, Advanced Materials 2014, 26, 436. [DOI] [PubMed] [Google Scholar]
- [193].Mauck RL, Yuan X, Tuan RS, Osteoarthritis Cartilage 2006, 14, 179. [DOI] [PubMed] [Google Scholar]
- [194].Patrascu J, Kruger J, Boss H, Ketzmar A, Freymann U, Sittinger M, Notter M, Endres M, Kaps C, Journal of Biomedical Materials Research Part B-Applied Biomaterials 2013, 101, 1310. [DOI] [PubMed] [Google Scholar]
- [195].Snyder TN, Madhavan K, Intrator M, Dregella RC, Park D, 2014, 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Huang H, Zhang X, Hu X, Dai L, Zhu J, Man Z, Chen H, Zhou C, Ao Y, Biomedical Materials 2014, 9, 035008. [DOI] [PubMed] [Google Scholar]
- [197].Fan C, Liao L, Zhang C, Liu L, Journal of Materials Chemistry B 2013, 1, 4251. [DOI] [PubMed] [Google Scholar]
- [198].Amini A, Nair L, Biomedical Materials 2012, 7, 024105; [DOI] [PubMed] [Google Scholar]; Liu M, Zeng X, Ma C, Yi H, Ali Z, Mou X, Li S, Deng Y, He N, Bone Research 2017, 5, 17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Li J, Chen G, Xu X, Abdou P, Jiang Q, Shi D, Gu Z, Regen Biomater 2019, 6, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Zhao Y, Li M, Liu B, Xiang J, Cui Z, Qu X, Qiu D, Tian Y, Yang Z, Journal of Materials Chemistry B 2018, 6, 1351. [DOI] [PubMed] [Google Scholar]
- [201].Boyer C, Figueiredo L, Pace R, Lesoeur J, Rouillon T, Le Visage C, Tassin J, Weiss P, Guicheux J, Rethore G, Acta Biomaterialia 2018, 65, 112. [DOI] [PubMed] [Google Scholar]
- [202].Stagnaro P, Schizzi I, Utzeri R, Marsano E, Castellano M, Carbohydrate Polymers 2018, 185, 56; [DOI] [PubMed] [Google Scholar]; Radhakrishnan J, Manigandan A, Chinnaswamy P, Subramanian A, Sethuraman S, Biomaterials 2018, 162, 82; [DOI] [PubMed] [Google Scholar]; Zhang N, Lock J, Sallee A, Liu H, Acs Applied Materials & Interfaces 2015, 7, 20987; [DOI] [PubMed] [Google Scholar]; Levett P, Hutmacher D, Malda J, Klein T, Plos One 2014, 9, e113216; [DOI] [PMC free article] [PubMed] [Google Scholar]; Pirinen S, Karvinen J, Tiitu V, Suvanto M, Pakkanen T, Journal of Applied Polymer Science 2015, 132, DOI: 10.1002/app.42272. [DOI] [Google Scholar]
- [203].Bouten C, Dankers P, Driessen-Mol A, Pedron S, Brizard A, Baaijens F, Advanced Drug Delivery Reviews 2011, 63, 221. [DOI] [PubMed] [Google Scholar]
- [204].Munoz-Pinto D, Jimenez-Vergara A, Gharat T, Hahn M, Biomaterials 2015, 40, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Deng C, Zhang P, Vulesevic B, Kuraitis D, Li F, Yang A, Griffith M, Ruel M, Suuronen E, Tissue Engineering Part a 2010, 16, 3099. [DOI] [PubMed] [Google Scholar]
- [206].Ye D, Yang P, Lei X, Zhang D, Li L, Chang C, Sun P, Zhang L, Chemistry of Materials 2018, 30, 5175. [Google Scholar]
- [207].Yao L, Liu J, Andreadis S, Pharmaceutical Research 2008, 25, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Clarkin O, Wu B, Cahill P, Brougham D, Banerjee D, Brady S, Fox E, Lally C, Carbohydrate Polymers 2019, 217, 152. [DOI] [PubMed] [Google Scholar]
- [209].Dong R, Zhao X, Guo B, Ma P, Acs Applied Materials & Interfaces 2016, 8, 17138. [DOI] [PubMed] [Google Scholar]
- [210].Steele A, Stapleton L, Farry J, Lucian H, Paulsen M, Eskandari A, Hironaka C, Thakore A, Wang H, Yu A, Chan D, Appel E, Woo Y, Advanced Healthcare Materials 2019, 8, 1801147. [DOI] [PubMed] [Google Scholar]
- [211].Smith A, Segar C, Nguyen P, MacEwan M, Efimov I, Elbert D, Acta Biomaterialia 2012, 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Kharaziha M, Nikkhah M, Shin S, Annabi N, Masoumi N, Gaharwar A, Camci-Unal G, Khademhosseini A, Biomaterials 2013, 34, 6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Khan M, Kharaghani D, Nishat N, Sanaullah A Shahzad T Yamamoto Y Inoue I Kim, Journal of Applied Polymer Science 2019, 136, 47222. [Google Scholar]
- [214].Camci-Unal G, Cuttica D, Annabi N, Demarchi D, Khademhosseini A, Biomacromolecules 2013, 14, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Shin S, Jung S, Zalabany M, Kim K, Zorlutuna P, Kim S, Nikkhah M, Khabiry M, Azize M, Kong J, Wan K, Palacios T, Dokmeci M, Bae H, Tang X, Khademhosseini A, Acs Nano 2013, 7, 2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Kageyama T, Kakegawa T, Osaki T, Enomoto J, Ito T, Nittami T, Fukuda J, Biofabrication 2014, 6, 025006. [DOI] [PubMed] [Google Scholar]
- [217].Walker B, Lara R, Mogadam E, Yu C, Kimball W, Annabi N, Progress in Polymer Science 2019, 92, 135; [DOI] [PMC free article] [PubMed] [Google Scholar]; Baei P, Jalili-Firoozinezhad S, Rajabi-Zeleti S, Tafazzoli-Shadpour M, Baharvand H, Aghdami N, Materials Science & Engineering C-Materials For Biological Applications 2016, 63, 131; [DOI] [PubMed] [Google Scholar]; Navaei A, Moore N, Sullivan R, Truong D, Migrino R, Nikkhah M, Rsc Advances 2017, 7, 3302. [Google Scholar]
- [218].Islam MM, Buznyk O, Reddy JC, Pasyechnikova N, Alarcon EI, Hayes S, Lewis P, Fagerholm P, He C, Iakymenko S, Liu W, Meek KM, Sangwan VS, Griffith M, NPJ Regen Med 2018, 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Meek KM, Knupp C, Prog Retin Eye Res 2015, 49, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Avadhanam V, Vol. 9 (Ed: H. Smith), Dovepress, Clinical Ophthalmology 2015, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Liu C, Paul B, Tandon R, Lee E, Fong K, Mavrikakis I, Throp S, Herold J, Brittain P, Francis I, Ferrett C, Hull C, Lloyd A, Green D, Franklin V, Tighe B, Fukuda M, Hamada S, Seminars in Opthalmology 2005, 20, 113. [DOI] [PubMed] [Google Scholar]
- [222].Avadhanam V, Liu C, British Journal of Ophthalmology 2015, 99, 878. [DOI] [PubMed] [Google Scholar]
- [223].Connon C, Procedia Engineering 2015, 110, 15. [Google Scholar]
- [224].Goodarzi H, Jadidi K, Pourmotabed S, Sharifi E, Aghamollaei H, International Journal of Biological Macromolecules 2019, 126, 620; [DOI] [PubMed] [Google Scholar]; Liu Y, Ren L, Wang Y, Mater Sci Eng C Mater Biol Appl 2013, 33, 196; [DOI] [PubMed] [Google Scholar]; Ghezzi C, Rnjak-Kovacina J, Kaplan D, Tissue Engineering Part B-Reviews 2015, 21, 278; [DOI] [PMC free article] [PubMed] [Google Scholar]; Oelker A, Grinstaff M, Ieee Transactions on Nanobioscience 2012, 11, 37; [DOI] [PubMed] [Google Scholar]; Islam M, Cepla V, He C, Edin J, Rakickas T, Kobuch K, Ruzele Z, Jackson W, Rafat M, Lohmann C, Valiokas R, Griffith M, Acta Biomaterialia 2015, 12, 70; [DOI] [PubMed] [Google Scholar]; Liu Y, Ren L, Wang Y, Applied Surface Science 2014, 301, 396. [Google Scholar]
- [225].Rafat M, Li F, Fagerholm P, Lagali N, Watsky M, Munger R, Matsuura T, Griffith M, Biomaterials 2008, 29, 3960. [DOI] [PubMed] [Google Scholar]
- [226].Tan X, Hartman L, Tan K, Poh R, Myung D, Zheng L, Waters D, Noolandi J, Beuerman R, Frank C, Ta C, Tan D, Mehta J, Journal of Materials Science-Materials in Medicine 2013, 24, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Myung D, Koh W, Bakri A, Zhang F, Marshall A, Ko J, Noolandi J, Carrasco M, Cochran J, Frank C, Ta C, Biomedical Microdevices 2007, 9, 911. [DOI] [PubMed] [Google Scholar]
- [228].Liu W, Merrett K, Griffith M, Fagerholm P, Dravida S, Heyne B, Scaiano JC, Watsky MA, Shinozaki N, Lagali N, Munger R, Li F, Biomaterials 2008, 29, 1147. [DOI] [PubMed] [Google Scholar]
- [229].Merrett K, Fagerholm P, McLaughlin CR, Dravida S, Lagali N, Shinozaki N, Watsky MA, Munger R, Kato Y, Li F, Marmo CJ, Griffith M, Invest Ophthalmol Vis Sci 2008, 49, 3887. [DOI] [PubMed] [Google Scholar]
- [230].Fagerholm P, Lagali NS, Merrett K, Jackson WB, Munger R, Liu Y, Polarek JW, Söderqvist M, Griffith M, Sci Transl Med 2010, 2, 46ra61. [DOI] [PubMed] [Google Scholar]
- [231].Fagerholm P, Lagali N, Carlsson D, Merrett K, Griffith M, Cts-Clinical and Translational Science 2014, 7, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Cong H, Wang P, Yu S, Chemistry of Materials 2013, 25, 3357. [Google Scholar]
- [233].Azevedo S, Costa AMS, Andersen A, Choi IS, Birkedal H, Mano JF, Adv Mater 2017, 29. [DOI] [PubMed] [Google Scholar]
- [234].Lin S, Liu X, Liu J, Yuk H, Loh HC, Parada GA, Settens C, Song J, Masic A, McKinley GH, Zhao X, Sci Adv 2019, 5, eaau8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Tong X, Du L, Xu Q, Journal of Materials Chemistry a 2018, 6, 3091. [Google Scholar]
- [236].Azevedo S, Costa AMS, Andersen A, Choi IS, Birkedal H, Mano JF, Adv Mater 2017, 29, 1700759; [DOI] [PubMed] [Google Scholar]; Jia H, Huang Z, Fei Z, Dyson P, Zheng Z, Wang X, Acs Applied Materials & Interfaces 2016, 8, 31339. [DOI] [PubMed] [Google Scholar]