

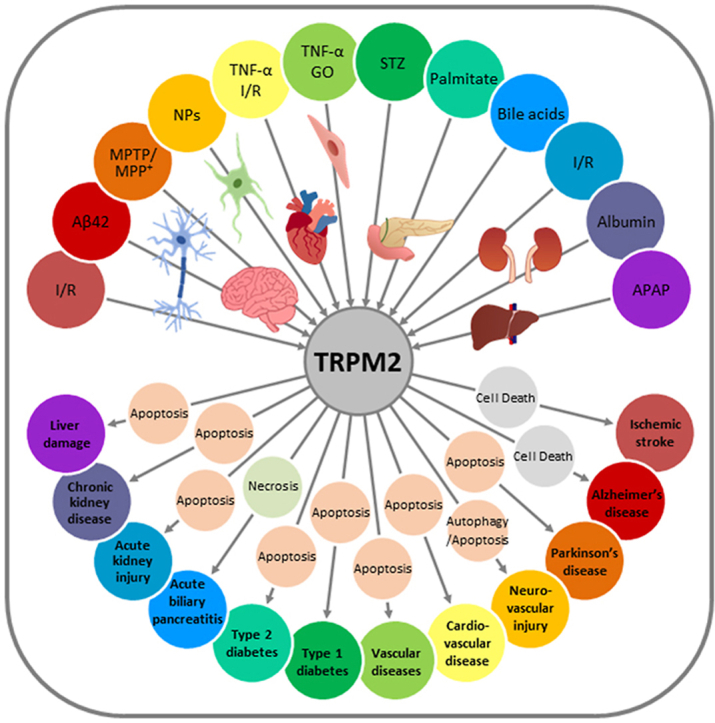
Abstract
Oxidative stress resulting from the accumulation of high levels of reactive oxygen species is a salient feature of, and a well-recognised pathological factor for, diverse pathologies. One common mechanism for oxidative stress damage is via the disruption of intracellular ion homeostasis to induce cell death. TRPM2 is a non-selective Ca2+-permeable cation channel with a wide distribution throughout the body and is highly sensitive to activation by oxidative stress. Recent studies have collected abundant evidence to show its important role in mediating cell death induced by miscellaneous oxidative stress-inducing pathological factors, both endogenous and exogenous, including ischemia/reperfusion and the neurotoxicants amyloid-β peptides and MPTP/MPP+ that cause neuronal demise in the brain, myocardial ischemia/reperfusion, proinflammatory mediators that disrupt endothelial function, diabetogenic agent streptozotocin and diabetes risk factor free fatty acids that induce loss of pancreatic β-cells, bile acids that damage pancreatic acinar cells, renal ischemia/reperfusion and albuminuria that are detrimental to kidney cells, acetaminophen that triggers hepatocyte death, and nanoparticles that injure pericytes. Studies have also shed light on the signalling mechanisms by which these pathological factors activate the TRPM2 channel to alter intracellular ion homeostasis leading to aberrant initiation of various cell death pathways. TRPM2-mediated cell death thus emerges as an important mechanism in the pathogenesis of conditions including ischemic stroke, neurodegenerative diseases, cardiovascular diseases, diabetes, pancreatitis, chronic kidney disease, liver damage and neurovascular injury. These findings raise the exciting perspective of targeting the TRPM2 channel as a novel therapeutic strategy to treat such oxidative stress-associated diseases.
Keywords: Oxidative stress, TRPM2 channel, Ca2+ and Zn2+ homeostasis, Cell death, Diseases
Graphical abstract
1. Introduction
Mammalian cells under physiological conditions generate a small quantity of reactive oxygen species (ROS) during normal cellular metabolism [1]. It is recognised that ROS at low to moderate concentrations serve to modulate various cellular signalling pathways and regulate cell fate and proliferation, thereby playing an important part in maintaining cellular and tissue homeostasis [1]. ROS are chemically reactive and can potently modify many biological macromolecules such as nucleic acids, proteins and lipids. Phagocytes thus produce a large amount of ROS via membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) as weaponry to destroy invading pathogens during immune responses [2,3]. Conceivably, exposure to high levels of ROS is also harmful to healthy cells and tissues, significantly disrupting their physiological functions and even causing them to die. Mammalian cells express multiple ROS-scavenging mechanisms that equip them with an antioxidant capacity [4]. However, a variety of pathological factors, both endogenous and exogenous, for example, reperfusion following transient ischemia, accumulation and aggregation of amyloid-β (Aβ) peptides, misfolded α-synuclein proteins, mitochondrial dysfunction, metabolism, infection, drug overdose, nanoparticles (NPs) and particulate matters (PMs) in polluted ambient air, are known to stimulate excessive generation of ROS, impair the antioxidant capacity of cells, or both [[5], [6], [7], [8], [9], [10], [11], [12], [13], [14]]. Such an imbalance can lead to the accumulation of ROS at high levels that induces a noxious condition termed oxidative stress (OS) [4]. A wealth of evidence exists to indicate that OS is an important pathological factor for, as well as a salient feature of, various diseases and conditions, including ischemia/reperfusion (I/R) damage, Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis, neurovascular dysfunction, cardiovascular disorders, diabetes, pancreatitis, chronic kidney disease, liver injury, hearing loss, inflammatory diseases, and cancers [3,6,7,10,[15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28]].
A salient and recurring feature in many, if not all, of these pathological processes is the dysregulation of ionic mechanisms, with a substantial impact on intracellular ion homeostasis, particularly Ca2+, which is well known for its ubiquitous role in determining the pathological processes as well as the physiological functions of cells, and also Zn2+, which is notorious for its ability to induce neurotoxicity. Mammalian cells express a repertoire of ion-transporting mechanisms, including numerous ion channels that are important to maintain and regulate intracellular ionic homeostasis. Transient receptor potential melastatin 2 (TRPM2) belongs to the transient receptor potential (TRP) channel superfamily and forms a non-selective Ca2+-permeable cation channel gated by intracellular ADP-ribose (ADPR) [29,30]. The TRPM2 channel is widely distributed throughout the body and is highly sensitive to activation by OS-inducing stimuli [[31], [32], [33]]. A large number of studies have been carried out over the past decades that provide compelling evidence to show that the TRPM2 channel is a key mechanism mediating OS-induced alteration in intracellular Ca2+ and Zn2+ homeostasis to disrupt various cellular functions and further support a significant role for aberrant TRPM2 channel activity in diverse OS-associated pathologies [[34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48]]. A well-established mechanism, among others, by which the TRPM2 channel contributes to these pathological processes, is to mediate OS-induced cell death. Considering such a prominent role for the TRPM2 channel, increasing research efforts are being devoted to understanding the mechanisms of TRPM2 channel activation and the consequences on intracellular ion homeostasis and downstream signalling pathways leading to cell death. In this review, we will give a brief introduction of the TRPM2 channel, and provide an overview of the current understanding of TRPM2-mediated cell death as a cellular mechanism that links miscellaneous OS-inducing pathological factors to associated pathological or diseased conditions (Table 1).
Table 1.
Summary of TRPM2-mediated cell death linking oxidative stress-inducing pathological factors to associated pathologies.
| Cell type | Stimulus | Death mechanism | Associated pathology | Key observations |
|---|---|---|---|---|
| Neurons | I/R | Unclear | Ischemic stroke |
|
| Aβ42 | Unclear | Alzheimer's disease |
|
|
| MPTP/MPP+ | Apoptosis | Parkinson's disease | ||
| Pericytes | ZnO-NPs | Autophagy/apoptosis | Neurovascular injury | |
| Cardiomyocytes | TNF-α, I/R | Apoptosis | Cardiovascular diseases | |
| Endothelial cells | TNF-α, GO | Apoptosis | Vascular diseases |
|
| Pancreatic β-cells | STZ | Apoptosis | Type 1 diabetes |
|
| Palmitate | Apoptosis | Type 2 diabetes |
|
|
| Pancreatic acinar cells | Bile acids | Necrosis | Acute biliary pancreatitis |
|
| Kidney tubular epithelial cells | I/R | Apoptosis | Acute kidney injury | |
| Kidney cortical collecting duct cells | Albumin | Apoptosis | Chronic kidney disease |
|
| Hepatocytes | APAP | Apoptosis | Liver damage |
Abbreviations: Aβ, amyloid-β; APAP N-acetyl-para-aminophenol; APP/PS1, amyloid precursor protein/presenilin 1; BCCAO/R, bilateral common carotid artery occlusion/reperfusion; BSA, bovine serum albumin; CDC, chenodeoxycholate; GO, glucose oxidase; IFN, interferon; IL, interleukin; I/R, ischemia/reperfusion; MCAO/R, middle cerebral artery occlusion/reperfusion; MPP+, 1-methyl-4-phenylpyridinium; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NPs, nanoparticles; OGD/R, oxygen-glucose deprivation/reoxygenation; SNpc, substantia nigra pars compacta; STZ, streptozotocin; TNF-α, tumour necrosis factor-α.
2. TRPM2 channel properties, activation mechanisms, and pharmacological inhibitors
2.1. Molecular, structural and functional properties
TRP channels were originally discovered in the genetic study of the visual signal transduction pathway in Drosophila melanogaster [49]. The subsequent search for the mammalian counterparts led to identification of a large superfamily, consisting of 28 members that are often grouped into TRPA (ankyrin), TRPC (canonical), TRPM (melastatin), TRPML (mucolipin), TRPP (polycystin) and TRPV (vanilloid) subfamilies, according to amino acid sequence relatedness [[50], [51], [52]]. All TRP proteins can form non-selective cation channels, mainly as homo-tetramers and also as hetero-tetramers, with most of them being Ca2+-permeable. Despite sharing similar structural arrangements, these channels exhibit distinct and frequently multi-modal activation mechanisms in response to a wide array of chemical, physical and biological stimuli, and serve an important role in a variety of physiological and pathophysiological processes [50,51].
The gene encoding the TRPM2 protein has been isolated from several mammalian species including humans. The human TRPM2 gene, located on chromosome 21q22.3, is around 90 kb in length and contains 32 exons [53]. Mammalian TRPM2 proteins are approximately 170 kDa in size and have a membrane-spanning domain comprising of six transmembrane segments (S1–S6) and exceptionally large N- and C-termini residing intracellularly [30,53] (Fig. 1A). Cryo-electron microscopy (cryo-EM) structures of the tetrameric human TRPM2 channel confirm that S5 and S6, together with the re-entrant loop between them, from all four subunits, form an aqueous pore through the centre of the protein complex that is permeable to Ca2+, Na+ and K+, while S1–S4 constitute a distinct structural module that is positioned peripherally to the pore-forming module [54,55]. The N-terminus of each subunit contains four TRPM homology regions (MHR1-4) [56] and the C-terminus has a conserved coiled-coil tetramerization domain [57,58] and a NUDT9-H domain that exhibits noticeable homology to the ADPR-metabolising enzyme NUDT9 (Fig. 1A). In mammalian TRPM2 channels, this NUDT9-H domain has little ADPR-degrading ability [59] but provides a critical ADPR-binding site and confers TRPM2 channel activation by intracellular ADPR [30,60,61].
Fig. 1.

The molecular and activation properties of the TRPM2 channel.
(A) A cartoon representation of the tetrameric TRPM2 channel and its subunit, which is comprised of 6 transmembrane domains (S1–S6) with a re-entrant loop between S5 and S6 that forms a pore permeable to Ca2+, K+ and Na+. The intracellular N-terminus contains four MHR (1–4), and the intracellular C-terminus contains aCC tetramerization domain and a NUDT9-H domain. The TRPM2 channel is activated by binding of intracellular ADPR at two sites, the MHR1/2 and NUDT9-H domain, and Ca2+ binding to the intracellular face of the transmembrane domain (not depicted). (B) ADPR and structurally related compounds that activate the TRPM2 channel, with the reported EC50 value shown in brackets where available. (C) Activation of the TRPM2 channel by ROS. Accumulation of intracellular ROS, resulting from exposure to extracellular ROS, increased intracellular ROS generation or decreased antioxidant capacity, induces activation of PARP and PARG enzymes in the nucleus that convert NAD+ to ADPR. ADPR diffuses into the cytosol and opens the TRPM2 channel, permitting Ca2+ influx to increase intracellular Ca2+ concentration, or activates the TRPM2 channel in intracellular organelles (not depicted). Abbreviations: ADPR, ADP-ribose; CC, coiled-coil; EC50, the concentration evoking 50% of the maximal response; MHR: TRPM homology regions; NAD+, nicotinamide adenine dinucleotide; NUDT9-H: NUDT9 homology; PARG, poly(ADPR) glycohydrolase; PARP, poly(ADPR) polymerase; ROS, reactive oxygen species.
In addition to ADPR, several ADPR analogues have been reported to activate the TRPM2 channel, including ADPR-2′-phosphate, 2′-O-acetyl-ADPR, 2′-deoxy-ADPR and cyclic ADPR (cADPR) [[62], [63], [64], [65], [66], [67]] (Fig. 1B). Whether cADPR is capable of activating the TRPM2 channel has been a matter of debate, mainly due to the fact that commercially available cADPR may be significantly contaminated with ADPR. In a recent study examining the effects of introducing point mutations in the NUDT9-H domain on the binding and activation of the human TRPM2 channel by purified cADPR, we provide strong evidence to favour cADPR as a TRPM2 channel activator [68]. Intracellular Ca2+ is critical for TRPM2 channel activation by ADPR [30,69,70]. The complex gating mechanism for the mammalian TRPM2 channel by ADPR and Ca2+ is still not fully delineated and nonetheless cryo-EM structures of the human TRPM2 channel in the absence or presence of ADPR, Ca2+ or both ligands have shed light on how the binding of ADPR and Ca2+ induces channel opening [54,55]. In the closed state, the NUDT9-H domain interacts with the MHR1/2 of the same subunit as well as with the MHR1/2 from a neighbouring subunit. ADPR binding primes the channel for opening by eliciting conformational changes that rotate the MHR1/2 and thereby disengage the inter-subunit interaction, and binding of Ca2+ to a site in the intracellular S2–S3 loop [54] or within the intracellular side of the TRPM2 channel [55] induces additional conformational changes that open the ion-conducting channel. Surprising, but important to mechanistic understanding of TRPM2 channel activation, is that a recent cryo-EM structural study has unambiguously revealed two ADPR-binding sites, the MHR1/2 as well as the NUDT9-H domain, and suggests that the MHR1/2 site is the orthostatic site while ADPR binding to the NUDT9-H domain elicits additional conformational changes for channel activation [55].
In addition to the full-length protein, several alternative splice variant isoforms of TRPM2 have been identified, including TRPM2-S made of only the N-terminus and the first two transmembrane segments, TRPM2-ΔN containing a deletion of 538–557 amino acid residues in the N-terminus, TRPM2-ΔC with a deletion of 1292–1325 residues in the C-terminus, TRPM2-SSF (striatal short form) lacking residues 1–214 in the N-terminus, and TRPM2-TE (tumour-enriched) comprised of a large part of the NUDT9-H domain [32,33,[71], [72], [73]]. The TRPM2-S protein is of particular interest in that while short of channel-forming ability, it is capable of co-assembling with the full-length TRPM2 protein or channel and imposing dominant negative inhibition of the TRPM2 channel. Due to limited pharmacological tools for investigating the TRPM2 channel as discussed below, this dominant negative attribute of TRPM2-S has been employed in in vitro studies to interrogate the role of the TRPM2 channel in mediating ROS-induced Ca2+ signalling and regulation of cellular functions by overexpression of TRPM2-S to suppress the formation or activity of functional channels by the endogenous full-length TRPM2 protein [33,[74], [75], [76], [77]].
Mammalian TRPM2 channels exhibit a widespread distribution, as has been documented in a number of tissues and cells including the brain, heart, blood vessels, kidney, liver, lung, pancreas and various immune cells [78]. The TRPM2 channel functions as a Ca2+-permeable channel in the plasma membrane of all cell types examined so far, with only the exception of dendritic cells where it is exclusively localised to the lysosomes and serves as a lysosomal Ca2+ release channel [79]. The TRPM2 channel is also present in the lysosomes of pancreatic β-cells and possibly endothelial cells [36,80,81] and in the mitochondria of hippocampal neurons [82]. There is increasing evidence to support an important role for the TRPM2 channel in mediating or regulating many physiological processes, such as insulin secretion, inflammatory cytokine production, phagosome maturation and temperature sensing, and interested readers can consult reviews on these topics [13,[83], [84], [85], [86], [87], [88]].
2.2. ROS-induced activation mechanisms
ROS are mostly short-lived, such as hydroxyl radicals (•OH), superoxide anions (O2•−) and peroxynitrite (OONO−). Hydrogen peroxide (H2O2) is relatively stable and thus widely used as an experimental paradigm in the study of OS signalling. While an early study suggested that H2O2 directly activated the TRPM2 channel, it is now generally accepted that H2O2-induced TRPM2 channel activation is indirect via mechanisms that generate ADPR from nicotinamide adenine dinucleotide (NAD+). One such widely-documented mechanism is activation of poly(ADPR)-polymerase (PARP), particularly PARP-1, and poly(ADPR)-glycohydrolase (PARG) enzymes in the nucleus (Fig. 1C), a common DNA damage repairing mechanism [73,74,[89], [90], [91]]. Some evidence also suggests that activation of mitochondrial NADase is another mechanism for H2O2-induced ADPR generation and TRPM2 channel activation [92]. Since early in vitro studies that established a role for the TRPM2 channel in conferring susceptibility to H2O2-induced cell death in human embryonic kidney (HEK) 293 cells expressing recombinant mammalian TRPM2 channels [31,33,89], extensive efforts have been directed towards examining the importance of endogenous TRPM2 channels in mammalian cells in coupling a wide spectrum of OS-inducing pathological factors, as discussed below, to their profound impacts on cellular functions and, furthermore, the pathogenesis and progression of associated pathological conditions [73,78,[83], [84], [85],87,[93], [94], [95], [96], [97], [98], [99], [100], [101], [102], [103], [104], [105]].
2.3. Pharmacological inhibitors
A number of low molecular weight compounds with different chemical structures have been found to inhibit ADPR- or H2O2-induced Ca2+ responses or ionic currents in cells that express recombinant mammalian TRPM2 channels (Table 2), providing useful pharmacological tools for in vitro and in vivo studies to investigate the role of the TRPM2 channel (Table 1). It is worth noting that inhibitors identified in earlier studies, including 2-aminoethoxydiphenyl borate (2-APB), N-(p-amycinnamoyl) anthranilic acid (ACA), antifungal agents such as clotrimazole (CTZ) and econazole and the non-steroidal anti-inflammatory flufenamic acid (FFA), while inhibiting the TRPM2 channel with concentrations producing a 50% inhibition (IC50) in the range of μM (Table 2) [[106], [107], [108], [109], [110], [111]], lack specificity towards the TRPM2 channel [73]. 8-Br-cADPR, an ADPR analogue, is reported to inhibit the TRPM2 channel by in vitro [62] and also in vivo studies [112] but, of notice, its inhibition has not been consistently observed [113]. Recent studies have tested various other ADPR-related analogues for their potential to selectively inhibit the TRPM2 channel in vitro [114,115], identifying multiple compounds such as 8-phenyl-2′-deoxy-ADPR, 7i and 8a (Table 2). A more recent study, by screening a large chemical library, has identified JNJ-28583113 as a potent TRPM2 antagonist with an IC50 of 10–100 nM (Table 2) that protects against TRPM2-mediated effects including ROS-induced cell death [116]. JNJ-28583113 was described to readily penetrate the blood brain barrier (BBB) but is metabolically unstable, limiting its use for in vivo studies. Several divalent cations, including Cu2+, Zn2+, Fe2+ and Pb2+, have been shown to act as extracellular TRPM2 inhibitors at micromolar concentrations, with Cu2+ being most potent with an IC50 of 2.6 μM [[117], [118], [119]]. It is worth mentioning that additional efforts have been made, focusing on the development of cell-permeable peptide inhibitors specific to the TRPM2 channel, such as tat-M2NX, a peptide corresponding to the NUDT9-H domain that is shown to prevent TRPM2 channel activation in vivo as well as in vitro [120]. Further efforts are necessary to develop specific, potent and metabolically stable TRPM2 inhibitors, particularly BBB-penetrable ones, given the increasing recognition of the important role of the TRPM2 channel in mediating various pathological conditions of the central nervous system (CNS).
Table 2.
Chemical structures and IC50 values of TRPM2 channel inhibitors.
| Inhibitor | Chemical structure | IC50 | Reference |
|---|---|---|---|
| 2-APB | 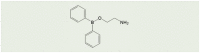 |
1.2 μMa | [109] |
| ACA | 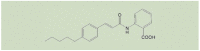 |
1.7 μMa,b | [108] |
| Clotrimazole (CTZ) | 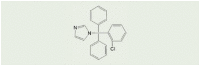 |
3-30 uMc | [107] |
| Econazole | 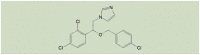 |
3-30 μMc | [107] |
| Flufenamic acid (FFA) | 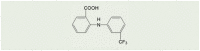 |
70 μMa 58.3 μMb |
[106,110,111] |
| 8-Br-cADPR | 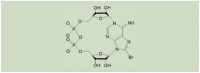 |
100 μMd | [62] |
| 7i | 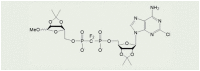 |
5.7 μMa | [115] |
| 8-phenyl-2′-deoxy-ADPR | 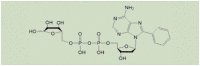 |
3 μMa | [114] |
| 8a | 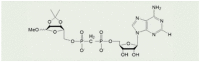 |
5.4 μMa | [115] |
| JNJ-28583113 | 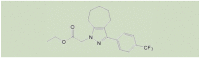 |
13 nMa 126 nMb |
[116] |
IC50, the concentration of the inhibitor inducing 50% inhibition of ADPR-induced currents a or H2O2-induced Ca2+ responses b; Where IC50 was not determined, concentrations showing inhibition of currents induced by ADPR c, cADPR d or H2O2d in HEK293 cells expressing the human TRPM2 channel.
As discussed above, the activity of nuclear PARP, particularly PARP-1, is crucial for the induction of ADPR generation and subsequent TRPM2 channel activation by ROS and other OS-inducing pathological stimuli. PARP inhibitors have been used in many studies as an alternative or additional means to prevent OS-induced TRPM2-mediated effects [36,39,48,74,75,82,[121], [122], [123], [124], [125], [126]], including cell death. Example PARP inhibitors include SB-750139, N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide (PJ34), 3-aminobenzamide (3-AB) and 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (DPQ). Considering the increasing availability of clinically tested and safe PARP inhibitors developed as anti-cancer treatments, it is attractive to repurpose PARP inhibitors as potential therapeutics to intervene with OS-induced PARP/TRPM2-mediated pathological conditions.
3. TRPM2 in neuronal cell death associated with ischemia/reperfusion brain damage, AD and PD
Neurons in the CNS, particularly those in the brain, are highly vulnerable to OS damage. OS-induced neuronal cell death has been strongly implicated in the pathogenesis of diverse conditions affecting the human brain and cognitive functions, including I/R-induced damage and neurodegenerative diseases such as AD and PD. TRPM2 channel expression has been documented in neurons in the cortex, hippocampus and striatum, the key brain regions in maintaining cognitive functions. The role of the TRPM2 channel in mediating OS-induced neuronal cell death has been extensively investigated in vitro using primary neuronal cultures from rodents and in vivo using rodent models of human diseases and conditions. In addition to the use of rodents and neuronal cultures derived from them, the role of the TRPM2 channel in OS-induced neuronal cell death has been examined using neuroblastoma cell lines, especially those of human origin. One such example is SH-SY5Y, a human neuroblastoma cell line widely used as a cell model to study the molecular mechanisms of neurodegeneration, particularly those related to PD [127]. In these studies, OS was introduced by exposure to H2O2 or pathological conditions or factors in vitro and in vivo, including I/R, Aβ peptides associated with AD pathogenesis, or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)/1-methyl-4-phenylpyridinium ions (MPP+) that induce PD-like pathologies.
3.1. Neuronal cell death induced by exposure to H2O2
It is well known that exposure to H2O2 imposes OS on neurons under in vitro conditions [128,129]. A number of studies have examined the role of the TRPM2 channel in mediating H2O2-induced neuronal cell death, using primary cortical, hippocampal or striatal neuronal cultures from rodents and also human SH-SY5Y cells. Fonfria et al. were the first to study the role of the TRPM2 channel in H2O2-induced neuronal cell death using rat striatal neurons [75]. Exposure to H2O2 (300 μM for 24 h) resulted in neuronal cell death, measured using SYTOX® Green staining. H2O2-induced cell death was decreased in striatal neurons transiently transfected with TRPM2-specific small interference RNA (TRPM2-siRNA) to reduce TRPM2 expression, or with a plasmid expressing TRPM2-S. H2O2-induced cell death in striatal neurons was also reduced by treatment with the PARP inhibitor SB-750139 [75]. These results suggest that PARP-dependent TRPM2 channel activation mediates H2O2-induced neuronal cell death. Another early study reported a similar role for the TRPM2 channel in H2O2-induced cell death in cultured rat cortical neurons [130]. In this study, neurons were exposed to H2O2 either at a very high concentration (1 mM for 20 min) or at a low and pathologically relevant concentration (50 μM for 6 h), with both conditions inducing substantial neuronal cell death, detected using the trypan blue exclusion method. H2O2-induced neuronal cell death was reduced by treatment with TRPM2-siRNA and also by elimination of extracellular Ca2+, suggesting that TRPM2-mediated Ca2+ influx is crucial for H2O2-induced cell death in cortical neurons. We have recently examined H2O2-induced cell death in cultured hippocampal neurons from wild-type (WT) and TRPM2-knockout (TRPM2-KO) mice and provided further evidence to demonstrate a key role for the TRPM2 channel in mediating H2O2-induced neuronal cell death [126]. Exposure of WT neurons to H2O2 (30–300 μM) for various durations (2–24 h) led to concentration-dependent and duration-dependent cell death, determined using propidium iodide (PI) staining. In contrast, there was negligible H2O2-induced cell death in TRPM2-KO neurons. H2O2-induced cell death in WT neurons was also attenuated in the absence of extracellular Ca2+ or by treatment with PJ34, supporting the importance of PARP-dependent TRPM2 channel activation and subsequent TRPM2-mediated Ca2+ signalling in H2O2-induced cell death in hippocampal neurons [126], as reported in cortical neurons [130]. In summary, accumulating evidence consistently supports a critical role for the TRPM2 channel and, more specifically, TRPM2-mediated Ca2+ influx-dependent Ca2+ signalling, in mediating H2O2-induced cell death in striatal, cortical and hippocampal neurons.
3.2. Delayed neuronal cell death associated with ischemic stroke and other I/R brain damage
It is known that cerebral ischemia, whether focal as it occurs in ischemic stroke or global under cardiac arrest, if severe or long lasting, can disrupt neuronal function and induce neuronal demise, whereas transient ischemia-induced brain damage can largely be attributed to neuronal cell death due to subsequent reperfusion, which is thus specifically referred to as delayed neuronal cell death [131]. Delayed neuronal cell death induced by OS resulting mainly from excessive ROS generation during reperfusion is an important cellular mechanism for the progressive decline in cognitive function associated with I/R-induced brain damage [[131], [132], [133], [134]]. The TRPM2 channel has been extensively scrutinised for its role in mediating delayed neuronal cell death induced by oxygen-glucose deprivation followed by reoxygenation (OGD/R), an in vitro I/R model, and brain damage and cognitive dysfunction by middle cerebral artery occlusion followed by reperfusion (MCAO/R), an in vivo model of ischemic stroke, or by bilateral common carotid artery occlusion followed by reperfusion (BCCAO/R) or cardiac arrest followed by resuscitation (CA-R) [100,105].
Two early studies examined the role of the TRPM2 channel in delayed neuronal cell death using cultured mouse cortical and hippocampal neurons [35,135]. Delayed neuronal cell death, induced by exposure to OGD for 2 h followed by reoxygenation for 24 h and determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) cell viability assay, was attenuated by treatment with 2-APB, ACA, FFA or CTZ, introduced during OGD, or by treatment with TRPM2-specific short-hairpin RNA (shRNA) to reduce TRPM2 expression prior to OGD. Consistently, subsequent studies showed that OGD/R-induced delayed neuronal cell death was much less severe in cultured cortical neurons or in CA1 pyramidal neurons in hippocampal slices that were prepared from TRPM2-KO mice [40,136]. In addition, OGD/R-induced delayed neuronal death in hippocampal neurons was effectively suppressed by treatment with CTZ after re-oxygenation [135]. These results suggest a significant role for the TRPM2 channel in mediating delayed neuronal cell death. It was noted that genetic or pharmacological intervention of the TRPM2 channel inhibited OGD/R-induced delayed neuronal cell death in cortical and hippocampal neurons from male mice, but not in those from female mice [35,135], suggesting a sexually dimorphic role of the TRPM2 channel in mediating delayed neuronal cell death. In contrast, delayed neuronal cell death in hippocampal neurons from both male and female mice, induced by exposure to H2O2 (50 μM for 2 h) followed by incubation in H2O2-free normal culture medium for a further 24 h, was inhibited to a similar level by treatment with CTZ applied during exposure to H2O2 [135], indicating that the dimorphism may arise from the cellular conditions rather than from OS itself.
Several studies have examined the role of the TRPM2 channel in mediating neuronal cell death measured by the number of live neurons and brain damage quantified by the infarct size in mice as a consequence of cerebral ischemia induced by MCAO/R. In an aforementioned early study, it was shown that intra-striatal injection of lentivirus expressing TRPM2-specific shRNA to reduce TRPM2 expression, introduced before occlusion, protected against MCAO/R-induced neuronal cell death and infarction in the striatum [35]. In addition, the infarct size in the cortex, striatum and across the entire brain hemisphere in mice subjected to MCAO/R, analysed 24 h after reperfusion, was attenuated by administration of CTZ during occlusion and again at the time of reperfusion [35]. Consistently, several subsequent studies showed independently that MCAO/R induced significantly less severe brain damage in TRPM2-KO mice as compared to WT mice [37,136,137]. However, there was no difference in infarction between WT and TRPM2-KO mice subjected to MCAO without ensuing reperfusion [37]. There was also less neuronal cell death in the neocortex of TRPM2-KO mice than that seen in WT mice after exposure to MCAO/R [137]. One of these studies showed subcutaneous injection of CTZ after reperfusion suppressed MCAO/R-induced brain damage in WT mice but was without effect on the residual brain damage in TRPM2-KO mice [136]. Similarly, a more recent study has reported that MCAO/R-induced infarction in WT mice was reduced by injection of the inhibitory tat-M2NX peptide prior to ischemia and such a protective effect was absent in TRPM2-KO mice [120]. These results are consistent with those from in vitro studies discussed above and together indicate the protective effects of CTZ and tat-M2NX in WT mice via specific targeting of the TRPM2 channel, which is in agreement with our finding of exclusive TRPM2 channel activation by OS-inducing reperfusion [40]. Collectively, these results support an important role for the TRPM2 channel in mediating brain damage by reperfusion after transient ischemia, rather than ischemia itself. Studies have also examined the role of the TRPM2 channel in delayed neuronal cell death and brain damage induced by reperfusion following global ischemia. We showed that BCCAO/R induced death of pyramidal neurons in the hippocampus CA1 region in male mice and impairment in learning and memory, both of which were largely rescued by TRPM2-KO [40]. Similarly, death of pyramidal neurons in the hippocampal CA1 region observed in male mice subjected to CA/R was suppressed by administration of CTZ after resuscitation [138], a finding that also supports the notion that the TRPM2 channel is critical for reperfusion-induced neuronal cell death. Of note, as shown for delayed neuronal cell death in cultured neurons by in vitro studies, there is evidence to suggest the role of the TRPM2 channel is sexually dimorphic in vivo. It has been proposed that sex-dependent differences in the mechanisms regulating the TRPM2 channel contribute to such a dimorphism [135,136,139,140], but the exact reason remains to be fully established.
In summary, a number of studies using various in vitro and in vivo I/R models have gathered substantial evidence to support a critical role for the TRPM2 channel in mediating neuronal cell death due to reperfusion, leading to I/R-induced brain damage. Importantly, as discussed above, studies provide a proof of concept that post-ischemia inhibition of the TRPM2 channel is a promising therapeutic strategy to mitigate ischemic stroke brain damage and cognitive deficits. We will further deliberate in detail below the underlying molecular and signalling mechanisms for TRPM2-mediated delayed neuronal cell death (Fig. 2A).
Fig. 2.

TRPM2-mediated signalling pathways in oxidative stress-induced neuronal death associated with ischemia/reperfusion brain damage, AD and PD.
(A) Delayed neuronal cell death associated with ischemia/reperfusion brain damage. OS resulting from reperfusion, based on examining H2O2-induced delayed neuronal cell death in human neuroblastoma SH-SY5Y cells, stimulates activation of PKC and NOX, and NOX-mediated ROS production. ROS induces activation of PARP and the TRPM2 channel, and TRPM2-mediated Ca2+ influx. ROS also induces lysosomal dysfunction and lysosomal Zn2+ release. TRPM2-dependent mitochondrial Zn2+ accumulation leads to mitochondrial dysfunction and cell death. The cell death mechanisms or pathways are unclear. Mitochondrial dysfunction also leads to ROS generation. (B) Aβ42-induced cell death in hippocampal neurons associated with AD. Exposure to Aβ42 at pathologically relevant concentrations induces a similar signalling pathway as described in (A) to cause mitochondrial dysfunction and release of cytochrome c. (C) MPP+-induced neuronal cell death related to PD, based on examining MPP+-induced cell death in SH-SY5Y cells. Exposure to MPP+ promotes ROS generation that activates the TRPM2 channel, likely via PARP, leading to intracellular Ca2+ increase, activation of calpain and pro-apoptotic proteins (Bak, Bid and Bad), cyt c release, and caspase-3 activation to initiate apoptotic cell death. Abbreviations: Aβ, amyloid-β peptide; ADPR, ADP-ribose; AD, Alzheimer's disease; cyt c, cytochrome c; MPP+, 1-methyl-4-phenylpyridinium; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NOX, NADPH oxidase; PARP, poly(ADPR) polymerase; PD, Parkinson's disease; PKC, protein kinase C; ROS, reactive oxygen species.
3.3. Aβ-induced neuronal cell death associated with AD
It is well documented that Aβ peptides, especially Aβ42, found plentifully in senile amyloid plaques that represent one of the histopathological hallmarks of human AD brains, are able to induce OS and cause neurotoxicity, particularly synapse loss and neuronal cell death, as has been posited in the amyloid cascade hypothesis of AD [141,142]. Efforts have thus been made to examine the role of the TRPM2 channel in mediating Aβ-induced neurotoxicity. In an aforementioned study examining H2O2-induced neuronal cell death, Fonfria et al. also showed that exposure to Aβ42 (20 μM for 48 h) led to cell death in cultured rat striatal neurons [75]. Similar to H2O2-induced neuronal cell death discussed above, Aβ42-induced neuronal cell death was reduced by treatment with TRPM2-siRNA, overexpression of TRPM2-S or treatment with SB-750139, supporting an important role for PARP-dependent TRPM2 channel activation in Aβ-induced neuronal cell death. The significance or relevance of this finding to AD pathogenesis however remained unclear, given the extremely high concentration of Aβ42 used. In a more recent study using hippocampal neurons, we have shown that exposure to Aβ42 at pathologically relevant concentrations (<100 nM-1 μM) for various durations (24–96 h) led to concentration-dependent and duration-dependent cell death in WT neurons, whereas exposure to Aβ42 (1 μM for up to 96 h) induced no or slight cell death in TRPM2-KO neurons [82]. In addition, Aβ42-induced cell death in WT neurons was attenuated by treatment with 2-APB or ACA and also by treatment with PJ34 or DPQ. The study by Ostapchenko et al. showed that neurotoxicity, indicated by synapse loss and age-related memory impairment, was salvaged by TRPM2-KO in APP/PS1 transgenic mice, which are widely used in the study of AD pathology due to excessive generation of Aβ peptides [42]. These in vitro and in vivo studies provide consistent evidence to demonstrate a critical role for the TRPM2 channel in Aβ-induced neurotoxicity. In the aforementioned in vitro study using cultured hippocampal neurons, we further showed that Aβ42-induced neuronal cell death was suppressed by treatment with the protein kinase C (PKC) inhibitor Gö6983, generic NOX inhibitors apocynin and diphenyleneiodonium (DPI), or NOX1/4-selective inhibitor GKT137831. These results suggest that exposure to Aβ42 promotes activation of PKC and NOX, and NOX-mediated ROS generation, leading to PARP-dependent TRPM2 channel activation and neuronal cell death. We will elaborate below the molecular and signalling mechanisms for Aβ42-induced TRPM2-mediated neuronal cell death (Fig. 2B).
3.4. MPTP/MPP+-induced neuronal cell death associated with PD
The pathological hallmark of PD is the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) of the human brain that gives rise to the disease symptoms [143,144]. MPTP is metabolised to MPP+ which, after penetrating the BBB into the brain, is selectively accumulated in dopaminergic neurons and induces mitochondrial dysfunction and ROS generation causing neurons to die. MPTP/MPP+ are thus known as neurotoxicants generating PD-like pathologies in humans, non-human primates and rodents [145]. A recent study has shown that TRPM2 protein expression was upregulated in the SNpc of human PD brains and the SNpc of MPTP-induced PD mouse brains [44]. As mentioned above, human SH-SY5Y cells are widely used in the study of the molecular mechanism for PD-related neurodegeneration [127]. TRPM2 protein expression was also upregulated in SH-SY5Y cells after extended exposure to MPP+ (6–24 h). Consistently, prior MPP+ exposure resulted in greater H2O2-induced currents and Ca2+ responses that were inhibited by treatment with FFA or ACA, further supporting MPP+-induced upregulation of TRPM2 channel expression. Exposure to H2O2 (10–400 μM) or MPP+ (125–500 μM) for various durations (2–24 h) caused duration-dependent and concentration-dependent cell death in SH-SY5Y cells, measured using the MTT assay. MPP+-induced cell death was suppressed by treatment with the antioxidant compounds resveratrol and N-acetylcysteine (NAC), consistent with the ability of MPP+ to induce ROS generation. MPP+-induced cell death was shown to occur via apoptosis by annexin V and PI co-staining. Moreover, exposure to MPP+ induced upregulation and activation of the Ca2+-sensitive protease calpain, pro-apoptotic proteins Bak, Bid and Bad, and caspase-3, as well as the release of mitochondrial cytochrome c, indicating that activation of the mitochondria-dependent intrinsic apoptotic pathway drives MPP+-induced TRPM2-mediated cell death. H2O2/MPP+-induced cell death in SH-SY5Y cells was also attenuated by treatment with FFA. Both MPP+-induced cell death and activation of the apoptotic pathways were also inhibited by treatment with TRPM2-siRNA [44]. In a more recent study using PI staining, we have shown that exposure to H2O2 (100–500 μM for 24 h) induced concentration-dependent cell death in SH-SY5Y cells and that H2O2-induced cell death was suppressed or abolished by treatment with 2-APB, PJ34 or DPQ [123]. Furthermore, by generating a stable SH-SY5Y cell line overexpressing the TRPM2 channel and comparing cell death following exposure to H2O2 (30–300 μM for 24 h), we further demonstrated that an increase in TRPM2 channel expression remarkably enhanced susceptibility to H2O2-induced cell death [123]. Taken together, these studies examining TRPM2 expression in the SNpc of PD brains, the SNpc of MPTP-induced PD mouse brains and also MPP+-exposed human SH-SY5Y cells, particularly the role of the TRPM2 channel in H2O2/MPP+-induced cell death in SH-SY5Y cells, suggest that PD-related OS induces TRPM2 channel expression upregulation and activation, resulting in altered intracellular Ca2+ homeostasis and subsequent activation of calpain and the intrinsic apoptotic pathway (Fig. 2C). It is worth mentioning that recent studies have reported that microglial cells, the CNS-resident immune cells, undergo cell death after exposure to OS induced by H2O2 [146] or MPP+ [147]. Further investigations are required to examine the role of these TRPM2-mediated cell death mechanisms in the pathogenesis of PD.
3.5. Molecular mechanisms driving OS-induced TRPM2-mediated neuronal cell death
An increasing volume of evidence from in vitro and in vivo studies, as discussed above, supports an important role for the TRPM2 channel in mediating neuronal cell death and brain damage following exposure to diverse notorious OS-inducing pathological factors. It remains relatively less well understood how TRPM2 channel activation drives neuronal cell death. Studies, by examining various signalling pathways, have proposed distinct mechanisms, and it should be recognised that these mechanisms are not necessarily mutually exclusive.
It is known that the N-methyl-d-aspartate (NMDA)-subtype glutamate receptors (NMDARs) participate in signalling pathways that determine neuronal cell death or survival [[148], [149], [150], [151]]. More specifically, expression of the GluN2B-containing NMDAR is important in activating cell death signalling, whereas expression of the GluN2A-containing NMDAR acts as a pro-survival signal through the activation of extracellular-regulated kinases (ERK) and Akt. In an aforementioned study, Alim et al. showed that TRPM2-KO led to downregulation of neuronal GluN2B expression and simultaneous upregulation of neuronal GluN2A expression and activation of downstream signalling molecules in mice subjected to MCAO/R [37]. These results have led to the proposal that the TRPM2 channel mediates ischemic stroke brain damage via modulation of neuronal NMDARs to promote neuronal cell death signalling pathways and weaken neuronal cell survival signalling pathways [37].
It is also well documented that altered intracellular Zn2+ homeostasis is a detrimental factor inducing neuronal cell death, particularly neuronal death associated with I/R-induced brain damage [94,152,153]. In an aforementioned study, we showed that BCCAO/R induced intracellular Zn2+ increase and delayed neuronal cell death in CA1 pyramidal neurons in the hippocampal region in WT mice, both of which were strongly attenuated or lost in TRPM2-KO mice [40]. Furthermore, by using time-lapse confocal imaging of intracellular Zn2+ concentration in cultured hippocampal neurons during exposure to OGD/R, we revealed that TRPM2-KO led to exclusive loss of intracellular Zn2+ increase during reperfusion, without effect on intracellular Zn2+ increase during ischemia. These results support the notion that reperfusion-induced TRPM2 channel activation alters intracellular Zn2+ homeostasis that triggers delayed neuronal cell death [40]. As described above in an early study showing H2O2-induced TRPM2-mediated delayed neuronal cell death in cultured mouse hippocampal neurons [135], our recent study has shown that H2O2 can induce similar delayed cell death in SH-SY5Y cells, with PI staining detecting negligible cell death immediately after exposure to H2O2 (100–300 μM for 2 h) but substantial cell death after cells were cultured in normal and H2O2‐free medium for a further 24 h [124]. H2O2‐induced delayed cell death in SH-SY5Y cells was also mitigated by treatment with PJ34, DPQ, 2-APB or ACA, and by removal of extracellular Ca2+, suggesting a critical role for PARP-dependent TRPM2 channel activation and subsequent TRPM2-mediated Ca2+ signalling in such delayed cell death. Furthermore, H2O2-induced delayed cell death was largely prevented by treatment with the Zn2+ chelators TPEN and clioquinol, indicating that altered intracellular Zn2+ homeostasis is vital for H2O2-induced delayed cell death, as discussed above in delayed neuronal death in hippocampal neurons induced by OGD/R in vitro and BCCAO/R in vivo. H2O2-induced delayed cell death in SH-SY5Y cells was inhibited by treatment with Gö6983, apocynin, DPI or GKT137831, indicating critical involvement of PKC/NOX-mediated ROS generation. We further revealed, by confocal imaging of the co-localization of Zn2+ indicator FluoZin-3 staining and intracellular organelle-specific fluorescent indicators, that the labile Zn2+ was located in the lysosomes, but not the mitochondria or ER under control conditions. Exposure to H2O2 induced lysosomal dysfunction indicated by loss of LysoTracker staining, a rise in intracellular Zn2+ concentration by an increase in FluoZin-3 staining, mitochondrial Zn2+ accumulation by an increase in mitochondrial Zn2+ indicator RhodZin-3 staining, mitochondrial fragmentation revealed by MitoTracker staining, and mitochondrial ROS generation by an increase in MitoTracker Red CM‐H2Xros staining. These H2O2-induced effects or events were all prevented by treatment with PJ34, 2-APB or TPEN, as well as apocynin, DPI or GKT137831. Intriguingly, we have found that lysosomal dysfunction induced by exposure to bafilomycin also resulted in intracellular Zn2+ increase, mitochondrial Zn2+ accumulation, mitochondrial fragmentation and mitochondrial ROS generation, all of which were also inhibited by treatment with PJ34, 2-APB or TPEN. TRPM2 protein was detected in isolated mitochondria, and ADPR induced Zn2+ accumulation in isolated mitochondria in the presence but not in the absence of Ca2+ [124]. Collectively, these results have led us to hypothesise a positive feedback mechanism that drives H2O2-induced delayed neuronal cell death in SH-SY5Y cells. As summarised in Fig. 2A, such a mechanism is comprised of multiple steps, including initial activation of PKC and NOX, NOX-mediated ROS generation, lysosomal dysfunction, lysosomal Zn2+ release, TRPM2-dependent mitochondrial Zn2+ accumulation, mitochondrial dysfunction and mitochondrial ROS generation.
We have gathered evidence to suggest that a similar vicious positive feedback mechanism drives neuronal cell death induced by sustained exposure to H2O2 and Aβ42 [[82], [126]]. For example, H2O2-induced cell death in cultured hippocampal neurons was inhibited by treatment with TPEN [126]. The labile Zn2+ in hippocampal neurons was also mainly detected in the lysosomes, but not in the mitochondria or ER under control conditions. Exposure to H2O2 induced intracellular Zn2+ increase and, furthermore, lysosomal dysfunction, lysosomal Zn2+ release, mitochondrial Zn2+ accumulation, mitochondrial fragmentation and mitochondrial ROS generation [126]. Similarly, Aβ42-induced cell death in hippocampal neurons was inhibited by treatment with TPEN [82]. Exposure to Aβ42 induced lysosomal dysfunction, lysosomal Zn2+ release, mitochondrial Zn2+ accumulation, mitochondrial fragmentation and mitochondrial ROS generation. These effects induced by H2O2 and Aβ42 were completely prevented by treatment with TPEN as well as by TRPM2-KO. Consistent with the notion that TRPM2 channel activation is required for mitochondrial Zn2+ accumulation found in SH-SY5Y cells, TRPM2 expression and ADPR-induced Ca2+-dependent Zn2+ accumulation were observed in mitochondria isolated from the hippocampus and cortex of WT mouse brains, but ADPR-induced Zn2+ accumulation did not occur in mitochondria isolated from the hippocampus and cortex of TRPM2-KO mouse brains [82]. Aβ42-induced lysosomal and mitochondrial dysfunction, as well as alteration in intracellular Zn2+ homeostasis, was also prevented by treatment with Gö6983, apocynin or GKT137831. Moreover, exposure to Aβ42 induced release of cytochrome c into the cytosol in hippocampal neurons from WT mice, but not from TRPM2-KO. Taken together, these results indicate that activation of the TRPM2 channel in mitochondria sets in motion the above-described vicious positive feedback mechanism to drive Aβ42-induced cell death in hippocampal neurons (Fig. 2B).
4. TRPM2 in pericyte death associated with neurovascular function injury
Pericytes are critical constituents of a functional and anatomical structure called the neurovascular function unit that connects the brain parenchyma to the cerebral vasculature, and is crucial in regulating cerebral blood flow and maintaining BBB function [154,155]. Loss of pericytes and their activity impairs neurovascular function, particularly BBB integrity, a mechanism that exacerbates numerous pathological conditions in the brain including ischemic stroke and AD [155,156]. It is known that ER stress is an important indicator of pericyte dysfunction that can itself potentiate cellular damage and induce cell death via autophagy [157,158]. Increasing evidence suggests that ultrafine PMs with a size of <100 nm commonly present in polluted air and diverse NPs widely used in nanomaterials are potent in inducing OS [13,48], ER stress and autophagy-mediated cell death [159,160]. We have recently examined the role of the TRPM2 channel in mediating zinc oxide-NPs (ZnO-NPs)-induced OS, ER stress and autophagy in brain vascular pericytes [161]. Exposure of cultured human brain vascular pericytes to ZnO-NPs (2.5–20 μg/ml for 6 h) resulted in a concentration-dependent increase in the level of LC3-II, indicative of autophagosome formation or initiation of autophagy. ZnO-NPs-induced increase in the level of LC3-II was prevented by treatment with the selective ER stress inhibitor salubrinal, supporting the role of ZnO-NPs-induced ER stress in the initiation of autophagy. In addition, ZnO-NPs induced a greater increase in the LC3-II level in pericytes treated with chloroquine to inhibit conversion of the autophagosome to an autolysosome and thereby stop autophagic flux. It is known that an acidic environment can quench the fluorescence of green fluorescent protein (GFP) but not that of red fluorescent protein (RFP), and expression of the tandem mRFP-GFP-LC3 protein followed by fluorescent imaging is widely used to probe autophagic flux in cells in vitro and in vivo [162]. We demonstrated by confocal imaging of pericytes expressing a mRFP-GFP-LC3 construct that exposure to ZnO-NPs enhanced autophagosome formation, indicated by an increase in the number of yellow puncta (emitting both RFP and GFP fluorescence), and also autolysosome formation, by an increase in the number of red puncta (due to quenching of GFP fluorescence by the acidic pH of autolysosomes). ZnO-NPs-induced effects on the level of LC3-II in control or chloroquine-treated cells were reduced by treatment with TRPM2-siRNA, suggesting an important role for the TRPM2 channel in ZnO-NPs-induced initiation of autophagy and increase in autophagic flux. We also showed using TUNEL staining, cell counting kit-8 cell viability assay or PI staining that exposure to ZnO-NPs (10 μg/ml for 6 h) induced cell death via apoptosis, which was suppressed by treatment with TRPM2-siRNA and also by siRNA-mediated knockdown of AGT5, a key autophagy-mediating protein. In addition, exposure to ZnO-NPs induced caspase-8 activation, which was also attenuated by treatment with TRPM2-siRNA or ATG5-siRNA. These results indicate that ZnO-NPs-induced TRPM2 channel activation triggers autophagy-mediated apoptotic cell death. Furthermore, we showed that exposure to ZnO-NPs (10 μg/ml for 2–24 h) resulted in a time-dependent increase in the levels of CHOP and phosphorylation of PERK and c-Jun N-terminal kinase (JNK), which are known to be critically involved in ER stress signalling. ZnO-NPs-induced ER stress signalling was attenuated by treatment with TRPM2-siRNA. Collectively, these results support the importance of the TRPM2 channel in mediating ZnO-NPs-induced ER stress signalling and subsequent induction of autophagy and apoptotic cell death in pericytes [161]. Exposure to ZnO-NPs induced pericytes to generate peroxynitrite, which was inhibited by treatment with the peroxynitrite scavenger uric acid (UA). ZnO-NPs-induced ER stress indicated by an increase in the levels of CHOP and JNK activation, autophagy by the increase in the level of LC3-II, and apoptotic cell death were suppressed by treatment with UA, the nitric oxide synthase (NOS) inhibitor N(G)-nitro-l-arginine methyl ester (l-NAME) or in combination. These results suggest that ZnO-NPs induce oxidative and nitrosative stress to cause ER stress and autophagy, triggering apoptosis in pericytes. In HEK293 cells expressing the human TRPM2 channel, exposure to ZnO-NPs (10 μg/ml) or the peroxynitrite donor 3-morpholinosydnonimine (SIN-1; 0.5 mM) attenuated ADPR-induced currents, which were rescued by treatment with UA or l-NAME. In contrast, exposure to ZnO-NPs or SIN-1 induced no effect on ADPR-induced current response in HEK293 cells expressing a mutant channel, in which the nitration site Tyr1485 in the intracellular C-terminus was mutated to serine (Y1485S). Consistently, both the increase in the levels of CHOP and activation of PERK and JNK (ER stress) and the increase in the level of LC3-II (autophagy) induced by exposure to ZnO-NPs or peroxynitrite were significantly attenuated in pericytes overexpressing the Y1485S mutant channel. These results have led to the notion that upregulation of TRPM2 channel activity as a result of nitration at Tyr1485 enhances ER stress and autophagy-mediated apoptosis in human pericytes (Fig. 3). In mice, introduction of ZnO-NPs (0.1 ml of 0.5 mg/ml) via tail vein injection induced loss of pericytes in the brain micro-vessels, assessed 24 h post-injection by double labelling of α-smooth muscle actin (α-SMA), a commonly used marker of pericytes, and laminin in vascular-specific basal membrane followed by confocal imaging [161]. Exposure to ZnO-NPs also resulted in increased autolysosome formation indicated by an increase in the number of red puncta in α-SMA-positive pericytes in micro-vessels, revealed by confocal imaging of the cortex of mice prior injected with adenovirus expressing the mRFP-GFP-LC3 reporter. ZnO-NPs-induced autophagy and loss of brain pericytes were attenuated in TRPM2-KO mice [161]. Taken together, these results from in vitro experiments on human brain vascular pericytes and in vivo studies using WT and TRPM2-KO mice provide consistent evidence to support an important role for the TRPM2 channel in mediating autophagy-mediated apoptotic cell death in brain vascular pericytes as a result of exposure to pathological stimuli inducing OS and ER stress (Fig. 3).
Fig. 3.

TRPM2-mediated signalling pathways in oxidative stress-induced pericyte death associated with neurovascular injury induced by exposure to nanoparticles.
Exposure to ZnO-NPs increases ROS generation to induce TRPM2 channel activation, possibly via PARP and also generation of peroxynitrite for TRPM2 nitration to upregulate the channel activity in pericytes. TRPM2 channel activation leads to intracellular Ca2+ increase and ER stress that induces autophagy and caspase-8 activation to cause pericyte death by apoptosis. Abbreviations: ADPR, ADP-ribose; ER, endoplasmic reticulum; PARP, poly(ADPR) polymerase; ROS, reactive oxygen species; ZnO-NPs, zinc oxide nanoparticles.
5. TRPM2 in cardiomyocyte death associated with myocardial I/R injury
Myocardial injury induced by reperfusion contributes to adverse cardiovascular outcomes after myocardial ischemia or cardiac arrest. OS, resulting from ROS generation mainly by mitochondria and also mediated by NOX, is one of the major causes of I/R-induced cardiomyocyte death and myocardial injury [[24], [25], [26], [27]]. An early in vitro study examined the role of the TRPM2 channel in mediating H2O2-induced cell death in cultured neonatal rat ventricular myocytes [163]. H2O2 (10–500 μM exposure for 1 h) concentration-dependently induced an increase in cell membrane permeabilisation assessed by PI staining and a reduction in intracellular ATP level measured immediately after exposure and, in addition, nuclear chromatin condensation shown by Hoechst staining of cells after incubation in normal and H2O2-free medium for a further 4.5 h. More specifically, in response to exposure to H2O2 (100 μM for 1 h), marked activation of caspase-3 was detected at 20 min, a reduction in intracellular ATP level at 40 min, an increase in PI staining at 1 h, and chromatin condensation after cells were cultured in the absence of H2O2 for a further 2 h. These results suggest that H2O2-induced OS causes cardiomyocytes to die via both apoptosis and necrosis, both of which are known to contribute to myocardial I/R injury. Exposure to H2O2 (100 μM for 1 h) also induced cytochrome c release and caspase-3 activation examined immediately after exposure, and DNA fragmentation revealed by TUNEL staining as well as chromatin condensation in cells after culturing in the absence of H2O2 for a further 4.5 h. H2O2-induced chromatin condensation and DNA fragmentation were attenuated by treatment with the caspase-3 inhibitor z-DEVD-FMK, supporting caspase-3-dependent apoptotic cell death. H2O2-induced chromatin condensation was also reduced by treatment with the mitochondrial Ca2+ uniporter (MCU) inhibitor RU360 or removal of extracellular Ca2+, and completely abolished by removal of both extracellular Ca2+ and Na+. Exposure to H2O2 (100 μM) induced increases in intracellular Ca2+ and Na+ concentrations that were prevented by treatment with CTZ. H2O2-induced cytochrome c release, caspase-3 activation, chromatin condensation and DNA fragmentation were attenuated by treatment with CTZ. H2O2-induced intracellular Ca2+ increase, chromatin condensation and DNA fragmentation were inhibited but only partially by treatment with 3-AB or DPQ. Collectively, these results suggest ROS induces cardiomyocyte apoptosis, mediated by a molecular mechanism which is composed of activation of PARP and the TRPM2 channel, TRPM2-mediated extracellular Ca2+ and Na+ influx and MCU-mediated mitochondrial accumulation of Ca2+ and Na+, leading to mitochondrial dysfunction, cytochrome c release and caspase-3 activation (Fig. 4A). In contrast, H2O2-induced membrane permeabilisation was not affected by treatment with CTZ, but almost completely abolished by treatment with 3-AB or DPQ, leading to the notion that H2O2-induced necrotic cell death results from PARP-dependent but TRPM2-indepdent depletion of ATP [163] (Fig. 4A). As mentioned above, CTZ has limited selectivity towards the TRPM2 channel and thus further studies using more specific pharmacological or genetic interventions are required to elaborate the role of the TRPM2 channel in ROS-induced neonatal cardiomyocyte death.
Fig. 4.

TRPM2-mediated signalling pathways in oxidative stress-induced cardiomyocyte death associated with myocardial ischemia/reperfusion injury.
(A) H2O2-induced neonatal cardiomyocyte death. Exposure to H2O2 induces apoptotic cell death via activation of PARP and the TRPM2 channel, TRPM2-mediated Na+ and Ca2+ influx and subsequent MCU-mediated mitochondrial Na+ and Ca2+ overloading, leading to mitochondrial dysfunction, cyt c release and caspase-3 activation to initiate apoptotic cell death. Exposure to H2O2 also induces necrotic cell death as a result of PARP-dependent and TRPM2-indpendent depletion of ATP. (B) TNF-α-induced adult cardiomyocyte death. Exposure to TNF-α induces caspase-8-dependent mitochondrial ROS generation, activation of PARP and the TRPM2 channel, TRPM2-mediated Ca2+ influx and cell death via unknown mechanism(s). (C) TRPM2 channel activation in adult cardiomyocytes has been proposed to play an opposing role by protecting against cardiomyocyte death induced by H2O2 or myocardial I/R. TRPM2-mediated Ca2+ influx activates Ca2+-dependent PYK2 kinase and ERK1/2 and Akt, downstream pro-survival signalling. PYK2 is also translocated to mitochondria and upregulates MCU function and MCU-mediated Ca2+ into mitochondria to maintain mitochondrial function and reduce mitochondrial ROS generation. Abbreviations: ADPR, ADP-ribose; cyt c, cytochrome c; ERK, extracellular signal-regulated kinase; MCU, mitochondrial Ca2+ uniporter; PARP, poly(ADPR) polymerase; PYK2, proline-rich tyrosine kinase 2; ROS, reactive oxygen species; TNF-α, tumour necrosis factor-α.
It is known that the proinflammatory cytokine TNF-α contributes to the exacerbation of myocardial I/R injury by inflammation via the induction of mitochondrial dysfunction and alteration in intracellular Ca2+ homeostasis in cardiomyocytes [164]. A recent study has examined, using ventricular myocytes isolated from adult mice, the TRPM2 channel in mediating TNF-α-induced cardiomyocyte death in relation to myocardial I/R injury [165]. Exposure to TNF-α (10 ng/ml for 1 h) induced mitochondrial ROS generation shown using MitoSOX Red, which was attenuated by treatment with the caspase-8 inhibitor z-IETD-FMK, indicating caspase-8-dependent mitochondrial ROS production. Prolonged exposure to TNF-α (10 ng/ml for up to 4 h) induced a cationic current that was reduced by treatment with NAC as well as z-IETD-FMK. TNF-α-induced current was abolished or reduced by treatment with 2-APB, CTZ, ACA, FFA or 3-AB. Exposure to H2O2 (100 μM) or application of intracellular cADPR (100 μM) evoked a similar cationic current that was reduced by treatment with 2-APB or CTZ. In addition, exposure to TNF-α for 4 h induced poly(ADPR) polymer formation. Together, these results support that TNF-α induces TRPM2 channel activation, dependent on ROS generation and PARP activation. In response to exposure to TNF-α, there was an initial increase in intracellular Ca2+ concentration followed by cell death detected by PI staining. Both the TNF-α-induced initial Ca2+ response and subsequent cell death were largely prevented by treatment with CTZ or an anti-TRPM2 blocking antibody [165]. These results in combination suggest that TNF-α induces cardiomyocyte death via caspase-8 activation, mitochondrial ROS generation, activation of PARP and the TRPM2 channel, and TRPM2-mediated Ca2+ signalling (Fig. 4B).
Hiroi et al. examined the role of the TRPM2 channel in I/R-induced cardiomyocyte death and myocardial injury in vivo using a mouse myocardial I/R model [166]. Exposure to ischemia for 45 min followed by reperfusion for 24 h led to considerable infarction in the myocardium of WT mice, which was reduced in TRPM2-KO mice. Myocardial I/R-induced increase in the plasma levels of troponin I, a marker of myocardial injury, measured 2 h post-reperfusion, was also reduced by TRPM2-KO. However, ischemia with various durations (45 min–24 h) without ensuing reperfusion resulted in a duration-dependent increase in infarct size that was comparable between WT and TRPM2-KO mice, suggesting exclusive involvement of the TRPM2 channel in reperfusion-induced myocardial injury, as reported for the TRPM2 channel in I/R-induced brain damage discussed above. Neutrophils are known to infiltrate the myocardium as a prominent inflammatory component of post-ischemic injury and an important source of proinflammatory mediators such as TNF-α and ROS contributing to myocardial I/R-induced injury. Neutrophil infiltration after myocardial I/R was attenuated in TRPM2-KO mice compared with that in WT mice. To elaborate the contribution of the TRPM2 channel in cardiomyocytes and infiltrating neutrophils in myocardial I/R injury, isolated hearts from WT and TRPM2-KO mice were perfused with polymorphonuclear leucocytes (PMNs) of each genotype before being subjected to ischemia via regional coronary ligation for 1 h followed by reperfusion for 2 h. I/R-induced infarction was less in WT hearts perfused with TRPM2-KO PMNs than with WT PMNs. The infarct size in TRPM2-KO hearts perfused with TRPM2-KO PMNs was smaller than that in TRPM2-KO perfused with WT PMNs. Moreover, the infarct size in TRPM2-KO hearts infused with WT PMNs was decreased by treatment with econazole. While the infarct size in WT mice with intravenous administration of TRPM2-KO PMNs, induced by ischemia for 45 min followed by reperfusion 2 h, was slightly but insignificantly attenuated compared to that in WT mice with WT PMNs, the infarct size in TRPM2-KO mice administrated with TRPM2-KO PMNs was smaller than in TRPM2-KO with WT PMNs. Furthermore, the maximal left ventricular pressure in TRPM2-KO mice was higher than that in WT mice after exposure to myocardial I/R, indicating that TRPM2 deficiency offers a protective effect on cardiac contractile function. These results support an important role for the TRPM2 channel, particularly the TRPM2 channel in neutrophils, in mediating myocardial I/R injury [166]. Overall, the findings from these in vitro and in vivo studies support the TRPM2 channel as an important molecular mechanism mediating I/R-induced cardiomyocyte death and myocardial injury.
Miller and colleagues provide evidence, however, that favours an opposing role of the TRPM2 channel in cardiomyocytes, namely, that the TRPM2 channel protects against ROS-induced cardiomyocyte death and myocardial I/R injury and thereby preserves cardiac function [96,[167], [168], [169], [170]]. As shown in an earlier study using ventricular myocytes isolated from adult mice [169], exposure to H2O2 (200 μM) increased intracellular Ca2+ concentration that was dependent on extracellular Ca2+ and reduced by treatment with CTZ. Such H2O2-induced Ca2+ response was smaller in myocytes isolated from TRPM2-KO mice. These results are overall similar to those reported in earlier studies by other groups using neonatal or adult ventricular myocytes [163,165], supporting a key role for the TRPM2 channel in mediating ROS-induced Ca2+ signalling in cardiomyocytes. However, the area at risk and infarct size were similar in hearts of WT and TRPM2-KO mice after exposure to ischemia for 30 min followed by reperfusion for 3 days. There was also no difference in I/R-induced reduction in systolic intracellular Ca2+ concentration and maximal contraction amplitude in myocytes from WT and TRPM2-KO mice. Nonetheless, both fractional shortening and the maximal first time derivative of the left ventricular pressure rise (+dP/dt), assessed on days 2–3 after myocardial I/R, were lower in TRPM2-KO hearts than in WT hearts, indicating TRPM2 deficiency worsens heart contractility. The expression of proteins that are important for excitation-contraction coupling was differentially altered in WT and TRPM2-KO mice after myocardial I/R, with the level of Na+/Ca2+ exchanger elevated and the level of Na+-K+-ATPase α1-subunit decreased in hearts from TRPM2-KO mice compared to WT mice. In addition, the action potential duration was prolonged in cardiomyocytes from TRPM2-KO mice relative to WT mice after exposure to myocardial I/R. Furthermore, ROS generation was higher in TRPM2-KO myocytes than in WT myocytes after exposure to hypoxia for 2 h followed by reoxygenation for 30 min. Consistently, the expression levels of superoxide dismutase 1 (SOD1) and SOD2, ROS-scavenging enzymes, and hypoxia-inducible factor-1α and forkhead box transcription factors, FoxO1 and FoxO3a, upstream regulators of SOD expression, were attenuated, whereas the NOX4 expression level was higher in hearts from TRPM2-KO mice exposed I/R. These results led to the conclusion that TRPM2 channel activation is protective against myocardial I/R injury by enhancing the ROS-scavenging capacity to reduce I/R-induced OS [169]. As reported in a subsequent study, exposure to myocardial I/R induced greater mitochondrial dysfunction in TRPM2-KO hearts than in WT hearts, suggesting a role for the TRPM2 channel in preserving mitochondrial function and thereby preventing mitochondrial ROS generation in cardiomyocytes [168]. Mice deficient in cardiac-specific TRPM2 expression, while showing similar I/R-induced infarction in the heart as WT mice, exhibited significantly reduced myocardial performance [167]. This study provided further evidence by expressing the mutant TRPM2 channel with impaired Ca2+ permeability in cardiomyocytes to show that TRPM2-mediated Ca2+ influx into cardiomyocytes is critical in the maintenance of mitochondrial function and protection against myocardial I/R injury. Moreover, as shown in a more recent study [170], exposure to H2O2 (200 μM for 15 min) increased the phosphorylation levels of the Ca2+-sensitive proline‐rich tyrosine kinase PYK2, and downstream pro-survival signalling molecules ERK1/2 and Akt in WT adult mouse cardiomyocytes, which was attenuated by TRPM2-KO and also by treatment with the intracellular Ca2+ chelator BAPTA-AM. In addition, exposure to H2O2 promoted the translocation of PYK2 from the cytosol to mitochondria in WT adult mouse cardiomyocytes. Exposure to myocardial I/R also elevated the levels of phosphorylation or activity of PYK2, ERK1/2 and Akt in WT hearts, which were attenuated in TRPM2-KO hearts. Furthermore, exposure to hypoxia for 30 min followed by reoxygenation for 30 min induced a greater level of mitochondrial ROS generation in TRPM2-KO myocytes than in WT myocytes. It has been proposed, therefore, that OS-induced TRPM2 channel activation protects against I/R-induced cardiomyocyte death and myocardial injury by a molecular and signalling mechanism, including TRPM2-mediated Ca2+ influx, Ca2+-dependent activation of PYK2 and subsequent activation of ERK1/2 and Akt and, in addition, translocation of PYK2 into mitochondria to upregulate the MCU function and MCU-mediated Ca2+ entry into the mitochondria to maintain mitochondrial function and reduce mitochondrial ROS generation (Fig. 4C).
It is clear that there are substantial discrepancies in the molecular and cellular mechanisms mediated by the TRPM2 channel and therefore it remains a matter of debate and further investigation whether the TRPM2 channel plays a beneficial or harmful role in myocardial I/R injury. Such a dichotomy in the role of the TRPM2 channel or TRPM2-mediated Ca2+ signalling in terms of cardiomyocyte function may be attributable to various factors [96], including the use of cardiomyocytes from mice of different ages (neonate versus adult), different experimental conditions and assessment methods, particularly different I/R models that are well known to be important in the study of myocardial I/R-induced injury and cardiovascular outcomes [171]. As such, further efforts with rigour and reproducibility in the design and execution of experiments and analysis of data may be required to clarify the physiological or pathological role of the TRPM2 channel in myocardial I/R injury.
6. TRPM2 in endothelial cell death associated with vascular dysfunction and cardiovascular diseases
Endothelial cells that line the interior surface of blood vessels are particularly susceptible to OS, mainly resulting from ROS production by neutrophils and other PMNs as well as by endothelial cells themselves [172]. It is well known that OS-induced vascular endothelial dysfunction plays a significant role in the pathophysiology of multiple cardiovascular diseases, including I/R injury, hypertension, heart failure, diabetes, pulmonary inflammation and atherosclerosis [23,24,28,[173], [174], [175], [176], [177], [178], [179]]. Hecquet et al. were the first to examine endothelial expression of the TRPM2 channel using human pulmonary artery endothelial cells [74]. Exposure to H2O2 (300 μM) elicited an inward cationic current that was attenuated by treatment with TRPM2-siRNA or with DPQ. Consistently, exposure to H2O2 (25–300 μM) induced a concentration-dependent increase in intracellular Ca2+ concentration, which was dependent on extracellular Ca2+. H2O2-induced Ca2+ response was inhibited by treatment with TRPM2-siRNA or an anti-TRPM2 blocking antibody, by overexpression of TRPM2-S, and by treatment with 3-AB or DPQ. These results support functional expression of the TRPM2 channel and PARP-mediated activation by ROS [74]. The study further showed a significant role for the TRPM2 channel in mediating H2O2-induced endothelial barrier dysfunction [74]. A separate study by Wang et al. reported that TRPM2-mediated Ca2+ signalling in endothelial cells plays an important role in coupling ultrafine PMs-induced ROS generation to activation of calpain and subsequent degradation of zonula occludens tight junction proteins to impair endothelial barrier function [180].
Sun et al. examined the role of the TRPM2 channel in mediating OS-induced endothelial cell death using H5V, a mouse endothelial cell line [181]. Exposure to 3 mM H2O2 resulted in a cationic current and Ca2+ influx, both of which were attenuated by treatment with TRPM2-shRNA or an anti-TRPM2 blocking antibody. Exposure to H2O2 (156–2500 μM for 1 h) resulted in a significant and concentration-dependent reduction in cell viability, measured using the MTT assay, which was attenuated by treatment with TRPM2-shRNA. The activity of caspase-3 and caspase-8 was elevated and the level of pro-caspase-9 was reduced in cells after exposure to H2O2 (3 mM for 6 h), and DNA fragmentation, and nuclear condensation and fragmentation was observed in cells after exposure to H2O2 (3 mM for 24 h). All of these effects were reduced by treatment with TRPM2-shRNA or an anti-TRPM2 blocking antibody. As shown in the same study, exposure to TNF-α (10 ng/ml for 36 h), which is proposed to induce ROS production and mediate endothelial cell apoptosis with a potential role in atherosclerosis development [182], reduced cell viability assessed using the MTT assay, which was attenuated by treatment with TRPM2-shRNA or an anti-TRPM2 blocking antibody [181]. These results support that OS, as a result of exposure to H2O2 or TNF-α-induced ROS generation, induces TRPM2 channel activation, leading to caspase-dependent apoptosis and thereby a reduction in endothelial cell viability (Fig. 5A). A recent study by Hecquet et al. has confirmed the role of the TRPM2 channel in H2O2-induced apoptosis in human pulmonary artery endothelial cells [77]. Exposure to H2O2 (25–300 μM for 24 h) caused a concentration-dependent increase in apoptotic cells detected by staining with annexin V-phycoerythrin and 7-aminoactinomycin D. Exposure to glucose oxidase (GO) together with glucose for 1.5 h, which is known to induce H2O2 generation, also induced endothelial cells to undergo apoptosis. Apoptotic cell death induced by both H2O2 and GO was attenuated by treatment with an anti-TRPM2 blocking antibody or TRPM2-siRNA. Perfusion of WT mice with H2O2 (300 μM) or GO (1–2.5 mU/mL) for 75 min caused apoptosis of endothelial cells in lung vessels, assessed by immunostaining of vascular endothelial-cadherin and cleaved-PARP due to activation of apoptosis-associated caspases, 3 h after perfusion with H2O2 or GO. Consistently, perfusion with H2O2 or GO resulted in caspase-3 activation and PARP cleavage in the lung tissue. These effects induced by H2O2 or GO were attenuated or prevented in the lungs of TRPM2-KO mice. As shown in human pulmonary artery endothelial cells, endothelial cells express both full-length TRPM2 and TRPM2-S isoforms. Interestingly, TRPM2-S interacted with TRPM2 and also with PKCα, shown by co-immunoprecipitation. Exposure to H2O2 (300 μM for up to 5 min) led to phosphorylation of TRPM2-S and PKCα, and also an increase in their interaction; both H2O2-induced phosphorylation and interaction of these two proteins were largely prevented by treatment with Gö6976, or the PKCα inhibitor peptide PKCαi, as well as by treatment with TRPM2-siRNA and PKCα-siRNA. Ca2+ influx-dependent intracellular Ca2+ increase induced by exposure to H2O2 (100 μM) was also attenuated by treatment with Gö6976, PKCαi, PKCα-siRNA or TRPM2-siRNA. Furthermore, H2O2-induced phosphorylation and interaction of TRPM2-S and PKCα, and Ca2+ response were abolished by overexpression of TRPM2-S with the phosphorylation site Ser39 mutated to alanine. Exposure to H2O2 (100 μM for up to 5 min) induced rapid dissociation of TRPM2-S from TRPM2. H2O2-induced dissociation of TRPM2-S and TRPM2 and also H2O2/GO-induced apoptotic cell death were attenuated by treatment with Gö6976 or PKCαi, or overexpression of TRPM2-S carrying the S39A mutation. H2O2-induced TRPM2-mediated Ca2+ influx and apoptotic cell death were reduced in endothelial cells from PKCα-KO mice. These results support a novel mechanism, by which TRPM2-S inhibits the TRPM2 channel and its role in mediating ROS-induced apoptosis in endothelial cells (Fig. 5B). TRPM2-S forms a complex with the TRPM2 channel under resting conditions. ROS activate PKCα that in turn interacts with TRPM2-S and phosphorylates Ser39, resulting in dissociation of TRPM2-S from the TRPM2 channel and loss of the inhibition, and ROS also activate PARP and the TRPM2 channel, inducing TRPM2-mediated Ca2+ influx to trigger apoptotic cell death (Fig. 5B). To illustrate the pathophysiological significance of such a molecular and cellular mechanism, endothelial cell apoptosis in mouse lungs and survival were examined in mice after intraperitoneal injection with lipopolysaccharide (LPS), a bacterial endotoxin known to produce ROS in endothelial cells. Endothelial cell apoptosis in the lungs, determined 4 h after injection of LPS by staining frozen lung tissues with a vascular endothelial-cadherin antibody and TUNEL, was observed in WT mice, but largely eliminated in PKCα-KO or TRPM2-KO mice that were prior transplanted with bone marrow cells from WT mice to restore the expression of PKCα or TRPM2 in immune cells. Consistently, the survival rate during the 96 h following LPS injection was significantly improved in TRPM2-KO and PKCα-KO chimeric mice compared to WT mice. Overall, these results suggest that OS-induced apoptotic cell death leads to endothelial dysfunction and increases the susceptibility to lung inflammation and damage. These findings gain further support from a more recent study by the same group using tamoxifen-inducible endothelial cell-specific conditional TRPM2-KO mice [183]. Endothelial cell-specific TRPM2-KO mice had a survival rate of 80%, in contrast with the survival of no WT mice, 96 h after intraperitoneal injection with LPS. Moreover, compared to WT mice, endothelial cell-specific TRPM2-KO mice had significantly lower myeloperoxidase activity at 6 and 24 h following treatment with LPS, and exhibited reduced infiltration of PMNs into lung tissues. It was further shown that PMNs-derived ROS generation is sufficient in inducing TRPM2-mediated increase in intracellular Ca2+ concentration in endothelial cells to trigger endothelial barrier dysfunction and trans-endothelial migration of PMNs [183]. It remains unclear whether such PMNs-derived ROS-induced TRPM2 channel activation results in endothelial cell apoptosis. In summary, studies have so far provided clear evidence to support an important role for the TRPM2 channel or TRPM2-mediated Ca2+ signalling in OS-induced endothelial cell death and endothelial barrier dysfunction, leading to increased extravasation of PMNs and vascular inflammation, a mechanism with potential significance in the pathophysiology of vascular diseases.
Fig. 5.

TRPM2-mediated signalling pathways in oxidative stress-induced endothelial cell death associated with endothelial barrier dysfunction and vascular diseases.
Two distinctive but overlapping signalling mechanisms mediate H2O2-induced endothelial death. (A) Exposure to H2O2 induces apoptotic cell death via activation of PARP and the TRPM2 channel, TRPM2-mediated Ca2+ influx and subsequent activation of caspase-3, caspase-8 and caspase-9 activation to initiate apoptotic cell death. Exposure to TNF-α also induces apoptotic cell death through the same signalling mechanism. (B) Exposure to H2O2 also induces PKCα activation, which in turn phosphorylates TRPM2-S and promotes dissociation of TRPM2-S with, and releases its inhibition of, the TRPM2 channel, resulting in increased TRPM2-mediated Ca2+ influx and caspase-3 activation. ROS generation by GO in the presence of glucose induces endothelial cell death via the signalling mechanisms presented in (B). Abbreviations: ADPR, ADP-ribose; GO, glucose oxidase; PARP, poly(ADPR) polymerase; PKCα, protein kinase Cα; ROS, reactive oxygen species; TNF-α, tumour necrosis factor-α.
7. TRPM2 in pancreatic cell death associated with diabetes and acute biliary pancreatitis
7.1. H2O2/STZ-induced pancreatic β-cell death associated with diabetes
Pancreatic β-cells make up 50–70% of the cells in human islets and are responsible for the generation of insulin. Insulin insufficiency due to loss of pancreatic β-cells and impairment of their function leads to hypoinsulinemia and hyperglycaemia or diabetes [184]. It is well recognised that pancreatic β-cells are weak in their antioxidant capacity and thus known to be highly susceptible to OS damage [185]. Streptozotocin (STZ) has the ability to induce ROS generation as well as DNA damage and therefore is used clinically as a chemotherapeutic agent in the treatment of pancreatic β-cell carcinoma, and is also used experimentally as a diabetogenic agent to induce diabetes-like pathologies in rodents [186]. It is worth mentioning that activation of nuclear PARP-1 is long known to be critical in mediating STZ-induced loss of pancreatic β-cells and diabetes in rodents [187,188]. An early study reported functional expression of a molecularly unidentified Ca2+-permeable channel, which is known as TRPM2 today, that mediated H2O2-induced Ca2+ signalling and cell death in CRI-G1, a rat insulin-secreting cell line [189]. A number of studies, using various insulin-secreting cell lines, rodent and human islets, and mouse models of diabetes, have examined the role of PARP-dependent TRPM2 channel activation in mediating pancreatic β-cell death induced by H2O2 or stimuli known to induce OS, with particular interest in the pathogenesis of both type 1 and type 2 diabetes (T1D and T2D). Ishii et al. showed that exposure to H2O2 (100 μM) elevated intracellular Ca2+ concentration in RIN-5F, a rat insulin-secreting pancreatic islet tumour cell line, which was attenuated by treatment with 3-AB or PJ34 [39]. In addition, exposure to H2O2 (300 μM for up to 4 h) induced cell death, shown using the trypan blue exclusion method. The onset of H2O2-induced cell death was delayed by treatment with 3-AB, and H2O2-induced cell death was attenuated by treatment with 3-AB or PJ34 [39]. Similarly, we showed using PI staining that exposure to H2O2 (200 μM for 2 h) resulted in cell death in INS-1 cells, an insulin-secreting β-cell line derived from a rat insulinoma, which was attenuated by treatment with PJ34 or 2-APB, supporting that PARP-dependent TRPM2 channel activation mediates H2O2-induced cell death [36]. H2O2-induced cell death was suppressed by treatment with the caspase-dependent apoptosis inhibitor Ac-DEVD-CHO, but not with the necrosis inhibitor IM-54 or the necroptosis inhibitor necrostatin-1, indicating apoptotic cell death, as is known for pancreatic β-cell death in diabetes [190]. Like H2O2-induced neuronal cell death discussed above, H2O2-induced cell death in INS-1 cells was prevented by treatment with TPEN or clioquinol, supporting that alteration in intracellular Zn2+ homeostasis is vital for such H2O2-induced cell death [36]. Consistently, exposure to H2O2 (200 μM for 2 h) resulted in intracellular Zn2+ increase shown using FluoZin-3 staining, which was suppressed by treatment with PJ34 or 2-APB. Furthermore, by examining the co-localization of FluoZin-3 staining and intracellular organelle-specific fluorescent indicators, we showed that labile Zn2+ was mainly present in the lysosomes, but not in the ER or mitochondria in INS-1 under control conditions and that exposure to H2O2 induced lysosomal Zn2+release, which was prevented by treatment with PJ34 or 2-APB [36]. T1D is a condition characterised by loss of pancreatic β-cells and to large extent can be recapitulated in mice administered with multiple low doses of STZ. We showed that mice injected with STZ (40 mg/kg, 6 times daily) exhibited extensive loss of pancreatic β-cells, revealed by immunostaining of insulin in the pancreas, and hyperglycaemia determined by blood glucose levels after overnight fasting. Importantly, these STZ-induced detrimental effects on pancreatic β-cells and the diabetes-associated phenotype were largely prevented by TRPM2-KO, indicating that TRPM2-mediated pancreatic β-cell death is a key mechanism in T1D pathogenesis. Consistently, exposure to STZ (2 mM for 48 h) led to considerable destruction of pancreatic islets isolated from WT mice, which was rescued by TRPM2-KO as well as treatment with PJ34 or TPEN [36]. Taken together, these results have led us to propose that STZ-induced PARP-dependent TRPM2 channel activation, TRPM2-mediated Ca2+ influx and TRPM2-mediated lysosomal Zn2+ release alter intracellular Ca2+ and Zn2+ homeostasis and drive apoptotic cell death in pancreatic β-cells and the development of T1D (Fig. 6A).
Fig. 6.

TRPM2-mediated signalling pathways in oxidative stress-induced pancreatic cell death associated with diabetes and acute biliary pancreatitis.
(A) STZ-induced cell death in pancreatic β-cells associated with type 1 diabetes. Exposure to STZ induces an increase in the ROS level and PARP-dependent TRPM2 channel activation, leading to cell death in a signalling pathway elucidated mainly based on examining H2O2-induced cell death in INS-1 insulin-secreting cells. Activation of the TRPM2 channel on the cell surface induces Ca2+ influx and intracellular Ca2+ increase, and activation of TRPM2 in the lysosomes results in lysosomal Zn2+ release. Alterations in intracellular Ca2+ and Zn2+ homeostasis lead to apoptosis. (B) Palmitate-induced cell death in pancreatic β-cells associated with type 2 diabetes. Exposure to palmitate triggers activation of NOX for ROS generation. ROS-induced PARP-dependent TRPM2 channel activation in the lysosomes results in lysosomal Zn2+ release, mitochondrial Zn2+ accumulation, recruitment of Drp1 to mitochondrial and Drp1-mediated mitochondrial fission, leading to mitochondrial dysfunction and apoptosis. (C) Bile acid-induced cell death in pancreatic acinar cells associated with acute biliary pancreatitis. Exposure to CDC induces ROS generation and TRPM2 channel activation likely via PARP, leading to intracellular Ca2+ increase and necrotic cell death. Abbreviations: ADPR, ADP-ribose; CDC, chenodeoxycholate; Drp1, dynamin-related protein 1; NOX, NADPH oxidase; PARP, poly(ADPR) polymerase; ROS, reactive oxygen species; STZ, streptozotocin.
It is known that accumulation of free fatty acids, a significant risk factor in T2D, promotes ROS generation and reduces insulin secretion by pancreatic β-cells [191]. A recent study by Li et al. using INS-1832/13 cells, a subclone of INS-1 cells, has investigated the role of the TRPM2 channel in pancreatic β-cell death in response to palmitate [121]. Exposure to palmitate (500 μM for 12 h) induced apoptotic cell death, shown by analysis of annexin V-GFP and PI staining. Like H2O2-induced cell death, palmitate-induced cell death was inhibited by treatment with PJ34 or TRPM2-siRNA, and also by treatment with TPEN. Consistently, extended exposure to palmitate (500 μM for 5 days) resulted in significant cell death shown using PI staining in pancreatic islets from WT mice but not from TRPM2-KO mice. It was of notice that human pancreatic islets, compared to mouse pancreatic islets, were relatively resistant to damage by palmitate alone. Nonetheless, extended exposure to palmitate (500 μM) in combination with IL-1β (5 ng/ml) and IFN-γ (5 ng/ml), cytokines known to be increased in obesity, for 7 days resulted in significant apoptotic cell death determined using TUNEL staining, which was reduced by treatment with PJ34, ACA or TRPM2-siRNA [121]. These results provide evidence to support a crucial role for the TRPM2 channel in mediating palmitate-induced alteration in intracellular Zn2+ homeostasis and ensuing apoptotic pancreatic β-cell death, a mechanism of potential relevance to T2D pathogenesis. This study went on to explore the underlying molecular and signalling mechanism [121]. Exposure to palmitate (500 μM for 4 h) led to mitochondrial fragmentation in INS-1832/13 cells that was prevented by treatment with apocynin or NOX-2 specific peptide gp91ds-tat and also by treatment with NAC, indicating critical engagement of NOX-mediated ROS generation. Palmitate-induced mitochondrial fragmentation was also inhibited by treatment with PJ34 or TRPM2-siRNA. Furthermore, exposure to palmitate (500 μM for 5 days) caused mitochondrial fragmentation in pancreatic islets from WT mice, which was not observed in pancreatic islets from TRPM2-KO mice [121]. These results support the importance of the TRPM2 channel in pancreatic β-cells in coupling palmitate-induced NOX-mediated ROS generation and PARP activation to mitochondrial dysfunction and apoptotic cell death. As shown for H2O2/STZ-induced cell death, alteration in intracellular Zn2+ homeostasis is critical for palmitate-induced TRPM2-mediated mitochondrial dysfunction and cell death. Exposure to palmitate (500 μM for 4 h) elevated the Zn2+ level in mitochondria that was inhibited by treatment with PJ34 or TRPM2-siRNA [121]. Extended exposure to palmitate induced mitochondrial membrane permeabilisation showed by the diffusion of MitoTracker into the cytoplasm, which was prevented by treatment with the Zn2+ chelators TPEN and TPA. Consistently, exposure to palmitate resulted in a reduction of mitochondrial membrane potential determined using JC-10 staining, which was also attenuated by treatment with PJ34, TRPM2-siRNA or TPEN. These results suggest that palmitate-induced PARP-dependent TRPM2 channel activation stimulates mitochondrial Zn2+ accumulation and thereby impairs mitochondrial function. Dynamin-related protein 1 (Drp1) is a dynamin-related GTPase with a key role in mitochondrial fission [192]. The study, by confocal imaging of the subcellular location of GFP-tagged Drp1, demonstrated that exposure to palmitate promoted the translocation of Drp-1 from the cytoplasm to mitochondria, which was attenuated by treatment with PJ34, TRPM2-siRNA or TPEN [121]. Taken together, these results show that palmitate induces apoptosis in pancreatic β-cells via a molecular and signalling mechanism comprised of NOX activation, NOX-mediated ROS generation, activation of PARP and the TRPM2 channel, TRPM2-mediated lysosomal Zn2+ release, mitochondrial Zn2+ accumulation, mitochondrial fragmentation via Drp1-mediated fission, and mitochondrial dysfunction (Fig. 6B).
7.2. Bile acid-induced pancreatic acinar cell death associated with acute biliary pancreatitis
Acute biliary pancreatitis (ABP) has a multifactorial and complex aetiology [193]. An important factor in ABP pathogenesis is the dysregulation of intracellular Ca2+ signalling in pancreatic acinar cells as a result of the excessive production of bile acids, and ensuing alteration in intracellular Ca2+ homeostasis leads to protease activation, mitochondrial dysfunction and ATP depletion that induces necrotic cell death, in addition to mitochondrial ROS generation that triggers the apoptotic pathway [[194], [195], [196], [197], [198]]. Fanczal et al. have recently examined the role of the TRPM2 channel in pancreatic acinar cells, particularly TRPM2-mediated Ca2+ signalling, in ABP pathogenesis [46]. Exposure to H2O2 (1 mM) induced intracellular Ca2+ increase in pancreatic acinar cells isolated from WT mice that were attenuated by removal of extracellular Ca2+, supporting H2O2-induced Ca2+ influx. Exposure to 250 μM chenodeoxycholate (CDC), a bile acid, induced intracellular Ca2+ increase and also ROS generation in pancreatic acinar cells, shown using H2DCFDA (also known as DCFH-DA), a fluorescent indicator of cellular ROS. CDC-induced Ca2+ responses were reduced in pancreatic acinar cells from TRPM2-KO mice, suggesting that TRPM2 channel activation is a significant mechanism for bile acid-induced Ca2+ signalling. Exposure to H2O2 (1 mM for 30 min) induced cell death in acinar cells isolated from WT mice mainly via necrosis, examined using DNA Nuclear Green DCS1 and Apopxin™ Deep Red that stain necrotic and apoptotic cells, respectively. Similarly, exposure to CDC (250 μM for 30 min) also induced necrosis in WT acinar cells. Cell death induced by H2O2 and CDC was attenuated in acinar cells from TRPM2-KO mice, providing strong evidence for TRPM2-mediated H2O2/CDC-induced necrotic cell death in pancreatic acinar cells. In addition, short exposure to H2O2 (1 mM) or CDC (250 μM) led to a reduction in mitochondrial membrane potential in WT acinar cells, measured using tertamethylrhodamine-methyl ester. Intriguingly, such an effect on mitochondrial function induced by H2O2, but not induced by CDC, was significantly attenuated by TRPM2-KO, indicating a TRPM2-independent mechanism for bile acid-induced mitochondrial dysfunction. The study further showed that intraductal infusion of mice with 4% sodium taurocholate to model necrotising pancreatitis induced similar interstitial oedema and leukocyte infiltration in WT and TRPM2-KO mice, examined using haematoxylin and eosin (H&E) staining. However, the serum amylase activity and the extent of necrosis in the pancreas were significantly reduced by TRPM2-KO [46]. Taken together, this study is the first to demonstrate an important role for the TRPM2 channel in mediating bile acid-induced Ca2+ signalling and necrotic cell death in pancreatic acinar cells (Fig. 6C), providing evidence to implicate the TRPM2 channel as a previously unrecognised mechanism of ABP pathogenesis.
8. TRPM2 in kidney cell death associated with acute kidney injury and chronic kidney disease
8.1. Ischemia/reperfusion-induced kidney cell death in acute kidney injury
Ischemic acute kidney injury (AKI) is a frequent complication that is caused by acute limitation or deprivation of arterial blood flow to the kidneys, particularly by restoration of renal blood supply, and AKI is causatively associated with the development of chronic kidney diseases (CKD) [[199], [200], [201]]. It has been proposed that excessive production of ROS and TNF-α as well as elevation in intracellular Ca2+ concentration in kidney cells represent important pathological factors, and that cell death, both apoptotic and necrotic, acts as an important cellular mechanism in the pathogenesis of AKI. Gao et al. examined the TRPM2 channel in renal I/R-induced kidney injury [38]. Bilateral kidney ischemia followed by reperfusion caused kidney injury in WT mice, examined histologically using periodic acid Schiff staining, which was attenuated in TRPM2-KO mice. I/R also led to impairment in kidney function in WT mice, indicated by increases in the blood levels of urea nitrogen and creatinine, which was reduced in TRPM2-KO mice. Such kidney damage and loss of function in WT mice were also reduced in mice injected with 2-APB prior to ischemia or after reperfusion. A more recent study has reported that I/R-induced kidney damage examined histologically using H&E staining in WT mice was prevented by treatment with 8-Br-cADPR to antagonise TRPM2 channel activation [112]. These studies provide consistent evidence to support an important role for the TRPM2 channel in I/R-induced acute kidney injury. In agreement with a role for inflammation, I/R resulted in considerable neutrophil infiltration into the kidneys of WT mice that was attenuated in TRPM2-KO mice. However, selective deletion of TRPM2 expression in parenchymal cells, but not in hematopoietic cells protected against I/R-induced kidney damage, indicating the TRPM2 channel in kidney cells as the major mediator [38]. It is known that I/R causes apoptosis of tubular epithelial cells in the kidney [202]. I/R resulted in apoptotic cell death examined using TUNEL staining 24 h post-reperfusion and, consistently, increases in activation of caspase-3 and caspase-9 and cleavage of PARP in kidney cells of WT mice, all of which were reduced in TRPM2-KO mice [38]. Conversely, the expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL were augmented in the kidney of TRPM2-KO mice subjected to I/R. These results support that TRPM2 channel activation leads to initiation of apoptotic pathways in kidney cells. I/R stimulated activation of NOX and increased levels of RAC1, a GTPase involved in the activation of NOX, in the kidney cells of WT mice, both of which were lessened in TRPM2-KO mice. Furthermore, RAC1 was found to interact with the TRPM2 channel, and such protein-protein interaction was stimulated by I/R-induced RAC1 activation, leading to an increase in the cell surface expression of the TRPM2 channel. Consistently, treatment with the RAC1 inhibitor NSC23766 before ischemia protected against I/R-induced damage to kidney function in WT mice. Such treatment also reduced the expression of OS biomarkers, NOX activity, PARP cleavage and caspase-3 activation, while increasing the level of Bcl-2 in mice subjected to I/R [38]. Collectively, these results support an important role for the TRPM2 channel in coupling OS resulting from I/R-induced NOX-mediated ROS generation to activation of apoptotic pathways to drive kidney cell death, leading to kidney injury (Fig. 7A).
Fig. 7.

TRPM2-mediated signalling pathways in oxidative stress-induced kidney cell death associated with acute kidney injury and chronic kidney disease.
(A) Ischemia/reperfusion-induced kidney cell death associated with acute kidney injury. Exposure to transient ischemia followed by reperfusion increases RAC1 activity that stimulates NOX activation and NOX-mediated ROS production, activation of PARP and the TRPM2 channel. TRPM2 channel activation leads to intracellular Ca2+ increase, downregulation of anti-apoptotic proteins (Bcl-2 and Bcl-xL) and activation of caspase-3 and caspase-9 to trigger apoptosis. (B) Albumin-induced cell death in cortical collecting duct cells related to chronic kidney disease, based on examining BSA-induced cell death in mpkCCDc14 murine cortical collecting duct cells. Exposure to BSA increases ROS production and reduces antioxidant capacity of the cell by downregulating GPx and GSH, increasing intracellular ROS level. PARP-dependent TRPM2 channel activation results in intracellular Ca2+ increase, contributing to mitochondrial dysfunction, activation of caspase-3 and caspase-9 and inducing apoptotic cell death. Abbreviations: ADPR, ADP-ribose; BSA, bovine serum albumin; GPx, glutathione peroxidase; GSH, glutathione; I/R, ischemia/reperfusion; NOX, NADPH oxidase; PARP, poly(ADPR) polymerase; ROS, reactive oxygen species.
8.2. BSA-induced kidney cortical collecting duct cell death associated with chronic kidney disease
Albumin functions as a carrier protein in the blood that transports various substances such as fatty acids and hormones around the body and is filtered by the glomeruli in the kidneys to be reabsorbed by epithelial cells. However, it is known that albumin excreted through urine following glomerular injury or tubular damage stimulates ROS production and elicits a proinflammatory response in cortical collecting duct cells that exacerbates CKD progression [203]. Nazıroğlu et al. have recently examined the TRPM2 channel in bovine serum albumin (BSA)-induced cellular damage within the kidneys [125]. In the mpkCCDc14 murine cortical collecting duct cell line, exposure to BSA (25 mg/ml) enhanced the ADPR-induced cationic current that was attenuated by treatment with ACA, and also caused an increase in intracellular Ca2+ concentration that was reduced by treatment with PJ34, DPQ or 2-APB. Prolonged exposure to BSA (25 mg/ml for 24 h) induced apoptotic cell death, shown using the APOPercentage apoptosis assay, which was reduced by treatment with ACA or the antioxidant compound curcumin and, conversely, augmented by treatment with the oxidising agent cumene hydroperoxide. Collectively, these results support that BSA induces apoptotic cell death via PARP-dependent TRPM2 channel activation. Exposure to BSA (25 mg/ml) stimulated ROS production shown using H2DCFDA, and also reduced the ROS-scavenging ability of cells indicated by a reduction in the levels of glutathione (GSH) peroxidase (GPx) and GSH, both of which were also rescued by treatment with curcumin. These results suggest both ROS generation and impairment in antioxidant capacity contribute to BSA-induced OS. Consistent with BSA-induced apoptotic cell death, exposure to BSA caused a reduction in mitochondrial membrane potential determined by JC-1 staining and an increase in the activation of caspase-3 and caspase-9, both of which were diminished by treatment with ACA or curcumin [125]. These results in combination suggest that exposure to albuminuria-associated conditions induces OS that in turn activates PARP and the TRPM2 channel, and TRPM2-mediated Ca2+ signalling initiates mitochondrial dysfunction and apoptotic cell death in kidney collecting duct cells (Fig. 7B). These findings provide new insights to albuminuria-associated injury to kidneys in the pathogenesis and progression of CKD.
9. TRPM2 in hepatocyte death associated with acetaminophen overdose-induced liver damage
Acetaminophen (or paracetamol) is a commonly used analgesic and antipyretic drug, though acetaminophen overdose has been well recognised as a major cause of acute liver damage worldwide by inducing hepatocyte death. OS has long been known as a key mediator of acetaminophen-induced hepatocyte death and liver damage [204,205]. Recent research has drawn attention to the TRPM2 channel for its role in acetaminophen-induced hepatocyte death and the underlying signalling mechanism [41,206]. Kheradpezhouh et al. showed that exposure to acetaminophen (10–15 mM for 1 h) evoked Ca2+ influx to increase intracellular Ca2+ concentration and a cationic current in cultured rat and mouse hepatocytes [41]. Both acetaminophen-induced Ca2+ influx and currents in rat hepatocytes were blocked by treatment with ACA, CTZ or TRPM2-siRNA. Acetaminophen-induced Ca2+ influx in rat hepatocytes was also inhibited by treatment with DPQ. Similarly, acetaminophen-induced Ca2+ responses in mouse hepatocytes were prevented by treatment with ACA or CTZ, and also by TRPM2-KO [41]. Together, these results support that acetaminophen induces Ca2+ signalling via PARP-dependent TRPM2 channel activation. Prolonged exposure to acetaminophen (10 mM for 16 h) led to cell death in rat hepatocytes assessed using the trypan blue exclusion method, which was inhibited by treatment with ACA [41]. In agreement with acetaminophen-induced hepatotoxicity in vitro, liver sections of WT mice injected with acetaminophen (500 mg/kg) exhibited extensive hepatic necrosis as well as infiltration of lymphocytes and haemorrhage, visualised using H&E staining. Furthermore, injection of acetaminophen caused liver damage in mice, indicated by an increase in the concentration of the liver enzymes alanine transaminase and aspartate transaminase within the blood determined 24 h following injection. These acetaminophen-induced detrimental effects were significantly reduced in TRPM2-KO mice. Collectively, this study provides evidence to show that TRPM2 channel activation acts as an important mechanism mediating liver damage resulting from acetaminophen overdose (Fig. 8).
Fig. 8.

TRPM2-mediated signalling pathways in oxidative stress-induced hepatocyte death associated with acetaminophen overdose-induced liver damage.
Exposure to high concentrations of acetaminophen leads to OS by increasing ROS production in hepatocytes, and subsequent PARP-dependent TRPM2 channel activation and intracellular Ca2+ increase. Ca2+ activates CaMKII that in turn phosphorylates BECN1 protein, thereby reducing autophagy and stimulating apoptosis. Abbreviations: ADPR, ADP-ribose; BECN1, Beclin-1; CaMKII, Ca2+/calmodulin-dependent protein kinase II; PARP, poly(ADPR) polymerase; ROS, reactive oxygen species.
A recent study by Wang et al. has confirmed that exposure to acetaminophen (15 mM) reduced the viability of cultured mouse hepatocytes, assessed using the MTT assay, which was largely suppressed by treatment with CTZ [206]. Intriguingly, it was found that exposure to acetaminophen induced cellular ROS generation in mouse hepatocytes shown using CM-H2DCFDA, which was also prevented by treatment with CTZ. The study also showed that administration of acetaminophen (500 mg/kg) in mice caused liver cell death shown by H&E staining, and damage to liver function indicated by increased serum levels of the liver enzymes alanine transaminase and aspartate transaminase. However, this study provides evidence to suggest that apoptosis rather than necrosis mediates hepatocyte death resulting from acetaminophen overdose. Exposure to acetaminophen (≥15 mM) reduced the level of LC3-II in cultured mouse hepatocytes, which was rescued by treatment with NAC or CTZ. Detailed examination of the effects of exposure to H2O2 (75 μM) on autophagy in HeLa cells (without TRPM2 expression) or HeLa cells overexpressing recombinant human TRPM2 channel led to identification of a novel TRPM2-mediated molecular and signalling mechanism for acetaminophen-induced cell death. As illustrated in Fig. 8, TRPM2-mediated Ca2+ signalling activates Ca2+/calmodulin-dependent kinase II (CaMKII) and CaMKII in turn phosphorylates BECN1 protein, which reduces the interaction of BECN1 with PIK3C3 to inhibit autophagy and, at the same time, increase the interaction of BECN1 with anti-apoptotic Bcl-2 to induce Bcl-2 dissociation from pro-apoptotic Bax protein and promote apoptosis. This finding is consistent with an early study showing that OS mediates acetaminophen-induced hepatocyte death by inhibiting autophagy [207]. Wang et al. have further shown that acetaminophen-induced decrease in the level of LC3-II in mouse hepatocytes was prevented by treatment with the calmodulin inhibitor W-13 or the CaMKII inhibitor KN-93 [206]. Similarly, acetaminophen-induced liver injury in WT mice, revealed by H&E staining of liver sections and by measuring serum liver enzymes, was mitigated by treatment with KN-93 [208]. These results support CaMKII as a key signalling molecule in the above-described TRPM2-mediated molecular and signalling mechanism that mediates acetaminophen-induced apoptotic cell death in hepatocytes and liver damage (Fig. 8).
10. Concluding remarks and perspectives
OS has been well recognised as a salient and common feature in, and more importantly a significant pathological factor for, various diseases and conditions. It is evident from the above discussion that the TRPM2 channel, expressed in a variety of cells and tissues, plays an important role in mediating OS-induced cell death and damage to the physiological functions of diverse cells and tissues via alteration of intracellular ion homeostasis, particularly Ca2+ and Zn2+, and disruption of diverse molecular and signalling mechanisms. As summarised in Table 1, increasing evidence supports such TRPM2-mediated cell death as an important cellular mechanism coupling OS induced by a diverse range of pathological factors to associated pathological conditions. It is known that distinct mechanisms direct cell death such as apoptosis, necrosis, necroptosis and autophagy [208]. Considerable progress has been made in developing a mechanistic understanding of how OS-induced TRPM2 channel activation triggers cell death via apoptosis, necrosis or autophagy, and nevertheless the cell death pathways are still not clearly defined in many cases. In addition, it remains less clear why TRPM2 channel activation in different cell types by pathology-specific factors initiates distinct signalling mechanisms and death pathways. Further efforts are required to understand whether such TRPM2-mediated cell death mechanisms are intrinsic to specific cell types, depend on the nature of OS-inducing pathological factors, or both. For example, ZnO-NPs-induced TRPM2 channel activation stimulates autophagy to induce apoptotic cell death in pericytes and, by contrast, acetaminophen overdose-induced TRPM2 channel activation subverts autophagy in order to enhance apoptotic cell death in hepatocytes. It is also noted that a number of studies show that the TRPM2 channel, besides acting as a sensor or mediator for OS, also participates in the induction of OS in diverse cell types upon exposure to various pathological factors. Our studies in hippocampal neurons and also in SH-SY5Y cells have led us to propose that TRPM2 channel activation is central to a vicious positive feedback mechanism driving ROS/Aβ-induced neuronal cell death [82,124]. Such a mechanism offers an appealing explanation to the intriguing observation that acetaminophen-induced ROS generation in mouse hepatocytes was prevented by inhibition of the TRPM2 channel with CTZ [206]. It is likely that a similar positive feedback mechanism is engaged in TRPM2-mediated cell death in other cell types discussed above. It is worthwhile to mention that activation of the TRPM2 channel in phagocytes can induce membrane depolarisation that suppresses the activity of membrane potential-sensitive NOX and thereby reduces NOX-mediated ROS generation, a negative feedback mechanism that is important for the role of the TRPM2 channel in protecting against LPS-induced lung inflammation [209]. Therefore, a better understanding of the role of the TRPM2 channel in the pathogenesis of OS-associated diseases is crucial for rationalising the strategies of developing therapeutics. Taking AD as an example, in addition to the positive feedback mechanism we have proposed for TRPM2-mediated neuronal cell death, numerous other feedback loops among the key events in the amyloid cascade hypothesis of AD have been identified to occur both at the level of generation and aggregation of Aβ peptides [210] and also at the level of interactions among different cell types in the brain [211]. These findings and new insights lead to the interesting proposal of the feedback hypothesis instead of the prevailing amyloid cascade hypothesis of AD. There is increasing evidence to suggest that breaking such positive feedback loops is a promising approach to intervene in the pathogenesis and progression of AD [212]. Currently, there are no effective treatments, or a substantial unmet clinical need, for many debilitating diseases or conditions of which the TRPM2 channel plays an important part in the pathogenesis and progression. The findings discussed above have raised an interesting and exciting perspective of the TRPM2 channel as a new target. Development of TRPM2-specific and metabolically stable ligands, including agonists, antagonists and modulators, is the prerequisite for testing the concept of targeting the TRPM2 channel as a novel avenue of developing treatments for OS-associated diseases and conditions.
Author contributions
Both authors contributed in literature analysis, discussion, writing and revision of manuscript.
Declaration of competing interest
We declare no conflict of interest.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (31471118), Wellcome Trust (072275) and Alzheimer's Research Trust (ART/PPG2009 A/2) for supporting our research discussed in this article. PM is supported by a PhD studentship from University of Leeds.
References
- 1.Bardaweel S.K. Reactive oxygen species: the dual role in physiological and pathological conditions of the human body. Eurasian J. Med. 2018;50:193–201. doi: 10.5152/eurasianjmed.2018.17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 3.Forrester S.J., Kikuchi D.S., Hernandes M.S., Xu Q., Griendling K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018;122:877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pizzino G. Oxidative stress: harms and benefits for human health. Oxid. Med. Cell Longev. 2017;2017:8416763. doi: 10.1155/2017/8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spooner R., Yilmaz O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011;12:334–352. doi: 10.3390/ijms12010334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dias V., Junn E., Mouradian M.M. The role of oxidative stress in Parkinson's disease. J. Parkinsons Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Granger D.N., Kvietys P.R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du K., Ramachandran A., Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol. 2016;10:148–156. doi: 10.1016/j.redox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lakey P.S. Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract. Sci. Rep. 2016;6:32916. doi: 10.1038/srep32916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheignon C. Oxidative stress and the amyloid β peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Underwood E. The polluted brain. Science. 2017;355:342–345. doi: 10.1126/science.355.6323.342. [DOI] [PubMed] [Google Scholar]
- 12.Bencsik A., Lestaevel P., Guseva Canu I. Nano- and neurotoxicology: an emerging discipline. Prog. Neurobiol. 2018;160:45–63. doi: 10.1016/j.pneurobio.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Wang L. Predisposition to Alzheimer's and age-related brain pathologies by PM2.5 exposure: perspective on the roles of oxidative stress and TRPM2 channel. Front. Physiol. 2020;11:155. doi: 10.3389/fphys.2020.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W., Zhao F., Ma X., Perry G., Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol. Neurodegener. 2020;15:30. doi: 10.1186/s13024-020-00376-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robles L., Vaziri N.D., Ichii H. Role of oxidative stress in the pathogenesis of pancreatitis: effect of antioxidant therapy. Pancreat. Disord. Ther. 2013;3:112. doi: 10.4172/2165-7092.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sosa V. Oxidative stress and cancer: an overview. Ageing Res. Rev. 2013;12:376–390. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Pollari E., Goldsteins G., Bart G., Koistinaho J., Giniatullin R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014;8:131. doi: 10.3389/fncel.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeller M., Cottin Y., Vergely C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta. 2014;1840:2709–2729. doi: 10.1016/j.bbagen.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Esterberg R. Mitochondrial calcium uptake underlies ROS generation during aminoglycoside-induced hair cell death. J. Clin. Invest. 2016;126:3556–3566. doi: 10.1172/JCI84939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gul F. A comprehensive study of oxidative stress in sudden hearing loss. Eur. Arch. Oto-Rhino-Laryngol. 2017;274:1301–1308. doi: 10.1007/s00405-016-4301-1. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran A., Jaeschke H. Oxidative stress and acute hepatic injury. Curr. Opin. Toxicol. 2018;7:17–21. doi: 10.1016/j.cotox.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daenen K. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019;34:975–991. doi: 10.1007/s00467-018-4005-4. [DOI] [PubMed] [Google Scholar]
- 23.Förstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017;120:713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 24.Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radical Biol. Med. 2018;117:76–89. doi: 10.1016/j.freeradbiomed.2018.01.024. [DOI] [PubMed] [Google Scholar]
- 25.Del Re D.P., Amgalan D., Linkermann A., Liu Q., Kitsis R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 2019;99:1765–1817. doi: 10.1152/physrev.00022.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bugger H., Pfeil K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2020;1866:165768. doi: 10.1016/j.bbadis.2020.165768. [DOI] [PubMed] [Google Scholar]
- 27.Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020 doi: 10.1038/s41569-020-0403-y. [DOI] [PubMed] [Google Scholar]
- 28.Zuchi C. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020;25:21–30. doi: 10.1007/s10741-019-09881-3. [DOI] [PubMed] [Google Scholar]
- 29.Sano Y. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 30.Perraud A.L. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 31.Hara Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 32.Wehage E. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 2002;277:23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- 33.Zhang W. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J. Biol. Chem. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 34.Xu C. TRPM2 variants and bipolar disorder risk: confirmation in a family-based association study. Bipolar Disord. 2009;11:1–10. doi: 10.1111/j.1399-5618.2008.00655.x. [DOI] [PubMed] [Google Scholar]
- 35.Jia J. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J. Cerebr. Blood Flow Metabol. 2011;31:2160–2168. doi: 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manna P.T. TRPM2-mediated intracellular Zn2+ release triggers pancreatic β-cell death. Biochem. J. 2015;466:537–546. doi: 10.1042/BJ20140747. [DOI] [PubMed] [Google Scholar]
- 37.Alim I., Teves L., Li R., Mori Y., Tymianski M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J. Neurosci. 2013;33:17264–17277. doi: 10.1523/JNEUROSCI.1729-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao G. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Invest. 2014;124:4989–5001. doi: 10.1172/JCI76042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishii M., Hagiwara T., Mori Y., Shimizu S. Involvement of TRPM2 and L-type Ca2+ channels in Ca2+ entry and cell death induced by hydrogen peroxide in rat β-cell line RIN-5F. J. Toxicol. Sci. 2014;39:199–209. doi: 10.2131/jts.39.199. [DOI] [PubMed] [Google Scholar]
- 40.Ye M. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 2014;5:e1541. doi: 10.1038/cddis.2014.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kheradpezhouh E., Ma L., Morphett A., Barritt G.J., Rychkov G.Y. TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. U. S. A. 2014;111:3176–3181. doi: 10.1073/pnas.1322657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostapchenko V.G. The transient receptor potential melastatin 2 (trpm2) channel contributes to β-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 2015;35:15157–15169. doi: 10.1523/JNEUROSCI.4081-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Rodriguez S. CD38 promotes pristane-induced chronic inflammation and increases susceptibility to experimental lupus by an apoptosis-driven and TRPM2-dependent mechanism. Sci. Rep. 2018;8:3357. doi: 10.1038/s41598-018-21337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y. TRPM2 promotes neurotoxin MPP+/MPTP-Induced cell death. Mol. Neurobiol. 2018;55:409–420. doi: 10.1007/s12035-016-0338-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller B.A. TRPM2 in cancer. Cell Calcium. 2019;80:8–17. doi: 10.1016/j.ceca.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fanczal J. TRPM2-mediated extracellular Ca2+ entry promotes acinar cell necrosis in biliary acute pancreatitis. J. Physiol. 2020;598:1253–1270. doi: 10.1113/JP279047. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Q. TRPM2 ion channel is involved in the aggravation of cognitive impairment and down regulation of epilepsy threshold in pentylenetetrazole-induced kindling mice. Brain Res. Bull. 2020;155:48–60. doi: 10.1016/j.brainresbull.2019.11.018. [DOI] [PubMed] [Google Scholar]
- 48.Wang M. Silica nanoparticles induce lung inflammation in mice via ROS/PARP/TRPM2 signaling-mediated lysosome impairment and autophagy dysfunction. Part. Fibre Toxicol. 2020;17:23. doi: 10.1186/s12989-020-00353-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cosens D.J., Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature. 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- 50.Clapham D.E. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 51.Venkatachalam K., Montell C. TRP channels. Annu. Rev. Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samanta A., Hughes T.E.T., Moiseenkova-Bell V.Y. Transient receptor potential (TRP) channels. Subcell. Biochem. 2018;87:141–165. doi: 10.1007/978-981-10-7757-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagamine K. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54:124–131. doi: 10.1006/geno.1998.5551. [DOI] [PubMed] [Google Scholar]
- 54.Wang L. Structures and gating mechanism of human TRPM2. Science. 2018;362 doi: 10.1126/science.aav4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Y., Roth B., Lu W., Du J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife. 2019;8 doi: 10.7554/eLife.50175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perraud A.L., Schmitz C., Scharenberg A.M. TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium. 2003;33:519–531. doi: 10.1016/s0143-4160(03)00057-5. [DOI] [PubMed] [Google Scholar]
- 57.Mei Z.Z., Xia R., Beech D.J., Jiang L.H. Intracellular coiled-coil domain engaged in subunit interaction and assembly of melastatin-related transient receptor potential channel 2. J. Biol. Chem. 2006;281:38748–38756. doi: 10.1074/jbc.M607591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujiwara Y., Minor D.L., Jr. X-ray crystal structure of a TRPM assembly domain reveals an antiparallel four-stranded coiled-coil. J. Mol. Biol. 2008;383:854–870. doi: 10.1016/j.jmb.2008.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iordanov I., Mihalyi C., Toth B., Csanady L. The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. eLife. 2016;5 doi: 10.7554/eLife.17600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuhn F.J., Luckhoff A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J. Biol. Chem. 2004;279:46431–46437. doi: 10.1074/jbc.M407263200. [DOI] [PubMed] [Google Scholar]
- 61.Yu P. Identification of the ADPR binding pocket in the NUDT9 homology domain of TRPM2. J. Gen. Physiol. 2017;149:219–235. doi: 10.1085/jgp.201611675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kolisek M., Beck A., Fleig A., Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 63.Beck A., Kolisek M., Bagley L.A., Fleig A., Penner R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006;20:962–964. doi: 10.1096/fj.05-5538fje. [DOI] [PubMed] [Google Scholar]
- 64.Grubisha O. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J. Biol. Chem. 2006;281:14057–14065. doi: 10.1074/jbc.M513741200. [DOI] [PubMed] [Google Scholar]
- 65.Lange I., Penner R., Fleig A., Beck A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium. 2008;44:604–615. doi: 10.1016/j.ceca.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toth B., Iordanov I., Csanady L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2'-phosphate. J. Gen. Physiol. 2015;145:419–430. doi: 10.1085/jgp.201511377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fliegert R. 2'-Deoxyadenosine 5'-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017;13:1036–1044. doi: 10.1038/nchembio.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu P. Direct gating of the TRPM2 channel by cADPR via specific interactions with the ADPR binding pocket. Cell Rep. 2019;27:3684–3695. doi: 10.1016/j.celrep.2019.05.067. [DOI] [PubMed] [Google Scholar]
- 69.McHugh D., Flemming R., Xu S.Z., Perraud A.L., Beech D.J. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J. Biol. Chem. 2003;278:11002–11006. doi: 10.1074/jbc.M210810200. [DOI] [PubMed] [Google Scholar]
- 70.Du J., Xie J., Yue L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7239–7244. doi: 10.1073/pnas.0811725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uemura T., Kudoh J., Noda S., Kanba S., Shimizu N. Characterization of human and mouse TRPM2 genes: identification of a novel N-terminal truncated protein specifically expressed in human striatum. Biochem. Biophys. Res. Commun. 2005;328:1232–1243. doi: 10.1016/j.bbrc.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 72.Orfanelli U. Identification of novel sense and antisense transcription at the TRPM2 locus in cancer. Cell Res. 2008;18:1128–1140. doi: 10.1038/cr.2008.296. [DOI] [PubMed] [Google Scholar]
- 73.Jiang L.H., Yang W., Zou J., Beech D.J. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin. Ther. Targets. 2010;14:973–988. doi: 10.1517/14728222.2010.510135. [DOI] [PubMed] [Google Scholar]
- 74.Hecquet C.M., Ahmmed G.U., Vogel S.M., Malik A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- 75.Fonfria E. Amyloid β-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 2005;95:715–723. doi: 10.1111/j.1471-4159.2005.03396.x. [DOI] [PubMed] [Google Scholar]
- 76.Hirschler-Laszkiewicz I. The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation. Am. J. Physiol. Cell Physiol. 2018;315:C571–C586. doi: 10.1152/ajpcell.00098.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hecquet C.M. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ. Res. 2014;114:469–479. doi: 10.1161/CIRCRESAHA.114.302414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamamoto S., Shimizu S. Targeting TRPM2 in ROS-coupled diseases. Pharmaceuticals. 2016;9 doi: 10.3390/ph9030057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sumoza-Toledo A. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011;25:3529–3542. doi: 10.1096/fj.10-178483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lange I. TRPM2 functions as a lysosomal Ca2+-release channel in β cells. Sci. Signal. 2009;2:ra23. doi: 10.1126/scisignal.2000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abuarab N., Munsey T.S., Jiang L.H., Li J., Sivaprasadarao A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aal4161. [DOI] [PubMed] [Google Scholar]
- 82.Li X., Jiang L.H. Multiple molecular mechanisms form a positive feedback loop driving amyloid β42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis. 2018;9:195. doi: 10.1038/s41419-018-0270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knowles H., Li Y., Perraud A.L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 2013;55:241–248. doi: 10.1007/s12026-012-8373-8. [DOI] [PubMed] [Google Scholar]
- 84.Uchida K., Tominaga M. The role of TRPM2 in pancreatic β-cells and the development of diabetes. Cell Calcium. 2014;56:332–339. doi: 10.1016/j.ceca.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 85.Syed Mortadza S.A., Wang L., Li D., Jiang L.H. TRPM2 channel-mediated ROS-sensitive Ca2+ signaling mechanisms in immune cells. Front. Immunol. 2015;6:407. doi: 10.3389/fimmu.2015.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tan C.H., McNaughton P.A. TRPM2 and warmth sensation. Eur. J. Physiol. 2018;470:787–798. doi: 10.1007/s00424-018-2139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malko P., Syed Mortadza S.A., McWilliam J., Jiang L.H. TRPM2 channel in microglia as a new player in neuroinflammation associated with a spectrum of central nervous system pathologies. Front. Pharmacol. 2019;10:239. doi: 10.3389/fphar.2019.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Islam M.S. Molecular regulations and functions of the transient receptor potential channels of the islets of langerhans and insulinoma cells. Cells. 2020;9 doi: 10.3390/cells9030685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fonfria E. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miller B.A. Inhibition of TRPM2 function by PARP inhibitors protects cells from oxidative stress-induced death. Br. J. Pharmacol. 2004;143:515–516. doi: 10.1038/sj.bjp.0705923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buelow B., Song Y., Scharenberg A.M. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008;283:24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perraud A.L. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 93.Ru X., Yao X. TRPM2: a multifunctional ion channel for oxidative stress sensing. Acta Physiol. Sin. 2014;66:7–15. [PubMed] [Google Scholar]
- 94.Li C., Meng L., Li X., Li D., Jiang L.H. Non-NMDAR neuronal Ca2+-permeable channels in delayed neuronal death and as potential therapeutic targets for ischemic brain damage. Expert Opin. Ther. Targets. 2015;19:879–892. doi: 10.1517/14728222.2015.1021781. [DOI] [PubMed] [Google Scholar]
- 95.Zhan K.Y., Yu P.L., Liu C.H., Luo J.H., Yang W. Detrimental or beneficial: the role of TRPM2 in ischemia/reperfusion injury. Acta Pharmacol. Sin. 2016;37:4–12. doi: 10.1038/aps.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller B.A., Cheung J.Y. TRPM2 protects against tissue damage following oxidative stress and ischaemia-reperfusion. J. Physiol. 2016;594:4181–4191. doi: 10.1113/JP270934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li J. TRPM2: a potential drug target to retard oxidative stress. Front. Biosci. 2017;22:1427–1438. doi: 10.2741/4551. [DOI] [PubMed] [Google Scholar]
- 98.Jiang L.H., Li X., Syed Mortadza S.A., Lovatt M., Yang W. The TRPM2 channel nexus from oxidative damage to Alzheimer's pathologies: an emerging novel intervention target for age-related dementia. Ageing Res. Rev. 2018;47:67–79. doi: 10.1016/j.arr.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 99.Sita G., Hrelia P., Graziosi A., Ravegnini G., Morroni F. TRPM2 in the brain: role in health and disease. Cells. 2018;7 doi: 10.3390/cells7070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Belrose J.C., Jackson M.F. TRPM2: a candidate therapeutic target for treating neurological diseases. Acta Pharmacol. Sin. 2018;39:722–732. doi: 10.1038/aps.2018.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Turlova E., Feng Z.P., Sun H.S. The role of TRPM2 channels in neurons, glial cells and the blood-brain barrier in cerebral ischemia and hypoxia. Acta Pharmacol. Sin. 2018;39:713–721. doi: 10.1038/aps.2017.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu X., Ong H.L., Ambudkar I. TRP channel involvement in salivary glands-some good, some bad. Cells. 2018;7 doi: 10.3390/cells7070074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Negri S., Faris P., Berra-Romani R., Guerra G., Moccia F. Endothelial transient receptor potential channels and vascular remodeling: extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2019;10:1618. doi: 10.3389/fphys.2019.01618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sakaguchi R., Mori Y. Transient receptor potential (TRP) channels: biosensors for redox environmental stimuli and cellular status. Free Radical Biol. Med. 2020;146:36–44. doi: 10.1016/j.freeradbiomed.2019.10.415. [DOI] [PubMed] [Google Scholar]
- 105.Mai C. TRPM2 channel: a novel target for alleviating ischaemia-reperfusion, chronic cerebral hypo-perfusion and neonatal hypoxic-ischaemic brain damage. J. Cell Mol. Med. 2020;24:4–12. doi: 10.1111/jcmm.14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hill K., Benham C.D., McNulty S., Randall A.D. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology. 2004;47:450–460. doi: 10.1016/j.neuropharm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 107.Hill K., McNulty S., Randall A.D. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. N. Schmied. Arch. Pharmacol. 2004;370:227–237. doi: 10.1007/s00210-004-0981-y. [DOI] [PubMed] [Google Scholar]
- 108.Kraft R., Grimm C., Frenzel H., Harteneck C. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br. J. Pharmacol. 2006;148:264–273. doi: 10.1038/sj.bjp.0706739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Togashi K., Inada H., Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB) Br. J. Pharmacol. 2008;153:1324–1330. doi: 10.1038/sj.bjp.0707675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Harteneck C., Frenzel H., Kraft R. N-(p-amylcinnamoyl)anthranilic acid (ACA): a phospholipase A2 inhibitor and TRP channel blocker. Cardiovasc. Drug Rev. 2007;25:61–75. doi: 10.1111/j.1527-3466.2007.00005.x. [DOI] [PubMed] [Google Scholar]
- 111.Chen G.L. Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. Br. J. Pharmacol. 2012;167:1232–1243. doi: 10.1111/j.1476-5381.2012.02058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eraslan E., Tanyeli A., Polat E., Polat E. 8-Br-cADPR, a TRPM2 ion channel antagonist, inhibits renal ischemia-reperfusion injury. J. Cell. Physiol. 2019;234:4572–4581. doi: 10.1002/jcp.27236. [DOI] [PubMed] [Google Scholar]
- 113.Partida-Sanchez S. Chemotaxis of mouse bone marrow neutrophils and dendritic cells is controlled by adp-ribose, the major product generated by the CD38 enzyme reaction. J. Immunol. 2007;179:7827–7839. doi: 10.4049/jimmunol.179.11.7827. [DOI] [PubMed] [Google Scholar]
- 114.Moreau C. Structure-activity relationship of adenosine 5'-diphosphoribose at the transient receptor potential melastatin 2 (TRPM2) channel: rational design of antagonists. J. Med. Chem. 2013;56:10079–10102. doi: 10.1021/jm401497a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luo X. Selective inhibition of TRPM2 channel by two novel synthesized ADPR analogues. Chem. Biol. Drug Des. 2018;91:552–566. doi: 10.1111/cbdd.13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fourgeaud L. Pharmacology of JNJ-28583113: a novel TRPM2 antagonist. Eur. J. Pharmacol. 2019;853:299–307. doi: 10.1016/j.ejphar.2019.03.043. [DOI] [PubMed] [Google Scholar]
- 117.Yang W. Zinc inactivates melastatin transient receptor potential 2 channels via the outer pore. J. Biol. Chem. 2011;286:23789–23798. doi: 10.1074/jbc.M111.247478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu W. Inactivation of TRPM2 channels by extracellular divalent copper. PloS One. 2014;9 doi: 10.1371/journal.pone.0112071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zeng B., Chen G.L., Xu S.Z. Divalent copper is a potent extracellular blocker for TRPM2 channel. Biochem. Biophys. Res. Commun. 2012;424:279–284. doi: 10.1016/j.bbrc.2012.06.107. [DOI] [PubMed] [Google Scholar]
- 120.Shimizu T. Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp. Neurol. 2016;283:151–156. doi: 10.1016/j.expneurol.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li F., Munsey T.S., Sivaprasadarao A. TRPM2-mediated rise in mitochondrial Zn2+ promotes palmitate-induced mitochondrial fission and pancreatic β-cell death in rodents. Cell Death Differ. 2017;24:1999–2012. doi: 10.1038/cdd.2017.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Syed Mortadza S.A., Sim J.A., Neubrand V.E., Jiang L.H. A critical role of TRPM2 channel in Aβ42-induced microglial activation and generation of tumor necrosis factor-α. Glia. 2018;66:562–575. doi: 10.1002/glia.23265. [DOI] [PubMed] [Google Scholar]
- 123.An X. Increasing the TRPM2 channel expression in human neuroblastoma SH-SY5Y cells augments the susceptibility to ros-induced cell death. Cells. 2019;8 doi: 10.3390/cells8010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X., Jiang L.H. A critical role of the transient receptor potential melastatin 2 channel in a positive feedback mechanism for reactive oxygen species-induced delayed cell death. J. Cell. Physiol. 2019;234:3647–3660. doi: 10.1002/jcp.27134. [DOI] [PubMed] [Google Scholar]
- 125.Naziroglu M. Albumin evokes Ca2+-induced cell oxidative stress and apoptosis through TRPM2 channel in renal collecting duct cells reduced by curcumin. Sci. Rep. 2019;9:12403. doi: 10.1038/s41598-019-48716-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li X., Yang W., Jiang L.H. Alteration in intracellular Zn2+ homeostasis as a result of TRPM2 channel activation contributes to ROS-induced hippocampal neuronal death. Front. Mol. Neurosci. 2017;10:414. doi: 10.3389/fnmol.2017.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xicoy H., Wieringa B., Martens G.J. The SH-SY5Y cell line in Parkinson's disease research: a systematic review. Mol. Neurodegener. 2017;12:10. doi: 10.1186/s13024-017-0149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Whittemore E.R., Loo D.T., Cotman C.W. Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. Neuroreport. 1994;5:1485–1488. doi: 10.1097/00001756-199407000-00019. [DOI] [PubMed] [Google Scholar]
- 129.Valencia A., Moran J. Reactive oxygen species induce different cell death mechanisms in cultured neurons. Free Radical Biol. Med. 2004;36:1112–1125. doi: 10.1016/j.freeradbiomed.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 130.Kaneko S. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J. Pharmacol. Sci. 2006;101:66–76. doi: 10.1254/jphs.fp0060128. [DOI] [PubMed] [Google Scholar]
- 131.Lipton P. Ischemic cell death in brain neurons. Physiol. Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 132.Love S. Oxidative stress in brain ischemia. Brain Pathol. 1999;9:119–131. doi: 10.1111/j.1750-3639.1999.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chan P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cerebr. Blood Flow Metabol. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 134.Sanderson T.H., Reynolds C.A., Kumar R., Przyklenk K., Huttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013;47:9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Verma S. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci. Lett. 2012;530:41–46. doi: 10.1016/j.neulet.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shimizu T. Androgen and PARP-1 regulation of TRPM2 channels after ischemic injury. J. Cerebr. Blood Flow Metabol. 2013;33:1549–1555. doi: 10.1038/jcbfm.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gelderblom M. Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke. 2014;45:3395–3402. doi: 10.1161/STROKEAHA.114.005836. [DOI] [PubMed] [Google Scholar]
- 138.Nakayama S., Vest R., Traystman R.J., Herson P.S. Sexually dimorphic response of TRPM2 inhibition following cardiac arrest-induced global cerebral ischemia in mice. J. Mol. Neurosci. 2013;51:92–98. doi: 10.1007/s12031-013-0005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Quillinan N., Grewal H., Klawitter J., Herson P.S. Sex steroids do not modulate TRPM2-mediated injury in females following middle cerebral artery occlusion. eNeuro. 2014;1 doi: 10.1523/ENEURO.0022-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shimizu K., Quillinan N., Orfila J.E., Herson P.S. Sirtuin-2 mediates male specific neuronal injury following experimental cardiac arrest through activation of TRPM2 ion channels. Exp. Neurol. 2016;275:78–83. doi: 10.1016/j.expneurol.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hardy J.A., Higgins G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 143.Trist B.G., Hare D.J., Double K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease. Aging Cell. 2019;18 doi: 10.1111/acel.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Poewe W. Parkinson disease. Nat. Rev. Dis. Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 145.Schildknecht S., Di Monte D.A., Pape R., Tieu K., Leist M. Tipping points and endogenous determinants of nigrostriatal degeneration by MPTP. Trends Pharmacol. Sci. 2017;38:541–555. doi: 10.1016/j.tips.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 146.Mortadza S.S., Sim J.A., Stacey M., Jiang L.H. Signalling mechanisms mediating Zn2+-induced TRPM2 channel activation and cell death in microglial cells. Sci. Rep. 2017;7:45032. doi: 10.1038/srep45032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yildizhan K., Naziroglu M. Glutathione depletion and parkinsonian neurotoxin MPP+-induced TRPM2 channel activation play central roles in oxidative cytotoxicity and inflammation in microglia. Mol. Neurobiol. 2020;57:3508–3525. doi: 10.1007/s12035-020-01974-7. [DOI] [PubMed] [Google Scholar]
- 148.Crowder R.J., Freeman R.S. Glycogen synthase kinase-3 β activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J. Biol. Chem. 2000;275:34266–34271. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- 149.Liu Y. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lai T.W., Zhang S., Wang Y.T. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 152.Stork C.J., Li Y.V. Rising zinc: a significant cause of ischemic neuronal death in the CA1 region of rat hippocampus. J. Cerebr. Blood Flow Metabol. 2009;29:1399–1408. doi: 10.1038/jcbfm.2009.64. [DOI] [PubMed] [Google Scholar]
- 153.Shuttleworth C.W., Weiss J.H. Zinc: new clues to diverse roles in brain ischemia. Trends Pharmacol. Sci. 2011;32:480–486. doi: 10.1016/j.tips.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sweeney M.D., Ayyadurai S., Zlokovic B.V. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 2016;19:771–783. doi: 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brown L.S. Pericytes and neurovascular function in the healthy and diseased brain. Front. Cell. Neurosci. 2019;13:282. doi: 10.3389/fncel.2019.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cheng J. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136:507–523. doi: 10.1007/s00401-018-1893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Niu M. Autophagy, endoplasmic reticulum stress and the unfolded protein response in intracerebral hemorrhage. Transl. Neurosci. 2017;8:37–48. doi: 10.1515/tnsci-2017-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thangaraj A. Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int. Rev. Cell Mol. Biol. 2020;350:285–325. doi: 10.1016/bs.ircmb.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 159.Johnson B.M. Acute exposure to ZnO nanoparticles induces autophagic immune cell death. Nanotoxicology. 2015;9:737–748. doi: 10.3109/17435390.2014.974709. [DOI] [PubMed] [Google Scholar]
- 160.Roy R. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol. Lett. 2014;227:29–40. doi: 10.1016/j.toxlet.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 161.Jiang Q. Nitration of TRPM2 as a molecular switch induces autophagy during brain pericyte injury. Antioxid. Redox Signal. 2017;27:1297–1316. doi: 10.1089/ars.2016.6873. [DOI] [PubMed] [Google Scholar]
- 162.Lopez A., Fleming A., Rubinsztein D.C. Seeing is believing: methods to monitor vertebrate autophagy in vivo. Open Biol. 2018;8 doi: 10.1098/rsob.180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang K.T. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ. 2006;13:1815–1826. doi: 10.1038/sj.cdd.4401813. [DOI] [PubMed] [Google Scholar]
- 164.Kleinbongard P., Schulz R., Heusch G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail. Rev. 2011;16:49–69. doi: 10.1007/s10741-010-9180-8. [DOI] [PubMed] [Google Scholar]
- 165.Roberge S. TNF-α-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc. Res. 2014;103:90–99. doi: 10.1093/cvr/cvu112. [DOI] [PubMed] [Google Scholar]
- 166.Hiroi T. Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2013;97:271–281. doi: 10.1093/cvr/cvs332. [DOI] [PubMed] [Google Scholar]
- 167.Hoffman N.E. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Heart Circ. Physiol. 2015;308:H637–H650. doi: 10.1152/ajpheart.00720.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Miller B.A. TRPM2 channels protect against cardiac ischemia-reperfusion injury: role of mitochondria. J. Biol. Chem. 2014;289:7615–7629. doi: 10.1074/jbc.M113.533851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Miller B.A. The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H1010–H1022. doi: 10.1152/ajpheart.00906.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Miller B.A. Trpm2 enhances physiological bioenergetics and protects against pathological oxidative cardiac injury: role of Pyk2 phosphorylation. J. Cell. Physiol. 2019;234:15048–15060. doi: 10.1002/jcp.28146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lindsey M.L. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018;314:H812–H838. doi: 10.1152/ajpheart.00335.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lum H., Roebuck K.A. Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 173.Cao Y. The toxicity of nanoparticles to human endothelial cells. Adv. Exp. Med. Biol. 2018;1048:59–69. doi: 10.1007/978-3-319-72041-8_4. [DOI] [PubMed] [Google Scholar]
- 174.Gimbrone M.A., Jr., García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Haybar H., Shahrabi S., Rezaeeyan H., Shirzad R., Saki N. Endothelial cells: from dysfunction mechanism to pharmacological effect in cardiovascular disease. Cardiovasc. Toxicol. 2019;19:13–22. doi: 10.1007/s12012-018-9493-8. [DOI] [PubMed] [Google Scholar]
- 176.Lawal A.O., Davids L.M., Marnewick J.L. Diesel exhaust particles and endothelial cells dysfunction: an update. Toxicol. Vitro. 2016;32:92–104. doi: 10.1016/j.tiv.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 177.Xu N. Reactive oxygen species in renal vascular function. Acta Physiol. 2020;229 doi: 10.1111/apha.13477. [DOI] [PubMed] [Google Scholar]
- 178.Yu Q., Chan S.Y. Mitochondrial and metabolic drivers of pulmonary vascular endothelial dysfunction in pulmonary hypertension. Adv. Exp. Med. Biol. 2017;967:373–383. doi: 10.1007/978-3-319-63245-2_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhang J., Wang Y., Liu X., Dagda R.K., Zhang Y. How AMPK and PKA interplay to regulate mitochondrial function and survival in models of ischemia and diabetes. Oxid. Med. Cell. Longev. 2017;2017:4353510. doi: 10.1155/2017/4353510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang T. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 2020;15:1245–1246. doi: 10.1186/1743-8977-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sun L. Role of TRPM2 in H2O2-induced cell apoptosis in endothelial cells. PloS One. 2012;7 doi: 10.1371/journal.pone.0043186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Deshpande S.S., Angkeow P., Huang J., Ozaki M., Irani K. Rac1 inhibits TNF-α-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB (Fed. Am. Soc. Exp. Biol.) J. 2000;14:1705–1714. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- 183.Mittal M. Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ. Res. 2017;121:1081–1091. doi: 10.1161/CIRCRESAHA.117.311747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cerf M.E. β cell dysfunction and insulin resistance. Front. Endocrinol. 2013;4:37. doi: 10.3389/fendo.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Newsholme P., Keane K.N., Carlessi R., Cruzat V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019;317:C420–C433. doi: 10.1152/ajpcell.00141.2019. [DOI] [PubMed] [Google Scholar]
- 186.Akbarzadeh A. Induction of diabetes by Streptozotocin in rats. Indian J. Clin. Biochem. 2007;22:60–64. doi: 10.1007/BF02913315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Burkart V. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic β-cell destruction and diabetes development induced by streptozocin. Nat. Med. 1999;5 doi: 10.1038/6535. [DOI] [PubMed] [Google Scholar]
- 188.Pieper A.A. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Herson P.S., Lee K., Pinnock R.D., Hughes J., Ashford M.L. Hydrogen peroxide induces intracellular calcium overload by activation of a non-selective cation channel in an insulin-secreting cell line. J. Biol. Chem. 1999;274:833–841. doi: 10.1074/jbc.274.2.833. [DOI] [PubMed] [Google Scholar]
- 190.Mathis D., Vence L., Benoist C. β-cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 191.Oprescu A.I. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56:2927–2937. doi: 10.2337/db07-0075. [DOI] [PubMed] [Google Scholar]
- 192.Detmer S.A., Chan D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 193.van Geenen E.J., van der Peet D.L., Bhagirath P., Mulder C.J., Bruno M.J. Etiology and diagnosis of acute biliary pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2010;7:495–502. doi: 10.1038/nrgastro.2010.114. [DOI] [PubMed] [Google Scholar]
- 194.Criddle D.N. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130:781–793. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 195.Gerasimenko J.V. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J. Biol. Chem. 2006;281:40154–40163. doi: 10.1074/jbc.M606402200. [DOI] [PubMed] [Google Scholar]
- 196.Huang W. Effects of the mitochondria-targeted antioxidant mitoquinone in murine acute pancreatitis. Mediat. Inflamm. 2015;2015:901780. doi: 10.1155/2015/901780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gerasimenko J.V., Peng S., Tsugorka T., Gerasimenko O.V. Ca2+ signalling underlying pancreatitis. Cell Calcium. 2018;70:95–101. doi: 10.1016/j.ceca.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 198.Pallagi P., Madacsy T., Varga A., Maleth J. Intracellular Ca2+ signalling in the pathogenesis of acute pancreatitis: recent advances and translational perspectives. Int. J. Mol. Sci. 2020;21:4005. doi: 10.3390/ijms21114005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cheung J.Y., Bonventre J.V., Malis C.D., Leaf A. Calcium and ischemic injury. N. Engl. J. Med. 1986;314:1670–1676. doi: 10.1056/NEJM198606263142604. [DOI] [PubMed] [Google Scholar]
- 200.Donnahoo K.K. Early kidney TNF-α expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am. J. Physiol. 1999;277:R922–R929. doi: 10.1152/ajpregu.1999.277.3.R922. [DOI] [PubMed] [Google Scholar]
- 201.Irazabal M.V., Torres V.E. Reactive oxygen species and redox signaling in chronic kidney disease. Cells. 2020;9:1342. doi: 10.3390/cells9061342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kaushal G.P., Basnakian A.G., Shah S.V. Apoptotic pathways in ischemic acute renal failure. Kidney Int. 2004;66:500–506. doi: 10.1111/j.1523-1755.2004.761_6.x. [DOI] [PubMed] [Google Scholar]
- 203.Dizin E. Albuminuria induces a proinflammatory and profibrotic response in cortical collecting ducts via the 24p3 receptor. Am. J. Physiol. Ren. Physiol. 2013;305:F1053–F1063. doi: 10.1152/ajprenal.00006.2013. [DOI] [PubMed] [Google Scholar]
- 204.Davidson D.G., Eastham W.N. Acute liver necrosis following overdose of paracetamol. Br. Med. J. 1966;2:497–499. doi: 10.1136/bmj.2.5512.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mitchell J.R. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Therapeut. 1973;187:185–194. [PubMed] [Google Scholar]
- 206.Wang Q. Mechanistic study of TRPM2-Ca2+-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy. 2016;12:1340–1354. doi: 10.1080/15548627.2016.1187365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ni H.M., Bockus A., Boggess N., Jaeschke H., Ding W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fricker M., Tolkovsky A.M., Borutaite V., Coleman M., Brown G.C. Neuronal cell death. Physiol. Rev. 2018;98:813–880. doi: 10.1152/physrev.00011.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Di A. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2011;13:29–34. doi: 10.1038/ni.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Doig A.J. Positive feedback loops in Alzheimer's disease: the Alzheimer's feedback hypothesis. J. Alzheim. Dis. 2018;66:25–36. doi: 10.3233/JAD-180583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.De Strooper B., Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 212.Park M.H. N,N'-Diacetyl-p-phenylenediamine restores microglial phagocytosis and improves cognitive defects in Alzheimer's disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 2019;116:23426–23436. doi: 10.1073/pnas.1916318116. [DOI] [PMC free article] [PubMed] [Google Scholar]