

Abstract
Vertebrate genomes replicate according to a precise temporal program strongly correlated with their organization into A/B compartments. Until now, the molecular mechanisms underlying the establishment of early‐replicating domains remain largely unknown. We defined two minimal cis‐element modules containing a strong replication origin and chromatin modifier binding sites capable of shifting a targeted mid‐late‐replicating region for earlier replication. The two origins overlap with a constitutive or a silent tissue‐specific promoter. When inserted side‐by‐side, these modules advance replication timing over a 250 kb region through the cooperation with one endogenous origin located 30 kb away. Moreover, when inserted at two chromosomal sites separated by 30 kb, these two modules come into close physical proximity and form an early‐replicating domain establishing more contacts with active A compartments. The synergy depends on the presence of the active promoter/origin. Our results show that clustering of strong origins located at active promoters can establish early‐replicating domains.
Keywords: chromatin accessibility, nuclear organization, promoter, replication origin, replication timing
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Two replicators carrying timing information can cooperate when located in proximity, and advance replication timing by forming higher‐order chromatin contacts with active “A” compartments.
Introduction
A precise, cell type‐specific temporal program governs the duplication of vertebrate genomes (Ryba et al, 2010). The genome‐wide mapping of replication origins with different methods has revealed a high density of efficient site‐specific origins in early‐replicated domains, whereas late regions are usually origin‐poor (Prioleau & MacAlpine, 2016). Replication timing (RT) domains are correlated with the organization of chromosomes into two main types of compartments: compartment A, which is accessible and replicated early, and compartment B, which is more condensed and replicated late (Ryba et al, 2010). These compartments, which display greater interaction within themselves rather than across them, were initially defined by the Hi‐C method, at a resolution of 1 megabase (Lieberman‐Aiden et al, 2009). The only major player in the RT program for which the mode of action has been elucidated is Rif1 (Cornacchia et al, 2012; Knott et al, 2012; Yamazaki et al, 2012). Rif1 has a global repressive effect on genome‐wide DNA replication. This effect is mediated by the recruitment of protein phosphatase 1, which opposes the Dbf4‐dependent kinase (DDK) activity required for origin firing (Hiraga et al, 2014). Consistent with this direct role, ChIP analyses of Rif1 throughout the mouse genome revealed an overlap between Rif1 associated domains and late replication (Foti et al, 2016). However, only regions associated with Rif1 but not with the nuclear lamina switch to early replication in a context of Rif1 depletion. This finding suggests that other major players, such as the nuclear lamina, are also involved in controlling late replication (Duriez et al, 2019). One key question that remains concerns whether some late‐replicating domains are so robustly constrained by their nuclear compartmentalization that the targeting of a very efficient origin associated with early timing control elements could not locally advance the timing of their replication. The underlying question is whether a late domain is defined by the deficiency of an early‐firing signal together with an accumulation of signals imposing the late‐firing of many potential initiation sites. A related question is whether early‐replicated domains are defined solely by the absence of a strong negative signal, such as association with the nuclear lamina and/or Rif1. Alternatively, early constant timing regions (CTRs) may result fortuitously from the more or less synchronous firing of a cluster of replicons, each with its own individual local early timing control elements. This hypothesis is supported by the high density of efficient origins mostly associated with transcription start sites (TSS) and thus proximal to sites associated with open chromatin marks in early‐replicated domains (Picard et al, 2014). In agreement with this model, a recent study showed that stem cell‐specific early‐replicating domains in mouse are controlled by stem cell‐specific cis‐elements located in promoter and enhancer regions (Sima et al, 2019). Moreover, several elements spanning altogether 30 kb had to be deleted so that the early domain switched from early to late, suggesting redundancy between these elements along early domains and their potential capacity to act remotely. Here we put forward the central role of constitutive promoters in the formation of constitutive early‐replicating domains and strongly suggests that spatial connections between strong initiation sites play a key role in this fundamental process.
Results
Cooperation between two minimal autonomous replicons impacts on RT at a large scale
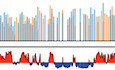
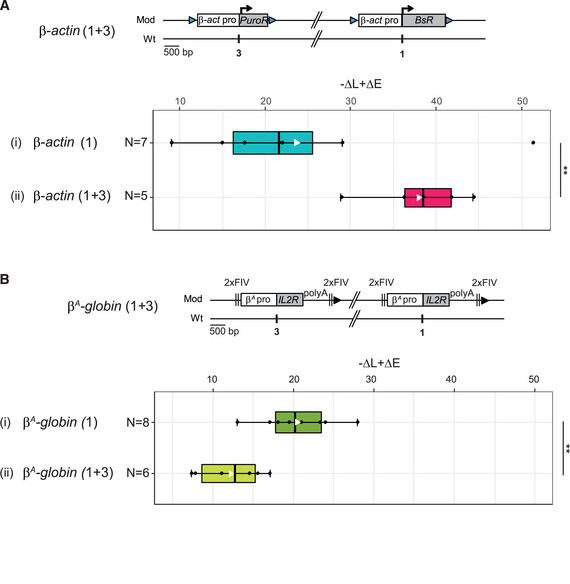
The current study is based on the method we developed to quantify the magnitude of the RT shift induced by the insertion of an ectopic DNA sequence into a specific mid‐late‐replicating locus (chr1:72,565,520 bp, galGal5) (Hassan‐Zadeh et al, 2012 and Appendix Fig S1). Here we confirmed our previous data on a much larger number of cell lines modified in the same targeted region, allowing a statistical quantification of RT shifts (Fig 1 and Appendix Table S1). The tissue‐specific β A ‐globin promoter containing a strongly active replication origin flanked by 2XFIV [Footprint IV of cHS4 insulator element is a binding site for the Upstream Stimulatory Factor (USF) (West et al, 2004)] significantly advanced RT (Fig 1 (ii), P‐value = 4.57E‐05) and to the same extent as the active β‐actin constitutive promoter containing an active replication origin (Figs EV1A and 1 (iii), P‐value = 3.38E‐03). Finally, the combination of these two minimal modules (β A ‐globin+β‐actin constructs) imposed a stronger shift to earlier replication at the inserted locus than the presence of a single minimal module alone (Fig 1 (iv), P‐value = 3.25E‐03 between (ii) and (iv); P‐value = 2.66E‐03 between (iii) and (iv) and Fig EV1 and Hassan‐Zadeh et al, 2012). These endogenous origins/promoters are naturally found in early‐replicating domains in DT40 cells (Appendix Fig S2) and therefore constitute excellent models to understand how early domains might be established. Insertion of this large construct on the two homologous chromosomes (2 × (β A ‐globin + β‐actin) cell line) allowed us to observe a 250 kb region displaying an advance RT compared to a WT cell line (Fig 2A and Appendix Fig S3). A zoom in centered on the site of insertion confirmed that this site is naturally a termination zone (TZ) flanked by two initiation zones (IZ) (Fig 2B). RT profiles obtained from S1 to S4 fractions showed that the IZ about 30 kb upstream of the site of insertion (IZ.1) is activated in S3 in WT cells, whereas the IZ located 60 kb downstream (IZ.2) is activated already in S2. IZ.1 and IZ.2 correspond to strong initiation sites detected by the short nascent strand (SNS) assay (Massip et al, 2019). Profiles observed at the same region in a cell line modified on both chromosomes (2 × (β A ‐globin + β‐actin) cell line) showed firing of the IZ.1 in S1, whereas the IZ.2 is unchanged compared to the WT cell line. Moreover, in this line, IZ.1 is then extended in S2 on the 3′ direction toward the insertion site, revealing the activation of origins brought by the inserted construct. These changes led to a 30 kb shift of the TZ downstream of the site of insertion. Overall, this result suggested that the large β A ‐globin + β‐actin construct impacted strongly on the RT profile of its surrounding regions in two different ways: It advanced significantly the RT of a 30 kb upstream strong endogenous origin from S3 to S1 and induced strong initiation in S2 due to efficient firing of replication origins present inside the construct.
Figure 1. Quantitative analysis of RT shifts reveals cooperation between two combinations of cis‐regulatory elements.
Distribution of RT shifts calculated with the –ΔL + ΔE method described in Appendix Fig S1. Different transgenes in a mid‐late‐replicating region are compared: the IL2R reporter gene under the control of the β A ‐globin promoter (β A pro) containing an inactive (i, N = 10) or an active origin (ii, N = 8) flanked by two copies of FIV (2xFIV), the blasticidin resistance gene (BsR) under the control of the β‐actin promoter (β‐act pro) (iii, N = 7), or a combination of these two transgenes (iv, N = 8). Blue and black triangles represent reactive loxP sites and recombined inactive loxP sites, respectively. Rectangle edges correspond to the 0.25 and 0.75 quartiles, the thick black lines represent the median, the white triangles represent the mean, and the whiskers extend to the smallest and largest –ΔL + ΔE values. Statistical analysis was performed with Wilcoxon nonparametric two‐tailed tests (ns, not significant; **P < 0.01; ***P < 0.001).
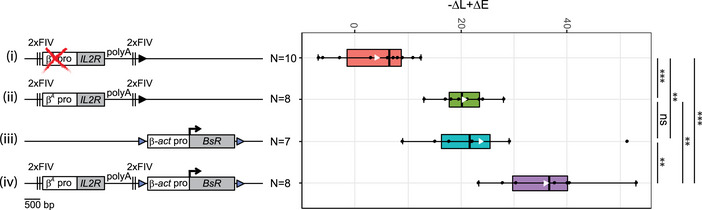
Figure EV1. Additional RT assays for transgenes shifting the timing of replication.
-
A, BRT profiles of each chromosomal allele are determined after targeted transgene integration using the allele‐specific analysis of RT method by real‐time PCR quantification described in Appendix Fig S1. Differences in –ΔL + ΔE values calculated at the target site following transgene integration are indicated. Blue triangles represent reactive loxP sites. Error bars correspond to the standard deviation for qPCR duplicates. Analysis of six β‐actin clonal cell lines (A) or two β A ‐globin + β‐actin clonal cell lines (B) described in Fig 1 is reported. Black vertical bars represent insertion sites.
Figure 2. One autonomous replicon inserted in the mid‐late region perturbs the RT over a 250 kb region through cooperation with an endogenous strong origin.
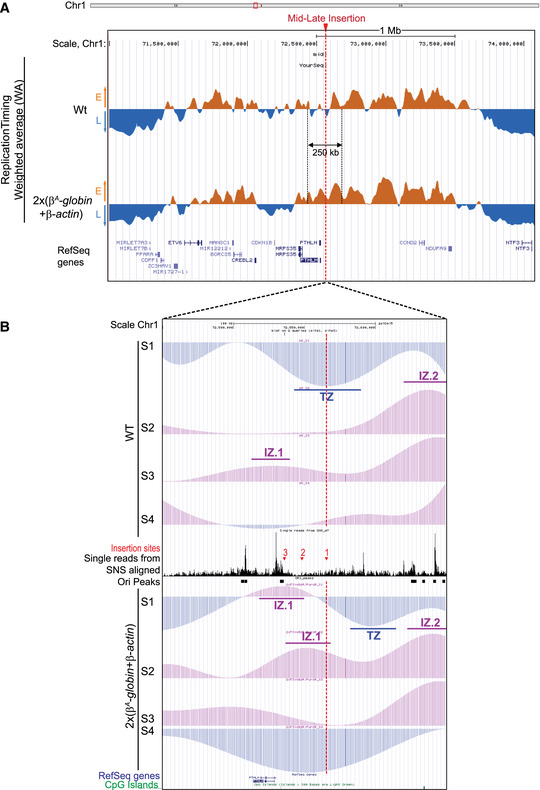
- UCSC genome browser visualization of the mid‐late insertion site of chromosome 1 (genomic positions: chr1:71,000,000–74,100,000 bp, 3.1 Mb; galGal5). RT‐weighted average (WA) values for the wt and the β A ‐globin + β‐actin cell lines are shown. Early‐replicated regions (E) are represented in orange and late‐replicating regions (L) in blue. Annotated genes (Ref Seq genes) are represented below. The mid‐late insertion site is indicated with a red arrow and dotted line.
- UCSC genome browser visualization of the mid‐late insertion site of chromosome 1 (genomic positions: chr1:72,450,000–72,650,000 bp; 200 kb; galGal5). Tracks of nascent strands (NS) enrichments in the four S‐phase fractions were represented separately (S1–S4) for the wt and the 2 × (β A ‐globin + β‐actin) cell lines. NS‐enriched and depleted regions for each fraction are represented in purple and blue, respectively. Single reads form SNS aligned and track of replication origins (Ori peaks) determined in (Massip et al, 2019) are reported in between. The three mid‐late insertion sites 1, 2, and 3 are indicated with red arrows. Annotated genes and CpG Islands are shown below. Initiation zones (IZ) and termination zones (TZ) are reported.
Two advanced replicons separated by 30 kb synergize to form a synthetic early‐replicating region that interacts more with A compartments
Our observation suggested that the large β A ‐globin + β‐actin construct might cooperate with endogenous replication origins located about 30 kb upstream. To further test this hypothesis, we inserted two large autonomous replicons 30 kb apart at sites 1 and 3 (insertion site 1; chr1:72,565,520 bp and insertion site 3; chr1: 72,536,061 bp, galGal5, Fig 2B) and tested their impact on RT. The autonomous replicon at site 3 was similar to the one at site 1 except that the gene used conferred puromycin‐N‐acetyltransferase resistance (PuroR, insertion site 3, Fig 3A). We assessed the impact of RT changes in the middle of our modified region, by introducing a reporter construct in a central position (Insertion site 2; chr1: 72,548,590 bp, galGal5, Fig 2B) between the two autonomous replicons (1 + 2 + 3) or on the other chromosome (1 + 3) (Fig 3A). The reporter construct consisted of the erythroid‐specific β A ‐globin origin/promoter linked to the green fluorescent protein (GFP) reporter gene and a 1.6 kb fragment of human chromosome 7 (h.K7; chr7:26,873,165–26,874,805 bp, hg38) containing no replication origin (Fig 3A). We found that the GFP reporter construct had no impact on RT when inserted alone at site 2 (−ΔL + ΔE = −5.1%, Fig EV2A). We then showed that the β A ‐globin + β‐actin construct inserted at site 1 had similar effects on RT when associated with GFP reporter construct insertion at site 2 on the same or the other chromosome (compare Figs EV1B with EV2B and C and Hassan‐Zadeh et al, 2012). These results confirm the neutral impact on RT of inserting the GFP reporter construct at site 2. We then investigated whether the insertion of a single autonomous replicon at site 3 was able to advance RT as efficiently as insertion at site 1. We confirmed that the β A ‐globin + β‐actin construct induced a substantial shift in RT when inserted at this new genomic position (−ΔL + ΔE = +26.8% for clone 1 and –ΔL + ΔE = +22% for clone 2, Fig EV2D). This shift was similar to the one observed in β A ‐globin + β‐actin clones after insertion at site 1 (Fig 3). As previously shown, the insertion of the GFP reporter construct at site 2 had no impact on RT when associated with a single autonomous replicon at site 3 on either the same or the other chromosome (Fig EV2, compare –ΔL + ΔE values between D and E). Insertion of two autonomous replicons at sites 1 and 3, with (1 + 2 + 3) or without (1 + 3) the central GFP reporter construct, resulted in larger shift in RT over the whole of the genomic region targeted (Fig EV3A and B, −ΔL + ΔE = +63.3 and +70.3% for 1 + 2 + 3 insertions and –ΔL + ΔE = +65.2, +84.2, +57.3%, for 1 + 3 insertions). A quantitative analysis revealed that the differences in RT between cell clones with one and two autonomous replicons were significant (P‐value = 0.01554 between (i) and (ii) Fig 3A). Overall, these results strongly suggest that two similar independent advanced replicons, 30 kb apart, can synergize to control the extent of the RT shift.
Figure 3. Two advanced replicons separated by 30 kb cooperate to form a synthetic early‐replicated region that is more associated with A compartments.
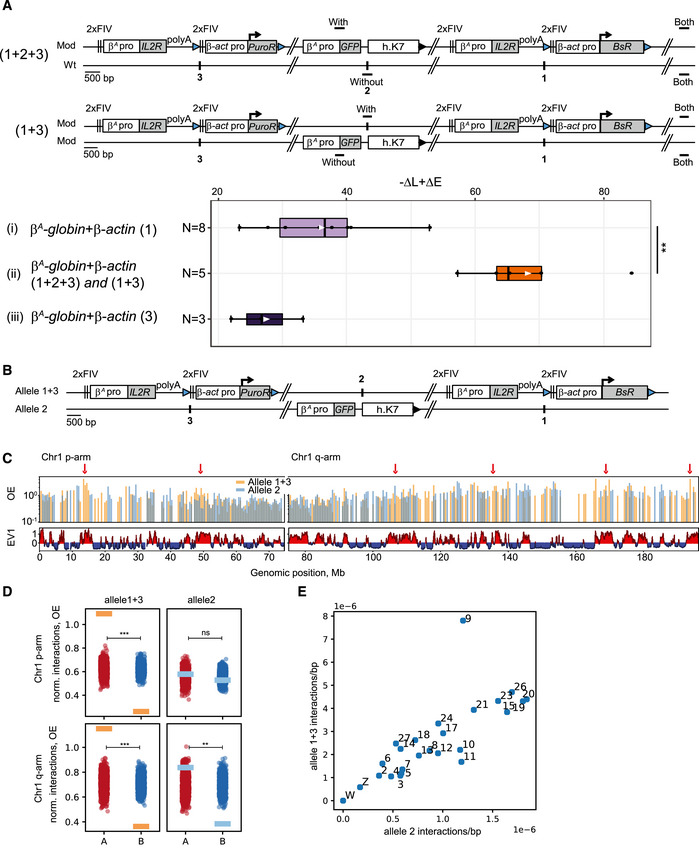
- Distribution of –ΔL + ΔE values for clonal cell lines containing one advanced replicon (β A ‐globin + β‐actin) inserted at site 1 (i, N = 8) or 3 (iii, N = 3) or at both sites on the same chromosome with one GFP reporter construct composed of the GFP reporter gene under the control of the β A ‐globin promoter/origin linked to a 1.6 kb fragment of human chromosome 7 (h.K7) inserted at site 2, either on the same chromosome as 1 and 3 (1 + 2 + 3), or on the other chromosome (1 + 3) (ii, N = 5). Blue and black triangles represent reactive loxP sites and recombined inactive loxP sites, respectively. Black vertical bars represent insertion sites. Rectangle edges correspond to the 0.25 and 0.75 quartiles, the thick black lines represent the median, the white triangles represent the mean and the whiskers extend to the smallest and largest –ΔL + ΔE values. Statistical analysis was performed with Wilcoxon nonparametric two‐tailed tests (**P < 0.01).
- Schematic representation of alleles 1 + 3 and 2. Blue and black triangles represent reactive loxP sites and recombined inactive loxP sites, respectively. Black vertical bars represent insertion sites.
- Interaction profiles of allele 1 + 3/2 with chr1 for combined Hi‐C libraries: distance‐corrected interactions, i.e., observed/expected (OE), binned at 500 kb with corresponding compartment (EV1) tracks. Red arrows represent the loci that show preferred interactions with allele 1 + 3 and that coincide with A compartment.
- OE interactions of allele 1 + 3 (orange rectangles) and allele 2 (blue rectangles) averaged over A and B compartments for p and q arms at 100 kb bin size. A random compartment status permutation test was made, and then, we calculated the one‐sided P‐value for the observed value of “difference allele 1 + 3/2 A − B”. Values corresponding to OE interactions of allele 1 + 3 and allele 2 averaged over shuffled A (red circles, N = 1,000) and B compartments (blue circles, N = 1,000) for p and q arms. ns, not significant; **P < 0.01; ***P < 0.001.
- Scatterplot of density of interactions between allele 1 + 3 and allele 2 with chromosomes larger than 5,000,000 bp.
Figure EV2. One advanced replicon inserted at site 1 or at site 3 directs a shift to earlier replication independently of the presence of a GFP reporter construct inserted at site 2.
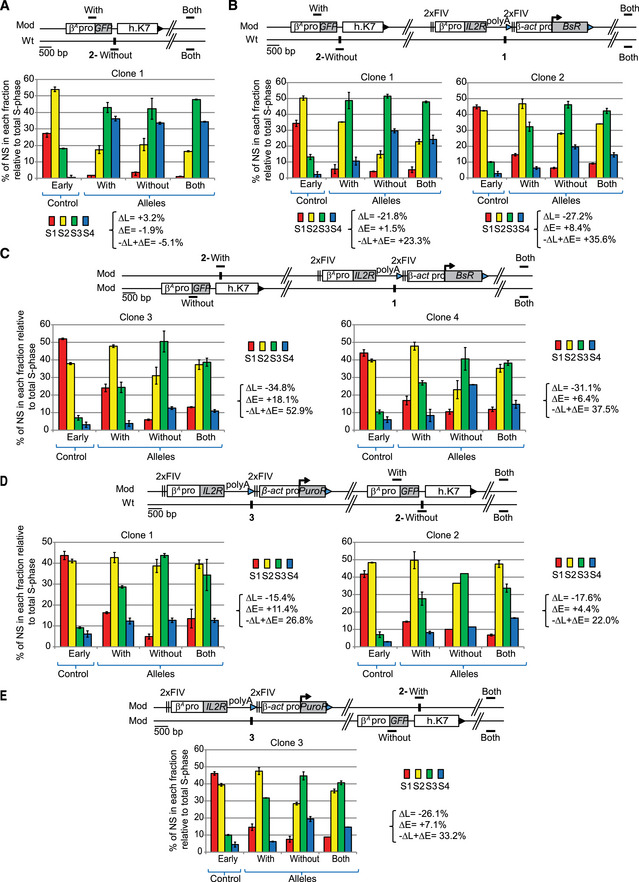
-
AAnalysis of one clonal cell line containing one GFP reporter construct composed of the GFP reporter gene under the control of the β A ‐globin promoter (β A pro) and linked to a 1.6 kb fragment of human chromosome 7 (h.K7) inserted at site 2.
-
B, CAnalysis of clonal cell lines containing one β A ‐globin + β‐actin construct described in Fig 1 inserted at site 1 and one GFP reporter construct inserted at site 2 on the same chromosome (B) or on the other chromosome (C).
-
D, EAnalysis of clonal cell lines containing one β A ‐globin+β‐actin construct inserted at site 3 and one GFP reporter construct inserted at site 2 on the same chromosome (D) or on the other chromosome (E).
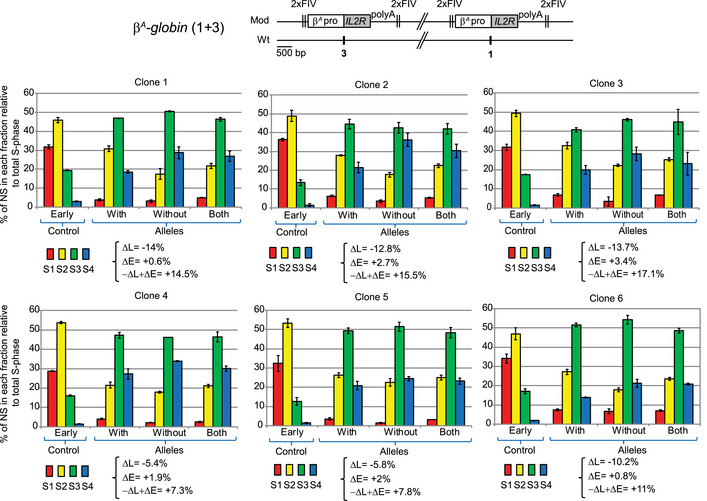
Figure EV3. Two β A ‐globin + β‐actin constructs as well as two β‐actin constructs inserted at sites 1 and 3 form an early‐replicated domain.
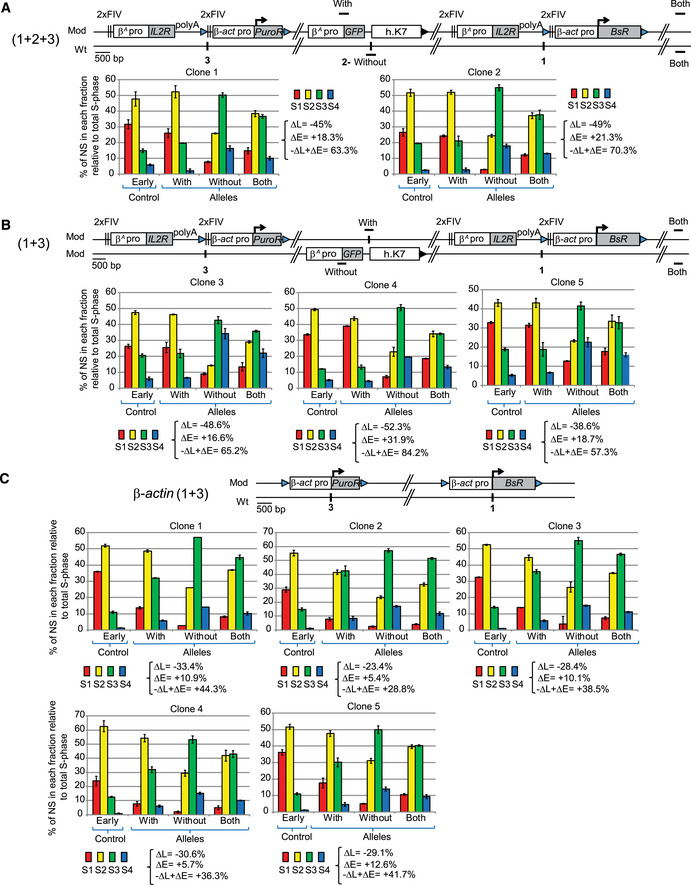
-
A–CRT profiles of each chromosomal allele are determined after targeted transgene integration using the allele‐specific analysis of RT method by real‐time PCR quantification described in Appendix Fig S1. Differences in −ΔL + ΔE values calculated at the target site following transgene integration are indicated. Error bars correspond to the standard deviation for qPCR duplicates. (A, B) Analysis of two 1 + 2 + 3 (A) and three 1 + 3 (B) clonal cell lines described in Fig 3A. (C) Analysis of five clonal cell lines containing two β‐actin constructs inserted at sites 1 and 3 on the same chromosome described in Fig 3B. Blue and black triangles represent reactive loxP sites and recombined inactive loxP sites, respectively. Black vertical bars represent insertion sites. Error bars correspond to the standard deviation for qPCR duplicates.
Genome‐scale studies have shown that RT is strongly correlated with A/B compartments in human cells, suggesting spatial proximity of regions of similar RT within the nucleus (Ryba et al, 2010). We tested whether the large change in RT of the modified chromosome is accompanied by a change in the way the inserted origins interact with A and B compartments. This system allows us to assess how the early‐replicating allele (allele 1 + 3) interacts with A and B compartments compared to the allele with the endogenous RT (allele 2) (Fig 3B). We conducted Hi‐C to assess compartment status genome wide and to measure long‐range chromatin interactions for allele 1 + 3 and allele 2 at the same time. We first examined interactions between inserted sequences at alleles 1 + 3 and 2 and chromosome 1 (chr1). The profile of interactions of allele 1 + 3 with chr1 and allele 2 with chr1 demonstrates alternating patterns in the p‐arm of chr1, whereas in the q‐arm the two profiles show more overlap (Fig 3C, and Appendix Fig S4A and D). Most of the loci that show preferred interactions with allele 1 + 3 coincide with loci located within A compartment domains (Fig 3C, and Appendix Fig S4A and D). To further quantify the observed preference, we calculated average distance‐corrected interactions between allele 1 + 3/allele 2 and regions of chr1 according to their compartment status (Fig 3D and Appendix Fig S4B and E). Averaged interactions confirm that allele 1 + 3 interacts more with A compartment than with B compartment both for p and q arms of chr1, unlike allele 2 which shows smaller preference for A compartment in the q‐arm, and no association with either A or B compartment in the p‐arm. Our observations regarding allele 2 are in line with the WT compartment status of the mid‐late insertion site, as shown by an eigenvector 1 (EV1) value near zero (see also Fig 7A below). We assessed the significance of the interaction preferences by re‐calculating the average interactions of allele 1 + 3/allele 2 and regions of chr1 with randomly shuffled compartment status (EV1) (Fig 3D, Appendix Fig S4B and E) (see Materials and Methods for details). We further looked if allele 1 + 3 and allele 2 demonstrated any preferential interactions with other chromosomes in the genome, by comparing their density of interactions (averaged interactions normalized by chromosome length, see Materials and Methods for details). The number of interactions with chr9 is disproportionately higher for allele 1 + 3 than for allele 2 compared to other chromosomes (Fig 3E, Appendix Fig S4C and F) showing that allele 1 + 3 interacts more with chr9. Our results show that allele 1 + 3 interacts more with specific A compartments located on chr1 and globally more with chr9. These specific interactions coincide with several strong replication origins and high gene density for chr9; however, precise mechanistic understanding will require further investigation.
The constitutive β‐actin promoter/origin is required for the synergic action of the two replicons separated by 30 kb
We have already shown that the large autonomous replicon used in this study is composed of two strong origins associated either with an active (β‐actin) or an inactive (β A ‐globin) promoter (Hassan‐Zadeh et al, 2012 and Appendix Fig S2 and Fig EV4A). MNase (micrococcal nuclease) titration analyses confirmed a differential chromosomal accessibility of the two minimal modules inserted separately at site 1 (Fig EV4B and C, and Appendix Fig S5). However, both minimal modules individually can induce a significant RT shift (Fig 1). To understand in more details the mechanisms involved in the synergy observed in the large β A ‐globin + β‐actin construct, we tested for each minimal construct the effect of a double insertion at sites 1 and 3 on the same chromosome. We observed that similarly to the large autonomous replicon, the β‐actin promoter induced a stronger shift in RT when inserted at 1 + 3 positions (Figs 4A and EV3C). By contrast, two inactive β A ‐globin promoters did not produce any advanced RT when inserted at 1 + 3 sites (Figs 4B and EV5). We observed for a subset of clones even a decrease in the intensity of the RT after the second insertion, a reproducible observation that cannot be explained yet. However, this last result showed that the minimal replicon containing an inactive promoter is not able to synergize when located at two remoted positions. Moreover, it showed that the synergy observed with constructs containing the active β‐actin promoter (small or large modules) did not result from the simple two‐fold increase in the number of active origins in the analyzed region. This result suggests that an unknown specific mechanism allowed two strong origins associated with an active TSS and separated by 30 kb to advance their RT.
Figure EV4. The open chromatin structure of the whole advanced replicon is imposed by the active β‐actin promoter.
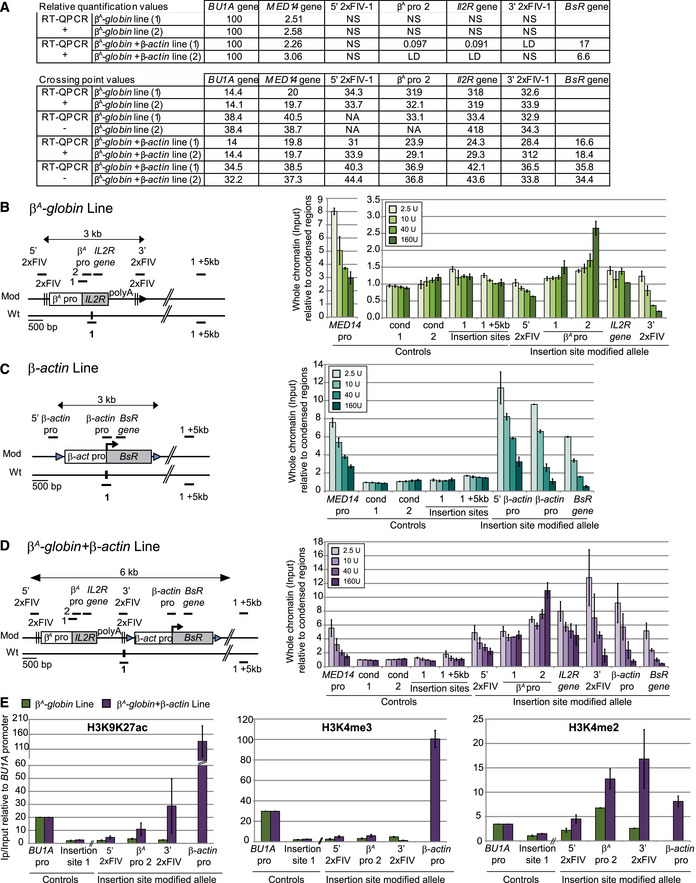
-
ARelative quantification, by real‐time qPCR, of mRNA expression levels (RT‐QPCR+) or background levels (RT‐QPCR−), was performed in the β A ‐globin and β A ‐globin + β‐actin clonal cell lines. For relative quantification, mRNA levels were normalized against BU1A mRNA levels arbitrarily set at 100 (first table). Crossing point (Cp) values for RT‐QPCR+ and RT‐QPCR− experiments are reported in the second table. NA corresponds to nonamplified samples, NS to nonspecific signals, and LD to the limit of detection.
-
B–DThe β A ‐globin (B), β‐actin (C), and β A ‐globin + β‐actin (D) transgenes are shown on the left, with the positions of the amplicons used for quantification (thick black lines with names indicated above). The endogenous active MED14 promoter and two genomic regions located within the condensed region upstream from the β‐globin locus (cond1 and cond2) were analyzed as controls. Quantification, by real‐time qPCR, of total chromatin (input) extracted from the β A ‐globin (B), β‐actin (C), and β A ‐globin + β‐actin (D) clonal cell lines after digestion with increasing concentrations of micrococcal nuclease (MNAse, 2.5 U, 10 U, 40 U, 160 U/ml). Error bars indicate the standard deviation for qPCR triplicates made on two independent clones.
-
EImmunoprecipitations of H3K9K27 acetylation (ac), H3K4 trimethylation (me3), or H3K4 dimethylation (me2) on formaldehyde‐cross‐linked chromatin extracted from the β A ‐globin (green) and β A ‐globin+β‐actin (purple) clonal cell lines. The endogenous active BU1A promoter (BU1A pro) was analyzed as a control. Data are presented as enrichments of immunoprecipitated material relative to input DNA and normalized against BU1A enrichment. Error bars indicate the standard deviation for at least qPCR duplicates made on two independent clones.
Figure 4. The constitutive β‐actin promoter/origin is necessary for the formation of the early‐replicating domain.
- Distribution of –ΔL +ΔE values for clonal cell lines containing one β‐actin construct at site 1 (i, N = 7) or at site 1 and 3 (ii, N = 5), on the same chromosome.
- Distribution of −ΔL + ΔE values for clonal cell lines containing one β A ‐globin construct at site 1 (i, N = 8) or at site 1 and 3 (ii, N = 6), on the same chromosome.
Figure EV5. Two β A ‐globin constructs inserted at sites 1 and 3 are not sufficient to form an early‐replicated domain.
RT profiles of each chromosomal allele are determined after targeted transgene integration using the allele‐specific analysis of RT method by real‐time PCR quantification described in Appendix Fig S1. Differences in –ΔL + ΔE values calculated at the target site following transgene integration are indicated. Error bars correspond to the standard deviation for qPCR duplicates. Analysis of six β A ‐globin (1 + 3) clonal cell lines described in Fig 4B.
The formation of the early‐replicating domain is associated with the spatial proximity of the two advanced replicons
We tested the hypothesis whether the synergy between the two advanced replicons leading to the formation of an early‐replicating domain involved spatial proximity between the replicons concerned. 3C experiments, after cross‐linking, digestion, ligation, and PCR with loci specific primers, measure the frequency of interactions between any two genomic loci. We thus could have used 3C to assess the interactions between the two advanced replicons. However, there are limitations inherent to our system: First, allele 1 + 3 and allele 2 share common sequences. There are two insertion sites on allele 1 + 3 and only one insertion site on allele 2 (Fig 3B). Therefore, we can only quantify the interactions within allele 1 + 3 (i.e., interactions between the two early replicons) without controls for allele 2. Second, the two early replicons located on allele 1 + 3 have almost similar sequences resulting in only few primers that would be 3C compatible. Third, 4C experiments could be another possibility but normalization is not trivial and interpretation of the data can be challenging in this case of different insertions on different alleles. Cre/loxP site‐specific recombination has been successfully used in yeast and bacteria to assess the relative probabilities of different regions of the genome colliding with each another (Hildebrandt & Cozzarelli, 1995; Burgess & Kleckner, 1999). Since Cre/loxP sites are inside our constructs, we used this elegant Cre/loxP system instead of 3C to evaluate the spatial proximity between the two autonomous replicons located 30 kb apart. We assessed the capacity of the 5′ recombination element located in the β A ‐globin + β‐actin construct at site 3 (loxP_RE, yellow triangle, Fig 5A) to recombine with the 3′ recombination element in the β A ‐globin + β‐actin construct at site 1 (loxP_LE, green triangle, Fig 5A). The percentage of chromosomes modified by this recombination (Fig 5C and Appendix Fig S6A, large 1 + 3 excision) was quantified in cell clones with 1 + 2 + 3 and 1 + 3 configurations, 1 h, 8 h, 24 h, and 48 h after the induction of the Cre recombinase. We evaluated the frequency of random contacts by constructing new clones in which the advanced replicon inserted at site 1 was replaced by a loxP_LE element alone (Fig 5A, loxP_LE (1) + β A ‐globin + β‐actin (3)). We quantified the large excision product of the modified chromosome by semi‐quantitative PCR on the whole cell population with a specific primer pair (Fig 5B, Up large excision and Low large excision). The percentage of cells containing this excision product increased steadily over time, from 6 to 14% after 1 h to 43 to 89% of the cells after 48 h of Cre recombinase induction (Fig 5C and Appendix Fig S6A). By contrast, only small amounts of this product were detected in three independent clones carrying the loxP_LE (1) + β A ‐globin+β‐actin (3) configuration, (from 3% after 1 h to 12% of the cells after 48 h of Cre recombinase induction; Fig 5C and Appendix Fig S6A). Recombination rates varied by five‐ to eight‐fold between the two types of configuration, as reported for yeasts harboring different combinations of loxP sites (Burgess & Kleckner, 1999). Since the loxP_LE element is located either at the end of the actively transcribed BsR gene or alone at site 1 in the two configurations, we investigated whether the difference in recombination efficiency might be due to differences in chromatin accessibility at this position. We therefore tested the chromatin accessibility of loxP_LE at site 1 in the two configurations by assessing the MNase sensitivity (Fig 6A and B). The heterochromatin region located upstream from the early‐replicating β‐globin locus was used as a control for the condensed state and the transcriptionally active MED14 promoter for the open state (Prioleau et al, 1999; Litt et al, 2001). We detected similar small amounts of DNA release from loxP_LE in both configurations, for all MNAse concentrations showing that the loxP_LE element had similar accessibility in both configurations. Since two β‐actin constructs can also synergize when inserted at sites 1 and 3 (Fig 4A), we tested their capacity to recombine after Cre recombinase induction. We observed a similar behavior as previously with the large β A ‐globin + β‐actin construct (Fig 5C and Appendix Fig S6B). Overall, our data strongly suggest the existence of molecular contacts between the two constructs synergistically acting on RT. The formation of the early‐replicating domain is, thus, linked to a spatial connection between the two advanced replicons containing an active promoter and located 30 kb apart. It suggests that the two advanced replicons might form a chromatin loop.
Figure 5. The formation of the early‐replicating domain is associated with the establishment of local interactions between the two advanced replicons separated by 30 kb.
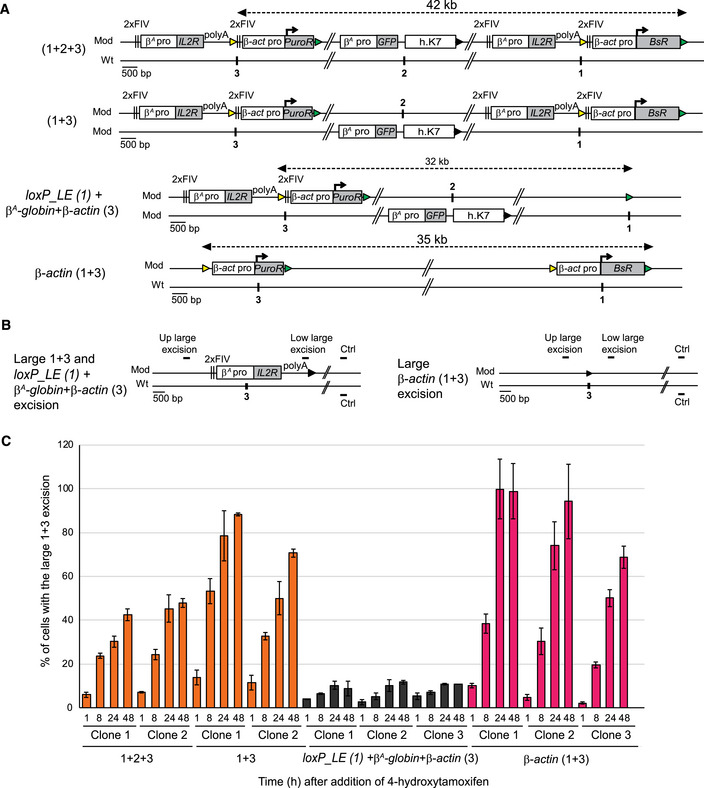
- Clones 1 + 2 + 3, 1 + 3, and β‐actin (1 + 3) described in Fig 3, together with control clones containing one β A ‐globin + β‐actin construct at site 3 and one loxP_LE sequence inserted at site 1 on the same chromosome with the GFP reporter construct inserted at site 2 on the other chromosome loxP_LE (1) + β A ‐globin + β‐actin (3), were tested for their capacity to recombine after induction of the Cre recombinase. Yellow and green triangles represent reactive loxP_RE and loxP_LE sites, respectively, and black triangles represent recombined inactive loxP sites. Black vertical bars represent insertion sites.
- Recombination between the upstream loxP_RE element (yellow triangle) inserted at site 3 and the downstream loxP_LE element (green triangle) inserted at site 1 leads to a large 1 + 3 and loxP_LE (1) + β A ‐globin+β‐actin (3) or β‐actin (1 + 3) excision product.
- After 1, 8, 24, and 48 h of 4‐hydroxytamoxifen treatment, genomic DNA was extracted and quantified by semi‐quantitative PCR (see Appendix Fig S6). Error bars correspond to the standard deviation for PCR duplicates. The percentages of cells with the large 1 + 3 and loxP_LE (1) + β A ‐globin + β‐actin (3) or β‐actin (1 + 3) excision at each time point are shown for different cell lines.
Figure 6. The close proximity of the two advanced replicons is not associated with an increase in chromatin accessibility.
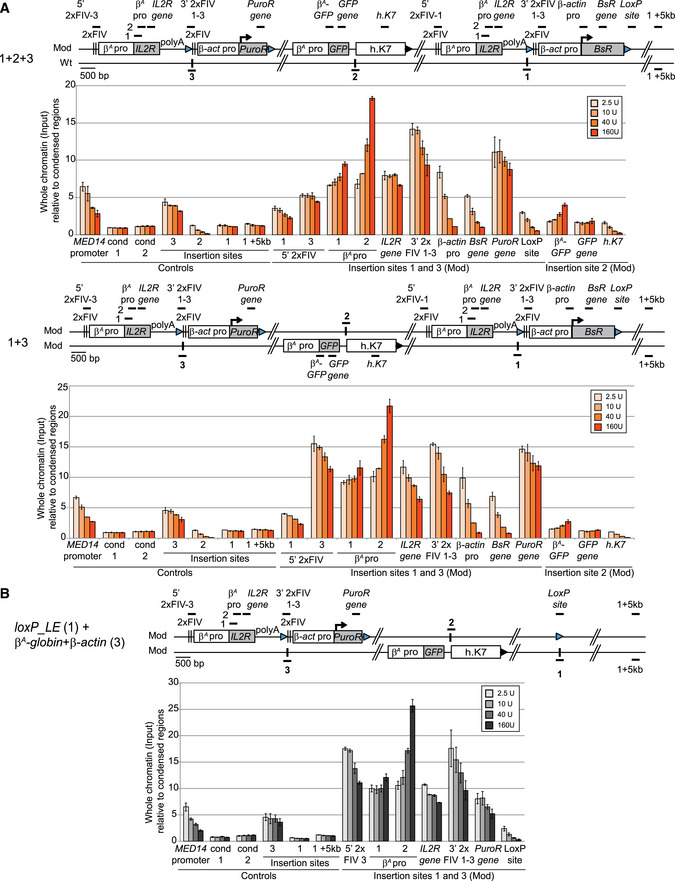
-
A, BClones 1 + 2 + 3, 1 + 3 (A), and loxP_LE (1) + β A ‐globin + β‐actin (3) (B) were used to assess chromatin accessibility within the transgenes. The positions of the amplicons used for quantification in each shifting construct individually (thick black lines; 5′ 2xFIV‐1 or 2xFIV‐1-3, β‐actin pro‐1 or pro‐3, BsR gene, PuroR gene, LoxP site), in both shifting constructs (β A pro 1 and 2, IL2R gene, 3′ 2xFIV1‐3) or in the GFP reporter construct (β A‐GFP,GFP gene, h.K7) are shown. The insertion sites on the wt chromosome (insertion sites 1, 2, and 3) and a genomic region located 5 kb downstream from the insertion site (insertion site + 5 kb) were used as controls for the targeted genomic region. The endogenous active MED14 promoter was analyzed as a control. Blue and black triangles represent reactive loxP sites and recombined inactive loxP sites, respectively. Black vertical bars represent insertion sites. Quantifications by real‐time qPCR of total chromatin (input) extracted from two 1 + 2 + 3, 1 + 3, and loxP_LE (1) + β A ‐globin + β‐actin (3) clonal cell lines, after digestion with micrococcal nuclease (MNAse, 2.5 U, 10 U, 40 U, 160 U/ml) and size selection were shown. Error bars indicate the standard deviation for qPCR triplicates, for two independent clones. Data are presented as total chromatin input relative to the condensed genomic regions (cond1 and cond2) used for the normalization.
The close spatial proximity of the two advanced replicons is not associated with an increase in chromatin accessibility
We then investigated whether the synergy of the two autonomous replicons associated with their physical proximity affected the chromatin accessibility of these replicons or of one of the genomic regions located within the early‐replicating domain. We addressed this question by quantifying, nuclease‐treated chromatin in 1 + 2 + 3 and 1 + 3 cell lines, by qPCR with specific primer sets specifically amplifying at either one or both autonomous β A ‐globin + β‐actin replicons, at the central GFP reporter transgene, at the three insertion sites or at control loci (Fig 6A and B). We compared chromatin accessibility between these cell lines and the one having only one β A ‐globin + β‐actin construct inserted at site 1 (Fig EV4D). We observed an increase in total nucleosome release at insertion site 3 relative to insertions sites 1 and 2 and condensed chromatin controls, for all MNAse concentrations. The chromatin of this insertion site is, therefore, more accessible than that at sites 1 and 2 (Fig 6, insertion site 3). The β‐actin module harbors the same pattern of accessibility as observed with one autonomous replicon (compare Figs 6B and EV4C). Nucleosomes embedded within the upstream 2xFIV element of the β A ‐globin+β‐actin construct at site 3 (5′ 2xFIV‐3) and within the PuroR gene were more susceptible to MNAse, regardless of MNAse concentration, than nucleosomes embedded at similar positions (5′ 2xFIV‐1 and BsR gene) in the β A ‐globin + β‐actin construct at site 1. The chromatin at this site is, therefore, also more exposed, probably reflecting the greater accessibility of the unmodified site 3. The GFP reporter construct in the middle of the early‐replicating domain (1 + 2 + 3) or on the other mid‐late‐replicating unmodified chromosome (1 + 3), released similar small amounts of material for all MNAse concentrations as were detected for heterochromatin regions or at the unmodified site 2. We can conclude from these results that two modules composed of 5.6 kb of accessible chromatin and containing efficient replicators separated by 30 kb are sufficient to create an early‐replicated domain when inserted into a naturally closed mid‐late‐replicating region. These two constructs can establish spatial connections without increasing the accessibility of their own chromatin or the accessibility of the chromatin located in the middle of the early‐replicating domain.
Two late‐replicating environments embedded into a B compartment are permissive to a shift toward earlier replication after the site‐specific insertion of a large autonomous replicon
It remains unknown whether late‐replicating regions are permissive to early timing control elements. One hypothesis is that the nuclear and chromatin organizations of late‐replicating domains impose robust constraints that prevent earlier firing. We addressed this question directly, by investigating whether the β A ‐globin + β‐actin construct behaves as efficiently when targeted to regions naturally displaying very late replication and located in a B compartment. We selected two late‐replicating regions from the genome‐wide RT profile obtained for wt DT40 cells. One of these regions was located 2 Mb upstream from the previously studied mid‐late‐replicating region on chromosome 1 (Late1 chr1:70,523,649 and mid‐late chr1:72,565,520 bp, galGal5; Fig 7A), and the other was located 105.4 Mb downstream from this mid‐late‐replicating region (Late2 chr1:177,936,192 bp, galGal5, Fig 7B). The late 2 locus is characterized by gene depletion over a 500 kb region in contrast to the late 1 locus (Fig 7). In addition, we know from a previous study that the two late loci are found at the nuclear periphery in DT40 cells. However, the late 2 locus is more tightly associated with nuclear lamina than the late 1 (fig 6 in Duriez et al, 2019). Altogether, it strongly suggests that the late 2 locus is a constitutive late domain whereas the late 1 locus might be a facultative late domain. We confirmed that these two regions were, indeed, replicated much later than the previously studied region (Fig 8A and B, Without). We calculated –ΔL + ΔE values for cell clones containing the β A ‐globin + β‐actin transgene inserted into these late‐replicating regions (Fig 8). We observed a strong impact on RT of transgene insertion at the late 1 site (Fig 8A). The RT shift was comparable with the one observed following insertions into the previously studied mid‐late‐replicating region (−ΔL + ΔE = +29.3% for clone 1 and +51.7% for clone 2, Fig 8A versus smallest and greatest –ΔL + ΔE values calculated for eight clones: +23.3 and 52.9%, respectively, in the mid‐late‐replicating region, Fig 1). However, in the late 2 region, we detected a slightly lower, but nevertheless significant shift in RT, than that induced in a mid‐late‐replicating region (−ΔL + ΔE = +16.1% for clone 1, +19.1% for clone 2 and +21.3% for clone 3, Fig 8B). Our findings confirm the robustness of the previously identified combination of cis‐elements to advance the RT. We show here that the β A ‐globin + β‐actin combination can impose local autonomous control over RT in at least three chromosomal regions, one replicating in mid‐late S‐phase and the other two displaying late replication among which one is a constitutive late domain (Late 2).
Figure 7. Genome‐wide overview of genomic features at the targeted insertion sites in DT40.
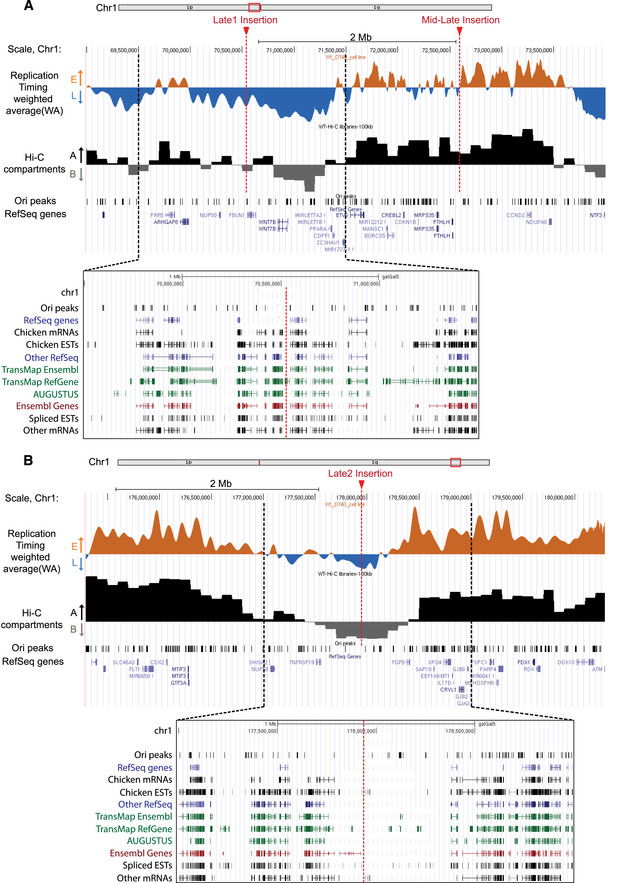
-
A, BUCSC genome browser visualization of two parts of chromosome 1 containing the insertion sites (genomic positions: chr1:69,000,000–74,200,000; 5.2 Mb (A) and chr1: 175,300,000–180,500,000; 5.2 Mb (B); galGal5). RT‐weighted average (WA) values obtained after cells sorting into four S‐phase fractions, BrdU pulse‐labeled nascent strands (NS) immunoprecipitation and sequencing are shown. Early‐replicated regions are represented in orange and late‐replicating regions in blue. Below, track of eigenvector 1 values corresponding to A/B compartments at 100 kb resolution after Hi‐C analysis in wt cells, track of replication origins determined in (Massip et al, 2019) (Ori peaks) and annotated genes are reported. The three sites chosen for insertion are indicated with red arrows and dotted lines (mid‐late, late 1 (A), and late 2 (B) insertion). At the bottom, an enlargement of late insertion sites (chr1: 69,500,000–71,500,000 (A), chr1: 177,000,000–179,000,000 (B); galGal5), displaying genes, mRNAs and ESTs for the chicken and all other species together with track of replication origins determined in (Massip et al, 2019) (Ori peaks) is shown. Tracks of RefSeq genes are represented in blue, tracks of Ensembl genes in red, tracks of AUGUSTUS ab initio gene predictions and TransMap alignements in green and tracks of mRNAs and ESTs in black. Insertion sites are indicated with red dotted lines, and black dotted lines determine the borders of the enlarged regions.
Figure 8. Two late‐replicating environments are permissive to a shift toward earlier replication after the site‐specific insertion of a large autonomous replicon.
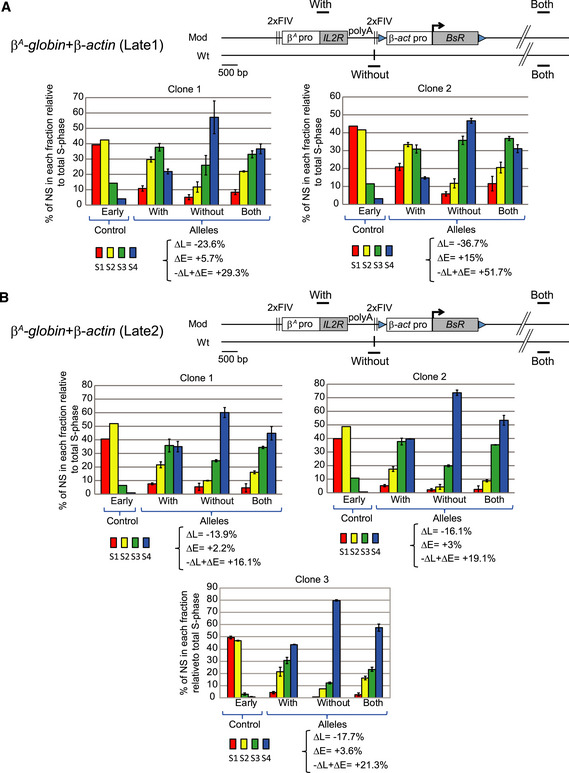
-
A, BRT profiles of chromosomal alleles following the targeted integration of a β A ‐globin + β‐actin construct into two late‐replicating loci, 1 (A) and 2 (B) in two or three clonal cell lines, respectively. Nascent strands (NS) were quantified by real‐time qPCR in four S‐phase fractions. Specific primer pairs determine the RT profile for the modified allele (With), the wt allele (Without), and both alleles (Both). The endogenous β‐globin locus was analyzed as an early‐replicated control. The modified (Mod) and wild‐type (wt) alleles are shown. Difference –ΔL + ΔE values calculated at the target site following transgene integration are indicated (see Appendix Fig S1). Error bars correspond to the standard deviation for qPCR duplicates. The black vertical bars represent the precise insertion position.
Discussion
The DNA RT program has emerged as a system that integrates genome regulation and the three‐dimensional organization of the genome. From these observations was proposed the replication domain model in which each RT domain acts as a regulatory unit, determining when the replicons within its boundaries can fire. We challenged this model, by disturbing naturally mid‐late‐ and late‐replicating domains after site‐specific insertion of constructs containing critical cis‐elements known to regulate origin firing. Based on the work described here, we identified five important principles underlying the establishment and maintenance of domains of early and late replication: (i) A strong origin embedded into an accessible chromatin structure can significantly advance the RT of a mid‐late‐replicating region; (ii) two replicators carrying timing information can act in cooperation if located in close proximity; (iii) strong autonomous replicons overlapping an active promoter can act in synergy on RT even when separated by 30 kb to form early‐replicated regions; (iv) the formation of an early‐replicating domain results locally in the formation of a spatial connection between the advanced replicons and on a larger scale in an increase of contacts with the A compartment; and (v) the RT of late‐replicating domains can be advanced locally by a strong autonomous replicon, but the final RT is also influenced by chromosomal context.
Molecular mechanisms shaping efficient replicons carrying timing information
Studies in Saccharomyces cerevisiae have suggested that the correct temporal activation of origins during S‐phase involves the activation of early‐firing origins, because these origins have a greater “accessibility” to firing factors, which are present in limiting amounts. After firing, the limiting factors are released and can bind to and activate less accessible origins, and so on (Douglas & Diffley, 2012). This theory suggests that the RT of a region directly reflects the accessibility to these limiting factors of specific regions bound by pre‐RCs. In vertebrate cells, there is a strong correlation between early replication and the accumulation of open chromatin marks, consistent with a role for the openness of chromatin in the recruitment of these potential limiting factors (Ryba et al, 2010; Picard et al, 2014). We previously identified a combination of cis‐elements constituting an independent replicon with the capacity to advance RT of a 250 kb mid‐late‐replicating region significantly (Hassan‐Zadeh et al, 2012 and Fig 2A). This construct contains only promoters and regulatory motifs naturally present in the chicken genome (one tissue‐specific promoter, one constitutive promoter and four FIVs elements), linked to either the IL2R reporter gene or the blasticidin gene selection cassette. Moreover, autonomous replicon activity is driven only by endogenously expressed trans‐factors. Investigation of the chromatin structure of each independent unit revealed a number of different features, highlighting the potential diversity of cis‐elements involved in RT control in vertebrates. One of the elements identified contains the β A ‐globin erythroid tissue‐specific promoter which cannot drive transcription in the DT40 lymphoid cell line. The other element contains the constitutive β‐actin promoter linked to a gene for selection. This strong origin/promoter facilitates the local opening of chromatin structure and the acquisition of H3K4me3 and H3K9K27ac marks at the inserted position (Fig EV4E). These findings are consistent with the observation that the synthetic activation of transcription, but not nuclear repositioning, shifts the RT of several mid‐late‐replicating loci to mid‐early in mouse ES cells (Therizols et al, 2014). Our first observation shows that, at least at this mid‐late‐replicated chromosomal position, origins located within the β A ‐globin and β‐actin promoters are highly efficient, because they were able to advance RT locally in a population‐based timing assay. We were unable to estimate the fraction of cells in which these two replicators were activated, but the significant shift in RT toward earlier replication strongly suggests that they are activated in a large proportion of cells. Based on genome‐wide studies and our recent genetic study, we suggest that most vertebrate origins consist of a G‐quadruplex motif linked to an as yet undefined cis‐element and that replicators overlapping a promoter tend to be the most effective (Besnard et al, 2012; Cayrou et al, 2012; Valton et al, 2014). Both the β A ‐globin and β‐actin promoters/origins contain several G‐quadruplex motifs and locally induce strong SNS enrichment when inserted ectopically, thereby satisfying the criteria for a “canonical strong origin” (Hassan‐Zadeh et al, 2012 and Appendix Fig S2).
Statistical analyses showed that these origins together with cis‐elements controlling chromatin structure could act in cooperation when located close together by providing more effective timing information. This result supports the hypothesis that early‐replicating domains result from the clustering of origins embedded within an open chromatin structure. Consistent with this hypothesis, genome‐wide mapping of replication origins by the SNS method in human and mouse cells has shown the clustering of efficient replication origins within early‐replicated domains, forming small initiation zones of several kilobases, mostly located around TSS containing a CpG island (CGI) (Picard et al, 2014; Cayrou et al, 2015). Our two linked RT shift‐inducing constructs are thus reminiscent of the organization of the endogenous structure of early‐replicated domains.
Early replicators can advance RT locally in late‐replicating domains
We investigated the possible dominance of late‐replicating domains over replicators carrying early timing information, by assessing the capacity of the same previously dissected combination of cis‐elements to function as an independent replicon in genomic regions that are naturally replicated in late S‐phase. This construct has the ability to advance significantly the RT at three chromosomal loci (one replicated in mid‐late S‐phase and two in late S‐phase), demonstrating the robustness of the signal embedded in this specific construct. With respect to the replication domain model, these findings suggest that any type of chromosomal environment, including a large constitutive late domain, is permissive for the formation of an autonomous advanced replicon. However, the observation that the final RT for the modified region was more advanced for the region normally replicated in mid‐late S‐phase than for that normally replicated in late S‐phase suggests that there may be two layers of regulation, at the local and large scales. Both these levels of regulation must be taken into account if we are to understand how large replication domains are constructed. Our data suggest that late‐replicating domains are also defined by a lack of signals associated with autonomous early replicons. Consistent with this hypothesis, one mechanism proposed for the regulation of late‐replicated regions associated with common fragile sites involves a low density of replication initiation events (Letessier et al, 2011). Less abundant but nevertheless efficient origins can also be detected in late‐replicating domains by the SNS method. A recent study investigated the role of H4K20me marks in controlling late‐firing origins. It showed that the conversion of K20me1 to K20me3 enhances ORCA recruitment and MCM loading at already defined origins required for the correct replication of heterochromatin (Brustel et al, 2017). Interestingly, the loss of Suv4‐20h, which is responsible for this methylation, delays RT for 15% of the mouse genome. Late origins are, therefore, also defined by a specific chromatin organization facilitating the coordination of late‐firing and counteracting the overall repressive effect of heterochromatic regions without providing early‐firing signals.
Robust early‐replicated domains can be formed by synergy between efficient early replicators separated by several tens of kilobases
We investigated the formation of early‐replicating domains further, by inserting two identical strong combinations of early origins into the same chromosome, at sites 30 kb apart. This distance is within the range of average spacing between strong initiation sites or zones detected by the SNS method in early‐replicated domains. To our surprise, we found that these two inserted constructs advanced RT to a much greater extent when inserted together than when inserted alone. We found that the presence of the two constructs favored physical contact between these two remote regions. We suspect that this spatial connection is required for the synergic effect on RT, although we have no definitive proof that this is the case. Interactions between active promoters have been observed by several methods, including the recent production of an ultrahigh‐resolution Hi‐C map during neural differentiation in mice (Li et al, 2012; Bonev et al, 2017). Moreover, a recent study proposed that origins of replication tend to cluster even more efficiently than promoters themselves (preprint: Jodkowska et al, 2019). These results are consistent with our observations, because our construct shown to act synergically contains the constitutive β‐actin promoter. In S. cerevisiae, the Fkh1/2 transcription factors have been shown to be directly involved in the early‐firing of a large group of origins (Knott et al, 2012). It has recently been shown that this function involves direct interaction with Dbf4, the regulatory unit of the essential DDK firing factor (Fang et al, 2017). Based on the dimerization capacity of Fkh1/2 and the observation that early‐firing origins associated with Fkh1/2 tend to cluster, it was suggested that this clustering might even increase the efficiency of Fkh1/2 for the local recruitment of Dbf4 to early replication factories (Knott et al, 2012). Recently, replication initiators containing intrinsically disordered regions (IDRs) have been described in Drosophila melanogaster and constitute a new class of phase separating elements that undergo liquid–liquid phase separation (LLPS) upon DNA binding in vitro. These elements may drive through IDR‐IDR interactions a spatial co‐localization of multiple active replication origins within a nuclear zone, which could also favor a better accessibility to firing factors (Parker et al, 2019). We thus suggest that the close proximity of two strong autonomous replicators carrying early timing information here locally increased the efficiency of limiting firing factor recruitment in a similar manner. Most efficient early‐firing origins are associated with active promoters containing CGI. This model is, therefore, consistent with genome‐wide observations, validating our synthetic approach.
Materials and Methods
Plasmid construction
The targeting vectors for homologous recombination in DT40 cells were constructed with the multisite Gateway Pro kit (Thermo Fischer Scientific #12537100), as previously described (Hassan‐Zadeh et al, 2012). The same entry vectors as before were used for the 5′ and 3′ target arms for specific insertion at site 1 in the mid‐late‐replicating region (chr1:72,565,520 bp, galGal5) (Hassan‐Zadeh et al, 2012). New arms were prepared for specific targeting at sites 2 (chr1: 72,548,589 bp, galGal5) and 3 (chr1: 72,536,060 bp, galGal5) in the mid‐late‐replicating region, and at sites 1 (Late1 chr1:70,523,649 bp, galGal5) and 2 (Late2 chr1:177,936,192 bp, galGal5) in the late regions. The 5′ and 3′ target arms for homologous recombination were amplified from DT40 genomic DNA with the primer pairs listed in Appendix Table S3.
We used four entry vectors to generate the new β A ‐globin + β‐actin construct inserted at site 1: two entry vectors containing the 5′ and 3′ target arms for specific insertion at site 1 (Appendix Table S3) and two new entry vectors. The first contained two copies of FIV linked to the β A ‐globin promoter (2xFIV_β A ‐globin). The 2XFIV_β A ‐globin entry vector was used as a template for PCR amplification with the following primers: 2XFIV_β A ‐globin‐Up/Low (Appendix Table S3). The second entry vector (2xFIV_β‐actin + BsR) contained the 2xFIV elements upstream from the β‐actin promoter linked with the blasticidin resistance gene (BsR), flanked by loxP sites. This construct was made by amplifying the β‐actin promoter from the pLoxBsR vector (Arakawa et al, 2001) such that XhoI and 2xFIV sequences were added upstream from the promoter with the following primers: 2XFIV_β‐actin‐Up/Low (Appendix Table S3). The PCR product was inserted into the previously described β‐actin+BsR entry vector (Hassan‐Zadeh et al, 2012), between the XhoI site located upstream from the β‐actin promoter and the BglII site at the end of the promoter by ligation with T4 DNA ligase (BioLabs #M0202S), and the 2xFIV_β‐actin+BsR entry vector was selected after sequence verification. The corresponding final vector was generated by recombining compatible att sites between the entry vectors, with LR clonase. For electroporation, the final vector was linearized with ScaI (NEB #R3122S).
We used four entry vectors to generate the β A ‐globin + β‐actin construct inserted at site 3. We used two entry vectors containing the 5′ and 3′ target arms for specific insertion at site 3 (Appendix Table S3), the previously described 2XFIV_β A ‐globin entry vector and a new entry vector (2xFIV_β‐actin+PuroR) containing the 2xFIV sequence upstream from the β‐actin promoter linked with the puromycin resistance (PuroR) gene, flanked by loxP sites. This construct was made by amplifying part of the entry vector from the previously described β A ‐globin+β‐actin vector such that the attB sites were flanked by the XhoI restriction site and 2xFIV sequences on one side of the recombined region and the XhoI sequence on the other, with the following primer pair: XhoI + 2xFIV‐Up/Low (Appendix Table S3). The β‐actin promoter associated with the PuroR gene cassette was produced by XhoI digestion of the pLoxPuro plasmid (Arakawa et al, 2001). The final 2xFIV_β‐actin+PuroR entry vector for insertion at site 3 was generated by XhoI digestion of the PCR product and ligation with T4 DNA ligase. The corresponding 2xFIV_β‐actin+PuroR entry vector was selected after sequence verification. The final vector was generated by recombining compatible att sites between the four entry vectors, with LR clonase. For electroporation, the final vector was linearized with PvuI (NEB #R3150S).
We used four entry vectors to generate the GFP reporter construct inserted at site 2. We used two entry vectors containing the 5′ and 3′ target arms for specific insertion at site 2 (Appendix Table S3), one entry vector previously described and containing a similar β‐actin+BsR cassette flanked by two loxP sites (Hassan‐Zadeh et al, 2012) and one new entry vector (β A ‐GFP‐h.K7). This entry vector contained the β A ‐globin promoter sequence amplified from the previously described 2xFIV_β A ‐globin entry vector with the following primer pair: β A‐Up/Low, the GFP gene sequence amplified from the peGFP plasmid (Clontech #6085‐1) with the following primer pair: GFP‐Up/Low and part of human chromosome 7 (chr7:26,873,165–26,874,805 bp, hg38) amplified from human genomic DNA with the following primer pair: h.K7‐Up/Low (Appendix Table S3). The β A‐Up and h.K7‐Low primers were associated with the corresponding attB sites for Gateway recombination. These three DNA fragments were combined with the In‐Fusion HD cloning plus kit (Takara #638909), introduced into the corresponding entry vector with BP clonase (Thermo Fisher Scientific #11789020) and selected after sequence verification. The final vector was generated by recombining compatible att sites between the four entry vectors, with LR clonase (Thermo Fisher Scientific #12538120). For electroporation, the final vector was linearized with PvuI (NEB #R3150S).
The loxP_LE sequence was inserted into site 1 by the insertion of a larger construct composed of the loxP_LE‐h.K7+ β‐actin + BsR elements followed by site‐specific Cre recombinase excision. This construct was generated by the association of two entry vectors containing the 5′ and 3′ target arms for specific insertion at site 1, a previously described entry vector containing a similar β‐actin + BsR selection cassette (Hassan‐Zadeh et al, 2012) and a new entry vector (loxP_RE‐h.K7). This entry vector contained part of the human chromosome 7 sequence amplified from human genomic DNA with the following primer pair containing the upstream loxP_RE sequence: loxP_RE‐h.K7‐Up/Low (Appendix Table S3). The loxP_RE‐h.K7‐Up and the loxP_RE‐h.K7‐Low primers were associated with the corresponding attB sites. The PCR products were introduced into the corresponding entry vector with BP clonase and selected after sequence verification. The final vector was generated by recombining compatible att sites between the four entry vectors, with LR clonase. For electroporation, the final vector was linearized with PvuII (NEB #R3151S).
We used four entry vectors to generate the new β A ‐globin +β ‐actin construct inserted at site late 1 and late 2: two entry vectors containing the 5′ and 3′ target arms for specific insertion at site late 1 or late 2 (Appendix Table S3) and the previously described β A ‐globin and β‐actin+BsR entry vectors (Hassan‐Zadeh et al, 2012).
We used three entry vectors to generate the β‐actin construct inserted at site 1 and 3: the previously described β‐actin + BsR or β‐actin + PuroR entry vector (Hassan‐Zadeh et al, 2012), one entry vector containing the 3′ target arm for specific insertion at site 1 or 3, and a new entry vector containing the 5′ target arm for specific insertion at site 1 or 3 flanked by att sequences necessary for Gateway recombination using three entry vectors (Appendix Table S3).
Cell culture and transfection
DT40 cells were grown in RPMI 1640 GlutaMAX (Thermo Fisher Scientific #61870010) containing 10% FBS, 1% chicken serum, 0.1 mM β‐mercaptoethanol, 200 U/ml penicillin, 200 μg/ml streptomycin, and 1.75 μg/ml amphotericin B, at 37°C, under an atmosphere containing 5% CO2. We transfected DT40 cells as previously described (Hassan‐Zadeh et al, 2012). Cell clones were selected on media containing a final concentration of 20 μg/ml blasticidin or 1 μg/ml puromycin, depending on the resistance gene carried by the transgene. Genomic DNA was extracted from cells in lysis buffer (10 mM Tris pH 8.0; 25 mM NaCl; 1 mM EDTA and 200 μg/ml proteinase K). Clones into which the plasmid DNA was integrated were screened by PCR with primer pairs designed to bind on one side of the insertion site such that one primer bound within the construct and the other primer bound just upstream or downstream from the arm used for recombination, as previously described (Hassan‐Zadeh et al, 2012; Appendix Fig S7 and Appendix Table S4). The insertion of constructs into the same chromosome or the other chromosome was determined by long‐range PCR (Appendix Fig S8). For each clone tested, genomic DNA was extracted with the DNeasy Blood & Tissue kit (Qiagen #69504). We designed primer pairs amplifying in the part of the genome between constructs to control for DNA extraction quality (Ctrl), and primer pairs amplifying the two tested constructs to test the insertions (Appendix Table S4). The LR 1 + 2 primer pair was used to amplify the h.K7 part of the GFP reporter construct inserted at site 2 and the IL2R gene within the β A ‐globin + β‐actin construct inserted at site 1. The LR 2 + 3 primer pair was used to amplify the PuroR gene within the β A ‐globin + β‐actin construct inserted at site 3 and the GFP gene within the GFP reporter construct inserted at site 2. The LR 1 + 3 primer pair was used to amplify the PuroR gene within the β A ‐globin + β‐actin construct inserted at site 3 and the IL2R gene within the β A ‐globin + β‐actin construct inserted at site 1, regardless of the presence or absence of the central construct. The LR loxP_LE(1) + β A ‐globin + β‐actin (3) primer pair was used to amplify the PuroR gene within the β A ‐globin + β‐actin construct inserted at site 3 and the loxP_LE element inserted at site 1, regardless of the presence or absence of the central construct. PCR was performed with the Long PCR Enzyme Mix (Thermo Fisher Scientific #K0181) under following conditions: initial denaturation at 94°C for 3 min, followed by 10 cycles of 95°C for 20 s, 68°C for 14 min, followed by 20 cycles of 95°C for 20 s, 68°C for 18 min and a final extension phase of 10 min at 68°C. Quantitative PCR (qPCR) was carried out for each clone, on 4 ng of genomic DNA, with a primer binding to the transgene (With on the GFP reporter construct, With on β A ‐globin + β‐actin construct, With on β‐actin and loxP_LE on the loxP site inserted at site 1), and a primer binding close to insertion site 1 and amplifying both alleles (Both or insertion site 1 + 5 kb), to confirm that only one copy of the transgene had been inserted (Appendix Tables S2 and S3).
Cre‐loxP excision
In this study, we used the DT40 Cre1 subclone, which constitutively expresses a tightly regulated Cre recombinase fused to a mutated estrogen receptor (Mer) (Arakawa et al, 2001). This inactive Mer‐Cre‐Mer fusion protein can be transiently activated in the presence of 4‐hydroxytamoxifen, resulting in the efficient excision of genomic regions flanked by two recombination signals (loxP sites) inserted in the same direction. We also used a modified inducible Cre recombination system involving two different mutant loxP sites (loxP_RE and loxP_LE). After Cre recombination, these two mutants are converted into a new nonfunctional loxP site (loxP_RE+LE) that is not recognized by the Cre recombinase, thereby preventing additional recombination events (Arakawa et al, 2001). For the excision of the genomic DNA flanked by loxP sites, we treated 3 × 105 cells with 5 μM 4‐hydroxytamoxifen (Sigma‐Aldrich #T176) for 2 days. Subclones were obtained by plating dilutions of the treated cell suspension at a density of 50, 150, and 1,500 viable cells per 10 ml in 96‐well flat‐bottomed microtiter plates. Genomic DNA was extracted from single subclones and analyzed by PCR with specific primer pairs (Appendix Table S4). We assessed the excision of the β‐actin + BsR selection cassette in clones with the GFP reporter construct inserted at site 2, with a primer pair amplifying the polyA sequence of the both BsR and GFP genes and the downstream part of the 3′ arm of insertion site 2 (Appendix Fig S7, primer pair #3‐4, BsR excision site 2, Appendix Table S4). We assessed the excision of the β‐actin + PuroR selection cassette in clones with the β A ‐globin + β‐actin construct inserted at site 3 and already containing the β A ‐globin construct at site 1 (Hassan‐Zadeh et al, 2012), with a primer pair amplifying the polyA sequence of the both IL2R and PuroR genes and the downstream part of the 3′ arm of insertion site 3 (Appendix Fig S7, primer pair #5‐6, PuroR excision at site 3, Appendix Table S4). All clones were cultured for 72 h in selective media containing the appropriate antibiotic, to confirm PCR results.
For the large 1 + 3 excision kinetic, 3 × 106 cells were treated with 5 μM 4‐hydroxytamoxifen for 1, 8, 24, 48 h before genomic DNA extraction with the DNeasy Blood & Tissue kit (Qiagen #69504). The large 1 + 3 excision was quantified by semi‐quantitative PCR with the large excision primer pair and the Ctrl primer pair used for normalization (Fig 5, Appendix Fig S6 and Appendix Table S4). Calibration was based on a standard curve generated with various amounts of genomic DNA (16–160 ng) obtained from a previously isolated clone in which the large recombination had occurred. For each set of excision kinetic conditions, 64 ng or 160 ng of genomic DNA was tested (for amplification with the Ctrl or the large excision primer pair, respectively). PCR was performed with the Herculase II fusion DNA polymerase (Agilent Technologies #600675) under the following conditions: initial denaturation at 98°C for 4 min, followed by 25 cycles of 98°C for 30 s, 57°C for 45 s, 72°C for 2 min, and a final extension phase for 3 min at 72°C. After migration of the PCR products on a 0.8% agarose gel and SYBR Safe staining, the mean intensity of each signal was quantified with ImageJ, corrected with the local background mean intensity and normalized with the mean intensity of the signal obtained in the control amplification.
RT analysis
The RT experiments presented in Figs 8, and EV1, EV2, EV3 and EV5, were performed as previously described (Hassan‐Zadeh et al, 2012). Briefly, about 107 exponentially growing cells were pulse‐labeled with 5‐Bromo‐2′‐deoxyuridine (BrdU, Sigma‐Aldrich #B9285) for 1 h and sorted into four S‐phase fractions, from early to late S‐phase. The collected cells were treated with lysis buffer (50 mM Tris pH 8.0; 10 mM EDTA pH 8.0; 300 mM NaCl; 0.5% SDS, 0.2 mg/ml of freshly added proteinase and 0.5 mg/ml of freshly added RNase A), incubated at 65°C for 2 h and stored at −20°C, in the dark. Genomic DNA was isolated from each sample by phenol–chloroform extraction and alcohol precipitation and sonicated four times for 30 s each, at 30 s intervals, in the high mode at 4°C in a Bioruptor water bath sonicator (Diagenode), to obtain fragments of 500 to 1,000 bp in size. The sonicated DNA was denatured by incubation at 95°C for 5 min. We added monoclonal anti‐BrdU antibody (BD Biosciences #347580) at a final concentration of 3.6 μg/ml in 1× IP buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 150 mM NaCl, 0.5% Triton X‐100, and 7 mM NaOH). We used 30 μl or 50 μl of protein‐G‐coated magnetic beads (from Ademtech #4342 or Thermo Fisher Scientific #10004D, respectively) per sample to pull down the anti‐BrdU antibody. Beads and BrdU‐labeled nascent DNA were incubated for 2–3 h at 4°C, on a rotating wheel. The beads were then washed once with 1× IP buffer, twice with wash buffer (20 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 250 mM NaCl, 0.25% Triton X‐100) and then twice with 1× TE buffer pH 8.0. The DNA was eluted by incubating the beads at 37°C for 2 h in 250 μl 1× TE buffer pH 8.0, to which we added 1% SDS and 0.5 mg/ml proteinase K. DNA was purified by phenol–chloroform extraction and alcohol precipitation and resuspended in 50 μl TE.
For Repli‐seq analyses, immunoprecipitated NS from the four S‐phase fractions collected by flow cytometry or from an asynchronous cell population were amplified by whole genome amplification (GenomePlex Complete Whole Genome Amplification kit #WGA2; Sigma) according to the manufacturer's recommendations to obtain sufficient DNA amount. After amplification, libraries were constructed with the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB #E7645S) following the manufacturer's instructions with minor modifications. For the adaptor ligation, undiluted adaptor and no size selection were used. The library amplification was performed using NEBNext Multiplex Oligos for Illumina (NEB #E7710S) with nine different NEBNext index primer and the NEBNext Universal PCR Primer (NEB #E6861A) with three PCR cycles. Library purification was performed with the SPRI‐select Reagent kit (Beckman Coulter #B23317), and the final elution step was reduced to 33 μl of 0.1 × TE. The mean size of the library molecules determined on an Agilent Bio‐analyser High Sensitivity DNA chip (Agilent technologies, #5067–4626) was 330–350 bp. Sequencing was performed on a NextSeq 500 Illumina sequencer with a High Output 150 cycles flow cell (paired‐end reads of 75 bp) according to standard procedures. For each sample, 15–20 M of reads were generated. All Illumina sequencing runs were performed at the GENOM'IC facility of the Cochin Institute.
Repli‐Seq data processing
Paired‐end sequencing data were mapped on the galGal5 chicken genome using bowtie2 (Langmead & Salzberg, 2012). For each timing fraction, replication timing was computed using 50 kb sliding windows at 10 kb intervals, normalized by the global and local genomic coverage of the asynchronous cell population, to normalize for total and local coverage variations. Then, the centered and standardized timing profiles were smoothed using cubic splines (smooth.spline function of R). In order to provide a single RT profile combining all fractions, we used the method proposed by (Du et al, 2019), by computing the weighted average WA = (0.917*G1) + (0.750*S1) + (0.583*S2) + (0.417*S3) + (0.250*S4) + (0*G2). An increase in WA indicates an earlier timing.
Flow cytometry analysis
After BrdU incorporation, DT40 cells were washed twice with PBS, fixed in 75% ethanol, and stored at −20°C. On the day of sorting, fixed cells were resuspended at a final concentration of 2.5 × 106 cells/ml in 0.1% IGEPAL in PBS (Sigma, #CA‐630), 50 μg/ml propidium iodide and 0.5 mg/ml RNase A, and incubated for 30 min at room temperature. Cells were sorted with an INFLUX 500 cell sorter (Cytopenia, BD Biosciences). Four fractions of S‐phase cells (S1–S4), each containing 5 × 104 cells, were collected and further treated for locus‐specific RT analyses. Two fractions of S‐phase cells (S1–S2), each containing 105 cells, were collected and further treated for genome‐wide RT analyses.
MNAse digestions
We cross‐linked 30 × 106 exponentially growing cells by incubation for 5 min with 1% (v/v) freshly prepared formaldehyde (Thermo Fisher Scientific, #28908) at room temperature. Fixation was stopped by adding glycine to a final concentration of 0.125 M and incubating for 5 min at room temperature, with stirring. Cells were then washed three times in cold PBS, and their nuclei were extracted in 3 ml of lysis buffer (10 mM Tris–HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.2% Triton X‐100, 0.5 mM EGTA pH 8.0, 1 mM DTT, 1× protease inhibitor cocktail (Sigma, #P8340)) for 5 min at 4°C, centrifuged, and resuspended in digestion buffer (10 mM Tris–HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 1× protease inhibitor cocktail (Sigma, #P8340)). Micrococcal Nuclease (MNase; Thermo Fisher Scientific, #EN0181) digestions were performed for 15 min at 37°C, using either a final concentration of 10 U/ml for ChIp analyses or a series of four exponentially increasing concentrations of MNase (2.5, 10, 40 and 160 U/ml) for chromatin accessibility analyses. The reaction was stopped by adding 0.1 volumes of stop buffer (200 mM EDTA pH 8.0, 40 mM EGTA pH 8.0). For ChIp analyses, MNase digestion conditions were established so as to produce mostly mono‐ to hexa‐nucleosome fragments of 150–1,000 bp. Chromatin was then diluted by a factor of two in 2× complement buffer (40 mM Tris–HCl pH 8.0, 300 mM NaCl, 2% Triton X‐100) and sonicated 20 times for 30 s each, at 30 s intervals in the high mode at 4°C in a Bioruptor water bath sonicator (Diagenode). For chromatin accessibility analyses, the lowest MNAse concentration generated a mixture of oligo‐, di‐, and mono‐nucleosomes, whereas the highest concentration produced mostly mono‐nucleosomes (Appendix Fig S5A). After digestion, DNA molecules were extracted in phenol–chloroform and precipitated with ethanol and were then subjected to a size selection process with the SPRI‐select Reagent kit (Beckman Coulter #B23317) using a 0.5× ratio to remove DNA molecules of more than 1,000 bp in length (Appendix Fig S5B). The MNAse titration approach can be used to determine whether nucleosome release requires low or high levels of MNAse. Low levels of MNAse release large numbers of nucleosomes in accessible regions, leading to higher levels of DNA molecules. Nucleosomes embedded in less accessible regions are less likely to be released with such low levels of MNAse. Moreover, this approach provides additional information about the accessibility of loci. Indeed, highly inaccessible or condensed regions are not sensitive to higher concentrations of MNAse, whereas the chromatin of accessible regions is rapidly digested under the same conditions.
Chromatin immunoprecipitation
Immunoprecipitation was performed overnight at 4°C, in a final volume of 200 μl of 1× IP buffer (20 mM Tris–HCl pH 8.0, 2 mM EDTA pH 8.0, 150 mM NaCl, 1% Triton‐X100 and 0.1% SDS) on an amount of chromatin corresponding to 10 μg of DNA, with anti‐trimethylated K4H3 (AbCam, #ab8580), anti‐dimethylated K4H3 (Millipore, #07‐030), or anti‐acetylated K9K27H3 (Millipore, #06‐599) antibodies, according to the manufacturer's recommendations. Immunocomplexes were pulled down with 50 μl of protein‐G‐coated magnetic beads (Thermo Fisher Scientific, Dynabeads Protein G, #10004D) per sample. Beads and immunocomplexes were incubated for 2 h at 4°C, on a rotating wheel. The beads were then washed once with 1× IP buffer, twice with wash B buffer (20 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 250 mM NaCl, 0.25% Triton X‐100) and then twice with 1× TE buffer pH 8.0. For the anti‐trimethylated K4H3 antibody, the wash buffer contained 500 mM NaCl, and an additional washing step was performed with buffer C (10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 1% sodium deoxycholate, 1% NP40, 250 mM LiCl). The DNA was eluted by incubating the beads for 2 h at 37°C with 250 μl 1× TE buffer pH 8.0, to which we added 1% SDS and 0.5 mg/ml proteinase K. Cross‐linking was reversed by overnight incubation at 65°C, and samples were further treated with 10 μg of RNase A for 15 min at 37°C, and with 20 μg of proteinase K for 1 h at 56°C. DNA was purified by phenol–chloroform extraction, precipitated in alcohol, and resuspended in 100 μl TE.
RNA extraction and reverse transcription
Total RNA were extracted from 5 × 106 cells with the NucleoSpin RNA kit (Macherey Nagel, #740955). The integrity of the extracted RNA was assessed with an Agilent 2100 bioanalyzer (Agilent RNA 6000 Nano Kit, #5067‐1511), and 20 μg of total RNA was then treated with 4 units of DNAse I (NEB, #M0303S) for 1 h at 37°C. The enzyme was inactivated by adding 5 mM EDTA and incubating the reaction mixture for 10 min at 75°C. The RNA was then purified by phenol–chloroform extraction and ethanol precipitation. Reverse transcription reactions (RT‐QPCR+) were then performed with 5 μg of RNA and random hexamers (NEB, S1330S), using the Superscript III Reverse Transcriptase (Thermo Fisher Scientific, #18080093) according to the manufacturer's instructions. Negative controls (RT‐QPCR−) were performed with the same procedure, but without the addition of reverse transcriptase. The comparison of RT‐QPCR+ and RT‐QPCR− samples was used to validate DNAseI treatment and the complete digestion of the genomic DNA in the RNA samples.
Real‐time PCR quantification of DNA
The Roche Light Cycler 2.0 detection system and the Absolute QPCR SYBR Green Capillary Mix (Thermo Fisher Scientific, #AB1285B) were used for the real‐time PCR quantification of BrdU‐labeled nascent strands (NS) or genomic DNA extracted from 4‐hydroxytamoxifen‐treated clonal cell lines. For all reactions, real‐time PCR was performed under the following cycling conditions: initial denaturation at 95°C for 15 min, followed by 50 cycles of 95°C for 15 s, 60°C for 30 s, 72°C for 20 s, and fluorescence measurement. Following PCR, a thermal melting profile was used for amplicon identification.
The Roche Light Cycler 480 detection system and the Light Cycler 480 SYBR Green I Master mix (Roche Applied Science, # 04707516001) were used for the real‐time PCR quantification of ChIp DNA, MNAse‐digested DNA, and cDNA. For all reactions, real‐time PCR was performed under the following cycling conditions: initial denaturation at 95°C for 5 min, followed by 50 cycles of 95°C for 10 s, 61°C for 20 s, 72°C for 20 s and fluorescence measurement. Following PCR, a thermal melting profile was used for amplicon identification.
For both quantification methods, standard curves were generated from four‐fold dilutions of the corresponding genomic DNA. Serial dilutions of plasmids containing the loci of interest were used as standards for cDNA quantification. Each reaction was performed at least in duplicate. The second‐derivative maximum method was used to quantify sequences, as described in the Light Cycler Software.
For BrdU‐labeled NS quantification, we used primer pairs binding to mitochondrial DNA to normalize enrichment in each S‐phase fraction (Mitochondrial DNA), an early‐replicated region (Early timing control) for the control of cell sorting, a genomic position located 5 kb downstream from insertion site 2 (Both), amplifying both alleles, for the control of accurate qPCR measurements and the studied regions in the transgenes. Specific primer pairs amplifying in the transgene (With) or the site of insertion in the wt allele (Without) were used (Appendix Table S4). In the case of clones with one or two autonomous replicons associated with the GFP reporter construct inserted on the other chromosome, we used a primer pair amplifying the GFP reporter construct to quantify the wt chromosome (Without on GFP reporter at site 2, Appendix Table S4) and a primer pair amplifying insertion site 2 to quantify the modified shifting chromosome (With at insertion site 2, Appendix Table S4).
Based on both the 1‐h BrdU pulse labeling used and the mean rate of fork progression, estimated at 1.5 kb/min in DT40 cells (Maya‐Mendoza et al, 2007), the resolution of our RT experiments cannot exceed a 90 kb window. Thus, for the analysis of RT within our 30 kb genomic region targeted with one, two or three constructs, we systematically used the With and Without primer pairs in central position 2. For the quantitative analyses, we analyzed independent clones. In Fig 1, N = 10 for (i) from Hassan‐Zadeh et al, 2012 and Valton et al, 2014, N = 8 for (ii) from Hassan‐Zadeh et al, 2012; Valton et al, 2014 and, N = 7 for (iii) from Hassan‐Zadeh et al, 2012 and this article Fig EV1A, and N = 8 for (iv) from Hassan‐Zadeh et al, 2012 and this article, Fig EV1B. In Fig 3, N = 8 for (i) the same clones as (iv) in Fig 3, N = 5 for (ii) from Fig EV3A and B, N = 3 for (iii) from this article Fig EV2D and E. For statistical analysis, the outliers values detected with the R outliers package were excluded from the analysis (clone 4 in Fig EV1A).
For ChIp DNA and MNAse‐digested DNA quantifications, we used primer pairs amplifying the wt allele at insertion sites 1, 2, and 3 (Wt allele insertion site 1, 2 or 3), the condensed region of the endogenous β‐globin locus, as a control for the heterochromatin state (cond 1 and 2), the BU1A promoter (BU1A pro), or the MED14 promoter (MED14 pro), as a control for the active chromatin state, at a genomic position located 5 kb downstream from insertion site 1 (1 + 5 kb), amplifying both alleles and the inserted transgenes, at the positions studied (Figs 6 and EV4, and Appendix Table S4).
For the quantification of MNAse‐digested DNA, data are presented as total chromatin input relative to condensed genomic regions (cond1 and cond2). For each clonal cell line, the mean values obtained for the two condensed genomic regions for each digestion were arbitrarily set to 1 and used to normalize independent clones for each transgenic line separately and between all transgenic lines.
For ChIp DNA quantifications, data are presented as Ip/Input signal relative to the signal for the BU1A promoter (BU1A pro) used for normalization between transgenic lines.
For cDNA quantification, we used primer pairs amplifying the second exon of the BU1A gene as a control for transcribed genes, the junction of exons 3–4 of the MED14 gene and the inserted transgenes, at the studied positions (Fig EV4 and Appendix Table S4).
Hi‐C methods
Hi‐C was performed as described previously (Belaghzal et al, 2017) with some minor modifications. Briefly, 10 million formaldehyde‐cross‐linked cells (1% formaldehyde for the wild types and 2% formaldehyde for the clones) were lysed and digested overnight at 37°C with 400 units of DpnII (NEB). Next, the DNA ends were marked with biotin‐14‐dATP (Thermo Fisher Scientific #19524016) for 4 h at 23°C and ligated for 4 h at 16°C. To remove proteins and reverse cross‐link, DNA was treated with proteinase K overnight at 65°C. DNA was then purified using phenol:chloroform and biotin was removed from unligated ends. Ligation products were fragmented by sonication to an average size of 200 bp and size selected with Ampure XP beads (Beckman Coulter, # A63880) to fragments ranging from 100 to 350 bp. End repair was performed on the DNA fragments to obtain blunt ends and biotinylated ligation products were purified using streptavidin beads (Thermo Fisher Scientific #65001). dA‐tailing was then performed, and the TrueSeq DNA LT kits Set A (Illumina, #15041757) was used for adapter ligation and PCR amplification. PCR primers were removed using Ampure XP beads. The libraries were sequenced using 50 bp paired‐end reads on Illumina HiSeq4000.
Hi‐C data processing
Hi‐C libraries were processed using the distiller pipeline (https://github.com/mirnylab/distiller-nf). Briefly, reads were mapped to the chicken reference assembly galGal5 using bwa mem to map fastq pairs in a single‐side fashion (−SP). The specific sequences of each inserted construct (allele 1 + 3 and allele 2) were pooled together and placed in the original galGal5 fasta file as two additional chromosomes to be able to map and balance them in the following procedure. Alignments were parsed, and pairs were classified using the pairtools package (https://github.com/mirnylab/pairtools) to generate pairs files. Uniquely mapped pairs were kept and duplicated pairs arising from PCR were removed. Pairs with high mapping quality scores on both sides (MAPQ > 30) were kept and aggregated into contact matrices in the cooler format using the cooler package (Abdennur & Mirny, 2020). Data were binned and stored into multiresolution cooler files (1, 2, 5, 10, 25, 50, 100, 250, 500 kb, 1 Mb). All contact matrices were normalized using the iterative correction normalization (Imakaev et al, 2012) after ignoring the first two diagonals to avoid short‐range ligation artifacts at a given resolution. Low‐coverage bins were excluded using the MADmax (maximum allowed median absolute deviation) filter on genomic coverage, described in (Schwarzer et al, 2017), using the default parameters. WT replicate 1 has 82,394,530 mapped reads with 37% cis interactions, WT replicate 2 has 139,022,250 mapped reads with 29% cis interactions, clone 4 has 541,023,662 mapped reads with 79.3% cis interactions, clone 5 has 542,918,613 mapped reads with 78.5% cis interactions (Appendix Table S5).
Additional filtering of Hi‐C interactions
For all of the quantification analyses, it is important to ignore the following regions: (i) large translocation on the p‐arm of chr1 chr1:9,000,000–9,500,000 (Appendix Fig S9A and B), (ii) smaller translocation on the p‐arm of chr1 chr1:32,270,000–32,770,000 (Appendix Fig S9A and B), (iii) beta‐A‐globin promoter region that is included in the allele 1 + 3 and allele 2 inserts chr1:194,569,000–194,572,000 (Appendix Fig S9C) beta‐actin promoter region that is included in the allele 1 + 3 insert chr14:4,194,000–4,197,000. Regions (iii) and (iv) were removed because we cannot properly assign the interactions that involved βA‐globin or β‐actin promoters to inserts or the endogenous loci.
A/B compartments
A and B compartments were assigned using an eigenvector decomposition procedure (Imakaev et al, 2012) implemented in the cooltools package (https://github.com/mirnylab/cooltools, https://doi.org/10.5281/zenodo.3553140). Eigenvector decomposition was performed on observed‐over‐expected cis contact matrices binned at 250 kb on each library and at 100 kb on the pooled libraries for a better resolution of compartments. The first eigenvectors (EV1) positively correlated with gene density were used to assign A and B compartment status to each bin. EV1 calculated at 100 kb was used for all quantitative analyses that involved compartment status, if not stated otherwise.
Hi‐C Allele‐specific analysis
In order to quantify preferential interactions of allele 1 + 3 and allele 2 with A and B compartments, we performed the following:
(i) We corrected for distance decay as the alleles were mapped as independent chromosomes (see above) but the insertion site is located on chr1. Thus, we divided the frequency of interactions of alleles 1 + 3/2 with chromosome 1 by the expected value for each bin based on genomic distance from the insertion site. (ii) We calculated the average interaction of allele 1 + 3/2 with compartment A and B on chr1 p and q arms, where compartment status A was assigned if eigenvector 1 value > 0 and compartment status B if eigenvector 1 value < 0. (iii) To test the significance of observed interaction preference, we randomly shuffled the A and B compartment status 1,000 times and calculated the difference between average interactions of a given allele with compartment A and compartment B “difference allele 1 + 3/2 A − B” for every iteration. We then calculated the P‐value for the observed value of “difference allele 1 + 3/2 A − B”.
In order to test if allele 1 + 3 and allele 2 demonstrate any preferential interactions with other chromosomes, i.e., trans‐interactions, we performed the following:
(i) Due to the sparsity and lack of distance decay for trans‐data, we averaged interactions on the scale of individual chromosomes and normalized them by corresponding chromosome lengths; we refer to this measure as density of interactions. (ii) We compared the density of interactions between allele 1 + 3 and allele 2 with chromosomes larger than 5,000,000 bp (Fig 3E, and Appendix Figs S4C and F, and S9D).
Author contributions
CB, BD, and M‐NP planned and supervised the project. FP performed bioinformatic analysis of the Repli‐seq data, A‐LV, SVV, and JD conceived the Hi‐C experiments, A‐LV conducted the Hi‐C experiments. A‐LV and SVV performed bioinformatic analysis of the Hi‐C data. For all other experiments, CB and M‐NP designed the methodology, CB, SC, AC, ML, CG conducted the experiments, CB, BD, and M‐NP analyzed and validate the data, CB provided visualization of the data. CB and M‐NP wrote the manuscript with input from FP, A‐LV and SVV.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors thank the members of the laboratory of M‐N.P for useful insights and discussions. We thank G. Felsenfeld and R. Veitia for critical reading of the manuscript. We thank J‐F Bercher for his help in defining the most accurate quantitative method for measuring RT shifts. We thank Griselda Wentzinger and Magali Fradet for performing cell sorting at ImagoSeine Institut Jacques Monod platform. Sequencing of replication timing data was performed by the GENOM'IC core facility at Institut Cochin‐Paris. We thank Ankita Nand for doing the initial mapping of the Hi‐C data. This work was supported by grants from the Association pour la Recherche sur le Cancer (Equipe Labellisée) and the Agence Nationale pour la Recherche (ANR‐15‐CE12‐0004‐01), and from the National Human Genome Research Institute (HG003143 to JD). M‐N.P and B.D are supported by Inserm. JD is an investigator of the Howard Hughes Medical Institute.
The EMBO Journal (2020) 39: e99520
Data availability
The datasets produced in this study have been deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE153566. Code for the Hi‐C allele‐specific analysis is available on github: https://github.com/dekkerlab/allelic-rt-supplement.git.
References
- Abdennur N, Mirny LA (2020) Cooler: scalable storage for Hi‐C data and other genomically labeled arrays. Bioinformatics 36: 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H, Lodygin D, Buerstedde JM (2001) Mutant loxP vectors for selectable marker recycle and conditional knock‐outs. BMC Biotechnol 1: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaghzal H, Dekker J, Gibcus JH (2017) Hi‐C 2.0: an optimized Hi‐C procedure for high‐resolution genome‐wide mapping of chromosome conformation. Methods 123: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin J‐M, Lemaitre J‐M (2012) Unraveling cell type‐specific and reprogrammable human replication origin signatures associated with G‐quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844 [DOI] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J‐P, Tanay A et al (2017) Multiscale 3D genome rewiring during mouse neural development. Cell 171: 557–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustel J, Kirstein N, Izard F, Grimaud C, Prorok P, Cayrou C, Schotta G, Abdelsamie AF, Déjardin J, Méchali M et al (2017) Histone H4K20 tri‐methylation at late‐firing origins ensures timely heterochromatin replication. EMBO J 36: 2726–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SM, Kleckner N (1999) Collisions between yeast chromosomal loci in vivo are governed by three layers of organization. Genes Dev 13: 1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M (2012) New insights into replication origin characteristics in metazoans. Cell Cycle 11: 658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau J‐C, van Helden J, Méchali M (2015) The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res 25: 1873–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy J‐P, Foti R, Tili F, Santarella‐Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SBC (2012) Mouse Rif1 is a key regulator of the replication‐timing programme in mammalian cells. EMBO J 31: 3678–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas ME, Diffley JFX (2012) Replication timing: the early bird catches the worm. Curr Biol 22: R81–R82 [DOI] [PubMed] [Google Scholar]
- Du Q, Bert SA, Armstrong NJ, Caldon CE, Song JZ, Nair SS, Gould CM, Luu P‐L, Peters T, Khoury A et al (2019) Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat Commun 10: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duriez B, Chilaka S, Bercher J‐F, Hercul E, Prioleau M‐N (2019) Replication dynamics of individual loci in single living cells reveal changes in the degree of replication stochasticity through S phase. Nucleic Acids Res 47: 5155–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Lengronne A, Shi D, Forey R, Skrzypczak M, Ginalski K, Yan C, Wang X, Cao Q, Pasero P et al (2017) Dbf4 recruitment by forkhead transcription factors defines an upstream rate‐limiting step in determining origin firing timing. Genes Dev 31: 2405–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti R, Gnan S, Cornacchia D, Dileep V, Bulut‐Karslioglu A, Diehl S, Buness A, Klein FA, Huber W, Johnstone E et al (2016) Nuclear architecture organized by Rif1 underpins the replication‐timing program. Mol Cell 61: 260–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan‐Zadeh V, Chilaka S, Cadoret J‐C, Ma MK‐W, Boggetto N, West AG, Prioleau M‐N (2012) USF binding sequences from the HS4 insulator element impose early replication timing on a vertebrate replicator. PLoS Biol 10: e1001277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt ER, Cozzarelli NR (1995) Comparison of recombination in vitro and in E. coli cells: measure of the effective concentration of DNA in vivo . Cell 81: 331–340 [DOI] [PubMed] [Google Scholar]
- Hiraga S‐I, Alvino GM, Chang F, Lian H‐Y, Sridhar A, Kubota T, Brewer BJ, Weinreich M, Raghuraman MK, Donaldson AD (2014) Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7‐mediated phosphorylation of the MCM complex. Genes Dev 28: 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA (2012) Iterative correction of Hi‐C data reveals hallmarks of chromosome organization. Nat Methods 9: 999–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodkowska K, Pancaldi V, Almeida R, Rigau M, Graña‐Castro O, Fernández‐Justel JM, Rodríguez‐Acebes S, Rubio‐Camarillo M, Pau ECS, Pisano D et al (2019) Three‐dimensional connectivity and chromatin environment mediate the activation efficiency of mammalian DNA replication origins. bioRxiv 10.1101/644971 [PREPRINT] [DOI] [Google Scholar]
- Knott SRV, Peace JM, Ostrow AZ, Gan Y, Rex AE, Viggiani CJ, Tavaré S, Aparicio OM (2012) Forkhead transcription factors establish origin timing and long‐range clustering in S. cerevisiae . Cell 148: 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachagès A‐M, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M (2011) Cell‐type‐specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470: 120–123 [DOI] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J et al (2012) Extensive promoter‐centered chromatin interactions provide a topological basis for transcription regulation. Cell 148: 84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO et al (2009) Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Recillas‐Targa F, Prioleau MN, Felsenfeld G (2001) Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J 20: 2224–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massip F, Laurent M, Brossas C, Fernández‐Justel JM, Gómez M, Prioleau M‐N, Duret L, Picard F (2019) Evolution of replication origins in vertebrate genomes: rapid turnover despite selective constraints. Nucleic Acids Res 47: 5114–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya‐Mendoza A, Petermann E, Gillespie DAF, Caldecott KW, Jackson DA (2007) Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 26: 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Bell M, Mir M, Kao JA, Darzacq X, Botchan MR, Berger JM (2019) A new class of disordered elements controls DNA replication through initiator self‐assembly. Elife 8: e48562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret J‐C, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau M‐N (2014) The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10: e1004282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prioleau MN, Nony P, Simpson M, Felsenfeld G (1999) An insulator element and condensed chromatin region separate the chicken beta‐globin locus from an independently regulated erythroid‐specific folate receptor gene. EMBO J 18: 4035–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prioleau M‐N, MacAlpine DM (2016) DNA replication origins‐where do we begin? Genes Dev 30: 1683–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM (2010) Evolutionarily conserved replication timing profiles predict long‐range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe‐Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L et al (2017) Two independent modes of chromatin organization revealed by cohesin removal. Nature 551: 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera‐Mulia JC, Trevilla‐Garcia C et al (2019) Identifying cis Elements for spatiotemporal control of mammalian DNA replication. Cell 176: 816–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therizols P, Illingworth RS, Courilleau C, Boyle S, Wood AJ, Bickmore WA (2014) Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 346: 1238–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton A‐L, Hassan‐Zadeh V, Lema I, Boggetto N, Alberti P, Saintomé C, Riou J‐F, Prioleau M‐N (2014) G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J 33: 732–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AG, Huang S, Gaszner M, Litt MD, Felsenfeld G (2004) Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol Cell 16: 453–463 [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H (2012) Rif1 regulates the replication timing domains on the human genome. EMBO J 31: 3667–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The datasets produced in this study have been deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE153566. Code for the Hi‐C allele‐specific analysis is available on github: https://github.com/dekkerlab/allelic-rt-supplement.git.