

SUMMARY
Mycobacterium tuberculosis (MTB) displays the remarkable ability to transition in and out of dormancy, a hallmark of the pathogen’s capacity to evade the immune system and exploit susceptible individuals. Uncovering the gene regulatory programs that underlie the phenotypic shifts in MTB during disease latency and reactivation has posed a challenge. We develop an experimental system to precisely control dissolved oxygen levels in MTB cultures in order to capture the transcriptional events that unfold as MTB transitions into and out of hypoxia-induced dormancy. Using a comprehensive genome-wide transcription factor binding map and insights from network topology analysis, we identify regulatory circuits that deterministically drive sequential transitions across six transcriptionally and functionally distinct states encompassing more than three-fifths of the MTB genome. The architecture of the genetic programs explains the transcriptional dynamics underlying synchronous entry of cells into a dormant state that is primed to infect the host upon encountering favorable conditions.
In Brief
Mycobacterium tuberculosis (MTB) persists within the host by counteracting disparate stressors including hypoxia. Peterson et al. report a transcriptional program that coordinates sequential state transitions to drive MTB in and out of hypoxia-induced dormancy. Among varied properties, this program encodes advanced preparedness to infect the host in favorable conditions.
Graphical Abstract
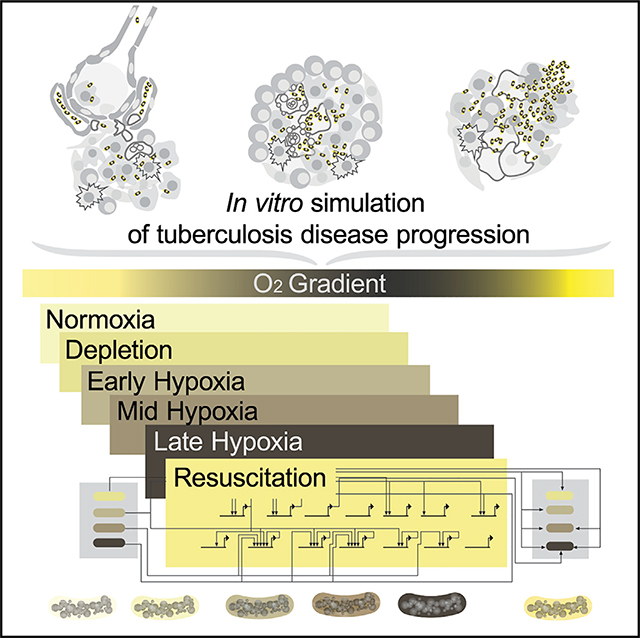
INTRODUCTION
Mycobacterium tuberculosis (MTB) kills more people than any other infectious agent, causing ~10 million new cases of active tuberculosis (TB) disease and 1.7 million deaths each year (Murray et al., 2014). TB remains a major human public health burden, in large part because of the sizable reservoir of latently infected individuals, who may relapse into active disease decades after acquiring the infection. MTB can persist in a stable, non-replicative (often termed dormant) state within the host for months or years without symptoms and then revive to initiate the production of lesions and active TB disease. Moreover, dormant cells may be responsible for the slow treatment response of patients with active TB. Elucidation of the factors that affect treatment outcome, latency, and activation requires a better characterization of functional states adopted by the pathogen during progression of the disease, as well as a mechanistic understanding of the genetic programs that orchestrate transitions between these states.
Hypoxia, an environmental stress encountered by MTB within granulomas (Tsai et al., 2006), is sufficient to shift the pathogen into a defined non-growing survival form, which can be reversed upon aeration of the culture (Chao and Rubin, 2010). Therefore, hypoxia has been leveraged as an in vitro approximation to study MTB dormancy and the underlying genetic programs. However, previous transcriptional analyses under in vitro hypoxic conditions (via the Wayne model, in which MTB cultures are sealed and gradually depleted of oxygen (O2) [Wayne and Sohaskey, 2001; Wayne and Hayes, 1996], or the defined hypoxia model, in which nitrogen (N2) gas is flowed into the headspace to rapidly deplete O2 [Kempner, 1939; Yuan et al., 1998]) were limited to either static snapshots or low-resolution time course studies (Rustad et al., 2008; Muttucumaru et al., 2004; Sherman et al., 2001). Moreover, deletion of previously identified transcriptional regulators thought crucial to hypoxia-induced dormancy conferred only mild growth defects under hypoxic conditions (Rustad et al., 2008, 2009; Boon and Dick, 2002), suggesting a genetic circuit architecture that has evolved to withstand environmental and genetic perturbations. Here, we developed an experimental platform to characterize MTB’s response to changing O2 levels in considerably more depth. We reveal detailed transcriptional dynamics and coordinated regulatory circuits that enable the pathogen’s transition into and out of hypoxia-induced dormancy.
RESULTS
To discern the genetic programs underlying hypoxia-induced dormancy in MTB, we needed to obtain accurate dynamic measurements of genome-wide expression over an O2 gradient. Previous experimental models to study O2 tension and growth arrest in MTB were not suitable for the accuracy and resolution of measurements needed. In particular, the Wayne model has difficulties with reproducibility, and the defined hypoxia model depletes O2 very quickly, thereby hindering high-resolution sampling during critical transition periods. Moreover, neither model has been performed with real-time monitoring of O2 levels to accurately relate the transcriptional state of MTB with a precise O2 measurement. Therefore, we designed a programmable multiplexed reactor system, the controlled O2 model, to precisely manipulate and monitor O2 levels within the growth medium, even during sampling (Figure 1A). The precise control engineered into the system enabled high-resolution sampling across a time course and an O2 gradient, with minimal disturbance to the bacteria and high reproducibility across culture replicates and experiments. Briefly, air and N2 gas lines were connected to separate mass flow controllers, which allowed programmable gradients of gas mixtures to be streamed into the headspace of spinner flasks containing MTB in media. Moreover, we used O2 sensor spots and fiber-optic technology to non-invasively measure the dissolved O2 (DO) content of the cultures. Both the mass flow controllers and O2 sensor spots were configured for remote management, advantageous for a biosafety level 3pathogen. With the controlled O2 model, we performed a time course experiment, which involved a steady depletion of DO over 2 days from full aeration (~80% DO) to hypoxia (0% DO). This steady depletion was achieved by programming the mass flow controllers to produce the desired mixture of air and N2. The cultures were maintained in hypoxia for 2 days by streaming only N2, then reaerated over 1 day by a programmed increase in air flow (Figure 1B). Over the time course, we harvested samples in triplicate, one each from three independent reactors, via sampling ports that prevented aeration of the culture. We sampled at high frequency during the period when cultures transitioned from 10% to 0% DO, as well as from 0% to 10% DO and at lower but regular frequency across the remaining 120 h experiment. An aliquot of the sample was used to measure growth by optical density (Figure 1C), and the remaining was flash frozen in liquid N2 and later processed for gene expression profiling by RNA sequencing.
Figure 1. Schematic, Dissolved Oxygen, and Growth Profiles of the Controlled O2 Model Reactor System.

(A) Schematic of controlled O2 model reactor system. Four reactors were multiplexed and individually monitored for DO levels to obtain biological replicates.
(B) DO levels across the 120 h time course.
(B and C) Points are the average of three biological replicates and error bars represent standard deviation; the yellow shading indicates the periods of controlled O2 depletion and reaeration, whereas the gray background indicates a sustained 2 day immersion in hypoxia.
(C) Growth of H37Rv in fully aerated cultures (triangles, dashed line) or cultures from the controlled O2 model reactor system (circles, solid line). Growth was monitored daily (aerated cultures) or at each sampling time points (controlled O2 model cultures) by optical density at 650 nm.
Over the course of the experiment, nearly 64% of all genes in the MTB genome were significantly differentially expressed with respect to time 0 (2,582 genes with adjusted p < 0.05 and estimated absolute log2 fold change > 1). The number of differentially expressed genes (DEGs) from our controlled O2 system is an order of magnitude greater than the number from earlier microarray studies using the Wayne (Muttucumaru et al., 2004) (299 genes) or defined hypoxia (Rustad et al., 2008) (274 genes) model. Nevertheless, there was significant overlap across gene sets among the three models of hypoxia-induced dormancy (Table S1A). Interestingly, the controlled O2 model significantly recapitulated differential expression observed from intracellular MTB (Peterson et al., 2019) (enrichment test p = 1.74e-30), whereas the other hypoxia models did not or had a low recall of the DEGs (Table S1B). These findings highlight the capability of our model to capture MTB’s transcriptional programs during hypoxic conditions that are relevant to MTB within host cells.
To further characterize the MTB transcriptional states over the time course and O2 gradient, we applied hierarchical (Suzuki and Shimodaira, 2006), k-means (Macqueen, 1967), and ensemble clustering (Sloutsky et al., 2013; Ronan et al., 2016) techniques that allowed us to define six groups of tightly clustered samples (Figure S1). Clustering was performed and evaluated (quantroStat p = 1e-4) (Hicks and Irizarry, 2015) using all 78 replicates, but for simplification, time point median values are shown in a two-dimensional t-distributed stochastic neighbor embedding (tSNE) plot (Figure 2A). Each cluster represents a distinct transcriptional state and was associated with sets of non-overlapping DEGs: normoxia (81 genes), depletion (446 genes), early hypoxia (328 genes), mid hypoxia (320 genes), late hypoxia (978 genes), and resuscitation (429 genes) (Data S1). Each DEG was assigned to the state in which it had the highest mean expression (see Table S2 for assessment of state gene assignment by mean squared residue, Pearson correlation, and Boruta recall). The average expression profiles for the gene sets reveal that the states transition from one to another and that transitions are O2 and time dependent (Figure 2B). As such, the six states were also defined with O2 and time intervals, with the exception of 46–49 h, when there was oscillation between early hypoxia and late hypoxia states as the culture went below ~3% DO (Figure 2). Although this intriguing “flicker” behavior could be experimental noise, these anomalous time points (measured roughly 1 h apart) clearly cluster with late hypoxia (Figure S1). Such oscillatory expression could be generated by inherent properties of the network structure, which we describe later.
Figure 2. The ControlledO2 Model Captures Distinct Cell States over Time Course and O2 Gradient.

(A) t-SNE visualization of all samples (time point median values) across the time course and hypoxia gradient.
(B) Average expression profiles for state-specific gene sets across the time course and hypoxia gradient. The yellow shading indicate the periods of controlled O2 depletion and reaeration, whereas the gray background indicates a sustained 2 day immersion in hypoxia. General theme of significant functional term clusters defined by DAVID (Huang et al., 2009) in each state is indicated. Asterisk indicates the most enriched term at the individual term level in the resuscitation state.
Genes associated with the depletion state (DO between 43% and 4%) were enriched for growth-related functions, including amino acid metabolism, oxidative phosphorylation, and translation (Figure 2B). In early hypoxia, ATP synthase and genes involved in electron transport chain and lipid metabolism were highly enriched and expressed, even more so than in normoxia. Furthermore, these metabolic genes were then significantly downregulated during late hypoxia. This result indicates that early hypoxia is a metabolically active state that may exist for MTB to prepare itself for an upcoming metabolically quiescent state (i.e., late hypoxia). Mid hypoxia genes were enriched in stress-response genes, indicating that the bacteria are sensing and adapting to the anaerobic environment. In late hypoxia, genes essential for MTB to infiltrate host cells were induced. Furthermore, genes for 32 proteins that belong to the prolineglutamic acid (PE) and proline-proline-glutamic acid (PPE) family, whose functions remain largely unknown (Bottai and Brosch, 2009), were upregulated. These PPE family proteins have been proposed to modulate the host’s immune response (Tiwari et al., 2012) and generate antigenic variation (Cole et al., 1998) and were shown to be secreted by MTB’s ESX-5 export system (Abdallah et al., 2008). Interestingly, genes encoding the components of ESX-5 export system were rapidly activated as soon as MTB shifted from late hypoxia into resuscitation, minutes after air was introduced back into the culture. It is possible that late hypoxia not only engenders quiescence in MTB but also sequesters a collection of PPE proteins in anticipation of resuscitation and ESX system production. In the resuscitation state, proteases, transposases, and insertion sequences were also enriched among activated genes. These functional groups suggest that MTB may strategically avert immune recognition through antigenic heterogeneity (via ESX secretion of PPE proteins) and simultaneously reorganize its genome (via transposases and insertion sequences) to increase its chances for survival and transmission to a new host upon resuscitation.
Using the gene expression data, we also analyzed changes in MTB metabolic pathways, as reconstructed by Kavvas et al. (2018), along the hypoxia time series. The expression of many metabolic pathways reiterated the state transitions described above (Figure S2). MTB adaptation to hypoxia involves rewiring of several metabolic pathways, indicating an evolutionarily learned and coordinated response to stresses that typically co-exist within the host environment, despite the singular in vitro perturbation.
The six-state model across the time course and O2 gradient revealed distinct patterns of expression suggestive of intriguing and coordinated regulatory programs. Several methods are available for reconstructing gene regulatory networks (GRNs) along time series expression data (Luscombe et al., 2004; Bromberg et al., 2008; Baugh et al., 2005). We selected Dynamic Regulatory Events Miner (DREM) 2.0 (Schulz et al., 2012), which has been successfully applied to various systems (e.g., fly [Roy et al., 2010], yeast [Ernst et al., 2007], E. coli [Ernst et al., 2008]) and is ideal for identifying dynamic transcriptional events over time and perturbations. DREM integrates time series and snapshots of the GRN of interest using an input-output hidden Markov model (Ernst et al., 2007). In so doing, DREM learns a dynamic GRN by identifying bifurcation points: places in the time series where a group of co-expressed genes begins to diverge. These bifurcation points are annotated with the proposed transcription factors (TFs) controlling the split, leading to a combined dynamic model. Using the hypoxia time course expression dataset and a TF-target gene network derived from the chromatin immunoprecipitation sequencing (ChIP-seq) assessment of 154 TFs overexpressed in MTB (Minch et al., 2015), DREM identified bifurcation points that coincide with transitions between the six states (Figure 3A; additional material to recreate the DREM output is provided in Data S2). The bifurcation points defined by DREM reinforce the importance of transcriptional regulation in the progression between states. DREM identified TFs that are known to mediate MTB’s response to hypoxia (e.g., DosR, Rv0081, Rv0324) (Galagan et al., 2013), along with additional TFs with potential roles in hypoxia. In particular, Rv1353c stood out for being the only TF linked to the time points that precede and mark the end of late hypoxia. In addition, DREM suggests that CsoR (Rv0967), a TF that controls MTB’s response to copper stress (Marcus et al., 2016), may also have an unappreciated role in hypoxia. Interestingly, DREM associated CsoR with the bifurcation points preceding early hypoxia and resuscitation. In the latter point, csoR transcriptional level transitioned from its lowest state value to its highest value. This suggests a potential bifunctional activity of CsoR in controlling MTB’s transcriptional response both in and out of hypoxia. In fact, 87% of DEGs (223 genes) from a csoR-knockout mutant (Marcus et al., 2016) were differentially expressed at some point during the time course and O2 gradient (p = 1.7e-16). Specifically, this set of DEGs was enriched with members of early hypoxia, mid hypoxia, and resuscitation, supporting the CsoR bifurcation points predicted by DREM. We propose further investigation to evaluate the consequences of perturbing Rv1353c and CsoR activity during hypoxia adaptation.
Figure 3. Transcriptional Circuits Controlling the Six Transcriptional States Adopted by MTB during Entry and Exit from Hypoxia.

(A) DREM output. Transcription factors (TFs) associated with selected branching points (white nodes) are shown. Each line represents a set of co-expressed genes with similar expression behavior until a branching point. Time points and TFs are colored on the basis of transcriptional states membership (TFs in black were not differentially expressed). See also Data S2.
(B) Heatmap with transcriptional profiles of 147 differentially expressed MTB TFs; number in parentheses indicates the number of TFs associated with each state.
(C) TF-TF network of differentially expressed TFs in the controlled O2 model. Only TFs with one or more differentially expressed targets were included in the diagram. For TFs in late hypoxia, outgoing edges to early and mid hypoxia (right gray box) are shown only for TFs with two or more TF targets in those states. The diagram was generated with Biotapestry (Paquette et al., 2016).
Although DREM implicated key TFs in state transitions with > 0% DO, its inability to associate more TFs could be attributed to the absence of bifurcations in co-expressed gene sets (e.g., purple path in Figure 3A) and the normoxic conditions used to map protein-DNA interactions. The set of TFs identified by DREM included only 16% of the 147 putative TFs differentially expressed across the experiment and assigned to the six-state model (Figure 3B). In fact, late hypoxia contains 32% of all differentially expressed TFs (47 TFs). The large number of differentially expressed TFs suggested that complex and combinatorial circuitry patterns could be involved in MTB’s adaptation to hypoxic conditions. Figure 3C shows the dense TF-TF connectivity within and between transcriptional states, according to available protein-DNA binding data (Minch et al., 2015). The key regulatory proteins of the TF-TF network were identified on the basis of betweenness centrality, which characterizes the connectivity of interacting nodes in the network. The top high-degree nodes were Rv0081, Rv3597c (Lsr2), Rv1990c, Rv2034, and Rv0023. As high-degree nodes, drugs targeting one or more of these regulatory hubs may have a major impact on MTB survival. In support of this, Bartek et al. (2014) showed that deletion of lsr2 significantly compromised adaptation of MTB to hypoxic conditions. Notably, Lsr2 had the second and third largest outdegree (number of TF targets) and indegree (number of transcriptional regulators), respectively. Lsr2 directly controls TFs from all other states (besides normoxia) and is itself regulated by mid hypoxia TFs and late hypoxia TFs. The critical role of Lsr2 in the coordination between hypoxia-related states offers an explanation for the known importance of Lsr2 in hypoxic conditions.
The high connectivity of the TF-TF network revealed regulatory hubs that activate one state while repressing another. Interestingly, DREM also identified bifurcation points in DO > 0% with downregulated late hypoxia TFs (Rv0324 and Rv1049), indicating a concurrent repression of late hypoxia regulators and activation of earlier states. Motivated by these findings, we evaluated the enrichment of regulons (on the basis of protein-DNA interaction data) of all differentially expressed TFs with members of each transcriptional state. Many TF regulons (n = 21) were significantly enriched with genes assigned to the same transcriptional state as the regulating TF was assigned to (permutation test p < 1e-3), while even more TF regulons (n = 49, permutation test p < 1e-3) were significantly enriched with members of other transcriptional states. In other words, TFs seem to activate members of their own state while repressing members of another state (Figure 3C). Such behavior by TFs, described as mutual inhibition (Gardner et al., 2000; Glass and Kauffman, 1973; Huang et al., 2007), proposes a mechanism for the coordination and cooperation between transcriptional states to achieve the proper timing and gene expression levels to successfully adapt to changes in O2.
Interactions among TFs can also form specific network motifs that perform defined dynamical functions in response to changing environmental conditions (Wu et al., 2011; Alon, 2007; Shiraishi et al., 2010). Network motifs, such as feedforward loops (FFLs), single-input modules, or bistable toggle switches are recurring gene patterns found within GRNs. To unbiasedly search for network motifs that may be involved in hypoxia adaptation, we analyzed the experimentally determined MTB TF-target gene interactions from ChIP-seq (Minch et al., 2015) using FANMOD (Wernicke and Rasche, 2006) in the MotifNet Webserver (Smoly et al., 2017). We found that the MTB ChIP-seq network is significantly enriched with FFLs, a common network motif composed of two input TFs, one of which regulates the other and both of which jointly regulate target gene(s) (Mangan and Alon, 2003). We ran extensive permutation tests to confirm the likelihood (p < 1e-3) of 1,690 FFL instances emerging in a random network with the same number of nodes and edges. Interestingly, Rv0081 is the most frequent regulator at the “top” of the FFLs (40.2% of all detected instances) and also has the highest degree connectivity. Rv0081 has been previously linked to MTB’s response to hypoxia (Prosser et al., 2017; Galagan et al., 2013) and is itself a target gene of the well-characterized regulator of dormancy survival, DosR (Boon and Dick, 2002; Sherman et al., 2001; Park et al., 2003). To evaluate the involvement of Rv0081-centered FFLs in the transcriptional changes observed during hypoxia, we explored some of the most frequent TF pairs found in FFL configuration). The top pair, Rv0081-Rv0324, controls 134 genes significantly enriched with late hypoxia genes (p = 6.4e-5). Rv0081 also frequently pairs with Rv3249c and controls 87 genes enriched with depletion state genes (p = 1.1e-5). Another frequent pair combines Rv0023 and Rv0324 to control 70 genes enriched in mid hypoxia genes (p = 3e-5). We explored the directionality of these state-specific FFL target genes using gene expression data from MTB TF overexpression (TFOE) strains in normoxia (Rustad et al., 2014) and a MTB Rv0081 gene deletion (ΔRv0081) strain in hypoxia (Sun et al., 2018). For example, the majority of depletion genes with differential expression in the ΔRv0081 strain were upregulated, suggesting a negative relationship with Rv0081 in hypoxia (Figure 4A). Moreover, we observed that depletion genes controlled by the Rv0081-Rv3249c FFL were predominantly downregulated in the Rv0081 TFOE strain (Figure 4B). In contrast, there is a positive relationship between Rv0081 and late hypoxia genes during hypoxia, as indicated by a largely decreased expression of late hypoxia genes in the ΔRv0081 strain (Figure 4C). Furthermore, late hypoxia genes controlled by the Rv0081-Rv0324 FFL were significantly upregulated (Figure 4D). Altogether, we generated a model of interlocking FFLs whose TFs together upregulate 231 genes corresponding to late hypoxia, while also coordinating the repression of mid hypoxia and depletion genes (Figure 4E). The overlapping sets of network motifs act to reinforce each other’s function and direct the complex physiological state transitions required to adapt to decreasing DO levels.
Figure 4. Data Supporting Rv0081-Controlled Interlocking FFLs and Overview of the Transcriptional Dynamics across the Six-State Model.

(A) The log2 fold change (FC) distribution of depletion genes with significant differential expression (adjusted p < 0.05, absolute log2 FC > 1) from ΔRv0081 strain in hypoxia.
(B) Boxplots representing log2 FC of various gene groups related to depletion state from Rv0081 overexpression. The number in parentheses indicates the number of genes evaluated in each group. Horizontal black lines in each box indicate median values. Boxes cover the 25th-75th percentile ranges.
(C) The log2 FC distribution of late hypoxia genes with significant differential expression (adjusted p < 0.05, absolute log2 FC > 1) from ΔRv0081 strain in hypoxia.
(D) Boxplots representing log2 FC of various gene groups related to late hypoxia state from Rv0081 overexpression. Horizontal black lines in each box indicate median values. Boxes cover the 25th-75th percentile ranges.
(E) Model of Rv0081-controlled interlocking FFLs that together upregulate a significant number of genes corresponding to late hypoxia, while also repressing mid hypoxia and depletion genes. The p values for enrichment of the FFL-controlled genes from each state are indicated below the state name, as evaluated using a hypergeometric test.
(F) Summary overview of the transcriptional dynamics, inferred key regulators, and regulatory circuits that were revealed from the high-resolution and longitudinal gene expression profiling across the O2 gradient.
See also Figures S2–S4. *p < 0.05 and **p < 0.01.
Three important hypotheses developed from identifying network motif topology. The first hypothesis is that Rv0081 plays a pivotal role in the adaptation of MTB to hypoxia, and its regulatory activity may be O2 dependent. The bifunctional activity of Rv0081 is based on comparison between the normoxic TFOE data and hypoxic ΔRv0081 data (Figure S3A). The regulatory targets of Rv0081 had very little concordance of fold change expression in these different conditions. Most genes that were significantly downregulated in the ΔRv0081 strain in hypoxia showed no fold change difference in the TFOE data. This observation supports recent work demonstrating that Rv0081 had altered DNA-binding ability under hypoxic conditions, with evidence that formate ion accumulation and/or post-translational modifications may be involved in the conditional regulatory activity (Kumar et al., 2019). Furthermore, dual activity of Rv0081 seems to be necessary for coordinating the repression of late hypoxia TFs in early hypoxic time points and their upregulation in late hypoxia (Figure S3B).
The second hypothesis is that Rv0081 may be involved in the state oscillations observed as the DO dropped below 3%. Under these low-O2 conditions, the MTB transcriptome oscillated between two states: late hypoxia genes were expressed (T8 and T9), then early hypoxia genes (T10), then back to late hypoxia (T11), before ultimately committing to early hypoxia (T12–T15). This “flicker” between early hypoxia and late hypoxia, measured roughly 1 h apart, could emerge from oscillatory mechanisms involving an Rv0081-directed incoherent FFL (I-FFL) (Geva-Zatorsky et al., 2006; Kholodenko, 2000; Novák and Tyson, 2008). In I-FFLs, one TF acts positively while the other TF acts negatively, resulting in a pulse of target gene(s) expression. Interestingly, the peak height between the first and second pulse of late hypoxia genes had roughly equal normalized expression (Figures S4A–S4C), suggesting a potential detection of fold change on the basis of the I-FFL, as described by Goentoro et al. (2009). Fitting Rv0081 and early hypoxia TFs (excluding Rv0081) between T7 and T20 with three configurations of I-FFLs (cooperative, independent, and exclusive; see STAR Methods), cooperative TF binding most closely modeled the observed average expression of late hypoxia genes (Figures S4A–S4C). Importantly, all three I-FFL configurations were able to reproduce the oscillatory expression of late hypoxia genes when DO dropped below 3%, concluding that the I-FFL motif can explain the observed “flicker” between early and late hypoxia states. Furthermore, we tested how well the I-FFL motif can replicate the experimental measurement across the three replicates during the “flicker” period. Using parameter optimization (see STAR Methods), we statistically compared the model simulations with the experimental data (Figures S4D and S4E). The I-FFL model significantly reflects the experimental finding across replicates, with the independent and cooperative models being the best configurations. Although this analysis does not exclude other network motifs from producing the “flicker” in late hypoxia gene expression, it does confirm that the oscillatory behavior is reproducible and significantly fits the inherent properties of an Rv0081-directed I-FFL. Although the role of the I-FFL as a fold change detector requires further exploration (parameters were not in the fold detection region according to Goentoro et al., 2009), it is intriguing to hypothesize that as the culture nears hypoxia, a potentially variable and “noisy” period of gene expression, the I-FFL circuit could serve to synchronize the hypoxic response across all cells in the population.
Finally, the third hypothesis is that the I-FFL controlled by Rv0081 also regulates the transition to late hypoxia. Late hypoxia accounts for the largest change in expression and the transcriptional state most characteristic of dormant (i.e., nonreplicating) MTB (Schnappinger et al., 2003; Voskuil et al., 2003). In addition to reproducing the “flicker,” all three I-FFL configurations modeled a delay in late hypoxia gene expression after entering hypoxia. The timescale of the delay element, about 40 h from entering 0% DO to late hypoxia transition, is consistent with delayed translation observed in slow-growing MTB in response to nitric oxide (Cortes et al., 2017). It was recently demonstrated that delayed regulatory interactions within I-FFLs (with mutual inhibition present) produced state transitions related to T cell exhaustion, after a fixed time post-stimulation (Bolouri et al., 2019). As such, the delay element may function in MTB to incorporate robustness into the hypoxic response, ensuring that late hypoxia (with large-scale expression changes) is not activated prematurely. Further investigation is required to determine whether the duration of the delay element is fixed or variable in a manner dependent on how MTB enters hypoxia. The shift to late hypoxia, after 40 h in hypoxia and following transition through two intermediate hypoxic states, is one of the most intriguing revelations from this study and required the development of a reactor system and the high-resolution profiling that was performed here. The elucidation of regulatory circuits that control the large, altered transcriptome of late hypoxia offers novel drug targets that could block the underlying mechanisms that contribute to replication suppression, alternative respiratory/metabolic pathways, and phenotypic tolerance associated with dormant MTB.
DISCUSSION
In this report, we present the high-resolution system-wide gene expression profiling of MTB across a 5 day time course of hypoxia and reaeration. A reactor system was designed to allow exquisite control and monitoring of O2 levels, thereby uncovering intermediate transcriptional states and dynamic expression patterns, not previously described. Gene expression profiling revealed that three-fifths of all genes in the MTB genome are differentially expressed and associated with six distinct transcriptional states as MTB enters into and exits from hypoxia. Moreover, there is strong evidence that the six-state model described in this paper relates to adaptations of MTB in vivo. The response to hypoxia is accompanied by other host-related stress mechanisms (e.g., alternative carbon use, iron limitation, copper stress), as a result of MTB’s evolutionary history as an intracellular pathogen. The large-scale expression changes demonstrate the importance of O2 as a major force in the evolution of MTB and reveals that the pathogen alters gene expression in anticipation of future conditions and challenges. For example, the increased production of PE/PPE proteins during late hypoxia in preparation for ESX export system production upon resuscitation foresees the benefit of PE/PPE protein secretion for dissemination to other host cells. Integrating high-resolution and longitudinal profiling with experimentally determined TF-gene interactions enabled inference of key regulators and intricate circuit architecture that explain how the state transitions unfold (Figure 4F). Regulatory programs with characteristic motifs and properties were identified that serve to incorporate robustness (e.g., time delay ensures state transition only upon proper conditions), synchronization (e.g., I-FFL might uniform response across all cells during “flicker” period), and coordination across the states (e.g., interlocking FFLs, bifunctional Rv0081, mutual inhibition) as MTB transitions across time and O2 gradient. This study reveals that MTB encodes abundant network motifs, presumably with functions that cannot be carried out by simpler circuits, to successfully tailor MTB physiology to stresses within the host environment. It is interesting to speculate that these regulatory interactions have evolved in MTB as an adaptive response to ineffective immunity and failure to clear the pathogen. One of the most important challenges for antibiotic research will be to overcome these overlapping and redundant regulatory mechanisms with novel combinatorial interventions. This study presents significant steps toward apprehending these genetic programs in MTB, paving the way for predictive and rational strategies to improve clinical outcomes of TB treatment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests may be directed to, and will be fulfilled by, the Lead Contact Nitin Baliga (Nitin.Baliga@isbscience.org).
Data and Code Availability
The datasets generated during this study are available at GEO: GSE116353.
Materials Availability
This study did not generate new unique reagents.
Experimental Model and Subject Details
Culturing conditions for in vitro system
Experiments were performed using Mycobacterium tuberculosis H37Rv (kind gift of David Sherman) grown at 37°C in Middlebrook 7H9 supplemented with ADC and 0.05% Tween in spinner flasks. For hypoxia time-course experiment, a 50 mL culture was grown to mid-log phase, and diluted in 700 mL 7H9 media within each bioreactor to a starting A600 of 0.01. Cultures were stirred throughout the experiment.
Method Details
Controlled O2 model design and operation
An Oxygen Sensor Spot (PreSens, Regensburg, Germany) was adhered within a 1L disposable spinner flask with two side arms (Corning, Corning, NY) using vacuum tweezers (Excelta, Buelton, CA). A velcro belt with a screw-on port for the fiber optic cable (PreSens) was wrapped around the flask. The fiber optic cable connected the Oxygen Sensor Spot to a Fibox 4 oxygen meter (PreSens). A gas line input was fastened on one arm of the flask, and a luer-lock/filter sampling port was connected to the other arm. Air and N2 gas lines were run into the Biological safety laboratory and connected to gas-specific mass flow controllers (Alicat Scientific, Tucson, AZ), whose outputs were connected downstream through a Y-connector that led into an incubator. Three separate flasks, all prepared as described above, were placed onto a stir plate inside an incubator at 37°C. The mixed gas line was split via additional Y-connecters, streamed through 0.2 um filters, and attached to the gas line inputs of each flask. Media was incubated overnight and checked for contamination before inoculated with MTB.
The mass flow controllers and oxygen sensor were linked to a computer, which could be remotely accessed and monitored in real-time. After inoculation, we programmed the mass flow controllers using Flow Vision software (Alicat Scientific) to achieve a changing gas mixture gradient, which allowed us creating a steady two-day depletion, followed by two-days of sustained hypoxia, and reaeration by flowing pure air into the headspace of the vessels and increasing the speed of the stir bars in each vessel. Adobe Illustrator CS6 and Inkscape 0.91 were used to generate figures including the diagram of the controlled O2 model in Figure 1A.
RNA isolation
Samples were collected by a luer-lock syringe to the sampling port. Sample volumes varied from 5 mL to 25 mL across the time course, depending on OD but were consistent across replicates of a time point. Samples were centrifuged at high speed for 5 min, supernatant was discarded and cell pellet was immediately flash frozen in liquid nitrogen. Cell pellets were stored at −80°C until all samples collected and then resuspended in 600 μL of fresh lysozyme solution in TE pH 8.0 (5 mg/mL). The resuspended cells were transferred to a tube containing Lysing Matrix B (MP Biomedicals, Santa Ana, CA) and incubated at 37°C for 30 min. Following incubation, 60 μL (1/10th volume of lysate volume) of 10% SDS was added and then tubes were vigorously shaken at max speed for 30 s in a FastPrep 120 homogenizer (MP Biomedicals) three times. Tubes were centrifuged for 1 min (max speed), then 66 μL of 3 M sodium acetate pH 5.2 added and mixed well. Acid phenol (pH 4.2) was added at 726 μL and tubes were inverted to mix well (~60 times). Samples were incubated at 65°C for 5 min, inverting tubes to mix samples every 30 s. Then, centrifuged at 14000 rpm for 5 min and upper aqueous phase was transferred to a new tube. 3M sodium acetate (pH 5.2) was added at 1/10th volume along with 3x volumes of 100% ethanol. Sample was mixed well and incubated at −20°C for 1 hr or overnight. Following incubation, samples were centrifuged at 14000 rpm for 30 min at 4°C, ethanol was discarded and 500 μL of 70% ethanol was added. Samples were centrifuged again at 14000 rpm for 10 min at 4°C, supernatant discarded, and any residual ethanol removed using pipet. Pellet was allowed to air dry, resuspended in 30–40 μL of RNase free water and quantified by Nanodrop (Thermo Scientific). This was followed by in solution genomic DNA digestion using RQ1 Dnase (Promega) following manufacturer’s recommendation. RNA quality was analyzed in a 2100 Bioanalyzer system (Agilent Technologies). Total RNA samples were depleted of ribosomal RNA using the Ribo-Zero Bacteria rRNA Removal Kit (Illumina, San Diego, CA).
Quantification and Statistical Analysis
Statistical details of particular analyses (including whether data met assumptions of statistical approaches) are detailed in the sections below.
Processing and analysis of RNA-seq data
Sample collection and RNA-extraction was performed as described above. Quality and purity of mRNA samples was determined with 2100 Bioanalyzer (Agilent, Santa Clara, CA). Samples were prepared with TrueSeq Stranded mRNA HT library preparation kit (Illumina, San Diego, CA) and multiplexed into a single run. All samples were sequenced on the NextSeq sequencing instrument in a high output 150 v2 flow cell. Paired-end 75 bp reads were checked for technical artifacts using Illumina default quality filtering steps. Raw FASTQ read data were processed using the R package DuffyNGS as described previously (Vignali et al., 2011). Briefly, raw reads were passed through a 3-stage alignment pipeline: (i) a prealignment stage to filter out unwanted transcripts, such as rRNA, mitochondrial RNA, albumin, and globin; (ii) a main genomic alignment stage against the genome(s) of interest; and (iii) a splice junction alignment stage against an index of standard and alternative exon splice junctions (Lakshmanan et al., 2015). Reads were aligned to M. tuberculosis H37Rv (ASM19595v2) with Bowtie2 (Langmead and Salzberg, 2012), using the command line option “verysensitive.” BAM files from stages (ii) and (iii) were combined into read depth wiggle tracks that recorded both uniquely mapped and multiply mapped reads to each of the forward and reverse strands of the genome(s) at single-nucleotide resolution. Gene transcript abundance was then measured by summing total reads landing inside annotated gene boundaries, expressed as both RPKM and raw read counts. Two stringencies of gene abundance were provided using all aligned reads and by just counting uniquely aligned reads.
Differential expression
We used the raw read counts, estimated with DuffyNGS as described above, as input for DESeq2 (Love et al., 2014). We compared the transcriptional profile of each time point respect to T0. Genes with adjusted P-value < 0.05 and estimated absolute log2 fold-change > 1 were considered differentially expressed.
Identification of transcriptional states adopted by MTB in the controlled O2 model
After normalizing the full transcriptional dataset with DESeq2 (Love et al., 2014), we used Principal Component Analysis (PCA) to reduce the dimensionality and to inspect the structure of our normalized data. In a three dimensional PCA plot, multiple groups of consecutive time points and similar O2 concentrations seemed to emerge (Figure S1A). To determine the transcriptional states adopted by MTB during entry to and exit from hypoxia, we first applied hierarchical clustering with bootstrapping, implemented in the Pvclust R package (Suzuki and Shimodaira, 2006), on the median (of each time point replicates) normalized transcriptional dataset. Clusters in the dendrogram were defined at the maximum height with 100% bootstrap support and more than two clusters were found. The second requirement was motivated by inspection of the PCA plot mentioned above. Six clusters were identified (Figure S1B). Then, we repeated this analysis using all replicates (Figure S1C). T15 had a total of 6 replicates, two from each reactor, taken 1 h apart. The average of the two samples from each reactor was used for T15. Each time point was assigned to the group that contained at least two (out of the three) replicates. Remarkably, there was significant overlap between the clusters generated with this approach in the full dataset and the six clusters previously defined on the median profiles. P-values associated with overlap hypergeometric tests are shown in parentheses in Figure S1C. To assess the robustness of the proposed six states model to clustering algorithm selection, we compared the predictions made with hierarchical clustering and k-means. We observed a strong agreement between both methods. In fact, five out of the six clusters identified by k-means with k = 6 were identical to the hierarchical clustering-based clusters (Figures S1C and S1D). In support of the six state model, we observed that k = 2 and k = 6 gave the highest quality clusters based on the Silhouette metric for k-means (Figure S1E). Notably, only nine replicates out of the 78 replicates (11.5%) did not cluster with other replicates of the same time point (in any of the two methods).
To measure the robustness of our six states definition to the (biological and technical) noise present in the collected transcriptional data, we used ensemble clustering as suggested by Naegle and collaborators (Sloutsky et al., 2013; Ronan et al., 2016). First, we used permutation sampling (5,000 permutations) to create time series (without replicates) with all 26 time points. To do so, for each time point, we randomly selected one of the three available replicates. Then, we applied k-means clustering (1 < k < 11) to group the time points in the generated time course. Next, we evaluated how often any pair of time points clustered together in the 45,000 k-means runs (9 different k values by 5,000 time courses; Figure S1F). From this analysis, we could confirm the presence of six clusters (pink/red squares, that represent co-clustering in 50% or more of the cases, in the heatmap) with significant overlap with our states’ definition (described in the previous paragraph). After confirming the presence of six clusters, we performed a second ensemble clustering (using the same bootstrapping strategy) with 200,000 permutations. This time we applied k-means with k = 6 on each generated time course (Figure S1G). 24 out of the 26 time points clustered more than 50% of the time with the expected time points (according to the six states model). T16 did not pass the 50% co-occurrence threshold with the other two members of Mid hypoxia (T17 and T18). Nonetheless, T17 and T18 are the two points that T16 most frequently clustered with (46.5% and 48.6%, respectively). So, the only time point that was not unequivocally assigned to any specific cluster was T9. The other two time points of the flicker (T8 and T11) did indeed cluster with the other Late hypoxia time points (T19, T20) as expected.
As more direct approach to compare within- and between-state variation, we used the quantro R Bioconductor package (Hicks and Irizarry, 2015). Quantro performs a statistical test (quantroStat) to compare the within- and between-groups variation. Using quantro in the expression dataset with all replicates, we obtained a quantroStat P-value < 1e-04. Overall, our observations indicated that noise is higher at the inter-state that within-state level. Finally, we visualized the six defined transcriptional states with the t-distributed stochastic neighbor embedding (tSNE) algorithm (Figure 2A).
Connecting differentially expressed genes with the six hypoxia-related transcriptional states of MTB
To understand the functional implications of the transcriptional states adopted by MTB during entry and exit from hypoxia, each differentially expressed gene was assigned to the state in which it had the highest average transcription level. In this step, we used the median normalized transcriptional profiles. We obtained similar gene assignment when we used the average among all replicates and time points of each cluster. As an unsupervised alternative, we used the Boruta R package (Kursa and Rudnicki, 2010), that implements random forest to select all features (in our case transcriptional profiles), to identify the genes that distinguish any given state from the rest. There was statistically significant overlap between the groups of genes associated to any given transcriptional state by the two approaches (Table S2). Because Boruta only selected 590 genes (out of 2,582 differentially expressed genes), we decided to use the average transcriptional profile based gene assignment. In this way we tried to capture the biological processes active in the different states without excluding any differentially expressed gene. To evaluate the quality of the resulting sets of genes, we computed the mean squared residue (MSR) of each gene cluster (Table S2). The MSR is widely used as a metric of performance of biclustering methods (which cluster both genes and conditions) (Reiss et al., 2006). A low MSR value indicates that individual gene profiles do not deviate from the average profile of the bicluster (in our case, the group of genes in the relevant time points/state). We also computed the mean Pearson correlation among the genes assigned to each transcriptional state (Table S2). In support of our gene assignment, the sets of genes associated to MTB transcriptional states had MSR values smaller than 0.1 In fact, for the Depletion and Late hypoxia genes the MSR values were smaller than 0.05. The average Pearson correlation values were equal to or greater than 0.4 for all gene clusters but Normoxia.
Metabolic pathway analysis
We mapped the measured gene expression data against the most recent genome-scale metabolic network construction of M. tuberculosis H37Rv iEK1011 (Kavvas et al., 2018) using COBRApy (Ebrahim et al., 2013). We used the subsystem definitions outlined in iEK1011 to explore pathway usage at the network level. See also Figure S2.
MTB ChIP-seq derived TF-gene network
The initial ChIP-seq derived MTB network consisted of 6,581 interactions occurring in the −150bp to +70bp region of genes promoters reported by Minch et al. (2015). We expanded that MTB ChIP-seq network by taking into account operon organizations. For a given TF-gene interaction, if the target gene is part of an operon, we included all other members of the operon as potential targets of the corresponding TF. The expanded MTB ChIP-seq network contained 12,188 interactions. The ChIP-seq derived protein-DNA interactions were used to establish connections between differentially expressed genes from the controlled O2 model using Biotapestry (Paquette et al., 2016).
Detection of network motifs in the MTB ChIP-seq network
To identify network motifs in the MTB transcriptional network, we used the MotifNet webserver (Smoly et al., 2017). We scanned for all potential three and four nodes motifs with maximum P-value ≤ 0.01 (in 1000 random networks) and with 100 or more instances in the analyzed network. For this analysis, we constrained the ChIP-seq derived network by excluding genes that were not differentially expressed in our time course. This network filtering was done to improve detection of motifs most relevant to the actual changes in transcript levels we observed.
DREM analysis
DREM2.0 (Schulz et al., 2012) was run with default parameters. The input TF-gene network was the MTB ChIP-seq network described above. The input expression data contained the median transcriptional profiles of the 2,582 differentially expressed genes. The minimum absolute expression change parameter was set to 0.75. The supplemental Data S2 contains all the files required to recreate the DREM output, without the need of running DREM again.
Permutation test for evaluating significance of overlap between TF regulons and sets of genes associated with identified transcriptional states
The 2,582 differentially expressed genes in the controlled O2 model were permutated 1000 times to generate shuffled gene clusters (corresponding to the six transcriptional states). In each permutation, the produced shuffled gene clusters had the same size as the original ones. Then, significance of the overlap between regulons of differentially expressed TFs and the shuffled gene sets was evaluated using a hypergeometric test. Hypergeometric test P-values below 0.05 were considered significant. The overall permutation test P-value was computed as the proportion of cases (out of 1000 permutations) in which the number of enriched regulons was equal or higher than the observed values in the original data.
Differential expression analysis of transcriptional data collected with the defined hypoxia model
We downloaded the transcriptional profile data of MTB at day 0, day 1, day 2, day 3, day 5, day 7 and day 8 (reaeration) collected by Galagan et al. (2013) using the defined hypoxia model (GEO accession number: GSE43466). We performed a Bayesian t test using Cyber-T (Baldi and Long, 2001) to compare the gene expression profiles at each time point respect to T0. Genes with adjusted P-value < 0.05 and absolute log2 fold-change > 1 were considered differentially expressed. See Tables S1A and S1B.
Modeling of Rv0081-directed I-FFL with Late hypoxia gene expression
The IFFL motif is commonly modeled by the following equations (Goentoro et al., 2009):
where Z is the output of the motif which in our case represents the average expression of Late hypoxia genes between T7-T20. X and Y represent the expression of Rv0081 and average expression of Early hypoxia TFs (excluding Rv0081), respectively. Goentoro et al. (2009) analyzed three possible models of the I-FFL motif representing three configurations in which Late hypoxia genes could be regulated. Their focus was to identify conditions for which the I-FFL motif works as fold change detection. The models correspond to exclusive binding (Figure S4A), independent binding (Figure S4B), and cooperative binding (Figure S4C) which are represented by the following equations (Goentoro et al., 2009):
where Ge, Gi, and Gc are functions that determine the rate change of Late hypoxia genes (Z); K1 is the binding rate between Rv0081 and Late hypoxia genes; K2 is the binding rate between Early hypoxia TFs and Late hypoxia genes; and K3 is the cooperative binding rate of Rv0081 and Early hypoxia TFs with the Late hypoxia genes. α2, β2, K1, K2, and K3 were estimated by an optimization procedure using the average experimental values of X, Y, Z at different time points. We used the Nelder-Mead simplex algorithm for optimization (Nelder and Mead, 1965) as implemented in MATLAB R2014a. The objective function used for minimization is the root-mean-squared deviation (RMSD) between experimental and estimated values of Late hypoxia gene expression is given by:
where Zexp(ti) is the average expression of Late hypoxia genes at time ti obtained experimentally, and Zmod(ti) is the corresponding model estimate. Similarly, α1 and β1 were estimated by a similar optimization procedure using the average experimental values of Y and X. The objective function in this case is given by:
where Yexp(ti) is the average expression of repressors at time ti obtained experimentally, and Ymod(ti) is the corresponding model estimate.
In addition, we also tested the reproducibility of the I-FFL model during the “flicker” period across the experimental replicates by including 6 additional parameters (in addition to the model parameters) in the fitting process, which correspond to the initial values of Y and Z for the three replicates. In total we optimized 12 parameters for the cooperative model, and 11 parameters for the independent and exclusive binding models. It is worth noting that the same values of the model parameters (β1,β2,α2,K1,K2and K3) are used to test a model with the three experimental replicates. By using this strategy, we find a mathematical model that includes the potential differences between the measurements of the replicates. Moreover, the strategy helps avoiding parameter over-fitting because a model of 12 (11) parameters is fitted to 48 experimental data points.
We used the routine pwFitBoost available in the MATLAB toolbox PottersWheel for parameter optimization (Maiwald and Timmer, 2008). pwFitBoost is a hybrid approach that combines a stochastic simulated annealing algorithm, which performs a global search of the parameter space, and a deterministic algorithm for local search. We used the following function as a measure of how good the IFFL models reproduces experimental gene expression:
Where R, M, T are the number of replicates (3), the number of observables (2 for Y and Z), and the number of time points (8 por the considered time window), respectively. θ is the parameter set which includes the model parameters (α2,β1, β2, K1, K2, and K3) and the initial conditions of Y and Z for the three experimental replicates. are the experimental data, are the values of the model at the times points when the experimental data are measured, are the measurement error of the experimental data at each time point. These errors were estimated as half the standard deviation of the gene expression of the set of Repressor and S2 genes. To ensure that we approach the global minimum of χ2 as close as possible, we repeated the parameter optimization 500 times varying the initial values of the parameters. The initial values of the parameters were selected randomly from a Latin Hypercube sampling using a range of 10×10−2 to 1×103 for α2, β1, β2, K1, K2, and K3, 7–12 for the initial Y in the three replicates, and 4–8 for the initial value of Z in the three replicates. The parameters of the best cooperative and independent models are presented in Figure S4D along with the summary statistics (Figure S4E). The p(N–d) value of the exclusive binding models was close to the rejection value of 0.05, therefore the parameter values are not reported.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| H37Rv (M. tuberculosis) | D. Sherman lab | N/A |
| Critical Commercial Assays | ||
| Ribo-Zero magentic kit bacteria | Illumina | Cat # MRZB12424 |
| TruSeq stranded mRNA library prep kit | Illumina | Cat # RS-122-2103 |
| Deposited Data | ||
| Hypoxia/reaeration RNA-Seq data | Peterson et al., 2019 | GEO: GSE116353 |
| TF-target gene network | Peterson et al., 2019 | http://networks.systemsbiology.net/mtb/data-center |
| Software and Algorithms | ||
| DuffyNGS | Vignali et al., 2011 | http://networks.systemsbiology.net/mtb/data-center |
| DESeq2 | Love et al., 2014 | https://github.com/mikelove/DESeq2 |
| DREM | Schulz et al., 2012 | https://sb.cs.cmu.edu/drem/ |
| Biotapestry | Paquette et al., 2016 | http://www.biotapestry.org/ |
| MATLAB R2014a | N/A | N/A |
| Adobe Illustrator CS6 | N/A | N/A |
| Inkscape 0.91 | N/A | N/A |
| Other | ||
| Fibox 4 oxygen meter with display | PreSens | Cat #200001478 |
| Polymer Optical Fiber for use with oxygen sensor | PreSens | Cat # 200000241 |
| Oxygen Sensor Spot | PreSens | Cat # 200000023 |
| Mass flow controller for Air | Alicat Scientific | Cat # MCW-100ACCM-D/5M,4IN, GAS: Air |
| Mass flow controller for Nitrogen | Alicat Scientific | Cat # MCW-100SCCM-D/5M,4IN, GAS: N2 |
| R notebook with scripts for performing computation analyses under “mtb hypoxia dormancy” | This paper | https://github.com/baliga-lab |
Highlights.
Reactor enables high-resolution transcriptomics of M. tuberculosis across O2 gradient
147 TFs drive MTB into and out of hypoxia-induced dormancy via distinct sub-states
Robustness, time delay, and anticipatory behavior encoded in transcriptional program
Rv0081-directed feedforward loops play a pivotal role in adaptation to hypoxia
ACKNOWLEDGMENTS
We thank members of the Baliga and Shmulevich labs for critical discussions, Lee Hiner (Airgas), and Seattle Children’s Research Institute for the BSL3 facilities. The granuloma formation shown in the graphical abstract was adapted from Ehlers and Schaible (2013). Funding was provided by the National Institute of Allergy and Infectious Diseases: R01AI128215, U19AI135976, and a pilot project grant awarded to Christopher Plaisier and E.J.R.P through U19AI106761. Funding was also provided by the National Science Foundation (DBI-1565166).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107577.
REFERENCES
- Abdallah AM, Savage ND, van Zon M, Wilson L, Vandenbroucke-Grauls CM, van der Wel NN, Ottenhoff TH, and Bitter W (2008). The ESX-5 secretion system of Mycobacterium marinum modulates the macrophage response. J. Immunol 181, 7166–7175. [DOI] [PubMed] [Google Scholar]
- Alon U (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet 8, 450–461. [DOI] [PubMed] [Google Scholar]
- Baldi P, and Long AD (2001). A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics 17, 509–519. [DOI] [PubMed] [Google Scholar]
- Bartek IL, Woolhiser LK, Baughn AD, Basaraba RJ, Jacobs WR Jr., Lenaerts AJ, and Voskuil MI (2014). Mycobacterium tuberculosis Lsr2 is a global transcriptional regulator required for adaptation to changing oxygen levels and virulence. MBio 5, e01106–e01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR, Hill AA, Claggett JM, Hill-Harfe K, Wen JC, Slonim DK, Brown EL, and Hunter CP (2005). The homeodomain protein PAL-1 specifies a lineage-specific regulatory network in the C. elegans embryo. Development 132, 1843–1854. [DOI] [PubMed] [Google Scholar]
- Bolouri H, Young M, Beilke J, Johnson R, Fox B, Huang L, Costa Santini C, Hill CM, van der Vuurst de Vries A-R, Shannon P, et al. (2019). Integrative network modeling reveals mechanisms underlying T cell exhaustion. Sci. Rep 10, 1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon C, and Dick T (2002). Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J. Bacteriol. 184, 6760–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottai D, and Brosch R (2009). Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol. Microbiol 73, 325–328. [DOI] [PubMed] [Google Scholar]
- Bromberg KD, Ma’ayan A, Neves SR, and Iyengar R (2008). Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science 320, 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MC, and Rubin EJ (2010). Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu. Rev. Microbiol 64, 293–311. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- Cortes T, Schubert OT, Banaei-Esfahani A, Collins BC, Aebersold R, and Young DB (2017). Delayed effects of transcriptional responses in Mycobacterium tuberculosis exposed to nitric oxide suggest other mechanisms involved in survival. Sci. Rep 7, 8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahim A, Lerman JA, Palsson BO, and Hyduke DR (2013). COBRApy: Constraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol 7, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S, and Schaible UE (2013). The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front. Immunol 3, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Vainas O, Harbison CT, Simon I, and Bar-Joseph Z (2007). Reconstructing dynamic regulatory maps. Mol. Syst. Biol 3, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Beg QK, Kay KA, Balázsi G, Oltvai ZN, and Bar-Joseph Z (2008). A semi-supervised method for predicting transcription factor-gene interactions in Escherichia coli. PLoS Comput. Biol 4, e1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, Gomes A, Rustad T, Dolganov G, Glotova I, et al. (2013). The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499, 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner TS, Cantor CR, and Collins JJ (2000). Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342. [DOI] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, Milo R, Sigal A, Dekel E, Yarnitzky T, Liron Y, Polak P, Lahav G, and Alon U (2006). Oscillations and variability in the p53 system. Mol. Syst. Biol 2, 2006.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass L, and Kauffman SA (1973). The logical analysis of continuous, nonlinear biochemical control networks. J. Theor. Biol 39, 103–129. [DOI] [PubMed] [Google Scholar]
- Goentoro L, Shoval O, Kirschner MW, and Alon U (2009). The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol. Cell 36, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks SC, and Irizarry RA (2015). quantro: a data-driven approach to guide the choice of an appropriate normalization method. Genome Biol 16, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Guo YP, May G, and Enver T (2007). Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev. Biol 305, 695–713. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Kavvas ES, Seif Y, Yurkovich JT, Norsigian C, Poudel S, Greenwald WW, Ghatak S, Palsson BO, and Monk JM (2018). Updated and standardized genome-scale reconstruction of Mycobacterium tuberculosis H37Rv, iEK1011, simulates flux states indicative of physiological conditions. BMC Syst. Biol 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempner W (1939). Oxygen tension and the tubercle bacillus. Am. Rev. Tuberc 40, 157–168. [Google Scholar]
- Kholodenko BN (2000). Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem 267, 1583–1588. [DOI] [PubMed] [Google Scholar]
- Kumar A, Phulera S, Rizvi A, Sonawane PJ, Panwar HS, Banerjee S, Sahu A, and Mande SC (2019). Structural basis of hypoxic gene regulation by the Rv0081 transcription factor of Mycobacterium tuberculosis. FEBS Lett 593, 982–995. [DOI] [PubMed] [Google Scholar]
- Kursa MB, and Rudnicki WR (2010). Feature selection with the Boruta package. J. Stat. Softw 36 (11). [Google Scholar]
- Lakshmanan V, Fishbaugher ME, Morrison B, Baldwin M, Macarulay M, Vaughan AM, Mikolajczak SA, and Kappe SH (2015). Cyclic GMP Balance Is Critical for Malaria Parasite Transmission from the Mosquito to the Mammalian Host. mBio 6 10.1128/mBio.02330-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, and Gerstein M (2004). Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431, 308–312. [DOI] [PubMed] [Google Scholar]
- Macqueen J (1967). Some methods for classification and analysis of multivariate observations. In Proceedings of the 5th Berkeley Symposium of Mathematical Statistics and Probability 1, 281–297. [Google Scholar]
- Maiwald T, and Timmer J (2008). Dynamical modeling and multi-experiment fitting with PottersWheel. Bioinformatics 24, 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, and Alon U (2003). Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. USA 100, 11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus SA, Sidiropoulos SW, Steinberg H, and Talaat AM (2016). CsoR is essential for maintaining copper homeostasis in Mycobacterium tuberculosis. PLoS ONE 11, e0151816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minch KJ, Rustad TR, Peterson EJ, Winkler J, Reiss DJ, Ma S, Hickey M, Brabant W, Morrison B, Turkarslan S, et al. (2015). The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun 6, 5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, Ortblad KF, Guinovart C, Lim SS, Wolock TM, Roberts DA, Dansereau EA, Graetz N, Barber RM, Brown JC, et al. (2014). Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 1005–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muttucumaru DG, Roberts G, Hinds J, Stabler RA, and Parish T (2004). Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberculosis (Edinb.) 84, 239–246. [DOI] [PubMed] [Google Scholar]
- Nelder JA, and Mead R (1965). A simplex method for function minimization. Comput. J 7, 308–318. [Google Scholar]
- Novák B, and Tyson JJ (2008). Design principles of biochemical oscillators. Nat. Rev. Mol. Cell Biol 9, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette SML, Leinonen K, and Longabaugh WJ (2016). BioTapestry now provides a web application and improved drawing and layout tools. F1000Res 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, Schoolnik GK, and Sherman DR (2003). Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol 48, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson EJ, Bailo R, Rothchild AC, Arrieta-Ortiz ML, Kaur A, Pan M, Mai D, Abidi AA, Cooper C, Aderem A, et al. (2019). Path-seq identifies an essential mycolate remodeling program for mycobacterial host adaptation. Mol. Syst. Biol 15, e8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser G, Brandenburg J, Reiling N, Barry CE 3rd, Wilkinson RJ, and Wilkinson KA (2017). The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect 19, 177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss DJ, Baliga NS, and Bonneau R (2006). Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformatics 7, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan T, Qi Z, and Naegle KM (2016). Avoiding common pitfalls when clustering biological data. Sci. Signal 9, re6. [DOI] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, et al. ; modENCODE Consortium (2010). Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad TR, Harrell MI, Liao R, and Sherman DR (2008). The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 3, e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad TR, Sherrid AM, Minch KJ, and Sherman DR (2009). Hypoxia: a window into Mycobacterium tuberculosis latency. Cell. Microbiol 11, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Rustad TR, Minch KJ, Ma S, Winkler JK, Hobbs S, Hickey M, Brabant W, Turkarslan S, Price ND, Baliga NS, and Sherman DR (2014). Mapping and manipulating the Mycobacterium tuberculosis transcriptome using a transcription factor overexpression-derived regulatory network. Genome Biol 15, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, and Schoolnik GK (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med 198, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz MH, Devanny WE, Gitter A, Zhong S, Ernst J, and Bar-Joseph Z (2012). DREM 2.0: Improved reconstruction of dynamic regulatory networks from time-series expression data. BMC Syst. Biol 6, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, and Schoolnik GK (2001). Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc. Natl. Acad. Sci. U S A 98, 7534–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi T, Matsuyama S, and Kitano H (2010). Large-scale analysis of network bistability for human cancers. PLoS Comput. Biol 6, e1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloutsky R, Jimenez N, Swamidass SJ, and Naegle KM (2013). Accounting for noise when clustering biological data. Brief. Bioinform 14, 423–436. [DOI] [PubMed] [Google Scholar]
- Smoly IY, Lerman E, Ziv-Ukelson M, and Yeger-Lotem E (2017). MotifNet: a web-server for network motif analysis. Bioinformatics 33, 1907–1909. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhang L, Jiang J, Ng M, Cui Z, Mai J, Ahn SK, Liu J, Zhang J, Liu J, and Li Y (2018). Transcription factors Rv0081 and Rv3334 connect the early and the enduring hypoxic response of Mycobacterium tuberculosis. Virulence 9, 1468–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, and Shimodaira H (2006). Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542. [DOI] [PubMed] [Google Scholar]
- Tiwari BM, Kannan N, Vemu L, and Raghunand TR (2012). The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages. PLoS ONE 7, e51686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, and Chan J (2006). Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell. Microbiol 8, 218–232. [DOI] [PubMed] [Google Scholar]
- Vignali M, Armour CD, Chen J, Morrison R, Castle JC, Biery MC, Bouzek H, Moon W, Babak T, Fried M, et al. (2011). NSR-seq transcriptional profiling enables identification of a gene signature of Plasmodium falciparum parasites infecting children. J. Clin. Invest 121, 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, and Schoolnik GK (2003). Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med 198, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, and Hayes LG (1996). An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun 64, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, and Sohaskey CD (2001). Nonreplicating persistence of mycobacterium tuberculosis. Annu. Rev. Microbiol 55, 139–163. [DOI] [PubMed] [Google Scholar]
- Wernicke S, and Rasche F (2006). FANMOD: a tool for fast network motif detection. Bioinformatics 22, 1152–1153. [DOI] [PubMed] [Google Scholar]
- Wu M, Liu L, and Chan C (2011). Identification of novel targets for breast cancer by exploring gene switches on a genome scale. BMC Genomics 12, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Crane DD, Simpson RM, Zhu YQ, Hickey MJ, Sherman DR, and Barry CE 3rd. (1998). The 16-kDa alpha-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc. Natl. Acad. Sci. U S A 95, 9578–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at GEO: GSE116353.