

Significance Statement
Kidney development is a complex process involving multiple interacting and transitioning cell types. Drop-seq single-cell technology, which measures gene expression from many thousands of individual cells, has been used to characterize these cellular differentiation changes that underlie organ development. However, the alternative splicing of many genes creates an additional layer of cellular heterogeneity that Drop-seq technology cannot measure. Therefore, in this study, full transcript length single-cell RNA sequencing was used to characterize alternative splicing in the mouse embryonic kidney, with particular attention to the identification of genes that are alternatively spliced during the transition from mesenchymal to epithelial cell states, as well as their splicing regulators. These results improve our understanding of the molecular mechanisms that underlie kidney development.
Keywords: kidney development, Single cell RNA sequencing, Alternative splicing, Mesenchymal to epithelial transition (MET), Wt1
Visual Abstract

Abstract
Background
During mammalian kidney development, nephron progenitors undergo a mesenchymal-to-epithelial transition and eventually differentiate into the various tubular segments of the nephron. Recently, Drop-seq single-cell RNA sequencing technology for measuring gene expression from thousands of individual cells identified the different cell types in the developing kidney. However, that analysis did not include the additional layer of heterogeneity that alternative mRNA splicing creates.
Methods
Full transcript length single-cell RNA sequencing characterized the transcriptomes of 544 individual cells from mouse embryonic kidneys.
Results
Gene expression levels measured with full transcript length single-cell RNA sequencing identified each cell type. Further analysis comprehensively characterized splice isoform switching during the transition between mesenchymal and epithelial cellular states, which is a key transitional process in kidney development. The study also identified several putative splicing regulators, including the genes Esrp1/2 and Rbfox1/2.
Conclusions
Discovery of the sets of genes that are alternatively spliced as the fetal kidney mesenchyme differentiates into tubular epithelium will improve our understanding of the molecular mechanisms that drive kidney development.
Kidney development is a complex process that involves multiple interacting cell types.1–5 It starts at the embryonic stage and continues from week 5 to week 36 of gestation in humans and from embryonic day (E) 10.5 to approximately day 3 after birth in mice. The process is initiated by signaling interactions between two lineages originating from the intermediate mesoderm: the ureteric duct and the metanephric mesenchyme (Figure S1 in Supplemental Appendix 1). These interactions invoke the ureteric duct to invade the metanephric mesenchyme and create a tree-like structure. Around the tip of each branch of this tree, the ureteric tip, cells from the metanephric mesenchyme are induced to condense and form the cap mesenchyme (CM), which is a transient nephron progenitor cell population. Next, cells from the CM undergo a mesenchymal-to-epithelial transition (MET) and progressively differentiate into early epithelial structures: pretubular aggregates, renal vesicles, and comma and S-shaped bodies. The S-shaped bodies further elongate and differentiate to form the various epithelial tubular segments of the fully developed nephron, whose main constituents are the podocytes (PODOs), the proximal tubule, the loop of Henle (LOH), and the distal tubule. At an early stage in their differentiation the distal tubules connect to the ureteric tips that form the collecting duct system for draining the nephrons. Meanwhile, the uninduced cells of the metanephric mesenchyme differentiate into other supporting cell types of the kidney such as interstitial fibroblasts, pericytes, and mesangial cells (Figure S1 in Supplemental Appendix 1). In the past few years, the various cell populations of the developing kidney were characterized,6–8 mainly using the Drop-seq single-cell RNA sequencing protocol that enables measuring of gene expression levels from many thousands of individual cells.9
A central process in kidney development is the MET that occurs during the differentiation of cells from the metanephric mesenchyme to the CM and then to nephron tubules. Similar transitions from mesenchymal to epithelial states and vice versa (epithelial-to-mesenchymal transition [EMT]) are thought to play a central role in development, as well as in pathologic processes such as cancer metastasis10 and organ fibrosis.11 There are significant structural and functional differences between mesenchymal and epithelial cells. Mesenchymal cells are typically loosely associated with each other, surrounded by an extracellular matrix, and have migratory capabilities, whereas epithelial cells are tightly interconnected by junctions and are polarized with distinct apical and basolateral membranes. Thus, epithelial cells can create two-dimensional surfaces and tubes with a clear in/out distinction that are capable of absorption and secretion. There are also large differences in gene expression: mesenchymal cells typically express Fibronectin (Fn1), Vimentin (Vim), and N-cadherin (Cdh2),12 as well as the transcription factors Snai1/2, Zeb1/2, and Twist1/2, whereas epithelial cells typically express other genes such as E-cadherin (Cdh1) and Epcam.
It was recently discovered that mesenchymal and epithelial cells also express alternative splice isoforms of genes that are expressed in both cell states. For example, in many systems both in vivo and in vitro, the genes Enah, Cd44, Ctnnd1, and Fgfr2 were found to be expressed in both mesenchymal and epithelial states, but with unique isoforms specific to each state.13–18 It was also found that RNA binding proteins, such as ESRP1/2, RBFOX1/2, RBM47, QKI, act as splicing regulators that promote splicing of specific mesenchymal or epithelial variants.12,17,19,20 mRNA splicing creates an additional layer of heterogeneity that, apart from specific genes,21–23 has not yet been comprehensively studied in the developing kidney.
Therefore, in this study we set to characterize the splice isoform switching events that occur during the transition between the mesenchymal and epithelial cellular states in the course of kidney development by comparing gene expression and alternative splicing in the various mesenchymal and epithelial cell populations. Because the kidney is a heterogeneous organ that is composed of numerous cell populations of widely varying proportions,6–8,24 it is difficult to isolate pure populations of mesenchymal and epithelial states. Moreover, typical sequencing depths from a single cell are not sufficient for splicing analysis. We therefore performed single-cell RNA sequencing on 576 individual cells from the kidneys of E18.5 mouse embryos using the Smartseq2 protocol for sequencing full transcript lengths.25,26 We first identified the main cell lineages that coexist in the nephrogenic zone of the fetal developing kidney: the uninduced metanephric mesenchyme, the CM, podocytes, epithelial cells, endothelial cells, and infiltrating immune cells (e.g., macrophages). We then merged the raw reads from all cells belonging to each population in order to create “bulk” in silico transcriptomes that represent each cell population. These bulk transcriptomes had sufficient sequencing depth to allow us to characterize splice isoform switching and to identify putative splicing regulators.
Methods
Tissue Collection, Dissociation, and Flow Cytometry
A wild-type female mouse was crossed with a male that was heterozygous for the Six2-GFP transgene.27 The female was euthanized at E18.5 of pregnancy and kidneys from 11 embryos were dissected, placed in PBS on ice, and examined under a fluorescence microscope to check for the presence of GFP. Kidneys from transgenic embryos showed a clear fluorescent pattern marking the CM (Figure 1A), whereas those from the nontransgenic embryos showed uniform background fluorescence. Seven out of the 11 embryos contained the Six2-GFP transgene. Keeping the transgenic and nontransgenic kidneys separate, each kidney was then cut into two to four small pieces using a surgical razor blade and placed in 1 ml of trypsin 0.25% (03–046–1A, Trypsin Solution B [0.25%]; Biologic Industries) using forceps. Initial tissue trituration was performed using a tissue grinder (D8938, Dounce, large clearance pestle) followed by up-down pipetting with a P1000 pipette. Tissue fragments were then incubated with trypsin at 37°C for 20 minutes, followed by additional trituration by pipetting. After visual confirmation that the majority of cells were fully dissociated, enzyme digestion was stopped by adding 2 ml of DMEM with 10% FBS and placed on ice. Cells were first filtered with 70-μm cell strainers (CSS-010–070; Lumitron) and then 40-μm cell strainers (732–2757; VWR International), pelleted by centrifugation for 5 minutes at 500×g, resuspended in 3 ml PBS, and kept on ice until FACS sorting.
Figure 1.
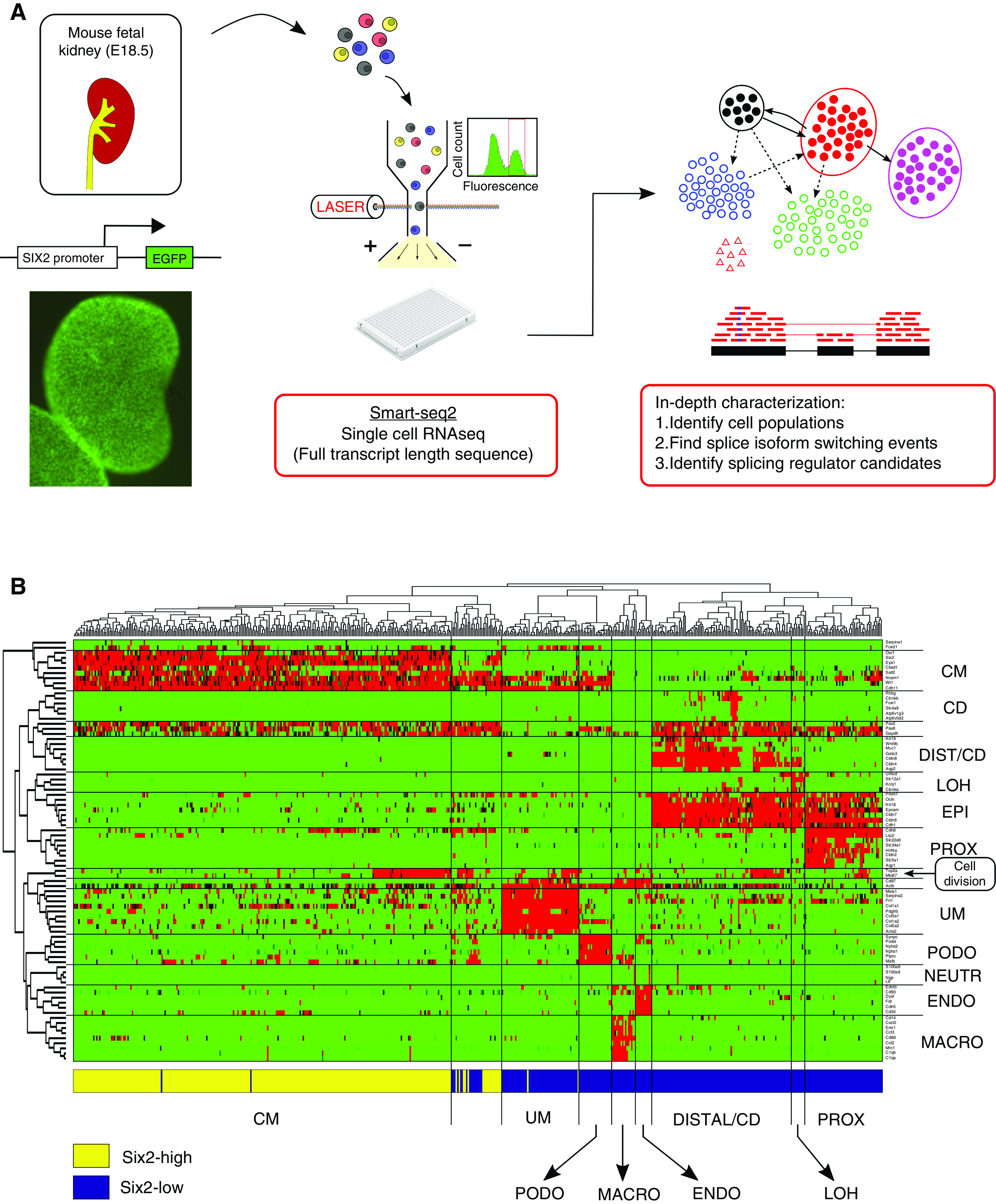
Single-cell RNA sequencing was used to identify and transcriptionally characterize the main cell lineages that coexist in the nephrogenic zone of the developing mouse fetal kidney. (A) Shown is the general outline of the experiment. Kidneys from transgenic mouse embryos with a Six2-GFP reporter gene were harvested at E18.5. The Six2-GFP reporter gene shows a clear fluorescent pattern marking the CM. After tissue dissociation, single cells from the Six2-high and Six2-low cell fractions were sorted into individual wells and their mRNA was sequenced for full transcript length using the Smartseq2 protocol. (B) A clustergram for 83 selected genes from the literature versus 544 cells shows the main cell lineages in the developing kidney. We manually classified the cells to lineages known to coexist in the nephrogenic zone of the developing fetal kidney, including the un-induced mesenchyme (UM), the cap mesenchyme (CM), podocytes (PODO), proximal tubular epithelial cells (PROX), the loop of Henle (LOH), distal tubular cells and collecting duct (DIST_CD), endothelial cells (ENDO), and infiltrating immune cells, mainly macrophages (MACRO) and a small number of neutrophils (NEUTR). The horizontal bar at bottom of the figure depicts the gate used by FACS to sort each cell (Six2-high or Six2-low). Consistent with the fact that Six2 is highly expressed in the CM, it can be seen that cells originating from the Six2-high fraction predominantly belong to the CM, whereas cells originating from the Six2-low fraction belong mostly to the other populations. The gene panel includes genes that were previously shown to be specific to the different populations, as well as general epithelial markers (EPI) and genes that are overexpressed in the S-G2-M phase of the cell cycle, indicating cell division. Notice that some cells (within the second cluster from the left) express a mixture of CM markers and epithelial markers (EPI, PODO), as well as cell division. These are presumably cells from early epithelial structures, presumably pretubular aggregates, renal vesicles, and C/S-shaped bodies (see Supplemental Appendix 1).
Single-cell sorting was done using a BD FACSAria III flow cytometer with an 85 μm nozzle. Forward and side scatter were used to filter out red blood cells and to select for live single cells. Gating on the basis of the negative control—the cells from the nontransgenic kidneys—was used to select for cells that were either positive (Six2-high) or negative (Six2-low) for expression of the Six2-GFP transgene.
All procedures were approved by the institutional Animal Care and Use Committee in Bar-Ilan University.
Single-Cell RNA Sequencing
For single-cell RNA sequencing, single cells were sorted into 96 individual wells from a 384-well plate that was prefilled with Smartseq2 cell lysis buffer, RNase inhibitor, oligo-dT primer, ERCC RNA spike-in mix, and dNTPs. Plates were spun for 1 minute to collect the liquid and cells at the bottom of the wells and immediately frozen. The Smartseq2 protocol was performed by the Israel National Center for Personalized Medicine (G-INCPM) as previously described,25 with 22 amplification cycles. Altogether, six plates were processed, each containing 96 individual cells (576 cells in total), with three plates containing Six2-high cells and three plates containing Six2-low cells. Each one of the six plates was separately sequenced (1×61 bases) on the Illumina HiSeq 2500 platform in the G-INCPM.
The GEO series record for the single-cell RNA sequencing data is GSE146988.
Bulk RNA Sequencing
Six2-high and Six2-low cells were sorted into separate 1.5 ml tubes, each containing RNA purification buffer. This was repeated in three experiments in which we used two types of RNA purification kits: the Norgen single-cell RNA Purification Kit (51800; Norgen Biotek) was used in one experiment where we sorted 50,000 Six2-high and 50,000 Six2-low cells, and the Direct-zol RNA MiniPrep Kit (R2050; Zymo Research) was used in two experiments where we sorted approximately 100,000 Six2-high cells and 400,000 Six2-low cells. Bulk total RNA was purified according to the manufacturer’s instructions and stored at −80°C. Altogether, six samples of total RNA were collected (three replicates, each containing total RNA from Six2-high and Six2-low cells).
Sequencing libraries were prepared using the G-INCPM in-house protocol for mRNA sequencing. Briefly, the poly-A fraction (mRNA) was purified from 80 to 280 ng of total RNA, followed by fragmentation and generation of double-stranded cDNA. Then, end repair, A-tailing, adapter ligation, and PCR amplification steps were performed. Libraries were evaluated using a Qubit (Thermo Fisher Scientific) and TapeStation (Agilent). Sequencing libraries were constructed with barcodes to allow for multiplexing of six libraries in a single lane. All six libraries were paired-end sequenced (2×126 bases) on an Illumina HiSeq 2500 platform in the G-INCPM. For each sample we obtained 35–42 million reads.
The GEO series record for the bulk RNA sequencing data is GSE147525.
Single-Cell RNA Sequencing Data Preprocessing and Gene Expression Analysis
Raw reads from 576 cells (6×96-well plates) were aligned by TopHat228 to the mouse mm10 genome. Aligned reads were counted by HTSeq.29 Data normalization and estimation of size factors was done by DESeq2,30 resulting in a matrix of normalized gene expression counts (Supplemental Table 1).
We then filtered out 11 cells that expressed zero levels of the housekeeping genes Gapdh or Actb, resulting in 565 cells. We chose highly variable genes using the method by Macosko et al.9 to select for genes whose variance exceeds those of other genes having a similar mean expression value (Figure S2A in Supplemental Appendix 1). This step resulted in 647 highly variable genes, to which we added a list of genes from the literature that were previously shown to be involved in kidney development (Supplemental Table 2). We also added an additional list of 48 genes from a previous single-cell quantitative PCR study that we previously conducted on human fetal kidney cells23 (Supplemental Table 2), which, in retrospect, were not crucial to the identification of the different cell populations. These steps resulted in a gene expression matrix of 677 genes × 565 cells. Each gene was then modified-log-transformed (log2[1+expression]) and standardized by subtracting the mean, dividing by the SD, and truncating to the range (−1,1).
We used tSNE31 to project the 565 single cells profiles into a two-dimensional plane (Figure S2E in Supplemental Appendix 1) and used genes that are known to mark different populations in the fetal kidney to identify the various populations, including a population of 21 low-quality cells that appears as a “mixture” of many cell types. This population of cells displayed low expression levels of the housekeeping genes Actb and Gapdh, as well as low DESeq size factors (Figure S3 in Supplemental Appendix 1). After removing these 21 low-quality cells, the process of selecting for highly variable genes (and adding known genes related to kidney development as described above) was then repeated for the remaining high-quality cells, resulting in a matrix of 728 genes × 544 cells, whose analysis is shown below. We note that once a sufficient number of highly variable genes are included, the ability to identify the different cell populations (Figure 2A) is not very sensitive to the exact choice of genes.
Figure 2.
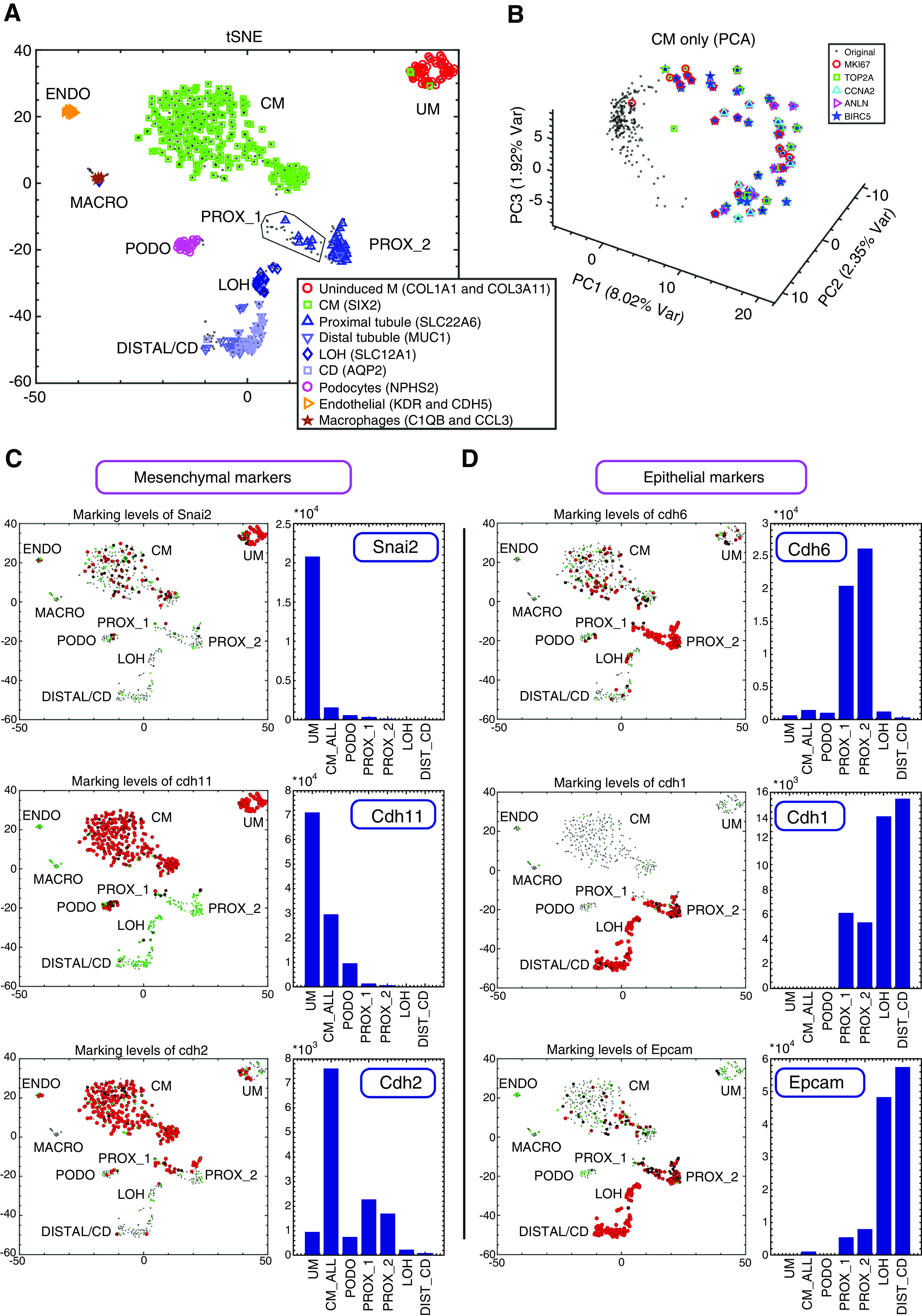
Single-cell gene expression analysis enables characterization of cellular heterogeneity, cell cycle dynamics, and the Mesenchymal to Epithelial Transition (MET) in the developing mouse fetal kidney. (A) Shown is a tSNE plot of 544 single-cell gene expression profiles, each consisting of 728 highly variable genes (see Methods). Each cell is represented by a dot. Cells overexpressing genes that were previously shown to mark different cell types are marked by additional symbols. (B) Cells of the CM create a circular manifold in gene expression space that corresponds to the cell cycle. Shown is a PCA figure of cells from the CM only. Each cell is represented by a dot. Cells overexpressing genes such as Top2a and Mki67 (genes that were shown to be overexpressed in the S-G2-M phases of the cell cycle) are marked by additional symbols. These cells are located in a specific segment of the circular manifold representing the S-G2-M segment of the cell cycle. (C and D) Expression levels of the mesenchyme related genes Snai2, Cdh11, and Cdh2 are typically higher in the UM and CM, whereas the epithelial genes Cdh6, Cdh1, and Epcam are typically higher in the PROX_1, PROX_2, LOH, and DIST_CD. Shown are tSNE plots and bar plots showing the expression levels of selected genes. The area of each circle in each tSNE plot is proportional to log2(1+expression) of the specific gene in that particular cell. The expression level in each cell is also encoded by the circle color (red, high expression; green, low expression). The bar plots show gene expression levels in bulk in silico cell transcriptomes representing the different populations (UM, CM, PODO, PROX_1, PROX_2, LOH, and DIST_CD) that were created by uniting raw reads from all cells belonging to each population. The annotations “CM_ALL” and “CM” are used interchangeably to represent all cells that were classified as belonging to the CM (see also Supplemental Appendix 1).
Creation of Bulk In Silico Transcriptomes Representing the Different Cell Populations
After identifying the different populations (Supplemental Table 3) according to known gene markers from the literature (e.g., Adam et al.6 and Magella et al.7), we created bulk in silico transcriptomes representing the different cell populations by merging reads (*.bam files) from all cells belonging to each population (using samtools merge). Again, aligned reads were counted by HTSeq29,32 and data normalization and estimation of size factors was done by DESeq2.30,33
Splicing Analysis: Identification of Splicing Events and Putative Splicing Regulators
rMATS34 was used to detect splice isoform switching events between the bulk in silico transcriptomes representing the different cell populations and to infer the inclusion levels of selected splicing events. In order to compare the mesenchymal populations (uninduced mesenchyme [UM] and CM) to the epithelial populations (early epithelial structures [PROX_1], proximal epithelial tubules [PROX_2], LOH, distal tubule and collecting duct [DIST_CD]), we performed the following comparisons: UM-PROX_1, UM-PROX_2, UM-LOH, UM-DIST_CD, CM-PROX_1, CM-PROX_2, CM-LOH, and CM-DIST_CD. Additionally, we compared the mesenchymal populations (UM and CM) to the PODOs: UM-PODO and CM-PODO (see Supplemental Tables 4–8). Selected splicing events (e.g., cassette exons) were visualized and validated using IGV35 and Sashimi plots,36 as well as with bar plots of inclusion levels inferred from rMATS34 or DEXSeq37 (see Supplemental Appendix 1).
Gene ontology enrichment of genes containing differential splicing events (Supplemental Table 9) was done with ToppGene (https://toppgene.cchmc.org).38 rMAPS (http://rmaps.cecsresearch.org/)39 was used to test for enrichment of binding motifs of RNA binding proteins in the vicinity of alternatively spliced cassette exons in order to identify putative splicing regulators. A list of 84 RNA binding proteins was obtained from the rMAPS website (http://rmaps.cecsresearch.org/Help/RNABindingProtein).39–41 Apart from the RNA binding motifs that are tested by the default settings in the rMATS website, we also tested additional UGG-enriched motifs that were previously found to be binding sites for the RNA binding proteins Esrp119,42 and Esrp243 (Supplemental Table 10). For the RNA binding proteins Rbfox1 and Rbfox2, following Yang et al.12 and the CISBP-RNA database40 (http://cisbp-rna.ccbr.utoronto.ca), we assumed that both proteins (Rbfox1 and Rbfox2) preferentially bind to the same motif ([AT]GCATG[AC]) on mRNA.
Results
Gene Expression Levels Were Used to Classify Each Cell into One of the Various Cell Types that Coexist within the Nephrogenic Zone of the Developing Mouse Fetal Kidney
We collected kidneys from transgenic mouse embryos that express GFP under the control of a Six promoter27 (Figure 1A). These mice have the advantage that cells from the CM—previously shown to express the transcription factor Six2—are fluorescently tagged and can be enriched by flow cytometry. The kidneys were collected at E18.5 of gestations because at this stage we expect to observe a still-active nephrogenic zone containing both early progenitor cell populations as well as fully developed nephrons.6,7 After kidney dissociation, we used flow cytometry to select 288 cells expressing high levels and 288 cells expressing low levels of the Six2-GFP transgene, and for each individual cell we performed full transcript length single-cell RNA sequencing using the Smartseq2 protocol.25,26 After discarding low-quality cells, this resulted in gene expression and sequence information for 544 individual cells, with approximately equal proportions of cells originating from the Six2-high and Six2-low fractions.
Using expression levels of selected genes from the literature that were previously shown to be specific to each cell population,24,44,45 genes from recent publications on single-cell profiling of the fetal kidney using the Drop-seq technology,6–8 as well as general epithelial markers and genes indicating cell division, we manually classified each cell into one of the major cell types that coexist in the nephrogenic zone of the developing kidney (Figures 1B and 2A, Figures S1 and S4–S9 in Supplemental Appendix 1). These include the un-induced mesenchyme (UM), the cap mesenchyme (CM), podocytes (PODO), early epithelial structures (PROX_1; presumably pretubular aggregates, renal vesicles, and C/S-shaped bodies), proximal epithelial tubules (PROX_2), the loop of Henle (LOH), distal tubule and collecting duct (DIST_CD), endothelial cells (ENDO), and infiltrating immune cells, mainly macrophages (MACRO). PROX_1 cells differ from PROX_2 cells in that they overexpress markers for early epithelial structures such as Mdk and Lhx1 (Figure S7 in Supplemental Appendix 1), as well as markers for actively dividing cells such as Mki67 and Top2a (Figure S10 in Supplemental Appendix 1). We note that we found it extremely difficult to distinguish between cells of the distal tubule and cells of the collecting duct in our data set, probably because of their transcriptional similarity as well as the relatively small number of cells in this experiment, and therefore we merged them into a single population.
After identifying the various cell subpopulations, we inspected the expression levels of the genes Mki67 and Top2a that are known to be overexpressed during cell division (Figure S10 in Supplemental Appendix 1). We found that the UM, CM, PROX_1, LOH, and DIST_CD each contain a substantial subset of dividing cells that overexpress these genes, whereas the PODOs and PROX_2 do not. Moreover, we found that the cells of the CM create a circular manifold (i.e., a high dimensional ring) in gene expression space, whose segments correspond to the different phases of the cell cycle (Figure 2B, Figure S11 in Supplemental Appendix 1). Using RNA velocity46—a computational tool for inferring a vector between the present and predicted future transcriptional state of each single cell by distinguishing between the spliced mRNA (present state) and yet-unspliced mRNA (future state)—we observed a consistent directional flow along this circular manifold (Figure S11 in Supplemental Appendix 1).
Next, we inspected the expression levels of genes that are known to be involved in the MET or that are known to be preferentially overexpressed in mesenchymal or epithelial lineages10,12,47 (Figure 2, C and D, Figures S12 and S13 in Supplemental Appendix 1). We observed high levels of mesenchyme-associated genes such as Snai2, Cdh11, and Cdh2 in the earlier developmental lineages: the UM and CM. Likewise, higher levels of epithelial genes such as Cdh6, Cdh1, and EpCAM were prevalent in the more differentiated lineages: the early epithelial structures (PROX_1), PROX_2, LOH, and DIST_CD. We noticed that the expression of Cdh11 showed a gradual decrease (Figure 2C, Figure S12 in Supplemental Appendix 1), with the highest expression levels being expressed in the UM, medium levels in the CM, lower levels in the PODOs, and very low levels in the epithelial lineages (PROX_1 and PROX_2). Likewise, when comparing Cdh6 and Cdh1 (Figure 2D, Figure S12 in Supplemental Appendix 1) we noticed that Cdh6 is higher in the early epithelial structures (PROX_1) and PROX_2, whereas Cdh1 is higher in the LOH and DIST_CD.47
rMATS Was Used to Characterize the Splice Isoform Switching Events that Occur during the Transition between the Mesenchymal and Epithelial Cellular States in the Course of Kidney Development
We next used the full transcript length sequence information to characterize the splice isoform switching that occurs during the MET in the course of kidney development. We first focused on identifying alternatively spliced exons (cassette/skipped exons) by searching for exons whose inclusion levels (defined as the fraction of transcripts that include the exon out of the total number of transcripts that either include the exon or skip over it) changes significantly between the mesenchymal and epithelial states. Because the coverage for each single cell was rather low for splicing analysis, we first merged the raw reads from all cells belonging to each population in order to create bulk in silico transcriptomes that represent each cell population, and then used the resulting bulk transcriptomes as input to rMATS34 (Supplemental Tables 4–8). We searched for cassette exons whose inclusion levels change significantly (FDR=0, difference in inclusion levels >0.2) between either of the mesenchymal populations, the UM or the CM, and all of the epithelial populations, PROX_1, PROX_2, LOH, and DIST_CD.
We found 57 cassette exons that were thus differentially expressed between the mesenchymal and epithelial lineages, out of which 55 could be validated by manual inspection in the IGV genome browser35 (Figure 3, A and C, selected examples in Figures S23–S41 in Supplemental Appendix 1, Supplemental Table 9). These exons include some known examples that were previously observed in EMT such as the epithelial-associated cassette exons in Map3k7,42,48,49 Dnm2,42,50,51 Pard3,50 and the mesenchymal-associated exons Plod2,12,18,52 Csnk1g3,18,49 and Ctnnd1.14,18 We next used ToppGene38 to perform gene ontology enrichment analysis (Figure 3B, Supplemental Table 9) and found that the genes containing these exons are related to epithelial characteristics (e.g., cadherin binding or cell junction) or mesenchymal characteristics (e.g., lamellipodium or cell leading edge, related to cellular motility). Moreover, the enrichment analysis pointed out that splicing in many of these genes is controlled by the splicing regulators Esrp1 and Esrp2, as was recently shown in developing mice.19
Figure 3.
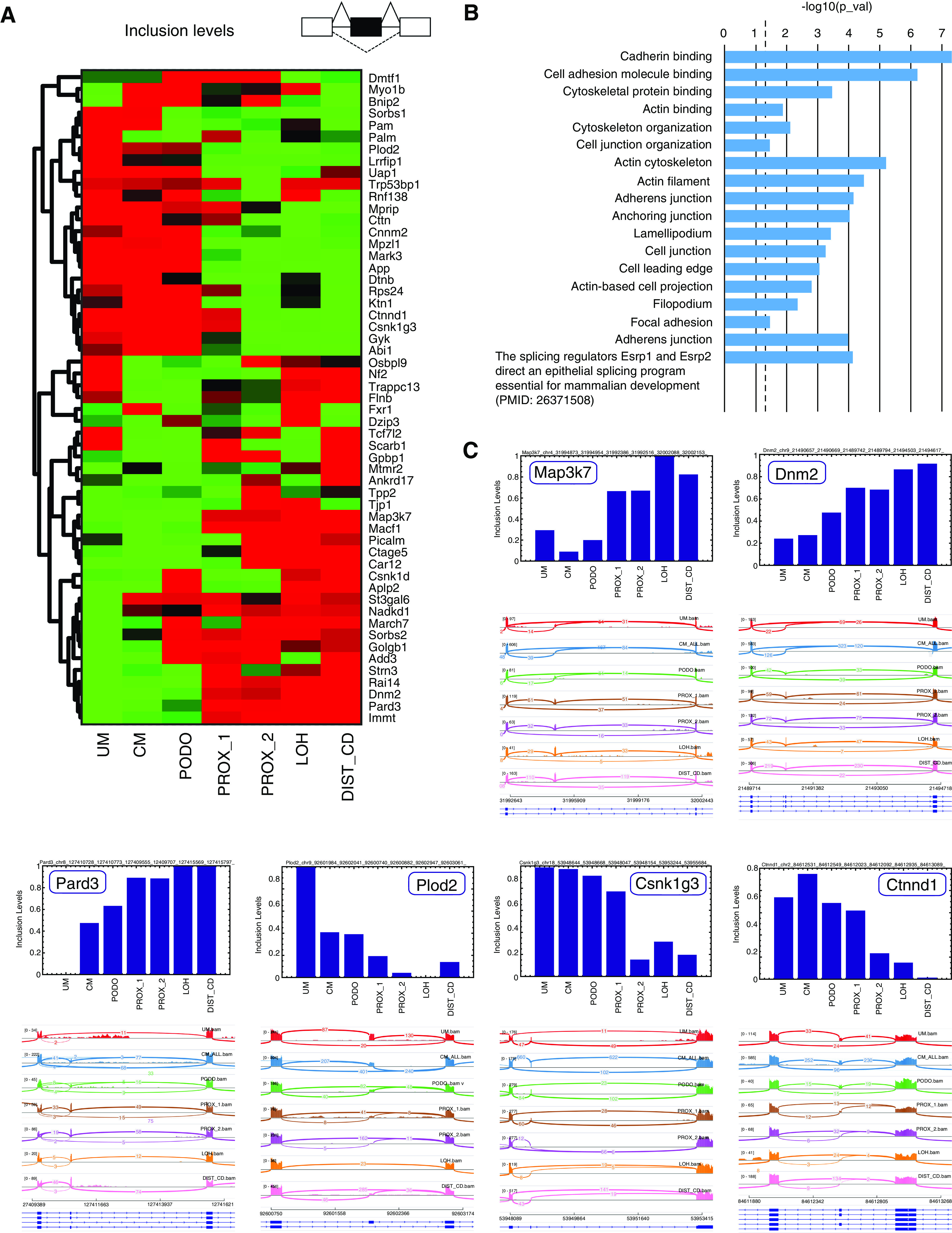
Characterization of splice isoform switching events that occur during the Mesenchymal to Epithelial transition (MET) in the course of kidney development. (A) Shown is a heatmap of inclusion levels of selected cassette exons that change significantly (FDR=0, difference in inclusion levels >0.2) between either of the mesenchymal populations (UM or CM) and all of the epithelial populations (PROX_1, PROX2, LOH, and DIST_CD). Exon inclusion levels were derived from in silico bulk transcriptomes representing the different cell populations (UM, CM, PODO, PROX_1, PROX_2, LOH, and DIST_CD) that were created by uniting raw reads from all cells belonging to each population. Colors indicate relative high (red) versus low (green) inclusion levels. The inclusion levels for each exon (row) were independently standardized by mean-centering and dividing by the SD. (B) Gene ontology (GO) enrichment analysis shows that the genes containing these differentially expressed cassette exons are related to structural and functional properties of epithelial cells (e.g., cadherin binding or cell junction) or to characteristics of mesenchymal cells (mainly cellular motility, e.g., lamellipodium and cell leading edge). (C) Sashimi plots and bar plots of inclusion levels in selected exons show gradual increase (Map3k7,42,48,49 Dnm2,42,50,51 and Pard350) or decrease (Plod2,12,18,52 Csnk1g3,18,49 and Ctnnd114,18) of inclusion levels during the transition from mesenchymal (UM and CM) to epithelial states (PROX_1, PROX_2, LOH, and DIST_CD; see also Supplemental Appendix 1).
Using similar analysis, we found additional MET-related alternative splicing events of types other than cassette/skipped exons (SE), although to a much smaller extent. These include mutually exclusive exons (MXE) such as Fgfr214,53 (Figure S15 in Supplemental Appendix 1), Actn1 (Figure S42 in Supplemental Appendix 1), and Tpm1 (Figure S43 in Supplemental Appendix 1); alternative 3′ splice sites (A3SS) such as Acsl4 (Figure S44 in Supplemental Appendix 1) and Bmp1 (Figure S45 in Supplemental Appendix 1); and alternative 5′ splice sites (A5SS) such as Polr2k (Figure S46 in Supplemental Appendix 1). We also found a retained intron (RI) in the gene Srsf1 (Figure S47 in Supplemental Appendix 1) that was expressed at higher levels in the PODOs with respect to other cell populations.
Supervised Analysis Identifies Additional Genes that Undergo Splice Isoform Switching during Kidney Development
Because the low coverage and bias of single-cell protocols limits the power of automated tools for discovering splice isoform switching, we searched the existing literature for additional genes for which different splice isoforms are expressed in mesenchymal versus epithelial cells or which are known to undergo splice isoform switching during EMT, and examined their alternative splicing between the different cell populations of the developing kidney. Indeed, we found that the genes Fgfr214,53 (Figure S15 in Supplemental Appendix 1, also found in our previous analysis), Epb41l517,51 (Figure S15 in Supplemental Appendix 1), Fat118,50 (Figure S16 in Supplemental Appendix 1), and Arhgef10l19,20,43 (Figure S17 in Supplemental Appendix 1) express their mesenchymal isoforms predominantly in the early mesenchymal populations (UM and CM), and their epithelial isoforms mostly in the more differentiated epithelial populations (LOH and DIST_CD). PROX_1 and PROX_2 express either the mesenchymal or the epithelial isoform or a mixture of both. Interestingly, we observed that the PODOs in many cases express the mesenchymal rather than the epithelial isoforms (Figure 3A, Figures S15–S17 in Supplemental Appendix 1). This reveals another aspect in which PODOs, which form a specialized type of epithelial tissue,5 are different from most other forms of epithelia.
The genes Enah and Cd44 are prominent examples of genes that undergo splice isoform switching during EMT.17,18 Both genes contain cassette exons that are expressed in epithelial cells only. However, this behavior was difficult to observe in our single-cell data set, probably because of the relative low coverage and bias of our single-cell protocol with respect to bulk RNA sequencing. We therefore performed additional bulk RNA sequencing on three replicates of sorted Six2-high and Six2-low cell fractions. Because Six2 is uniquely expressed in the CM, the cell fraction that was gated Six2-high is predominantly composed of cells originating from the CM, whereas the cell fraction gated for Six2-low contains a mixture of all of the other mesenchymal and epithelial populations. Nevertheless, by manually comparing Sashimi plots from these two cell fractions we were able to observe the alternatively spliced cassette exons in the genes Enah and Cd44 (Figure S18 in Supplemental Appendix 1).
A different form of alternative splicing, not related to EMT, was observed in gene Cldn10, which is an important component of epithelial tight junctions in the kidney and provides a barrier that permits selective paracellular transport.54 Two isoforms of the gene Cldn10 were previously found to coexist in the kidney, one being highly expressed in the cortex and the other in the medulla.55 These alternatively spliced isoforms are thought to generate different permselectivities in different segments of the nephron. In our data set we observed that the cortical isoform was indeed overexpressed in PROX_1 and PROX_2, which are located in the cortex, whereas the medullary isoform was overexpressed in the LOH, which is predominantly located in the medulla (Figure S19 in Supplemental Appendix 1). This confirmed previous in situ hybridization measurements55 also at the single-cell transcriptomic level.
We also observed two known splice isoform switching events in the gene Wt1, a central gene in kidney development4–8: cassette exon 5 is low in the mesenchymal populations and increases gradually during MET, and the KTS+:KTS− isoform ratio in exon 9 converges to 60:40 in the epithelial populations56–65 (see Figure S20 in Supplemental Appendix 1).
Differential Expression of RNA Binding Proteins and RNA Binding Motif Enrichment Analysis Suggest that Esrp1/2 and Rbfox1/2 Are Splicing Regulators of the MET that Occurs during Kidney Development
We next used rMAPS39 to identify putative RNA binding proteins that act as splicing regulators for splice isoform switching between the mesenchymal and epithelial states during renal development. We first compared the mean expression levels of 84 known RNA binding proteins39–41 (Supplemental Table 10) between the mesenchymal (UM, CM) and epithelial populations (PROX_1, PROX_2, LOH, DIST_CD) and found several putative splicing regulators that were differentially expressed (Figure 4A, Figure S21 in Supplemental Appendix 1).
Figure 4.
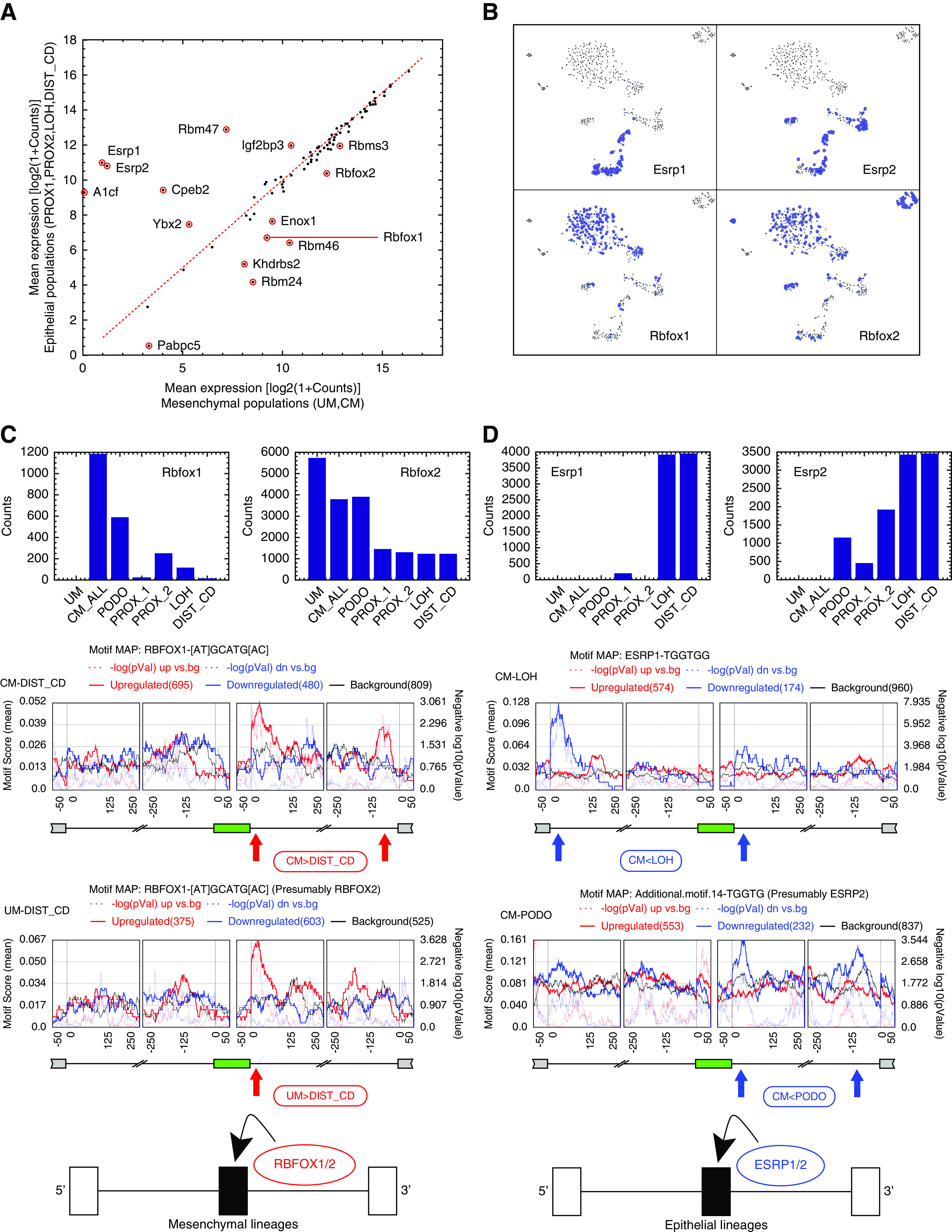
Differential expression of RNA binding proteins and RNA binding motif enrichment analysis suggest that Esrp1/2 and Rbfox1/2 are splicing regulators of the Mesenchymal to Epithelial transition (MET) that occurs during kidney development. (A) Shown is a comparison between mesenchymal and epithelial states of the mean expression levels of 84 RNA binding proteins (RBPs) that are known from the literature to regulate splicing through binding of mRNA transcripts. We used expression levels from bulk in silico transcriptomes that represent each cell population. (B) A tSNE plot of the single-cell profiles, highlighting cells that express the putative splicing regulators Rbfox1/2 and Esrp1/2. It can be seen that Rbfox1 and Rbfox2 are highly expressed in the mesenchymal populations, whereas Esrp1 and Esrp2 are highly expressed in the epithelial populations. The area of each circle is proportional to log2(1+expression) of each gene in that particular cell. (C and D) Cassette exons that are over-expressed in the mesenchymal populations contain a significant enrichment of Rbfox1/2 binding motifs at their downstream introns.12,40,66 Likewise, cassette exons that are over-expressed in the epithelial populations contain a significant enrichment of Esrp1/2 binding motifs in their downstream introns,19,42,43 and in some cases (CM versus LOH), also in the far 5′ end of their upstream introns (as previously observed in EMT42). This indicates that Rbfox1/2 and Esrp1/2 are splicing regulators12 involved in the MET that occurs during kidney development. The annotations “CM_ALL” and “CM” are used interchangeably to represent all cells that were classified as belonging to the CM (see also Methods and Supplemental Appendix 1).
Of these differentially expressed RNA binding proteins, we found Rbfox1 and Rbfox2 to be overexpressed in the mesenchymal cells, whereas Esrp1 and Esrp2 are overexpressed in the epithelial cells (Figure 4B). Likewise, we found that Rbfox1/2 and Esrp1/2 have RNA binding sites (motifs) that are enriched in the 5′ or 3′ neighboring introns of the cassette exons that are differentially expressed between mesenchymal and epithelial states (Figure 4, C and D, Figure S22 in Supplemental Appendix 1).12,19,42,43 This indicates that RBFOX1/2 and ESRP1/2 are splicing regulators involved in MET during kidney development, similar to what was previously observed in EMT12: in the mesenchymal cell populations Rbfox1 and/or Rbfox2 bind to mRNA in the downstream 3′-flanking introns of the mesenchymal-associated cassette exons and promote their inclusion,12,40,66 whereas in the epithelial cell populations Esrp1 and/or Esrp2 bind to the downstream 3′-flanking introns19,42,43 (and in some cases also to an additional site at the far 5′ end of the upstream flanking intron42) of the epithelial-associated cassette exons and promote their inclusion.
Discussion
In this study we used the Smartseq2 protocol25,26 for full transcript length single-cell RNA sequencing to characterize the splice isoform switching events that occur during the MET in the course of kidney development. Such splicing information is not obtainable using 3′-end digital counting protocols such as Drop-seq,6–9 apart from splicing events located at the very 3′ end of mRNA transcripts. These results highlight the importance of combining 3′-end digital counting technologies for transcriptional profiling of many thousands of cells, with full transcript length RNA sequencing for deeper analysis of selected cell populations, in order to obtain a detailed understanding of the molecular mechanisms involved in kidney development and disease.
Because the Smartseq2 protocol that we used is practically limited to a few hundreds of cells, we were unable to detect very small immune populations67 or to discern between all cell subtypes. For example, we were unable to discern subpopulations within the very early epithelial structures (PROX_1) or stromal subtypes within the UM.6 Nevertheless, the Smartseq2 protocol does have the advantage of being able to measure expression levels more precisely. For example, we were able to discern high, medium, and low expression levels of genes such as Cdh11 (Figure S12 in Supplemental Appendix 1) or Wt1 (Figure S20 in Supplemental Appendix 1).
Each cell in our analysis was sequenced at roughly 1–2 million reads per cell. Because splicing analysis typically requires roughly 20–40 million reads, we merged the raw reads from all cells belonging to each population in order to create bulk in silico transcriptomes that represent each cell population and then performed splicing analysis on these. We note, however, that splicing analysis can also be done for individual cells,68 but because of the small number of reads the inferred inclusion levels for most genes will be less accurate and have wide margins of error, except for the most highly expressed genes.
We note that the motif enrichment analysis for RNA binding proteins often gave nonspecific results, such as candidate splicing regulators that were not even expressed in some populations. We hypothesize that this stems from the fact that the binding motifs are not very specific because they are typically only a few bases long. We therefore based our identification of the splicing regulators Esrp1/2 and Rbfox1/2 on the existence of two additional criteria apart from RNA binding motif enrichment: first, the expression levels of Esrp1/2 and Rbfox1/2 differ significantly between the mesenchymal and epithelial cell states; and second, there is much previous evidence for similar functionality in other developing organs and in vitro systems.12,14,17,42,43,48,51 The marked differences in expression between mesenchymal and epithelial populations of other RNA binding proteins such as Cpeb2, Rbm47,12 Msi1, Rbms3, and others (Figure 4A, Figure S21 in Supplemental Appendix 1) indicate that they might also be involved in renal MET splicing regulation. However, we did not observe a consistent enrichment of known binding motifs for these genes as we did for Esrp1/2 and Rbfox1/2. Likewise, there may be additional splicing regulators whose expression does not change and whose RNA binding activity is modulated by protein modification. One example is QK (also known as QKI),12 but we did not find significant motif enrichment for this gene in the MET-associated differentially expressed cassette exons.
In a recent study it was shown that ablation of Esrp1 in mice, alone or together with Esrp2, resulted in reduced kidney size, fewer ureteric tips, reduced nephron numbers, and a global reduction of epithelial splice isoforms in the transcriptome of ureteric epithelial cells.69 We believe that our results provide a detailed picture at the single-cell level that complements the above study. Moreover, the fact that kidneys still develop under the ablation of Esrp1/2, taken with our results, suggests that there are multiple splicing regulators acting combinatorically thus creating a bypass mechanisms so that one splicing regulator compensates, although partially, for lack of another. Because it is infeasible to create transgenic mice containing knockouts of the many possible combinations of multiple regulators with current technology, we suggest using kidney organoids as a model system along with single-cell analysis for future functional studies.
Disclosures
T. Kalisky reports grants from the Israel Cancer Research Fund (ICRF), grants from The Israel Ministry of Science, and other funds from TaGeza Biopharmaceuticals (for consulting services provided), outside the submitted work. T. Kalisky has a patent application #US-20100255471, “Single cell gene expression for diagnosis, prognosis and identification of drug targets,” with Michael F. Clarke, Stephen R. Quake, Piero D. Dalerba, Huiping Liu, Anne A. Leyrat, and Maximilian Diehn with royalties paid to Stanford University; and a patent application #US-20130225435, “Methods and systems for analysis of single cells,” with Michael F. Clarke, Stephen R. Quake, Piero D. Dalerba, Huiping Liu, Anne A. Leyrat, Maximilian Diehn, Michael Rothenberg, Jianbin Wang, and Neethan Lobo with royalties paid to Stanford University. All remaining authors have nothing to disclose.
Funding
Y. Wineberg, T.H. Bar-Lev, N. Ben-Haim, S. Oriel, E. Bucris, Y. Yehuda, and T. Kalisky were supported by the Israel Science Foundation (Israeli Center of Research Excellence [I-CORE] no. 1902/12 and grants 1634/13 and 2017/13), the Israel Cancer Association (grant 20150911), the Israel Ministry of Health (grant 3-10146), and the European Union’s Seventh Framework Programme (Marie Curie International Reintegration grant 618592). N. Pode-Shakked and B. Dekel were supported by the Israel Science Foundation (grants 2071/17 and 910/11). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
Acknowledgments
We thank Nelly Komorovsky for assistance in fetal kidney collection. Likewise, we thank Jordan Kreidberg, Steve Potter, Oded Volovelsky, Morris Nehama, Tal Shay, Rotem Karni, and all members of our laboratories for useful comments and suggestions.
Dr. Benjamin Dekel, Dr. Achia Urbach, and Dr. Tomer Kalisky were responsible for study initiation and conception. Dr. Tali Hana Bar-Lev, Anna Futorian, Nissim Ben-Haim, Dr. Leah Armon, Dr. Efrat Bucris, Dr. Achia Urbach, and Dr. Tomer Kalisky were responsible for fetal kidney collection and dissociation. Dr. Debby Ickowicz was responsible for FACS sorting. Dr. Tali Hana Bar-Lev handled C1 experiments (data not used). Dr. Sarit Oriel handled Biomark experiments (data not used). Anna Futorian, Dr. Leah Armon, and Dr. Achia Urbach handled bulk RNA extraction. Dr. Tali Hana Bar-Lev handled bulk RNA quality checks. Dr. Shlomit Gilad and Dr. Sima Benjamin were responsible for Smartseq2 and RNA sequencing. Nissim Ben-Haim was responsible for bulk and single-cell RNA sequence preprocessing. Yishay Wineberg, Nissim Ben-Haim, and Dr. Tomer Kalisky were responsible for single-cell gene expression and splicing analysis. Dr. Yishai Yehuda, Dr. Naomi Pode-Shakked, and Dr. Peter Hohenstein made other intellectual contributions. Yishay Wineberg and Dr. Tomer Kalisky were responsible for manuscript writing.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Two Tales of Single-Cell RNA Sequencing: Gene Expression and Alternative Splicing in Mouse Kidney Development,” on pages 2234–2236.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019080770/-/DCSupplemental.
Supplemental Appendix 1. Supplemental text and figures.
Supplemental Table 1. Single-cell gene expression values (raw and normalized counts).
Supplemental Table 2. A list of genes from the literature that were previously shown to be involved in kidney development and an additional list of 48 genes from a previous single-cell quantitative PCR study that we previously conducted on human fetal kidney cells.23 We note that once a sufficient number of genes are included, the ability to identify the different populations (Figures 1B and 2A) is not very sensitive to the exact choice of genes.
Supplemental Table 3. Lists of cells in each population.
Supplemental Table 4. rMATS tables for cassette/skipped exons (SEs) that are alternatively spliced between the bulk in silico transcriptomes representing the different populations.
Supplemental Table 5. rMATS tables for mutually exclusive exons (MXEs) that are alternatively spliced between the bulk in silico transcriptomes representing the different populations.
Supplemental Table 6. rMATS tables for alternative 3′ splice sites (A3SS) that are alternatively spliced between the bulk in silico transcriptomes representing the different populations.
Supplemental Table 7. rMATS tables for alternative 5′ splice sites (A5SS) that are alternatively spliced between the bulk in silico transcriptomes representing the different populations.
Supplemental Table 8. rMATS tables for retained introns (RIs) that are alternatively spliced between the bulk in silico transcriptomes representing the different populations.
Supplemental Table 9. Gene ontology (GO) enrichment analysis results from ToppGene for genes containing alternatively spliced exons.
Supplemental Table 10. A list of RNA binding proteins and binding sites used for identifying putative splicing regulators.
Supplemental Table 11. DEXSeq counts. This table contains the number of reads that align to each exon within each bulk in silico transcriptome representing each cell population.
Program. A compressed directory containing a short MATLAB program and data sets for single-cell data visualization.
References
- 1.Hohenstein P, Pritchard-Jones K, Charlton J: The yin and yang of kidney development and Wilms’ tumors. Genes Dev 29: 467–482, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert SF: Intermediate mesoderm Developmental Biology, 6th Ed, Sunderland, MA, Sinauer Associates, 2000 [Google Scholar]
- 3.Little MH, McMahon AP: Mammalian kidney development: Principles, progress, and projections. Cold Spring Harb Perspect Biol 4: a008300, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Little M, Georgas K, Pennisi D, Wilkinson L: Kidney development: Two tales of tubulogenesis. In: Current Topics in Developmental Biology, edited by Chevalier RL, Thornhill BA, Amsterdam, The Netherlands, Elsevier, 2010, pp 193–229 [DOI] [PubMed] [Google Scholar]
- 5.Schell C, Wanner N, Huber TB: Glomerular development--shaping the multi-cellular filtration unit. Semin Cell Dev Biol 36: 39–49, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Adam M, Potter AS, Potter SS: Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: A molecular atlas of kidney development. Development 144: 3625–3632, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magella B, Adam M, Potter AS, Venkatasubramanian M, Chetal K, Hay SB, et al.: Cross-platform single cell analysis of kidney development shows stromal cells express Gdnf. Dev Biol 434: 36–47, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindström NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, et al.: Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J Am Soc Nephrol 29: 806–824, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al.: Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Weinberg RA: Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front Med 12: 361–373, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al.: Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21: 998–1009, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Park JW, Bebee TW, Warzecha CC, Guo Y, Shang X, et al.: Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial-to-mesenchymal transition. Mol Cell Biol 36: 1704–1719, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Modugno F, Iapicca P, Boudreau A, Mottolese M, Terrenato I, Perracchio L, et al.: Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc Natl Acad Sci U S A 109: 19280–19285, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP: ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591–601, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al.: CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest 121: 1064–1074, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sneath RJ, Mangham DC: The normal structure and function of CD44 and its role in neoplasia. Mol Pathol 51: 191–200, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warzecha CC, Shen S, Xing Y, Carstens RP: The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 6: 546–562, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al.: An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 7: e1002218, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, Rohacek AM, et al.: The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 4: 1–27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bangru S, Arif W, Seimetz J, Bhate A, Chen J, Rashan EH, et al.: Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat Struct Mol Biol 25: 928–939, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunskill EW, Park J-S, Chung E, Chen F, Magella B, Potter SS: Single cell dissection of early kidney development: Multilineage priming. Development 141: 3093–3101, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pode-Shakked N, Pleniceanu O, Gershon R, Shukrun R, Kanter I, Bucris E, et al.: Dissecting stages of human kidney development and tumorigenesis with surface markers affords simple prospective purification of nephron stem cells. Sci Rep 6: 23562, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pode-Shakked N, Gershon R, Tam G, Omer D, Gnatek Y, Kanter I, et al.: Evidence of in vitro preservation of human nephrogenesis at the single-cell level. Stem Cell Reports 9: 279–291, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, et al.: Atlas of gene expression in the developing kidney at microanatomic resolution [published correction appears in Dev Cell 16: 482, 2009]. Dev Cell 15: 781–791, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R: Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9: 171–181, 2014. [DOI] [PubMed] [Google Scholar]
- 26.Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R: Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10: 1096–1098, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, et al.: Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL: TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Pyl PT, Huber W: HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Der Maaten LJP, Hinton GE: Visualizing high-dimensional data using t-SNE. J Mach Learn Res 9: 2579–2605, 2008 [Google Scholar]
- 32.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, et al.: Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol 11: R106, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, et al.: rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111: E5593–E5601, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorvaldsdóttir H, Robinson JT, Mesirov JP: Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katz Y, Wang ET, Silterra J, Schwartz S, Wong B, Thorvaldsdóttir H, et al.: Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 31: 2400–2402, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anders S, Reyes A, Huber W: Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Bardes EE, Aronow BJ, Jegga AG: ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37: W305–W311, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park JW, Jung S, Rouchka EC, Tseng YT, Xing Y: rMAPS: RNA map analysis and plotting server for alternative exon regulation. Nucleic Acids Res 44: W333–W338, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, et al.: A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson ES, Lin CH, Xiao X, Stoilov P, Burge CB, Black DL: The cardiotonic steroid digitoxin regulates alternative splicing through depletion of the splicing factors SRSF3 and TRA2B. RNA 18: 1041–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, et al.: Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol 32: 1468–1482, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, et al.: ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun 6: 8768, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, et al.; GUDMAP project: GUDMAP: The genitourinary developmental molecular anatomy project. J Am Soc Nephrol 19: 667–671, 2008. [DOI] [PubMed] [Google Scholar]
- 45.Harding SD, Armit C, Armstrong J, Brennan J, Cheng Y, Haggarty B, et al.: The GUDMAP database--an online resource for genitourinary research. Development 138: 2845–2853, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al.: RNA velocity of single cells. Nature 560: 494–498, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho EA, Patterson LT, Brookhiser WT, Mah S, Kintner C, Dressler GR: Differential expression and function of cadherin-6 during renal epithelium development. Development 125: 803–812, 1998. 9449663 [Google Scholar]
- 48.Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al.: An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J 29: 3286–3300, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venables JP, Brosseau J-P, Gadea G, Klinck R, Prinos P, Beaulieu J-F, et al.: RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol 33: 396–405, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braeutigam C, Rago L, Rolke A, Waldmeier L, Christofori G, Winter J: The RNA-binding protein Rbfox2: An essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 33: 1082–1092, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Warzecha CC, Carstens RP: Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin Cancer Biol 22: 417–427, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeowell HN, Walker LC: Tissue specificity of a new splice form of the human lysyl hydroxylase 2 gene. Matrix Biol 18: 179–187, 1999. [DOI] [PubMed] [Google Scholar]
- 53.Hovhannisyan RH, Warzecha CC, Carstens RP: Characterization of sequences and mechanisms through which ISE/ISS-3 regulates FGFR2 splicing. Nucleic Acids Res 34: 373–385, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denker BM, Sabath E: The biology of epithelial cell tight junctions in the kidney. J Am Soc Nephrol 22: 622–625, 2011. [DOI] [PubMed] [Google Scholar]
- 55.Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM: Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol 291: F1288–F1299, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Hohenstein P, Hastie ND: The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet 15: R196–R201, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Ozdemir DD, Hohenstein P: Wt1 in the kidney--a tale in mouse models. Pediatr Nephrol 29: 687–693, 2014. [DOI] [PubMed] [Google Scholar]
- 58.Hastie ND: The genetics of Wilms’ tumor--a case of disrupted development. Annu Rev Genet 28: 523–558, 1994. [DOI] [PubMed] [Google Scholar]
- 59.Lefebvre J, Clarkson M, Massa F, Bradford ST, Charlet A, Buske F, et al.: Alternatively spliced isoforms of WT1 control podocyte-specific gene expression. Kidney Int 88: 321–331, 2015. [DOI] [PubMed] [Google Scholar]
- 60.Haber DA, Sohn RL, Buckler AJ, Pelletier J, Call KM, Housman DE: Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc Natl Acad Sci U S A 88: 9618–9622, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laity JH, Dyson HJ, Wright PE: Molecular basis for modulation of biological function by alternate splicing of the Wilms’ tumor suppressor protein. Proc Natl Acad Sci U S A 97: 11932–11935, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsson SH, Charlieu JP, Miyagawa K, Engelkamp D, Rassoulzadegan M, Ross A, et al.: Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell 81: 391–401, 1995. [DOI] [PubMed] [Google Scholar]
- 63.Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F, Kuttenn F, et al.: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17: 467–470, 1997. [DOI] [PubMed] [Google Scholar]
- 64.Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, et al.: Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 106: 319–329, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Menke AL, Schedl A: WT1 and glomerular function. Semin Cell Dev Biol 14: 233–240, 2003. [DOI] [PubMed] [Google Scholar]
- 66.Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, et al.: A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J 22: 905–912, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al.: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al.: Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498: 236–240, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bebee TW, Sims-Lucas S, Park JW, Bushnell D, Cieply B, Xing Y, et al.: Ablation of the epithelial-specific splicing factor Esrp1 results in ureteric branching defects and reduced nephron number. Dev Dyn 245: 991–1000, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.