

Abstract
The design of peptides that assemble in membranes to form functional ion channels is challenging. Specifically, hydrophobic interactions must be designed between the peptides and at the peptide–lipid interfaces simultaneously. Here, we take a multi-step approach towards this problem. First, we use rational de novo design to generate water-soluble α-helical barrels with polar interiors, and confirm their structures using high-resolution X-ray crystallography. These α-helical barrels have water-filled lumens like those of transmembrane channels. Next, we modify the sequences to facilitate their insertion into lipid bilayers. Single-channel electrical recordings and fluorescent imaging of the peptides in membranes show monodisperse, cation-selective channels of unitary conductance. Surprisingly, however, an X-ray structure solved from the lipidic cubic phase for one peptide reveals an alternative state with tightly packed helices and a constricted channel. To reconcile these observations, we perform computational analyses to compare the properties of possible different states of the peptide.
The de novo design of water-soluble proteins has progressed rapidly1,2. In comparison, membrane protein design is less developed. Indeed, relatively few de novo membrane proteins have been defined in structural detail: helical bundles have been specified by computationally designed hydrogen-bond networks3 or side-chain packing4, and a Zn2+/H+ antiporter has been achieved5. Moreover, many natural membrane proteins respond to chemical and physical cues; for example, G-protein coupled receptors, ligand- and voltage-gated ion channels, and water-soluble toxins that form membrane-spanning pores. The de novo design of such multi-state systems is particularly challenging6,7.
The self-assembly of de novo α-helical peptides presents an attractive route to address the challenge of designing transmembrane proteins. This is because established structural principles for α-helical folding and assembly provide starting points for design2,8. Nonetheless, there have been relatively few successful de novo designs of functional transmembrane peptides such as ion channels9,10. Advancing the design field to deliver such targets requires solutions to three interlinked design challenges: first, helix–helix interactions must be specified to maintain discrete oligomers; second, this has to be done in competition with interactions needed to interface the structures with lipids; third, the assemblies must have accessible, water-filled lumens lined with polar residues, which may compromise helix–helix packing and structural stability. Moreover, a particular challenge is that this all has to be encoded within approximately 30-residue peptides, the length required for an α helix to span a lipid bilayer11,12.
Here, we combine rational and computational design to produce peptides that self-assemble into water-soluble α-helical barrels (αHBs). These have polar lumens wetted by dynamic networks of water molecules. With insights from these structures, we introduce outwardly projecting hydrophobic residues to generate membrane-spanning peptides. These form well-defined ion channels that conduct approximately 108 ions per second. These are examples of modifying water-soluble, self-assembling de novo peptides to facilitate membrane insertion. Moreover, in membranes, the peptides form channels with lifetimes that are unusual for designed systems, allowing their dynamics to be imaged directly. We present a crystal structure for one peptide assembly from the lipidic cubic phase (LCP). This reveals an unanticipated alternate state with tightly packed helices. This is almost certainly a non-conducting state. To reconcile these data, we use computational analyses to develop a model for the peptide’s structure and activity. We posit that the multiple states observed can be understood in terms of the relative energetics of helix-packing interactions in water-soluble and membrane proteins, and possibly the influence of vectorial insertion of peptides into membranes. This study highlights the challenges of designing discrete membrane-spanning ion channels from self-assembling peptides, which we discuss and place in a wider context.
Results and discussion
Water-soluble α-helical barrels with solvated channels can be designed.
To start the design process, we targeted αHB coiled-coil peptides. These have ‘Type-2’ heptad sequence repeats of hydrophobic (h) and polar (p) residues, hpphhph (labelled abcdefg), which assemble into bundles of five or more α helices13,14. Previously, we have developed design principles and methods for water-soluble and membrane-spanning αHBs4,15. These exploit tight inter-helical packing of mostly hydrophobic residues to specify the association states and internal radii of the assemblies. This packing stabilizes the structures, but it precludes their use as ion channels. Therefore, we sought to place polar residues capable of favourable interactions with water into the lumens of αHBs.
We took a canonical water-soluble αHB (CC-Hept (ref. 15), systematically named CC-Type2-(LaId)4), with the following features (Fig. 1a and Table 1): hydrophobic Leu at position ‘a’ and Ile at position ‘d’ point into the lumen but also stabilize interactions between neighbouring helices; small hydrophobic Ala residues at ‘e’ and ‘g’ positions supplement the helical interfaces to specify high-order bundles; and interactions between charged Lys and Glu side chains at ‘b’ and ‘c’ positions contribute further to the interfaces and provide water solubility. We explored placing neutral polar side chains at the lumenal ‘a’ and ‘d’ sites in this background (Fig. 1, Table 1 and Supplementary Table 1).
Fig. 1 |. Computational design and characterization of water-soluble αHBs with solvated lumens.
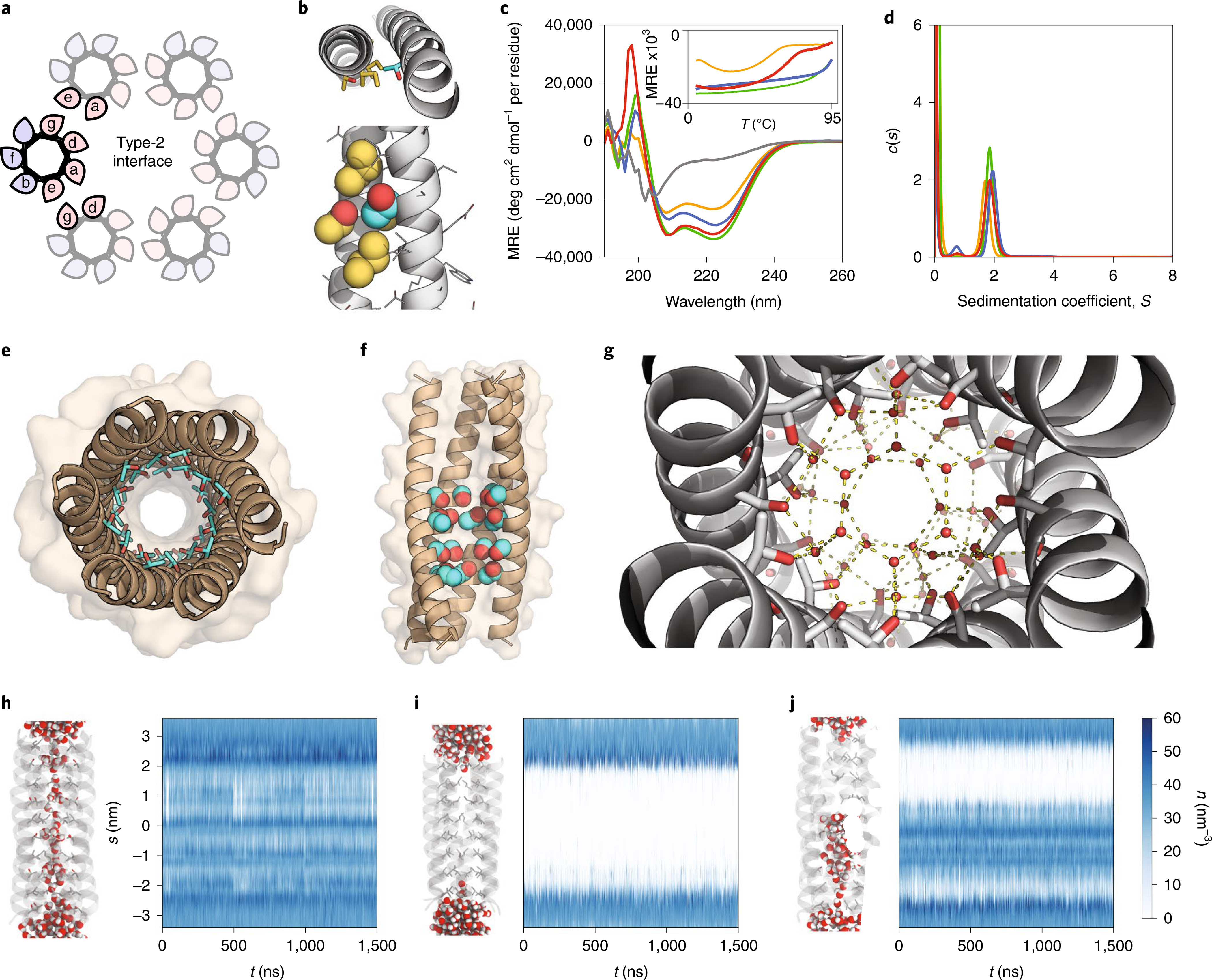
a, Helical wheels for parallel Type-2 coiled-coil interfaces with hydrophobic and polar residues shaded pink and blue, respectively. b, CCBuilder 2.0 model of CC-Type2-(LaId)4 with Thr residues at one ‘a’ site. Backbone parameters were from CC-Type2-(LaId)4 (PDB code: 4PNA). Knob and holes residues are coloured cyan and gold, respectively. c,d, CD spectra at 20 °C (c) with thermal denaturation profiles (inset) and sedimentation velocity AUC c(s) distributions (d) for CC-Type2-(TaId)5 (orange), CC-Type2-(LaTd)5 (green), CC-Type2-(SaId)5 (grey), CC-Type2-(LaSd)5 (blue) and CC-Type2-(TaSd)2 (red). MRE, mean residue ellipticity. e, X-ray crystal structure (1.9 Å resolution) of CC-Type2-(TaId)5 with internal Thr side chains (cyan) shown as sticks with hydroxyl groups (red) highlighted. f, X-ray crystal structure (1.9 Å resolution) of CC-Type2-(TaSd)2 with Thr and Ser side chains space-filled, and front two helices removed. g, Hydrogen-bonding water (red spheres) network in the lumen of the crystal structure of CC-Type2-(TaSd)2; hydrogen bonds (yellow dashes) are for O–O distances of <3 Å. h–j, MD simulations of water ingress into the cavities of CC-Type2-(TaId)5 (h), CC-Type2-(LaIdSg)4 (i) and CC-Type2-(TaSd)2 (j). Left-hand images: representative snapshots after equilibration, with water molecules space-filled and the N termini of the helices at the top. Plots: water number density (n) profiles for the channels generated using the CHAP computational tool. Colour scale applies to h–j.
Table 1 |.
Selected de novo designed peptide sequences
| Name | Sequence | Helical | Binds DPH | Oligomer | PDB code | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Water-soluble | cdefgab | cdefgab | cdefgab | cdefgab | cdefgab | ||||||
| CC-Type2-(LaId)4 | G | EIAKALK | EIAKALK | EIAWALK | EIAKALK | G | Yes | Yes | 7 | 4PNA | |
| CC-Type2-(LaIdSg)4 | G | EIAKSLK | EIAKSLK | EIAWSLK | EIAKSLK | G | Yes | Yes | 6 | 4PN9 | |
| CC-Type2-(TaId)2 | G | EIAQALK | EIAKATK | EIAWATK | EIAQALK | G | Yes | ND | 6 | 6YB2 | |
| CC-Type2-(TaId)5 | G | EIAQATK | EIAQATK | EIAKATK | EIAWATK | EIAQATK | G | Yes | Yes | 6 | 6YAZ |
| CC-Type2-(LaTd)5 | G | ETAQALK | ETAQALK | ETAKALK | ETAWALK | ETAQALK | G | Yes | No | 5–6 | ND |
| CC-Type2-(SaId)5 | G | EIAQASK | EIAQASK | EIAKASK | EIAWASK | EIAQASK | G | No | ND | ND | ND |
| CC-Type2-(LaSd)5 | G | ESAQALK | ESAQALK | ESAKALK | ESAWALK | ESAQALK | G | Yes | No | 5–6 | ND |
| CC-Type2-(TaSd)2 | G | EIAQALK | EIAQALK | ESAKATK | ESAWATK | EIAQALK | G | Yes | Yes | 6 | 6YB0 |
| Membrane-soluble | cdefgab | cdefgab | cdefgab | cdefgab |
Positions ‘a’ and ‘d’ line channel/pack core. Positions ‘e’ and ‘g’ are interfacial (interior) and help specify the packing of hexamers. Positions ‘b’ and ‘c’ are interfacial (exterior) and have a minor role in defining packing. Positions ‘b’, ‘c’ and ‘f’ are exterior facing and are thus polar or apolar in water-soluble or membrane-soluble designs, respectively. |
||||||
| CCTM-LbLc | KKKKGSG | ISAWATL | LSALATL | LSALATL | LSAWATL | G | |||||
| CCTM-VbIc | KKKKGSG | ISAWATV | ISALATV | ISALATV | ISAWATV | G | |||||
| K2-CCTM-VbIc | GKK | SAWATV | ISALATV | ISALATV | ISAWATV | G | |||||
| CCTM-IbIc | KKKKGSG | ISAWATI | ISALATI | ISALATI | ISAWATI | G | |||||
| CCTM-IbLc | KKKKGSG | ISAWATL | ISALATL | ISALATL | ISAWATL | G | |||||
Polar core residues are highlighted in bold. All peptides had N-terminal acetyl and C-terminal amide caps. Green shading indicates a membrane-spanning region. See Supplementary Table 1 for a complete list of peptides used in this study. CC-Type2-(LaId)4 and CC-Type2-(LaIdSg)4 are described previously as CC-Hept and CC-Hex2. ND, not determined.
Because β-branched side chains at ‘a’ and ‘d’ positions help maintain open barrels16, first we explored β-branched and polar Thr residues at these sites. Computational design17 indicated that Thr at an ‘a’ position was compatible with the knobs-into-holes packing of coiled coils18 and that the β-hydroxyl group projected into the lumen (Fig. 1b), whereas Thr at ‘d’ could not assume this arrangement. To test this, we synthesized two peptides with Thr at five consecutive ‘a’ or ‘d’ positions, CC-Type2-(TaId)5 and CC-Type2-(LaTd)5 (Table 1 and Supplementary Figs. 2 and 3). We examined their folding and assembly by far-ultraviolet circular dichroism (CD) spectroscopy and analytical ultracentrifugation (AUC); and we probed the binding of the environment-sensitive dye 1,6-diphenyl-1,3,5-hexatriene (DPH), which binds to open barrels but not to collapsed bundles16. Both peptides formed helical oligomers (Fig. 1c,d, Table 1 and Supplementary Figs. 19, 20, 32 and 33), but only CC-Type2-(TaId)5 bound DPH (Supplementary Fig. 38).
We determined a 1.9-Å-resolution X-ray crystal structure for CC-Type2-(TaId)5 (Fig. 1e and Supplementary Table 2), and another for the related CC-Type2-(TaId)2 to 1.2 Å. These revealed parallel hexameric αHBs with internal Thr and Ile side chains in alternating layers. The hydroxyl groups of Thr stabilize small clusters of water molecules (Supplementary Fig. 40). CC-Type2-(TaId)5 is a coiled coil with just one large hydrophobic residue per heptad repeat, and with an associated internal water network.
We explored the stability of this water using all-atom molecular dynamics (MD) simulations, analysed with the CHAP computational tool19, starting from a fully hydrated channel (Fig. 1h–j). The channel remained solvated through 1.5 μs of simulations (Fig. 1h). Moreover, water molecules exchanged freely between the channel and bulk solvent (Supplementary Figs. 41–51 and Supplementary Video 1). By comparison, in similar simulations for a hexamer with all Leu at ‘a’ positions and all Ile at ‘d’ positions (CC-Hex2, CC-Type2-(LaIdSg)4 (ref. 15)) the introduced water was immediately expelled from the channel, leaving it predominantly dry (Fig. 1i, Supplementary Figs. 41–51 and Supplementary Video 2).
Despite the mobility of water in the channel, we anticipated that the Ile layers of CC-Type2-(TaId)5 might present barriers to the passage of hydrated ions.20 Therefore, we tested other small polar residues at the lumenal sites (Supplementary Table 1); for example, Ser at ‘a’ and ‘d’ to give CC-Type2-(SaId)5 and CC-Type2-(LaSd)5 (Table 1). The former was unfolded. The latter assembled into a helical oligomer (Fig. 1c,d, Table 1 and Supplementary Figs. 22 and 34). Thus, Ser residues appear better accommodated at ‘d’ sites. We could not crystallize CC-Type2-(LaSd)5. Next, we made peptides with Thr and Ser at consecutive ‘a’ at ‘d’ sites. A peptide with two such heptads braced by Leu/Ile-based heptads, CC-Type2-(TaSd)2, formed stable helical hexamers in solution (Fig. 1c,d and Table 1). This crystallized as a hexameric αHB (Fig. 1f) with a large cavity (diameter ≈ 7 Å, length ≈ 20 Å) occupied by an unusually high number (~38) of ordered water molecules21. To our knowledge, this is the largest such cavity built into a de novo protein. The internal water molecules formed an intricate hydrogen-bonded network involving hydroxyl groups of the Ser and Thr side chains and lumen-facing backbone carbonyl groups (Fig. 1g). In MD simulations the hydrated channel was stable, and water freely exchanged in and out of it (Fig. 1j, Supplementary Figs. 41–51 Supplementary Video 3).
The water-soluble barrels can be converted into membrane-spanning assemblies.
To convert these water-soluble αHBs into transmembrane ion channels, we extended the Thr/Ser-based lumen of CC-Type2-(TaSd)2 over a length required to span a membrane and increased the hydrophobicity of the exterior residues. We reasoned that multiple Thr/Ser at the ‘a’/’d’ positions might give stable membrane-spanning barrels because these residues promote helix–helix interactions in membranes22. In the resulting ‘CCTM’ designs, the Ala residues were retained at the ‘e’ and ‘g’ positions, and the exterior ‘f’ positions were made Trp or Leu to match preferences for locating to headgroup and hydrocarbon regions of lipid bilayers, respectively23 (Fig. 2a). Initially, the ‘b’ and ‘c’ positions were made Leu. This peptide, CCTM-LbLc (Table 1), had an N-terminal tetra-Lys tag and a C-terminal hydrophobic residue to promote C-terminal insertion into the planar lipid bilayers (PLBs)10. Encouragingly, CCTM-LbLc was α helical in n-dodecyl β-d-maltoside (DDM) solutions (Fig. 2b and Supplementary Fig. 26).
Fig. 2 |. Engineering and characterization of transmembrane ion-channel peptides.
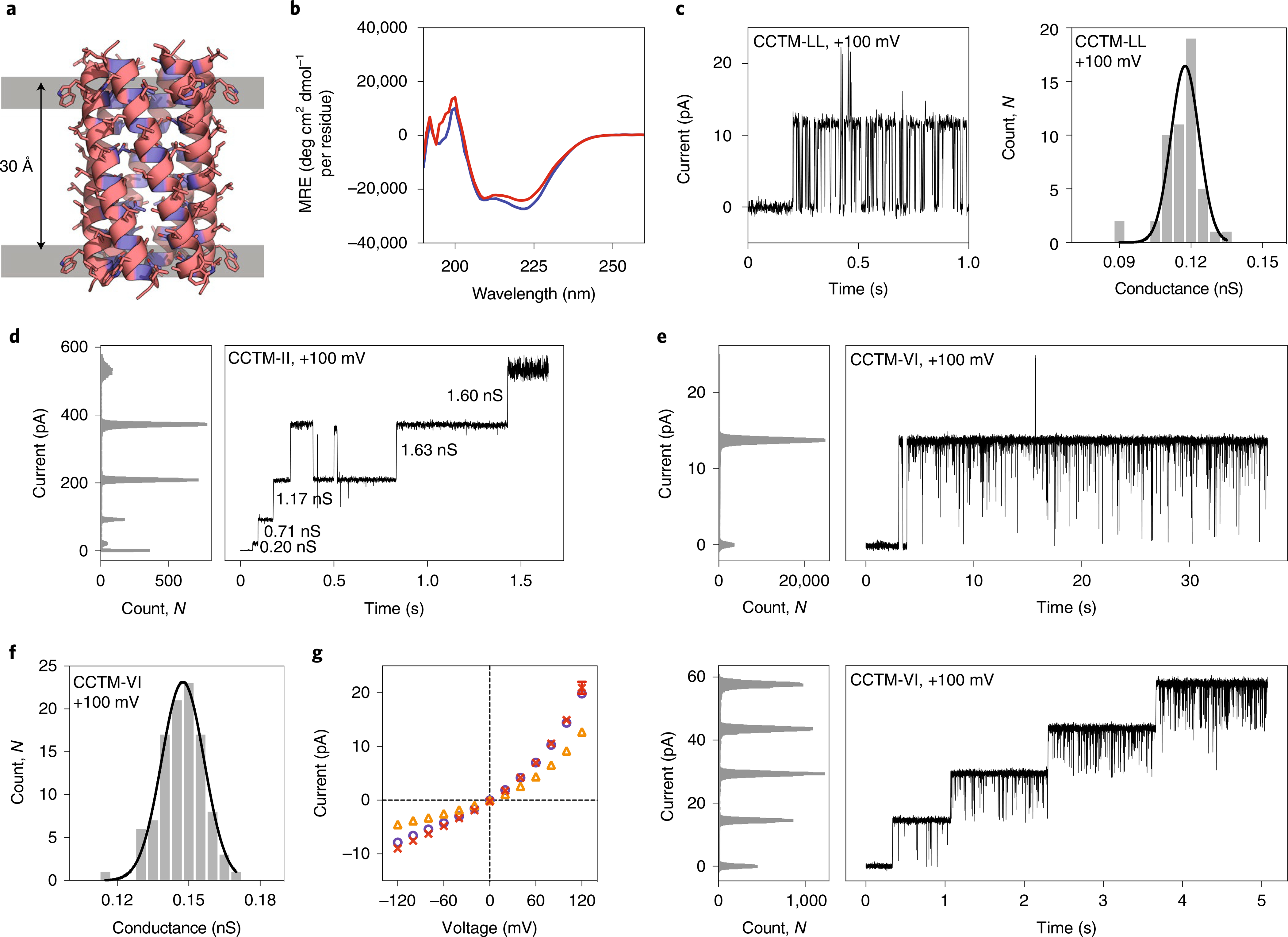
a, CC-Builder2 model of CCTM-LbLc with hydrophobic (Trp, Leu, Ala) and polar (Thr, Ser) residues coloured pink and blue, respectively. The Trp–Trp Cα distance is shown. b, CD spectra in 0.05% DDM at 20 °C for CCTM-LbLc (blue) and CCTM-VbIc (red). c, Insertion of CCTM-LbLc into a DPhPC PLB (left) and frequency distributions (right) with fitted Gaussian for this conductance (N = 51, μ = 0.12 nS, σ = 0.01 nS) at +100 mV potential in 1 M KCl. d, CCTM-IbIc channels recorded at +100 mV in 1 M KCl, with the conductance values of different events labelled. e, Single (top) and quadruple insertions (bottom) of CCTM-VbIc at +100 mV. f, Frequency distribution histogram for CCTM-VbIc channels (N = 104, μ = 0.15 nS, σ = 0.01 nS) at +100 mV in 1 M KCl. g, Current–voltage curves for single CCTM-VbIc channels. Electrolyte: 1 M each of KCl (purple), NaCl (orange) or CsCl (red). Buffer: 10 mM Tris-HCl, pH 8.0. For all experiments, peptide concentrations in the cis compartment were 50 nM, and signals were acquired at 10 kHz and low-pass filtered at 2 kHz.
To test for functional ion channels, we used single-channel electrical recordings across 1,2-diphytanoyl-sn-glycero-3-p hosphocholine (DPhPC) PLBs at +100 mV in 1 M KCl. CCTM-LbLc was added from micellar solutions to the cis (ground) side of the bilayer. This diluted the solutions below their critical micelle concentration. The formation of individual channels was evident from uniform steps in ionic current of ~12 pA (Fig. 2c, left), corresponding to ~7.5 × 107 ions per second. Based on this, conductance histograms (Fig. 2c, right) confirmed homogeneous channels with a unitary conductance of μ ≈ 0.12 nS.
Using a computational coiled-coil design method15, we explored the effect on ion-channel properties of placing different hydrophobic residues at ‘b’ and ‘c’ positions to optimize helix–helix packing4. The top-scoring sequences had Val/Ile, Ile/Ile and Leu/Ile combinations at the ‘b’/’c’ positions (Supplementary Table 3). In single-channel PLB experiments, the peptides with Ile/Ile and Leu/Ile at ‘b’/’c’ formed channels that no longer had a single conductance but showed increasingly large steps in current (Fig. 2d). This is reminiscent of alamethicin pores, which increase oligomeric state by sequential additions of peptide24. By contrast, channels formed by CCTM-VbIc (with Val/Ile at ‘b’/c’; Fig. 2e,f) were monodisperse, longer-lived than for CCTM-LbLc or foregoing de novo peptide channels9, and exhibited less gating (Fig. 2 and Supplementary Fig. 52).
All of the CCTM peptides were α helical in DDM solutions (Supplementary Figs. 26–30). In AUC experiments, the two variants of CCTM-VbIc (Table 1) clearly associated in detergent solutions (Supplementary Figs. 36 and 37). However, the sizes of the oligomers formed depended on the variant and the detergent used. In our understanding, AUC of pore-forming peptides with association states larger than four or five is notoriously difficult, and there are no reports of such experiments that reveal the assembly of an active barrel-stave-pore state.
CCTM-VbIc forms antiparallel helical bundles in the lipidic cubic phase.
Despite the difficulties encountered with AUC, having derived a CCTM design with favourable ion-channel properties we sought to solve its structure. A 2.1 Å resolution X-ray structure was determined for K2-CCTM-VbIc (Table 1) crystallized from the LCP25. This confirmed a bundle of amphipathic α-helices with Type-2 knobs-into-holes packing involving most ‘a’, ‘d’, ‘e’ and ‘g’ positions (Fig. 3a,b and Supplementary Table 4). Surprisingly, however, the structure had two antiparallel four-helix bundles associated laterally to form an octamer (Fig. 3a,b). While at odds with our target of a parallel αHB, we sought to reconcile this observation with sequence features of the CCTM-VbIc.
Fig. 3 |. LCP crystal structure of K2-CCTM-VbIc and modelling of alternate states.
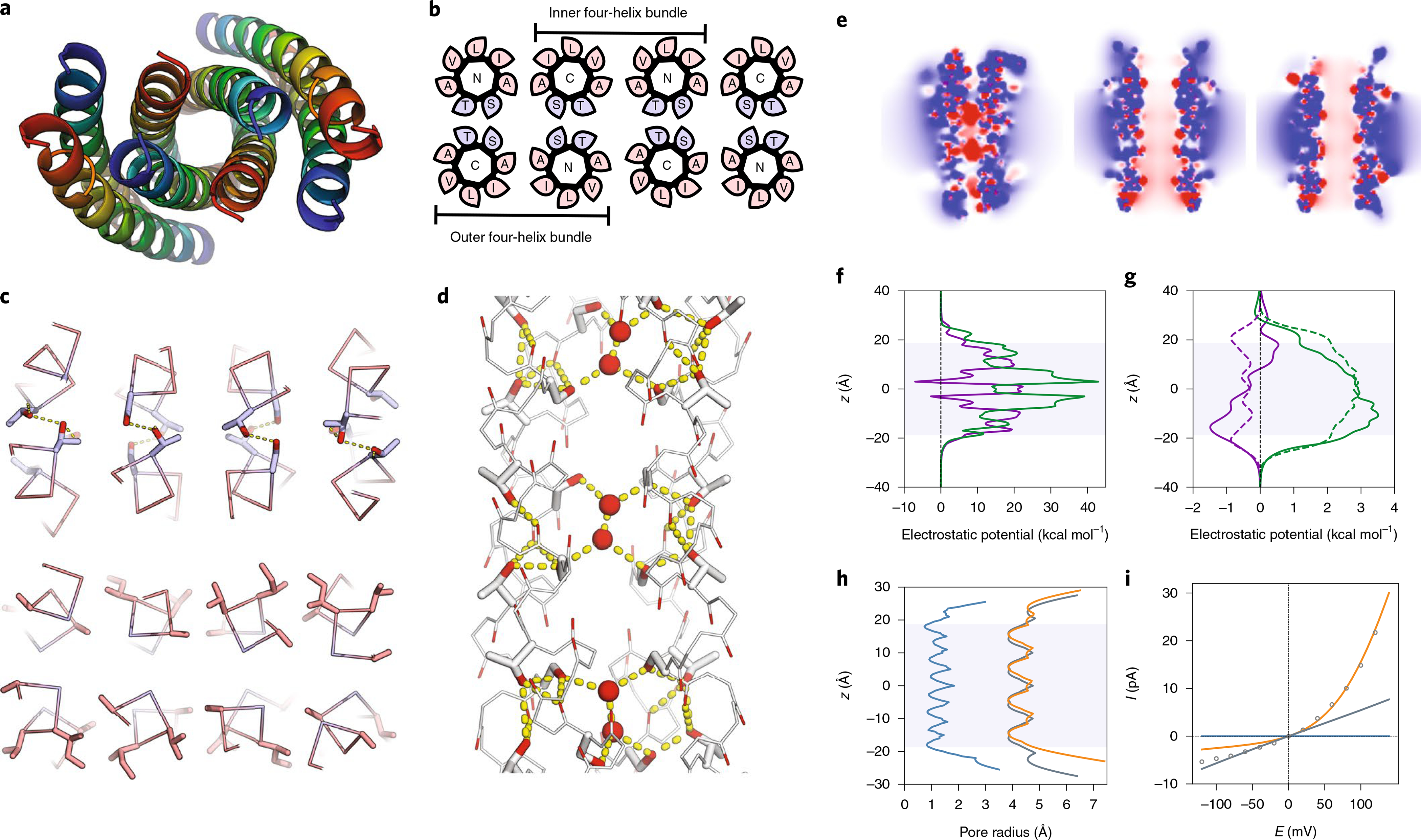
This structure is of a variant with a Lys2 rather than a Lys4 N-terminal tag; both peptides had similar biophysical and conductance properties (Supplementary Fig. 53). a, Axial view of the octameric assembly. N to C termini are coloured from blue to red. b, Corresponding helical wheels showing two distinct four-helix bundles. The inner bundle has a hydrophobic interior of methyl groups from Ala and Thr; whereas, the outer bundles are hydrophilic, lined with hydroxyl groups of Ser and Thr. Blue and pink represent polar and apolar residues, respectively. c, A hydrogen-bonded layer of Thr/Ser (blue) at ‘a’/’d’ (top) and the same layer showing the hydrophobic interfaces with Ala at ‘e’ and ‘g’, and Val/Ile (pink) at ‘b’/’c’ (bottom). Hydroxyl groups are shaded red. d, Hydrogen-bonding network within the outer bundle involving isolated water molecules (red spheres), Thr/Ser side chains and backbone carbonyls. Hydrogen bonds (yellow dashes) are for O–O distances of <3 Å. e, Slices through the isosurfaces, in the absence of an applied potential, of the outer four-helix bundle of the experimental octamer (left) and for models of a parallel hexamer (centre) and an antiparallel hexamer (right) of K2-CCTM-VbIc. These are coloured from red to blue for −8 kBT/ec to 8kBT/ec, respectively, and indicate internal negative electrostatic potentials consistent with cation selectivity. kB, Boltzmann constant; T, temperature; ec, charge of an electron. f,g, Calculated electrostatic potential energies for moving K+ (purple) and Cl− (green) through the channels of the outer four-helix bundle (f) and the hexameric models (g; parallel, solid lines; antiparallel, dashed lines). h, Calculated channel radii for the outer four-helix bundle of the octamer (blue) and the hexameric models (parallel, orange; antiparallel, grey). i, Calculated I–V curves for the four-helix bundle (blue) and the hexamers (parallel, orange; antiparallel, grey), with the experimental data shown as points.
The CCTM-VbIc sequence has small Ala and Ser residues each spaced seven residues apart. These are the hallmarks of Ala and Ser zippers, which have tightly packed antiparallel helices26–28. The observed structure appears to maximise the number of these zippers (Fig. 3c), and it allows the Ser and Thr residues to form an extensive hydrogen-bonded network. However, this leaves space for only a few isolated pairs of water molecules (Fig. 3d). The lack of a more substantial pore hints that this conformation is not the active ion-conducting state of CCTM-VbIc (see below), but an alternative state accessible on the free-energy landscape for the peptide sequence. Similar antiparallel structures are known. For example, the water-soluble peptide CC-Hex-L24E crystallizes as either a parallel six-helix αHB or an antiparallel tetramer, depending on the conditions29. Hence, both structures are accessible to CCTM-VbIc, and likely close in energy. In addition, the few available X-ray structures of natural membrane-spanning pore-forming α-helical peptides have antiparallel helices30–32, though these are not necessarily the active states33.
CCTM-VbIc forms well-defined ion channels in planar-lipid membranes.
The apparent contradiction between the crystal structure of K2-CCTM-VbIc and its channel properties in lipid bilayers led us to make further electrical recordings.
Single CCTM-VbIc channels had a unitary conductance, μ, of 0.15 ± 0.01 nS (+100 mV, 1 M KCl, Fig. 2f), corresponding to ~9 × 107 ions per second, a high conductance for synthetic peptide channels9. This is consistent with a channel diameter of ~7 Å (ref. 34), and far closer to the conductance predicted for the water-soluble hexamer, CC-Type2-(TaSd)2, than to the four-helix bundles of the octamer (see below). Reversal potential measurements showed that the channels were cation-selective, with a permeability ratio of ~5:1 for K+ versus Cl− (Supplementary Fig. 54), consistent with the solvation of cations by hydroxyl groups of Ser/Thr-lined channels. CCTM-VbIc also conducted Na+ and Cs+, and current–voltage curves obtained in different electrolytes were non-linear with marked current rectification (Fig. 2g). Such rectification arises from asymmetric charge distributions35,36. This is consistent with a parallel assembly of helices with N-terminal Lys tags, but not with the antiparallel arrangement seen in the LCP structure of K2-CCTM-VbIc (see below).
In addition, we performed simultaneous electrical and optical single-channel recording, oSCR24,37,38, monitoring the flux of calcium ions through individual CCTM-VbIc channels inserted into DPhPC droplet interface bilayers (DIBs; Fig. 4 and Supplementary Figs. 55 and 56)39. Electrically, as in PLBs, CCTM-VbIc showed discrete unitary conductance in DIBs (Fig. 4b). Optically, the calcium-flux imaging showed multiple mobile spots diffusing in the plane of the bilayer, indicative of transmembrane channels (Fig. 4c and Supplementary Video 4). The fluorescence from these isolated channels showed single distinct open and closed states, further demonstrating the unitary conductance of the CCTM-VbIc channels (Fig. 4d,e).
Fig. 4 |. CCTM-VbIc channels in DIBs.
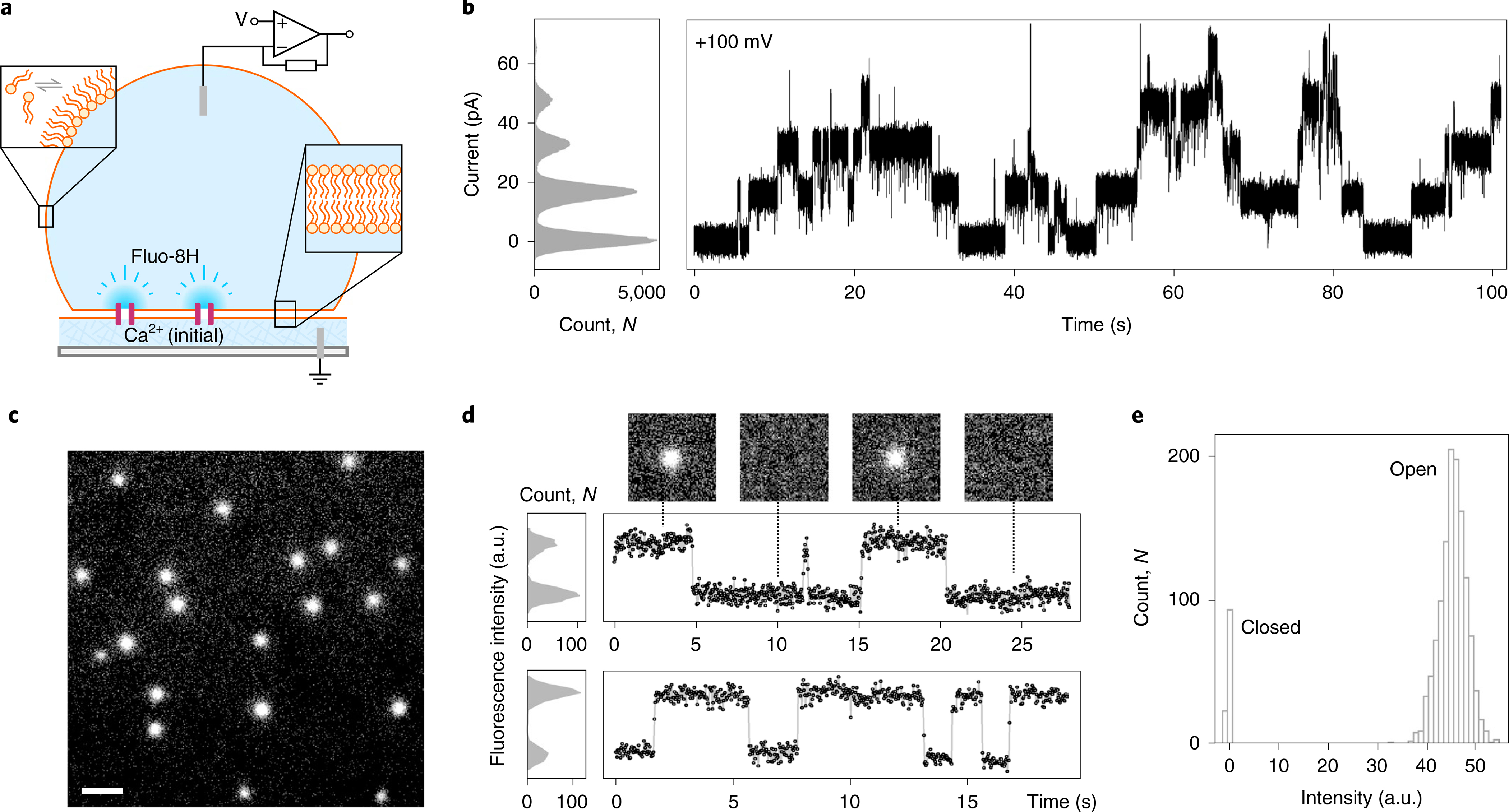
a, Schematic of a DIB formed between an aqueous droplet and a hydrogel substrate spun onto a coverslip (grey), both in the presence of a lipid-in-oil solution. The DIB has Ca2+ ions in the hydrogel and the Ca2+-sensitive dye Fluo-8H in the droplet. Channels (purple blocks) formed in the bilayer allow passage of Ca2+ ions into the droplet to generate plumes of Ca2+-bound dye (depicted in darker blue) that can be imaged using total internal reflection fluorescence microscopy. b, Conductance steps from multiple insertions of CCTM-VbIc in a DIB at +100 mV. Voltages are trans relative to cis (peptide). c, oSCR for CCTM-VbIc channels in a DPhPC membrane at +100 mV. This is a single 30 ms exposure; 100 nM CCTM-VbIc; scale bar, 10 μm. d, Fluorescence intensity versus time traces for two CCTM-VbIc channels. The y-axes represent mean pixel values of bilayer patches containing the channels. The upper trace is annotated with 30 ms frames (17.8 × 17.8 μm) from the respective oSCR image stack. e, Spot intensities extracted from eight CCTM-VbIc channels in the same bilayer, which represent 40 s of open-channel time, show a unitary open state and a closed state. For all experiments, the droplet peptide concentrations were between 25 and 100 nM.
Direct imaging of a Cyanine-5-labelled peptide, Cy5-CCTM-VbIc, showed that it diffused across the entire membrane, including regions away from the channels seen by oSCR (Supplementary Fig. 56). These species had a mean lateral diffusion coefficient (Dlat) of 2.56 ± 0.99 μm2 s−1, similar to monomeric transmembrane helices in related membranes40. However, the Ca2+-conducting channels diffused more slowly (Dlat = 1.05 ± 0.26 μm2 s−1, similar to multi-peptide alamethicin pores24). Using these Dlat values in the Saffman–Delbrück equation41, we estimated an approximate four-fold increase in diameter between the individual peptides and the conductive channels (Supplementary Fig. 55). Although this carries assumptions, it is consistent with the relative sizes of helical monomers and αHBs15.
Computational analyses of possible antiparallel and parallel states of CCTM-VbIc.
Finally, to help distinguish between the possible states that CCTM-VbIc might form in the membrane, we performed simulations and in silico calculations on the K2-CCTM-VbIc structure, that is the dimer of antiparallel four-helix bundles, and with the peptide modelled as parallel and antiparallel hexamers (Fig. 3e–i). Hexamers were chosen to match the oligomeric state of the water-soluble parent and are consistent with the conductance data. The models were generated in CCBuilder 2.0 (ref. 17) using parameters from the CC-Type2-(TaSd)2 X-ray crystal structure.
In contrast to simulations for the water-soluble barrels (Fig. 1h–j), MD studies of the K2-CCTM-VbIc octamer in DPhPC bilayers indicated that very few water molecules traversed the channel or exchange with bulk solvent (Supplementary Figs. 57–60 and Supplementary Video 5). Rather, and consistent with the ordered crystallographic water, water molecules remained bound to internal Ser and Thr side chains of the outer four-helix bundles for hundreds of nanoseconds (Supplementary Table 5). In short, these interiors were rarely solvated across their full lengths. Moreover, ions included in different simulations (for example, Ca2+, Na+, K+ and Cl−) did not enter these spaces over a cumulative 3.2 μs of simulation. The interior of the inner four-helix bundle remained dry in all simulations.
Poisson–Boltzmann electrostatic calculations using the graphical interface APBSmem42 supported these findings (Fig. 3e and Supplementary Figs. 61–67). The calculated electrostatic potential energy barriers for the outer four-helix bundle of the octameric crystal structure were too high to conduct K+ or Cl− (Fig. 3f). However, the barriers calculated for the parallel hexameric model were an order of magnitude lower and similar to those calculated for natural ion channels (Fig. 3g)43. We attribute this to the different diameters of the cavities in the four- and six-helix models (Fig. 3h). The K+/Cl− selectivity observed experimentally was manifest in the calculations as a lower energy profile for K+ compared with Cl− for the hexamers (Fig. 3g), which we attribute to the hydroxyl groups lining the channel. Finally, we modelled the current–voltage relationship using Nernst–Planck electrodiffusion theory34. The computed I–V curve for the parallel hexameric model agreed qualitatively with the current rectification observed experimentally (Figs. 2g and 3i and Supplementary Figs. 61–67). By contrast, the calculated I–V plot for the antiparallel hexameric model was linear; and no current was predicted for the octameric crystal structure across the voltage range. From these analyses, we propose an all-parallel barrel-like structure, possibly a hexamer, as the most likely open-channel state.
Conclusion
We have shown that multiple polar residues can be incorporated within the lumens of water-soluble, de novo αHBs with a→g heptad-repeat sequences. The combination of Thr at ‘a’ positions and Ser at ‘d’ positions generates large cavities that sequester water molecules that freely exchange with bulk water. This background can be used to generate ion-conducting membrane-spanning peptides by introducing hydrophobic residues at the ‘b’, ‘c’ and ‘f’ positions. One of these CCTM peptides forms discrete and stable ion channels in membranes. Designed peptide channels with this degree of stability and monodispersity are unprecedented and are reminiscent of those formed by the engineered natural peptides cWza and pPorA (refs. 10,44). This demonstrates how consideration of helix–helix packing and lumen design can deliver membrane-spanning assemblies with defined ion-channel activities.
That said, the peptides that we have made access multiple states. One of our CCTM peptides crystallizes from the LCP as a dimer of an antiparallel tetramer, while in membranes under voltage both monomeric peptides and assembled channels are observed. The antiparallel tetramers are unlikely to be the channel state: although they have polar interiors, these are too narrow to conduct ions, and their dihedral symmetry is inconsistent with rectification observed for the conducting state. Given this, we propose the following model for CCTM activity, though we recognize that different membrane mimics may result in different assemblies. In the LCP, the antiparallel structure is a low-energy, readily accessible state. By contrast, in PLBs the peptide forms an equilibrium of monomeric and oligomeric species. The voltage applied to record channel currents may aid in establishing the channels, either by increasing the local peptide concentration or thinning the membrane. However, in DIBs, peptide concentrations of 100–350 nM give currents from hundreds of channels even at low voltage (10–20 mV), suggesting that channels insert readily. For the conducting state, we propose a parallel αHB-stave structure with a solvent-accessible central channel similar to those seen in the water-soluble parent peptides. This multi-state behaviour mimics natural channel-forming peptides, the structures of which are often condition-dependent33.
Of course, without a high-resolution structure of the conducting state, we cannot be certain of this model. Given the apparent structural plasticity of the system, it may prove difficult to obtain such a structure. It may be possible to stabilize the active state through templating45, or to employ negative design to destabilize the alternate state(s)46. However, the generation of stable barrel-stave structures of peptide helices may continue to prove challenging. Indeed, we find that water-soluble αHBs collapse without key sequence features16. We observe something similar in the LCP structure of CCTM, where close packing of the Ala, Ser and Thr core residues leads to the dimer of four-helix bundles. Furthermore, it is interesting that although nature has evolved stable membrane-spanning αHBs that are crystallizable, these are usually templated by extra-membranous domains or buttressed by concentric rings of helices in the membrane47,48. Indeed, recently Xu, Baker and colleagues have successfully built membrane-spanning pores using precisely this strategy49. Interestingly, in early iterations of these designs (WSHC6 and TMHC6) an outer ring of helices buttresses a central six-helix barrel based on the de novo designed αHB CC-Hex2 (ref. 15) in which the original barrel-specifying lumenal and inter-helical hydrophobic residues are retained. Further still, many natural membrane pores are assembled from multiple β-hairpin units47,50. Although extremely large barrels appear to be accessible through this mode of assembly, again they are often templated by large water-soluble domains. Efforts to design even water-soluble β-barrel systems are in their infancy51 and carry the added challenge of avoiding amyloid states.
Thus, whilst our work and examples of natural membrane- spanning αHBs offer hope for designing such assemblies, they do not completely solve the problem of how to specify peptide–peptide interactions to generate barrel-stave structures that are stable over broad conditions. Nonetheless, we have achieved considerable control over self-assembly in water and function in membranes. Improving on this further would increase understanding of natural-channel and pore-forming peptides, provide further principles for designing peptide ion channels, and, ultimately, unlock applications for these in biotechnology50.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557–021-00688–0.
Methods
Computational modelling and simulation methodology is described in the Supplementary Information.
Solid-phase peptide synthesis.
Amino acids protected with the 9-fluorenyl-methoxycarbonyl (Fmoc) protecting group, synthesis-grade dimethylformamide (DMF) and coupling agents were purchased from Cambridge Reagents. Fmoc-protected pseudoproline dipeptides and MBHA resin were supplied by Novabiochem (Merck Millipore). Peptides were synthesized on a 0.1 mmol scale using a Liberty Blue automated peptide synthesizer (CEM) equipped with inline UV monitoring and using standard Fmoc-protected amino acid chemistry as previously described15. The activators used were N,N′ -diisopropylcarbodiimide (DIC) and 6-chloro-1-hydroxybenzotriazole (Cl-HOBt). Rink amide MBHA resin was used, which after cleavage yielded peptides with a C-terminal amide group. Peptides were N-terminally acetylated using acetic anhydride and pyridine. For CCTM peptides, Fmoc-Ala–Thr(ψMe,Mepro)–OH was used at all Ala–Thr positions.
Semi-preparative HPLC.
Crude peptides were purified using a Jasco 2000 series HPLC system. Crude peptide solution (1 ml, 6 mg ml−1, 20:80% v/v MeCN:H2O) was then injected onto a semi-preparative reversed-phase HPLC column and eluted with a 3 ml min−1 linear gradient of H2O:MeCN or H2O:IPA (IPA is isopropyl alcohol) over 30 min. All HPLC mobile phases were modified with 0.1% TFA (trifluoroacetic acid). Elution of the peptide was detected with inline UV monitoring at 220 and 280 nm wavelengths simultaneously.
For water-soluble peptides, a C18 column (Phenomenex Kinetex, 5 μm, 100 Å, 10 mm inner diameter (i.d.) × 150 mm length) with a 20–80% or 40–100% MeCN:H2O gradient was used. For more hydrophobic peptides, a C8 (Phenomenex Luna, 5 μm, 100 Å, 10 mm i.d. × 250 mm length) or C5 (Phenomenex Luna, 5 μm, 100 Å, 10 mm i.d. × 250 mm length) column was used, often with H2O:IPA as the mobile phase. When required, a column oven (50 °C) was employed to improve separation. Pure fractions of peptide, as determined by analytical HPLC and matrix-assisted laser desorption/ionization–time of flight (MALDI–TOF) mass spectrometry, were combined and freeze dried.
Fluorescent capping procedure.
To fluorescently label CCTM-VbIc, the peptide was capped via DIC/Cl-HOBt coupling with 3-(tritylthio)propionic acid and cleaved/deprotected and purified. To this peptide (0.05 μmol) in buffer (500 μl, 10% DMF, 10 mM HEPES, pH 6.5) was added tris(2-carboxyethyl)phosphine (1.25 mg, 0.5 μmol) followed by sulfo-Cyanine5 maleimide (Lumiprobe) in dimethyl sulfoxide (0.402 mg, 0.5 μmol, 10 mg ml−1). The mixture was rocked overnight at 20 °C, and the dye-labelled peptide purified by reversed-phase HPLC.
Analytical HPLC.
To determine peptide purity (relative to other peptide side products), a small quantity (approx. 10 nmol) of peptide was dissolved in MeCN:H2O (20:80 v/v, 50 μl). This was injected onto an analytical scale C18 column (Phenomenex Kinetex, 5 μm, 100 Å, 4.6 mm i.d. × 100 mm length) and eluted with a linear gradient of 0–60%, 20–80% or 40–100% MeCN:H2O with 0.1% TFA at a flow rate of 1 ml min−1. CCTM peptides were injected onto a C4 column (Phenomenex Aeris Widepore) of the same dimensions and eluted with a 40–100% gradient of IPA:MeCN:H2O (60:30:10% v/v/v) with 0.1% TFA. Elution of peptide was monitored at 220 and 280 nm wavelengths simultaneously. All peptides were >95% pure, as determined by integration of their chromatograms.
Matrix-assisted laser desorption/ionization–time of flight mass spectrometry.
MALDI–TOF mass spectra were obtained using a Bruker Ultraflex mass spectrometer, operating in positive-ion reflector mode. Peptides were spotted on a ground steel target plate with either dihydroxybenzoic acid or α-cyano-4-hydroxycinnamic acid dissolved in MeCN:H2O:TFA (49.95:49.95:0.1% v/v/v) as the matrix. The instrument was calibrated by the ‘nearest neighbour’ method, using Bruker Peptide Calibration Standard II as reference masses.
Circular dichroism spectroscopy.
CD spectra were recorded using either a Jasco J-810 or J-815 spectropolarimeter. Sample temperatures were controlled using a Jasco Peltier thermostatted cell holder. Typically, CD spectra were recorded between 190 and 260 nm wavelength, with a 1 nm interval, 1 nm bandwidth and 1 s response time. A 1 mm path length quartz cuvette was used. PBS buffer refers to 8.2 mM sodium phosphate, 1.8 mM potassium phosphate, 137 mM sodium chloride and 2.7 mM potassium chloride.
For thermal denaturation experiments, peptide unfolding and refolding were monitored at 222 nm wavelength. Unless otherwise stated, a temperature range of 5–95 °C and a temperature ramp rate of 60 °C per hour were used. Baseline/blank CD spectra were recorded using a buffer and cuvette matched to that of the peptide sample experiment and subtracted from that of the peptide sample. Peptide CD signals are expressed in units of mean residue ellipticity ([θ], deg cm2 dmol−1 per residue).
Analytical ultracentrifugation.
All AUC experiments were performed using either a Beckman Optima XL-A or XL-I and an An-50 or An-60 titanium rotor. All experiments were conducted at 20 °C. For sedimentation velocity experiments, a two-channel epon velocity cell centrepiece equipped with quartz windows was used. The sample channel was filled with 310 μl of sample, while the reference channel was filled with 320 μl of buffer. Unless otherwise stated, the sample was made to a concentration such that its absorbance A280 nm (1.2 cm path length) value was unity. A typical method involved centrifuging at 50,000 revolutions per minute (r.p.m.) and taking 120 scans, 5.8–7.3 cm radially, at five minute intervals. A radial calibration was performed before every new run. Data for each sample were fitted to a continuous c(s) distribution model using the Sedfit software package, at a 95% confidence level52. The buffer density, buffer viscosity and protein partial specific volume were all calculated using the Sednterp package (Biomolecular Interaction Technology Centre). The residuals bitmaps give a visual representation of fit quality. Good fits are uniformly grey without major dark or light streaks.
Sedimentation equilibrium experiments were conducted using a six-channel equilibrium cell centrepiece. For absorbance and interference experiments, quartz and sapphire windows were used, respectively. The sample channels were filled with 110 μl of sample and the reference channels filled with 120 μl of buffer. Unless otherwise stated, the sample was made to a concentration such that its A280–nm (1.2 cm path length) value was 0.5. Where a detergent was used, the micelles and buffer were density matched using D2O to remove the detergent’s contribution to the peptide–detergent complex buoyant mass. A typical method involved centrifuging from 18,000–36,000 r.p.m. (in 3,000 r.p.m. increments), with scans at eight and nine hours after each new speed was reached. A radial calibration was performed before every new run. Data were fitted to a single ideal species model using Ultrascan II (http://www.ultrascan2.aucsolutions.com/). If the model was deemed appropriate (as stated in the software fitting report), 95% confidence intervals for the fit were obtained via Monte Carlo analysis.
Crystallography (water-soluble barrels).
Diffraction-quality peptide crystals were grown using a sitting-drop vapour-diffusion method. Freeze-dried peptides were dissolved in ultrapure water and diluted to 10 mg ml−1. Hydrophobic peptides were dissolved in detergent solution. For water-soluble peptides, commercially available sparse matrix screens were used (Morpheus, JCSG plus, Structure Screen 1 + 2, Pact Premier, ProPlex; Molecular Dimensions), and the drops were dispensed using a robot (Oryx8; Douglas Instruments). For each well of an MRC 2 drop plate, 0.3 μl of peptide solution and 0.3 μl of reservoir solution were mixed and the plate was incubated at 20 °C. Crystals generally formed within two weeks, and after looping were soaked in reservoir solution containing 25% glycerol as a cryoprotectant. Diffraction data for the crystals were obtained at the Diamond Light Source on beamlines I03, I04 or I24. For specific wavelengths see Supplementary Table 2.
Diffraction images were processed using either an automated pipeline (xia2, https://xia2.github.io/) or manually (iMOSFLM (ref. 53)). In the latter case, data were reduced using POINTLESS (ref. 54), AIMLESS (ref. 55) and CTRUNCATE (ref. 56). Data were phased by the molecular replacement method, using Phaser-MR, integrated into the PHENIX software suite57. Phasing was achieved using polyalanine search models of the likely structures, with the number of helices present in the asymmetric unit inferred using the Matthews coefficient. CCBuilder 2.0 (ref. 17) was used to generate these polyalanine models. Models were refined with phenix.refine and PDB-REDO (ref. 58).
Crystallography (K2-CCTM-VbIc).
K2-CCTM-VbIc was crystallized with the LCP method. The methods described for LCP crystallization are described in detail in Caffrey and Cherezov59. Briefly, lyophilized peptide was weighed and dissolved in ethanol. Monoolein (60 mg; Sigma-Aldrich) was weighed and combined with the peptide solution with vortexing until clear. The solution containing the peptide and monoolein was then dried under a stream of nitrogen and lyophilized overnight. The LCP was prepared by first heating the monoolein–peptide mixture to 42 °C until it became liquid, and subsequently extruded ~200 times with 2/3 volume of Milli-Q water in coupled gastight Hamilton syringes at room temperature. Successful LCP formation was confirmed as the solution became clear and was not birefringent in the cross-polarizer. The final concentration of the peptide in LCP was estimated to be around 32 mg ml−1.
For crystallization, 50 nl of the LCP mixture was dispensed onto 96-well Laminex plastic sandwich plates (Molecular Dimensions) with 1 μL of precipitant solution using the TTP Labtech LCP Mosquito robot at room temperature. Plates were sealed using plastic coverslips (Molecular Dimensions) before storage and imaging in the Formulatrix RockImager at 20 °C. Plates were monitored every day for crystal formation. The initial hit from the sparse matrix screens (0.05 M glycine, pH 9.0, 55% v/v PEG400) was expanded and optimized for the final crystallization condition of 0.05 M glycine (Sigma-Aldrich) pH 10.0 and 55% v/v PEG400 (Qiagen Sciences).
Crystals were harvested from the crystallization plates and immediately frozen in liquid nitrogen without additional cryoprotection. Crystals were mounted under a cryostream and data were collected at the 8.3.1 beamline at the Advanced Light Source at a wavelength of 1.11582 Å. The data were processed with the XDS package60 and reduced with AIMLESS (ref. 55) in the CCP4 packages. The structure was determined by molecular replacement with ARCIMBOLDO Lite (ref. 61), using a single idea helix as the search model. The structure was refined with PHENIX (ref. 57) and model rebuilding was done with Coot (ref. 62). The data processing and structural refinement statistics are listed in Supplementary Table 2.
1,6-Diphenyl-1,3,5-hexatriene binding assays.
Binding experiments with DPH were performed at constant ligand concentrations of 0.1 μM or 1 μM with 5% v/v dimethyl sulfoxide. Peptide concentrations were varied from 1 to 500 μM. Datapoints from concentrations corresponding to unfolded peptide were discarded. Samples were equilibrated by rocking for two hours at 20 °C. Experiments were conducted using a BMG Labtech Clariostar plate reader. Samples were excited at 350 nm wavelength and emission was monitored at 455 nm wavelength. Measurements were made in triplicate/quadruplet and averaged to give binding curves, which were analysed using Graphpad Prism 7.
Data were fitted to the following quadratic tight-binding equation
where y is the fluorescence intensity, x the peptide concentration, Bmax the fitted maximum fluorescence value, c0 the ligand (DPH) concentration and KD the dissociation constant.
Planar lipid bilayer recordings.
PLB single-channel recordings were made using a custom Delrin chamber, with two 1 ml compartments separated by a 20-μm-thick Teflon film (Goodfellow) with an ~80 μm diameter aperture63. Electrical signals were recorded using a Molecular Devices Axopatch 200B amplifier, equipped with a CV 203BU headstage. The chamber and headstage were placed in a Faraday cage. Signals were digitized using a Molecular Devices Digidata 1550 digitizer and recorded using the pCLAMP 10.7 software. Data were analysed and plotted using Clampfit 10 and custom Python scripts.
The procedure outlined is based on the Montal–Mueller method of forming folded lipid bilayers64. The aperture was pre-painted on both sides with hexadecane in n-pentane (3 μl, 10 mg ml−1) using a glass micropipette. The pentane was allowed to dry for 15 min, and the cis and trans compartments filled with buffer solution (typically 10 mM Tris-HCl, 1 M KCl, pH 8). A drop of DPhPC in n-pentane (~6 μl, 5 mg ml−1) was added to the buffer surfaces on both sides, and the pentane allowed to evaporate for 15 min. After this time, buffer solution was removed slowly such that the meniscus fell below the aperture and was gently re-added. This was repeated on both sides of the aperture until a bilayer of the appropriate capacitance had formed. Peptides were added (2 μl, 25 μM, solubilized in 0.05% DDM) to the cis compartment to give a final concentration of approximately 50 nM. Voltages were applied between the cis (ground) and trans (active) compartments using Ag/AgCl electrodes connected to each compartment with salt bridges (3 M KCl, 2% agar). The Ag/AgCl electrodes were prepared by leaving silver wire in sodium hypochlorite solution (5%) overnight.
Reversal potentials (Supplementary Fig. 54) were calculated using asymmetric cis/trans electrolytes and a rearranged Goldman–Hodgkin–Katz equation, as described previously65.
Droplet interface bilayer electrical and optical recordings.
DPhPC was purchased from Avanti Polar Lipids. The Ca2+-sensitive dye Fluo-8H was purchased from AAT Bioquest. All aqueous solutions were made using ultrapure water.
DIBs were prepared as described previously39. Briefly, 0.75% (w/v) ultra-low gelling temperature agarose solution was homogenised at 90 °C, and 140 μl of this substrate agarose was spun onto a plasma-cleaned coverslip (Menzel-Gläzer, ThermoFisher Scientific). The coverslip was affixed to the underside of a custom-made poly(methyl methacrylate) device featuring 16 wells, 1 mm in diameter. Agarose solution (1.3% (w/v)) containing 1 M KCl (or 500 mM CaCl2 for oSCR experiments), buffered with 10 mM Tris-HCl at pH 8, was flowed into the device, where it made contact with and hydrated the substrate agarose but did not cover it. The wells were filled with hexadecane containing DPhPC at 10 mg ml−1 and the device was allowed to rest for one hour to allow for monolayer formation on the substrate. Meanwhile, aqueous droplets (~100 nl) were incubated in the same lipid-in-oil solution, again for monolayer formation. The droplets contained 1 M KCl, 10 mM Tris-HCl, 375 μM EDTA and 46 μM Fluo-8H, in addition to unlabelled (20–100 nM) and N-terminally labelled CCTM-VbIc (~50–100 pM) as required. After incubation, droplets were pipetted into the wells of the device where they sank onto the substrate, forming bilayers by the contact of the droplet and agarose-associated monolayers.
The Ag/AgCl electrodes inserted into the hydrating agarose, and the top of the droplet via a micromanipulator, allowed electrical access. Voltages were applied and bilayer currents recorded using an Axopatch 200B patch-clamp amplifier and headstage (Axon Instruments, Molecular Devices), with signals recorded and filtered at 1 kHz. Devices were placed within a Faraday cage atop an inverted microscope (TiE Eclipse, Nikon). Fluorescence imaging was carried out using a ×60 total internal reflection fluorescence objective lens (oil-immersion, 1.45 NA; Nikon). Excitation of the bilayers and collection of fluorescent signals was carried out by the same lens. For excitation of Ca2+-bound Fluo-8H, a 473 nm continuous-wave laser beam was used (the power at the back focal plane of the objective was 40–80 μW; Vortran Laser Technology), with fluorescence and excitation signals (λFluo-8H emission max = 514 nm) separated by a dichroic mirror (ZT 488/640; Chroma), and the collected signal from the bilayer transmitted through an emission filter (Brightline bandpass 550/88 nm; Semrock) to eliminate stray excitation wavelengths. Cy5-CCTM-VbIc was excited using a 644 nm continuous-wave laser beam (the power at the back focal plane of the objective was 450–840 μW; Vortran Laser Technology), with the emitted fluorescence passing through a 690/50 nm bandpass emission filter (Semrock). Images were recorded at 31–50 Hz using an electron-multiplying charged-coupled device camera (iXon+; Andor Technology). All experiments were conducted at room temperature of around 21 °C.
DIB data processing and analysis.
Electrical data were recorded using WinEDR electrophysiology software (John Dempster, Strathclyde University). Image analyses were carried out in Fiji image-processing software66; single-particle tracking was performed using the TrackMate plugin, using its Laplacian of Gaussian algorithm67. Numerical data and mean squared displacement versus time analyses were performed in Igor Pro (Wavemetrics) using custom-written procedures.
Using the measured Dlat values and the Saffma–Delbrück equation68, we estimated the ratio of the sizes of the assembled channels versus the monomer, rchannel/rmonomer, as 3.9 ± 1.3. This consistent with the relative cross-sections of monomeric α-helices and hexameric αHBs, which are ~1:3 (ref 15). This makes the assumption that the monomers are folded as single-pass transmembrane helices (diameter ≈ 1 nm), which may not be the case. In addition, we recognize that αHBs with five, six and seven helices have similar diameters in the range 3–3.5 nm.
For this estimation of rchannel/rmonomer, the following values were used: membrane thickness, h = 3.63 nm (ref. 69); membrane viscosity, μm = 0.08 Pa s (ref. 68); surrounding fluid viscosity, μf = 1 mPa s (ref. 68); temperature, T = 294 K, Dlat, channel = 1.05 μm2 s−1; and Dlat, monomer = 2.56 μm2 s−1.
Values for the membrane viscosity vary in the literature; the value of μm = 0.08 Pa s was chosen as one that has been shown to produce good agreement between experimentally measured diffusion coefficients and the Saffman–Delbrück model68, and is close to the value of 0.1 Pa s often quoted in the literature.
The error in rchannel/rmonomer was determined by the propagation of errors, assuming that the only contributions were from Dlat, channel, Dlat, monomer and μm, the other parameters in the Saffman–Delbrück equation being constant. Whilst not measured in our experiments, μm was included as a variable in the propagation of errors because a range of values are reported in the literature. The error values for each parameter (which were then used to compute the total error by the propagation of errors) were the standard errors (standard deviation/√n) of the lateral diffusion coefficients (0.0176 and 0.0612 μm2 s−1 for the pore and monomer respectively), and for the error in μm a value of 0.02 Pa s was chosen as it has been shown that the Saffman–Delbrück model applies in the range of μm = 0.08 ± 0.02 Pa s for a bilayer of similar thickness68. Furthermore, μm = 0.08 ± 0.02 Pa s covers the range of membrane viscosities often used in the literature.
Data availability
X-ray crystal structures have been submitted to the RCSB Protein Data Bank with accession codes 6YAZ, 6YB0, 6YB1 and 6YB2. Data supporting the results and conclusions are available within this paper and the Supplementary Information. Additional raw data are available at Figshare, https://doi.org/10.6084/m9.figshare.14406419.
Supplementary Material
Acknowledgements
A.J.S. thanks Diamond Light Source for a place on the CCP4 Data Collection and Structure Solution Workshop 2017. E.J.M.L. thanks S. Rao and G. Klesse (University of Oxford) for support with CHAP and F. Marcoline (UCSF) for support with APBSmem. A.J.S. was funded by the Bristol Chemical Synthesis Centre for Doctoral Training funded by the EPSRC (EP/G036764/1). A.N. and K.R.M. were supported by a BBSRC grant to R.L.B., H.B. and D.N.W. (BB/J009784/1). W.M.D., A.N., A.J.S., A.R.T. and D.N.W. were funded by ERC Grants to D.N.W. (340764 and 787173). E.J.M.L. was in the BBSRC/EPSRC-funded Synthetic Biology Research Centre, BrisSynBio (BB/L01386X/1). M.I.W. was funded by the BBSRC (BB/R001790/1). W.F.D. was supported by NIH (R35 GM122603), NSF (1709506) and US Air Force (1709506) grants. H.T.K. was supported by the NIH Ruth L. Kirschstein NRSA Postdoctoral Fellowship (F32 GM125217). M.M. is supported by the Howard Hughes Medical Institute Gilliam Fellowship. D.N.W. held a Royal Society Wolfson Research Merit Award (WM140008). Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15–328599, the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc. and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02–05CH11231, Office of Basic Energy Sciences.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41557–021-00688–0.
Peer review information Nature Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Huang PS, Boyken SE & Baker D The coming of age of de novo protein design. Nature 537, 320–327 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Korendovych IV & DeGrado WF De novo protein design, a retrospective. Q. Rev. Biophys 53, e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu PL et al. Accurate computational design of multipass transmembrane proteins. Science 359, 1042–1046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mravic M et al. Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 363, 1418–1423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joh NH et al. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 346, 1520–1524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davey JA, Damry AM, Goto NK & Chica RA Rational design of proteins that exchange on functional timescales. Nat. Chem. Biol 13, 1280–1285 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Chen K-YM, Keri D & Barth P Computational design of G Protein-Coupled Receptor allosteric signal transductions. Nat. Chem. Biol 16, 77–86 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Woolfson DN Coiled-coil design: updated and upgraded. Subcell. Biochem 82, 35–61 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Lear JD, Wasserman ZR & Degrado WF Synthetic amphiphilic peptide models for protein ion channels. Science 240, 1177–1181 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Mahendran KR et al. A monodisperse transmembrane α-helical peptide barrel. Nat. Chem 9, 411–419 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Bowie JU Helix packing in membrane proteins. J. Mol. Biol 272, 780–789 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Hong H Toward understanding driving forces in membrane protein folding. Arch. Biochem. Biophys 564, 297–313 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Liu J et al. A seven-helix coiled coil. Proc. Natl Acad. Sci. USA 103, 15457–15462 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaccai NR et al. A de novo peptide hexamer with a mutable channel. Nat. Chem. Biol 7, 935–941 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomson AR et al. Computational design of water-soluble α-helical barrels. Science 346, 485–488 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Rhys GG et al. Maintaining and breaking symmetry in homomeric coiled-coil assemblies. Nat. Commun 9, 4132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood CW & Woolfson DN CCBuilder 2.0: powerful and accessible coiled-coil modeling. Protein Sci. 27, 103–111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walshaw J & Woolfson DN SOCKET: a program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol 307, 1427–1450 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Klesse G, Rao SL, Sansom MSP & Tucker SJ CHAP: a versatile tool for the structural and functional annotation of ion channel pores. J. Mol. Biol 431, 3353–3365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aryal P, Sansom MSP & Tucker SJ Hydrophobic gating in ion channels. J. Mol. Biol 427, 121–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carugo O Statistical survey of the buried waters in the Protein Data Bank. Amino Acids 48, 193–202 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Dawson JP, Weinger JS & Engelman DM Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol 316, 799–805 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Hessa T et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Harriss LM, Cronin B, Thompson JR & Wallace MI Imaging multiple conductance states in an alamethicin pore. J. Am. Chem. Soc 133, 14507–14509 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Landau EM & Rosenbusch JP Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl Acad. Sci. USA 93, 14532–14535 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gernert KM, Surles MC, Labean TH, Richardson JS & Richardson DC The Alacoil: a very tight, antiparallel coiled-coil of helices. Protein Sci. 4, 2252–2260 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adamian L & Liang J Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins 47, 209–218 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Zhang SQ et al. The membrane- and soluble-protein helix-helix Interactome: similar geometry via different interactions. Structure 23, 527–541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhys GG et al. Navigating the structural landscape of de novo α-helical bundles. J. Am. Chem. Soc 141, 8787–8797 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Song C et al. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl Acad. Sci. USA 110, 4586–4591 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayouka Z et al. Quasiracemate crystal structures of magainin 2 derivatives support the functional significance of the phenylalanine zipper motif. J. Am. Chem. Soc 137, 11884–11887 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurgan KW et al. Retention of native quaternary structure in racemic melittin crystals. J. Am. Chem. Soc 141, 7704–7708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sansom MS The biophysics of peptide models of ion channels. Prog. Biophys. Mol. Biol 55, 139–235 (1991). [DOI] [PubMed] [Google Scholar]
- 34.Hille B Ionic Channels of Excitable Membranes (Oxford Univ. Press, 2001). [Google Scholar]
- 35.Kienker PK, DeGrado WF & Lear JD A helical-dipole model describes the single-channel current rectification of an uncharged peptide ion channel. Proc. Natl Acad. Sci. USA 91, 4859–4863 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noskov SY, Im W & Roux B Ion permeation through the α-hemolysin channel: theoretical studies based on Brownian dynamics and Poisson-Nernst-Plank electrodiffusion theory. Biophys. J 87, 2299–2309 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang SQ, Song LS, Lakatta EG & Cheng HP Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 410, 592–596 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Heron AJ, Thompson JR, Cronin B, Bayley H & Wallace MI Simultaneous measurement of ionic current and fluorescence from single protein pores. J. Am. Chem. Soc 131, 1652–1653 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Leptihn S et al. Constructing droplet interface bilayers from the contact of aqueous droplets in oil. Nat. Protoc 8, 1048–1057 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Ramadurai S, Duurkens R, Krasnikov VV & Poolman B Lateral diffusion of membrane proteins: consequences of hydrophobic mismatch and lipid composition. Biophys. J 99, 1482–1489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saffman PG & Delbrück M Brownian motion in biological membranes. Proc. Natl Acad. Sci. USA 72, 3111–3113 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callenberg KM et al. APBSmem: a graphical interface for electrostatic calculations at the membrane. PLoS One 5, e12722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roux B, Allen T, Berneche S & Im W Theoretical and computational models of biological ion channels. Q. Rev. Biophys 37, 15–103 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Krishnan RS et al. Autonomously assembled synthetic transmembrane peptide pore. J. Am. Chem. Soc 141, 2949–2959 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Spruijt E, Tusk SE & Bayley H DNA scaffolds support stable and uniform peptide nanopores. Nat. Nanotechnol 13, 739–745 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Grigoryan G, Reinke AW & Keating AE Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 458, 859–864 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peraro MD & van der Goot FG Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol 14, 77–92 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Niitsu A, Heal JW, Fauland K, Thomson AR & Woolfson DN Membrane-spanning α-helical barrels as tractable protein-design targets. Philos. Trans. Royal Soc. B 372, 20160213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu C et al. Computational design of transmembrane pores. Nature 585, 129–134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Howorka S Building membrane nanopores. Nat. Nanotechnol 12, 619–630 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Dou JY et al. De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuck P Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J 78, 1606–1619 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Battye TGG, Kontogiannis L, Johnson O, Powell HR & Leslie AGW iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D 67, 271–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans P Scaling and assessment of data quality. Acta Crystallogr. D 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans PR An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D 67, 282–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joosten RP, Long F, Murshudov GN & Perrakis A The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caffrey M & Cherezov V Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc 4, 706–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kabsch W XDS. Acta Crystallogr. D 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sammito M et al. ARCIMBOLDO_LITE: single-workstation implementation and use. Acta Crystallogr. D 71, 1921–1930 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maglia G, Heron AJ, Stoddart D, Japrung D & Bayley H Analysis of single nucleic acid molecules with protein nanopores. Methods Enzymol. 475, 591–623 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montal M & Mueller P Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc. Natl Acad. Sci. USA 69, 3561–3566 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu LQ et al. Reversal of charge selectivity in transmembrane protein pores by using noncovalent molecular adapters. Proc. Natl Acad. Sci. USA 97, 3959–3964 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tinevez JY et al. TrackMate: an open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Ramadurai S et al. Lateral diffusion of membrane proteins. J. Am. Chem. Soc 131, 12650–12656 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Kučerka N, Nieh M-P & Katsaras J Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta Biomembr 1808, 2761–2771 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
X-ray crystal structures have been submitted to the RCSB Protein Data Bank with accession codes 6YAZ, 6YB0, 6YB1 and 6YB2. Data supporting the results and conclusions are available within this paper and the Supplementary Information. Additional raw data are available at Figshare, https://doi.org/10.6084/m9.figshare.14406419.