

Summary
Quiescence is a state of reversible proliferative arrest in which cells are not actively dividing, and yet retain the capacity to reenter the cell cycle upon receiving an appropriate stimulus. Quiescent cells are remarkably diverse—they reside in different locations throughout the body, serve distinct roles, and are activated by a variety of signals. Despite this diversity, all quiescent cells must be able to persist in a non-dividing state without compromising their proliferative potential, which requires changes to core cellular programs. How drastically different cell types are able to implement extensive changes to their gene expression programs, metabolism, and cellular structures to induce a common cellular state is a fascinating question in cell and developmental biology. In this review, we explore the diversity of quiescent cells and highlight the unifying characteristics that define the quiescent state.
Introduction
Quiescence is a state of reversible growth arrest in which cells have exited the cell cycle but remain capable of renewed division upon stimulation. Entry into quiescence allows cells to persist in a non-dividing state over extended periods of time and enact mechanisms to protect themselves from damage. Although quiescent cells display some similarities to other non-dividing cell states, such as senescence and terminal differentiation, quiescence possesses unique characteristics and functions. In particular, whereas senescent and terminally differentiated cells arrest permanently and are unable to proliferate further, quiescent cells are defined by their ability to reenter the cell cycle. This broad definition of quiescence encompasses a wide range of diverse cell types in an organism. Quiescent cells include tissue-resident adult stem cells, such as hematopoietic, muscle, and neural stem cells, as well as differentiated cells, including fibroblasts, hepatocytes, lymphocytes, and oocytes (Bangru et al., 2018; Hwang et al., 2020; Liang et al., 2020; Mitra et al., 2018; Sampath et al., 2018; Swartz et al., 2019; Urban et al., 2019; Yi, 2017) (Figure 1). Quiescence maintains these cells in a poised state—non-proliferative, but ready to re-enter the cell cycle when confronted with the appropriate stimulus.
Figure 1. The diversity of quiescent cell types in an organism.
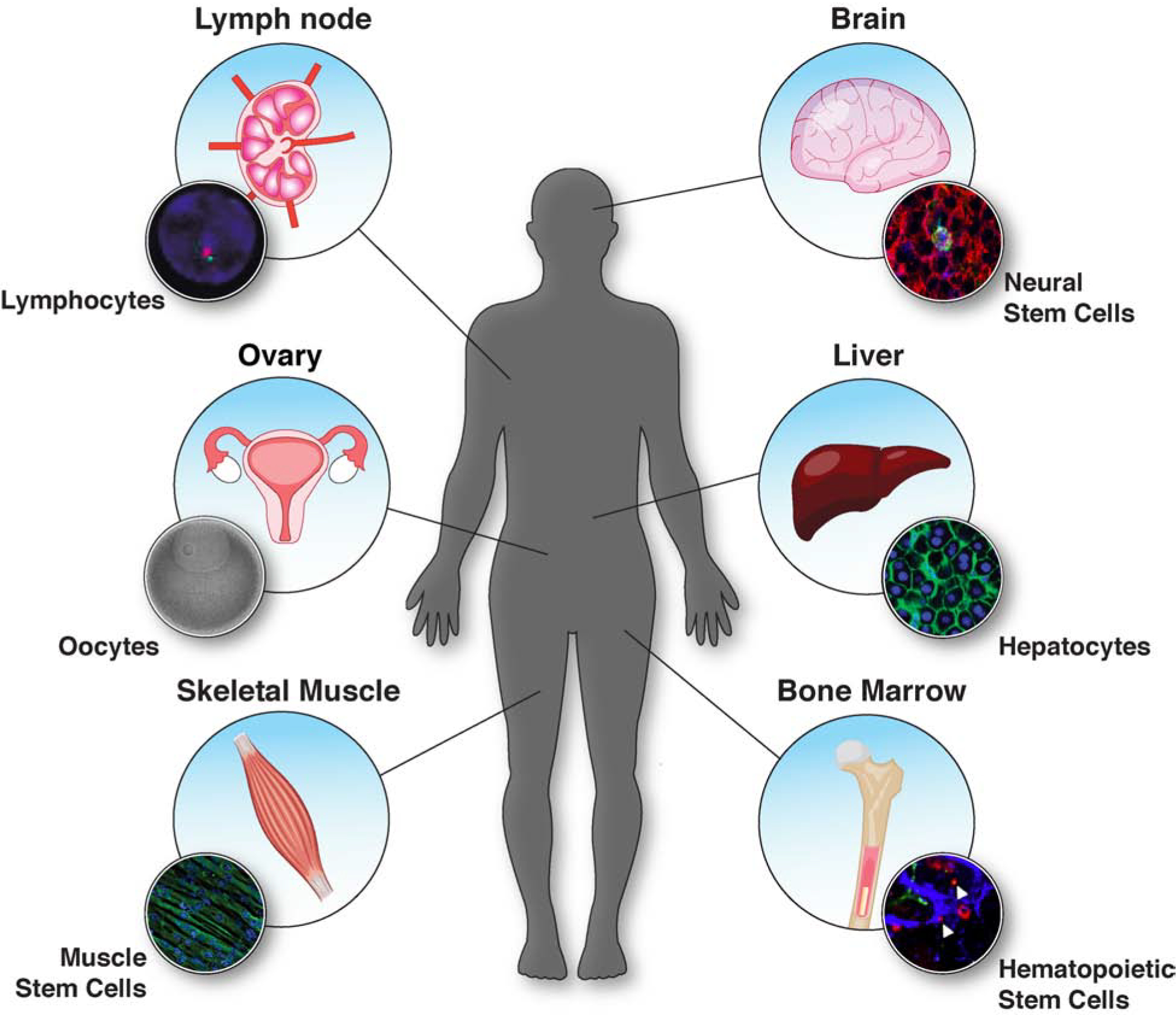
Diagram of key tissues and organs with their resident quiescent cell populations. Quiescent cell types are diverse and found throughout the body where they play roles in tissue repair, fertility, and immunity. Muscle stem cell and oocytes images were obtained from the Cheeseman lab. Hepatocyte image was kindly provided by Kristin Knouse, with permission. Lymphocyte image are from Grogan et al. (Grogan et al., 2001). Neural stem cell image are from Llorens-Bobadilla et al. (Llorens-Bobadilla et al., 2015). Hematopoietic stem cell image are from Wei et al. (Wei et al., 2020).
The maintenance of quiescence and the regulated reentry of a cell into the cell cycle are crucial for the functions of quiescent cells in tissue repair, immunity, and reproduction. For example, muscle stem cells (satellite cells) reside in a quiescent state until injury stimulates their renewed proliferation to regenerate surrounding damaged muscle tissue (de Morree et al., 2017; Goel et al., 2017; Yue et al., 2017). Similarly, other stem cells, as well as differentiated fibroblasts and hepatocytes, also exit quiescence and proliferate during wound healing (Bangru et al., 2018; Fabris et al., 2019; Urban et al., 2019). In T lymphocytes, quiescence and the timely exit from arrest upon binding of a cognate antigen to the T cell receptor are essential for mounting an appropriate immune response (Hwang et al., 2020). Quiescence is also important in oocytes for the maintenance of reproductive capacity. Oocytes in female mammals become arrested at Prophase I of meiosis, in contrast to other cell types that enter quiescence in G0 (described below). This arrest occurs prenatally and oocytes are maintained in a quiescent state that can persist for decades (Adhikari et al., 2010; Kim and Kurita, 2018). Exit from quiescence and the continuation of meiosis occurs during each reproductive cycle for only a small number of oocytes (Arroyo et al., 2020).
Thus, quiescent cells are diverse and perform various functions throughout the organism. To fulfill these functions, cells must regulate their entry into and exit from quiescence and enact multiple changes to their gene expression, metabolism, and cellular organization. In the following sections, we draw on examples from this diversity of quiescent cell types, focusing on mammalian systems, to highlight the importance of quiescence for organismal fitness, and address the functions, regulation, and features of this fascinating cell state.
Quiescence and the control of cell cycle progression
To enable a reversible state of arrest, cells must tightly control the entry into quiescence, the maintenance of this state, and the exit of cells from quiescence. These steps require the action of key cell cycle regulators, which in turn respond to inputs from upstream factors, including extracellular and intracellular signals.
Quiescent cells are not actively proliferating and thus must temporarily halt their progression through the cell cycle. For most quiescent cells, this arrest takes place in G0, a resting phase outside of the cell cycle that occurs prior to S phase, but is distinct from the G1 phase observed in cycling cells (Figure 2). Whether a cell enters G0 or proceeds through G1 to continue cycling is dictated by the regulation of several key factors, including cyclins and cyclin-dependent kinases (CDKs), CDK inhibitors, and the retinoblastoma protein (Rb) (Figure 2). Cyclin-CDK complexes drive cell cycle progression through the phosphorylation of proteins involved in each of the cell cycle phases (Malumbres, 2014). Cyclin D-CDK4/6 and cyclin E-CDK2 complexes promote G1 progression (Aktas et al., 1997). Thus, high levels of cyclin D/E and CDK4/6 increase proliferation by driving passage through G1. Conversely, stimuli that decrease the abundance and activity of these proteins induce quiescence (Aktas et al., 1997).
Figure 2. Cell cycle regulation of the quiescent state.
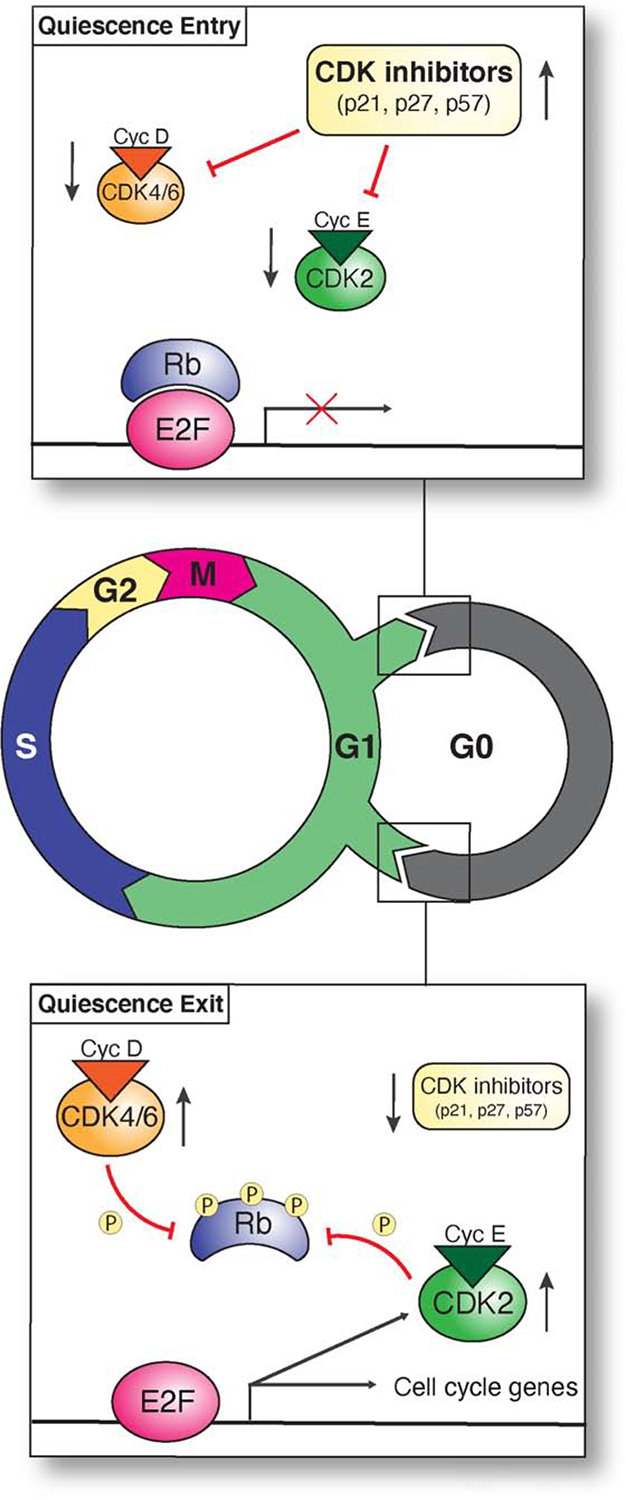
Diagram of the cell cycle, showing the proliferating and G0 quiescent states. Figure includes the key regulatory and transcription factors required to enact each state, including CDK inhibitors, cyclin/CDK complexes, and Rb.
An important mechanism by which cyclin-dependent kinases are regulated is through the expression of CDK inhibitors, including p21 (CDKN1A), p27 (CDKN1B), and p57 (CDKN1C) (Figure 2). CDK inhibitors promote quiescence and quiescent cells typically display high levels of these proteins (Aktas et al., 1997; Arora et al., 2017; Barr et al., 2017; Cheung and Rando, 2013; Fujimaki et al., 2019; Li et al., 2019; Wang et al., 2020). For instance, p27 is highly expressed during quiescence in cultured fibroblasts (Oki et al., 2014). Decreases in the activity or levels of CDK inhibitors can lead to exit from quiescence and re-entry into the cell cycle (Arora et al., 2017; Barr et al., 2017; Wang et al., 2020) (Figure 2). In human epithelial cell lines, genetic deletion of CDKN1A (p21) limits a cell’s ability to enter quiescence, as these cells are unable to achieve sufficiently low levels of CDK2 activity (Arora et al., 2017; Barr et al., 2017; Spencer et al., 2013). Thus, due to the critical regulatory role of these cell cycle factors, quiescence-inducing signals typically act to decrease cyclin/CDK activity or increase CDK inhibitor levels.
A major target of CDK4/6 phosphorylation and a central player in the proliferation-quiescence decision is the retinoblastoma protein (RB1). Rb inhibits proliferation by binding to and inactivating E2F1, a key transcriptional activator for cell cycle and cell division genes (Cheung and Rando, 2013; Yao et al., 2008) (Figure 2). The phosphorylation of Rb by cyclin D-Cdk4 prevents its ability to repress E2F, allowing for cell cycle entry (Yao et al., 2008) (Figure 2). E2F activity is sufficient to induce quiescent cells to enter the S phase, whereas preventing E2F activation inhibits cell cycle reentry (Yao et al., 2008). Thus, the Rb-E2F pathway acts as a key bistable switch that integrates graded growth signals into a binary decision for proliferation versus quiescence (Kwon et al., 2017; Wang et al., 2017; Yao et al., 2008).
Interestingly, Rb can also function in maintaining quiescence outside of the E2F pathway. In quiescent cells, hypophosphorylated Rb associates with the Cdh1-bound Anaphase Promoting Complex/cyclosome (APC/CCdh1) to target Skp2, a negative regulator of the p27 CDK inhibitor, for degradation (Binne et al., 2007). This allows p27 to accumulate and further promote the quiescent arrest (Binne et al., 2007). Indeed, APC/CCdh1 activity is necessary to maintain hepatocytes in a quiescent state and its loss results in re-entry of these cells into the cell cycle and subsequent liver failure (Wirth et al., 2004).
Thus, multiple cell cycle and transcriptional factors are involved in a cell’s decision to enter or exit quiescence. As discussed below, intra- and extracellular signals converge to influence the levels of these key regulators to control the quiescent state.
Signals controlling entry to and exit from quiescence
Extracellular signals and the in vivo niche.
Extracellular signals play important roles in regulating cellular quiescence (Figure 3). These signals are often provided by a niche—the in vivo microenvironment in which cells reside. The niche is comprised of surrounding cells, extracellular matrix, and blood vessels, which interact with quiescent cells using soluble factors or through direct contact (Fiore et al., 2018) (Figure 3). Growth factors (also known as mitogens) promote proliferation by inducing signaling cascades that ultimately act to increase cyclin levels or decrease the abundance of CDK inhibitors (Aktas et al., 1997). Niche-derived soluble growth factors are essential for the activation and proliferation of quiescent stem cells following tissue injury. For example, skeletal muscle injury induces the activation and release of hepatocyte growth factor from the surrounding extracellular matrix (ECM), which primes muscle stem cells for proliferation (Rodgers et al., 2014; Rodgers et al., 2017). Conversely, the absence of growth factors in uninjured tissue is important to maintain quiescence and the niche must prevent growth factor release to avoid the loss of quiescence and depletion of stem cell pools (Chakkalakal et al., 2012). In addition to withholding growth factors, the niche prevents inappropriate proliferation by producing quiescence-inducing soluble factors, including TGF-β1 (Batard et al., 2000; Ducos et al., 2000), Wnt4 (Eliazer et al., 2019), IL-6 cytokines (Sampath et al., 2018), and other factors (Delgado et al., 2014; Kalamakis et al., 2019; Sato et al., 2017). Quiescence can also be reinforced through cell-cell contacts (Goel et al., 2017; Liu et al., 2018; Porlan et al., 2014; Rozo et al., 2016; Urban et al., 2019). For instance, cadherin-mediated adhesion between stem cells and their niche maintains stem cell quiescence, whereas disrupting adherens junctions leads to stem cell activation. During injury, the physical disruption of cell-cell adhesion junctions relieves contact inhibition and allows for the proliferation of quiescent stem cells needed for tissue repair (Goel et al., 2017). Thus, proper regulation of signals arising from the niche is essential for maintaining quiescence in various physiological contexts.
Figure 3. Signals controlling quiescence.
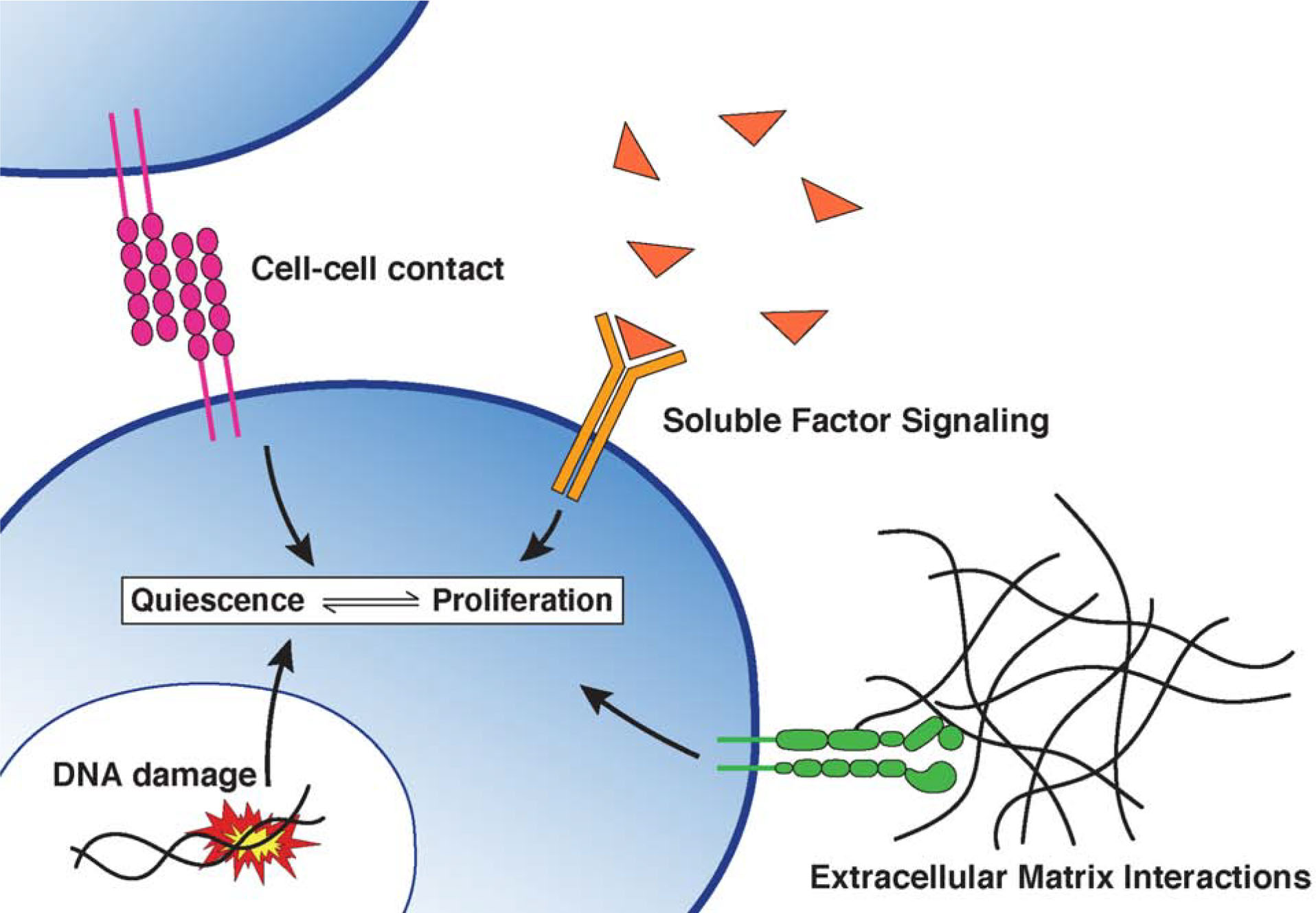
Model showing the extracellular and intracellular signals that contribute to a cell’s quiescence-proliferation decision. Contact-dependent interactions, DNA damage, and certain soluble factors promote quiescence, whereas growth factor signaling and extracellular matrix interactions stimulate proliferation.
Extracellular signals in vitro.
As with quiescent cells in the body, cultured cells integrate a variety of extracellular cues from their surroundings to control cell state decisions. These signals underlie the experimental strategies used to induce quiescence in vitro, such as serum starvation, the loss of adhesion, or contact inhibition (Coller et al., 2006; Mitra et al., 2018). Serum is commonly used as a supplement to cell growth medium and contains a combination of growth factors (Barr et al., 2017; Coller et al., 2006; Mitra et al., 2018). Just as the absence of growth factor secretion from the niche reinforces quiescence, growth factor deprivation through serum starvation induces quiescence by preventing the activation of proliferative growth factor signaling pathways (Barr et al., 2017; Coller et al., 2006; Mitra et al., 2018). Anchorage loss also causes the loss of pro-proliferative signaling. Many cell types rely on adhesion to the extracellular matrix (ECM) to grow and survive. The interaction of cell-surface integrins with ECM proteins activates signaling to promote cell cycle progression (Dike and Farmer, 1988; Fiore et al., 2018; Schwartz and Assoian, 2001). The loss of cell adhesion that occurs by placing cells in suspension culture therefore removes proliferative signals and promotes cell cycle exit (Dike and Farmer, 1988; Fiore et al., 2018; Subramaniam et al., 2014). In contrast, contact inhibition functions directly as an anti-proliferative signal. Even in the presence of serum-derived growth factors, cellular contact in confluent cultures causes a growth arrest (Fiore et al., 2018; Gos et al., 2005). For example, cadherin-mediated contact inhibition can arrest growth through signaling that elevates the levels of CDK inhibitors (Levenberg et al., 1999). In each of these cases, restoring growth-promoting signals can reactivate cells from quiescence. Plating suspended cells onto ECM proteins, adding growth factors to serum-starved cells, or sub-culturing cells to a lower population density promotes renewed division.
Serum deprivation, the loss of adhesion, and contact inhibition induce quiescence with varying degrees of efficiency depending on the cell type. Some cells, such as fibroblasts, enter quiescence is response to all three cues (Coller et al., 2006). However, other cell types may undergo permanent arrest or cell death under certain conditions. For instance, mitogen deprivation in cultured myoblasts triggers an irreversible cell cycle arrest and differentiation (Subramaniam et al., 2014). A reversible quiescent arrest is only achieved if myoblasts are grown in mitogen-rich suspension culture, which triggers quiescence through loss of adhesion (Subramaniam et al., 2014). Conversely, nonadherent culture leads to cell death in epithelial cells, which instead require contact inhibition for quiescence. In human bone marrow mesenchymal stem cells, contact inhibition is inefficient in suppressing proliferation, but these cells can be forced into quiescence by serum starvation (Li et al., 2019). Thus, in vitro extracellular signals induce quiescence in cultured cells, with different cues having distinct effects on cell state depending on the cell type.
Intracellular signals.
Even in the presence of proliferative extracellular cues, a subset of cells may still enter quiescence due to intracellular signals (Figure 3). Indeed, 20–30% of cultured MCF10A cells grown in full growth media exit the cell cycle and enter a transient quiescent state immediately following mitosis (Arora et al., 2017; Spencer et al., 2013). This spontaneous arrest can be attributed to unresolved endogenous DNA replication stress inherited from the S phase of the previous cell cycle (Arora et al., 2017; Barr et al., 2017). Accordingly, the subset of cells that enters quiescence displays higher levels of DNA damage than those that proceed through the cell cycle (Arora et al., 2017). In addition, the amount of time cells spend in quiescence correlates with the extent of inherited DNA damage (Arora et al., 2017). Thus, quiescence may be induced by the transmission of replication damage across generations of cells, allowing daughter cells to prepare for DNA damage repair and to maintain genomic stability (Arora et al., 2017; Barr et al., 2017).
Integrating signals.
To specify the quiescence-proliferation decision, it was originally thought that signals were sensed and integrated only during the G1 phase of the current cell cycle. However, recent work points towards the emerging view that cell state is not determined solely during this short window of time (Arora et al., 2017; Min et al., 2020; Wang et al., 2017). Not only are signals integrated from other phases of the cell cycle, but information can also be transmitted across several generations to influence a cell’s decision to enter quiescence. For example, as detailed above, DNA damage incurred in previous cell cycles can affect a daughter cell’s decision to enter quiescence (Arora et al., 2017; Barr et al., 2017). In addition, recent work has shown that growth factor signaling is integrated throughout the entire cell cycle of a mother cell and can influence the proliferation-quiescence decision of the next generation of cells (Min et al., 2020). This study demonstrated that inhibition of mitogenic signaling for as little as one hour, during any phase of the mother cell cycle, reduces the fraction of proliferating daughter cells by regulating the rate of Cyclin D translation (Min et al., 2020). Thus, both intracellular and extracellular signals can exert intergenerational effects on cell state and a cell integrates memory of events from its history to influence its quiescence-proliferation decision (Arora et al., 2017; Min et al., 2020; Wang et al., 2017).
Transcriptional and post-transcriptional control of quiescence
Quiescent cells must enact multiple changes to their cell state (Figure 4). In this section, we will describe the transcriptional and post-transcriptional programs required to create these changes.
Figure 4. Changes to cell state in quiescence.
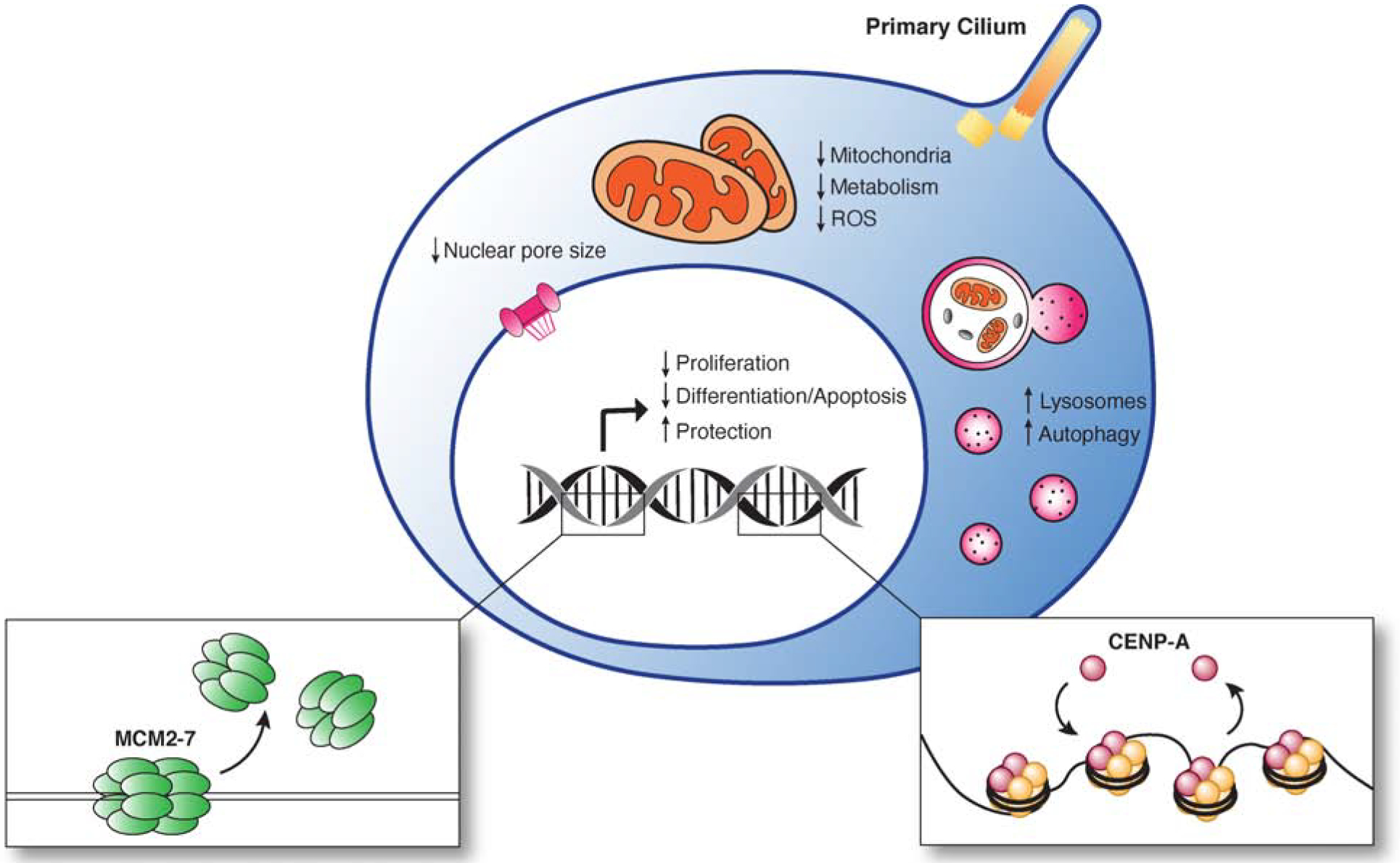
Diagram showing the transcriptional, metabolic, and physical changes to quiescent cells, including those that enable them to preserve their proliferative potential and ensure the reversibility of cell cycle arrest.
The quiescence transcriptional program.
The transcriptional program for quiescence must first and foremost halt the cell cycle and prevent proliferation (Figure 4). Entry into quiescence is accompanied by the downregulation of genes that directly promote cell cycle progression, including several cyclins, and of genes involved in mitogenic signal transduction, such as kinases that act in growth factor pathways (Coller et al., 2006). Reciprocally, genes that promote a cell cycle arrest, such as Rb or CDK inhibitors, are upregulated upon the induction of quiescence (Coller et al., 2006; Fukada et al., 2007). In addition to decreasing cell division, changes in gene expression also alter other cellular functions (discussed below). For instance, quiescent gene expression programs decrease biosynthetic activity, reducing the expression of genes required for DNA, RNA, and protein synthesis, as well as those crucial for lipid and carbohydrate metabolism (Cheung and Rando, 2013; Coller et al., 2006; Liang et al., 2020). Conversely, genes involved in intercellular signaling, such as cell-cell adhesion molecules and cell surface receptors, are upregulated in quiescent cells (Cheung and Rando, 2013; Coller et al., 2006; Fukada et al., 2007; Johnson et al., 2017; Min and Spencer, 2019; Subramaniam et al., 2014). Furthermore, the quiescence transcriptional program helps prevent progression into other cell states, such as terminal differentiation (Coller et al., 2006; Fukada et al., 2007; Subramaniam et al., 2014) (Figure 4). For example, muscle stem cells upregulate the expression of genes involved in preventing myogenic differentiation (Fukada et al., 2007; Subramaniam et al., 2014). Gene regulation in quiescent cells also prevents apoptosis and protects cells from accumulating damage over time (Coller et al., 2006; Min and Spencer, 2019). Accordingly, genes required to induce cell death display decreased expression, whereas those that protect cells from free radicals and environmental chemicals are upregulated (Cheung and Rando, 2013; Coller et al., 2006; Min and Spencer, 2019; Subramaniam et al., 2014) (Figure 4).
Heterogeneity in quiescent gene expression.
Although the transcriptional program outlined above is generally shared across quiescent cell types, there is also substantial heterogeneity in gene expression across quiescent cells (Coller et al., 2006; Gos et al., 2005; Kwon et al., 2017; Min and Spencer, 2019; Rodgers et al., 2014; Urban et al., 2019; Wang et al., 2017). Qualitative heterogeneity arises in response to different quiescence-inducing signals. Cells of the same cell type driven into quiescence through mitogen deprivation, contact inhibition, or loss of adhesion each display distinct, signal-dependent gene expression patterns (Barr et al., 2017; Coller et al., 2006; Gos et al., 2005; Min and Spencer, 2019). Conversely, cells of different cell types arrested using the same signal can possess cell-type specific differences in gene expression. For instance, suspension-induced quiescent myoblasts and fibroblasts share a core transcriptional profile for quiescence (Subramaniam et al., 2014), but also display a significant number of unique genes that are enriched in each cell type (Subramaniam et al., 2014). For example, myoblasts induce components of the Wnt signaling pathway during quiescence, whereas fibroblasts do not (Subramaniam et al., 2014).
Quantitative heterogeneity may also arise in quiescent cells based on the “depth” of their quiescence. Cells that have been in quiescence for different periods of time show differences in gene expression (Coller et al., 2006; Fujimaki et al., 2019; Kwon et al., 2017). For instance, cells arrested for 20 days show greater differences in gene expression levels from proliferating cells than do cells arrested for 4 days (Coller et al., 2006). Increasing the time of contact inhibition or serum deprivation “deepens” the quiescent state of fibroblasts by controlling the levels of genes that mediate E2F activation, such as Rb or Cyclin D (Kwon et al., 2017; Wang et al., 2017). Deeper quiescent cells require stronger growth stimulation and a longer time to reenter the cell cycle than cells arrested for shorter periods (Kwon et al., 2017), as greater gene expression changes are needed to reactivate the cell.
Post-transcriptional regulation of quiescence.
In addition to quiescence transcriptional programs, recent work has highlighted a role for post-transcriptional regulation in controlling the quiescent state. For many genes, the rates of mRNA decay differ widely between proliferating and quiescent cells (Johnson et al., 2017). For example, in fibroblasts, the mRNAs of genes involved in RNA processing or ribosome biogenesis show faster rates of decay during quiescence than in proliferation (Johnson et al., 2017). These changes in mRNA stability can be attributed partially to the action of miRNAs, with the levels of specific miRNAs helping to mediate cell state (Cheung et al., 2012; Crist et al., 2012; Johnson et al., 2017; Suh et al., 2012). For example, in muscle stem cells, miR-489 expression reinforces quiescence by suppressing inducers of proliferation. Furthermore, deletion of the miRNA processing enzyme, Dicer, causes spontaneous exit from quiescence (Cheung et al., 2012). Like miRNAs, RNA binding proteins can also promote mRNA decay (de Morree et al., 2017; Galloway et al., 2016; Hausburg et al., 2015; Hwang et al., 2020). For example, in B cells, the ZFP36L RNA binding protein binds to and subsequently decays target mRNAs that encode cell cycle promoting factors, including several cyclins and cyclin-dependent kinases (Galloway et al., 2016). Deletion of these RNA binding proteins in quiescent cells results in increased levels of target mRNAs and uncontrolled progression into the cell cycle (Galloway et al., 2016; Hwang et al., 2020).
Alternative splicing also plays a role in the post-transcriptional regulation of gene expression during quiescence (Bangru et al., 2018; Yue et al., 2020). Specifically, intron-retained transcripts appear to be enriched in quiescent adult stem cells (Yue et al., 2020). In satellite cells, retained introns are prevalent during quiescence, and may serve to dampen the translation of transcripts required for proliferation. Indeed, genes exhibiting intron retention during quiescence include regulators of cell proliferation, splicing, transcription, translation, and metabolism. Importantly, retained introns are rapidly spliced following exit from quiescence and the downregulation of factors involved in intron removal reduces a stem cell’s ability to proliferate (Yue et al., 2020). Such post-transcriptional mechanisms are well suited to regulate quiescence-to-activation transitions, as they allow for more rapid changes in protein production than would occur at the level of transcription alone. In this way, quiescent cells are primed for activation and able to rapidly express the proteins required to proliferate following stimulation. Interestingly, intron retention is increased only in quiescent cells and not in terminally differentiated or senescent cells (Yue et al., 2020). These irreversibly arrested cells may not require intron-retained transcripts to be preserved, as they do not have a similar requirement to rapidly transition to proliferation as do quiescent cells. Thus, intron retention may be a unique way in which quiescent cells maintain their non-dividing, but rapidly reversible state.
Changes to cell state in quiescence
The entry of a cell into quiescence is accompanied by substantial changes to cellular metabolism and physical changes to the cell and its structures. Physical modifications to cellular structures not only directly impact the quiescence-proliferation decision and enforce quiescence, but are also essential in regulating other characteristic aspects of the quiescent state, such as the decrease in metabolic activity or protein synthesis.
Quiescent cell metabolism.
Proliferating cells double their total mass every cell cycle, whereas quiescent cells are no longer dividing. This results in vastly different metabolic needs between quiescent and proliferating cells and requires corresponding metabolic changes. Indeed, the quiescent state is characterized by a substantial decrease in basal metabolic activity, energy production, and biosynthesis (Figure 4). Cellular ATP concentrations are significantly reduced during quiescence (Ho et al., 2017; Rodgers et al., 2014; Zhang et al., 2018), and some quiescent cell types decrease oxidative phosphorylation to instead rely on glycolysis as their primary metabolic pathway (Ho et al., 2017; Mohrin et al., 2018; Mu et al., 2020; Subramaniam et al., 2014; Zhang et al., 2018). Accordingly, both mitochondrial number and activity are reduced in quiescent cells (Figure 4), leading to decreased oxidative metabolism (Ho et al., 2017; Liang et al., 2020; Mohrin et al., 2018; Rodgers et al., 2014; Zhang et al., 2018). For many quiescent cell types, the clearance of mitochondria through autophagy (mitophagy) is responsible for decreasing metabolism and is important for maintaining quiescence (Ho et al., 2017; Zhang et al., 2018). The loss of mitophagy in hematopoietic stem cells (HSCs) results in increased mitochondrial number, higher overall metabolic activity, and aberrant cell cycle re-entry (Ho et al., 2017). Reciprocally, the transition from quiescence to proliferation is accompanied by metabolic upregulation and an increase in mitochondrial biogenesis to meet the increased energy demands of proliferating cells (Ho et al., 2017; Mohrin et al., 2018; Mu et al., 2020). In addition to changes in energy production, activation from quiescence requires increased consumption of extracellular nutrients and anabolic biosynthesis to support cell growth. For instance, proliferating HSCs uptake three times more glucose than quiescent HSCs (Liang et al., 2020), whereas T cells recently activated from quiescence express enzymes that promote nucleotide, lipid, cholesterol, and amino acid biosynthesis (Chapman et al., 2020). Thus, the activation of cells from quiescence is associated with an increase in metabolic activity, consistent with the energetically and biosynthetically demanding processes required for proliferation.
Another metabolic adjustment that occurs during quiescence is an overall decrease in the rate of translation. In the bone marrow, quiescent hematopoietic stem cells synthesize significantly less protein per hour than their cycling counterparts (Signer et al., 2014; Signer et al., 2016). Similarly, muscle stem cells lower their rates of translation during quiescence (Zismanov et al., 2016). This general translational repression is achieved through the inhibitory phosphorylation of translation initiation factors (Zismanov et al., 2016) or the repression of translation elongation factor expression levels (Oulhen et al., 2017). For example, mutant satellite cells unable to phosphorylate eIF2α increase their translation rates and subsequently exit quiescence (Zismanov et al., 2016). The acidification of the cytoplasm resulting from the decrease in oxidative phosphorylation and mitochondrial activity that occurs during quiescence may additionally contribute to reductions in translation (Oulhen et al., 2017).
Although decreased metabolic activity is detectable in most quiescent cells, not all cell types display this behavior. Quiescent fibroblasts are a notable exception and remain highly metabolically active, with comparable rates of glucose consumption and glycolysis to proliferating fibroblasts (Lemons et al., 2010). This deviation can be attributed to the cell-type specific function of fibroblasts as primary synthesizers of extracellular matrix (ECM). Whereas most other quiescent cells lack biosynthetic function, fibroblasts must constantly secrete proteins and other molecules needed for ECM formation (Lemons et al., 2010). Therefore, quiescent fibroblasts must expend energy and increase their biosynthetic activity to produce ECM components. Consistent with this function, ECM-related genes are upregulated in contact-inhibited quiescent fibroblasts (Johnson et al., 2017). Thus, despite residing in the same cell state, quiescent cells can possess diverse changes in metabolism depending on the unique functions and specific requirements of each cell type.
Cellular structures: Membrane-bound organelles.
Quiescence is also accompanied by changes in key cellular structures, including alterations in the abundance and activity of membrane-bound organelles. As mentioned above, quiescent cells show decreased mitochondrial number and activity. Conversely, many quiescent cell types have larger and more abundant lysosomes than their proliferative counterparts, and upregulate many lysosome-associated genes (Fujimaki et al., 2019; Kobayashi et al., 2019; Leeman et al., 2018; Liang et al., 2020; Min and Spencer, 2019) (Figure 4). However, the role of lysosomes in quiescence remains to be fully established. Some researchers have proposed that lysosomes act to degrade growth factor receptors and remove mitogenic signaling to maintain cells in a quiescent state (Kobayashi et al., 2019), or perhaps may play a protective role during quiescence through ROS reduction, mitochondrial degradation, or the elimination of toxic aggregates (Fujimaki et al., 2019; Leeman et al., 2018; Liang et al., 2020). Alternatively, others hypothesize that lysosomes may instead be important for cell cycle reentry. Lysosomal sequestration of cargo in quiescent cells may provide a source of carbon mass, which can then be degraded upon cell cycle entry to provide a burst of energy and resources for proliferation (Leeman et al., 2018; Liang et al., 2020).
Cellular structures: Centrioles, centrosomes, and the primary cilium.
In addition to alterations in membrane-bound organelles, quiescent cells also undergo changes to protein-based physical structures. These include changes to the centrioles, microtubule-based structures that play varying functions depending on cell state (Breslow and Holland, 2019). In proliferating cells, centrioles assemble into centrosomes— icrotubule-organizing centers that regulate spindle assembly during mitosis (Breslow and Holland, 2019). In contrast, in non-dividing cells including quiescent cells, centrioles form the base for the primary cilium (Figure 4), a structure that assembles at the plasma membrane to act as a key sensory structure for cell signaling (Breslow and Holland, 2019; Jaafar Marican et al., 2016; Venugopal et al., 2020). Induction of quiescence triggers centriole migration to the apical surface to initiate formation of the primary cilium, whereas exit from quiescence is accompanied by the shortening and resorption of the cilium (Pitaval et al., 2017; Pugacheva et al., 2007). Overexpression of ciliary disassembly factors induces premature entry into the cell cycle and increases the proportion of proliferating cells in culture (Goto et al., 2017). Reciprocally, failure to dismantle the cilium can delay cell cycle progression and act as a brake to retain cells in quiescence (Goto et al., 2017; Inaba et al., 2016). One way in which primary cilia have been proposed to regulate quiescence is by sequestering the centrioles, thus preventing centrosome formation in mitosis (Goto et al., 2017; Snell and Golemis, 2007; Venugopal et al., 2020). However, this model does not explain the failure of ciliated cells to progress through prior stages of the cell cycle where centrioles do not play functional roles. The observed suppression of cell division by the primary cilium could occur through cilia-mediated dampening of proliferative signaling pathways (Venugopal et al., 2020), as the cilium functions as a key signaling hub for multiple pathways, including the Hedgehog, PDGF, mTOR, Notch, and Wnt signaling pathways (Breslow and Holland, 2019). Cilia may also prevent cell cycle reentry by controlling the levels of the CDK inhibitor, p27, to maintain ciliated cells in a quiescent state (Izawa et al., 2015). The replacement of centrosomes with the centriole-derived primary cilium is a striking example of how a cell can physically adapt to enable transitions between cell states.
Cellular structures: Nuclear pore complexes.
Nuclear pores reside in the nuclear envelope where they function to regulate the transport of molecules between the cytoplasm and the nucleoplasm. Nuclear pore complexes are typically turned over during mitosis (D’Angelo et al., 2009). As arrested cells no longer undergo mitosis, the nuclear pores of quiescent cells are not renewed. Despite this, the overall nuclear pore number remains high in quiescent cells, as the existing complexes are stable, long-lived cellular structures (D’Angelo et al., 2009). However, the rate of nuclear transport decreases in quiescent cells and the size of their nuclear pore transport channels are also reduced compared to proliferating cells (Feldherr and Akin, 1991, 1993) (Figure 4). Indeed, the diameter of the nuclear pore channels in quiescent fibroblast (3T3) cells is less than half that of proliferating cells, measuring around 110 Å and 230 Å respectively (Feldherr and Akin, 1991, 1993). Such a change in pore diameter could reduce the nuclear export of mRNP particles and ribosomal subunits, thereby contributing to the overall decrease in cellular activity observed in quiescence (Feldherr and Akin, 1991, 1993). In addition to decreases in nuclear pore size, quiescent cells redistribute their nuclear pore density along the nuclear envelope, as evidenced by uneven pore distribution and the formation of “pore-free” regions on the envelope that are indicative of low pore density (Maeshima et al., 2006). Such pore-deficient areas on the nuclear envelope could create nuclear regions with reduced nuclear-cytoplasmic transport. The return to the cell cycle following serum stimulation reverses the observed changes in nuclear pore size and density (Feldherr and Akin, 1991, 1993; Maeshima et al., 2006). These changes may decrease nuclear transport during quiescence, which could contribute to the general decrease in cellular activity that occurs during growth arrest.
Ensuring the reversibility of quiescence
Despite remaining arrested in a non-dividing state for extended periods of time, quiescent cells maintain both their viability and their capacity to proliferate upon stimulation. To do so, they must protect themselves from accumulating damage over time and must actively preserve the key structures needed for cell division.
Protection from damage during quiescence.
Quiescent cells typically have low levels of reactive oxygen species (Coller, 2019; Liang et al., 2020; Loeffler et al., 2019; Zhang et al., 2018). Reactive oxygen species (ROS) can damage proteins, nucleic acids, lipids, membranes, and organelles (Redza-Dutordoir and Averill-Bates, 2016). As ROS are generated by the electron transport chain (Redza-Dutordoir and Averill-Bates, 2016), quiescent cells are able to limit the amounts that they produce by lowering their overall metabolism and favoring glycolysis over oxidative phosphorylation (see above) (Figure 4). Furthermore, antioxidant genes, such as superoxide dismutase 3 and peroxiredoxin 4, are upregulated during quiescence to protect cells from ROS-induced damage (Coller et al., 2006; Zhang et al., 2018). Autophagic flux also plays a role in lowering ROS levels and serves to eliminate damaged proteins and dysfunctional organelles during quiescence (Fujimaki et al., 2019; Garcia-Prat et al., 2016) (Figure 4). Inhibiting autophagy in quiescent cells can result in the accumulation of toxic cellular waste and elevated ROS levels, which can induce entry into senescence (Fujimaki et al., 2019; Garcia-Prat et al., 2016). Impaired autophagy in older satellite cells has also been shown to underlie the loss of quiescence and the reduction of stem cell pools in aged muscles (Garcia-Prat et al., 2016).
Preserving proliferative capacity.
In addition to protecting themselves from cellular damage, quiescent cells must safeguard their ability to proliferate upon activation. Despite the absence of DNA replication and chromosome segregation, quiescent cells must enact programs to ensure that both of these events can occur successfully once a cell reenters the cell cycle. The suppression of DNA replication during quiescence requires the downregulation of the replication origin licensing system (Kingsbury et al., 2005; Orr et al., 2010; Xouri et al., 2004), a complex of proteins needed to initiate DNA replication at specific sites throughout the chromosome (Figure 4). These proteins include members of the mini-chromosome maintenance complex (MCM2–7), the replicative helicase that unwinds DNA, and the factors required for its loading at replication origins, such as Cdc6 (Orr et al., 2010; Xouri et al., 2004). Geminin acts as a repressor to block MCM loading onto chromatin (Xouri et al., 2004). Although such a replication inhibitor would be expected to be highly expressed in quiescent cells, Geminin is instead present at low levels (Kingsbury et al., 2005; Xouri et al., 2004). The suppression of Geminin is essential for the reversibility of quiescence, as renewed MCM binding to chromatin in the absence of Geminin is necessary for DNA replication upon the return to the cell cycle (Orr et al., 2010). Ectopic Geminin expression in quiescent cells prevents the re-acquisition of DNA replication during cell cycle reentry (Kingsbury et al., 2005). Therefore, to successfully return to the cell cycle, quiescent cells must actively suppress Geminin (Kingsbury et al., 2005). Repression of Geminin is achieved through the activity of the Cdh1-bound Anaphase Promoting Complex/cyclosome (APC/CCdh1), which is active during G0 and targets Geminin for degradation (Skaar and Pagano, 2008). Interestingly, APC/CCdh1 also mediates the degradation of Cdc6, an essential licensing factor (Mailand and Diffley, 2005). Thus, both an inhibitor of origin licensing (Geminin) and a licensing factor (Cdc6) are substrates of APC/CCdh1-dependent degradation in quiescent cells. However, during activation from quiescence, Cdc6 phosphorylation by CyclinE-CDK2 protects it from APC/CCdh1, thus allowing for the licensing factor to accumulate while Geminin inhibitor levels remain low (Mailand and Diffley, 2005). This staggering allows for the appropriate licensing of origins for DNA replication as cells exit quiescence (Mailand and Diffley, 2005).
Quiescent cells must also preserve their ability to segregate the chromosomes during mitosis. Chromosome segregation is mediated by microtubule attachment to a defined region of the chromosome known as the centromere (McKinley and Cheeseman, 2016). The maintenance of centromeres during quiescence is essential to ensure the correct segregation of chromosomes once a cell reenters the cell cycle. The specification of the centromere region on each chromosome is achieved through the continued presence of protein-based epigenetic marks. As de novo centromere formation is exceptionally rare, loss of these protein factors would prevent any subsequent chromosome segregation. The key players in marking this site on each chromosome are specialized nucleosomes containing the histone H3 variant, CENP-A. Prior work assumed that CENP-A nucleosomes were indefinitely stable, which would create a stringent requirement for quiescent cells to maintain these structures and prevent their damage during quiescence. In contrast, our recent work found that quiescent cells slowly, but continuously incorporate new CENP-A at centromeric regions (Swartz et al., 2019) (Figure 4). This ongoing CENP-A deposition maintains centromere identity throughout indefinitely long periods of arrest, ensuring proper genome inheritance once cells begin to divide again. A similar process of chromatin turnover may occur for other epigenetically-defined loci. Consequently, loss of CENP-A deposition compromises centromere identity in quiescent RPE1 cells and results in chromosome segregation defects upon cell cycle re-entry (Swartz et al., 2019). Similarly, quiescent oocytes also require the maintenance of centromere identity during their extended Prophase I arrest. Blocking new CENP-A deposition in oocytes abrogates their ability to properly segregate their genetic material following hormone-induced activation, resulting in increased chromosome misalignment during meiosis (Swartz et al., 2019). In contrast, terminally differentiated cells, which are permanently arrested and do not undergo further division after exiting the cell cycle, do not maintain centromere identity. For example, C2C12 muscle myotubes in culture and heart muscle cells in adult mice are depleted for CENP-A, indicating that centromeres are not maintained in these terminally differentiated cells (Swartz et al., 2019). Thus, active centromere maintenance occurs specifically in quiescent cells to preserve their capacity for future division. This difference in CENP-A levels could potentially be used a biomarker to distinguish quiescent cells from other non-dividing cell states. Together, these measures allow a quiescent cell to remain undamaged and preserve the structures and capabilities necessary for future cell cycle progression, protecting the reversibility of cell cycle arrest.
Dysregulation of quiescence
As quiescent cells are essential and widespread throughout the body (Figure 1), the maintenance of the quiescent state and exit of cells from arrest at the appropriate time are vital for the health of an organism. Dysregulation of this delicate balance between quiescence and proliferation can have drastic pathological consequences. For example, inappropriate loss of quiescence in muscle stem cells can lead to spontaneous exit from the quiescent state and depletion of the muscle stem cell pool, ultimately resulting in defective muscle regeneration following injury (Cheung et al., 2012; Garcia-Prat et al., 2016; Wang et al., 2018; Yue et al., 2017). Limiting spontaneous exit from quiescence is also essential in T cells. The loss of negative immune regulators that maintain T cells in a quiescent state makes them overly sensitive to activation signals, which leads to decreased self-tolerance and increased autoimmune disease (ElTanbouly et al., 2020; Hwang et al., 2020). Similarly, deregulation of the signaling pathways that maintain quiescence in mouse oocytes causes global activation of primordial follicles and depletion of the entire oocyte pool in mice, causing premature ovarian failure and infertility (Adhikari et al., 2010). Reciprocally, the inability to exit quiescence is also problematic. For instance, suppressing quiescence exit in muscle stem cells results in severely impaired stem cell proliferation and decreased muscle regeneration (Wang et al., 2018). A loss of the balance between quiescence and proliferation can thus lead to dysfunction in tissue regeneration, immunity, or fertility.
Quiescence in aging.
The capacity for dysregulation of quiescence to lead to pathology is exemplified in aging. Muscle fibers in geriatric individuals undergo age-related degradation, as dysregulation of quiescence in tissue-resident muscle stem cells inhibits regeneration. The exact means by which quiescence is disrupted in aging satellite cells is unclear. Satellite cells have been proposed to become unable to exit quiescence with age, losing the ability to reverse their arrest and essentially becoming senescent (Garcia-Prat et al., 2016; Sousa-Victor et al., 2014). In this model, aged muscle stem cells fail to activate upon injury and cannot proliferate to renew injured tissue (Sousa-Victor et al., 2014). Conversely, other studies have observed excessive satellite cell reentry into the cell cycle during aging. Such widespread exit from quiescence would cause depletion of the reserve stem cell pool in elderly muscle and has been hypothesized to arise from unique age-related epigenetic modifications (Bigot et al., 2015) or changes in the surrounding muscle niche (Chakkalakal et al., 2012). Similarly, quiescent cell depletion may also underlie hair thinning during aging (Lay et al., 2016; Matsumura et al., 2016; Wang et al., 2016). Recent work in mice has shown that hair loss can occur as a result of excessive hair follicle stem cell (HFSC) activation, which in turn leads to loss of stem cell numbers and a delay in the regeneration of new hairs (Lay et al., 2016; Wang et al., 2016). Dysregulation of quiescence has also been implicated in the cognitive decline that occurs with age (Kalamakis et al., 2019). Neural stem cells (NSCs) in the aging brain show a decreased ability to exit quiescence, and thus are more resistant to injury-induced activation and are less capable of neurogenesis and repair (Kalamakis et al., 2019).
Another quiescent cell type that is affected during aging is the oocyte. Oocytes are arrested in Prophase I prior to birth and must remain quiescent for up to 50 years. However, during female reproductive aging, oocytes gradually begin to lose the capacity to successfully segregate their chromosomes, such that the rate of erroneous chromosome segregation and aneuploidy as they reenter meiosis increases markedly (Chiang et al., 2010). These errors underlie the well-documented relationship between the incidence of developmental disorders and maternal age, as well as the increases in miscarriages and infertility in older females. The age-related decline in oocyte function can be attributed to the loss of certain cellular structures. For example, the cohesin protein complex is essential to connect sister chromatids during chromosome segregation. During oocyte aging, cohesin proteins are gradually lost from the chromosomes (Chiang et al., 2010; Chiang et al., 2012; Lister et al., 2010). As a result, older quiescent oocytes lose chromosome cohesion, which can result in premature chromosome separation or misorientation of spindle attachments during meiosis (Chiang et al., 2012). In addition, loss of cohesin can also lead to decompaction of the centromeres and fragmentation of the kinetochores in aged oocytes (Zielinska et al., 2019). Kinetochore fragmentation, in turn, results in incorrect microtubule attachments during meiosis, increasing chromosome mis-segregation (Zielinska et al., 2019). These examples highlight the importance of preserving cellular structures to the reversibility of quiescent arrest and how the failure to do so can result in physiological consequences.
Quiescence in cancer.
Although typically considered to be highly proliferative cells, many cancer cells can also enter a quiescent state. During tumorigenesis, cancer cells can leave the primary tumor to colonize distant tissues (Goddard et al., 2018). These cells, known as disseminated tumor cells (DTCs), can remain quiescent in tissues for up to 20 years before reentering the cell cycle to initiate metastatic growth when conditions become favorable for their proliferation (Goddard et al., 2018). Non-dividing quiescent cancer cells are also resistant to most chemotherapies, which typically target features of actively proliferating cells (Chen et al., 2016). Thus, the entry of a small number of cancer cells into a quiescent state can negatively affect disease outcome, as drug-resistant dormant cancer cells can survive therapy and exit quiescence at later times to seed new tumor formation (Chen et al., 2016). To circumvent this problem, recent therapeutic strategies have attempted to manipulate quiescence to decrease the rate of cancer recurrence. “Lock out” strategies force cancer cells out of quiescence, which renders them susceptible to other chemotherapies (Chen et al., 2016; Saito et al., 2010; Takeishi et al., 2013). Conversely, “lock in” treatments seek to block quiescent cell reentry into the cell cycle to limit tumor outgrowth (Chen et al., 2016). Therefore, a combination of drugs that influence quiescence and classical chemotherapeutics could be used to reduce the overall rate of relapse arising from quiescent cancer cells. Given the important roles of quiescent cells in the body and the effects of their dysregulation, a deeper understanding of the quiescent state is a critical step towards the development of therapies that could fundamentally improve human health.
Future Directions and Concluding Remarks
The reversible growth arrest of diverse quiescent cell types throughout the body and the regulation of quiescence are critical for organismal health and reproduction. However, despite recent work, some areas of quiescence remain poorly understood. An important current challenge in studying quiescent cells and monitoring changes in cell state is the lack of quiescence-specific markers. Methods to identify quiescent cells typically rely on characteristics that distinguish non-dividing from proliferating cells, such as label retention, or the expression of certain cell cycle regulators. For instance, an mVenus-p27 fluorescent construct or a live-cell CDK2 sensor can be used to mark quiescent cells, which exhibit characteristic expression levels or activities of these proteins (Oki et al., 2014; Spencer et al., 2013). However, although these reporters can separate quiescent from proliferating cells, a molecular marker that would accurately distinguish between quiescence and other non-dividing cell states, such as senescence or terminal differentiation, has yet to be established. Further investigation of the factors necessary for renewed proliferation after arrest, such as the centromere protein CENP-A, may be a fruitful strategy to develop candidate markers. Factors required for the reversibility of quiescence, when combined with previously developed molecular markers, may offer a more thorough identification of quiescent cells.
Second, much of the current knowledge on the quiescent state comes from work conducted in tissue culture cells. Although tissue culture systems have been crucial towards our understanding of quiescence, they cannot encompass the complexity found in an organism, including the myriad of signals that arise from the niche. Thus, some of the findings highlighted in this review still need to be substantiated in complex systems, such as whole animals or organoids. For example, recent work found that cells retain information from past cell cycles to influence their quiescence decision in vitro (Min et al., 2020). It would of interest to evaluate whether this also occurs in vivo. Similarly, much of the work detailing the heterogeneity of gene expression in cultured quiescent fibroblasts should be re-evaluated in an organism.
In addition to considerations regarding the markers and systems used to study quiescence, several key concepts in the field remain to be investigated. Post-transcriptional mechanisms undoubtedly play an important role in the regulation of the quiescent state, as they allow for rapid changes in protein levels necessary for rapid changes in cell state. Thus, it will be of interest to uncover other post-transcriptional mechanism at work in quiescent cells. For example, intron retention has recently been implicated in regulating quiescence (Yue et al., 2020). However, it is still unknown whether other forms of alternative splicing or varied use of isoforms are present. Similarly, it is important to determine whether post-translational modifications impact the quiescent state. With respect to the physiology of quiescent cells themselves, little attention has been devoted to discovering the changes to physical structures that occur during quiescence, a key consideration for their function and ability to persist in a non-dividing state. Finally, a defining feature of quiescence that renders it unique is the ability of quiescent cells to return to the cell cycle. Yet, the mechanisms that preserve the processes necessary for renewed proliferation are still mostly unknown. The field would thus greatly benefit from uncovering how quiescent cells are able to successfully reverse their state of arrest.
Together, these methodological and conceptual advancements would contribute to a greater understanding of this crucial cell state.
Acknowledgments
The authors thank Sabrina Spencer, Humza Ashraf, Kara McKinley, Kristin Knouse, Zak Swartz, and Sean-Luc Shanahan for critical reading of the manuscript and helpful discussions, and Kristin Knouse and Zak Swartz for generously sharing images. Work in the Cheeseman laboratory is supported by grants from The Harold G & Leila Y. Mathers Charitable Foundation to IMC, the NIH/National Institute of General Medical Sciences (R35GM126930), the Gordon and Betty Moore Foundation, and the Global Consortium for Reproductive Longevity and Equity (GCRLE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Iain Cheeseman is a member of the Editorial Advisory Board for Developmental Cell.
References
- Adhikari D, Zheng W, Shen Y, Gorre N, Hamalainen T, Cooney AJ, Huhtaniemi I, Lan ZJ, and Liu K (2010). Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet 19, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas H, Cai H, and Cooper GM (1997). Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol 17, 3850–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora M, Moser J, Phadke H, Basha AA, and Spencer SL (2017). Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep 19, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo A, Kim B, and Yeh J (2020). Luteinizing Hormone Action in Human Oocyte Maturation and Quality: Signaling Pathways, Regulation, and Clinical Impact. Reprod Sci 27, 1223–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangru S, Arif W, Seimetz J, Bhate A, Chen J, Rashan EH, Carstens RP, Anakk S, and Kalsotra A (2018). Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat Struct Mol Biol 25, 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novak B, and Bakal C (2017). DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun 8, 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batard P, Monier MN, Fortunel N, Ducos K, Sansilvestri-Morel P, Phan T, Hatzfeld A, and Hatzfeld JA (2000). TGF-(beta)1 maintains hematopoietic immaturity by a reversible negative control of cell cycle and induces CD34 antigen up-modulation. J Cell Sci 113 (Pt 3), 383–390. [DOI] [PubMed] [Google Scholar]
- Bigot A, Duddy WJ, Ouandaogo ZG, Negroni E, Mariot V, Ghimbovschi S, Harmon B, Wielgosik A, Loiseau C, Devaney J, et al. (2015). Age-Associated Methylation Suppresses SPRY1, Leading to a Failure of Re-quiescence and Loss of the Reserve Stem Cell Pool in Elderly Muscle. Cell Rep 13, 1172–1182. [DOI] [PubMed] [Google Scholar]
- Binne UK, Classon MK, Dick FA, Wei W, Rape M, Kaelin WG Jr., Naar AM, and Dyson NJ (2007). Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat Cell Biol 9, 225–232. [DOI] [PubMed] [Google Scholar]
- Breslow DK, and Holland AJ (2019). Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu Rev Biochem 88, 691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal JV, Jones KM, Basson MA, and Brack AS (2012). The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Boothby MR, and Chi H (2020). Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol 20, 55–70. [DOI] [PubMed] [Google Scholar]
- Chen W, Dong J, Haiech J, Kilhoffer MC, and Zeniou M (2016). Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int 2016, 1740936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, and Rando TA (2012). Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 482, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, and Rando TA (2013). Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 14, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Duncan FE, Schindler K, Schultz RM, and Lampson MA (2010). Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr Biol 20, 1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Schultz RM, and Lampson MA (2012). Meiotic origins of maternal age-related aneuploidy. Biol Reprod 86, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA (2019). The paradox of metabolism in quiescent stem cells. FEBS Lett 593, 2817–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA, Sang L, and Roberts JM (2006). A new description of cellular quiescence. PLoS Biol 4, e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crist CG, Montarras D, and Buckingham M (2012). Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell 11, 118–126. [DOI] [PubMed] [Google Scholar]
- D’Angelo MA, Raices M, Panowski SH, and Hetzer MW (2009). Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Morree A, van Velthoven CTJ, Gan Q, Salvi JS, Klein JDD, Akimenko I, Quarta M, Biressi S, and Rando TA (2017). Staufen1 inhibits MyoD translation to actively maintain muscle stem cell quiescence. Proc Natl Acad Sci U S A 114, E8996–E9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado AC, Ferron SR, Vicente D, Porlan E, Perez-Villalba A, Trujillo CM, D’Ocon P, and Farinas I (2014). Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron 83, 572–585. [DOI] [PubMed] [Google Scholar]
- Dike LE, and Farmer SR (1988). Cell adhesion induces expression of growth-associated genes in suspension-arrested fibroblasts. Proc Natl Acad Sci U S A 85, 6792–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducos K, Panterne B, Fortunel N, Hatzfeld A, Monier MN, and Hatzfeld J (2000). p21(cip1) mRNA is controlled by endogenous transforming growth factor-beta1 in quiescent human hematopoietic stem/progenitor cells. J Cell Physiol 184, 80–85. [DOI] [PubMed] [Google Scholar]
- Eliazer S, Muncie JM, Christensen J, Sun X, D’Urso RS, Weaver VM, and Brack AS (2019). Wnt4 from the Niche Controls the Mechano-Properties and Quiescent State of Muscle Stem Cells. Cell Stem Cell 25, 654–665 e654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElTanbouly MA, Zhao Y, Nowak E, Li J, Schaafsma E, Le Mercier I, Ceeraz S, Lines JL, Peng C, Carriere C, et al. (2020). VISTA is a checkpoint regulator for naive T cell quiescence and peripheral tolerance. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabris G, Dumortier O, Pisani DF, Gautier N, and Van Obberghen E (2019). Amino acid-induced regulation of hepatocyte growth: possible role of Drosha. Cell Death Dis 10, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldherr CM, and Akin D (1991). Signal-mediated nuclear transport in proliferating and growth-arrested BALB/c 3T3 cells. J Cell Biol 115, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldherr CM, and Akin D (1993). Regulation of nuclear transport in proliferating and quiescent cells. Exp Cell Res 205, 179–186. [DOI] [PubMed] [Google Scholar]
- Fiore A, Ribeiro PF, and Bruni-Cardoso A (2018). Sleeping Beauty and the Microenvironment Enchantment: Microenvironmental Regulation of the Proliferation-Quiescence Decision in Normal Tissues and in Cancer Development. Front Cell Dev Biol 6, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimaki K, Li R, Chen H, Della Croce K, Zhang HH, Xing J, Bai F, and Yao G (2019). Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc Natl Acad Sci U S A 116, 22624–22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto H, Miyagoe-Suzuki Y, and Takeda S (2007). Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 25, 2448–2459. [DOI] [PubMed] [Google Scholar]
- Galloway A, Saveliev A, Lukasiak S, Hodson DJ, Bolland D, Balmanno K, Ahlfors H, Monzon-Casanova E, Mannurita SC, Bell LS, et al. (2016). RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science 352, 453–459. [DOI] [PubMed] [Google Scholar]
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. [DOI] [PubMed] [Google Scholar]
- Goddard ET, Bozic I, Riddell SR, and Ghajar CM (2018). Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol 20, 1240–1249. [DOI] [PubMed] [Google Scholar]
- Goel AJ, Rieder MK, Arnold HH, Radice GL, and Krauss RS (2017). Niche Cadherins Control the Quiescence-to-Activation Transition in Muscle Stem Cells. Cell Rep 21, 2236–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gos M, Miloszewska J, Swoboda P, Trembacz H, Skierski J, and Janik P (2005). Cellular quiescence induced by contact inhibition or serum withdrawal in C3H10T1/2 cells. Cell Prolif 38, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Inaba H, and Inagaki M (2017). Mechanisms of ciliogenesis suppression in dividing cells. Cell Mol Life Sci 74, 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, and Locksley RM (2001). Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity 14, 205–215. [DOI] [PubMed] [Google Scholar]
- Hausburg MA, Doles JD, Clement SL, Cadwallader AB, Hall MN, Blackshear PJ, Lykke-Andersen J, and Olwin BB (2015). Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. Elife 4, e03390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegue E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SS, Lim J, Yu Z, Kong P, Sefik E, Xu H, Harman CCD, Kim LK, Lee GR, Li HB, et al. (2020). mRNA destabilization by BTG1 and BTG2 maintains T cell quiescence. Science 367, 1255–1260. [DOI] [PubMed] [Google Scholar]
- Inaba H, Goto H, Kasahara K, Kumamoto K, Yonemura S, Inoko A, Yamano S, Wanibuchi H, He D, Goshima N, et al. (2016). Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein-Aurora A pathway. J Cell Biol 212, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa I, Goto H, Kasahara K, and Inagaki M (2015). Current topics of functional links between primary cilia and cell cycle. Cilia 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaafar Marican NH, Cruz-Migoni SB, and Borycki AG (2016). Asymmetric Distribution of Primary Cilia Allocates Satellite Cells for Self-Renewal. Stem Cell Reports 6, 798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EL, Robinson DG, and Coller HA (2017). Widespread changes in mRNA stability contribute to quiescence-specific gene expression patterns in a fibroblast model of quiescence. BMC Genomics 18, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamakis G, Brune D, Ravichandran S, Bolz J, Fan W, Ziebell F, Stiehl T, Catala-Martinez F, Kupke J, Zhao S, et al. (2019). Quiescence Modulates Stem Cell Maintenance and Regenerative Capacity in the Aging Brain. Cell 176, 1407–1419 e1414. [DOI] [PubMed] [Google Scholar]
- Kim SY, and Kurita T (2018). New Insights into the Role of Phosphoinositide 3-Kinase Activity in the Physiology of Immature Oocytes: Lessons from Recent Mouse Model Studies. Eur Med J Reprod Health 3, 119–125. [PMC free article] [PubMed] [Google Scholar]
- Kingsbury SR, Loddo M, Fanshawe T, Obermann EC, Prevost AT, Stoeber K, and Williams GH (2005). Repression of DNA replication licensing in quiescence is independent of geminin and may define the cell cycle state of progenitor cells. Exp Cell Res 309, 56–67. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Piao W, Takamura T, Kori H, Miyachi H, Kitano S, Iwamoto Y, Yamada M, Imayoshi I, Shioda S, et al. (2019). Enhanced lysosomal degradation maintains the quiescent state of neural stem cells. Nat Commun 10, 5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JS, Everetts NJ, Wang X, Wang W, Della Croce K, Xing J, and Yao G (2017). Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep 20, 3223–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay K, Kume T, and Fuchs E (2016). FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc Natl Acad Sci U S A 113, E1506–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, Dulken BW, Zhao X, Yeo RW, Ho TT, et al. (2018). Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, and Coller HA (2010). Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol 8, e1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenberg S, Yarden A, Kam Z, and Geiger B (1999). p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene 18, 869–876. [DOI] [PubMed] [Google Scholar]
- Li B, Sun C, Sun J, Yang MH, Zuo R, Liu C, Lan WR, Liu MH, Huang B, and Zhou Y (2019). Autophagy mediates serum starvation-induced quiescence in nucleus pulposus stem cells by the regulation of P27. Stem Cell Res Ther 10, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, et al. (2020). Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 26, 359–376 e357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y, et al. (2010). Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol 20, 1511–1521. [DOI] [PubMed] [Google Scholar]
- Liu YF, Zhang SY, Chen YY, Shi K, Zou B, Liu J, Yang Q, Jiang H, Wei L, Li CZ, et al. (2018). ICAM-1 Deficiency in the Bone Marrow Niche Impairs Quiescence and Repopulation of Hematopoietic Stem Cells. Stem Cell Reports 11, 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, and Martin-Villalba A (2015). Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell 17, 329–340. [DOI] [PubMed] [Google Scholar]
- Loeffler D, Wehling A, Schneiter F, Zhang Y, Muller-Botticher N, Hoppe PS, Hilsenbeck O, Kokkaliaris KD, Endele M, and Schroeder T (2019). Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573, 426–429. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Yahata K, Sasaki Y, Nakatomi R, Tachibana T, Hashikawa T, Imamoto F, and Imamoto N (2006). Cell-cycle-dependent dynamics of nuclear pores: pore-free islands and lamins. J Cell Sci 119, 4442–4451. [DOI] [PubMed] [Google Scholar]
- Mailand N, and Diffley JF (2005). CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 122, 915–926. [DOI] [PubMed] [Google Scholar]
- Malumbres M (2014). Cyclin-dependent kinases. Genome Biol 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura H, Mohri Y, Binh NT, Morinaga H, Fukuda M, Ito M, Kurata S, Hoeijmakers J, and Nishimura EK (2016). Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science 351, aad4395. [DOI] [PubMed] [Google Scholar]
- McKinley KL, and Cheeseman IM (2016). The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 17, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Rong Y, Tian C, and Spencer S (2020). Temporal integration of mitogen history in mother cells controls proliferation of daughter cells. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, and Spencer SL (2019). Spontaneously slow-cycling subpopulations of human cells originate from activation of stress-response pathways. PLoS Biol 17, e3000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra M, Ho LD, and Coller HA (2018). An In Vitro Model of Cellular Quiescence in Primary Human Dermal Fibroblasts. Methods Mol Biol 1686, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Widjaja A, Liu Y, Luo H, and Chen D (2018). The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell 17, e12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu WC, Ohkubo R, Widjaja A, and Chen D (2020). The mitochondrial metabolic checkpoint in stem cell aging and rejuvenation. Mech Ageing Dev 188, 111254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, Sakaue-Sawano A, Niida A, Miyano S, Aburatani H, et al. (2014). A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0-G1 transition. Sci Rep 4, 4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr SJ, Gaymes T, Ladon D, Chronis C, Czepulkowski B, Wang R, Mufti GJ, Marcotte EM, and Thomas NS (2010). Reducing MCM levels in human primary T cells during the G(0)-->G(1) transition causes genomic instability during the first cell cycle. Oncogene 29, 3803–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Swartz SZ, Laird J, Mascaro A, and Wessel GM (2017). Transient translational quiescence in primordial germ cells. Development 144, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitaval A, Senger F, Letort G, Gidrol X, Guyon L, Sillibourne J, and Thery M (2017). Microtubule stabilization drives 3D centrosome migration to initiate primary ciliogenesis. J Cell Biol 216, 3713–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porlan E, Marti-Prado B, Morante-Redolat JM, Consiglio A, Delgado AC, Kypta R, Lopez-Otin C, Kirstein M, and Farinas I (2014). MT5-MMP regulates adult neural stem cell functional quiescence through the cleavage of N-cadherin. Nat Cell Biol 16, 629–638. [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, and Golemis EA (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redza-Dutordoir M, and Averill-Bates DA (2016). Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 1863, 2977–2992. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai CR, et al. (2014). mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature 510, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Schroeder MD, Ma C, and Rando TA (2017). HGFA Is an Injury-Regulated Systemic Factor that Induces the Transition of Stem Cells into GAlert. Cell Rep 19, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozo M, Li L, and Fan CM (2016). Targeting beta1-integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat Med 22, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, et al. (2010). Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol 28, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath SC, Sampath SC, Ho ATV, Corbel SY, Millstone JD, Lamb J, Walker J, Kinzel B, Schmedt C, and Blau HM (2018). Induction of muscle stem cell quiescence by the secreted niche factor Oncostatin M. Nat Commun 9, 1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Uchida Y, Hu J, Young-Pearse TL, Niikura T, and Mukouyama YS (2017). Soluble APP functions as a vascular niche signal that controls adult neural stem cell number. Development 144, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, and Assoian RK (2001). Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci 114, 2553–2560. [DOI] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Qi L, Zhao Z, Thompson D, Sigova AA, Fan ZP, DeMartino GN, Young RA, Sonenberg N, and Morrison SJ (2016). The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev 30, 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, and Pagano M (2008). Cdh1: a master G0/G1 regulator. Nat Cell Biol 10, 755–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell WJ, and Golemis EA (2007). A ciliary timer for S-phase entry. Nat Rev Mol Cell Biol 8. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. (2014). Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321. [DOI] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Sreenivas P, Cheedipudi S, Reddy VR, Shashidhara LS, Chilukoti RK, Mylavarapu M, and Dhawan J (2014). Distinct transcriptional networks in quiescent myoblasts: a role for Wnt signaling in reversible vs. irreversible arrest. PLoS One 8, e65097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh EJ, Remillard MY, Legesse-Miller A, Johnson EL, Lemons JM, Chapman TR, Forman JJ, Kojima M, Silberman ES, and Coller HA (2012). A microRNA network regulates proliferative timing and extracellular matrix synthesis during cellular quiescence in fibroblasts. Genome Biol 13, R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz SZ, McKay LS, Su KC, Bury L, Padeganeh A, Maddox PS, Knouse KA, and Cheeseman IM (2019). Quiescent Cells Actively Replenish CENP-A Nucleosomes to Maintain Centromere Identity and Proliferative Potential. Dev Cell 51, 35–48 e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, and Nakayama KI (2013). Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell 23, 347–361. [DOI] [PubMed] [Google Scholar]
- Urban N, Blomfield IM, and Guillemot F (2019). Quiescence of Adult Mammalian Neural Stem Cells: A Highly Regulated Rest. Neuron 104, 834–848. [DOI] [PubMed] [Google Scholar]
- Venugopal N, Ghosh A, Gala H, Aloysius A, Vyas N, and Dhawan J (2020). The primary cilium dampens proliferative signaling and represses a G2/M transcriptional network in quiescent myoblasts. BMC Mol Cell Biol 21, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhu H, Situ C, Han L, Yu Y, Cheung TH, Liu K, and Wu Z (2018). p110alpha of PI3K is necessary and sufficient for quiescence exit in adult muscle satellite cells. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jin S, Dai P, Zhang T, Shi Y, Ai G, Shao X, Xie Y, Xu J, Chen Z, et al. (2020). p57(Kip2) is a master regulator of human adipose derived stem cell quiescence and senescence. Stem Cell Res 44, 101759. [DOI] [PubMed] [Google Scholar]
- Wang L, Siegenthaler JA, Dowell RD, and Yi R (2016). Foxc1 reinforces quiescence in self-renewing hair follicle stem cells. Science 351, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fujimaki K, Mitchell GC, Kwon JS, Della Croce K, Langsdorf C, Zhang HH, and Yao G (2017). Exit from quiescence displays a memory of cell growth and division. Nat Commun 8, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Nakahara F, Asada N, Zhang D, Gao X, Xu C, Alfieri A, Brodin NP, Zimmerman SE, Mar JC, et al. (2020). Snai2 Maintains Bone Marrow Niche Cells by Repressing Osteopontin Expression. Dev Cell 53, 503–513 e505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth KG, Ricci R, Gimenez-Abian JF, Taghybeeglu S, Kudo NR, Jochum W, Vasseur-Cognet M, and Nasmyth K (2004). Loss of the anaphase-promoting complex in quiescent cells causes unscheduled hepatocyte proliferation. Genes Dev 18, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xouri G, Lygerou Z, Nishitani H, Pachnis V, Nurse P, and Taraviras S (2004). Cdt1 and geminin are down-regulated upon cell cycle exit and are over-expressed in cancer-derived cell lines. Eur J Biochem 271, 3368–3378. [DOI] [PubMed] [Google Scholar]
- Yao G, Lee TJ, Mori S, Nevins JR, and You L (2008). A bistable Rb-E2F switch underlies the restriction point. Nat Cell Biol 10, 476–482. [DOI] [PubMed] [Google Scholar]
- Yi R (2017). Concise Review: Mechanisms of Quiescent Hair Follicle Stem Cell Regulation. Stem Cells 35, 2323–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Bi P, Wang C, Shan T, Nie Y, Ratliff TL, Gavin TP, and Kuang S (2017). Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat Commun 8, 14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Wan R, Luan S, Zeng W, and Cheung TH (2020). Dek Modulates Global Intron Retention during Muscle Stem Cells Quiescence Exit. Dev Cell. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhang X, Wang K, Xu X, Li M, Zhang J, Zhang Y, Hao J, Sun X, Chen Y, et al. (2018). Newly Generated CD4(+) T Cells Acquire Metabolic Quiescence after Thymic Egress. J Immunol 200, 1064–1077. [DOI] [PubMed] [Google Scholar]
- Zielinska AP, Bellou E, Sharma N, Frombach AS, Seres KB, Gruhn JR, Blayney M, Eckel H, Moltrecht R, Elder K, et al. (2019). Meiotic Kinetochores Fragment into Multiple Lobes upon Cohesin Loss in Aging Eggs. Curr Biol 29, 3749–3765 e3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, Koromilas AE, and Crist C (2016). Phosphorylation of eIF2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell 18, 79–90. [DOI] [PubMed] [Google Scholar]