

Abstract

Carbonic anhydrases from Vibrio cholerae (VchCAs) play a significant role in bacterial pathophysiological processes. Therefore, their inhibition leads to a reduction of gene expression virulence and bacterial growth impairment. Herein, we report the first ligand-based pharmacophore model as a computational tool to study selective inhibitors of the β-class of VchCA. By a virtual screening on a collection of sulfonamides, we retrieved 9 compounds that were synthesized and evaluated for their inhibitory effects against VchCAβ as well as α- and γ-classes of VchCAs and selectivity over human ubiquitous isoforms hCA I and II. Notably, all tested compounds were active inhibitors of VchCAs. The N-(4-sulfamoylbenzyl)-[1,1′-biphenyl]-4-carboxamide (20e) stood out as the most exciting inhibitor toward the β-class (Ki = 95.6 nM), also showing a low affinity against the tested human isoforms. By applying docking procedures, we described the binding mode of the inhibitor 20e within the catalytic cavity of the modeled open conformation of VchCAβ.
Keywords: Carbonic anhydrase inhibitors, Vibrio cholerae, ligand-based modeling, sulfonamides, molecular docking
The Gram-negative bacterium Vibrio cholerae (Vch) is the causative agent of cholera, a severe diarrheal disease that is endemic in various Southeast Asian and African countries as well as regions of South America.1,2 This pathology can lead to severe dehydration, metabolic acidosis, and death in the absence of therapeutic intervention. It is well-known that Vch colonizes gastro-intestinal lumen and causes pathological effects by producing virulence factors related to transcriptional regulator ToxT.3−5 Consequently, an emerging challenge is to fight cholera by using antivirulence drug candidates in place of antibiotics.6 It has been established that the ToxT activation is regulated by ion bicarbonate (HCO3–) as intestinal pH buffer secreted by epithelial cells.7 The bicarbonate production is mediated by carbonic anhydrases (CAs, EC 4.2.1.1) that are metalloenzymes catalyzing the reversible hydration of CO2. CAs are a superfamily of enzymes belonging to several classes (α-, β-, γ-, δ-, ζ-, ε-, θ-, and ι-classes) that are diffused in vertebrates, protozoa, algae, and bacteria.8,9 Specifically, the Vch genome encodes α-, β-, and γ-classes named VchCAα, VchCAβ, and VchCAγ, which share a low structural homology with each other.9 The α- and β-CAs are Zn(II) metalloenzymes, whereas γ-CA classes employ Fe(II) in the catalytic site, even if they are also active with Zn(II) or Co(II) metal ions. In detail, the metal ion is coordinated by three His residues in the α- and γ-classes and one His and two Cys residues in the β-class.10
The ability to inhibit VchCA isozymes has been demonstrated by a large series of compounds bearing a zinc binder group (ZBG). Among them, acetazolamide (AAZ) and ethoxzolamide (EZA) (Figure 1) proved to significantly inhibit VchCA isozymes.9,11 Furthermore, EZA decreases the bicarbonate-mediated virulence gene expression and reduces the growth of pathogen; this latter evidence paved the way for the development of VchCA inhibitors as potential therapeutics for treatment of cholera.9,12,13
Figure 1.

Chemical structures of well-known VchCA inhibitors: acetazolamide (AAZ) and ethoxzolamide (EZA).
The design of selective inhibitors targeting the medium/small cavity of VchCA isozymes is a very intriguing challenge. Actually, the best active inhibitors displayed good affinity in the low nanomolar range.11,12,14−21 However, they generally display low selectivity over human off-target α-class CA isoforms (hCA I and hCA II), thus reducing their potential therapeutic interest in humans. The CA inhibitors possess the ZBG linked to a lipophilic cap-group through a suitable “spacer”. Apart from that crucial ZBG moiety, structural studies have suggested that the cap-group can establish relevant contacts with the rim of the CA cavity thus controlling CA isozyme selectivity.
Seeking selective inhibitors targeting the medium/small cavity of VchCA, we focused our interest on β-CA classes which exert catalytic activity as a crucial event for bacterial survival. It has been established that β-CAs are structurally unique from human CAs, so that it has been proposed that the selective β-CA inhibitors might be promising innovative antibacterial agents.22,23 To better understand the binding mode of VchCA inhibitors, we have previously investigated the pose of prototype AAZ bound to the hypothetical cavity located in the interface of a dimeric VchCAβ, that displays a tetrameric composition as a dimer of dimers. In detail, we have modeled the open active site on the basis of the cocrystal structure of AAZ in complex with β-CA from the green algae Coccomyxa (PDB Code: 3UCJ).24 This in silico study suggested that the deprotonated sulfonamide moiety is anchored to zinc ion, which is coordinated by residues Cys42, His98, and Cys101 (chain A, blue in Figure 2); a hydrogen bonding interaction was found between the oxygen atom of Gly102 (chain A) and the exocyclic nitrogen atom of the acetamide moiety; finally, the nitrogen atom of the thiadiazole ring establishes H-bond contact with the hydroxyl group of Tyr83 (chain B, wheat in Figure 2), for which a π/π stacking with the thiadiazole nucleus might reinforce the binding within the catalytic site.
Figure 2.
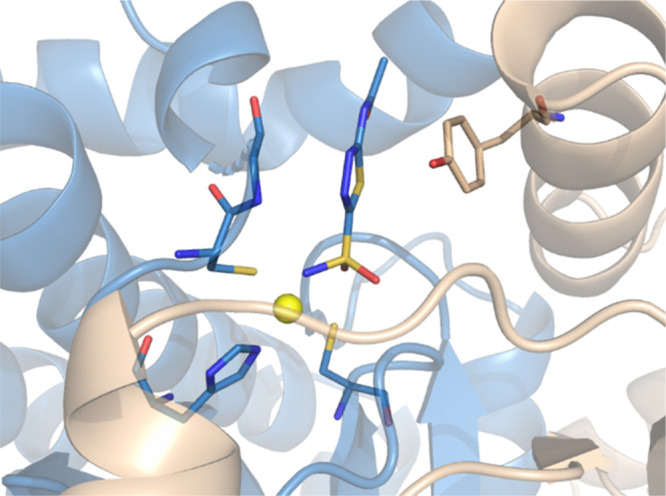
Binding site analysis of the modeled dimeric VchCAβ open cavity bound to acetazolamide (AAZ).
Continuing our efforts aimed to the identification of selective CA inhibitors,14,25−36 in this work we report a computationally driven design of small molecules which possess the minimal structural requirements to occupy the tight cleft of the β-CA cavity and establish favorable contacts with crucial residues of VchCA isozymes, thus anchoring the catalytic site as well as hydrophobic/hydrophilic walls of the CA cavity.
Our study began with a ligand-based strategy to obtain a three-dimensional pharmacophore model, that was validated to establish its robustness as a valuable tool to identify a compound having affinity to the β-class of VchCA. Then, a structure-based virtual screening of 3D-databases allowed us to select hypothetical drug-like sulfonamides able to establish interaction with enzymatic cavity. Finally, nine compounds were synthesized and screened to establish the reliability of our computational study and reach new information about the SAR for selective and potent VchCAβ inhibitors over hCA I and hCA II. Then, the hypothetical binding pose was suggested by docking studies. The following sections describe the step-by-step procedure to achieve our goal.
We initially constructed our pharmacophore model for CA inhibitors targeting VchCAβ isozymes. To assemble the data set, we selected from the literature14,21 19 known inhibitors (compounds 1–19, Figure 3) that possess the RSO2NHR chemical moiety as well-established ZBG; to guarantee the best reliability of the pharmacophore hypotheses, we selected a homogeneous series of inhibitors that have been assayed by employing the stopped-flow carbon dioxide hydrase assay; compounds 1–19 displayed Ki values ranging from 68 to 6000 nM as active compounds. Then, the data set compounds were distributed in two subsets of compounds: the training set (Figure 3, compounds 1–9) and test set (Figure 3, compounds 10–19). By employing the above-mentioned two data sets of sulfonamides, a collection of ten pharmacophore models was generated by LigandScout;37 then, the pharmacophore hypothesis with the best score value (72.7165) was considered from the 10 generated models. To validate this pharmacophore model, we established its discrimination power by considering the enrichment factor and the area under the curve (AUC) of the receiver operating characteristic (ROC) curve (for details see Figure 6 in the Supporting Information); the model displays a preference for active compounds with an AUC value of 0.97 and EF of 11.5.
Figure 3.

Chemical structures of compounds 1–19.
As shown in Figure 4A the best pharmacophore model consists of one aromatic ring feature (blue), one hydrophobic feature (yellow), three hydrogen bond acceptors (red), two hydrogen bond donors (green), one negative ionizable (red star), and 31 exclusion volumes (gray).
Figure 4.
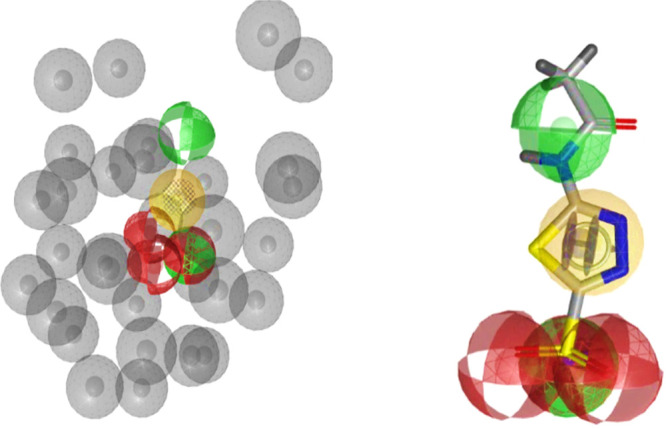
(A) Best pharmacophore model: one aromatic ring feature (blue) overlapped with the hydrophobic feature (yellow), three hydrogen bond acceptors (red), two hydrogen bond donors (green), one negative ionizable (red star), and 31 exclusion volumes (gray). (B) AAZ mapped into the pharmacophore model.
The alignment of the 3D coordinates of the active inhibitor AAZ (Ki value of 451 nM) onto the pharmacophore (see Figure 4B) highlighted that the deprotonated form of sulfonamide moiety (RSO2NH–) is defined by two oxygen atoms as hydrogen bond acceptors as well as one nitrogen atom corresponding to a hydrogen bond donor or a negative ionizable group; in addition, the heteroaromatic ring describes a hydrophobic/aromatic ring feature. Furthermore, the other hydrogen bond donor feature corresponds to the nitrogen atom of the amide portion. Notably, the above-described features resulted in good agreement with previously reported contacts found for AAZ docked into our “modeled” open conformation of VchCAβ catalytic cavity (cf. Figure 2).
Encouraged by this strong matching between the pharmacophore model displayed in Figure 4A and docking results for AAZ, we employed this plausible model to carry out a virtual screening (VS) in order to identify new of VchCAβ inhibitors. The second step of our computational study involved the construction of a plausible database of VchCAβ inhibitors through the collection of para-benzenesulfonamide derivatives retrieved from the SciFinder chemical database (https://scifinder.cas.org). We employed specific filters to select both drug-likeness and commercially available compounds; therefore, we collected 8,208 molecules that were screened by our best pharmacophore model, thus obtaining 661 compounds. Among them we selected 118 molecules having fit-score values greater than 72.86. By visual inspection we focused our interest on 40 compounds, that were docked into our modeled β-CA cavity by Gold software.38 Therefore, docking analysis afforded 9 sulfonamides that were selected on the basis of a very simple synthetic procedure to obtain them.
As depicted in Figure 5 the selected compounds 20a–i are characterized by the canonical ZBG linked to the lipophilic cap-group by an amide spacer as a crucial motif to bind the VchCAβ catalytic cavity through the requested H-bond donor group; additionally, the cap group might furnish a selective interaction with VchCAs over other CA classes.
Figure 5.
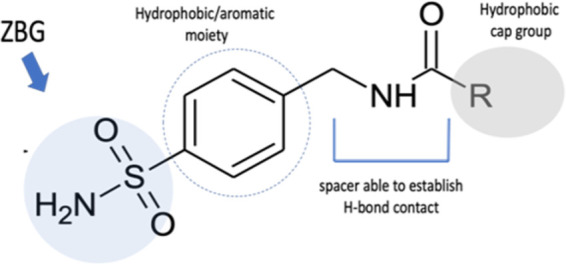
Schematic representation of structural moieties shared by sulfonamides 20a–i
By coupling the 4-aminomethylbenzenesulfonamide (21) with commercially available carboxylic acids or aroyl chlorides, we obtained in good yields the small series of desired N-(4-sulfamoylbenzyl)amide derivatives 20a–i (Scheme 1). The chemical characterization of compounds 20a–i was supported by elemental analyses and 1H and 13C NMR spectroscopic measurements.
Scheme 1. Synthetic Route for Desired N-(4-Sulfamoylbenzyl)amide Derivatives 20a–i.

Reagents and conditions: (i) (A) RCOCl, DIPEA, DCM/DMF (2:1, v/v), MW, 25 °C, 10 min; (B) RCO2H, HBTU, DIPEA, DCM/DMF (2:1, v/v), MW, 25 °C, 25 min.
The N-(4-sulfamoylbenzyl)amide derivatives 20a–i were assayed for their inhibitory activity against VchCAα, -β, and -γ by means of a stopped-flow carbon dioxide hydrase assay. The obtained results are summarized in Table 1 and compared with Ki values of AAZ as reference compound. For comparative purposes the inhibitory profiles against the ubiquitous hCA I and hCA II are reported in Table 1.
Table 1. Inhibitory Effects against VchCAα, VchCA-β, VchCA-γ, hCA I, and hCA II of Compounds 20a–i and Reference Compound Acetazolamide (AAZ).
|
Ki (nM) |
|||||
|---|---|---|---|---|---|
| VchCAα | VchCβ | VchCAγ | hCA I | hCA II | |
| 20a | 45.0 | 6442.0 | 56.1 | 60.7 | 3.3 |
| 20b | 9.1 | 626.7 | 250.8 | 65.9 | 5.1 |
| 20c | 8.8 | 3596.0 | 722.4 | 77.8 | 31.6 |
| 20d | 18.1 | 179.2 | 98.4 | 95.0 | 63.0 |
| 20e | 11.6 | 95.6 | 174.6 | 2113.0 | 919.7 |
| 20f | 12.1 | 586.1 | 657.4 | 98.3 | 54.4 |
| 20g | 6.2 | 553.9 | 593.0 | 269.3 | 26.3 |
| 20h | 10.0 | 200.4 | 775.0 | 44.2 | 83.8 |
| 20i | 7.7 | 538.5 | 79.6 | 571.5 | 69.5 |
| AAZ | 6.8 | 451.0 | 470.0 | 250.0 | 12.1 |
Errors in the range of ±10% of the reported value, from 3 different assays.
All computationally inspired compounds 20a–i affected the carbon dioxide hydrase activity of VchCA classes showing Ki values ranging from 6.2 to 6442 nM. The screening toward VchCA α evidenced that the R substituent did not significantly affect the inhibitory potency of tested compounds 20a–i. Notably, the 1-(biphenyl-4-yl)-substituted compound 20e was the most active VchCA β inhibitor (Ki value of 95.6 nM). Compounds 20b, 20f, 20g, and 20i were about 5-fold less active VchCA β inhibitors when compared with analogue 20e (Ki value of 95.6 nM). The presence of m-tolyl or 2,5-chlorophenyl as hydrophobic group was critical for VchCA β affinity and dramatically reduced the activity toward VchCA β of compounds 20a and 20c, respectively. On the contrary, compounds 20d and 20h were more active when compared with 20a and 20c, whereas they were weakly active with respect to the most interesting inhibitor 20e. All these data evidenced how changes in the size and/or shape of the hydrophobic fragment can impact the inhibitory profile toward VchCA β. The inhibitory trend toward VchCA γ revealed that compounds 20a, 20d, and 20i were active at low nanomolar concentration; the remaining compounds of the series resulted weak inhibitors. Taken together these data confirmed that VchCA α is more able to accommodate the various R-substituents compared to the other tested isozymes VchCA-β and VchCA-γ. Overall, the most relevant result was the identification of compound 20e as a potent and selective VchCA-β inhibitor that displayed very low affinity toward human CA isoforms hCA I and hCA II (Ki values of 2113.0 and 919.7 nM, respectively).
A further step of our study was to analyze the hypothetical orientations into the VchCAβ cavity for synthesized compounds by docking analysis, that were performed by means of Gold Software.38 In detail, we used the crystal structure of the dimeric (chains A and B) VchCAβ retrieved from the Protein Data Bank (PDB Code: 5CXK),10 that has been “modeled” in open conformation on the basis of β-CA from the green algae Coccomyxa(24) (PDB Code: 3UCJ) as previously reported and shown in Figure 2 for AAZ.14 The docking results confirmed that N-(4-sulfamoylbenzyl)amide derivatives 20a–i adopted the canonical orientation of sulfonamide-based CAIs for which for the deprotonated form of the sulfonamide moiety is anchored to the zinc ion coordinated by residues Cys42, His98, and Cys101 (chain A, colored in blue). As expected, the aromatic ring of the benzenesulfonamide portion is stabilized through a π–π interaction with Tyr83 (chain B, colored in wheat). In addition, the -NH- group of the amide spacer establishes H-bond interaction with the oxygen atom of the Gly102 backbone. Our studies suggested that the cap-group might be involved in a network of interactions with a cluster of residues Thr105, Ala106, Ala139, and Ile108 lining the hydrophobic subpocket along the rim of chain A. The network of above-mentioned interactions is displayed in Figure 6 for the most active inhibitor N-(4-sulfamoylbenzyl)biphenyl-4-carboxamide (20e, Ki value of 96.5 nM).
Figure 6.
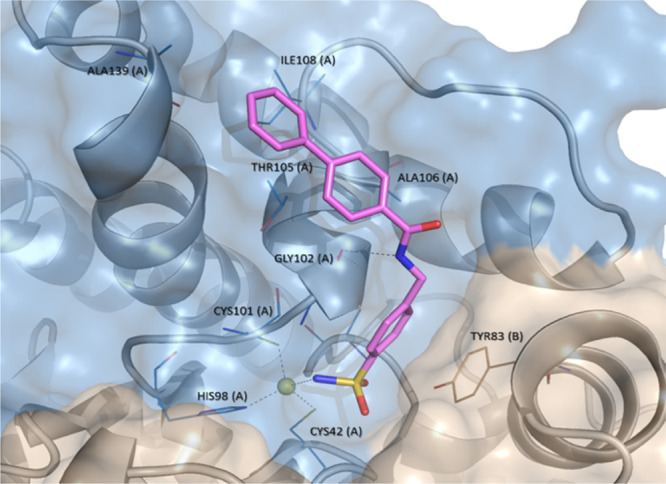
Plausible binding mode of 20e into our “modeled” open conformation of VchCAβ. Dark gray dashed lines represent hydrogen bond interaction. Zinc ion is depicted as a yellow sphere. The interactions were examined by using LigandScout software.37 The images were created by means of PyMOL software (https://pymol.org).
In conclusion a ligand based virtual screening strategy led to the identification of compound 20e as active VchCA-β inhibitor (Ki value of 95.6 nM) that combined high affinity with a surprising selectivity over the human off-target isoform. The screening and docking efforts established that this compound might be a promising lead compound for further biological studies, HTS screening, and structural optimization aimed to the identification of novel anti-infective agents characterized by a peculiar mechanism of action, in order to overcome the global threat of antibiotic resistance.
Glossary
Abbreviations
- CAs
carbonic anhydrases
- Vch
Vibrio cholerae
- AAZ
acetazolamide
- EZA
ethoxzolamide
- HCT
hydrochlorothiazide
- BZA
benzolamide
- SAC
saccharine
- BRZ
brinzolamide
- SLP
sulpiride
- SLT
sulthiame
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00417.
Experimental details (synthetic experimental details, analytical and 1H and 13C spectral data, biochemical assays, and computational studies) (PDF)
Author Contributions
All authors contributed to data interpretation. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Fondo di Ateneo per la Ricerca (PRA grant number ORME09SPNC - Università degli Studi di Messina). PO FSE Regione Siciliana 2014-2020 doctoral scholarship for F.M. (XXXIII Cycle – University of Messina, Italy).
The authors declare no competing financial interest.
Supplementary Material
References
- Ramamurthy T.; Mutreja A.; Weill F. X.; Das B.; Ghosh A.; Nair G. B. Revisiting the Global Epidemiology of Cholera in Conjuction With the Genomics of Vibrio cholerae. Front Public Health 2019, 7, 203. 10.3389/fpubh.2019.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil A. A.; Ryan E. T. Cholera: recent updates. Curr. Opin. Infect. Dis. 2018, 31, 455–461. 10.1097/QCO.0000000000000474. [DOI] [PubMed] [Google Scholar]
- Peterson K. M.; Gellings P. S.. Multiple intraintestinal signals coordinate the regulation of Vibrio cholerae virulence determinants. Pathog. Dis. 2018, 76, 10.1093/femspd/ftx126. [DOI] [PubMed] [Google Scholar]
- Cruite J. T.; Kovacikova G.; Clark K. A.; Woodbrey A. K.; Skorupski K.; Kull F. J. Structural basis for virulence regulation in Vibrio cholerae by unsaturated fatty acid components of bile. Commun. Biol. 2019, 2, 440. 10.1038/s42003-019-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose K. E. Regulation of virulence in Vibrio cholerae. Int. J. Med. Microbiol. 2001, 291, 81–8. 10.1078/1438-4221-00104. [DOI] [PubMed] [Google Scholar]
- Davoodi S.; Foley E. Host-Microbe-Pathogen Interactions: A Review of Vibrio cholerae Pathogenesis in Drosophila. Front. Immunol. 2020, 10, 3128. 10.3389/fimmu.2019.03128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuaita B. H.; Withey J. H. Bicarbonate Induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 2009, 77, 4111–20. 10.1128/IAI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 28, 709–712. 10.1080/13543776.2018.1523897. [DOI] [PubMed] [Google Scholar]
- Capasso C.; Supuran C. T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets 2015, 19, 1689–704. 10.1517/14728222.2015.1067685. [DOI] [PubMed] [Google Scholar]
- Ferraroni M.; Del Prete S.; Vullo D.; Capasso C.; Supuran C. T. Crystal structure and kinetic studies of a tetrameric type II beta-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 2449–56. 10.1107/S1399004715018635. [DOI] [PubMed] [Google Scholar]
- Del Prete S.; Vullo D.; De Luca V.; Carginale V.; Osman S. M.; AlOthman Z.; Supuran C. T.; Capasso C. Comparison of the sulfonamide inhibition profiles of the alpha-, beta- and gamma-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg. Med. Chem. Lett. 2016, 26, 1941–6. 10.1016/j.bmcl.2016.03.014. [DOI] [PubMed] [Google Scholar]
- Del Prete S.; Vullo D.; De Luca V.; Carginale V.; di Fonzo P.; Osman S. M.; AlOthman Z.; Supuran C. T.; Capasso C. Anion inhibition profiles of alpha-, beta- and gamma-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg. Med. Chem. 2016, 24, 3413–7. 10.1016/j.bmc.2016.05.029. [DOI] [PubMed] [Google Scholar]
- Capasso C.; Supuran C. T. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin. Ther. Pat. 2013, 23, 693–704. 10.1517/13543776.2013.778245. [DOI] [PubMed] [Google Scholar]
- Gitto R.; De Luca L.; Mancuso F.; Del Prete S.; Vullo D.; Supuran C. T.; Capasso C. Seeking new approach for therapeutic treatment of cholera disease via inhibition of bacterial carbonic anhydrases: experimental and theoretical studies for sixteen benzenesulfonamide derivatives. J. Enzyme Inhib. Med. Chem. 2019, 34, 1186–1192. 10.1080/14756366.2019.1618292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bua S.; Osman S. M.; Del Prete S.; Capasso C.; AlOthman Z.; Nocentini A.; Supuran C. T. Click-tailed benzenesulfonamides as potent bacterial carbonic anhydrase inhibitors for targeting Mycobacterium tuberculosis and Vibrio cholerae. Bioorg. Chem. 2019, 86, 183–186. 10.1016/j.bioorg.2019.01.065. [DOI] [PubMed] [Google Scholar]
- Angeli A.; Pinteala M.; Maier S. S.; Del Prete S.; Capasso C.; Simionescu B. C.; Supuran C. T. Inhibition of bacterial alpha-, beta- and gamma-class carbonic anhydrases with selenazoles incorporating benzenesulfonamide moieties. J. Enzyme Inhib. Med. Chem. 2019, 34, 244–249. 10.1080/14756366.2018.1547287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bua S.; Berrino E.; Del Prete S.; Murthy V. S.; Vijayakumar V.; Tamboli Y.; Capasso C.; Cerbai E.; Mugelli A.; Carta F.; Supuran C. T. Synthesis of novel benzenesulfamide derivatives with inhibitory activity against human cytosolic carbonic anhydrase I and II and Vibrio cholerae alpha- and beta-class enzymes. J. Enzyme Inhib. Med. Chem. 2018, 33, 1125–1136. 10.1080/14756366.2018.1467901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli A.; Abbas G.; Del Prete S.; Capasso C.; Supuran C. T. Selenides bearing benzenesulfonamide show potent inhibition activity against carbonic anhydrases from pathogenic bacteria Vibrio cholerae and Burkholderia pseudomallei. Bioorg. Chem. 2018, 79, 319–322. 10.1016/j.bioorg.2018.05.015. [DOI] [PubMed] [Google Scholar]
- Angeli A.; Abbas G.; Del Prete S.; Carta F.; Capasso C.; Supuran C. T. Acyl selenoureido benzensulfonamides show potent inhibitory activity against carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg. Chem. 2017, 75, 170–172. 10.1016/j.bioorg.2017.09.016. [DOI] [PubMed] [Google Scholar]
- Vullo D.; Del Prete S.; De Luca V.; Carginale V.; Ferraroni M.; Dedeoglu N.; Osman S. M.; AlOthman Z.; Capasso C.; Supuran C. T. Anion inhibition studies of the beta-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg. Med. Chem. Lett. 2016, 26, 1406–10. 10.1016/j.bmcl.2016.01.072. [DOI] [PubMed] [Google Scholar]
- Del Prete S.; Vullo D.; De Luca V.; Carginale V.; Ferraroni M.; Osman S. M.; AlOthman Z.; Supuran C. T.; Capasso C. Sulfonamide inhibition studies of the beta-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg. Med. Chem. 2016, 24, 1115–20. 10.1016/j.bmc.2016.01.037. [DOI] [PubMed] [Google Scholar]
- Lomelino C. L.; Andring J. T.; McKenna R. Crystallography and Its Impact on Carbonic Anhydrase Research. Int. J. Med. Chem. 2018, 2018, 9419521. 10.1155/2018/9419521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso C.; Supuran C. T. Inhibition of Bacterial Carbonic Anhydrases as a Novel Approach to Escape Drug Resistance. Curr. Top. Med. Chem. 2017, 17, 1237–1248. 10.2174/1568026617666170104101058. [DOI] [PubMed] [Google Scholar]
- Huang S.; Hainzl T.; Grundstrom C.; Forsman C.; Samuelsson G.; Sauer-Eriksson A. E. Structural studies of beta-carbonic anhydrase from the green alga Coccomyxa: inhibitor complexes with anions and acetazolamide. PLoS One 2011, 6, e28458 10.1371/journal.pone.0028458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buemi M. R.; Di Fiore A.; De Luca L.; Angeli A.; Mancuso F.; Ferro S.; Monti S. M.; Buonanno M.; Russo E.; De Sarro G.; De Simone G.; Supuran C. T.; Gitto R. Exploring structural properties of potent human carbonic anhydrase inhibitors bearing a 4-(cycloalkylamino-1-carbonyl)benzenesulfonamide moiety. Eur. J. Med. Chem. 2019, 163, 443–452. 10.1016/j.ejmech.2018.11.073. [DOI] [PubMed] [Google Scholar]
- De Luca L.; Mancuso F.; Ferro S.; Buemi M. R.; Angeli A.; Del Prete S.; Capasso C.; Supuran C. T.; Gitto R. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur. J. Med. Chem. 2018, 143, 276–282. 10.1016/j.ejmech.2017.11.061. [DOI] [PubMed] [Google Scholar]
- Bruno E.; Buemi M. R.; Di Fiore A.; De Luca L.; Ferro S.; Angeli A.; Cirilli R.; Sadutto D.; Alterio V.; Monti S. M.; Supuran C. T.; De Simone G.; Gitto R. Probing Molecular Interactions between Human Carbonic Anhydrases (hCAs) and a Novel Class of Benzenesulfonamides. J. Med. Chem. 2017, 60, 4316–4326. 10.1021/acs.jmedchem.7b00264. [DOI] [PubMed] [Google Scholar]
- Bruno E.; Buemi M. R.; De Luca L.; Ferro S.; Monforte A. M.; Supuran C. T.; Vullo D.; De Sarro G.; Russo E.; Gitto R. Vivo Evaluation of Selective Carbonic Anhydrase Inhibitors as Potential Anticonvulsant Agents. ChemMedChem 2016, 11, 1812–8. 10.1002/cmdc.201500596. [DOI] [PubMed] [Google Scholar]
- Buemi M. R.; De Luca L.; Ferro S.; Bruno E.; Ceruso M.; Supuran C. T.; Pospíšilová K.; Brynda J.; Řezáčová P.; Gitto R. Carbonic anhydrase inhibitors: Design, synthesis and structural characterization of new heteroaryl-N-carbonylbenzenesulfonamides targeting druggable human carbonic anhydrase isoforms. Eur. J. Med. Chem. 2015, 102, 223–232. 10.1016/j.ejmech.2015.07.049. [DOI] [PubMed] [Google Scholar]
- De Luca L.; Ferro S.; Damiano F. M.; Supuran C. T.; Vullo D.; Chimirri A.; Gitto R. Structure-based screening for the discovery of new carbonic anhydrase VII inhibitors. Eur. J. Med. Chem. 2014, 71, 105–11. 10.1016/j.ejmech.2013.10.071. [DOI] [PubMed] [Google Scholar]
- Gitto R.; Damiano F. M.; Mader P.; De Luca L.; Ferro S.; Supuran C. T.; Vullo D.; Brynda J.; Rezacova P.; Chimirri A. Synthesis, structure-activity relationship studies, and X-ray crystallographic analysis of arylsulfonamides as potent carbonic anhydrase inhibitors. J. Med. Chem. 2012, 55, 3891–9. 10.1021/jm300112w. [DOI] [PubMed] [Google Scholar]
- Mader P.; Brynda J.; Gitto R.; Agnello S.; Pachl P.; Supuran C. T.; Chimirri A.; Rezacova P. Structural basis for the interaction between carbonic anhydrase and 1,2,3,4-tetrahydroisoquinolin-2-ylsulfonamides. J. Med. Chem. 2011, 54, 2522–6. 10.1021/jm2000213. [DOI] [PubMed] [Google Scholar]
- Gitto R.; Damiano F. M.; De Luca L.; Ferro S.; Vullo D.; Supuran C. T.; Chimirri A. Synthesis and biological profile of new 1,2,3,4-tetrahydroisoquinolines as selective carbonic anhydrase inhibitors. Bioorg. Med. Chem. 2011, 19, 7003–7. 10.1016/j.bmc.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Gitto R.; Agnello S.; Ferro S.; Vullo D.; Supuran C. T.; Chimirri A. Identification of potent and selective human carbonic anhydrase VII (hCA VII) inhibitors. ChemMedChem 2010, 5, 823–6. 10.1002/cmdc.201000044. [DOI] [PubMed] [Google Scholar]
- Gitto R.; Agnello S.; Ferro S.; De Luca L.; Vullo D.; Brynda J.; Mader P.; Supuran C. T.; Chimirri A. Identification of 3,4-Dihydroisoquinoline-2(1H)-sulfonamides as potent carbonic anhydrase inhibitors: synthesis, biological evaluation, and enzyme--ligand X-ray studies. J. Med. Chem. 2010, 53, 2401–8. 10.1021/jm9014026. [DOI] [PubMed] [Google Scholar]
- Gitto R.; Ferro S.; Agnello S.; De Luca L.; De Sarro G.; Russo E.; Vullo D.; Supuran C. T.; Chimirri A. Synthesis and evaluation of pharmacological profile of 1-aryl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-sulfonamides. Bioorg. Med. Chem. 2009, 17, 3659–64. 10.1016/j.bmc.2009.03.066. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–9. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–48. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.