

Abstract
Exoribonuclease-resistant RNAs (xrRNAs) are discrete elements that block the progression of 5′ to 3′ exoribonucleases using specifically folded RNA structures. A recently discovered class of xrRNA is widespread in several genera of plant-infecting viruses, within both noncoding and protein-coding subgenomic RNAs. The structure of one such xrRNA from a dianthovirus revealed three-dimensional details of the resistant fold but did not answer all questions regarding the conservation and diversity of this xrRNA class. Here, we present the crystal structure of a representative polerovirus xrRNA that contains sequence elements that diverge from the previously solved structure. This new structure rationalizes previously unexplained sequence conservation patterns and shows interactions not present in the first structure. Together, the structures of these xrRNAs from dianthovirus and polerovirus genera support the idea that these plant virus xrRNAs fold through a defined pathway that includes a programmed intermediate conformation. This work deepens our knowledge of the structure–function relationship of xrRNAs and shows how evolution can craft similar RNA folds from divergent sequences.
Keywords: RNA structure, viral RNA, exoribonuclease resistance, RNA pseudoknot, X-ray crystallography
INTRODUCTION
RNA structure-dependent exoribonuclease resistance is now well-established as a means of subgenomic RNA production or maintenance by many viruses (Slonchak and Khromykh 2018). This mechanism depends on discrete, specifically folded RNA elements in viral genomes called xrRNAs (for “exoribonuclease-resistant RNAs”). xrRNAs block the 5′ to 3′ progression of processive cellular exoribonucleases using only a folded RNA element, without accessory proteins (Chapman et al. 2014a). In so doing, they protect downstream RNA from degradation. xrRNAs were discovered in flaviviruses and have since been identified in other viruses, including plant viruses of the Luteoviridae and Tombusviridae families (Iwakawa et al. 2008; Pijlman et al. 2008; Steckelberg et al. 2018a). In flaviviruses, xrRNAs have been found exclusively at the beginning of the 3′ untranslated region (UTR) of the viral genome where they function in the generation of viral noncoding RNAs, whereas in Luteoviridae and Tombusviridae, xrRNAs are associated with both noncoding and protein-coding regions of the viral genome (Fig. 1A; Steckelberg et al. 2018b). Despite this evidence that xrRNAs are widespread we are only beginning to understand the principles of xrRNA folding that lead to structures capable of blocking host cell exoribonucleases.
FIGURE 1.

A brief overview of plant xrRNAs. (A) Depending on the plant virus family, xrRNAs are located within intergenic regions and/or at the beginning of the 3′ UTR. (B) Consensus secondary structure for xrRNALTs. The 95th percentile (η.95) is indicated for all variable regions. Covariation analysis was expanded and updated from Steckelberg et al. (2018b). (C) Alignment of xrRNALT sequences from select plant viruses. Bold sequences (PLRV and SCNMV) indicate a crystal structure is available; *, xrRNA confirmed to be resistant to Xrn1 in vitro; gray highlight in L2ACN; the gray shaded nucleotide aligns 2 nt downstream in three dimensions, as indicated by the arrow pointing to the “=” sign. The G–C pair involving J1/2 and L2B is labeled. The complete alignment is shown in Supplemental Figure S1.
Crystal structures of xrRNAs from flaviviruses revealed that two pseudoknots and other long-range interactions create a ring-like fold that wraps around the 5′ end of the xrRNA (Chapman et al. 2014a; Akiyama et al. 2016; Jones et al. 2020). Mechanistic studies suggest that this ring protects downstream RNA by “bracing” against the surface of the exoribonuclease, acting as a physical block to the 5′ to 3′ progression of the enzyme (Chapman et al. 2014a; Akiyama et al. 2016; MacFadden et al. 2018). A recent crystal structure of an xrRNA from sweet clover necrotic mosaic virus ([SCNMV]; a dianthovirus from the Tombusviridae family) showed a different three-dimensional fold conferring exoribonuclease resistance (Steckelberg et al. 2018a). Specifically, the SCNMV xrRNA was captured in a folding intermediate state that contains a Watson–Crick base-paired helix (P1) that must be unwound to allow the formation of a pseudoknotted ring structure. Single-molecule Förster resonance energy transfer (smFRET) experiments revealed that the SCNMV xrRNA's intermediate state can be remodeled by the helicase activity of the arriving exoribonuclease to favor the resistant pseudoknotted fold (Steckelberg et al. 2018a). While a ring encircling the 5′ end seems to be thus far a defining characteristic of all xrRNAs, the degradation-induced structural remodeling seen in SCNMV's xrRNA appears to be unique. Overall, the xrRNAs of the flavivirus-type (xrRNAF) and the dianthovirus-type (xrRNAD) are distinguishable based on their different underlying three-dimensional folds.
The discovery of intramolecular interactions within xrRNAD that could not be predicted from the sequence informed subsequent bioinformatic-based approaches that identified >40 new examples of this class of xrRNA (Steckelberg et al. 2018b). These xrRNAs pervade the Luteoviridae and Tombusviridae families in general, and the Umbravirus and Polerovirus genera in particular (polerovirus sequences make for ∼2/3 of this alignment). Some of the deadliest viruses in agriculture contain an xrRNA, such as potato leafroll virus ([PLRV], leading responsible virus for worldwide potato yield loss [Wale et al. 2008]), maize chlorotic mottle virus ([MCMV], responsible for 90% maize/corn yield loss in sub-Saharan Africa [Mahuku et al. 2015]), and maize yellow dwarf virus-RMV ([MYDV-RMV], formerly BYDV-RMV [Krueger et al. 2013], responsible for 30% yield loss in affected winter wheat fields [Perry et al. 2000]). All newly identified xrRNAs in Luteoviridae are found in intergenic regions, whereas xrRNAs from Tombusviridae are generally found at the beginning of the 3′ UTR (Steckelberg et al. 2018b). Because they are widespread in the Luteoviridae and Tombusviridae, we now refer to this class as xrRNALT in this manuscript rather than xrRNAD. Interestingly, the many newly discovered xrRNALTs contain conserved sequence elements absent from the crystallized SCNMV xrRNA (Fig. 1B,C; Steckelberg et al. 2018b). This raised the questions of whether the newly identified xrRNALTs form the same fold as the dianthovirus xrRNA and, if so, how they accommodate these different sequence elements.
To address the universality of the interactions necessary to support an xrRNALT fold as first seen in SCNMV, we explored the structure and function of the xrRNALT from PLRV using X-ray crystallography and in vitro assays of exoribonuclease resistance. In the crystal, the PLRV xrRNALT adopts a folding intermediate conformation similar to that of the SCNMV xrRNALT. But as the PLRV xrRNALT is ∼10% longer than the SCNMV xrRNALT, our structure reveals how extra nucleotides leading to extended helices and loops generate interactions and motifs not seen in the SCNMV structure. Using site-directed mutagenesis coupled with functional assays, we show how these interactions are critical for exoribonuclease resistance. Together, our results rationalize sequence conservation patterns of xrRNALT, thereby providing insights into the mechanism of programmed exoribonuclease resistance in the viral world.
RESULTS AND DISCUSSION
Crystallization of a polerovirus xrRNALT
The previously reported structure of the SCNMV xrRNALT revealed how an RNA sequence of only 44 nt adopts a specific fold to generate an exoribonuclease-resistant structure (Steckelberg et al. 2018a). Because the dianthovirus sequences in the original xrRNALT alignment were not representative of the broader diversity in the expanded alignment (Fig. 1C; Supplemental Fig. S1), the SCNMV structure could not fully explain the consensus secondary structure (Fig. 1B). In particular, all xrRNALTs contain a guanosine in the J1/2 internal loop between the P1 and P2 stems (G8 in PLRV; loop previously referred to as L1; Figs. 1C and 2A), except for SCNMV, which contains an adenosine. In SCNMV, the Hoogsteen side of A8 interacts with the sugar edge of G33 in J2/1, an interaction that could not be reproduced by swapping the A and the G as in the majority of the sequences in the alignment. Additionally, xrRNALT sequences are longer for poleroviruses than for dianthoviruses, with extra nucleotides in P2 and in the less-conserved regions within the apical loop L2A (including the part involved in the pseudoknot) and the adjoining region J1/3 (Fig. 1B,C). Examining the SCNMV structure did not suggest how these extra nucleotides could be accommodated. Finally, the SCNMV xrRNALT was crystallized in a state that appeared to be a necessary folding intermediate. Additional structural information was needed to confirm this as an authentic intermediate or to visualize the final pseudoknotted state.
FIGURE 2.
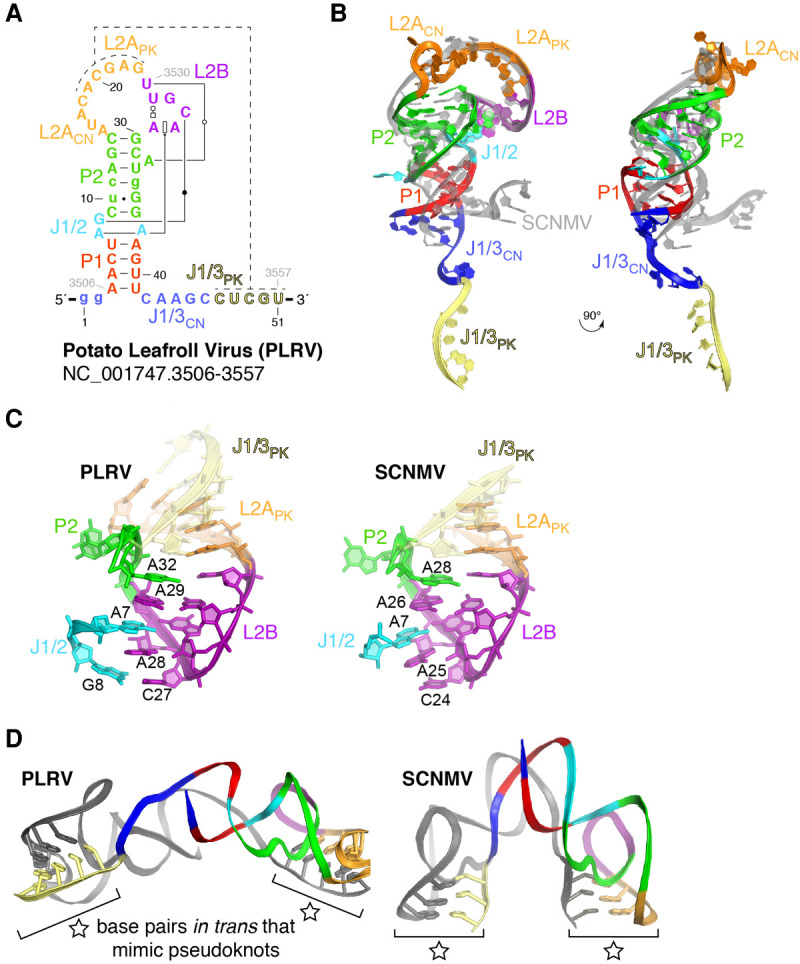
Structure of the PLRV xrRNA. (A) Secondary structure of the crystallized PLRV RNA, with matching colors to Figure 1B,C. Engineered nucleotides (see Materials and Methods) are shown in lower case. Non-Watson–Crick base pairs are according to the Leontis–Westhof annotation (Leontis and Westhof 2001). (B) Ribbon representation of the PLRV xrRNALT structure in colors to match (A), over that from SCNMV in gray (two orthogonal views). (C) Side-by-side comparison of the conserved structural core involving J1/2, L2B, and P2. (D) The PLRV and SCNMV xrRNALTs both crystallize as a domain-swap dimer, in which the intramolecular pseudoknot is replaced by intermolecular base-pairing (indicated by a star).
To allow a structural comparison with the SCNMV xrRNALT, we selected several diverse xrRNALTs that had the conserved G, expanded regions in P2, L2A, and J1/3, and various pseudoknot lengths (4–5 bp). Of these, the xrRNALT from PLRV yielded crystals that diffracted to 5–7 Å on our home source but that we could not reproduce (construct #1, Supplemental Fig. S2A). Diffraction was improved to ∼3 Å upon adding of 100 mM guanidinium hydrochloride during crystallization (see Materials and Methods) and introducing a G·U wobble base pair “phasing module” at various locations to aid with phasing using iridium(III) hexammine (Supplemental Fig. S2A; Keel et al. 2007). Introducing the phasing module in constructs #1–4 did not alter the ability of the RNA to resist exoribonuclease-catalyzed degradation (Supplemental Fig. S2B). The structure of construct #3 was eventually solved to 2.6 Å from combined data sets collected at a synchrotron using single anomalous dispersion phasing at the L-I edge of iridium (Supplemental Table S1; Supplemental Fig. S3; Batey and Kieft 2016).
A common intermediate fold for xrRNALT elements
The global structure of the PLRV xrRNALT is similar in shape and architecture to that of the SCNMV xrRNALT. Like SCNMV, the PLRV xrRNALT folds as a “bent-over” stem–loop in which L2B creates a docking point for long-range tertiary interactions involving nucleotides located in J1/2 (Fig. 2A,B). The all-atom root-mean square deviation (RMSD) is 0.9 Å for a superimposition of L2APK (the section of L2A involved in pseudoknot formation; to be distinguished from L2ACN, the 5′ part of L2A connecting P2 to L2APK) and L2B from SCNMV and PLRV, indicating that the two RNAs share a similar “core” structure (Fig. 2C). The improvement in diffraction quality upon addition of guanidinium is likely due to binding of four guanidinium molecules around J1/2 and J2/1, at sites where iridium(III) hexammine complexes bind in the SCNMV structure (Supplemental Fig. S4A). Three of the guanidinium molecules stack to form a spine in the deep groove of three consecutive guanines (G34–G36, Supplemental Fig. S4A,B). Stacking is a known property of guanidinium (Di Tondo and Pritchard 2012; Vazdar et al. 2018), but it had not been observed to that extent within a complex with RNA. Guanidiniums interact with the RNA in a manner reminiscent of that seen in guanidinium riboswitches (Breaker et al. 2017; Nelson et al. 2017); compare, for example, guanidinium bound to G36–G39 in PLRV to the guanidinium binding site within a guanidine-II riboswitch (Supplemental Fig. S4C; Reiss and Strobel 2017). While guanidinium stacking improved crystal diffraction, it did not alter the biochemical behavior of the RNA. Specifically, the addition of low concentrations of guanidine hydrochloride did not change the equilibrium between the monomeric and a dimeric state of the RNA in native gel electrophoresis (Supplemental Fig. S5A,B), nor did it impair Xrn1 resistance in in vitro RNA degradation assays (Supplemental Fig. S5C).
As in the SCNMV xrRNALT, the PLRV xrRNALT was crystallized in a “domain-swap dimer” conformation, in which the intramolecular pseudoknot required for activity was replaced by interactions in trans between the two molecules in the asymmetric unit (Supplemental Fig. S3A–C). Five Watson–Crick base pairs formed between the L2APK region of one molecule and J1/3PK of the second molecule (Fig. 2D). Hence, the PLRV RNA was captured in a similar folded state to the SCNMV RNA (Steckelberg et al. 2018a), although the relative angle between the two molecules is about three times wider for PLRV (Fig. 2D). Because the P1 stem formed, but not the pseudoknot, in both SCNMV and PLRV xrRNALT, this “open” state is likely an authentic intermediate conformation adopted by diverse xrRNALTs to ensure that the correct topology is ultimately achieved (Supplemental Fig. S3C; Steckelberg et al. 2018a). This conclusion is further supported by previous smFRET and functional studies that showed that mutations favoring this intermediate state are not deleterious to exoribonuclease resistance, while those destabilizing this state reduce exoribonuclease resistance (Steckelberg et al. 2018a). Taking into account the sequence and secondary structure conservation of known xrRNALTs (Fig. 1B,C; Supplemental Fig. S1), we can expect that every xrRNALT generally follows a similar folding strategy, in which the initial formation of P1 positions the 3′ end relative to the 5′ end of the RNA, and thereby ensures that adoption of the final “closed” pseudoknotted form creates a protective ring encircling the 5′ end (Supplemental Fig. S3C). As previously proposed, this closed form, not yet directly visualized, would be promoted by the helicase activity of the approaching exoribonuclease, which unwinds P1 (Steckelberg et al. 2018a).
An unexpected G–C base pair contributes to the “bent-over” conformation
The PLRV and SCNMV xrRNA architectures are globally similar (Supplemental Fig. S6); however, several local structural differences account for the diversity in xrRNALT sequences. Although A8 is paired with G34 in SCNMV, the >97% conserved G8 and A37 at the equivalent positions in PLRV and all other xrRNALTs identified are both bulged out from the stem (Fig. 3A). A37 forms crystal contacts (Supplemental Fig. S3D), but G8 pairs with C27 in L2B (equivalent to C24 in SCNMV; Figs. 2C and 3A). The G8–C27 pair caps a stack of purines from L2B, J1/2, and P2, which is otherwise conserved between SCNMV and PLRV (Fig. 2C). Because the overall conformation of L2B relative to P1 and P2 is similar between the two structures, this additional base pair could be viewed as stabilizing but not altering the xrRNALT conformation (Figs. 2C and 3A). The local changes at J1/2 and L2B seen in the PLRV structure may seem minimal, but they contribute to explaining the >97% sequence conservation at J1/2 and L2B in a manner that the SCNMV structure could not.
FIGURE 3.

Structural diversity of the xrRNALT fold. (A) Sequence differences at J1/2 lead to the formation of the G8–C27 pair with L2B in PLRV (left; color coding as in Fig. 1C). Similar view for SCNMV (right). (B) In vitro Xrn1 degradation assay of PLRV xrRNALT WT and G8–C27 mutants. (C) Architecture of the L2ACN and L2APK regions in PLRV (left) and SCNMV (right). (D) In vitro Xrn1 degradation assay of PLRV xrRNA WT and L2ACN mutants.
The high conservation of the G8–C27 pair suggests it is important for exoribonuclease resistance. To test this, we generated two mutant RNAs that each disrupted the ability of these two bases to form a Watson–Crick base pair: G8A and C27U (Supplemental Fig. S7). When challenged with the recombinant 5′ to 3′ exoribonuclease Xrn1 in our established exoribonuclease resistance assay (Chapman et al. 2014b), both mutant RNAs were degraded, indicating they are unable to block progression of the nuclease (Fig. 3B, lanes 3–6). A double-mutant G8A + C27U was designed to restore the ability of the Watson–Crick pair to form (Supplemental Fig. S7). Yet, this mutant was also unable to block progression of the exoribonuclease (Fig. 3B, lanes 7 and 8), suggesting that the specific identity and configuration of the base pair is important. This result is consistent with the sequence alignment, which shows no covariation at these positions (Supplemental Fig. S1). Substitution at these positions likely results in local structural changes that prevent formation of this tertiary base pair. Hence, the conservation of G8 and C27 serves not only to create a base pair, but also to promote structures that make these two bases available to pair. The fact that this pair is not present in SCNMV suggests it may not be required in all contexts, as evolution can craft compensatory stabilizing interactions.
PLRV's longer sequence is accommodated within the active fold
The P2 stem and the predicted pseudoknot are longer by one base pair in PLRV compared to SCNMV. L2ACN is also longer (by one nucleotide) in PLRV. Finally, the J1/3 connector (J1/3CN) is longer by two nucleotides in PLRV (Fig. 1C; next section). The PLRV xrRNA structure shows how these additional nucleotides are accommodated. In the SCNMV structure, L2ACN has a 5′-AUC-3′ sequence, while PLRV has 5′-AUAC-3′, which leads to a significant structural adjustment. For SCNMV, A14 and U15 stack against P2, and C16 stacks against the pseudoknot-like structure formed in trans. For PLRV, the equivalent A15 and U16 flip out in opposite directions, and C18, equivalent to C16 from SCNMV, stacks against the elongated P2 (Fig. 3C; Supplemental Fig. S3D). Spatially, C14 from the C–G pair extending P2 and C18 in PLRV are in similar locations to A14 and U15 in SCNMV. The protrusion comprising A15–A17 in PLRV is accommodated by stacking of A15 and A17 against L2APK (via A19; Fig. 3C) and A37 from a symmetry-related molecule (Supplemental Fig. S3D). U16 is in the vicinity of another symmetry-related molecule, although the poor quality of the map for that base suggests it is dynamic (Supplemental Fig. S3D). This side-by-side comparison of the xrRNALT structures from SCNMV and PLRV reveals extensive differences that were not apparent from sequence alignments but that show how differences in the sequences are accommodated in a globally similar fold.
To test whether the alternative stacking strategy against L2APK involving a distorted backbone of L2ACN is required for resistance, we tested xrRNA mutants that alter L2ACN (Supplemental Fig. S7). Removal of nucleotides 16–18 in PLRV to return to an SCNMV-like L2ACN does not affect resistance, though additionally removing A15 does (Fig. 3D, lanes 3–6). Most likely, removal of A15 would make L2ACN too short to support the proper conformation of L2APK and L2B. Replacing A15 with a U in the 16–18 deletion mutant does not affect resistance (Fig. 3D, lanes 7 and 8), suggesting that the presence of a nucleotide at that position is more important than its identity. Notably, xrRNALT tolerates the presence of an extra nucleotide in L2APK after A15, but not after C18 (Fig. 3D, lanes 9–12). A straightforward interpretation of these results is that stacking of A17 and C18 is important to support the active fold, while stacking of A15 may only occur due to the presence of A37 from a symmetry-related molecule. Overall, this analysis helps to make sense of the generally longer L2ACN and P2 stems in poleroviruses compared to dianthoviruses (Supplemental Fig. S1).
Conserved interactions in the domain-swap dimer have a functional role
PLRV and SCNMV xrRNALT both crystallized in open conformations in which the pseudoknot is replaced by similar base pairs in trans (“domain-swapped dimer state”; Fig. 2D; Supplemental Fig. S3C). The intermolecular dimer contacts likely created favorable crystal packing, and dimerization is favored at high RNA concentrations used for crystallization. Using electrophoretic mobility shift assays (EMSAs), we showed that at the RNA concentration of 150 µM used for crystallization, around 50% of the RNA molecules form dimers. At a lower RNA concentration used for functional in vitro assays (∼6 µM), RNA molecules are predominantly monomeric, suggesting that dimerization is a crystallization artifact and not the functional state of the RNA (Supplemental Fig. S5A,B). Supporting this conclusion, the relative orientation of the two RNA molecules in a dimer is different in PLRV and SCNMV xrRNA crystals (Fig. 2D). The intermolecular base pairs involving L2APK of one molecule and J1/3PK of the other are nonetheless similarly mimicking the pseudoknot in both structures (Fig. 2C,D).
The interactions involving J1/3CN (nucleotides 42–46 in PLRV; Fig. 2A) immediately adjacent to J1/3PK are also conserved in the PLRV and SCNMV structures (Fig. 4A–D). Examination of our sequence alignment reveals that these nucleotides are highly conserved, with >97% conservation of A44 and G45 (A40 and G41 in SCNMV) and the conserved presence of a pyrimidine at position 46 (Fig. 1C). A44, G45, and U46 all stack together and help bridge the bent-over appearance of this hairpin. In particular, A44 interacts with the Hoogsteen side of A7 and is part of an interaction network involving U25 and G26 from L2B. G45 stacks on A44 and bridges A7 to the conserved A29–U25 pair (Fig. 4C). Finally, C46/U42 points toward a negatively charged region formed by the backbone of P1 and P2, where iridium(III) hexammine complexes have been assigned in both PLRV and SCNMV (Fig. 4D). The absence of a purine at this position across xrRNALT sequences (Supplemental Fig. S1) could be justified by the steric clashes or the disturbance of potential physiological metal ion binding sites that a purine would cause.
FIGURE 4.
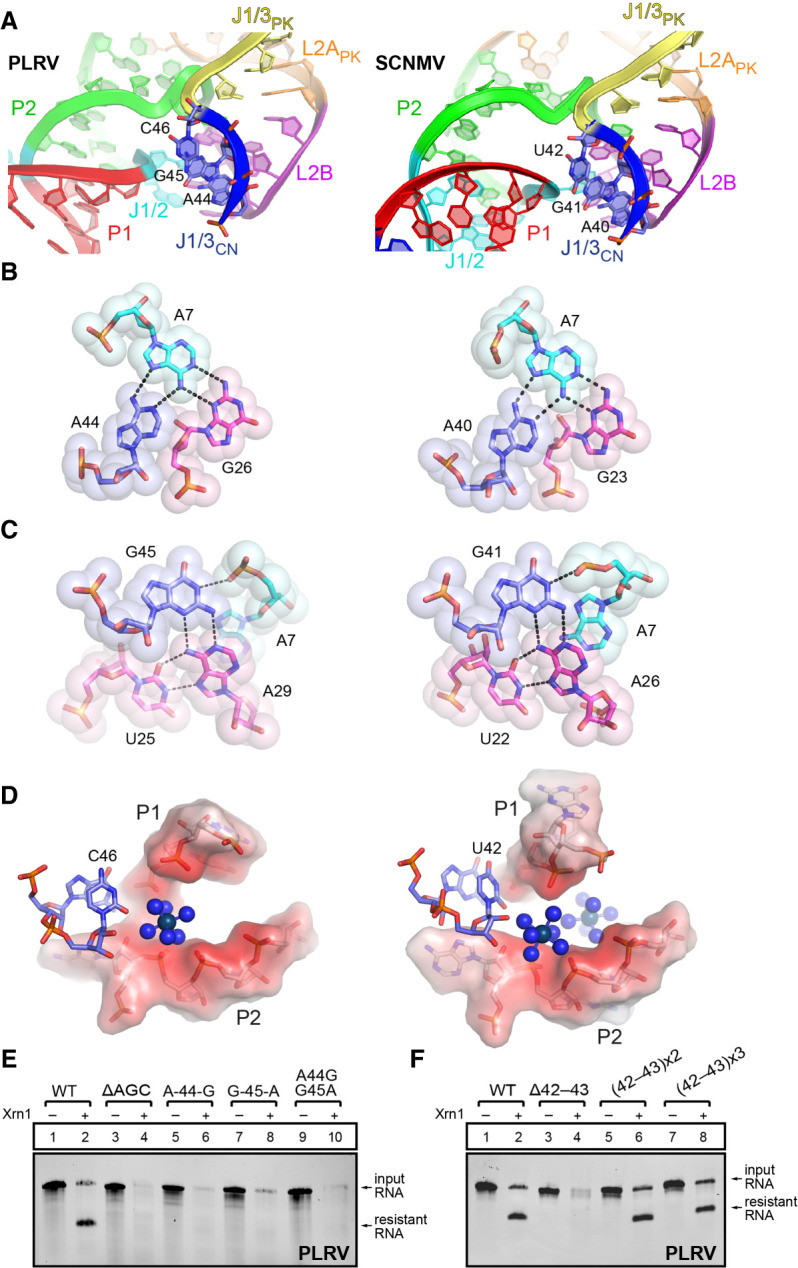
Conserved interactions involving J1/3CN at the dimer interface. (A) Close-up on the region involving J1/3CN (A44, G45, C46 in PLRV, and A40, G41, U42 in SCNMV) in both structures. For panels A–D, the PLRV structure on the left side is compared to the SCNMV on the right side. (B) Interaction network involving PLRV A44 (left) and SCNMV A40 (right). (C) Interaction network involving PLRV G45 (left) and SCNMV G41 (right). (D) C46 (PLRV)/U42 (SCNMV) are in the vicinity of a negatively charged region (in red) formed by the backbone atoms of P1 and P2. Iridium(III) hexammines are shown as blue spheres. (E,F) In vitro Xrn1 degradation assays of PLRV xrRNA WT and J1/3CN mutants.
To interrogate the importance of the conservation at J1/3CN, we generated mutants to disrupt the interaction involving nucleotides 44–46 (Supplemental Fig. S7) and tested them for exoribonuclease resistance. As expected, deletion of A44–C46 (ΔAGC) removes the ability to resist degradation by Xrn1 (Fig. 4E, lanes 3 and 4). But even purine to purine substitutions at positions 44 and/or 45 are sufficient to disrupt tertiary interactions, leading to complete degradation by Xrn1 (Fig. 4E, lanes 5–10). In conclusion, the conserved 5′-AGY-3′ sequence in J1/3CN is involved in tertiary interactions that are key for resistance against Xrn1 degradation. This suggests that the interactions occurring in trans within the dimer probably occur in cis to stabilize the active fold.
The equivalent of J1/3CN in flavivirus xrRNA is part of the ring-like structure that wraps around the 5′ end (Chapman et al. 2014a; Steckelberg et al. 2018a), which suggests its length would be important for proper ring formation and hence resistance. Because J1/3CN shows variation in length across xrRNALT elements beyond the more frequent length of 6 nt (Supplemental Fig. S1), we aimed to test the effect on resistance of decreasing or increasing J1/3CN. As for P2 and L2ACN discussed above, J1/3CN is on the shorter side for SCNMV (three nucleotides vs. five in PLRV; Fig. 1C). Shortening the J1/3CN in the PLRV xrRNALT as with the ΔAGC mutant affected function (Fig. 4E), although that effect is more likely due to the disruption of the associated interaction networks (Fig. 4A–D). Upon shortening J1/3CN by two nucleotides upstream of the AGC stretch (deleting C42 and A43), resistance to Xrn1 was lost (Fig. 4F, lanes 3 and 4). This result suggested that this mutant may no longer be able to form a ring that would prevent it from being degraded. In contrast, extending J1/3CN by 2 nt [(42–43) × 2] or 4 nt [(42–43) × 3] did not significantly reduce exoribonuclease resistance (Fig. 4F, lanes 5–8; Supplemental Fig. S7), indicating a tolerance for a somewhat longer J1/3CN, in line with the observed variation across xrRNALT sequences.
Conclusions
The presence of xrRNAs in a growing list of viruses from several major superfamilies, combined with emerging structural information, continues to expand our insight into their diversity and evolution. The work presented here further shows that all xrRNA structures solved thus far have the potential to form a ring-like fold, but there are different ways to achieve it. In that sense, visualizing the closed conformation for an xrRNALT—that is, with a ring encircling the 5′ end—remains a priority, as it would enable a thorough comparison across the currently known xrRNA active folds. The fact that xrRNAs from flaviviruses and dianthoviruses/poleroviruses are variations on the same theme suggests they may have evolved from a common precursor. Indeed, an emerging hypothesis in biology is that all viruses have a common ancestor (Wolf et al. 2018). Thus, continuing to expand our understanding of the relatedness of xrRNA folds will not only inform computational searches for additional versions, but will open the possibility to use the similarity of structured RNA elements with conserved functional roles as another criterion for virus relatedness, evolution, and taxonomy.
MATERIALS AND METHODS
DNA templates and mutagenesis
DNA templates for in vitro transcription were gBlocks ordered from IDT, cloned into pUC19 and verified by sequencing. RNA constructs for Xrn1 degradation assays contained the xrRNA sequence plus 34 nt of the endogenous upstream sequence of the viral genome (“leader sequence”) to allow loading of the exoribonucleases. Mutations were introduced using custom DNA primers (IDT) during PCR amplification of DNA templates (see “In vitro transcription” below). Nucleotide numbering throughout the manuscript is in reference to the crystallized RNA construct. Transcription with T7 RNA polymerase was started with two guanosine nucleotides for enhanced transcription efficiency.
In vitro transcription
DNA templates for in vitro transcription were amplified by PCR using custom DNA primers (IDT) and Phusion Hot Start polymerase (New England BioLabs). An amount of 2.5 mL transcription reactions were assembled using 1000 µL PCR reactions as template (∼0.2 µM template DNA), 6 mM each NTP, 60 mM MgCl2, 30 mM Tris pH 8.0, 10 mM DTT, 0.1% spermidine, 0.1% Triton X-100, T7 RNA polymerase, and 2 µL RNasin RNase inhibitor (Promega) and incubated overnight at 37°C. After inorganic pyrophosphates were precipitated by centrifugation, the reactions were ethanol precipitated and purified on a 7 M urea 8% denaturing polyacrylamide gel. RNAs of the correct size were excised, eluted overnight at 4°C into ∼40 mL of diethylpyrocarbonate (DEPC)-treated milli-Q filtered water (Millipore) and concentrated and washed using Amicon Ultra spin concentrators (Millipore).
Protein expression and purification
The expression vector for Kluyveromyces lactis Xrn1 (Chang et al. 2011) (residues 1–1245) was a gift of Prof. Liang Tong at Columbia University and the expression vector for Bdellovibrio bacteriovorus RppH was a gift of Joel Belasco at NYU (Messing et al. 2009). All recombinant proteins were 6XHis-tagged, expressed in E. coli BL21 cells and purified using Ni-NTA resin (Thermo), followed by size exclusion with either a Superdex 75 or Superdex 200 column in an AKTA pure FPLC (GE Healthcare). The final product was stored in buffer containing 20 mM Tris pH 7.3, 300 mM NaCl, 1 mM DTT, or 2 mM BME, and 10% glycerol at −80°C. The purity of the recombinant proteins was verified by SDS–PAGE and Coomassie staining.
5′–3′ exoribonuclease degradation assay
An amount of 4 µg RNA was resuspended in 40 µL 100 mM NaCl, 10 mM MgCl2, 50 mM Tris pH 7.5, and 1 mM DTT and refolded at 90°C for 3 min then 20°C for 5 min. An amount of 3 µL recombinant RppH (0.5 µg/µL stock) was added and the samples were split into two 20 µL reactions (±exoribonuclease). An amount of 1.5 µL of the recombinant Xrn1 (0.8 µg/µL stock) was added where indicated. All reactions were incubated for 1.5 h at 30°C using a thermocycler. The degradation reactions were resolved on a 7 M urea 10% denaturing polyacrylamide gel and stained with ethidium bromide. Where indicated, guanidine hydrochloride (1, 10, or 100 mM final concentration) was added to the RNA folding buffer before heat-denaturing the RNA at 90°C.
32P-5′ end-labeling of RNA
RNA was dephosphorylated using rApid Alkaline Phosphatase (Millipore) to convert 5′-triphosphate to 5′-monophosphate ends. To this end, 100 pmol RNA was incubated in a 20 µL reaction containing 2 µL rApid Alkaline phosphatase buffer, 2 µL rApid Alkaline phosphatase, 1 µL RNasin Ribonuclease Inhibitor (Sigma-Aldrich), and incubated for 30 min at 37°C. The enzyme was heat-inactivated for 2 min at 75°C. The RNA was subsequently 5′ end-labeled with [γ32-P] ATP (PerkinElmer) using T4 polynucleotide kinase (PNK) (New England Biolabs). This was done by adding 1 µL 100 mM MgCl2, 4 µL PNK Buffer, 2 µL of 5 mCi [γ32-P] ATP, and 2 µL of PNK in a total volume of 40 µL. The reaction mix was incubated for 30 min at 37°C and purified using Micro Bio P-30 spin columns (Bio-Rad).
Electrophoretic mobility shift assay
A 1:1 serial dilution of unlabeled RNA ranging from 600 µM to 9.4 µM was diluted in 6 µL 1× TH-loading buffer (66 mM Tris-HCl, 34 mM HEPES, 5 mM MgCl2, 10% glycerol, xylene cyanol, bromophenol blue) supplemented with trace amounts of 32P-labeled RNA, heated for 3 min at 85°C, followed by 5 min at 20°C, cooled to 4°C, and loaded on an 8% 1× TH-polyacrylamide native gel (8% polyacrylamide/bis solution 29:1, 66 mM Tris-HCl, 34 mM HEPES, 5 mM MgCl2). The gels were run at 70V at 4°C in 1× TH buffer (66 mM Tris-HCl, 34 mM HEPES, 5 mM MgCl2) until the bromophenol blue dye front reached the lower quarter of the gel (∼3 h), then imaged using a phosphor screen and Typhoon 9400 scanner (GE Life Sciences). Where indicated, 100 mM guanidine hydrochloride was added to the TH-loading buffer, the TH-polyacrylamide gel, and the TH-running buffer.
RNA crystallization and diffraction data collection
RNA for crystallization was prepared as described above. The sequence used for in vitro transcription of the RNA that was used for structure determination was
5′-ggAACTAGCtcAGCATACACGAGTTGCAAGCATgGGAAGTTCAAGCCTCGTGGGCGGCAT
GGTCCCAGCCTCCTCGCTGGCGCCGCCTGGGCAACATGCTTCGGCATGGCGAATGGGACC-3′
where the underlined sequence belongs to a hepatitis delta ribozyme that was used to generate homogenous 3′ ends. Lower case letters represent sequences altered to facilitate transcription (two additional Gs on the 5′ end) and the G·U phasing module. Ribozyme cleavage was induced at the end of the transcription reaction by adding MgCl2 (final conc. 120 mM) and incubating for 10 min at 65°C. Ribozyme-cleaved RNA was purified on a 7 M urea 8% polyacrylamide gel as described above. 5 mg/mL RNA was refolded at 65°C for 3 min in a buffer containing 30 mM HEPES pH 7.5, 20 mM MgCl2, and 100 mM KCl. Crystal Screens I and II, Natrix I and II, and the Nucleic Acid Mini Screen (all from Hampton Research) were used to perform initial screens at 20°C with sitting-drop vapor diffusion crystallization. Initial hits were optimized using custom screens and the Hampton Research Additive Screen.
The RNAs used for the final structural determination were crystallized in drops of 1 µL RNA solution (5 mg/mL RNA in 30 mM HEPES pH 7.5, 20 mM MgCl2, 100 mM KCl) + 1 µL crystallization solution (10% 2-methyl-2,4-pentanediol, 40 mM Sodium Cacodylate pH 6.0, 12 mM Spermine, 150 mM KCl, 100 mM Guanidinium HCl) over a reservoir of 500 µL of 30% 2-methyl-2,4-pentanediol and 100 mM Guanidinium HCl at 20°C with sitting-drop vapor diffusion crystallization. Crystals appeared overnight and grew over the course of several days to a final size of 200–300 µm in their longest dimension. Crystals were buffer-exchanged into freezing solution (30% 2-methyl-2,4-pentanediol, 40 mM Sodium Cacodylate pH 6.0, 12 mM Spermine, 150 mM KCl, 100 mM Guanidinium HCl), with or without 5 mM iridium(III) hexammine to obtain derived crystals for experimental SAD phasing, and then flash-frozen in liquid nitrogen for X-ray diffraction. Diffraction data were collected at Advanced Light Source Beamline 4.2.2 using “shutterless” collection at the Iridium L-I edge (0.9234 Å) at 100K and using a CMOS RDI 8M detector. Two native and three derivative 180° data sets were collected with 0.2° oscillation images. Data were indexed, integrated, and scaled using XDS (Kabsch 2010). Three data sets collected on the same crystal were used for SAD phasing.
Structure determination and refinement
Ten iridium(III) hexammine sites were identified and used in SAD phasing within the AutoSol function of Phenix v. 1.13_2998 on Mac OS (overall FOM = 0.48) (Liebschner et al. 2019). The map was used to manually build an initial model, which was improved through iterative rounds of model building and refinement (simulated annealing, Rigid-body, B-factor refinement) using Coot (Emsley et al. 2010) and Phenix (Liebschner et al. 2019). The final model contains two RNA molecules in the asymmetric unit and all 51 nt are resolved.
Peaks in the anomalous map disappearing around 5 sigma were assigned to cacodylate molecules (f″ (As) ∼ 3 electrons at 0.9234 Å), also taking geometry into account. The model was refined in Phenix v. 1.17.1_3660 on Mac OS, using the graphic user interface, down to R/Rfree of 0.25/0.28. The GTP introduced upon transcription at the 5′ end and the 2′,3′ cyclic UMP introduced during ribozyme cleavage at the 3′ end were linked to the RNA chain using JLigand in CCP4i v. 7.0 (Winn et al. 2011; Lebedev et al. 2012). Map calculation in Refmac v. 5.8.0257 within CCP4i (Winn et al. 2011) led to a higher-quality map with clear density peaks in both 2|Fo| − |Fc| and |Fo| − |Fc| for the guanidinium ligands. The structure was further refined in Refmac to R/Rfree 0.23/0.27. The deposited structure (R/Rfree of 0.225/0.254) was obtained from phenix.refine using default parameters for TLS refinement, occupancy refinement, optimization of X-ray/stereochemistry and X-ray/B-factor weights, and ligand CIF files that were adjusted from the Phenix default geometry parameters and partial charges (available as Supplemental Material). In particular, C–N distances within guanidinium were changed from 1.252 Å (double) and 1.452 Å (single) to 1.37 Å (each C–N bond is delocalized) (Adams and Small 1974; Sawinski et al. 2013). Charges were distributed as follows: +0.3 on the C atom and +0.233 on each N atom. For iridium(III) hexammine, the Ir-N distance was changed from 2.035 Å to 2.1 Å (Gorol et al. 2000), a +1 charge was placed at each amine, and a −3 charge was placed at the Ir atom. Together, these changes led to better interpretation of the electron density, as reflected by drops in B-factors and overall Rfree. Finally, the geometry of the PLRV xrRNALT structure was validated using the MolProbity web server (Williams et al. 2018).
DATA DEPOSITION
The coordinates have been deposited in the Protein Data Bank with accession code 7JJU.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank Pascal Auffinger for helpful suggestions during refinement, and Jack Harrowfield for discussions about iridium(III) hexammine. Support was provided by the National Institutes of Health (grants R35GM118070 and R01AI133348 to J.S.K.) and the Deutsche Forschungsgemeinschaft Fellowship STE2509/201 to A.-L.S.). Beamline 4.2.2 at the Advanced Light Source is partially funded by the National Institutes of Health (P30GM124169-01) and operated under contract with the U.S. Department of Energy (DE-AC02-05CH11231). The University of Colorado Anschutz Medical Campus X-ray crystallography facility is supported by the National Institutes of Health (grants P30CA046934 and S10OD012033).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.076224.120.
REFERENCES
- Adams JM, Small RWH. 1974. The crystal structure of guanidinium carbonate. Acta Cryst B30: 2191–2193. [Google Scholar]
- Akiyama BM, Laurence HM, Massey AR, Costantino DA, Xie X, Yang Y, Shi P-Y, Nix JC, Beckham JD, Kieft JS. 2016. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 354: 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey RT, Kieft JS. 2016. Soaking hexammine cations into RNA crystals to obtain derivatives for phasing diffraction data. Methods Mol Biol 1320: 219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker RR, Atilho RM, Malkowski SN, Nelson JW, Sherlock ME. 2017. The biology of free guanidine as revealed by riboswitches. Biochemistry 56: 345–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JH, Xiang S, Xiang K, Manley JL, Tong L. 2011. Structural and biochemical studies of the 5′→3′ exoribonuclease Xrn1. Nat Struct Mol Biol 18: 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman EG, Costantino DA, Rabe JL, Moon SL, Wilusz J, Nix JC, Kieft JS. 2014a. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 344: 307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman EG, Moon SL, Wilusz J, Kieft JS. 2014b. RNA structures that resist degradation by Xrn1 produce a pathogenic dengue virus RNA. eLife 3: e01892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tondo P, Pritchard RG. 2012. Pentaguanidinium monohydrogendiphosphopentamolybdate(VI) 2.5-hydrate, (CH6N3)5[HP2Mo5O23].2.5H2O: hydrogen bonding and pi-stacking in guanidinium cations. Acta Crystallogr C 68: i50–i52. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorol M, Mösch-Zanetti NC, Noltemeyer M, Roesky HW. 2000. Water-soluble and halogen-free hexaammine complexes of metal ions of group 9—synthesis, crystal structures, and vibrational spectra. Zeitschrift für anorganische Chemie 626: 2318–2324. [Google Scholar]
- Iwakawa H-O, Mizumoto H, Nagano H, Imoto Y, Takigawa K, Sarawaneeyaruk S, Kaido M, Mise K, Okuno T. 2008. A viral noncoding RNA generated by cis-element-mediated protection against 5′→3′ RNA decay represses both cap-independent and cap-dependent translation. J Virol 82: 10162–10174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RA, Steckelberg AL, Vicens Q, Szucs MJ, Akiyama BM, Kieft JS. 2020. Different tertiary interactions create the same important 3-D features in a distinct flavivirus xrRNA. RNA 10.1261/rna.077065.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keel AY, Rambo RP, Batey RT, Kieft JS. 2007. A general strategy to solve the phase problem in RNA crystallography. Structure 15: 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger EN, Beckett RJ, Gray SM, Miller WA. 2013. The complete nucleotide sequence of the genome of Barley yellow dwarf virus-RMV reveals it to be a new Polerovirus distantly related to other yellow dwarf viruses. Front Microbiol 4: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. 2012. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr 68: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, Westhof E. 2001. Geometric nomenclature and classification of RNA base pairs. RNA 7: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. 2019. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75: 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFadden A, O'Donoghue Z, Silva PAGC, Chapman EG, Olsthoorn RC, Sterken MG, Pijlman GP, Bredenbeek PJ, Kieft JS. 2018. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat Commun 9: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahuku G, Lockhart BE, Wanjala B, Jones MW, Kimunye JN, Stewart LR, Cassone BJ, Sevgan S, Nyasani JO, Kusia E, et al. 2015. Maize lethal necrosis (MLN), an emerging threat to maize-based food security in sub-saharan Africa. Phytopathology 105: 956–965. [DOI] [PubMed] [Google Scholar]
- Messing SAJ, Gabelli SB, Liu Q, Celesnik H, Belasco JG, Piñeiro SA, Amzel LM. 2009. Structure and biological function of the RNA pyrophosphohydrolase BdRppH from Bdellovibrio bacteriovorus. Structure 17: 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JW, Atilho RM, Sherlock ME, Stockbridge RB, Breaker RR. 2017. Metabolism of free guanidine in bacteria is regulated by a widespread riboswitch class. Mol Cell 65: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry KL, Kolb FL, Sammons B, Lawson C, Cisar G, Ohm H. 2000. Yield effects of Barley yellow dwarf virus in soft red winter wheat. Phytopathology 90: 1043–1048. [DOI] [PubMed] [Google Scholar]
- Pijlman GP, Funk A, Kondratieva N, Leung J, Torres S, van der Aa L, Liu WJ, Palmenberg AC, Shi P-Y, Hall RA, et al. 2008. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 4: 579–591. [DOI] [PubMed] [Google Scholar]
- Reiss CW, Strobel SA. 2017. Structural basis for ligand binding to the guanidine-II riboswitch. RNA 23: 1338–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawinski PK, Meven M, Englert U, Dronskowski R. 2013. Single-crystal neutron diffraction study on guanidine, CN3H5. Crystal Growth Design 13: 1730–1735. [Google Scholar]
- Slonchak A, Khromykh AA. 2018. Subgenomic flaviviral RNAs: what do we know after the first decade of research. Antiviral Res 159: 13–25. [DOI] [PubMed] [Google Scholar]
- Steckelberg A-L, Akiyama BM, Costantino DA, Sit TL, Nix JC, Kieft JS. 2018a. A folded viral noncoding RNA blocks host cell exoribonucleases through a conformationally dynamic RNA structure. Proc Natl Acad Sci 115: 6404–6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckelberg A-L, Vicens Q, Kieft JS. 2018b. Exoribonuclease-resistant RNAs exist within both coding and noncoding subgenomic RNAs. MBio 9: e02461-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazdar M, Heyda J, Mason PE, Tesei G, Allolio C, Lund M, Jungwirth P. 2018. Arginine “Magic”: guanidinium like-charge ion pairing from aqueous salts to cell penetrating peptides. Acc Chem Res 51: 1455–1464. [DOI] [PubMed] [Google Scholar]
- Wale SJ, Platt HW, Cattlin ND. 2008. Virus and phytoplasma diseases. In Diseases, pests and disorders of potatoes: a color handbook, pp. 75–92. Manson Publishing, London. [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. 2018. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27: 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Kazlauskas D, Iranzo J, Lucía-Sanz A, Kuhn JH, Krupovic M, Dolja VV, Koonin EV. 2018. Origins and evolution of the global RNA virome. MBio 9: e02329–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.