

Abstract
Ferroptosis is a form of cell death resulting from the catastrophic accumulation of lipid reactive oxygen species (ROS). Oncogenic signaling elevates lipid ROS production in many tumor types and is counteracted by metabolites derived from the amino acid cysteine. Here we show that the import of oxidized cysteine (cystine) via system xC– is a critical dependency of pancreatic ductal adenocarcinoma (PDAC), a leading cause of cancer mortality. Cysteine was used to synthesize glutathione and coenzyme A which combined to control ferroptosis. Studying genetically engineered mice, we found that deletion of a system xC– subunit, Slc7a11, induced tumor-selective ferroptosis and inhibited PDAC growth. This was replicated through administration of cyst(e)inase, a drug that depletes cysteine/cystine, demonstrating a translatable means to induce ferroptosis in PDAC.
One sentence summary:
Genetic and pharmacological targeting of cystine import induces cancer-selective ferroptosis in pancreatic tumors of genetically engineered mice.
Pancreatic ductal adenocarcinoma (PDAC) is a deadly cancer that is resistant to traditional therapies. Over 90% of PDAC cases harbor mutations in KRAS that both promote proliferation and alter cellular metabolism. A byproduct of mutant KRAS signaling is the increased production of reactive oxygen species (ROS), which can damage cellular components. To compensate, PDAC cells upregulate metabolic programs that detoxify ROS using cysteine-derived metabolites such as glutathione (GSH) (1). Most cellular cysteine is acquired through the system xC– antiporter, which exchanges extracellular, oxidized cysteine (cystine) for intracellular glutamate. Yet, germline deletion of the system xC– gene, Slc7a11, is well tolerated in unstressed mice (2), suggesting normal cells have low basal cystine import requirements. We hypothesized that cystine import is a critical dependency of PDAC that may be selectively targeted as an anticancer therapy.
To investigate the role of cysteine metabolism in PDAC, we measured the viability of human PDAC cell lines cultured for 24 hours in media with varying concentrations of cystine or the system xC– inhibitor imidazole ketone erastin (IKE) (Fig. 1, A and B) (3). In four of five PDAC lines, cystine starvation reduced cell viability by >80%; this was largely prevented by addition of the lipophilic antioxidant Trolox. Cystine-starved cells underwent catastrophic destabilization of their plasma membranes, without visual evidence of nuclear fragmentation (movie S1). IKE treatment mimicked the effects of cystine withdrawal, quickly killing most cells from the four sensitive PDA lines in a manner visually identical to that of cystine starvation, but distinct from staurosporine-induced apoptosis (fig. S1A, movies S2 and S3). Neither cystine starvation nor system xc– inhibition (collectively referred to as “cysteine depletion”) induced caspase 3 cleavage (fig. S1B), indicating the cell death was not apoptotic. Rather, the oxidative cell death resembled ferroptosis, a form of iron-dependent, non-apoptotic cell death previously associated with system xC– inhibition (4). We found that co-treatment of human PDAC cells with either deferoxamine (DFO, an iron chelator), ferrostatin-1 (Fer1, a ferroptosis inhibitor), or N-acetylcysteine (NAC, a cell permeable analog of cysteine) significantly reduced cell death from cysteine depletion, whereas inhibitors of apoptosis or necroptosis had little impact on cell death, consistent with previous reports (Fig. 1C; fig. S1, C to E) (5). Autophagy inhibition had variable effects in different lines, likely reflecting the known impact of ferritinophagy on ferroptosis (6). Using the fluorescent probe C11-BODIPY, we observed a large increase in lipid oxidation (a hallmark of ferroptosis) prior to cell death in response to cysteine depletion; this was prevented by co-treatment with Trolox, Fer-1, NAC, and DFO (Fig. 1D; fig. S2, A and B). By contrast, elevated total ROS levels induced by cysteine depletion were not prevented by these agents, arguing against a more general oxidative process (Fig. S2C). We conclude from these experiments that most PDAC lines rely on cysteine to prevent ferroptotic cell death. An analysis of SLC7A11 expression across human datasets revealed a modest overexpression in PDAC versus normal tissues, enrichment in the malignant epithelial compartment of PDAC, and an association with signatures of redox stress (fig. S3). Across multiple human cancers, SLC7A11 was frequently overexpressed and associated with reduced survival (fig. S4).
Fig. 1. Pancreatic cancer cells require exogenous cystine to avert ferroptosis.
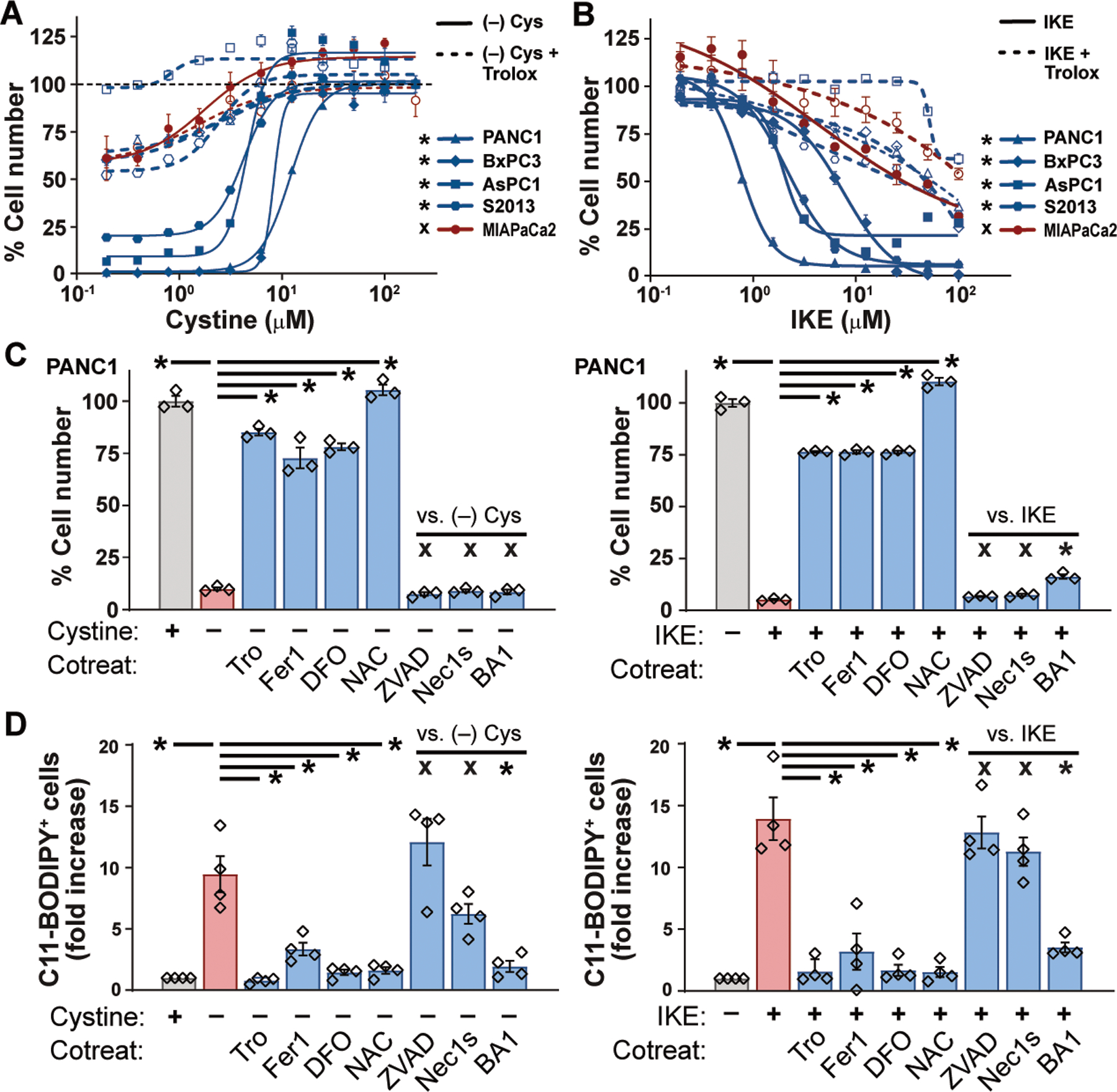
(A and B) Viability of human PDAC lines after 24 hours culture in varying concentrations of cystine (A) or IKE (B), alone or in combination with 100 μM Trolox. Student’s t-test comparing maximal cytotoxicity ± Trolox. (C) Viability of PANC-1 cells cultured for 24 hours in cystine-free media or treated with 10 μM IKE, alone or in combination with 100 μM Trolox (Tro), 500 nM ferrostatin-1 (Fer1), 100 μM deferoxamine (DFO), 1mM N-acetyl cysteine (NAC), 50 μM ZVAD-FMK, 1 nM Bafilomycin A1 (BA1) or 10 μM Necrostatin-1s (Nec1s). Tukey test. (D) Flow cytometry of C11-BODIPY fluorescence in PANC-1, AsPC-1, BxPC-3, and S-2013 cells after 6–8 hours of treatment with conditions from panel C. Tukey test. All data are means ±SEM of three independent experiments. * p < 0.05, x = no significant difference.
To learn whether pancreatic tumors in mice depend on system xc– for survival, we employed a dual recombinase genetic engineering strategy based on the “KPC” mouse model (7). KrasFSF.G12D/+; Tp53R172H/+; Pdx1FlpOtg/+; Slc7a11Fl/Fl; Rosa26CreERT2/+ (KPFSR) mice (fig. S5) spontaneously develop PDAC driven by FlpO-dependent activation of mutant Kras and germline expression of mutant Tp53. These tumors are identical in genotype and phenotype to the KPC model, but administration of tamoxifen induces systemic deletion of Slc7a11 through the action of Cre recombinase expressed from the Rosa26 locus (figs. S5 and S6, A to C). We randomized KPFSR mice bearing 4–7mm tumors to receive six daily doses of vehicle or tamoxifen and monitored tumor growth by ultrasound (8). We found that deletion of Slc7a11 in established tumors of KPFSR mice nearly doubled median survival compared to vehicle treatment (Fig. 2A, p = 0.0295, n = 20). Most recombined tumors exhibited a period of stable disease or partial response—one underwent a complete regression; these responses were never observed in vehicle-treated mice (Fig. 2B; figs. S6, D to F and S7A). Critically, addition of NAC to the drinking water of tamoxifen-treated mice restored baseline survival and eliminated tumor responses, supporting a link to cysteine metabolism (Fig. 2, A and B). At necropsy, escaped tumors exhibited evidence of incomplete Slc7a11 recombination by PCR and restored protein expression by Western blotting, suggesting the outgrowth of unrecombined tumor cells (fig. S7, B to C).
Fig. 2. Deletion of Slc7a11 in KPC mice induces tumor ferroptosis and extends survival.
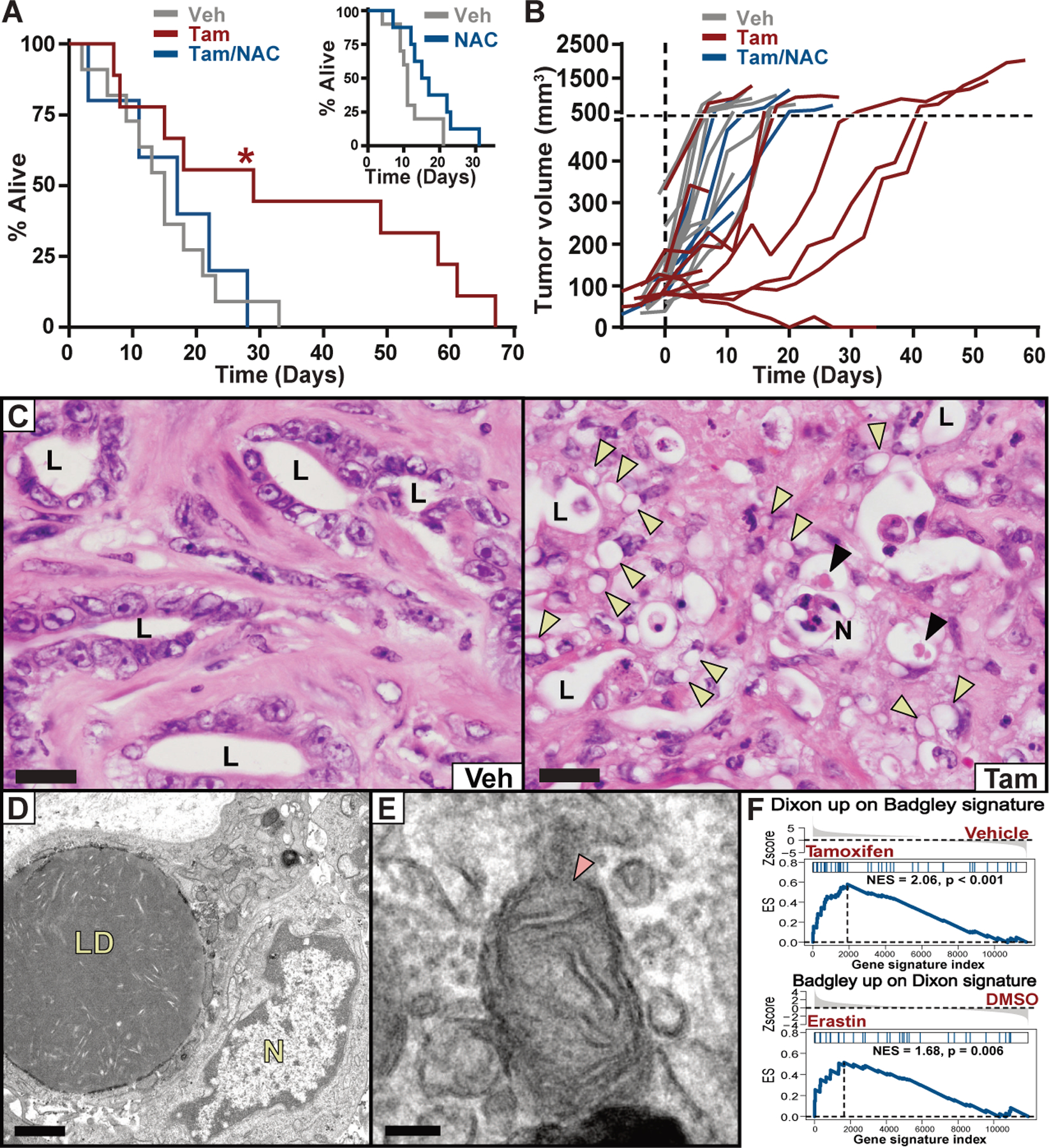
(A) Survival of KPFSR mice treated with vehicle (n = 11, median 15 days), tamoxifen (n = 9, median 29 days), or tamoxifen/NAC (n = 5, median 17 days). * p < 0.0295, log-rank. INSET: Survival of KPC mice treated with NAC alone (n = 8, median = 16 days) versus historical saline-treated controls (n = 10, median = 11 days). (B) Growth curves for each KPFSR tumor. (C) Hematoxylin and eosin (H&E) stained sections of tumor tissue from KPFSR mice treated with vehicle or tamoxifen. L = lumen of malignant epithelium, N = necrosis, yellow arrowheads = lipid droplets, black arrowheads = megamitochondria. Scale = 20 μm. (D and E) TEM images from tamoxifen-treated KPFSR tumors. LD = lipid droplets, N = nucleus, arrow indicates damaged mitochondrion. (D) scale = 1μm; (E) scale = 100 nm. (F) Gene set enrichment analysis. Top panel depicts enrichment of a published ferroptosis expression signature (Dixon) among genes differentially expressed in tamoxifen-treated KPFSR epithelia (Badgley) (p < 0.001). Bottom panel depicts the reciprocal comparison (p < 0.006).
The study of in vivo ferroptosis has been hindered by the lack of a validated, selective biomarker and the absence of a histopathological characterization of the phenomenon in tissues. Within tamoxifen-treated KPFSR tumors, we observed numerous lesions of ballooned epithelial cells with lipid droplet-like structures and intermittent megamitochondria, often juxtaposed to necrotic regions—a phenotype only occasionally observed in vehicle-treated KPFSR and untreated KPC tumors (Fig. 2, C to D; fig. S7C). These lesions exhibited no alterations in apoptosis or proliferation markers (fig. S8, A and B), but did display accumulation of 4-hydroxynonenal (4HN) (fig. S8, C to E), a byproduct of lipid peroxidation, making them candidates for in vivo ferroptosis. Critically, no pathologies were observed in non-pancreatic tissues of tamoxifen-treated KPFSR mice, indicating a tumor-selective phenotype. Transmission electron microscopy (TEM) and Oil Red O staining of tamoxifen-treated KPFSR tumors confirmed the presence of abnormally large lipid droplets (Fig. 2D and fig. S8F). TEM also revealed structural aberrations in mitochondria of malignant epithelial cells, including disrupted cristae and compromised membrane integrity (Fig. 2E), consistent with the results of prior in vitro studies (9). Finally, we performed laser capture microdissection and RNA-sequencing to isolate malignant epithelial cells from KPFSR tumors and found that genes upregulated in response to Slc7a11 deletion were enriched in a ferroptotic expression signature from erastin-treated HT-1080 cells (Fig. 2F) (10); apoptotic gene sets were not enriched (table S1). We conclude that the phenotype observed in tamoxifen-treated KPFSR tumors is a histologically identifiable, in vivo manifestation of ferroptosis.
Prior studies have indicated that cysteine regulates ferroptosis primarily through the synthesis of glutathione, a critical cofactor for the lipid peroxide-detoxifying enzyme GPX4 (3). Indeed, we found that cysteine depletion rapidly reduced GSH levels in two human PDAC cell lines (Fig. S9A). Furthermore, co-treatment with the membrane-permeable GSH analog glutathione ethyl ester (GSH-EE) prevented lipid oxidation and ferroptosis (fig. S9, B to E). However, inhibition of GSH biosynthesis using buthionine sulfoximine (BSO) (fig. S9F) did not induce lipid ROS or reduce cell viability (Fig. 3, A and B), demonstrating that GSH loss is not sufficient to induce ferroptosis in PDAC cells (11). To investigate whether additional cysteine-derived metabolites contribute to the regulation of ferroptosis, we traced the metabolism of exogenous cystine by using 13C-labelled cystine and measuring labelled metabolites by mass spectrometry. In addition to exhibiting rapid flux into glutathione pools, cystine was also converted to coenzyme A (CoA) over 24 hours (Fig. 3 C and D); no flux was observed into taurine, lactate, citrate, or glutamate (fig. S9G). CoA is synthesized from cysteine via the pantothenate pathway and plays a role in many metabolic pathways, particularly lipid metabolism. Both CoA and its derivative coenzyme Q10 (CoQ10) have been shown to impact sensitivity to ferroptosis (12, 13). We found that system xC– inhibition reduced CoA levels and increased levels of pantothenate (Fig. 3E), a metabolite upstream of cysteine incorporation in CoA synthesis. Moreover, treatment of PDAC cells with exogenous CoA (14) prevented IKE-induced ferroptosis (Fig. 3F) whereas pantothenate kinase inhibition with PANKi sensitized cells to IKE (fig. S10, A to C). Strikingly, PANKi combined synergistically with BSO to induce ferroptosis (Fig. 3G and fig. S10D). Co-treatment with idebenone (a membrane-permeable analog of CoQ10) or a monounsaturated fatty acid blocked BSO/PANKi induced ferroptosis, whereas saturated or poly-unsaturated fatty acids did not (fig. S10E), similar to prior observations with IKE (15). Together, these experiments demonstrate that CoA and GSH cooperate to regulate ferroptosis in human PDAC cells (fig. S10H).
Fig. 3. Combination GSH and CoA inhibition induces ferroptosis in human PDAC cells.
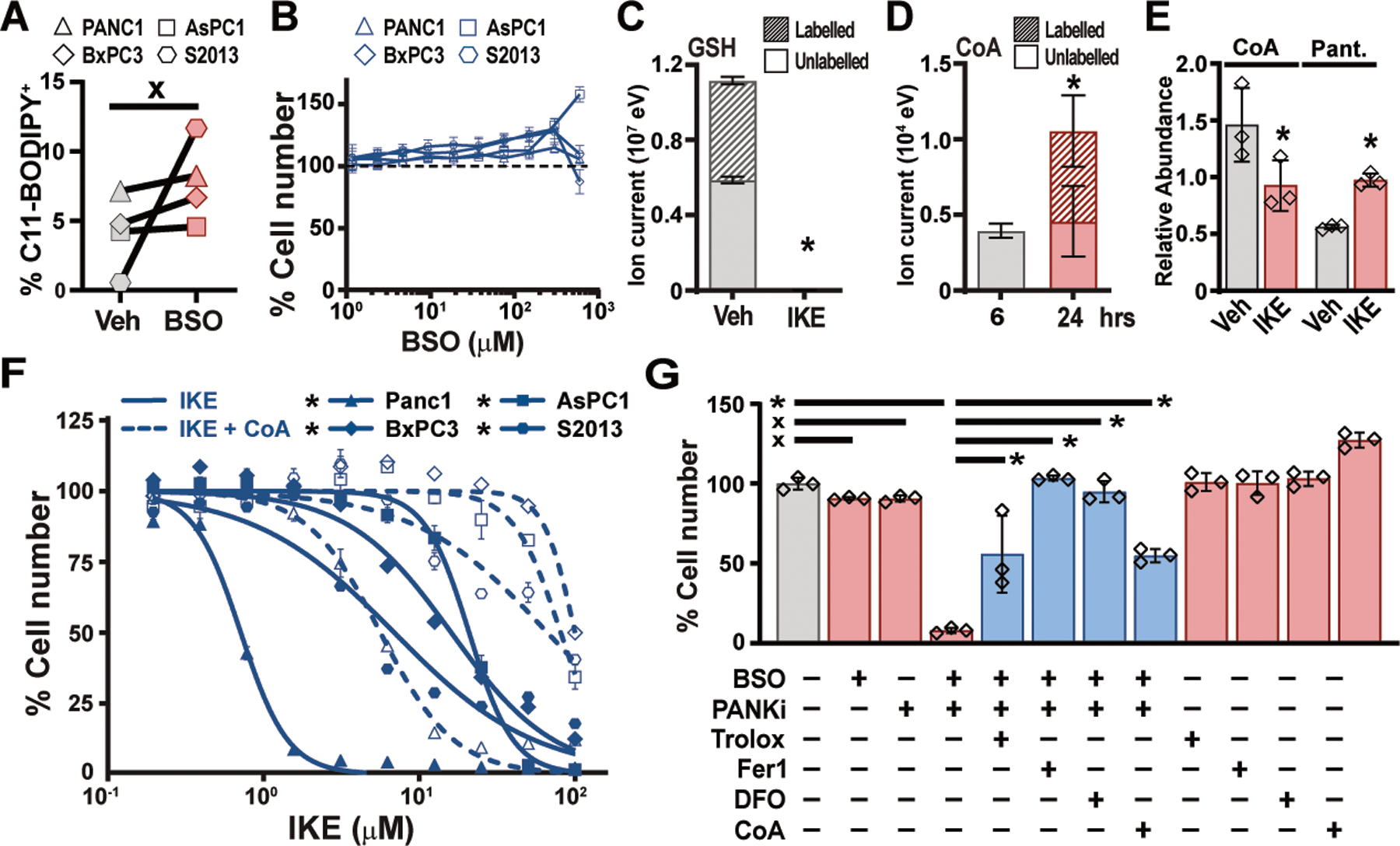
(A) Flow cytometry for C11-BODIPY fluorescence in four human PDAC lines treated for 6 hours with 150 μM BSO. Paired t-test. (B) Viability of human PDAC cells treated for 24 hours with indicated concentrations of BSO. (C) Liquid chromatography time-of-flight mass spectrometry (LC-TOF-MS) analysis of GSH in PANC-1 cells labelled for 6 hours with 13C-cystine combined with vehicle or 5μM IKE. Student’s t-test. (D) LC-TOF-MS analysis of CoA in PANC-1 cells labelled for 6 hours with 13C-cystine, after 6 or 24 hours. Student’s t-test. (E) Liquid chromatography triple quadrupole tandem mass spectrometry (LC-QqQ-MS) measurements of CoA and pantothenate (Pant.) levels in Panc-1 cells treated with vehicle and IKE for six hours. Student’s t-test. (F) Viability of human PDAC cell lines treated with IKE, alone or in combination 200 μM CoA. Student’s t-test comparing maximal cytotoxicity ± CoA. (G) PANC-1 cells treated for 24 hours with combinations of 300 μM BSO and 5 μM PANKi along with Trolox, Fer-1, DFO, or CoA as described in Fig. 1C. Tukey test. In all panels, * p < 0.05, x = not significantly different. In panels A, B, E, and F, data are means ±SEM from three independent experiments. In panels C and D, data are means ± SD from three biological replicates. In (E), * p < 0.05 comparing maximal cytotoxicity in CoA treated vs. untreated conditions for each line, Student’s t-test. In (F), * p < 0.05, one-way ANOVA with posthoc Tukey test.
Finally, we sought a pharmacological means to target cysteine metabolism in pancreatic tumors. Drug delivery is compromised in PDAC due to the effects of fibrosis on tissue perfusion (16). Although current system xC– inhibitors are not optimized for the PDAC microenvironment, the engineered enzyme cyst(e)inase is well-tolerated in mammals, has a long half-life, and potently degrades both cystine and cysteine in circulation (17). In vitro, cyst(e)inase treatment induced lipid oxidation and reduced the viability of IKE-sensitive PDAC lines; this was largely prevented by co-treatment with ferroptosis inhibitors (Fig. 4, A to C; fig. S11, A and B). To determine the effects of cyst(e)inase on pancreatic tumors in vivo, we treated tumor-bearing KPC mice for 10 days with vehicle, low-dose cyst(e)inase, or high-dose cyst(e)inase (n=2 each). Histopathological examination of cyst(e)inase-treated tumors revealed a severe ferroptosis phenotype, with extensive lipid droplet formation, stromal disruption, decompressed blood vessels, and necrosis (Fig. 4D and E; fig. S12, A to B). TEM revealed enlarged lipid droplet formation, extracellular lipid droplets, and mitochondrial defects, preferentially in cyst(e)inase-treated KPC tumors (Fig. 4F to I). Ferroptotic lesions were generally 4HN positive and cleaved caspase 3 negative (fig. S13A). Finally, four additional KPC mice were treated with high-dose cyst(e)inase and their tumor growth was monitored by ultrasound. Notably, all four tumors exhibited stabilizations or regressions whereas historical vehicle-treated controls never stabilized (Fig. 4J). Thus, we conclude that the therapeutic depletion of cysteine/cystine can induce ferroptosis in Kras/p53 mutant pancreatic tumors in mice.
Fig. 4. Cyst(e)inase treatment induces tumor-selective ferroptosis in KPC mice.
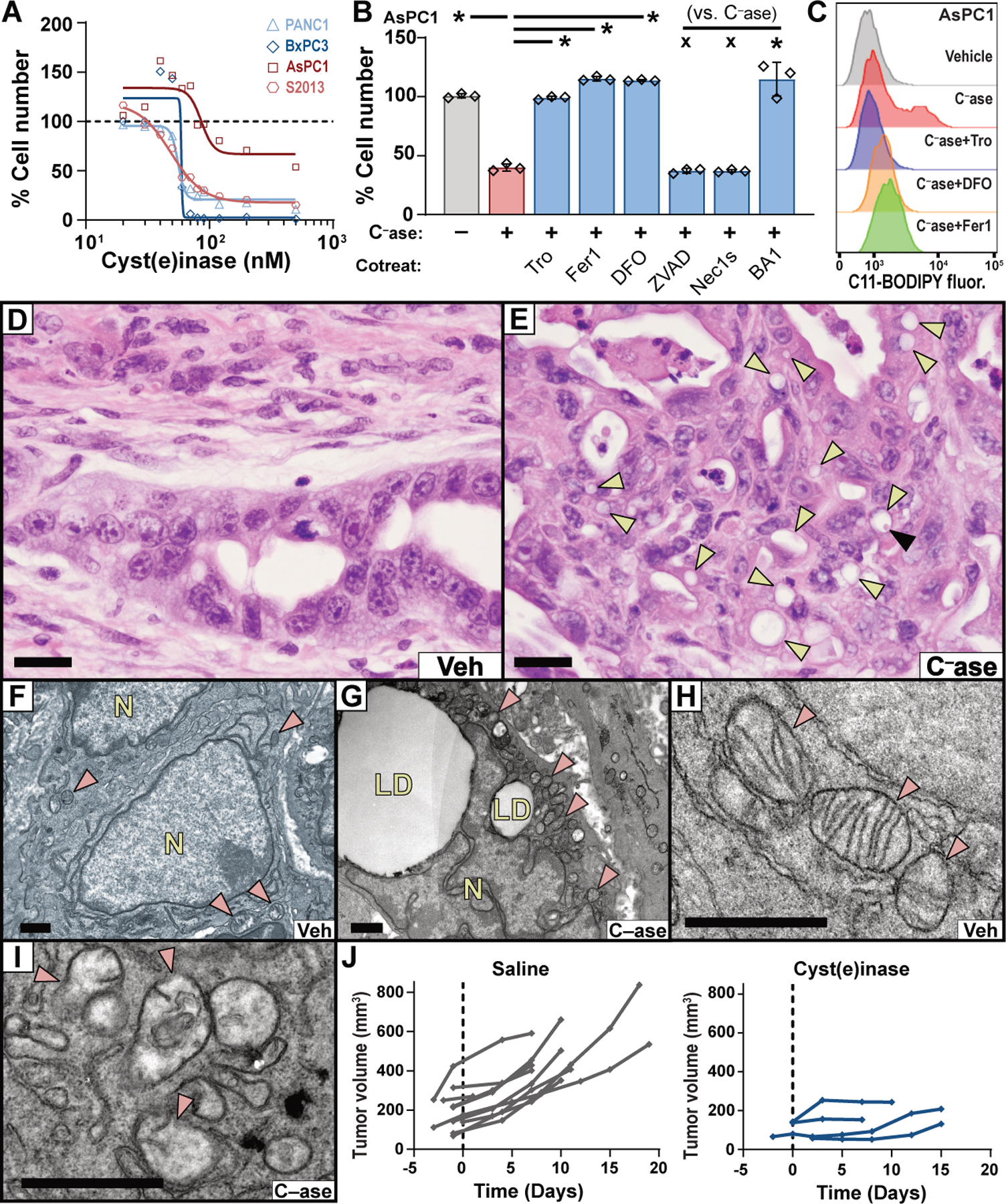
(A) Viability of human PDAC lines cultured with varying concentrations of cyst(e)inase for 48 hours (AsPC1) or 72 hours (PANC-1, BxPC3, S2–013). (B) Viability of AsPC1 cells treated with 90 nM cyst(e)inase (C–ase) for 72 hours, alone or in combination with indicated agents, under conditions described in Fig. 1C. (C) C11-BODIPY fluorescence was measured by flow cytometry in AsPC-1 cells after 24 hours of treatment with 90 nM cyst(e)inase, alone or in combination with indicated agents, under conditions described in Fig. 1C. (D and E) H&E stained sections of pancreatic tumors from KPC mice treated with vehicle or cyst(e)inase. Yellow arrowheads indicate lipid droplets. Black arrowhead indicates megamitochondrion. Bars = 20 μm. (F–I) TEM of pancreatic tumors from the KPC model treated with vehicle (F and H) or cyst(e)inase (G and I). Red arrowheads indicate mitochondria; LD = lipid droplets; N = nucleus. Bars = 1 μm. (J) Tumor growth curves from KPC mice treated with saline (historical controls) or 100 mg/kg cyst(e)inase, q.2.d., i.p.. For panels A–B, data depict mean of 3 biological replicates. * p < 0.05, x = not significant.
In summary, our data add to a growing body of evidence showing that certain cancers, including PDAC, rely on cysteine metabolism to avert ferroptosis. Previously, SLC7A11 deletion via CRISPR-Cas9 was shown to induce ferroptosis in cultured PDAC cells and slow xenograft engraftment and growth (18), and system xC– inhibition was shown to limit the growth of lymphoma xenografts, inducing a lipid oxidative signature and other indicators of ferroptosis (15). However, cysteine depletion in a PDAC xenograft model had little effect on tumor growth (19), perhaps indicating that the nutrient-deprived, hypoxic microenvironment of autochthonous pancreatic tumors may contribute to the tumor-selective cysteine dependency we observed in genetically engineered mouse models of PDAC. While it is not yet known whether human PDAC is also susceptible to ferroptosis from cysteine depletion, the clinical development of cyst(e)inase for treatment of the metabolic disorder cystinuria provides a pathway for future translation of this concept.
Supplementary Material
Acknowledgements:
We thank members of the Olive, Lyssiotis, and Stockwell labs for technical advice and thoughtful critiques. We also thank K.L. Olive for manuscript editing and S.W. Novak for advice on electron microscopy interpretation.
Funding: This work was supported by the NIH/NCI Cancer Center Support Grant P30CA013696 and utilized the Confocal and Specialized Microscopy, Molecular Pathology, Flow Cytometry Core, and OPTIC shared resources. M.A.B was supported by a training grant (T32 A009503) and a pre-doctoral fellowship (F31 CA180738). D.M.K was supported by a Department of Education GAANN fellowship through The University of Michigan Program in Chemical Biology. K.P.O was supported by the Lustgarten Foundation for Pancreatic Cancer Research (2011 Innovator Award) and the NIH/NCI (1R01CA215607). C.A.L was supported by a Pancreatic Cancer Action Network/AACR Pathway to Leadership award (13-70-25-LYSS), Dale F. Frey Award for Breakthrough Scientists from the Damon Runyon Cancer Research Foundation (DFS-09-14), Junior Scholar Award from The V Foundation for Cancer Research (V2016-009), Kimmel Scholar Award from the Sidney Kimmel Foundation for Cancer Research (SKF-16-005), and by NIH grant U24-DK097153. V.P. was supported by a PRCRP Horizon award by the Department of Defense (W81XWH-17-1-0497). L.R.A. and U.M. were supported by NCI CCSG (CA014195) and the Waitt Foundation. B.R.S. is supported by the NCI/NIH (R35CA209896 and P01CA087497). G.M.W. and K.E.D.G. were funded by a Cancer Center Core Grant (CA014195), NIH/NCI (R35 CA197687), and funding from the Freeberg Foundation and the Hirschberg Foundation. K.E.D.G. was also funded by the Salk Women and Special Science Award.
Footnotes
Competing interests: B.R.S. holds equity in and serves as a consultant to Inzen Therapeutics and is an inventor on patents and applications related to ferroptosis, including the following U.S. patents and corresponding applications and patents in other countries, all submitted by Columbia University: 10,259,775, 10,233,171, 9,580,398, 20190292135, 20190135782, 20170233370, 20160297748, 20150079035, and 20080299076, 9,938,245, 20160332974, 9,695,133, 20150175558, 8,546,421, 20100081654, 8,535,897, 20110008803, 8,518,959, 20090214465, 8,124,365, 7,358,262, 20080220454, 7,615,554, 20190315681, and 20070161644. G.G. and E.S. have an equity interest in Aeglea Biotherapeutics, a company that has licensed the commercial development of Cyst(e)inase, and are inventors on U.S. patents and corresponding applications and patents in other countries, all submitted by the University of Texas, related to L-cyst(e)inase: 10,363,311, 20180327734. The other authors declare no competing financial interests.
Data and Materials availability: Cyst(e)inase is available from G.G. and E.S. under a material transfer agreement with the University of Texas. RNA seq datasets are available at GEO (Accession number GSE119628). All other data is available in the text or the supplementary materials. Mouse strains comprising the KPC and KPFSR models (other than Pdx1-FlpO) were provided through material transfer agreements with EUCOMM (Slc7a11Flox) and MIT (all others).
Supplementary Materials:
Materials and Methods
Figures S1-S13
Table S1
Movies S1-S3
References (20–46)
References and Notes
- 1.DeNicola GM et al. , Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato H et al. , Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem 280, 37423–37429 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Yang WS et al. , Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dixon SJ et al. , Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao M et al. , Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedmann Angeli JP et al. , Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hingorani SR et al. , Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Sastra SA, Olive KP, Quantification of murine pancreatic tumors by high-resolution ultrasound. Methods Mol. Biol 980, 249–266 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao M et al. , Role of Mitochondria in Ferroptosis. Mol. Cell 73, 354–363 e353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ et al. , Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris IS et al. , Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 29, 1166–1181 e1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimada K et al. , Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol 12, 497–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leu JI, Murphy ME, George DL, Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl. Acad. Sci. U S A 116, 8390–8396 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srinivasan B et al. , Extracellular 4’-phosphopantetheine is a source for intracellular coenzyme A synthesis. Nat. Chem. Biol 11, 784–792 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y et al. , Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol 26, 623–633 e629 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olive KP et al. , Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cramer SL et al. , Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23, 120–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daher B et al. , Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 79, 3877–3890 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Kshattry S et al. , Enzyme-mediated depletion of l-cyst(e)ine synergizes with thioredoxin reductase inhibition for suppression of pancreatic tumor growth. NPJ Precis. Oncol 3, 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R. C. Team, R: A Language and Environment for Statistical Computing. (2016). [Google Scholar]
- 23.Davis S, Meltzer PS, GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Donahue TR et al. , Integrative Survival-Based Molecular Profiling of Human Pancreatic Cancer. Clinical Cancer Research 18, 1352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiraoka N et al. , CXCL17 and ICAM2 Are Associated With a Potential Anti-Tumor Immune Response in Early Intraepithelial Stages of Human Pancreatic Carcinogenesis. Gastroenterology 140, 310–321. e314. [DOI] [PubMed] [Google Scholar]
- 26.Pei H et al. , FKBP51 Affects Cancer Cell Response to Chemotherapy by Negatively Regulating Akt. Cancer Cell 16, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S et al. , A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Research 76, 3838 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moffitt RA et al. , Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet 47, 1168–1178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruckert F et al. , Co-expression of KLK6 and KLK10 as prognostic factors for survival in pancreatic ductal adenocarcinoma. Br. J. Cancer 99, 1484–1492 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grützmann R et al. , Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia 6, 611–622 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F, A Model-Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association 99, 909–917 (2004). [Google Scholar]
- 32.Samur MK, RTCGAToolbox: A New Tool for Exporting TCGA Firehose Data. PLOS ONE 9, e106397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey P et al. , Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Ritchie ME et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hänzelmann S, Castelo R, Guinney J, GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 1–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viechtbauer W, Conducting Meta-Analyses in R with the metafor Package. Journal of Statistical Software, Articles 36, 1−−48 (2010). [Google Scholar]
- 38.Therneau TM, Grambsch PM, Modeling Survival Data: Extending the Cox Model. Statistics for Biology and Health (Springer; New York, 2013). [Google Scholar]
- 39.Hingorani SR et al. , Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Raymond CS, Soriano P, High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS One 2, e162 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hingorani SR et al. , Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Rhim AD et al. , EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M, Acute pancreatitis accelerates initiation and progression to pancreatic cancer in mice expressing oncogenic Kras in the nestin cell lineage. PLoS One 6, e27725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M, Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys. Res. Commun 382, 561–565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eberle-Singh JA et al. , Effective Delivery of a Microtubule Polymerization Inhibitor Synergizes with Standard Regimens in Models of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res 25, 5548–5560 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Sastra SA, Olive KP, Quantification of murine pancreatic tumors by high-resolution ultrasound. Methods Mol. Biol 980, 249–266 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.