

Abstract
Mutasynthesis of pyrichalasin H from Magnaporthe grisea NI980 yielded a series of unprecedented 4′‐substituted cytochalasin analogues in titres as high as the wild‐type system (≈60 mg L−1). Halogenated, O‐alkyl, O‐allyl and O‐propargyl examples were formed, as well as a 4′‐azido analogue. 4′‐O‐Propargyl and 4′‐azido analogues reacted smoothly in Huisgen cycloaddition reactions, whereas p‐Br and p‐I compounds reacted in Pd‐catalysed cross‐coupling reactions. A series of examples of biotin‐linked, dye‐linked and dimeric cytochalasins was rapidly created. In vitro and in vivo bioassays of these compounds showed that the 4′‐halogenated and azido derivatives retained their cytotoxicity and antifungal activities; but a unique 4′‐amino analogue was inactive. Attachment of larger substituents attenuated the bioactivities. In vivo actin‐binding studies with adherent mammalian cells showed that actin remains the likely intracellular target. Dye‐linked compounds revealed visualisation of intracellular actin structures even in the absence of phalloidin, thus constituting a potential new class of actin‐visualisation tools with filament‐barbed end‐binding specificity.
Keywords: cytochalasins, molecular tools, mutasynthesis, semi-synthesis
Molecular toolbox: Unprecedented cytochalasins functionalised at the 4′‐position were produced by mutasynthesis. These compounds were then chemically diversified to produce a range of molecular tools without the need for protecting strategies. As a pertinent example the Texas‐red‐modified compound was synthesised in a single step from 4′‐azidocytochalasin and used to visualise sub‐micrometer actin microstructures in immobilised U2OS cells.
Cytochalasans are a family of fungal secondary metabolites that bind to actin and modify its polymerisation in eukaryotic cells. [1] In particular, cytochalasans [2] selectively bind the barbed ends of filamentous (F) actin.[ 3 , 4 ] Many of the biological roles of the cytochalasans can be traced to this function, but antiviral [5] and antibiofilm [6] properties point to the likelihood of other targets in eukaryotes and prokaryotes, and characterisation of the inhibition of glucose transport in eukaryotes [7] suggests that their targets may be more diverse. Cytochalasans are generally produced by fermentation, and although hundreds of individual structures have been reported, [8] relatively few are available commercially. Total syntheses of several cytochalasans have been reported, but their scope and practicality are limited, and the syntheses have not been extended to the production of functionalised analogues. Surprisingly, almost no investigations have been reported on the chemical modification of cytochalasans, or attempts to functionalise them for more targeted use in chemical biology. Here, we reveal a simple and effective series of methods for the functionalisation of cytochalasans based on the manipulation of the biosynthesis of pyrichalasin H (1 a).
Pyrichalasin H (1 a) is produced by the fungus Magnaporthe grisea NI980 [9] and is the 4′‐OMe substituted analogue of the better‐known cytochalasin H (1, X=H, Scheme 1). [10] We recently reported the discovery and characterisation of the biosynthetic gene cluster (BGC) encoding the biosynthesis of 1 a.[ 11 , 12 ] Knockout experiments showed that the biosynthesis starts with the O‐methylation of l‐tyrosine 2 to give 3 a (Scheme 1), which is the substrate for a hybrid polyketide synthase non‐ribosomal peptide synthetase (PKS‐NRPS, PyiS) that, in association with auxiliary proteins, [13] produces the cytochalasin skeleton 4 a. This undergoes various oxygenation [14] and acylation reactions to give 1 a. Crucially, KO of pyiA, which encodes the construction of O‐methyl tyrosine 3 a, prevents biosynthesis of 1 a. Mutasynthesis experiments [15] with 4′‐fluoro, 4′‐chloro and 4′‐bromo phenylalanines (3 b–3 d) then gave the corresponding 4′‐halo‐cytochalasins (1 b–d) in titres similar to that of 1 a production in wild‐type (WT) strains (ca 60 mg L−1). This indicates that the adenylation (A) domain of the NRPS component of PyiS is relatively unselective.
Scheme 1.
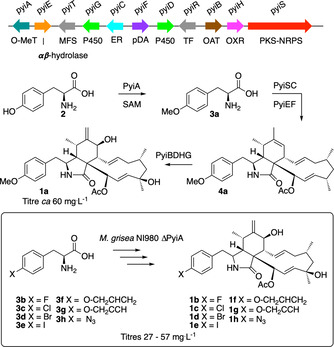
Biosynthesis of pyrichalasin H (1 a) and production of 4′‐substituted cytochalasins 1 b–1 h by mutasynthesis. Abbreviations: O‐MeT=O‐methyltransferase, MFS=major facilitator superfamily (transporter), P450=cytochrome P450 oxygenase, ER=trans‐acting enoyl reductase, pDA=putative Diels Alderase, TF=transcription factor, OAT=O‐acetyl transferase, OXR=oxidoreductase, PKS‐NRPS=polyketide synthase‐nonribosomal peptide synthetase.
We next investigated the substrate tolerance of the PyiS A domain by feeding a wider range of substituted phenylalanines to the M. grisea ΔpyiA strain. These experiments showed that 4′‐iodo (3 e), 4′‐O‐allyl (3 f), 4′‐O‐propargyl (3 g) and 4′‐azido (3 h) phenylalanines are all good substrates, with feeding experiments producing the corresponding 4′‐functionalised cytochalasins 1 e–1 h in purified titres of 30–60 mg L−1 (Scheme 1). All compounds were fully characterised.
These functionalised cytochalasins were then used as starting materials for further derivatisation. Reduction of 4′‐azidocytochalasin (1 h) by using triscarboxyethylphosphine (TCEP) led to the expected but unprecedented 4′‐aminocytochalasin (1 i) in good yield (Scheme 2). 4′‐Azidocytochalasin (1 h) was also a good substrate for azide–alkyne Huisgen cycloaddition [16] (click) [17] reactions with a series of acetylenes giving the expected triazole adducts 5 a–5 c in good yields (Scheme 2). Having shown the success of these reactions we also tested biotin‐propargyl esters 6 a, biotin‐homopropargyl ester 6 b and biotin‐(4‐pentynyl) ester 6 c and showed that the expected cytochalasin‐biotin adducts 7 a–7 c were formed as expected.
Scheme 2.
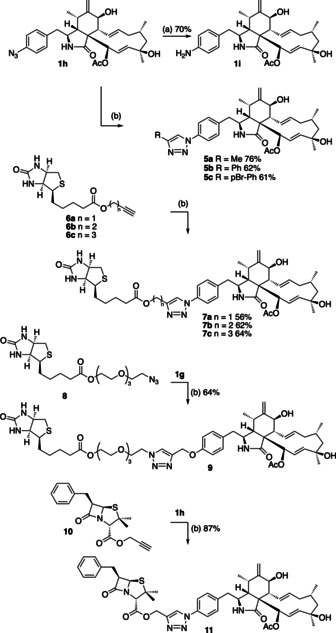
Synthesis of functionalised cytochalasins. Reagents and conditions: a) dioxane, H2O, tris(2‐carboxyethyl)phosphine (TCEP), RT, 1 h; b) N,N‐diisopropylethylamine (DIPEA), DMF, CuI (0.05 equiv), RT, 12 h.
Similar chemistry was achieved by coupling the biotin‐PEG‐azide (PEG=polyethylene glycol) 8 with 4′‐O‐propargylcytochalasin (1 g) to create 9 featuring a longer and more hydrophilic linker. We also experimented with the production of hybrid metabolites. For example, the propargyl ester of penicillin G 10 reacted smoothly with 1 h to give 11. All compounds were synthesised in 5–10 mg scale using standard procedures, purified and fully characterised.
Fluorescent tags were also easily added to the cytochalasin skeleton by using click chemistry (Scheme 3). For example, 4′‐azidocytochalasin (1 h) was reacted with the perylene‐linked acetylene 12 to give the expected adduct 13 in high yield. Likewise, the acetylene‐linked coumarin dye 14 also couples smoothly to give the expected 15. Texas Red‐16 and AF488‐18 based dyes were also linked using the same methodology to afford 17 and 19, respectively. Finally, the benzoxadiazole dye 20 [18] was added to 1 h to form the expected 21.
Scheme 3.
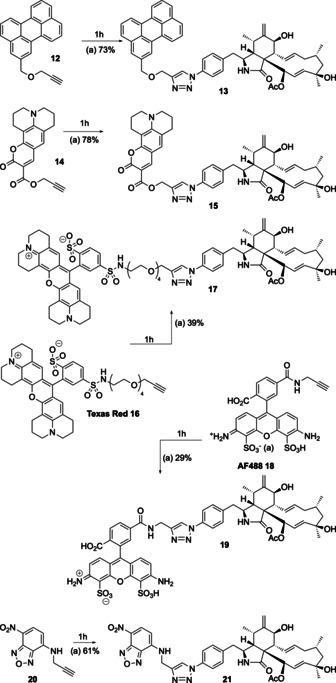
Synthesis of dye‐linked cytochalasins. Reagents and conditions: a) DIPEA, DMF, CuI (0.05 equiv), RT, 12 h.
The 4′‐bromo (1 d) and 4′‐iodo (1 e) cytochalasins were tested for their ability to act as substrates in Pd‐catalysed cross‐coupling reactions. In initial experiments, the brominated derivative 1 d did not react smoothly in attempted Sonogashira cross‐coupling reactions. However, the unprotected 4′‐iodo substrate 1 e reacted cleanly, for example to give the 4′ acetylene 22 (Scheme 4).
Scheme 4.

Sonogashira reaction of 4′‐iodocytochalasin (1 e). Reagents and conditions: CuI (0.05 equiv), [Pd(PPh3)4] (0.009 equiv).
We also attempted the synthesis of dimeric cytochalasins (Scheme 5). 4′‐Bromocytochalasin (1 d) reacted with bis(pinacolato)diboron in the presence of [Pd(dppf)Cl2] (dppf=bis(diphenylphosphino)ferrocene) to form the expected 4′‐borolated cytochalasin 23. Subsequent reaction with 1 d in a separate reaction catalysed by [Pd(PPh3)4], in the presence of the X‐phos ligand, [19] then yielded the unprecedented 4′‐4′′‐cytochalasin H symmetrical dimer 24. In a simpler reaction sequence, 1 g and 1 h were directly coupled via Huisgen chemistry to form the unsymmetrical dimer 25 with a simple hydroxymethyl triazole linker. Finally, a dimer featuring a longer and more polar linker was formed by dual Huisgen coupling between the bis‐propargyl‐PEG linker 26 and 1 h to give 27.
Scheme 5.
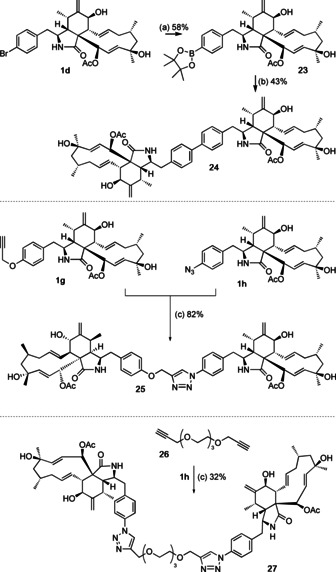
Synthesis of dimeric cytochalasins. Reagents and conditions: a) [Pd(dppf)Cl2] (0.1 equiv), CH3CO2K, dioxane, 110 °C, 8 h; b) 1 d, [Pd(PPh3)4] (0.1 equiv), X‐Phos (0.2 equiv), dioxane, H2O, 110 °C, 2 h; c) DIPEA, DMF, CuI (0.05 equiv), RT, 12 h.
The synthetic compounds were tested for their antifungal activities and for cytotoxicity (see the Supporting Information, section 3 for full results). Compounds 1 a–1 i, 5 a–5 c, 7 a–7 c, 9, 11, 13, 15, 17, 19, 21–25 and 27 were tested in a standard dilution assay (minimum inhibitory concentration, MIC) for their activity towards Schizosaccharomyces pombe, Pichia anomala, Mucor hiemalis, Candida albicans, and Rhodotorula glutinis. Most of the compounds did not show significant antifungal activity. However, moderate activity was observed for 1 a versus S. pombe, M. hiemalis and R. glutinis (30–60 μm). Notably, the 4′‐iodo (1 e) and 4′‐azido (1 h) compounds were 5–10 times more potent versus S. pombe and R. glutinis than 1 a, but the 4′‐amino analogue 1 i showed no observable activity versus any of the organisms. Likewise, compounds 5, 7, 9, 11 and 13 were also inactive. Coumarin‐linked 15 displayed among the best activity of any compound with an IC50 of approximately 5 μm versus the opportunistic pathogen C. albicans, but moderate or weak activity against the other organisms.
Standard cytotoxicity assays were performed versus mouse (L929) and human (KB3.1, PC3, A549, MCF‐7, A431 and SKOV‐3) cell lines (see the Supporting Information for details). Compounds 1 e–1 h showed similar cytotoxicity to 1 a itself (e.g. ca. 30–1000 nm). However, 4′‐aminocytochalasin (1 i) was significantly less cytotoxic (400–6000 nm). Larger substituents at the 4′‐position (e.g., 5 a–5 c) also showed attenuated cytotoxicity (1200–10 000 nm). Compounds with yet‐larger modifications, such as biotins (7 a–7 c), penicillin (11) and perylene (12) were also much less cytotoxic. Surprisingly, dimeric compounds 24 and 25 showed better than expected cytotoxicity (e.g., 120 nm for 25 vs. KB3.1 cells) whereas acetylene (22) and borate (23) also had relatively good potency (e.g., 300 nm for 23 vs. KB3.1 cells). However, Texas‐red dyed compound 17 showed the most potent cytotoxicity of all with IC50 values as low as 20 nm versus the KB3.1 cell line.
Selected compounds were investigated for their actin disruption properties in vivo. Here, osteosarcoma U2OS cells were fixed and stained with 4′,6‐diamidino‐2‐phenylindole (DAPI), [20] to highlight the nucleus, and phalloidin‐atto‐594, [21] which selectively stains filamentous actin. The well‐characterized and commercially available compound cytochalasin B [22] clearly causes the collapse of actin structures at 10 μg mL−1 versus DMSO control after one hour of treatment (Figure 1 A). Pyrichalasin H (1 a) shows the same activity at this concentration (Figure 1).
Figure 1.
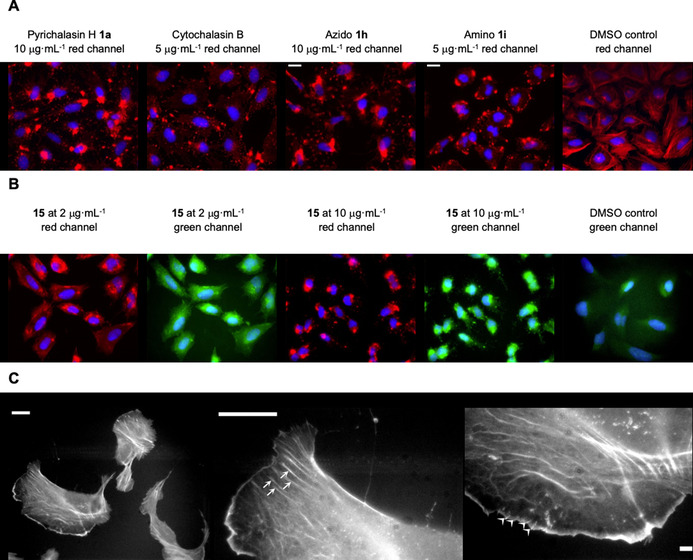
Cell treatment and staining assays with cytochalasin derivatives. A) Actin disruption assay with selected compounds (or DMSO as control), treated at concentrations as indicated and for 1 h, followed by cell fixation and staining of the actin cytoskeleton with fluorescently labelled phalloidin (red) and DAPI for cell nuclei (blue). B) Actin cytoskeleton disruption with the coumarin‐tagged cytochalasin derivative (compound 15) at different concentrations, as indicated. Panels show coumarin fluorescence (pseudo‐coloured in green) and phalloidin (red) as well as DAPI in blue. Note significant co‐localization of the coumarin derivative with phalloidin‐stained actin aggregates. C) Single‐staining experiment with 17 after fixation of non‐treated U2OS cells. The left panel shows an overview image of several cells, whereas the middle and right panels display close‐ups of parts of a selected cell in the left panel. Stress fibres (middle panel) and microspike bundles embedded into lamellipodia (right) are highlighted with arrows and arrowheads, respectively. All scale bars in A)–C) correspond to 20 μm.
Azidocytochalasin (1 h) and the 4′/4′′ dimer 24 (Supporting Information, Table 3.2) also show similar activity at 5 μg mL−1, but perylene 13 and biotin‐linked 7 a compounds show limited or no activity towards intracellular actin at this concentration, consistent with their previously determined lower cytotoxicities (Supporting Information, Table 3.2). However, the biotinylated compound 7 a does cause collapse of actin structures at higher (>10 μg mL−1) concentrations (Supporting Information, Table 3.2).
Compounds 13 and 15 feature fluorescent dyes, which allows the exploration of their interaction sites with the cytoskeleton while simultaneously exerting their inhibitory functions. For instance, the coumarin‐linked 15 induces collapse of the actin cytoskeleton at 10 μg mL−1, causing the formation of large, phalloidin‐stainable actin aggregates (Figure 1 B, phalloidin red channel). The response at lower concentrations (2 μg mL−1) was slightly less pronounced, but already reflected by the formation of unusual, rod‐shaped structures. Fluorescence in the coumarin‐channel reveals compound 15 to prominently co‐localize with actin aggregates induced with both concentrations (Figure 1 B). Moreover, the perylene‐linked compound 13 showed a much more diffuse localization within cells, and thus less prominent co‐localization with F‐actin (see Supplementary Information, Figures 3.2–3.3). We hypothesize that this relates to the increased hydrophobicity of the perylene unit itself, mediating pronounced association with membranes.
Due to both the specific association of some of these actin‐inhibitory compounds with actin aggregates during or following cell treatment and their co‐localization with phalloidin, as exemplified by compound 15 in Figure 1 B, we wondered whether such derivatives might also be employed as a simple staining tool of fixed, non‐treated cells. Indeed, this was observable, as exemplified, for instance, for the hydrophilic Texas red‐linked 17, which showed highly selective F‐actin binding, revealing key structures of the actin cytoskeleton at sub‐micrometer resolution.
To our surprise, staining with 17 revealed prominent subcompartments of the actin cytoskeleton such as microspikes, stress fibres and lamellipodia [23] (Figure 1 C), and the staining patterns within these structures were equally continuous to the patterns obtained with phalloidin (see the Supporting Information, Figures 3.2–3.3). These results suggest the continuous distribution of actin filament barbed ends throughout those structures composed of filaments with variable length, at least if assuming barbed end specificity of actin filament binding of this novel compound. This will be characterized in further detail in future studies.
Thus, mutasynthesis followed by chemical derivatisation of unprotected intermediates rapidly yields a range of new cytochalasins with interesting and useful properties. This significantly extends limited previous studies where precursor‐directed biosynthesis was used to produce halogenated chaetoglobosins in the 1.4–10.4 mg L−1 range. [24] In our experiments, modification of cytochalasins at the 4′‐position appears not to significantly impede actin binding in many cases and thus allows functional modifications such as fluorescence‐tagging. Compounds such as the biotin‐linked adducts 7 and 9 should therefore prove useful in combination with streptavidin‐linked molecular tools for target discovery. Fluorescently tagged compounds such as 17 should find significant utility in the study of actin structure and dynamics where its ability to selectively bind to barbed ends of actin differs to the ability of current tools such as phalloidin and jasplakinolide, which bind along the entire length of F‐actin structures. [25] Likewise, dimeric cytochalasins such as 24, 25 and 27 may induce formation of new types of actin structure in vivo. Future work will involve the detailed study of these new compounds and possibilities. We have also recently developed methods, which result in the engineering of the tailoring steps involved in cytochalasin biosynthesis [14] and methods for the directed reprogramming of the polyketide moiety of fungal polyketide–peptide hybrids. For example, we demonstrated the rational manipulation of polyketide chain length in related PKS‐NRPS systems. [26] The combination of these methods is expected to yield unprecedented diversity of available cytochalasins and related molecular tools.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Chinese Scholarship Council [C.W. (201608310143)] and the German Research Foundation (DFG) [grants numbers: CO 1328/ 2‐1, INST 187/ 621‐1, INST 187/ 686‐1)]. T.E.B.S. and K.R. acknowledge support through intramural funding from the Helmholtz Society. Open access funding enabled and organized by Projekt DEAL.
C. Wang, C. Lambert, M. Hauser, A. Deuschmann, C. Zeilinger, K. Rottner, T. E. B. Stradal, M. Stadler, E. J. Skellam, R. J. Cox, Chem. Eur. J. 2020, 26, 13578.
References
- 1. Skellam E. J., Nat. Prod. Rep. 2017, 34, 1252–1263. [DOI] [PubMed] [Google Scholar]
- 2.Cytochalasans are the wider family of chaetoglobosins (which are derived from tryptophan) and compounds derived from other amino acids (cytochalasins and zygosporins etc.). See references [1] and [8].
- 3. Nair U. B., Joel P. B., Wan Q., Lowey S., Rould M. A., Trybus K. M., J. Mol. Biol. 2008, 384, 848–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonder E., Mooseker M., J. Cell Biol. 1986, 102, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang X.-Q., Guan F.-F., Li D.-B., Wang C.-Y., Shao C.-L., Chem. Nat. Compd. 2017, 53, 109–113. [Google Scholar]
- 6. Yuyama K., Wendt L., Surup F., Kretz R., Chepkirui C., Wittstein K., Boonlarppradab C., Wongkanoun S., Luangsa-ard J., Stadler M., et al., Biomolecules 2018, 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kapoor K., Finer-Moore J., Pedersen B., Caboni L., Waight A., Hillig R., Bringmann P., Heisler I., Müller T., Siebeneicher H., Stroud R. M., Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scherlach K., Boettger D., Remme N., Hertweck C., Nat. Prod. Rep. 2010, 27, 869–886. [DOI] [PubMed] [Google Scholar]
- 9. Nukina M., Agric. Biol. Chem. 1987, 51, 2625–2628; [Google Scholar]; Nukina M., Namai T., Agric. Biol. Chem. 1991, 55, 1899–1900; [Google Scholar]; Tsurushima T., Don L. D., Kawashima K., Murakami J., Nakayashiki H., Tosa Y., Mayama S., Mol. Plant Pathol. 2005, 6, 605–613. [DOI] [PubMed] [Google Scholar]
- 10. Beno M. A., Christoph G. G., J. Chem. Soc. Chem. Commun. 1976, 344–345. [Google Scholar]
- 11. Wang C., Hantke V., Cox R. J., Skellam E. J., Org. Lett. 2019, 21, 4163–4167. [DOI] [PubMed] [Google Scholar]
- 12. Hantke V., Wang C., Skellam E. J., Cox R. J., RSC Adv. 2019, 9, 35797–35802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hantke V., Skellam E. J., Cox R. J., Chem. Commun. 2020, 56, 2925–2928. [DOI] [PubMed] [Google Scholar]
- 14. Wang C., Becker K., Pfütze S., Kuhnert E., Stadler M., Cox R. J., Skellam E. J., Org. Lett. 2019, 21, 8756–8760. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Weissman K., Trends Biotechnol. 2007, 25, 139–142; [DOI] [PubMed] [Google Scholar]
- 15b. Kirschning A., Hahn F., Angew. Chem. Int. Ed. 2012, 51, 4012–4022; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4086–4096. [Google Scholar]
- 16. Huisgen R., Proc. Chem. Soc. Lond. 1961, 357–369. [Google Scholar]
- 17. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075. [Google Scholar]
- 18. Zhuang Y., Chiang P., Wang C., Tan K., Angew. Chem. Int. Ed. 2013, 52, 8124–8128; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8282–8286. [Google Scholar]
- 19. Bruno N., Tudge M., Buchwald S., Chem. Sci. 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kapuscinski J., Biotech. Histochem. 1995, 70, 220–233. [DOI] [PubMed] [Google Scholar]
- 21. Panchuk-Voloshina N., Haugland R. P., Bishop-Stewart J., Bhalgat M. K., Millard P. J., Mao F., Leung W.-Y., Haugland R. P., J. Histochem. Cytochem. 1999, 47, 1179–1188. [DOI] [PubMed] [Google Scholar]
- 22. Aldridge D. C., Armstrong J. J., Speake R. N., Turner W. B., Chem. Commun. 1967, 26–27. [Google Scholar]
- 23. Small J., Stradal T., Vignal E., Rottner K., Trends Cell Biol. 2002, 12, 112–120. [DOI] [PubMed] [Google Scholar]
- 24. Ge H. M., Yan W., Guo Z. K., Luo Q., Feng R., Zang L. Y., Shen Y., Jiao R. H., Xu Q., Tan R. X., Chem. Commun. 2011, 47, 2321–2323. [DOI] [PubMed] [Google Scholar]
- 25. Pospich S., Merino F., Raunser S., Structure 2020, 28, 437–449. [DOI] [PubMed] [Google Scholar]
- 26. Yang X., Friedrich S., Yin S., Piech O., Williams K., Simpson T. J., Cox R. J., Chem. Sci. 2019, 10, 8478–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary