

Abstract
Aims
Chronic adventitial and medial infiltration of immune cells play an important role in the pathogenesis of abdominal aortic aneurysms (AAAs). Nicotinic acid (niacin) was shown to inhibit atherosclerosis by activating the anti-inflammatory G protein-coupled receptor GPR109A [also known as hydroxycarboxylic acid receptor 2 (HCA2)] expressed on immune cells, blunting immune activation and adventitial inflammatory cell infiltration. Here, we investigated the role of niacin and GPR109A in regulating AAA formation.
Methods and results
Mice were supplemented with niacin or nicotinamide, and AAA was induced by angiotensin II (AngII) infusion or calcium chloride (CaCl2) application. Niacin markedly reduced AAA formation in both AngII and CaCl2 models, diminishing adventitial immune cell infiltration, concomitant inflammatory responses, and matrix degradation. Unexpectedly, GPR109A gene deletion did not abrogate the protective effects of niacin against AAA formation, suggesting GPR109A-independent mechanisms. Interestingly, nicotinamide, which does not activate GPR109A, also inhibited AAA formation and phenocopied the effects of niacin. Mechanistically, both niacin and nicotinamide supplementation increased nicotinamide adenine dinucleotide (NAD+) levels and NAD+-dependent Sirt1 activity, which were reduced in AAA tissues. Furthermore, pharmacological inhibition of Sirt1 abrogated the protective effect of nicotinamide against AAA formation.
Conclusion
Niacin protects against AAA formation independent of GPR109A, most likely by serving as an NAD+ precursor. Supplementation of NAD+ using nicotinamide-related biomolecules may represent an effective and well-tolerated approach to preventing or treating AAA.
Keywords: Abdominal aortic aneurysm, Nicotinic acid, Nicotinamide, GPR109A, NAD+, Sirt1
1. Introduction
Abdominal aortic aneurysms (AAAs) are characterized by localized structural deterioration of the aortic wall, leading to progressive aortic dilation and rupture.1 Chronic adventitial and medial inflammation plays a key role in the pathogenesis of AAA through production of pro-inflammatory cytokines [i.e. monocyte chemoattractant protein 1 (MCP)-1, tumour necrosis factor (TNF)-α, and interleukin (IL)-6], reactive oxygen species, and matrix metalloproteinases (MMPs), which promote degradation of the vascular wall.2 Targeting immune pathways can prevent the formation of experimental AAA,3,4 suggesting a promising strategy for AAA treatment in humans. An ideal anti-inflammatory agent would not only be effective but safe, well-tolerated, and inexpensive for long-term use. In this regard, HMG Co-A reductase inhibitors (statins), which favourably modulate inflammation and oxidative stress, are safe and effective at reducing the complications of atherosclerosis.5 While statins may be of modest benefit in AAA, they have not been proven to diminish the need for intervention, thereby exposing patients to complex invasive vascular procedures.6
Nicotinic acid (niacin) was the first hypolipidaemic agent shown to reduce mortality in humans7 and remains in use as an alternative lipid-lowering agent in statin-intolerant patients, despite its poor tolerability. Niacin possesses numerous biological properties that suggest it may be highly efficacious in AAA, including elevation of high-density lipoprotein (HDL) cholesterol8 and adiponectin levels,9 inhibiting monocyte/macrophage inflammatory responses,10,11 and modulating oxidative stress by supplying reducing equivalents, up-regulating heme oxidase-1,12 and inhibiting nicotinamide adenine dinucleotide phosphate oxidase, which has been linked to the pathogenesis of AAA.13,14 Additionally, niacin is an important precursor of nicotinamide adenine dinucleotide (NAD+), a central metabolic cofactor that is required for the activity of multifunctional enzymes such as sirtuin-1,15 deficiency of which in vascular smooth muscle cells was reported to promote aortic aneurysm.16 However, to our knowledge, niacin has not been systematically studied in human or experimental models of AAA disease.
In a collar injury-induced atherosclerosis model, niacin was shown to inhibit adventitial vascular inflammatory cell infiltration and accumulation of myeloperoxidase (MPO),17 which we recently reported to contribute to the pathogenesis of AAA.18 The anti-atherosclerotic effects of niacin have been attributed to the activation of the G protein-coupled receptor GPR109A [also known as hydroxycarboxylic acid receptor 2 (HCA2)] expressed on immune cells, independent of its lipid-modifying effects.19 GPR109A is highly expressed on adipocytes and inflammatory cells,20 including neutrophils, which are critical to the pathogenesis of AAA.21 GPR109A activation promotes neutrophil apoptosis and inhibits MPO release, thereby suppressing oxidative stress.22 Thus, we hypothesized that GPR109A activation by niacin might also inhibit AAA formation by reducing vascular inflammation and matrix degradation.
Here, we investigated the role of niacin in preventing AAA using two different murine models: angiotensin II (AngII) infusion in low-density lipoprotein receptor (LDLR) knockout (KO−/−) mice and calcium chloride (CaCl2) application in wild-type (WT) mice. Niacin effectively inhibited AAA formation, but surprisingly, these effects were independent of GPR109A. Nicotinamide, which does not activate GPR109A, phenocopied the effects of niacin, which were mechanistically linked to restoration of aortic NAD+ levels and Sirt1 activity. These results suggest that elevating NAD+ levels and concomitant Sirt1 activity by nicotinamide-related biomolecules, which are safe and well-tolerated clinically, is a promising therapeutic strategy for AAA treatment and prevention.
2. Methods
2.1 Animals
Animal experimental protocols were approved by the Institutional Animal Care and Use Committee at the Medical College of Georgia at Augusta University and complied with National Institute of Health guidelines. LDLR KO mice were purchased from Jackson Laboratory (Bar Harbor, ME). GPR109A KO mice were obtained from Dr Stefan Offermanns (Max Planck Institute for Heart and Lung Research, Germany). GPR109A KO mice were identified by PCR-based genotyping, and PCR confirmed that GPR109A gene expression was undetectable in whole bone marrow cells isolated from GPR109A KO mice (Supplementary material online, Figure S1).20 Mice were anaesthetized by isoflurane vaporizer (0.5–1.0 L/min for oxygen flowmeter, 4–5% for induction, and 1–3% for maintenance, EZ Anesthesia Systems) and buprenorphine SR (1.2 mg/kg body weight) was administered subcutaneously prior to making the incision to relieve additional pain. Mice were euthanized with intraperitoneal pentobarbital 150 mg/kg, inhaled anaesthesia (isoflurane, 5%) followed by carbon dioxide (CO2) narcosis followed by cervical dislocation or bilateral thoracotomy, in accordance with AVMA Panel 2007 recommendations and institutional IACUC guidelines.
2.2 AngII-induced AAA model
AngII (Enzo Life Sciences, 1000 ng/kg/min) was infused into male LDLR KO mice via osmotic mini-pumps (ALZET Model 2004) as described.20 Mice were treated with oral niacin (0.3% wt/vol) or nicotinamide (0.1% or 0.4% wt/vol, purchased from Sigma-Aldrich)8,19,23,24 supplemented in drinking water for 2 days prior to mini-pump implantation and continued throughout the study. In some experiments, mice were co-treated with 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (EX-527, 20 mg/kg body weight) to inhibit Sirt1 activity.25 Blood pressure measurements were obtained using a validated tail-cuff method (Coda 6, Kent Scientific).18 Serum cholesterol and triglyceride were quantified using commercial assays (Wako Pure Chemical Industries). Two to four weeks after mini-pump implantation, mice were euthanized, aortic outer diameter was measured, and tissues were collected for analysis.
2.3 CaCl2-induced AAA model
Male C57Bl/6 mice received water with or without nicotinic acid (0.3% wt/vol) or nicotinamide (0.1% or 0.4% wt/vol) for 2 days prior to CaCl2 induction of AAA using 0.5 mol/L of CaCl2 (Sigma-Aldrich), as described previously.18 After 3 weeks, animals were anaesthetized, aortic outer diameter was measured, and tissues were collected for analysis.
2.4 Gelatin zymography
Zymography to detect matrix metalloproteinase (MMP) activity was performed as previously described.18 Briefly, protein lysate (600 µg) was placed in a non-reducing zymogram buffer and applied without boiling to a 10% zymogram gel (Bio Rad). Gels were incubated in 2% Triton X 100 at room temperature for 30 min, and then rinsed in distilled water for 5 min. Gels were incubated overnight at 37°C with gentle agitation and proteins were stained with Coomassie Brilliant Blue R-250 solution (Bio Rad) and de-stained with a solution containing 40% methanol, 10% acetic acid, and 50% water.
2.5 Immunohistochemistry
Paraffin-embedded cross sections (5 µm) were stained with haematoxylin and eosin (H&E), Verhoeff-van Gieson (VVG), or Alizarin red (Ricca Chemical Company), MPO (Abcam), Mac-3 (BD Pharmingen), MCP-1 (Novus Biologicals), and matrix metalloproteinase (MMP)-2/MMP-9 (Calbiochem) antibodies were used with the HistoMouse-SP kit (Invitrogen) or DAB Substrate kit (Vector Labs). The number of elastin breaks was counted in three sections per mouse to quantify elastin degradation.26 Vascular calcification was detected by Alizarin Red staining. To quantify the immunostaining data, total stained area in the media and adventitial region was analysed using Image-Pro Plus Software.27
2.6 Quantitative PCR
Total RNA was extracted from aortic tissues with QIAzol Lysis Reagent, and purified with RNeasy Lipid Tissue Mini Kit (Qiagen). Real-time quantification of mRNA levels of the genes of interest was performed using Brilliant II SYBR Green QPCR Master Mix (Agilent Technologies) per manufacturer’s instructions. Normalized Ct values were subjected to statistical analysis and fold difference was calculated by ΔΔCt method as described previously.28 Primers used in this study are listed in Supplementary material online, Table S1.
2.7 Measurement of respiratory burst in neutrophilic cells
Respiratory burst assay was performed as previously described.29 In brief, human promyelocytic NB4 cells were differentiated into neutrophilic cells by treating with all-trans retinoic acid ATRA (1 µM, Sigma-Aldrich) for 5 days. Cells (5 × 104/well in 96 well plates) were incubated in phenol-free medium containing L-012 for 10 min, and phorbol 12-myristate 13-acetate (PMA, 100 nM) was applied in the presence or absence of niacin (50 µM or 300 µM). Luminescence was quantified using a POLARstar luminometer (BMG Labtech).
2.8 Measurement of vascular smooth muscle cell (VSMC) viability
Primary aortic VSMCs were grown to confluence, and serum-starved for 24 h, followed by pretreatment with niacin (10 µM) for 6 h. Then, cells were exposed to hydrogen peroxide (H2O2, 100 μM) and cell viability was assessed by trypan blue exclusion method. In brief, cells were rinsed with PBS after H2O2 treatment, and a mixture of 0.4% trypan blue and fresh culture media (1:5 ratio) was added. Cell viability in three randomly selected areas was evaluated by counting the number of dead cells per total number of cells.
2.9 Enzyme-linked immunosorbent assay (ELISA)
Serum levels of MCP-1 were determined by Mouse CCL2/JE/MCP-1 DuoSet ELISA kit per manufacturer’s instructions (R&D Systems).
2.10 Measurement of tissue levels of NAD+ and Sirt1 activity
Aortic NAD+ levels were determined using NAD/NADH Quantitation Colorimetric Kit (BioVision), and Sirt1 activity was assayed using Sirt1 Activity Assay Kit (Abcam) per manufacturer’s instructions.
2.11 Western blotting
Protein extraction and western blotting were performed as described previously.20 Sirt1 (Cell Signaling Technology) and GAPDH (Ambion) antibodies were used.
2.12 Statistical analysis
All statistical analysis was performed using GraphPad Software (GraphPad Software, Inc., USA). Results are expressed as mean ± SEM unless otherwise noted. Differences between two groups were analysed by Student’s t-test. Multiple group datasets were evaluated for normality, and differences were analysed by one-way ANOVA followed by Bonferroni post hoc analysis. P-values <0.05 were considered to be significant.
3. Results
3.1 Niacin protected against AngII-induced AAA formation
To investigate whether niacin protects against AAA formation, LDLR KO mice were treated with niacin (0.3% in drinking water) during AngII infusion for 2 weeks. This dose equates to approximately 2.5 g/day in a 70 kg person given the 10-fold greater metabolism of niacin in mice vs. humans.8,19 Niacin supplementation decreased AAA incidence (Figure 1A), aortic diameter (Figure 1B) and plasma MPO levels (Figure 1C). Aortic MPO accumulation, macrophage infiltration, and MCP-1 expression were also reduced by niacin treatment (Figure 1D, Supplementary material online, Figure S2). Furthermore, niacin prevented elastin degradation (VVG staining in Figure 1D and E), in conjunction with reduced aortic MMP-2 and MMP-9 immunostaining (Figure 1D, Supplementary material online, Figure S2) and activity (zymography, Figure 1F). MMP-2/-9 and MCP-1 mRNA expression also showed a trend towards reduction, while TNFα was significantly decreased, in aortic tissues of niacin-treated mice (Figure 1G). In vitro, niacin inhibited the respiratory burst produced by neutrophilic NB4 cells (Supplementary material online, Figure S3) and enhanced VSMC viability following treatment with 100 µM H2O2 (Supplementary material online, Figure S4). In contrast, niacin did not alter systolic blood pressure in AngII-infused mice (Supplementary material online, Figure S5), nor did it significantly reduce plasma cholesterol or triglyceride levels during the 2-week course of this study (Supplementary material online, Figure S6).
Figure 1.

Niacin protected against AngII-induced AAA formation. AngII was infused via osmotic mini-pump in LDLR KO mice, which were treated with or without niacin in the drinking water. (A) AAA incidence (n = 12–13). (B) Aortic diameter (n = 5–9). (C) Plasma MPO levels (ELISA, n = 3). (D) Representative histology; H&E, MPO, Mac-3 (macrophages), VVG (elastin fragmentation), MMP-2/-9, and MCP-1. (E) Elastin break count (n = 4–6). (F) Representative zymogram (upper panel) and quantified data (bottom panel) for aortic MMP-2 and MMP-9 activities (n = 3–4). (G) mRNA expression of MMP-2 (n = 4–5), MMP-9 (n = 4–5), MCP-1 (n = 4–7), and TNFα (n = 6–7). Data were analysed using one-way ANOVA followed by Bonferroni post hoc analysis. AngII, angiotensin II; H&E, haematoxylin & eosin; L, lumen; Mac, macrophage; MCP-1, monocyte chemoattractant protein 1; MMP, matrix metalloproteinase; MPO, myeloperoxidase; NA, nicotinic acid; TNFα, tumour necrosis factor α; VVG, Verhoeff-van Gieson.
3.2 Niacin treatment reduced CaCl2-induced AAA formation
We next tested the effects of niacin using the CaCl2-induced AAA model, which exhibits similar pathological characteristics found in human AAA, including calcification, vascular smooth muscle cell apoptosis, marked adventitial inflammatory cell infiltration, and more severe elastin degradation as compared to the AngII-induced AAA model.30 Moreover, unlike the AngII model, the CaCl2 model employs normolipidaemic, non-atherosclerotic mice, which eliminates potentially confounding systemic influences of niacin therapy. Niacin efficiently prevented CaCl2-induced AAA formation (Figure 2A), in association with diminished elastin degradation (H&E and VVG staining), aortic macrophage infiltration and MMP-2/-9 expression (Figure 2B and Supplementary material online, Figure S2). Thus, niacin effectively prevented AAA formation in both animal models of AAA.
Figure 2.
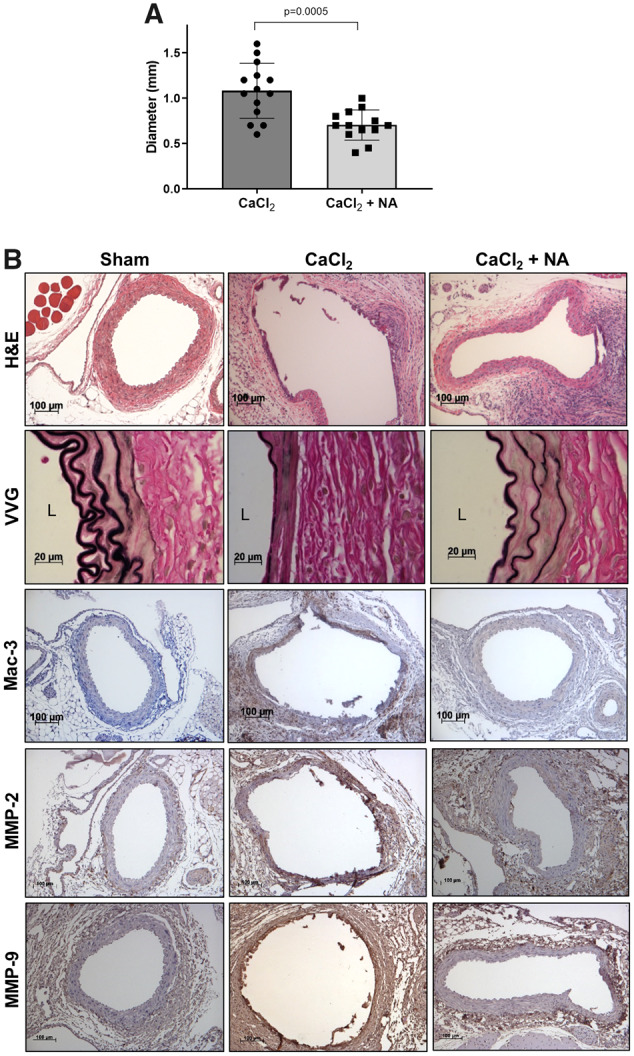
Niacin protected against CaCl2-induced AAA formation. CaCl2 was applied to the infrarenal aortae of C57Bl/6 mice. (A) AAA diameter (n = 13–14). (B) Representative histology; H&E, VVG (elastin fragmentation), Mac-3 (macrophages), and MMP-2/-9. Data were analysed using one-way ANOVA followed by Bonferroni post hoc analysis. CaCl2, calcium chloride; H&E, haematoxylin & eosin; L, lumen; Mac, macrophage; NA, nicotinic acid; VVG, Verhoeff-van Gieson.
3.3 GPR109A gene deletion did not abrogate the beneficial effects of niacin against AAA formation
GPR109A activation in immune cells was reported to mediate the anti-atherosclerotic effects of niacin.19 To investigate whether the protective effects of niacin against AAA formation are also mediated by GPR109A, GPR109A KO mice were treated with niacin, and AAA was induced by CaCl2 application. Unexpectedly, GPR109A gene deletion did not impact the efficacy of niacin to protect against AAA formation (Figure 3A). Similarly, GPR109A deficiency did not alter niacin’s ability to blunt aortic macrophage infiltration (Mac-3), MPO accumulation, or MCP-1 expression (Figure 3B, Supplementary material online, Figure S2). We also assayed MCP-1 levels in plasma of WT and GPR109A KO mice. Interestingly, there was a trend towards increased plasma MCP-1 levels in GPR109A KO mice as compared to WT mice in the absence of AAA induction (Sham, Figure 3C). AAA induction was associated with increased plasma MCP-1, and niacin treatment tended to reduce MCP-1 levels in both WT and GPR109A KO mice, although the data were not statistically significant. These results suggest that niacin exerts anti-inflammatory effects independent of GPR109A in the context of AAA.
Figure 3.

GPR109A gene deletion did not abrogate the protective effects of niacin against AAA formation in the CaCl2 model. (A) AAA diameter (n = 6–15). (B) Representative histology; H&E, Mac-3 (macrophages), VVG (elastin fragmentation), MPO, and MCP-1. (C) Plasma MCP-1 levels (ELISA, n = 4–6). Data were analysed using one-way ANOVA followed by Bonferroni post hoc analysis. H&E, haematoxylin & eosin; L, lumen; Mac, macrophage; MCP-1, monocyte chemoattractant protein 1; MPO, myeloperoxidase; NA, nicotinic acid.
3.4 Nicotinamide treatment protected against AngII-induced AAA formation
Since niacin’s protective effects against AAA formation were found to be independent of GPR109A, we next tested nicotinamide, which exhibits similar metabolic effects as niacin but does not activate GPR109A on inflammatory cells,31 and does not have lipid-lowering effects,15 in the AngII infusion model. Interestingly, nicotinamide treatment (0.4%) also significantly reduced AAA formation (Figure 4A) and phenocopied the effects of niacin with regard to suppression of macrophage infiltration, elastin degradation, MPO accumulation and MCP-1 expression (Figure 4B and C, Supplementary material online, Figure S2). MMP-2 and -9 activities were also significantly reduced by nicotinamide treatment (zymography, Figure 4D). Like niacin, nicotinamide did not alter systolic blood pressure in AngII-infused mice (Supplementary material online, Figure S5). As expected, plasma lipid levels were not affected by nicotinamide treatment (Supplementary material online, Figure S6).
Figure 4.

Nicotinamide reduced AngII-induced AAA formation. AngII was infused in LDLR KO mice via osmotic mini-pump, and mice were treated without or with nicotinamide in the drinking water. (A) AAA diameter (n = 3–10). (B) Representative histology; H&E, Mac-3 (macrophages), VVG (elastin fragmentation), MPO, and MCP-1. (C) Elastin break count (n = 6). (D) Representative zymogram (upper panel) and quantified data (bottom panel) for aortic MMP-2 and MMP-9 activities (n = 3). Data were analysed using one-way ANOVA followed by Bonferroni post hoc analysis. AngII, angiotensin II; H&E, haematoxylin & eosin; L, lumen; Mac, macrophage; MMP, matrix metalloproteinase; Nam, nicotinamide; VVG, Verhoeff-van Gieson.
3.5 Nicotinamide treatment reduced CaCl2-induced AAA formation
We next tested the effects of nicotinamide on AAA formation using the CaCl2 application model. Nicotinamide (0.1% and 0.4%) was highly effective at inhibiting AAA formation induced by CaCl2 application (Figure 5A), and these protective effects were accompanied by reduced MMP-2/-9 activities (Figure 5B), macrophage infiltration, and elastin degradation (Figure 5C, Supplementary material online, Figure S2). The CaCl2 application model is also associated with aortic calcification, which is frequently observed in human AAAs.30 Thus, we performed Alizarin red staining to quantify calcification,27 which was found to be significantly reduced by nicotinamide treatment (Figure 5D). Taken together, these results demonstrate that nicotinamide, like niacin, strongly protected against AAA formation in both animal models, further suggesting a GPR109A-independent mechanism of action.
Figure 5.

Nicotinamide inhibited CaCl2-induced AAA formation. (A) AAA diameter (n = 5–6). (B) Representative zymogram (upper panel) and quantified data (bottom panel) for aortic MMP-2 and MMP-9 activities (n = 3). (C) Representative histology; H&E, Mac-3 (macrophages), and VVG (elastin fragmentation). (D) Alizarin red (left panel, aortic calcification) and quantified data (right, n = 3). Data were analysed using one-way ANOVA followed by Bonferroni post hoc analysis. CaCl2, calcium chloride; H&E, haematoxylin & eosin; L, lumen; Mac, macrophage; Nam, nicotinamide; VVG, Verhoeff-van Gieson.
3.6 Niacin and nicotinamide restored aortic NAD+ levels and Sirt1 activity
Since both niacin and nicotinamide function as NAD+ precursors, we next examined whether NAD+ levels are altered during AAA formation in the absence or presence of treatment with niacin or nicotinamide. Intriguingly, NAD+ levels were markedly reduced in both AngII- and CaCl2-induced AAA tissues and similarly restored by either niacin or nicotinamide treatment (Figure 6A and B). Sirt1 is a key NAD+ consuming enzyme that regulates cellular metabolism,32 and deletion of Sirt1 in vascular smooth muscle cells augmented aortic aneurysm formation.16 Thus, we examined whether Sirt1 activity is regulated by niacin or nicotinamide in the context of AAA formation. Interestingly, we detected significantly reduced Sirt1 activity in aortic tissues in both experimental models that was similarly restored by either niacin or nicotinamide treatment (Figure 6C and D). Treatment with niacin or nicotinamide alone, without inducing AAA formation, did not significantly increase NAD+ levels or Sirt1 activity. In contrast, Sirt1 protein expression was not significantly altered in either the CaCl2 or the AngII model in the absence or presence of niacin or nicotinamide treatment (Figure 6E and F, Supplementary material online, Figure S7). These results suggest that niacin/nicotinamide restore Sirt1 activity by primarily by repleting NAD+ rather than by regulating Sirt1 protein expression.
Figure 6.

Aortic NAD+ levels and Sirt1 activity were reduced in AAA and restored by either niacin or nicotinamide supplementation. Aortic NAD+ levels in AngII-induced AAA model (A, n = 3) and CaCl2-induced AAA model (B, n = 3). Sirt1 activities in AngII-induced AAA model (C, n = 3) and CaCl2-induced AAA model (D, n = 3). Sirt1 protein expression levels in AngII-induced (E, n = 3) and CaCl2-induced (F, n = 6) AAA models. Data were analysed using one-way ANOVA followed by Bonferroni post hoc analysis. AngII, angiotensin II; CaCl2, calcium chloride; NA, nicotinic acid; NAD, nicotinamide adenine dinucleotide; Nam, nicotinamide; N.S., not significant; Sirt, sirtuin.
3.7 Inhibition of Sirt1 activity abolished the protective effects of NAD+ repletion on AAA formation
To investigate whether NAD+ repletion is functionally dependent on Sirt1 activity, AngII-infused mice were treated with EX-527, a selective Sirt1 inhibitor. We first confirmed that EX-527 did not significantly influence the incidence of AngII-induced AAA in the absence of treatment with nicotinamide (AngII vs. AngII + EX-527, Figure 7A). Also, when administered along with a lower dose of AngII (500 ng/kg/min), EX-527 did not significantly increase the incidence of AAA (not shown) or aortic diameter (Supplementary material online, Figure S8). However, when co-administered to mice treated with 1000 ng/kg/min AngII, EX-527 blocked the ability of nicotinamide to prevent AAA formation (Figure 7A and B), elastin degradation, MPO accumulation and macrophage infiltration (Figure 7D, Supplementary material online, Figure S2). As expected, EX-527 prevented the increase in aortic Sirt1 activity in mice treated with nicotinamide (Figure 7C). Furthermore, MCP-1 and TNFα gene expression was significantly reduced by nicotinamide that was abrogated by co-treatment with EX-527 (Figure 7E), suggesting that the protective effects of nicotinamide against AAA formation are mediated by NAD+ repletion/Sirt1 activation.
Figure 7.

Protective effects of nicotinamide against AAA formation were abrogated by Sirt1 inhibition. AngII was infused via osmotic mini-pump in LDLR KO mice treated with or without nicotinamide ± EX-527, a selective Sirt1 inhibitor. (A) AAA incidence (n = 11–19). (B) Aortic diameter (n = 6–19). (C) Aortic Sirt1 activity (n = 2–4). (D) Representative histology; VVG (elastin fragmentation), MPO, and Mac-3 (macrophages). (E) Pro-inflammatory gene (MCP-1, left panel and TNFα, right, n = 6–9) expression. Data were analysed using one-way ANOVA followed by Bonferroni post-hoc analysis. AngII, angiotensin II; L, lumen; Mac, macrophage; MCP-1, monocyte chemoattractant protein 1; MPO, myeloperoxidase; Nam, nicotinamide; TNFα, tumour necrosis factor α; VVG, Verhoeff-van Gieson.
4. Discussion
Niacin is a lipid-lowering agent that also exhibits anti-inflammatory and anti-oxidative effects in many cell types. Using two distinct animal models of AAA, we report for the first time that niacin can effectively protect against AAA formation, in conjunction with reduced aortic immune cell infiltration, inflammation, and matrix degradation. Unexpectedly, these inhibitory effects of niacin were found to be independent of GPR109A signalling and shared by nicotinamide, which like niacin restored aortic NAD+ levels and Sirt1 activity. These results suggest that nicotinamide and related biomolecules, which are safe, well-tolerated, and inexpensive, can potentially restore aortic NAD+ levels and concomitant Sirt1 activity to protect against AAA formation.
4.1 Novel GPR109A-independent role of niacin-mediated AAA protection
GPR109A is a G protein-coupled receptor highly expressed on inflammatory cells (neutrophils and macrophages) that can be activated by various agonists, including niacin, to elicit anti-inflammatory effects. In neutrophils, GPR109A activation promotes apoptosis and inhibits release of MPO.17 Deletion of GPR109A in bone marrow-derived cells prevented the ability of niacin to inhibit macrophage recruitment and atherosclerosis progression,19 suggesting a critical role for this receptor in transducing niacin’s vasculoprotective effects. We observed a trend toward increased expression of GPR109A mRNA in the aortas of AngII-infused mice (Supplementary material online, Figure S9), in conjunction with enhanced inflammatory cell infiltration (Figures 1D and 4B), which were reduced by niacin or nicotinamide treatment. However, deletion of GPR109A did not abrogate niacin’s ability to prevent AAA or diminish aortic inflammation. These disparate findings with niacin in atherosclerosis versus AAA models are likely consistent with the unique pathophysiological features of these two vascular inflammatory disorders. Interestingly, Zhang et al.33 reported that macrophage-specific Sirt1 deficiency enhanced MCP-1 expression and promoted AAA formation, suggesting that niacin-induced Sirt1 activation in macrophages could potentially contribute to the GPR109A-independent effects of niacin in preventing aneurysm formation. VSMC apoptosis, likely triggered in part by oxidative stress, is a major pathogenic feature of AAA.3 Here, we demonstrated that niacin protected against H2O2-induced cytotoxicity in VSMCs, which do not express GPR109A (Supplementary material online, Figure S4). Watson et al.16 also demonstrated that nicotinamide riboside, which likewise does not activate GPR109A, augmented VSMC survival, suggesting that the protective effects of niacin against AAA formation could be mediated in part by NAD+ repletion in VSMC. However, whether niacin acts specifically on VSMC, macrophages, and/or other cell types to prevent AAA formation remains to be definitively established.
4.2 Protective effects of NAD+ repletion on AAA formation
Our data indicate that nicotinamide, which does not activate GPR109A, also inhibited AAA formation and phenocopied the effects of niacin, further supporting the hypothesis that GPR109A-independent mechanisms contribute to niacin’s protective effects against AAA formation. These results led us to investigate NAD+ metabolism in AAA since both niacin and nicotinamide are dietary precursors of NAD+. NAD+ levels are depleted with aging,34 which is also an important risk factor for AAA formation. Although 16 mg of daily niacin is reportedly sufficient to meet the minimum requirement of NAD+ synthesis in humans,35 mounting evidence suggests that substantially greater NAD+ levels could be beneficial to protect against cardiovascular disease.15 NAD+ is a substrate of NAD+-consuming enzymes (i.e. sirtuins), and reduced NAD+ levels and sirtuin activity with age are implicated in the pathogenesis of a variety of cardiovascular and metabolic diseases.36 Moreover, expression of nicotinamide phosphoribosyltransferase (Nampt), an NAD+ producing enzyme, in vascular smooth muscle cells was shown to protect against thoracic aortic aneurysm by maintaining genome integrity and preventing senescence in VSMCs,16 further suggesting that NAD+ metabolism is important to maintain vascular homeostasis and prevent aneurysm disease. In both AngII-infusion and CaCl2 application models of AAA, we demonstrate reduced aortic NAD+ levels, which were efficiently restored by either niacin or nicotinamide treatment, suggesting that NAD+ depletion per se may contribute to the pathogenesis of AAA.
Dysfunctional perivascular adipose tissue (PVAT) regulates vascular pathobiology and may be involved in AAA formation. Visfatin, a PVAT-derived adipokine, was reported to promote VSMC proliferation by increasing nicotinamide nucleotide synthesis.37 Furthermore, high-fat diet feeding in mice reduced the expression of Nampt, an enzyme that biosynthesizes NAD+, in PVAT,38 suggesting a mechanistic linkage between NAD+ and PVAT-mediated vascular inflammation. In this study, we did not examine whether NAD+ supplementation by niacin or nicotinamide specifically regulates PVAT function to modulate aneurysm formation.
4.3 Sirt1 is a downstream regulator of NAD+-mediated AAA protection
Sirtuins are NAD+-dependent deacetylases that play various roles in cellular energy metabolism, DNA repair, inflammatory signalling, redox regulation, and regulation of transcription factors (i.e. p53, NFκB, FOXOs, PGC1α, and PARP1)25,39 Interestingly, Sirt1 expression and activity are significantly decreased in human AAA tissues, and age-associated Sirt1 reduction in VSMCs increases susceptibility to AAA in mice.40 Moreover, macrophage-specific Sirt1 expression was reported to contribute to AAA protection.33 Sirt1 reduction also promoted vascular cell senescence, p21 up-regulation, and inflammatory cell recruitment.33 In total, these studies suggest that Sirt1 reduction might be mechanistically linked to AAA pathogenesis. Consistent with this notion, we observed reduced Sirt1 activity, but not protein expression, during experimental AAA formation. Notably, nicotinamide restored Sirt1 activity and protected against AAA formation, which was abrogated by a Sirt1 inhibitor, suggesting that Sirt1 is a downstream mediator of nicotinamide’s effects. In this study, we employed a pharmacological Sirt1 inhibitor, EX-527, which potently and selectively inhibits Sirt1 activity (200–500-fold more potent against Sirt1 than Sirt2 or Sirt3) via a unique NAD+-dependent deacetylation mechanism.41 Mice in which Sirt1 has been genetically deleted have been developed, but inbred strains of these mice exhibited developmental defects and prenatal death,42 while outbred mice led to viable but smaller mice with metabolic abnormalities.43
Our data suggest that nicotinamide prevents AAA formation by preserving Sirt1 activity, which can be blocker by the Sirt1 inhibitor EX-527. However, EX-527, when administered without nicotinamide, did not significantly augment AAA formation induced by either 1000 or 500 ng/kg/min of AngII (Figure 7A, Supplementary material online, Figure S9). The reason for these findings is unclear. However, it is noteworthy that Sirt1 activity was markedly suppressed (i.e. by approximately two-thirds) in the aortas of AngII-infused and CaCl2-treated mice (Figure 6C and D). Under these conditions, it is reasonable to presume that an enzymatic inhibitor of Sirt1 might have little biological impact.
4.4 NAD+ precursors as novel AAA therapeutic agents?
Niacin, when added to statin therapy, failed to reduce atherosclerotic events in the AIM-HIGH and HPS2-THRIVE trials.44,45 These data cannot be extrapolated to AAA, which is a distinct disease process from atherosclerosis. Considering that NAD+ repletion, rather than GPR109A activation, appears to account for niacin’s beneficial effects against experimental AAA formation, our data suggest that nicotinamide or related biomolecules that elevate NAD+ levels yet lack the flushing side effects of niacin (which are specifically linked to GPR109A activation) merit further investigation. In this regard, several recent studies indicate that nicotinamide riboside is highly bioavailable, safe and effective at elevating NAD+ levels in adults.46,47
4.5 Study limitations
Although we used two different mouse models of AAA to investigate the role of niacin and nicotinamide in the pathogenesis of AAA, neither of these animal models perfectly mimics the pathology of AAA in humans. Moreover, while we demonstrated that niacin and nicotinamide can prevent AAA formation, we did not test their efficacy to limit the growth of existing AAA. Thus, while these findings should stimulate further research, they cannot be extrapolated to treatment of AAA in humans.
5. Conclusions
We provide novel evidence that niacin protects against AAA formation in two distinct animal models. Mechanistically, these effects are independent of GPR109A, shared by nicotinamide, and likely mediated through NAD+ repletion leading to Sirt1 activation. These results suggest that boosting NAD+ levels and Sirt1 activity by supplementation with nicotinamide or related biomolecules may represent an effective and well-tolerated approach to treating AAA.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Author's contributions
Conception and design of the study: A.L.B., N.L.W., and H.W.K. Performed experiments and collection of data: T.H., M.O., M.M., D.K., S.P., N.G., L.R., T.W.B., J.P., S.A., A.T., N.R., A.M., A.E., S.B., and H.W.K. Data analysis: T.H., A.L.B., M.O., S.P., N.G., T.W.B., A.T., N.R., A.M., A.E., S.B., N.L.W., and H.W.K. Interpretation of data and discussion: T.H., A.L.B., B.K.S., Y.H., D.J.F., G.A., N.S., S.O., N.L.W., and H.W.K. Manuscript preparation: A.L.B., N.L.W., and H.W.K.
Conflict of interest: none declared.
Funding
This work was supported by the National Institutes of Health [HL124097, HL126949, HL134354, and AR070029 to N.L.W.].
Supplementary Material
Time for primary review: 17 days
Translational perspective
Abdominal aortic aneurysms (AAAs) are associated with pronounced adventitial and medial inflammation leading to matrix degradation and progressive aortic expansion. We report that niacin blunts aortic inflammation and matrix degradation, thereby suppressing AAA formation. These effects are independent of the niacin receptor GPR109A and mimicked by nicotinamide, which does not induce flushing. These results suggest that nicotinamide and related biomolecules that replete cellular NAD+ may be an effective medical therapy for AAA.
References
- 1. Thompson RW, Lee JK, Curci JA. The pathobiology of abdominal aortic aneurysms In Gewertz BL. (ed) Surgery of the Aorta and Its Branches. Philadelphia, PA: W.B. Saunders, 2000. pp. 75–106. [Google Scholar]
- 2. Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol 2015;35:1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCormick ML, Gavrila D, Weintraub NL. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2007;27:461–469. [DOI] [PubMed] [Google Scholar]
- 4. Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res 2007;75:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mihos CG, Salas MJ, Santana O. The pleiotropic effects of the hydroxy-methyl-glutaryl-CoA reductase inhibitors in cardiovascular disease: a comprehensive review. Cardiol Rev 2010;18:298–304. [DOI] [PubMed] [Google Scholar]
- 6. Takagi H, Yamamoto H, Iwata K, Goto S, Umemoto T; ALICE (All-Literature Investigation of Cardiovascular Evidence) Group. Effects of statin therapy on abdominal aortic aneurysm growth: a meta-analysis and meta-regression of observational comparative studies. Eur J Vasc Endovasc Surg 2012;44:287–292. [DOI] [PubMed] [Google Scholar]
- 7. Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in Coronary Drug Project patients; long term benefit with niacin. J Am Coll Cardiol 1986;8:1245–1255. [DOI] [PubMed] [Google Scholar]
- 8. van der Hoorn JWA, de Haan W, Berbée JFP, Havekes LM, Jukema JW, Rensen PCN, Princen HMG. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol 2008;28:2016–2022. [DOI] [PubMed] [Google Scholar]
- 9. Wanders D, Graff EC, White BD, Judd RL. Niacin increases adiponectin and decreases adipose tissue inflammation in high fat diet-fed mice. PLoS One 2013;8:e71285.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JM, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I, Digby JE, Bannister T, Handa A, Wiesmann F, Durrington PN, Channon KM, Neubauer S, Choudhury RP. Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function: a randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol 2009;54:1787–1794. [DOI] [PubMed] [Google Scholar]
- 11. Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai J, Wamil M, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler Thromb Vasc Biol 2012;32:669–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu BJ, Chen K, Barter PJ, Rye KA. Niacin inhibits vascular inflammation via the induction of heme oxygenase-1. Circulation 2012;125:150–158. [DOI] [PubMed] [Google Scholar]
- 13. Ganji SH, Qin S, Zhang L, Kamanna VS, Kashyap ML. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis 2009;202:68–75. [DOI] [PubMed] [Google Scholar]
- 14. Thomas M, Gavrila D, McCormick ML, Miller FJ Jr, Daugherty A, Cassis LA, Dellsperger KC, Weintraub NL. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 2006;114:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 2008;28:115–130. [DOI] [PubMed] [Google Scholar]
- 16. Watson A, Nong Z, Yin H, O’Neil C, Fox S, Balint B, Guo L, Leo O, Chu MWA, Gros R, Pickering JG. Nicotinamide phosphoribosyltransferase in smooth muscle cells maintains genome integrity, resists aortic medial degeneration and is suppressed in human thoracic aortic aneurysm disease. Circ Res 2017;120:1889–1902. [DOI] [PubMed] [Google Scholar]
- 17. Wu BJ, Yan L, Charlton F, Witting P, Barter PJ, Rye KA. Evidence that niacin inhibits acute vascular inflammation and improves endothelial dysfunction independent of changes in plasma lipids. Arterioscler Thromb Vasc Biol 2010;30:968–975. [DOI] [PubMed] [Google Scholar]
- 18. Kim HW, Blomkalns AL, Ogbi M, Thomas M, Gavrila D, Neltner BS, Cassis LA, Thompson RW, Weiss RM, Lindower PD, Blanco VM, McCormick ML, Daugherty A, Fu X, Hazen SL, Stansfield BK, Huo Y, Fulton DJ, Chatterjee T, Weintraub NL. Role of myeloperoxidase in abdominal aortic aneurysm formation: mitigation by taurine. Am J Physiol Heart Circ Physiol 2017;313:H1168–H1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest 2011;121:1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 2003;9:352–355. [DOI] [PubMed] [Google Scholar]
- 21. Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR Jr. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 2005;112:232–240. [DOI] [PubMed] [Google Scholar]
- 22. Kostylina G, Simon D, Fey MF, Yousefi S, Simon HU. Neutrophil apoptosis mediated by nicotinic acid receptors (GPR109A). Cell Death Differ 2008;15:134–142. [DOI] [PubMed] [Google Scholar]
- 23. Stratford MR, Dennis MF. Pharmacokinetics and biochemistry studies on nicotinamide in the mouse. Cancer Chemother Pharmacol 1994;34:399–404. [DOI] [PubMed] [Google Scholar]
- 24. Petley A, Macklin B, Renwick AG, Wilkin TJ. The pharmacokinetics of nicotinamide in humans and rodents. Diabetes 1995;44:152–155. [DOI] [PubMed] [Google Scholar]
- 25. Zhang T, Kraus WL. SIRT1-dependent regulation of chromatin and transcription: linking NAD+ metabolism and signaling to the control of cellular functions. Biochim Biophys Acta Proteins Proteomics 2010;1804:1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daugherty Lab, Saha Cardiovascular Research Center, University of Kentucky. Quantification of elastin fragmentation in the ascending aorta [Online]. 2018. http://cvrc.med.uky.edu/sites/default/files/Elastin_Break_Counting_UK.pdf (14 March 2019, date last accessed).
- 27. Yamanouchi D, Morgan S, Stair C, Seedial S, Lengfeld J, Kent KC, Liu B. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vas Surg 2012;56:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3:1101.. [DOI] [PubMed] [Google Scholar]
- 29. Qian J, Chen F, Kovalenkov Y, Pandey D, Moseley MA, Foster MW, Black SM, Venema RC, Stepp DW, Fulton DJ. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Rad Biol Med 2012;52:1806–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Krishna S, Golledge J. The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis 2013;26:29–39. [DOI] [PubMed] [Google Scholar]
- 31. Olsson AG. Nicotinic acid and derivatives In Schettler G, Habenicht AJR (eds). Principles and Treatment of Lipoprotein Disorders. Handbook of Experimental Pharmacology. Springer, Berlin, Heidelberg, 1994. p109. [Google Scholar]
- 32. Cantó C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD+? Pharmacol Rev 2012;64:166–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z, Xu J, Liu Y, Wang T, Pei J, Cheng L, Hao D, Zhao X, Chen HZ, Liu DP. Mouse macrophage specific knockout of SIRT1 influences macrophage polarization and promotes angiotensin II-induced abdominal aortic aneurysm formation. J Genet Genomics 2018;45:25–32. [DOI] [PubMed] [Google Scholar]
- 34. Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015;350:1208–1213. [DOI] [PubMed] [Google Scholar]
- 35. Yates AA, Schlicker SA, Suitor CW. Dietary reference intakes: the new basis for recommendations for calcium and related nutrients, B vitamins, and choline. J Am Diet Assoc 1998;98:699–706. [DOI] [PubMed] [Google Scholar]
- 36. Kane AE, Sinclair DA. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ Res 2018;123:868–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovas Res 2009;81:370–380. [DOI] [PubMed] [Google Scholar]
- 38. Xia N, Weisenburger S, Koch E, Burkart M, Reifenberg G, Förstermann U, Li H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br J Pharmacol 2017;174:3443–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol 2014;24:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, Tang X, Fu WY, Lu J, Yan YF, Wang XM, Han L, Zhang ZQ, Zhang R, Zou MH, Liu DP. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res 2016;119:1076–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Nat Acad Sci U S A 2013;110:E2772–E2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng HL, Mostoslavsky R, Saito SI, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Nat Acad Sci U S A 2003;100:10794–10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, Xuan J, Evans M, Harper ME, McBurney MW. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One 2008;3:e1759.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011;365:2255–2267. [DOI] [PubMed] [Google Scholar]
- 45.HPS2-Thrive Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 2014;371:203–212. [DOI] [PubMed] [Google Scholar]
- 46. Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M, Seals DR. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat Commun 2018;9:1286.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 2016;7:12948.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.