

Abstract
Chemists have to a large extent gained their knowledge by doing experiments and thus gather data. By putting various data together and then analyzing them, chemists have fostered their understanding of chemistry. Since the 1960s, computer methods have been developed to perform this process from data to information to knowledge. Simultaneously, methods were developed for assisting chemists in solving their fundamental questions such as the prediction of chemical, physical, or biological properties, the design of organic syntheses, and the elucidation of the structure of molecules. This eventually led to a discipline of its own: chemoinformatics. Chemoinformatics has found important applications in the fields of drug discovery, analytical chemistry, organic chemistry, agrichemical research, food science, regulatory science, material science, and process control. From its inception, chemoinformatics has utilized methods from artificial intelligence, an approach that has recently gained more momentum.
Keywords: artificial neural networks, drug design, chemoinformatics, chemometrics, prediction of data
Artificial Intelligence (AI) methods learn from data and thus find an ideal application field in chemistry. AI can thereby build on 60 years of developments in chemoinformatics on computer methods for the representation and analysis of chemical information such as database management, property prediction, synthesis design, and structure analysis.
1. Introduction
Artificial Intelligence (AI) has entered many domains of society, and artificial intelligence methods are used for such diverse tasks as human speech recognition, successfully competing with experts in strategic games (like chess and GO), and autonomously operating cars. These methods essentially derive their power by learning from data and are sometimes called machine learning or even deep learning.
Chemistry has from the very beginning derived its knowledge from data. Chemists have run experiments to obtain data on chemical or physical properties, on chemical reactions, or on biological activities. These data were then used to make predictions by analogy or to derive models for the principles that underly the data. To foster an understanding of chemistry in students Rolf Huisgen has written a chapter “Mesomerie‐Lehre” for a textbook on laboratory experiments.1 It should, however, be recognized that the concepts of inductive and resonance effect contained in this chapter were not derived from any theory but were an attempt to order the observations and data on product distributions and reaction rates in electrophilic aromatic substitution.
By doing experiments, chemists have amassed a huge amount of data on chemical structures and their properties. In 1971, about one million substances were registered in the Chemical Abstracts Service STN database and our supervisor Rolf Huisgen gave us the impression that he knew whether a compound was known or not–and we could not find a case where he was wrong. While this may have been feasible with one million compounds it is definitely not possible any more now with 160 million organic and inorganic substances and 68 million protein and nucleic acid sequences in the CAS database.
This review will show how the methods of chemoinformatics have made accessible this huge amount of data and information and how these data can be converted into knowledge to increase our understanding of chemistry and to accelerate chemical innovation. It will further be shown where new methods from artificial intelligence are introduced into various fields of chemistry to further assist in understanding chemical data.
2. Learning in Chemistry
Fortunately, concomitant with this vast increase in chemical data, computer technology arrived and rapidly became more and more powerful. Thus, computers could be used to make mathematical operations solving equations such as those encountered in quantum mechanics (QM), the theory that underlies chemistry. This allowed the calculation of physical and chemical data by QM methods of increasing complexity. This is deductive learning, learning from a theory to produce data.
However, it was also realized that a computer operates on a bit level and thus can be used for logical operations. Furthermore, software can be developed that allow the processing of data and information. Thus, computers can be used for inductive learning (Figure 1): data can be put together to generate information and many pieces of information can be generalized to produce knowledge.
Figure 1.
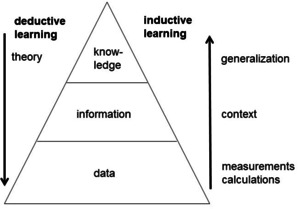
Deductive and inductive learning.
As an example, the measurement of the biological activity of a compound is only of much use when the structure of the compound is known; this is then information, putting the activity data in the context of the chemical structure. Several sets of structures and their corresponding biological activities can then be analyzed and generalized to produce an understanding, knowledge, of the relationships between structure and biological activity.
2.1. Chemoinformatics
Starting in the 1960s, computer methods were developed that allowed one to perform inductive learning in chemistry, a field that later became known as Chemoinformatics.2, 3 First, methods had to be developed for the computer representation of chemical structures and reactions. Then, procedures had to be utilized or developed for inductive learning, a field that was coined as chemometrics and encompassed methods from statistics and pattern recognition. Chemometrics methods were applied to the analysis of data from analytical chemistry.4 However, work was also initiated to tackle quite difficult tasks such as those embedded in the fundamental questions of a chemist:
What structure do I need for a desired property?
How can I synthesize this structure?
What is the outcome of my reaction?
For answering a question on property predictions quantitative structure property/activity relationships (QSPR and QSAR) were, and still are being, established.5, 6 The challenge of computer‐assisted synthesis design was taken up early on.7 The third question needs automatic procedures for structure elucidation.8, 9
These fundamental questions of a chemist have been the driving forces of a lot of work in chemoinformatic in the last few decades which will be reported in some of the following chapters.
2.2. Artificial Intelligence
It was realized from early on that the development of systems for property prediction, synthesis design, or structure elucidation are quite demanding tasks and would require a lot of conceptual work and state of the art computer technology. Therefore, emerging methods from computer science found their early applications in chemistry. This is true for methods that were subsumed under the name of artificial intelligence and publications with titles such as “Applications of Artificial Intelligence for Chemical Inference” appeared in the context of the DENDRAL project at Stanford University.10 The DENDRAL project developed methods for predicting the structure of a compound from its mass spectrum. In spite of the collaboration of some highly reputed chemists and computer scientists and a lot of work put into its development, the DENDRAL project was eventually discontinued. Many reasons might be found for that decision, not the least that the field of artificial intelligence had lost its promise and reputation in the late 1970s. In recent years, a renaissance of artificial intelligence in general and of its application in chemistry, in particular, can be observed. Several reasons have contributed to this development: availability of large amounts of data, increase in computer power, and new methods for processing these data. As these methods all are based on computer processing this field has also often been referred to “machine learning”. There is no clear distinction between these two terms although the term artificial intelligence seems to be the more comprehensive one.
3. Databases
In the beginning, various forms of computer‐readable chemical structure representations were explored as a basis for processing chemical structures and reactions and for building databases. Linear notations were favored because of their concise nature but they required the learning of a sizeable set of rules for encoding. With the rapid development of computer technology computer storage space became more easily and cheaper available. This allowed the coding of chemical structures in a manner that opened many desirable possibilities for structure processing and manipulations.
Eventually the representation of chemical structures by a connection table, i.e., by lists of atoms and lists of bonds became the rule. This allows the representation of structure information with atomic resolution and provides access to each bond in a molecule. One linear code, however, the SMILES notation11 is still in widespread use as it can easily be converted into a connection table and is optimum for sharing chemical structure information on the internet.
A molecular structure is essentially a mathematical graph. For an appropriate storing and retrieving of chemical structures many graph theoretical problems had to be solved such as unique and unambiguous numbering of the atoms of a molecule, ring perception, perception of tautomers, etc.12 A connection table representation of a molecule allowed the development of methods for full structure, substructure, and similarity searching.
A major step forward in the processing of chemical structures and the building of databases was the development of methods that allowed the communication of structure information with the computer in the language of the chemist, i.e., in the form of 2D drawings of structures. So‐called molecule editors and molecule viewers have been developed that permit the graphical input of chemical structures and reactions.12
With this arsenal of methods available, a variety of all‐important databases containing chemical information have been built.13 Here only the most outstanding databases will be mentioned. All new substances are registered in the Chemical Abstracts Service Registry System presently containing 160 million organic and inorganic structures and 68 million biosequences.14 Physical and chemical (e.g. reaction) data on chemical substances are stored in Reaxys, which combines three former databases: Beilstein DB, Gmelin DB, and Patent Chemistry DB.15 This database contains about 500 million experimental properties. A freely available database on chemical reactions has been obtained by text mining from United States patents (https://doi.org/106084/m9.figshare.5104873.v1). An important database for drug design and development is the Cambridge Structural Database (CSD) containing data on experimentally determined 3D structures of organic and organometallic compounds presently comprising slightly more than one million structures.16 Large as this number may sound, it is minute compared to the number of known compounds (less than 1 % !). However, in an early application of chemoinformatics it was shown how the known data can be used to generate a method for the prediction of the 3D structure of any organic molecule. The data in CSD (in 1990: 230,000 structures as compared to 22,000,000 molecules in CAS Registry at that time) were used to generate a procedure, CORINA, for the calculation of the 3D structure of any organic molecule (more than 99 % success rate).17
It is clear that these massive amounts of data that have been accumulated by researchers can only be managed by computer methods. Thus, access to databases is an essential prerequisite for any planning of laboratory work ‐ and any analysis of data. Without databases modern chemical research cannot be imagined anymore. It would have been worth the effort of developing computer methods in chemoinformatics if it had only resulted in databases. However, chemoinformatics has achieved ‐ and will achieve ‐ much more as will be demonstrated in the following chapters.
4. Prediction of Properties
Many data on chemical compounds are quite difficult or even impossible to calculate. This is particularly true for biological data. In this situation an indirect approach has to be taken to predict such data. In a two‐step approach, first molecular descriptors are calculated to represent a molecular structure.
Then, a series of known pairs of the descriptors of a molecule and the property of interest for the chemical compound are submitted to a mathematical procedure to develop a model that can then be used for the prediction of properties of additional molecules (Figure 2). This approach is known as Quantitative Structure Property/Activity Relationship (QSPR, QSAR).
Figure 2.
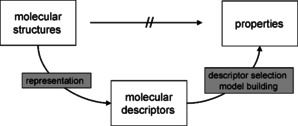
The QSPR/QSAR approach.
Many different methods for the calculation of structure descriptors have been developed.18 They are representing molecules with increasing detail: 1D, 2D, 3D descriptors, representations of molecular surface properties, and even taking account of molecular flexibility.
Also, quite a variety of mathematical methods for modeling the relationship between the molecular descriptors and the property of a compound are available. These are the inductive learning methods and are sometimes subsumed by names like data analysis methods, machine learning, or data mining.19 They comprise methods like a simple multi‐linear regression analysis, a variety of pattern recognition methods, random forests, support vector machines, and artificial neural networks. Artificial neural networks (ANN) try to model the information processing in the human brain and offer much potential for studying chemical data.20, 21
Figure 3 shows a simple artificial neural network consisting of six input units for providing the molecular descriptors, four neurons in the so‐called hidden layer and one neuron for the output of the result, in this case a property of the molecule.20
Figure 3.
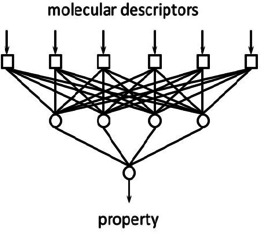
A two‐layer artificial neural network.
For establishing a relationship between the molecular descriptors and the property, values, so‐called weights, have to be attributed to the connections between the neurons. This is most often achieved by the so‐called backpropagation algorithm22 by repeatedly presenting pairs of molecular descriptors and their properties; these iterations quite often go into the ten‐thousands and more. An ANN has the advantage that the mathematical relationship between the input units and the output need not be specified or known; it is implicitly laid down in the weights and can also comprise non‐linear relationships.
It was tempting to envisage that with artificial neural networks the field of artificial intelligence was awakening again (cf. also title of ref.22). And in fact, in recent years terms like deep learning or deep neural networks have appeared in many a field including chemistry that provide a renaissance to the domain of artificial intelligence.
Deep neural networks (DNN) have some quite complex architectures with several hidden layers (Figure 4). This has the consequence that many weights for the connections have to be determined in order to avoid overfitting. Novel approaches and algorithms had to be developed to obtain networks that have true predictivity.23, 24 Deep neural networks need a large amount of data for training in order to obtain truly predictive models. Most applications of DNN have been made in drug design and in analysing reaction data (vide infra).
Figure 4.
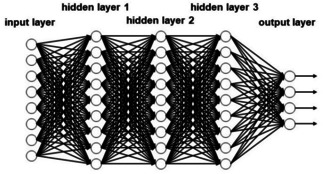
A deep neural network (observe that the network has been rotated by 90 degrees compared to the one in Figure 3).
5. Analytical Chemistry
The characterization of chemical objects is the domain of analytical chemistry. The objects can be compounds, samples from archeology, food samples, explosives, medical plants, urine samples, etc. These objects are investigated by a variety of methods such as chromatography, spectroscopy, etc. generating a host of data. Already in the 1960s, computer methods were developed, mainly from the domain of pattern recognition, to study such data.4 These developments established the field of chemometrics which has led to numerous studies and still retains in many cases its name25 although it should now be considered to be part of chemoinformatics. Already in the early days of chemometrics the term artificial intelligence was also used26 and in 1995 suggestions were made for the use of artificial intelligence in the clinical laboratory.27
Figure 5 shows the results of a study for finding out from which of nine areas a sample of an Italian olive oil came from. Each olive oil was characterized by its content of eight different fatty acids. 250 samples of the available 572 samples were used to train a self‐organizing neural network (SONN). From the additional 322 samples, 312 could correctly be predicted by the SONN.28 It should be emphasized that a SONN is an unsupervised learning method; no information from where the oil sample came from was used in the training. The SONN only projected the data from an eight‐dimensional space into two dimensions to generate the map of Figure 5. Closer inspection of this map shows that it reproduces the map of Italy, separating areas of northern Italy from southern Italy and even isolating the island of Sardegna. Thus, new information, the geography of Italy, was found because this is inherently contained in the data, emphasizing the benefits of unsupervised learning.
Figure 5.
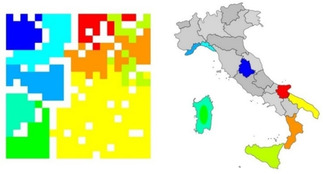
Classification of Italian olive oils.
6. Computer‐Assisted Structure Elucidation (CASE)
It has already been mentioned that the DENDRAL project stood as the first application of artificial intelligence in chemistry.10 Concomitant with the work at Stanford, two groups in Japan and the USA worked on the development of a general automatic system for the prediction of a chemical structure from spectroscopic data.8, 9 Work on this CHEMICS and the SESAMI system was continued for several decades, achieving remarkable results and progress. However, it is fair to say that these two systems have not found universal usage. On the other hand, work by Elyashberg29, 30 concentrating on the most recent developments in NMR spectroscopy has ended up with a system that is made commercially available by ACD/Labs and has made remarkable achievements in structure prediction.31 Applications of this system have led to the revision of the structures for highly complex natural products and the elucidation of structures deemed “undecipherable” by classical NMR methods.32, 33
Thus, CASE has matured to a point where it has become a valuable tool for the experimental chemist solving problems that would take him/her a very long time or even be unsolvable to him/her (is that artificial intelligence?) One reason for this is that a computer can exhaustively explore all possibilities whereas a chemist tends to find a (non‐optimum) solution as rapidly as possible.
7. Reaction Prediction and Computer‐Assisted Synthesis Design (CASD)
The prediction of the outcome of a chemical reaction is one of the fundamental questions of a chemist (see Section 2.1). Quantum mechanical methods can be used to calculate transition states and thus predict the course of a reaction. However, influences on reactions by solvents or temperature are hard if at all to calculate. In this situation recourse is being made to searching in reaction databases to find the reaction of interest or a similar reaction. With the largest reaction databases CASREACT34 and REAXYS35 containing 123 million reactions and 49 million reactions, respectively, there is always a fair possibility that the desired reaction or a closely related one is found. Reaction prediction is also an important task in computer‐assisted synthesis design (CASD) a fundamental question for an organic chemist (see Section 2.1). In a CASD system a target structure is given and a reaction that can produce this molecule has to be suggested.
CASD had been taken up as a challenge from the very beginning of chemoinformatics.7 Several research groups had embarked on the task of designing a CASD system: Ugi,36 Gelernter,37 Hendrickson,38 Gasteiger,39, 40 Funatsu.41 Although many interesting results were obtained none of the systems came into widespread use. Clearly, organic chemists were at that time not yet prepared to accept computers for a domain they liked to do by themselves.
Several decades had to pass until new attempts were made for the development of CASD systems.42 These were largely due to the availability of large reaction databases and the development of novel search procedures.
Grzybowski realized that his approach of automatically extracting reaction rules from a reaction database led in many cases to suboptimum syntheses. He and his research group therefore manually coded 20,000 reaction transforms and introduced new network search algorithm. The resulting Chematica program can successfully suggest new and interesting syntheses schemes.43
A deep learning approach based on a natural language processing architecture (Transformer) was taken to extract reaction rules from a database of reactions of the US‐Patent Office and integrate this into RXN for Chemistry a program that is made freely available by IBM Research in Zürich.44, 45
Segler and coworkers46 applied a combination of three different neural networks and a Monte Carlo search to extract reaction transforms from a database of 12.4 million reactions from Reaxys. The approach automatically generated successful multistep syntheses.
Thus, recent work based on the combination of large reaction databases with novel data processing algorithms has provided CASD systems that are mature enough to assist organic chemists in planning laboratory work.
An important question in organic synthesis is how easy or difficult it is to synthesize a compound. This is particularly important in drug discovery where de novo design methods generate many novel structures. It is then up to the medicinal chemist to select those molecules for further investigation that are more easily synthesizable. Methods for calculating values for concepts like synthetic accessibility47 or current complexity48 have been developed that allow the chemist to make selections which molecules should preferably be made.
8. Biochemical Pathways and Metabolic Engineering
Reaction databases of particularly high interest are those that gather the reaction and pathways occurring in living species. BioPath.Explore49, 50, 51 has stored all the reactions and pathways contained on the poster distributed by Roche52 in computer‐readable form as connection tables and with reaction sites marked. Thus, a variety of standard chemoinformatics searches can be performed on biochemical pathways.
Furthermore, interesting studies of high chemical significance can be performed.50 Of particular interest are those where chemoinformatics and bioinformatics methods are simultaneously used. Thus in a study combining information on annotated genomes in the PEDANT database53 with information extracted from the BioPath database and phenotypic traits from diseases interesting insights were obtained (Figure 6).54 As an example, the major pathways considered for periodontal disease were found.54
Figure 6.
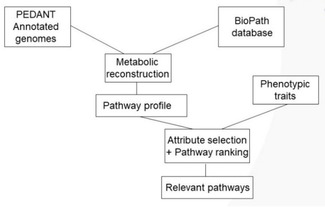
Combining information on genes with pathways and phenotypic traits.
In another approach, a chemical systems biology approach for Reverse Pathway Engineering (RPE) was established by combining chemoinformatics with bioinformatics methods. In an application for the study of flavor‐forming pathways in cheese by lactic acid bacteria, the known and some novel pathways could be derived.55
Much interest is centered on the redesign of pathways by redirecting the action of enzymes to produce basic organic chemicals. As an example, an optimized methanol assimilation pathway was developed by utilizing promiscuous formaldehyde‐condensing aldolases in E. coli.56 Novel artificial intelligence methods such as deep learning architectures are applied to metabolic pathway prediction.57
9. Drug Discovery
Drug discovery is presently the most prominent field for the application of chemoinformatics methods. All major drug companies have divisions of chemoinformatics–whatever their name–and drug companies are the largest employers of chemoinformatics specialists. Furthermore, all the drugs developed in the last few years have benefitted from the use of chemoinformatics methods in one way or other.
The field of computer‐based drug discovery is far too huge to be covered here. An overview is available.58 Quite many books have appeared that deal with computer‐assisted drug discovery and development; in fact, so many excellent books have been published that none will here be picked out for recommendation. The reader is advised to search the internet for a book that will meet his/her interest most.
The drug discovery and development process starts with the identification and validation of a protein target, then has to select a lead structure which has to be optimized and has to go through preclinical testing to make sure that properties like adsorption, solubility, distribution, excretion, and metabolism (ADME) as well as toxicity have acceptable values. Only then can a candidate be submitted to clinical development. The chemoinformatics methods used in drug discovery can be classified into ligand‐based methods (when the 3D structure of the protein target is not known) and structure‐based methods (when the protein structure is known) (Figure 7).
Figure 7.
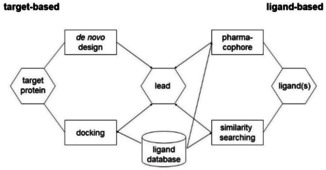
Some of the more important ligand‐based and target‐based methods in lead searching.
A more in‐depth modeling of the biological activity of a molecule needs its 3D structure, both for ligand‐based and for structure‐based methods; fortunately, CORINA17 can also generate 3D models for virtual (not yet synthesized) molecules. The methods include similarity search, pharmacophore searching, virtual screening, de novo design, docking and scoring, active site identification, xenobiotic metabolism prediction, ADME properties and toxicity prediction.
Databases play an important role in the drug design process, both databases of the compounds available in the company as well as those of virtually generated structures. Massive amounts of publicly available bioactivity data are collected by the National Institutes of Health in the PubChem database presently containing data on 103 million compounds and 259 million bioactivity values.59 With the drug design and development process being so demanding both bioinformatics and chemoinformatics methods should be used. Figure 8 is illustrating this idea and should emphasize that there is not a distinct separation between the bioinformatics and chemoinformatics methods. Often, the same mathematical procedures are used both in bioinformatics and in chemoinformatics. And, it should not be forgotten that, in the end, a gene is a chemical compound and the representation of a gene may eventually benefit from representing it by chemoinformatics methods.
Figure 8.
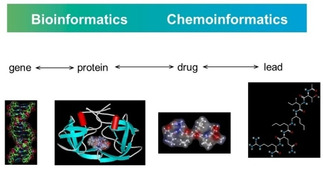
The role of bioinformatics and chemoinformatics in drug discovery.
In recent years various artificial intelligence methods have been introduced into drug development; a review has been published.60 The importance of artificial intelligence in drug discovery may be emphasized by the fact that 230 startup companies61 have been founded that use AI in drug discovery ‐ and the list is further growing. Major drug companies such as
Merck62 are also using AI. The AI company Exscientia and Sumitomo Dainippon Pharma Co. have jointly developed a drug candidate for obsessive‐ compulsive disorder that is now entering phase 1 clinical trials. The development took only one year as against an estimated 4.5 years with conventional research techniques; this amounts to massive savings.63
10. Agricultural Research
Over the last decades, the requirements for the compounds that are entered into the market as agrochemicals have continuously increased. Society expects these compounds to have low toxicity against humans and beneficial animals, low bioaccumulation, and quite specific activities. This has led to a situation where the development of a new agrochemical uses much the same methods as drug discovery.64, 65 Thus, both ligand‐based and structure‐based methods are used. Furthermore, adverse effects like toxicity have to be estimated without using animal testing. This has to be done not only for the compound of interest but also for its metabolites and for products of degradation in the environment. This has led to the introduction of cell‐based methods and the use of in silico methods. Commercial programs, so‐called expert systems, such as DEREK66 have been developed to assist chemists and toxicologists in estimating important properties of new compounds to be used as agrochemicals. Even companies like Bosch have entered the field of providing artificial intelligence techniques and services in agriculture.67 To summarize, chemoinformatics methods now play a major role in the development of a new agrochemical.
11. Food Science
The quality of food has become of increasing concern to society. The origin of food, the safety of food, the appearance of food, the flavor or taste, and the contents of food additives are some of the major topics investigated. Quite some chemoinformatics methods have been applied to investigate these topics. The field has grown to an extent that even the term “food informatics” has been coined.68, 69 First, a variety of databases have been built on properties of chemicals found or of interest in food. An important term is to classify a compound as GRAS (Generally Recognized As Safe). It was then investigated how the space of GRAS compounds overlaps with EAFUS (Everything Added to Food in the US) compounds and with the space of drugs as well as inhibitors of DNA methyltransferase 1 (DNAMT1), showing that these inhibitors are well separated from GRAS and EAFUS compounds and are therefore not to be expected to be expected in food.69, 70
The classification of Italian olive oils as an example for the important identification of the origin of a food sample has been mentioned in Chapter 5.28
Sensory properties of food such as taste (e.g. bitter or sweet), flavor or scent are of high interests and models have been built for the optimization of these properties. Furthermore, the properties of food additives (antioxidants, preservation compounds, or coloring agents) have to be closely monitored in order to make sure that no compounds with adverse effects are introduced into food. The available information has been analyzed with chemoinformatics methods to build predictive models for the properties of these food additives.
Machine Learning and AI methods are used in food industry along the entire manufacturing chain including new recipes.71
12. Cosmetics Products Discovery
In recent years, chemoinformatics and bioinformatics methods that have established their value in drug discovery such as molecular modeling, structure‐based design, molecular dynamics simulations and gene expression have also been utilized in the development of new cosmetics products.72 Thus, novel skin moisturizers and anti‐aging compounds have been developed.
Legislation has been passed in the European Union with the Cosmetics Directive73 that no chemicals are any longer allowed to be added to cosmetics products that have been tested on animals. This has given a large push to the establishment of computer models for the prediction of toxicity of chemicals to be potentially included in cosmetics products.74
13. Regulatory Science
In addressing the concern of society about the harmful effects of chemicals, the European Union has not only issued the Cosmetics Directive (see chapter 12,73) but has also passed the REACH legislation on the Regulation, Evaluation, Authorization and restriction of Chemicals.75 The REACH legislation requires companies that want to introduce large‐scale production chemicals into the market in Europe to provide a dossier that shows that these chemical are not harmful to humans or animals. Legislation similar to REACH has been introduced in other countries such as Canada, USA, Japan, China. These laws ask, among other things, for a lot of toxicity testing which is time‐consuming and costly. In this situation many approaches to the prediction of toxicity, bioaccumulation, and degradation in the environment have been and still are developed.76, 77 Expert systems for the prediction of toxicity have been around for quite a while but the availability of new high‐quality data has allowed the building of new models on toxicity prediction of much higher quality and predictivity. Not surprisingly, methods of machine learning and artificial intelligence have been applied to toxicity prediction.78, 79 Ref. 79presents a review of the use of AI in toxicity prediction. Interestingly, the authors showed that the predictive accuracy can be increased by augmenting the chemical structure descriptors with human transcriptome data.
14. Material Science
The prediction of the properties of materials is probably the most active area of chemoinformatics. The properties that are investigated range from properties of nanomaterials, materials from regenerative medicine, solar cells, homogeneous or heterogeneous catalysts, electrocatalysts, phase diagrams, ceramics, or the properties of supercritical solvents, and a few reviews have appeared.80, 81 In most cases, the chemical structure of the material investigated is not known and therefore other types of descriptors have to be chosen to represent a material for a QSAR study. Materials could be represented by physical properties such as refractive index or melting point, spectra, the components or the conditions for the production of the material, etc. Use of chemoinformatics methods in material science are particularly opportune as in most cases the properties of interest depend on many parameters and cannot be directly calculated. A QSAR model would allow the design of new materials with the desired property.
As the properties of materials are so hard to predict it is not surprising that in many recent studies artificial intelligence techniques have been applied in material science. For reviews see refs. 24 and 82.
15. Process Control
The problem of the detection of faults in chemical processes and process control have benefitted quite early on from the application of artificial neural networks.20, 21, 83 An overall course on the application of artificial intelligence in process control has been developed by six European universities.84 Chemical processes rapidly generate a host of data on flow of chemicals, concentration, temperature, pressure, product distribution, etc. These data have to be used to recognize potential faults in the process and rapidly bring the process back to optimum. The relationships between the various data produced by sensors and the amount of desired product cannot be explicitly given, making it an ideal case for the application of powerful data modeling techniques. Quite a few excellent results have been obtained for such processes as petrochemical or pharmaceutical processes, water treatment, agriculture, iron manufacture, exhaust gas denitration, distillation column operation, etc.85
16. Recent Publications
The field of artificial intelligence in chemistry is presently in very active development as underscored by editorials that have collected in the last few weeks publications on that topic. Ref 86, entitled “Computational Chemistry for Systems Chemistry”, offers a Special Collection of nine publications. The Editorial “Artificial intelligence in chemistry and drug design”87 is followed by eight pertinent articles. The Editorial “Artificial Intelligence in Chemistry”88 collects six papers on that topic.
Not surprisingly, the advent of the CORONA virus has led to a flurry of applications of artificial intelligence to data obtained from the COVID‐19 pandemia as a simple Google search will indicate. AI techniques have also been used to discover drugs that might be used against COVID‐19 targets.89, 90
17. Conclusions and Outlook
Learning from data has always been a cornerstone of chemical research. In the last sixty years computer methods have been introduced in chemistry to convert data into information and then derive knowledge from this information. This has led to the establishment of the field of chemoinformatics that has undergone impressive developments over the last 60 years and found applications in most areas of chemistry from drug design to material science. Artificial intelligence techniques have recently seen a rebirth in chemistry and will have to be optimized to also allow us to understand the basic foundations of chemical data. It is clear that computer methods will increase in playing a fundamental role in chemistry as emphasized by the Swedish Academy of Sciences on the occasion of awarding in 2013 the Nobel Prize in Chemistry to Martin Karplus, Michael Levitt and Arieh Warshel: “Today the computer is just as important a tool for chemists as the test tube.”91
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Johann Gasteiger has studied chemistry at the Ludwig‐Maximilians‐University of Munich and the University of Zürich. He obtained his PhD in 1971 at the University of Munich under the guidance of Prof. Dr. Rolf Huisgen with studies on the chemistry of cyclooctatetraene and homotropylium ions. He then went as a postdoc to Prof. Andrew Streitwieser Jr. at the University of California in Berkeley where he did ab initio calculations on carbanions. In 1972 he joined the research group of Prof. Ivar Ugi at the Technical University of Munich, working on a computer program for the planning of organic syntheses. In independent work he ventured into diverse areas of the development of computer programs for solving chemical problems leading to his Habilitation in 1978. In 1993 he moved to the University of Erlangen‐Nuremberg founding a Computer‐Chemie‐Centrum together with Prof. P. von R. Schleyer and PD Dr. Tim Clark. Prof. Gasteiger established the field of chemoinformatics in Germany. In his research he developed computer programs for the representation of chemical structures and reactions, the prediction of reactions, the design of syntheses, the simulation of spectra and a variety of methods for drug discovery. His work was recognized by several awards such as the Gmelin‐Beilstein medal of the GDCh, awards from the Division of Chemical Information of the American Chemical Society (ACS) and of the Division of Computers in Chemistry of the ACS, as wells as an award of the Chemical Structure Association.

Acknowledgements
Professor Rolf Huisgen advanced in me the urge for a deeper understanding of chemistry, particularly of chemical reactions. After ventures into experimental and theoretical chemistry I was fortunate to be able to develop the field of chemoinformatics together with inspiring colleagues. I also want to thank my able coworkers that embarked with me into the exciting field of chemoinformatics.
J. Gasteiger, ChemPhysChem 2020, 21, 2233.
Dedicated to the memory of Professor Rolf Huisgen who passed away on March 26, 2020, shortly before his 100th birthday.
References
- 1. Huisgen R. in Die Praxis des organischen Chemikers, L. Gattermann, H. Wieland, Th. Wieland, Walter de Gruyter & Co, Berlin, 1959, pp. 377–395. [Google Scholar]
- 2. Chemoinformatics–Basic Concepts and Methods, (Eds. T. Engel, J. Gasteiger), Wiley-VCH, Weinheim, 2018. [Google Scholar]
- 3. Applied Chemoinformatics–Achievements and Future Opportunities, (Eds. T. Engel, J. Gasteiger), Wiley-VCH, Weinheim, 2018. [Google Scholar]
- 4. Kowalski B. R., Bender C. F., J. Am. Chem. Soc. 1972, 94, 5632–5639. [Google Scholar]
- 5. Hansch C., Fujita T., J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar]
- 6.W. Sippl, D. Robaa, in ref. [3], pp. 9–52.
- 7. Corey E. J., Wipke W. T., Science 1969, 166, 178–192. [DOI] [PubMed] [Google Scholar]
- 8. Sasaki S. I., Abe H., Ouiki T., Sakamoto M., Ochiai S., Anal. Chem. 1968, 40, 2220–2223. [Google Scholar]
- 9. Munk M. E., Sodano C. S., McLean R. L., Haskell T. H., J. Am. Chem. Soc. 1967, 89, 4158–4165. [DOI] [PubMed] [Google Scholar]
- 10. Lederberg J., Sutherland G. L., Buchanan B. G., Feigenbaum E. A., Robertson A. V., Duffield A. M., Djerassi C., J. Am. Chem. Soc. 1969, 91, 2973–2976. [Google Scholar]
- 11. Weininger D., J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar]
- 12.T. Engel, in ref. [2], pp. 43–119.
- 13.E. Zass, T. Engel, in ref. [2], pp. 185–230.
- 14.https://www.cas.org/support/documentation/chemical-substances, last accessed June 6, 2020.
- 15.https://www.elsevier.com/solutions/reaxys, last accessed June 6, 2020.
- 16.https://www.ccdc.cam.ac.uk/solutions/csd-system/components/csd/, last accessed June 6, 2020.
- 17. Sadowski J., Gasteiger J., Chem. Rev. 1993, 93, 2567–2581. [Google Scholar]
- 18.L. Terfloth, J. Gasteiger in ref. [2], pp. 349–396.
- 19.K. Varmuza, in ref. [2], pp. 397–437.
- 20. Gasteiger J., Zupan J., Angew. Chem. 1993, 105, 510–536; [Google Scholar]; Angew. Chem. Int. Ed. 1993, 32, 503–527. [Google Scholar]
- 21. Zupan J., Gasteiger J., Neural Networks in Chemistry and Drug Design, Second Edition, Wiley-VCH, Weinheim, 1999. [Google Scholar]
- 22. Rumelhart D. E., Hinxton G. E., Williams R. J., in Parallel Distributed Processing: Explorations into the Microstructures of Cognition, Vol. 1 (Eds.: D. E. Rumelhart, J. L. McClelland), MIT Press, Cambridge, MA, USA, 1986, pp. 318–352. [Google Scholar]
- 23. Goodfellow I., Bengo Y., Courville A., Deep Learning, MIT Press, Cambridge, MA, USA: 2017. [Google Scholar]
- 24. Wei J., Chu X., Sum X.-Y., Xu K., Deng H.-X., Chen J., Wei Z., Lei M., Info Chim. Mag. 2019, 1, 338–358. [Google Scholar]
- 25.A. Rátz, D. Bajusz, K. Héberger, in ref. [3], pp. 471–499.
- 26. Hippe Z., Anal. Chim. Acta 1983, 150, 11–21. [Google Scholar]
- 27. Place J. F., Truchaud A., Ozawa K., Pardue H., Schnipelski P., J. Autom. Chem. 1995, 17, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zupan J., Novic M., Li X., Gasteiger J., Anal. Chim. Acta 1994, 292, 219–234. [Google Scholar]
- 29. Elyashberg M., Blinov K., Molodtsov S., Smurny Y., Williams A. J., Churanova T., J. Cheminf. 2009, 1, 3–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elyashberg M. E., Williams A. J., Computer-Based Structure Elucidation from Spectral Data, Springer, Berlin, 2015. [Google Scholar]
- 31.https://www.acdlabs.com/products/com_iden/elucidation/struc_eluc/, last accessed June 6, 2020.
- 32. Elyashberg M. E., Blinov K. A., Molodtsov S. G., Williams A. J., Magn. Reson. Chem. 2012, 50, 22–27. [DOI] [PubMed] [Google Scholar]
- 33.J. Aires de Sousa, in ref. [3], pp. 133–163.
- 34.https://www.cas.org/support/documentation/reactions, last accessed June 6, 2020.
- 35.https://www.reaxys.com/#/search/quick, last accessed June 6, 2020.
- 36. Blair J., Gasteiger J., Gillespie C., Gillespie P. D., Ugi I., Tetrahedron 1974, 30, 1845–1859. [Google Scholar]
- 37. Gelernter H. L., Sridharan N. S., Hart A. J., Yen S.-C., Top. Curr. Chem. 1973, 41, 113–150. [Google Scholar]
- 38. Hendrickson J. B., J. Am. Chem. Soc. 1971, 93, 6847–6854. [Google Scholar]
- 39. Gasteiger J., Jochum C., Top. Curr. Chem. 1978, 74, 93–126. [Google Scholar]
- 40. Ihlenfeldt W. D., Gasteiger J., Angew. Chem. 1995, 107, 2807–2829; [Google Scholar]; Angew. Chem. Int. Ed. 1995, 34, 2613–2633. [Google Scholar]
- 41. Funatsu K., Sasaki S., Tetrahedron Comput. Methodol. 1988, 127–137. [Google Scholar]
- 42. Osterath B., Nachr. Chem. 2019, 67, 35–37. [Google Scholar]
- 43. Szymkuc S., Gajewska E. P., Klucznik T., Molga K., Dittwald P., Startek M., Bajczyk M., Grzybowski B. A., Angew. Chem. 2016, 128, 6004–6040; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2016, 55, 5904–5937. [DOI] [PubMed] [Google Scholar]
- 44. Schwaller P., Gaudin T., Lányi D., Bekasa C., Laino T., Chem. Sci. 2018, 9, 6091–6098; [DOI] [PMC free article] [PubMed] [Google Scholar]; Schwaller P., Petraglia R., Zullo V., Nair V. H., Haeuselmann R. A., Pisoni R., Bekas C., Iuliano A., Laino T., Chem. Sci. 2020, 11, 3316–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.https://rxn.res.ibm.com/, last accessed June 7, 2020.
- 46. Segler M. H. S., Preuss M., Waller M. P., Nature 2018, 555, 604–610. [DOI] [PubMed] [Google Scholar]
- 47. Boda K., Seidel T., Gasteiger J., J. Comput.-Aided Mol. Des. 2007, 21, 311–325. [DOI] [PubMed] [Google Scholar]
- 48. Li J., Eastgate M. D., Org. Biomol. Chem. 2015, 13, 7164–7176. [DOI] [PubMed] [Google Scholar]
- 49. Reitz M., Sacher O., Tarkhov A., Trümbach D., Gasteiger J., Org. Biomol. Chem. 2004, 2, 3226–3237. [DOI] [PubMed] [Google Scholar]
- 50.O. Sacher, J. Gasteiger, in ref. [3], pp. 106–131.
- 51.https://v12.chemtunes.com/biopath3/, last accessed June 8, 2020.
- 52.http://biochemical-pathways.com/#/map/1, last accessed June 8, 2020.
- 53.https://www.biomax.com/product/pedant-pro-sequence-analysis-suite/, last accessed June 8, 2020.
- 54. Kastenmüller G., Schenk M. E., Gasteiger J., Mewes H.-W., Genome Biol. 2009, 10, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu M., Bienfait B., Sacher O., Gasteiger J., Siezen R. J., Nauta A., Geurts J. M. W., PLoS One 2014, 9, e84769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. He H., Höper R., Dotzenhöft M., Marliere P., Bar-Even A., Metab. Eng. 2020, 60, 1–13. [DOI] [PubMed] [Google Scholar]
- 57. Baranwal M., Magner A., Elvati P., Saldinger J., Violi A., Hero A. O., Bioinf. India 2019, 36, 2547–2553. [DOI] [PubMed] [Google Scholar]
- 58.L. Terfloth, S. Spycher, J. Gasteiger, in ref. [3], pp. 165–194; see also the Sections 6.2–6.13 in ref. [3], pp. 195–416 that follow in ref. [3].
- 59.https://pubchem.ncbi.nlm.nih.gov/, last accessed June 8, 2020.
- 60. Mak K.-K., Pichika M. R., Drug Discovery Today 2019, 24, 773–780. [DOI] [PubMed] [Google Scholar]
- 61. https://blog.benchsci.com/startups-using-artificial-intelligence-in-drug-discovery, last accessed, June 8, 2020.
- 62.https://www.merckgroup.com/en/research/science-space/envisioning-tomorrow/precision-medicine/generativeai.html, last accessed June 8, 2020.
- 63.https://www.exscientia.ai/news-insights/sumitomo-dainippon-pharma-and-exscientia-joint-development, last accessed June 8, 2020.
- 64. Schleifer K. J., in Modern Methods in Crop Protection Research (Eds. P. Jeschke, W. Krämer, U. Schirmer, W. Witschel), Wiley-VCH, Weinheim, 2012, pp. 21–41. [Google Scholar]
- 65.K. J. Schleifer, in ref. [3], pp. 417–438.
- 66.https://www.lhasalimited.org/products/derek-nexus.htm, last accessed June 9, 2020.
- 67.https://www.bosch.com/stories/greenhouse-guardian-ai-in-agriculture/, last accessed June 9, 2020.
- 68. Martinez-Mayorga K., Medina-Franco J. L., Food Informatics: Application of Chemical Information to Food Chemistry, Springer, New York, 2015, 251 pp. [Google Scholar]
- 69.A. Pena-Castillo, O. Méndez-Lucio, J. R. Owen, K. Martinez-Mayorga, J. L. Medina-Franco, in ref. [3], pp. 501–525.
- 70. Owen J. R., Nabey I. T., Medina-Franco J. L., López-Vallejo F., J. Chem. Inf. Model. 2011, 51, 1552–1563. [DOI] [PubMed] [Google Scholar]
- 71.https://spd.group/machine-learning/machine-learning-and-ai-in-food-industry/, last accessed June 10, 2020.
- 72.S. Anzali, F. Pflücker, L. Heider, A. Jonczyk, in ref. [3], pp. 527–546.
- 73.https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32009R1223, last accessed June 10, 2020.
- 74. Anzali S., Berthold M. R., Fioravanzo E., Neagu D., Péry A. R. R., Worth A. P., Yang C., Cronin M. T. D., Richarz A. N., IFSCC Magazine 2012, 15, 249–55. [Google Scholar]
- 75.https://ec.europa.eu/environment/chemicals/reach/reach_en.htm, last accessed June 10, 2020.
- 76.C. Yang, J. F. Rathman, A. Tarkhov, O. Sacher, T. Kleinöder, J. Liu, T. Magdziarz, A. Mostraq, J. Marusczyk, D. Mehta, C. Schwab, B. Bienfait, in ref. [3], pp. 439–470.
- 77. Yang C., Hasselgren C. H., Boyer S., Arvidson K., Aveston S., Dierkes P., Benigni R., Benz R. D., Contrera J., Kruhlak N. L., Matthews E. J., Han X., Jaworska J., Kemper R. A., Rathman J. F., Richard A. M., Toxicol. Mech. Methods 2008, 18, 277–295. [DOI] [PubMed] [Google Scholar]
- 78. Luechtefeld T., Marsh D., Rowlands C., Hartung T., Toxicol. Sci. 2018, 165, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu T., Wang G., Int. J. Mol. Sci. 2018, 19, 2358–2378. [Google Scholar]
- 80. Le T., Epa V. C., Burden F. R., Winkler D. A., Chem. Rev. 2012, 112, 2889–2919. [DOI] [PubMed] [Google Scholar]
- 81.T. C. Le, D. A. Winkler, in ref. [3], 547–569..
- 82. Sha W., Guo Y., Yuan Q., Tang S., Zhang X., Lu S., Guo X., Cao Y.-C., Cheng S., Adv. Intern. Med. 2020, 2, 1900143. [Google Scholar]
- 83. Hoskins J. S., Himmelblau D. M., Comput. Chem. Eng. 1988, 12, 881–890. [Google Scholar]
- 84. Application of Artificial Intelligence in Process Control (Eds. L. Boullart, R. Krijgsman, R. A. Vingerhoeds), Pergamon Press, Oxford, 1992. [Google Scholar]
- 85.K. Funatsu, in ref. [3], pp.571–584.
- 86.https://chemistry-europe.onlinelibrary.wiley.com/doi/toc/10.1002/(ISSN)2570-4206.Computational-Chemistry-for-Systems-Chemistry, last accessed June 13, 2020.
- 87. Brown N., Ertl P., Lewis R., Luksch T., Reker D., Schneider N., J. Comp.-Aided Mol. Design 2020, 34, 709–715. [DOI] [PubMed] [Google Scholar]
- 88. Cancilla J. C., Torrecilla J. S., Proestos C. V., Valderrama J. O., Frontier 2020, 8, 275–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Amin S. A., Ghosh K., Gayen S., Jha T., J. Biomol. Struct. Dyn. 2020, https://DOI: 10.1080/07391102.2020.1780946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Battisti V., Wieder O., Garon A., Seidel T., Urban E., Langer T., Mol. Inf. 2020, https:// DOI: 10.1002/minf.202000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.https://www.nobelprize.org/uploads/2018/06/press-22.pdf, last accessed June 12, 2020.