

Abstract
While microbial communities in the human body (microbiota) are now commonly associated with health and disease in industrialised populations, we know very little about how these communities co-evolved and changed with humans throughout history and deep prehistory. We can now examine these communities by sequencing ancient DNA preserved within calcified dental plaque (calculus), providing insights into the origins of disease and their links to human history. Here, we examine ancient DNA preserved within dental calculus samples and their associations with two major cultural periods in Japan: the Jomon period hunter–gatherers approximately 3000 years before present (BP) and the Edo period agriculturalists 400–150 BP. We investigate how human oral microbiomes have changed in Japan through time and explore the presence of microorganisms associated with oral diseases (e.g. periodontal disease, dental caries) in ancient Japanese populations. Finally, we explore oral microbial strain diversity and its potential links to ancient demography in ancient Japan by performing phylogenomic analysis of a widely conserved oral species—Anaerolineaceae oral taxon 439. This research represents, to our knowledge, the first study of ancient oral microbiomes from Japan and demonstrates that the analysis of ancient dental calculus can provide key information about the origin of non-infectious disease and its deep roots with human demography.
This article is part of the theme issue ‘Insights into health and disease from ancient biomolecules’.
Keywords: Japan, microbiome, ancient DNA, phylogenomics, palaeomicrobiology
1. Introduction
Microbiota within the human body possess functions that can influence the development, physiology, behaviour and the health of their hosts [1–8]. Therefore, altering these functions can lead to disease, compromising the health of the host [9,10]. Microbiota alterations result from a range of factors, including the use of antibiotics [11,12], changes in diet [13], infection by pathogens [14,15] and the adoption of lifestyles associated with industrialization [16]. Evidence suggests that specific microbes within the microbiota can be vertically inherited [17–20] and have been co-speciating with humans throughout hominid evolution [21,22]. Consequently, different human populations can have distinct microbiota as a result of their unique evolutionary and demographic histories [16,23–25].
Understanding the factors that drive microbial variation and examining how these microbial communities have changed and adapted over time with humans can provide key insights into human health. However, little is known about how the human microbiome has adapted and evolved in human history, especially during cultural admixture (e.g. when Europeans first met the peoples of the Americas). Such cultural admixtures could disrupt long-term relationships between microbiomes and host, and potentially contribute to microbiome disturbances that could influence host health [26]. Additionally, microbial lineage replacement owing to cultural/population admixture could also shape the microbiome in distinct ways; for example, ‘signatures’ of past human interaction and population replacement (e.g. loss of particular species or strains) may still be present in living populations today [27].
Recently, ancient human calcified dental plaque (calculus) was identified as a robust source of ancient human-associated microbial DNA [28–31] and now allows researchers to examine human-associated microbial communities from the past. Dental calculus is the result of a microbial biofilm that grows on teeth and undergoes periodic mineralization events that lock oral microorganisms in place within a robust calcium phosphate matrix [32]. The direct association of dental calculus on human teeth, coupled with its robust nature, provides an unprecedented opportunity to examine the bioarchaeological record of past human oral microbiomes, allowing researchers to identify factors that have altered the oral microbiome through time [28–30]. For example, dental calculus research has correlated shifts in the European microbiota community composition to large-scale dietary and lifestyle changes (e.g. from hunting–gathering to an agricultural lifestyle in Europe) [28]. Dental calculus is, therefore, a tool that can be used to sample the oral microbiome of past human populations and explore how the microbiome adapts and evolves following major cultural and demographic shifts.
Japan is an ideal location to examine human-associated microbiota change and evolution, as Japan has experienced large shifts in diet, culture, and demography over time and is geographically isolated from mainland Asia. The Japanese Archipelago was largely inhabited by the Jomon culture from approximately 16 000 to 3000 years before present (BP) [33,34]. Agriculture-bearing migrants from continental Asia came to the Japanese Archipelago and admixed with the Jomon during the early Yayoi period around 3000 BP [35–37]. Both modern and ancient DNA studies suggest that the admixture was weighted towards migrants, with modern estimates of Jomon contribution to mainland Japanese populations being less than 20% [37,38]. Before this admixture, a mitochondrial divergence estimate suggests that over 22 000 years of separation existed between the Jomon and continental Asian populations [39], which, coupled with their putatively disparate lifestyles (e.g. hunter–gatherer versus agriculturalist), may have resulted in divergent coevolution of their microbiomes. Here, we examine bacterial DNA preserved within ancient dental calculus from the Jomon (approx. 3000 BP) and Edo periods (400–150 BP) in Japan to investigate how and why microbial communities changed in the past.
2. Methods
(a). Ancient dental calculus samples
Ancient dental calculus samples (5 = Jomon, approx. 2400–3000 BP; 10 = Edo, 400–150 BP) (figure 1) were collected from the Natural Museum of Nature and Science in Tsukuba, Ibaraki, Japan. Of the Jomon samples, one was from the Ebishima shell mound in Iwate prefecture [40]. One was from the Ikenohata Shichikencho site in Ikawazu, Aichi prefecture, with radiocarbon dates of associated skeletal remains being 2440–3070 cal BP. [41]. Three were from a site in Miyano, Iwate prefecture [42]. The Edo period samples originated from the Ikenohata-Shichikencho site [43], which is located in Taito-ku, Tokyo. The excavation of this site was undertaken between 1993 and 1995 and yielded about six hundred graves which belong to the period from the late seventeenth to the nineteenth centuries [43]. The graves represented samurai and townsmen, known from the fact that the burials contained ceramic coffins (kamekan) and wooden coffins (mokkan) that were used for samurai and commoners, respectively.
Figure 1.

Map of Japan illustrating sites where ancient dental calculus samples were collected. EB, Ebishima; IK, Ikawazu; IS, Ikenohata Shichikencho, MI , Miyano. Yellow arrow in top right box denotes black teeth painting (Ohaguro). Yellow arrow in bottom right box denotes dental calculus.
Dental calculus was removed from specimens as previously described [44]. Briefly, a sterile dental pick was used to carefully remove dental calculus from one side of one tooth, and the specimen was placed in a sterile plastic bag for transport at room temperature to the Australian Centre for Ancient DNA at the University of Adelaide. Accompanying metadata was also collected at this time (electronic supplementary material, table S1).
(b). DNA extraction and library preparation
As authentic ancient DNA can be contaminated by modern DNA, steps to minimize and monitor the introduction of contaminant DNA were used [45]. All sample processing and molecular biology procedures prior to polymerase chain reaction (PCR) amplification were carried out at the Australian Centre for Ancient DNA facility at the University of Adelaide. These experiments were performed within a specialized ancient DNA laboratory, which maintains positive air pressure, HEPA filtered air, daily ultraviolet (UV)-treatment, regular 3% bleach cleanings, and work in isolated still-air hoods located in isolated rooms to limit the introduction of modern contaminant DNA. All technicians entered the facility using a dedicated entry room and wore full-body clean suits, gloves, and facemasks. Dental calculus samples were decontaminated to minimize environmental contamination by UV-irradiation for 15 min on each side, following by soaking in 2 ml of 5% sodium hypochlorite for 3 min, rinsing in 80% ethanol for 1 min, and drying at room temperature for 2 min. Immediately post-decontamination, dental calculus samples were crushed on the side of plastic tubes with sterile tweezers, and DNA was extracted using an in-house silica-based method described previously [46]. Extraction blank controls were included to monitor microbial DNA background signals throughout this process; one extraction blank control was analysed for each DNA extraction batch (1 control: 10 samples).
Shotgun metagenomic libraries were constructed as previously described [47], using unique combinations of 7 bp forward and reverse barcodes. Thirteen cycles of PCR were used for the first amplification with P5/P7 barcoded adapters (Platinum™ Taq HiFi Polymerase), followed by an additional 13 cycles for the addition of GAII-index and sequencing primers. Metagenomic shotgun libraries were cleaned using Ampure XP, quantified using an Agilent TapeStation, and pooled at equimolar concentrations prior to initial sequencing on the Illumina NextSeq (2 × 150 bp), and further sequencing on an Illumina HiSeq (2 × 150 bp). All samples were sequenced together in the same sequencing pool.
(c). Data used from other previously published studied
Eighteen modern dental plaque samples from the Human Microbiome Project (HMP) [48] were downloaded (SRS011098, SRS011126, SRS011152, SRS011255, SRS011343, SRS012285, SRS013170, SRS013252, SRS013533, SRS013723, SRS013836, SRS013949, SRS014476, SRS014578, SRS014690, SRS014894, SRS015044, SRS015063). Because MALT does not have a paired-end alignment mode, only the R1 files were used. The R1 files were randomly subsampled to a depth of 1 500 000 sequences using Seqtk with a seed of 666 https://github.com/lh3/seqtk. Modern and ancient dental calculus DNA sequences were obtained from a previous study [30] (https://www.oagr.org.au/experiment/view/65/).
(d). Data processing and taxonomic composition analyses
The resulting data converted into FASTQ format using Illumina's bcl2fastq software, before being demultiplexed, trimmed and merged using AdapterRemoval 2 based on unique P5/P7 barcodes [49]. Only merged reads were used for subsequent analyses. Merged reads from separate sequencing runs were concatenated per sample. Taxonomic composition was determined using MEGAN Alignment Tool (MALTn) v. 0.3.8 [50], whereby DNA reads from samples were aligned (default settings) against the RefSeqGCS database (2017) [51] containing 47 713 archaeal and bacterial genome assemblies from the NCBI Assembly database [52]. The resulting blast-text files were converted into RMA files via the blast2rma script included in the program MEGAN v.6.11.1 [53], with the following lowest common ancestor (LCA) parameters: weighted-LCA = 80%, minimum bitscore = 44, minimum E-value = 0.01, minimum support per cent = 0.1 [51].
Samples were first assessed for ancient DNA authenticity by comparison to extraction blank controls. Subtractive filtering was used to remove species found in the extraction blank controls from ancient dental calculus samples. For analysis in QIIME [54], the filtered, species-level taxonomic composition was exported from MEGAN into BIOM format, and imported into QIIME 1.9.1 and rarefied to 86 267 species-level reads per sample. SourceTracker (v.0.9.8) analysis [55] was also carried out on this rarefied BIOM table to help examine exogenous contamination using various, previously published well-characterized source sample types: soil [56], skin [57,58], gut [25,59,60], saliva [61,62], dental plaque [63] and ancient dental calculus [30,31] (see the electronic supplementary material, table S7 for more information and accession numbers). Raw sequences from these studies were downloaded and processed in the same manner as the samples in the present study. PERMANOVA was used to test for statistical significance in composition between groups using the compare_categories.py script with 999 permutations. Differential abundance of species between groups was tested using the Kruskal–Wallis test with Bonferroni-correction in the group_significance.py script. The rarefied table was imported into STAMP [64] to calculate and plot the Welch's t-test of Methanobrevibacter oralis relative abundance (figure 4).
Figure 4.
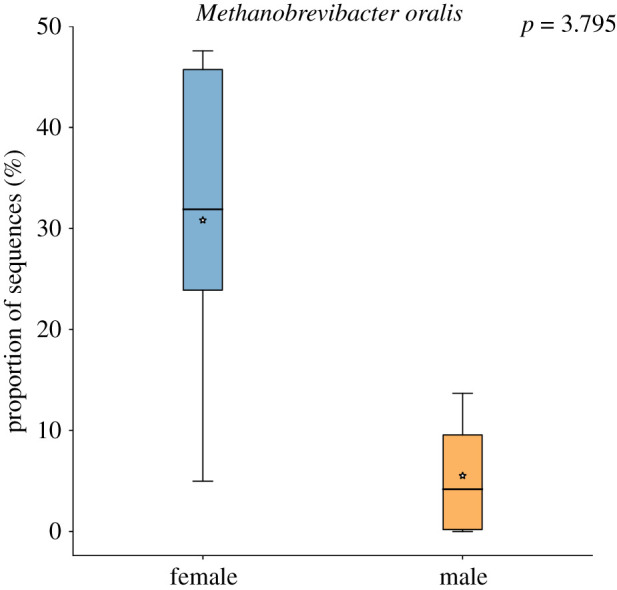
Box-plot of Methanobrevibacter oralis abundance in Edo period individuals. Females n = 5 (blue) versus males n = 5 (orange). Welch's t-test uncorrected p-value = 0.028, Benjamini–Hochberg false discovery rate corrected p-value 3.795.
(e). Whole-genome phylogenetic analysis
Genomic sequences were assembled by mapping reads to the Anaerolineaceae sp. oral taxon 439 genome (RefSeq accession: GCF_001717545.1) using BWA-aln [65] with the seed disabled, as recommended for ancient DNA [66]. The resulting BAM files were filtered to remove reads with mapping quality of less than 1 (keeping reads that only have 1 best hit) using SAMtools [67], and duplicates were removed using DeDup [68]. Estimation of cytosine deamination was performed using MapDamage2 [69] using the Anaerolineaceae sp. oral taxon 439 reference genome (RefSeq accession: GCF_001717545.1). Edit distances were calculated using BAMstats (https://github.com/guigolab/bamstats). Coverage visualizations were created using Anvi'o [70]. Samples with fewer than 100 000 mapped reads (electronic supplementary material, table S6) were excluded from phylogenetic analyses (A18017_Japan_Jomon_3, A18019_Japan_Jomon_5, and A18022_Japan_Edo_3). Variant calling was performed using the SNIPPY pipeline (https://github.com/tseemann/snippy), which uses FreeBayes [71]. The pipeline was adjusted to use a FreeBayes –ploidy of ‘1’. Using a .bed file, we masked 16S rRNA and tRNA gene regions and putative phage regions identified using PHASTER [72] (electronic supplementary material, file 5). To account for cytosine deamination, a minimum depth of 3 sequences was required to call a variant. A minimum fraction of 90% (i.e. greater than 90% of nucleotides at a site had to be the same) was used to ensure the dominant variant was used. Missing data (depth less than 3) were labelled as N's. Phylogenetic reconstruction was performed on the masked whole genome single nucleotide polymorphism alignment (.full.aln SNIPPY suffix) using RAxML [73], with the GTR-GAMMA substitution model and autoMRE bootstrapping (raxmlHPC-PTHREADS-SSE3 -f a -x 12345 -p 12345 -# autoMRE -m GTRGAMMA -s ‘alignment’ -o Elsidron1). Trees were visualized and annotated using FigTree (https://github.com/rambaut/figtree).
3. Results
(a). Authentic ancient microbial DNA was isolated from dental calculus
We applied metagenomic shotgun sequencing to 15 ancient Japanese dental calculus samples: five male Jomon period (approx. 3000 BP) and 10 (five male; five female) from the Early Edo period 400–300 BP) (figure 1). An average of 8 992 067 sequences per sample was obtained, with the fragment length distributions being as expected for ancient DNA (average size 78 bp; electronic supplementary material, table S2). We used MALTn (MEGAN Alignment Tool) to align DNA sequences to a reference database containing 47 713 archaeal and bacterial genome assemblies, and as expected for ancient dental calculus studies [51]; an average of 49.8% (±10.1%) of DNA sequences in each sample could be assigned taxonomy. The ancient Japanese calculus samples looked similar to previously published ancient calculus samples (figure 2) and were distinct from extraction blank controls (EBCs) (figure 2). Additionally, there were phyla present in the ancient calculus samples that were absent in modern plaque samples from the HMP and included Synergistetes, Chloroflexi, Candidatus Sacchararibacteria, and Euryarchaeota (figure 2). These phyla contain several species that can be associated with periodontal disease in modern populations, such as Synergistetes: Fretibacterium fastidiosum [74]; Chloroflexi: Anaerolineaceae sp. oral taxon 439 [75]; Candidatus Sacchararibacteria: TM7x [76]; and Euryarchaeota: Methanobrevibacter oralis [77]. Therefore, the absence of these phyla from the modern plaque samples might be associated with disease-state, as all HMP samples were taken from healthy individuals [27], but a current lack of information regarding the microbiome in health status of ancient individuals makes this difficult to classify [78].
Figure 2.

Phylum level taxonomic composition of samples and EBCs.
As background DNA contamination can influence ancient microbiome studies [27,79,80], we next assessed oral and contaminant DNA levels in the samples by ordinating Bray Curtis dissimilarity in a principal coordinates analysis (PCoA) (figure 3), which included EBCs, ancient Japanese samples, previously published ancient calculus specimens [30], and modern healthy plaque samples from the HMP [27]. Ancient Japanese calculus specimens clustered with published ancient calculus specimens and were dissimilar to EBCs, as expected (figure 3). Except for one Edo calculus specimen (A18022), ancient Japanese samples were distinct from modern plaque samples from the HMP (figure 3). We took a conservative approach and removed any species found in the EBCs (electronic supplementary material, table S3) from the Japanese calculus samples to help eliminate the contributions of contaminant DNA [81]; an average of 94.8% species-assigned sequences remained after filtering, highlighting the robust preservation of the specimens (electronic supplementary material, table S4). Species present in ancient Japanese samples after filtering by EBCs were largely previously identified in other oral microbiome studies [27,82] and have entries in the Human Oral Microbiome Database (HOMD) (electronic supplementary material, table S5), [83]. Lastly, we ran SourceTracker on the filtered dental calculus samples, which, of the source sample types used (skin, soil, gut, saliva, modern plaque and ancient dental calculus), predicted dental calculus was the most likely source, with the exception of sample A18022, which was predominantly modern plaque (electronic supplementary material, figure S8).
Figure 3.

PCoA ordination of species-level Bray-Curtis distances. Extraction blank controls (EBCs, grey crosses), cluster separately from the rest of the ancient and modern oral samples. Ancient Japanese dental calculus samples (black circles) cluster with the El Sidron Neanderthal samples (yellow triangles). Modern dental plaque samples from the Human Microbiome Project (blue squares), and a modern dental calculus sample (blue triangle) cluster separately from ancient dental calculus samples, with the exception of one Edo period Japanese sample (18022_Japan_Edo_3). One Jomon sample (A18019_Japan_Jomon_5) is also pulled towards the EBCs (asterisk).
Upon closer taxonomic investigation of our samples, we noted that one sample (A18019_Japan_Jomon_5) was more contaminated and poorly preserved than the others. This sample was pulled towards the EBCs in the PCoA (figure 3) and had 8 out of 41 species classified that were of oral origin (HOMD) after filtering by EBCs (electronic supplementary material, figure S1). As samples with poorer preservation typically yield lower quantities of DNA, which can lead to higher percentages of duplicates, we assessed the percentage of duplicate sequences in this sample using BBMap's dedupe2 (sourceforge.net/projects/bbmap/) and found that 81.9% of sequences were duplicates. In the light of these findings, we removed this sample from subsequent compositional analyses.
(b). Comparing the oral microbiota of Jomon and Edo periods Japan
As the Jomon and Edo cultures are associated with distinct diets and lifestyles, we wanted to explore the similarities and differences between the microbiomes found in both cultures. We found no significant differences in alpha diversity between Jomon and Edo period Japanese samples (Shannon and observed species non-parametric t-test p-values greater than 0.05), which probably reflects the difficulty in obtaining clear diversity signatures in ancient calculus specimens [84]. We also found no significant differences in composition (PERMANOVA of Bray-Curtis and Binary Jaccard distance p-values greater than 0.05) or differentially abundant species between the Edo period or Jomon samples (Kruskal–Wallis; Bonferroni-correction p-values greater than 0.05). However, one Edo period sample clustered closely with the modern dental plaque samples in figure 3 (A18022) and had 10 classified species that were not present in any other sample: Rothia aeria, Corynebacterium durum, Actinomyces johnsonii, Actinomyces sp. HPA0247, Haemophilus parainfluenzae, Neisseria meningitidis, Neisseria sicca, Neisseria sp. HMSC072F04, Fusobacterium hwasookii and Porphyromonas sp. KLE 1280.
(c). Oral microbiota correlates with disease status and sex in the Edo period Japan
All of the female specimens in this study had evidence of periodontal disease and had their teeth dyed black, which was a common cultural practice of females in the Edo period (called Ohaguro). Therefore, we wanted to test if the female samples had different microbiota to male samples in our study. Again, we found no significant differences in alpha diversity (Shannon and observed species p-values greater than 0.05), as expected. However, we did find a significant difference in microbiota composition (PERMANOVA of Bray–Curtis distances p-value 0.028, test statistic 2.35) in females versus males, although this was not observed in non-abundance weighted metric (PERMANOVA of Binary Jaccard p-value greater than 0.05). No species were significantly differentially abundant between males and females with periodontal disease (Kruskal–Wallis test with Bonferroni-correction p-values greater than 0.05). We also tested for signatures of periodontal disease [85–87] in female Edo individuals. No species were significantly associated with caries prevalence or periodontitis (Kruskal–Wallis test with Bonferroni-corrected p-values greater than 0.05), including members of the periodontitis-associated ‘red-complex’ (Treponema denticola, Tannerella forsythia, Porphyromonas gingivalis) [88]. However, the abundance of the periodontitis-associated archaeon, Methanobrevibacter oralis [77,89,90], was substantially higher in the females (mean abundance in females = 32%, mean abundance in males = 5%) (figure 4), although this difference was not statistically significant when controlling for multiple comparisons (Welch's t-test Benjamini-Hochberg false discovery rate corrected p-value 3.795). These results suggest that the oral microbiota composition in Japanese Edo women, who both practiced Ohaguro and suffered from periodontal disease, is distinct from Edo men.
(d). Phylogenomic analysis
To further explore factors that drive microbial variation in ancient Japan, we performed phylogenomic analysis to explore strain diversity present in both periods. To find suitable candidates for phylogenomic analysis, we determined the core oral microbiome in ancient Japan (i.e. species present in every sample). We found Actinomyces sp. oral taxon 414, Actinomyces dentalis, Anaerolineaceae sp. oral taxon 439, and Olsenella sp. oral taxon 807 to be present in all samples.
The oral bacterium Anaerolineaceae sp. oral taxon 439 was chosen for phylogenetic analysis owing to its high mean relative abundance within calculus samples (16.5%), which yielded a greater depth of coverage and higher quality variant calls for our fairly low coverage sequencing data (electronic supplementary material, table S6). This bacterium is present at low abundance in healthy human plaque and higher abundance in individuals with periodontal disease [75]. This bacterium also has a high-quality, complete genome assembly needed for phylogenomic reconstructions, although it remains the only human-associated Anaerolineaceae genome currently publicly available. Sequences mapped against the Anaerolineaceae sp. oral taxon 439 genome had terminal cytosine deamination typical of ancient DNA (electronic supplementary material, figures S2 and S3), with the sequences mapping from Jomon and El Sidron Neanderthal samples having higher levels of cytosine deamination at terminal ends (13.9%) compared to the more recent (400–150 year old) Edo samples (6%), as expected with the increasing age of the samples [91].
We then used a conservative approach to examine Anaerolineaceae genomic variants in all individuals. DNA sequences mapped evenly across the genome (figure 5), and whole-genome phylogenetic reconstruction found strong support for a distinct Jomon clade (figure 6), which clustered separately from Edo period samples (figure 6). This suggests that at least two distinct lineages of Anaerolineaceae strains existed in ancient Japan.
Figure 5.

Read mapping results against the Anaerolineaceae sp. oral taxon 439 genome. Each bin represents a gene, with the magnitude of the bar representing the mean depth of coverage of that gene (maximum 3). *Putative phage region that is present in modern reference.
Figure 6.

Maximum-likelihood phylogenetic tree of reconstructed Anaerolineaceae sp. oral taxon 439 genomes. Node labels represent percentage support of 300 bootstrap replicates. Elsidron 1 Neanderthal set as an outgroup, with two separate clades containing Jomon (orange), or Edo period Japanese samples (black). Elsidron1 = Elsidron Neanderthal, Reference genome = Anaerolineaceae sp. oral taxon 439 (ASM171754v1).
4. Discussion
This study is, to our knowledge, the first to explore oral microbiomes from ancient Japanese individuals, providing evidence for past microbial changes in response to disease and changes in human demography. While we did not observe major differences between Jomon and Edo period microbiome compositions, differences between male and female Edo period Japanese individuals were apparent, although the contributions of cultural practices and periodontal disease need further investigation. Finally, phylogenomic investigations revealed at least two distinct Anaerolineaceae sp. lineages between Jomon and Edo periods.
The switch to agricultural lifestyles from hunting and gathering has been associated with a compositional change in oral microbiota [28,30]. Here, we assessed dental calculus in both hunter–gatherers and agriculturalists in ancient Japan. Archaeological evidence suggests that Jomon hunter–gatherers relied on both terrestrial and marine resources, including nuts, deer, boar, marine fishes and shellfish [92]. Carbon isotope ratios of human teeth also suggest that C3 plants and terrestrial mammals were major dietary resources for the Jomon people [41]. This is in stark contrast to individuals from the Edo period, who led a predominantly agricultural subsistence [43]. Contrary to published research conducted in Europe, we did not detect a statistically significant difference in microbiome composition between Jomon (hunter–gatherer) and Edo (agricultural) period Japanese. Furthermore, no microbial species were found to be differentially abundant between cultures, which could suggest that the classifiable oral microbiome composition did not drastically change in Japan from Jomon to Edo periods. This supports findings that oral microbiota are highly stable through time [93,94] and maybe minimally influenced by certain dietary changes [95,96]. This is also consistent with other studies comparing modern hunter–gatherer populations to industrialized populations, which failed to see large changes in oral microbiota between these two lifestyles, despite findings changes in gut microbiota [16]. However, there are several alternative explanations. First, the limited sample size of our study (four Jomon and 10 Edo period) could have prevented the detection of such differences. Additionally, we were not able to control for tooth type owing to our small sample size, which has been shown to influence the microbial composition of modern plaque [97] and ancient dental calculus samples [98]. Bioinformatically, we may have also experienced biases in our species identifications, as the species that we classified maybe biased towards core oral taxa that are stable through time in Europeans, given that most modern oral reference genomes are more commonly generated from European and American isolates [93,94,97,99]. Furthermore, we were only able to classify on average approximately 49.8% of sequences from the ancient Japanese samples, consistent with other ancient calculus studies (e.g. [51]), and therefore, perhaps missed some of the microbial diversity present in these ancient samples that were unique to each culture or labile to dietary changes. Future improvements of analytical tools and further sampling of oral microbial genomes from broader human populations could allow for classification of the unclassified portion of our data and potentially provide enhanced bio-archaeological information from ancient dental calculus.
We found a significant difference between the microbiome composition of female and male Edo period Japanese. A potential driver of this difference is oral disease status, as all of the female samples had evidence of periodontal disease, which has been demonstrated in modern populations to impact microbiome composition [85,87,100]. In particular, we found the periodontal disease-associated archaeon, Methanobrevibacter oralis [77,89,90], to be generally more abundant in females versus males Edo period Japanese, although members of the periodontitis-associated ‘red-complex’ were not found to be differentially abundant in females versus males [88]. However, this is unsurprising given recent recognition that periodontal disease is of complex aetiology, not the result of a handful of periopathogens [101]. Interestingly, one Edo period sample (A18022) was also compositionally distinct from others and had 10 species classified that were not found in any other sample. This sample clustered with the modern healthy HMP plaque samples on the PCoA plot and in SourceTracker analysis and could represent a ‘healthy’ ancient sample. Future studies with larger sample sizes including both periodontal-positive and negative individuals are needed to determine the influence of periodontal disease on the male/female split we observed in Edo period Japanese. Overall, our findings suggest that periodontal disease is an important factor to examine when comparing microbial composition in ancient dental calculus studies, and future studies should aim to control for periodontal disease when making cultural comparisons.
It is also possible that the use of Ohaguru may have also influenced the female oral microbiota in ancient Japan. The practice of Ohaguru was common in higher-status women until the end of the Edo period, when the practice was outlawed in 1870 [102]. Women would paint their teeth with a black paste, called kanemizu, which was typically a mixture of iron (ferric) acetate and vegetable tannins; for example, kanemizu can be created by dissolving iron filings in vinegar and then adding in tannins from tea [103]. It is plausible that introduction of both iron and vegetable tannins using this method influenced the oral microbiota. For example, iron availability in an in vitro model of salivary microbiota had a significant impact on the microbiota composition [104]. Further, tea drinking has been shown to significantly alter both salivary oral microbiota and that surveyed by systematic oral brushing [105,106]. Regardless, this practice probably impacted the oral microbiota via access to these micronutrients, as previously reported isotope data from the skeletons found no significant differences in the general dietary intake between male and female samples from the Early Edo period [43]. Surprisingly, the practice of Ohaguru was thought to protect teeth from dental decay [103]; however, we find it associated with evidence of periodontal disease, raising questions about its health benefits. Future studies could empirically test the impact of kanemizu on oral microbiota using in vitro models or examining the impact of other tooth blackening processes on oral microbiota and health, as tooth blackening was practised historically and is still practised today in Oceania [107]. Nevertheless, microbiome studies may provide further information into how cultural practices influenced oral health in the past and today.
It is widely accepted that the modern Japanese population is the result of admixture between indigenous Jomon and later migrants from continental Asia during and after the Yayoi period [37]. Here, we observed a separation between Edo-associated Anaerolineaceae lineages and those found in ancient Jomon samples. While it is unclear how these two distinct clades originated, one potential hypothesis is that the Edo-period Anaerolineaceae strain originated in Japan through human demographic processes. For example, continental Asian Anaerolineaceae lineage/s could have been brought to Japan by migrants who arrived in Japan from mainland Asia. It also remains unclear if either strain still exists today, or if the prevalence of the Jomon-period strain was diminished by the Edo strain. This later scenario is plausible if the continental Asian contribution to modern Japanese was larger than the Jomon, resulting in the loss of the lineage in a fashion analogous to genetic drift. Current estimates of Jomon genetic contribution to modern Japanese is less than 20%, supporting this scenario [37]. However, another possibility for this finding is that the Jomon lineage has survived to this day, but that we did not detect it owing to the small sample size of our study and lack of comparable modern metagenomic data. Future studies investigating modern individuals from across Japan could test for the presence of the Jomon Anaerolineaceae lineage and try to pinpoint the source of the Edo strains. Spatially diverse sampling will be important, as it has been shown that genetic contribution from Jomon varied among populations across the Japanese Archipelago [35–37]. Further studies using ancient dental calculus could also assist in learning more about the source/s of Yayoi admixture, or the diversity of Jomon strains prior to the arrival of mainland migrants, which remain undetermined. Future DNA sequencing efforts will allow for the phylogenetic reconstruction of other human-associated microorganisms and permit investigations into how these genomes have changed through time, potentially yielding insights into mechanisms of co-speciation with humans.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank Sterling Wright for completing the SourceTracker analysis used in this study. We also thank the reviewers for providing valuable feedback and improving the manuscript.
Ethics
Ethics approval for this study was obtained from the University of Adelaide Human Research Ethics Committee (H-2012-108).
Data accessibility
The untrimmed, demultiplexed paired-end sequences generated in this study are available at the NCBI SRA under project PRJNA608555
Authors' contributions
R.E. carried out the molecular laboratory work, data analysis, participated in the design of the study and drafted the manuscript; L.S.W., H.K.K. and K.S. participated in the design of the study, collected the samples and critically revised the manuscript. All authors gave final approval for publication and agree to be held accountable for the work performed therein.
Competing interests
We declare we have no competing interests.
Funding
We received no funding for this study.
References
- 1.de Agüero MG, et al. 2016. The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302. ( 10.1126/science.aad2571) [DOI] [PubMed] [Google Scholar]
- 2.Gensollen T, Iyer SS, Kasper DL, Blumberg RS. 2016. How colonization by microbiota in early life shapes the immune system. Science 352, 539–544. ( 10.1126/science.aad9378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157, 121–141. ( 10.1016/j.cell.2014.03.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, Tuohy K. et al. 2018. Gut microbiota functions: metabolism of nutrients and other food components. Eur. J. Nutr. 57, 1–24. ( 10.1007/s00394-017-1445-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yano JM, et al. 2015. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276. ( 10.1016/j.cell.2015.02.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sampson TR, Mazmanian SK. 2015. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 17, 565–576. ( 10.1016/j.chom.2015.04.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1131. ( 10.1038/nature05414) [DOI] [PubMed] [Google Scholar]
- 8.Ravussin Y, Koren O, Spor A, LeDuc C, Gutman R, Stombaugh J, Knight R, Ley RE, Leibel RL. 2012. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 20, 738–747. ( 10.1038/oby.2011.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamboli CP, Neut C, Desreumaux P, Colombel JF. 2004. Dysbiosis in inflammatory bowel disease. Gut 53, 1–4. ( 10.1136/gut.53.1.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. 2015. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 26, 26191 (accessed 18 March 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croswell A, Amir E, Teggatz P, Barman M, Salzman NH. 2009. Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric salmonella infection. Infect. Immun. 77, 2741–2753. ( 10.1128/IAI.00006-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blaser MJ. 2016. Antibiotic use and its consequences for the normal microbiome. Science 352, 544–545. ( 10.1126/science.aad9358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown K, DeCoffe D, Molcan E, Gibson DL. 2012. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 4, 1095–1119. ( 10.3390/nu4081095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pham TAN, Lawley TD. 2014. Emerging insights on intestinal dysbiosis during bacterial infections. Curr. Opin. Microbiol. 17, 67–74. ( 10.1016/j.mib.2013.12.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beatty JK, et al. 2017. Giardia duodenalis induces pathogenic dysbiosis of human intestinal microbiota biofilms. Int. J. Parasitol. 47, 311–326. ( 10.1016/j.ijpara.2016.11.010) [DOI] [PubMed] [Google Scholar]
- 16.Clemente JC, et al. 2015. The microbiome of uncontacted Amerindians. Sci. Adv. 1, e1500183 ( 10.1126/sciadv.1500183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. et al. 2010. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl Acad. Sci. USA 107, 1 1971–11 975. ( 10.1073/pnas.1002601107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stahringer SS, Clemente JC, Corley RP, Hewitt J, Knights D, Walters WA, Knight R, Krauter KS. 2012. Nurture trumps nature in a longitudinal survey of salivary bacterial communities in twins from early adolescence to early adulthood. Genome Res. 22, 2146–2152. ( 10.1101/gr.140608.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw L, Ribeiro ALR, Levine AP, Pontikos N, Balloux F, Segal AW, Roberts AP, Smith AM, Fraser CM. 2017. The human salivary microbiome is shaped by shared environment rather than genetics: evidence from a large family of closely related individuals. mBi 8, e01237-17 ( 10.1128/mBio.01237-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korpela K, Costea P, Coelho LP, Kandels-Lewis S, Willemsen G, Boomsma DI, Segata N, Bork P. 2018. Selective maternal seeding and environment shape the human gut microbiome. Genome Res. 28, 561–568. ( 10.1101/gr.233940.117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falush D. et al. 2003. Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585. ( 10.1126/science.1080857) [DOI] [PubMed] [Google Scholar]
- 22.Moeller AH. et al. 2016. Cospeciation of gut microbiota with hominids. Science 353, 380–382. ( 10.1126/science.aaf3951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yatsunenko T. et al. 2012. Human gut microbiome viewed across age and geography. Nature 486, 222–227. ( 10.1038/nature11053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schnorr SL. et al. 2014. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5, 3454 ( 10.1038/ncomms4654) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rampelli S. et al. 2015. Metagenome sequencing of the Hadza hunter-gatherer gut microbiota. Curr. Biol. 25, 1682–1693. ( 10.1016/j.cub.2015.04.055) [DOI] [PubMed] [Google Scholar]
- 26.Blaser MJ. 2006. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 7, 956–960. ( 10.1038/sj.embor.7400812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominguez-Bello MG, Blaser MJ. 2011. The human microbiota as a marker for migrations of individuals and populations. Annu. Rev. Anthropol. 40, 451–474. ( 10.1146/annurev-anthro-081309-145711) [DOI] [Google Scholar]
- 28.Adler CJ. et al. 2013. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet. 45, 450–455. ( 10.1038/ng.2536) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warinner C. et al. 2014. Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 46, 336–344. ( 10.1038/ng.2906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weyrich LS. et al. 2017. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544, 357–361. ( 10.1038/nature21674) [DOI] [PubMed] [Google Scholar]
- 31.Mann AE. et al. 2018. Differential preservation of endogenous human and microbial DNA in dental calculus and dentin. Sci. Rep. 8, 9822 ( 10.1038/s41598-018-28091-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroeder HE, Shanley D. 1969. Formation and inhibition of dental calculus. J. Periodontol. 40, 643–646. ( 10.1902/jop.1969.40.11.643) [DOI] [PubMed] [Google Scholar]
- 33.Habu J. 2004. Ancient Jomon of Japan (Vol. 4). Cambridge, UK: Cambridge University Press. [Google Scholar]
- 34.Imamura K. 2016. Prehistoric Japan: new perspectives on insular east Asia. Honolulu, Hawaii: University of Hawaii Press; Routledge. [Google Scholar]
- 35.Jinam T, et al. 2012. The history of human populations in the Japanese Archipelago inferred from genome-wide SNP data with a special reference to the Ainu and the Ryukyuan populations. J. Hum. Genet. 57, 787–795. ( 10.1038/jhg.2012.114) [DOI] [PubMed] [Google Scholar]
- 36.Jinam TA, Kanzawa-Kiriyama H, Inoue I, Tokunaga K, Omoto K, Saitou N. 2015. Unique characteristics of the Ainu population in Northern Japan. J. Hum. Genet. 60, 565–571. ( 10.1038/jhg.2015.79) [DOI] [PubMed] [Google Scholar]
- 37.Kanzawa-Kiriyama H. et al. 2017. A partial nuclear genome of the Jomons who lived 3000 years ago in Fukushima, Japan. J. Hum. Genet. 62, 213–221. ( 10.1038/jhg.2016.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanzawa-Kiriyama H, et al. 2019. Late Jomon male and female genome sequences from the Funadomari site in Hokkaido, Japan. Anthropol Sci. 127, 83–110. [Google Scholar]
- 39.Adachi N, Shinoda K, Umetsu K, Kitano T, Matsumura H, Fujiyama R, Sawada J, Tanaka M. 2011. Mitochondrial DNA analysis of Hokkaido Jomon skeletons: remnants of archaic maternal lineages at the southwestern edge of former Beringia. Am. J. Phys. Anthropol. 146, 346–360. ( 10.1002/ajpa.21561) [DOI] [PubMed] [Google Scholar]
- 40.Matsu’ ura S. 1984. Fluorine test on human skeletal remains from the Ebishima shell mound in Iwate Prefecture (In Japanese with English summary). Mem. Natn Sci. Mus. Tokyo 17, 189–192. [Google Scholar]
- 41.Kusaka S, Uno KT, Nakano T, Nakatsukasa M, Cerling TE. 2015. Carbon isotope ratios of human tooth enamel record the evidence of terrestrial resource consumption during the Jomon period, Japan. Am. J. Phys. Anthropol. 158, 300–311. ( 10.1002/ajpa.22775) [DOI] [PubMed] [Google Scholar]
- 42.Hayashi K, Yamaguchi B, Dodo Y, Hiramoto Y. 1981. The middle of middle Jomon to the end of final Jomon: a preliminary report of the survey of localities B and C in the Miyano shell-mound: with reference to the human skeletal remains. Report of the research supported by the Grant-in-Aid for Scientific Research of the Ministry of Education, Science and Culture, Japan (In Japanese).
- 43.Tsutaya T, Nagaoka T, Kakinuma Y, Kondo O, Yoneda M. 2016. The diet of townspeople in the city of Edo: carbon and nitrogen stable isotope analyses of human skeletons from the Ikenohata-Shichikencho site. Anthropol. Sci. 124, 17–27. ( 10.1537/ase.150914) [DOI] [Google Scholar]
- 44.Weyrich LS, Dobney K, Cooper A. 2015. Ancient DNA analysis of dental calculus. J. Hum. Evol. 79, 119–124. ( 10.1016/j.jhevol.2014.06.018) [DOI] [PubMed] [Google Scholar]
- 45.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. 2019. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. ( 10.1016/j.tim.2018.11.003) [DOI] [PubMed] [Google Scholar]
- 46.Brotherton P. et al. 2013. Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans. Nat. Commun. 4, 1764 ( 10.1038/ncomms2656) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 ( 10.1101/pdb.prot5448) [DOI] [PubMed] [Google Scholar]
- 48.The Human Microbiome Project Consortium et al. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. ( 10.1038/nature11234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schubert M, Lindgreen S, Orlando L. 2016. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 1–7. ( 10.1186/s13104-016-1900-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herbig A, Maixner F, Bos KI, Zink A, Krause J, Huson DH. 2016. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv, 050559 ( 10.1101/050559) [DOI]
- 51.Eisenhofer R, Weyrich LS. 2019. Assessing alignment-based taxonomic classification of ancient microbial DNA. PeerJ 7, e6594 ( 10.7717/peerj.6594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Leary NA. et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. ( 10.1093/nar/gkv1189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh H-J, Tappu R, Poisot T. 2016. MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12, e1004957 ( 10.1371/journal.pcbi.1004957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caporaso JG. et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. ( 10.1038/nmeth.f.303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. 2011. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763. ( 10.1038/nmeth.1650) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin X. et al. 2014. Microbial metabolic potential for carbon degradation and nutrient (nitrogen and phosphorus) acquisition in an Ombrotrophic peatland. Appl. Environ. Microbiol. Am. Soc. Microbiol. 80, 3531–3540. ( 10.1128/AEM.00206-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oh J, Byrd AL, Park M, Kong HH, Segre JA. 2016. Temporal stability of the human skin microbiome. Cell 165, 854–866. ( 10.1016/j.cell.2016.04.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tirosh O, Conlan S, Deming C, Lee-Lin S-Q, Huang X, NISC comparative sequencing program et al. 2018 Expanded skin virome in DOCK8-deficient patients. Nat. Med. 24, 1815–1821. ( 10.1038/s41591-018-0211-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ormerod KL. et al. 2016. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 36 ( 10.1186/s40168-016-0181-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gopalakrishnan V. et al. 2018. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. ( 10.1126/science.aan4236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lassalle F, Spagnoletti M, Fumagalli M, Shaw L, Dyble M, Walker C, Thomas MG, Bamberg Migliano A, Balloux F. 2018. Oral microbiomes from hunter-gatherers and traditional farmers reveal shifts in commensal balance and pathogen load linked to diet. Mol. Ecol. 27, 182–195. ( 10.1111/mec.14435) [DOI] [PubMed] [Google Scholar]
- 62.Aleti G. et al. 2019. Identification of the bacterial biosynthetic gene clusters of the oral microbiome illuminates the unexplored social language of bacteria during health and disease. mBio 10 (Accessed 2 March 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lloyd-Price J. et al. 2017. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66. ( 10.1038/nature23889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. 2014. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. ( 10.1093/bioinformatics/btu494) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. ( 10.1093/bioinformatics/btp324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schubert M, Ginolhac A, Lindgreen S, Thompson JF, AL-Rasheid KA, Willerslev E, Krogh A, Orlando L. 2012. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13, 178 ( 10.1186/1471-2164-13-178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinforma Oxf. Engl. 25, 2078–2079. ( 10.1093/bioinformatics/btp352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peltzer A, Jäger G, Herbig A, Seitz A, Kniep C, Krause J, Nieselt K. 2016. EAGER: efficient ancient genome reconstruction. Genome Biol. 17, 60 ( 10.1186/s13059-016-0918-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jónsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L. 2013. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinforma Oxf. Engl. 29, 1682–1684. ( 10.1093/bioinformatics/btt193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. 2015. Anvi'o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 ( 10.7717/peerj.1319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garrison E, Marth G.2012. Haplotype-based variant detection from short-read sequencing. ArXiv, 12073907 Q-Bio [Internet]. (Accessed 4 September 2019).
- 72.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. ( 10.1093/nar/gkw387) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. ( 10.1093/bioinformatics/btu033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marchesan JT, Morelli T, Moss K, Barros SP, Ward M, Jenkins W, Aspiras MB, Offenbacher S. 2015. Association of synergistetes and cyclodipeptides with periodontitis. J. Dent. Res. 94, 1425–1431. ( 10.1177/0022034515594779) [DOI] [PubMed] [Google Scholar]
- 75.Campbell AG, Schwientek P, Vishnivetskaya T, Woyke T, Levy S, Beall CJ, Griffen A, Leys E, Podar M. 2014. Diversity and genomic insights into the uncultured Chloroflexi from the human microbiota. Environ. Microbiol. 16, 2635–2643. ( 10.1111/1462-2920.12461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brinig MM, Lepp PW, Ouverney CC, Armitage GC, Relman DA. 2003. Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl. Environ. Microbiol. 69, 1687–1694. ( 10.1128/AEM.69.3.1687-1694.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, Relman DA. 2004. Methanogenic Archaea and human periodontal disease. Proc. Natl Acad. Sci. USA 101, 6176–6181. ( 10.1073/pnas.0308766101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Velsko IM. et al. 2019. Microbial differences between dental plaque and historic dental calculus are related to oral biofilm maturation stage. Microbiome 7, 102 ( 10.1186/s40168-019-0717-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eisenhofer R, Cooper A, Weyrich LS. 2017. Reply to Santiago-Rodriguez et al. 2017 proper authentication of ancient DNA is essential. FEMS Microbiol. Ecol. 93 (Accessed 27 June 2017). [DOI] [PubMed] [Google Scholar]
- 80.Eisenhofer R, Weyrich LS. 2018. Proper authentication of ancient dna is still essential. Genes 9, 122 ( 10.3390/genes9030122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salter SJ. et al. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 ( 10.1186/s12915-014-0087-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu W-H, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J. Bacteriol. 192, 5002–5017. ( 10.1128/JB.00542-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen T, Yu W-H, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. 2010. The human oral microbiome database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, baq013 ( 10.1093/database/baq013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ziesemer KA. et al. 2015. Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification. Sci. Rep. 5, 16498 ( 10.1038/srep16498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Y. et al. 2014. Phylogenetic and functional gene structure shifts of the oral microbiomes in periodontitis patients. ISME J. 8, 1879–1891. ( 10.1038/ismej.2014.28) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Camelo-Castillo AJ, Mira A, Pico A, Nibali L, Henderson B, Donos N, Tomás I. 2015. Subgingival microbiota in health compared to periodontitis and the influence of smoking. Front. Microbiol. 6, 119 ( 10.3389/fmicb.2015.00119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boutin S, Hagenfeld D, Zimmermann H, El Sayed N, Höpker T, Greiser HK, Becher H, Kim T-S, Dalpke AH. 2017. Clustering of subgingival microbiota reveals microbial disease ecotypes associated with clinical stages of periodontitis in a cross-sectional study. Front. Microbiol. 8, 340 ( 10.3389/fmicb.2017.00340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Socransky Ss, Haffajee Ad, Cugini Ma, Smith C, Kent RL. 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144. ( 10.1111/j.1600-051X.1998.tb02419.x) [DOI] [PubMed] [Google Scholar]
- 89.Nguyen-Hieu T, Khelaifia S, Aboudharam G, Drancourt M. 2012. Methanogenic archaea in subgingival sites: a review. APMIS 121, 467–477. ( 10.1111/apm.12015) [DOI] [PubMed] [Google Scholar]
- 90.Horz H-P, Conrads G. 2011. Methanogenic Archaea and oral infections – ways to unravel the black box. J. Oral Microbiol. 3, 5940 (Accessed 26 April 2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. 2012. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 7, e34131 ( 10.1371/journal.pone.0034131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tomioka N. 2010. Animal resources and subsistence range during the Jomon period. Jomon archaeology, vol. 4, relationship between humans and animals [In Japanese]. Tokyo, Japan: Doseisha. [Google Scholar]
- 93.Utter DR, Mark Welch JL, Borisy GG. 2016. Individuality, stability, and variability of the plaque microbiome. Front. Microbiol. 7, 564 ( 10.3389/fmicb.2016.00564) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hall MW, Singh N, Ng KF, Lam DK, Goldberg MB, Tenenbaum HC, Neufeld JD, G. Beiko R, Senadheera DB. 2017. Inter-personal diversity and temporal dynamics of dental, tongue, and salivary microbiota in the healthy oral cavity. Npj Biofilms Microbiomes 3, 2 ( 10.1038/s41522-016-0011-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Belstrøm D, Holmstrup P, Nielsen CH, Kirkby N, Twetman S, Heitmann BL, Klepac-Ceraj V, Paster BJ, Fiehn N-E. 2014. Bacterial profiles of saliva in relation to diet, lifestyle factors, and socioeconomic status. J. Oral Microbiol. 6, 23609 ( 10.3402/jom.v6.23609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keller MK, Kressirer CA, Belstrøm D, Twetman S, Tanner ACR. 2017. Oral microbial profiles of individuals with different levels of sugar intake. J. Oral. Microbiol. 9, 1355207 (Accessed 22 December 2017) ( 10.1080/20002297.2017.1355207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Proctor DM, Fukuyama JA, Loomer PM, Armitage GC, Lee SA, Davis NM, Ryder MI, Holmes SP, Relman DA. 2018. A spatial gradient of bacterial diversity in the human oral cavity shaped by salivary flow. Nat. Commun. 9, 681 ( 10.1038/s41467-018-02900-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Farrer AG, Bekvalac J, Redfern R, Gully N, Dobney K, Cooper A, Weyrich L. 2018. Biological and cultural drivers of oral microbiota in Medieval and Post-Medieval London, UK. bioRxiv 343889 ( 10.1101/343889) [DOI]
- 99.Cameron SJS, Huws S, Hegarty MJ, Smith DPM, Mur LAJ. 2015. The human salivary microbiome exhibits temporal stability in bacterial diversity. FEMS Microbiol. Ecol. 91, fiv091 ( 10.1093/femsec/fiv091) [DOI] [PubMed] [Google Scholar]
- 100.Kilian M, Chapple ILC, Hannig M, Marsh PD, Meuric V, Pedersen AML, Tonetti MS, Wade WG, Zaura E.. 2016. The oral microbiome – an update for oral healthcare professionals. Br. Dent. J. 221, 657 ( 10.1038/sj.bdj.2016.865) [DOI] [PubMed] [Google Scholar]
- 101.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 27, 409–419. ( 10.1111/j.2041-1014.2012.00663.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ezure T, et al. 1987. A report of clinical example concerning the tooth black (Japanese with English summary). Dent. J. Iwate Med. Univ. 12, 217–221. [Google Scholar]
- 103.Ai S, Ishikawa T, Seino A. 1965. ‘Ohaguro’ traditional tooth staining custom in Japan. Int. Dent. J. 15, 426–441. [PubMed] [Google Scholar]
- 104.Wang R, Kaplan A, Guo L, Shi W, Zhou X, Lux R, He X. 2012. The influence of iron availability on human salivary microbial community composition. Microb. Ecol. 64, 152–161. ( 10.1007/s00248-012-0013-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peters BA, McCullough ML, Purdue MP, Freedman ND, Um CY, Gapstur SM, Hayes RB, Ahn J. 2018. Association of coffee and tea intake with the oral microbiome: results from a large cross-sectional study. Cancer Epidemiol. Biomark Prev. 27, 814–821. ( 10.1158/1055-9965.EPI-18-0184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schueller K, Riva A, Pfeiffer S, Berry D, Somoza V. 2017. Members of the oral microbiota are associated with il-8 release by gingival epithelial cells in healthy individuals. Front. Microbiol. 8, 416. (Accessed 18 February 2020) ( 10.3389/fmicb.2017.00416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zumbroich T. 2015. ‘We Blacken Our Teeth with Oko to Make Them Firm’: teeth blackening in Oceania. Anthropologica 57, 539–555. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The untrimmed, demultiplexed paired-end sequences generated in this study are available at the NCBI SRA under project PRJNA608555