

Abstract
Severe congenital neutropenia (SCN) is a monogenic disorder. SCN patients are prone to recurrent life-threatening infections. The main causes of SCN are autosomal dominant mutations in the ELANE gene that lead to a block in neutrophil differentiation. In this study, we use CRISPR-Cas9 ribonucleoproteins and adeno-associated virus (AAV)6 as a donor template delivery system to repair the ELANEL172P mutation in SCN patient-derived hematopoietic stem and progenitor cells (HSPCs). We used a single guide RNA (sgRNA) specifically targeting the mutant allele, and an sgRNA targeting exon 4 of ELANE. Using the latter sgRNA, ∼34% of the known ELANE mutations can in principle be repaired. We achieved gene correction efficiencies of up to 40% (with sgELANE-ex4) and 56% (with sgELANE-L172P) in the SCN patient-derived HSPCs. Gene repair restored neutrophil differentiation in vitro and in vivo upon HSPC transplantation into humanized mice. Mature edited neutrophils expressed normal elastase levels and behaved normally in functional assays. Thus, we provide a proof of principle for using CRISPR-Cas9 to correct ELANE mutations in patient-derived HSPCs, which may translate into gene therapy for SCN.
Keywords: CRISPR-Cas9, ribonucleoprotein, RNP, adeno-associated virus, AAV, non-homologous end joining, NHEJ, homology-directed repair, HDR, hematopoietic stem and progenitor cells, HSPCs, gene therapy, ELANE gene, neutrophil elastase, NE, severe congenital neutropenia
Graphical Abstract
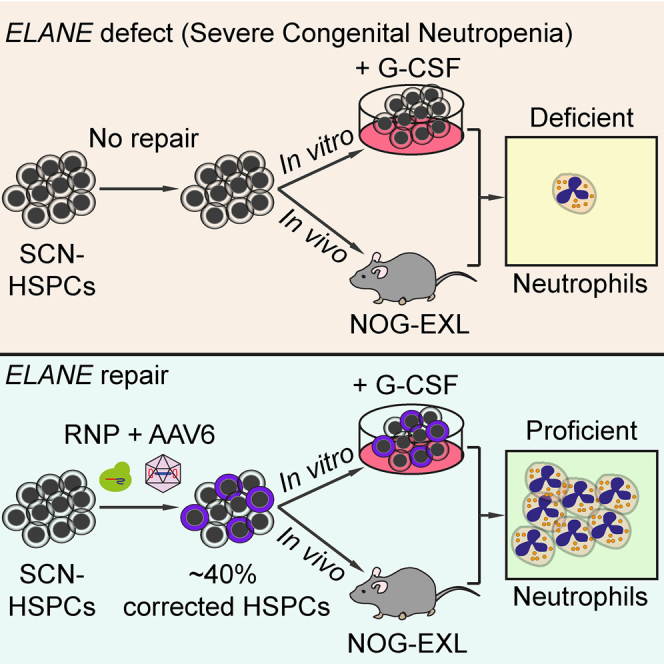
Using CRISPR-Cas9 and AAV6 donor vectors, Tran et al. achieve efficient gene correction of ELANE mutations in severe congenital neutropenia patient-derived hematopoietic stem and progenitor cells (HSPCs). ELANE-repaired HSPCs restored neutrophil differentiation in vitro and in vivo. Mature edited neutrophils show normal elastase expression and neutrophil functions.
Introduction
Severe congenital neutropenia (SCN), a rare inherited monogenic disease, is diagnosed when the number of mature neutrophils in the peripheral blood as the absolute neutrophil count (ANC) is below 0.5 × 109/L for months or years.1, 2, 3, 4, 5, 6, 7 SCN patients tend to develop myelodysplastic syndromes (MDSs) or acute myeloid leukemia (AML). This MDS/AML conversion is often associated with acquired mutations in the CSF3R gene (encoding the granulocyte colony-stimulating factor [G-CSF] receptor), and subsequently other leukemia-associated genes such as RUNX1.8, 9, 10, 11 The treatment of children suffering from SCN with G-CSF reduces the risk of sepsis and infections by increasing the ANC in the majority of SCN patients. However, SCN patients treated with G-CSF are at high risk of MDS/AML development, especially in the case of G-CSF poor responders.4,7,11 For SCN patients who do not respond to G-CSF or develop MDS/AML, the only available treatment option is allogeneic hematopoietic stem cell transplantation with frequent adverse effects and high mortality (17%).12,13
Autosomal dominant mutations in the ELANE gene, encoding neutrophil elastase (NE), are associated with more severe forms of disease1 and account for 60% of the genetic defects causing SCN.14, 15, 16 To date, more than 200 different ELANE mutations have been identified in SCN patients. These mutations are predominantly located in exon 4 (34%) and exon 5 (26%), and to a lesser extent in exon 1 (1.6%), exon 2 (16%), exon 3 (15%), and intron sequences (7%).14,17,18 The pathophysiological mechanisms by which ELANE mutations cause SCN are complex and only partially understood.18,19 ELANE mutations cause an arrest of granulocytic differentiation at the promyelocyte stage and a strongly impaired production of mature neutrophils. According to the prevailing view, they can lead to an accumulation of mislocalized or misfolded NE proteins, which may trigger an unfolded protein response and apoptosis of granulocytic precursors,20, 21, 22, 23 although this scenario has been challenged.24
Recently, Nasri et al.25 have shown that knock out (KO) of the ELANE gene in SCN patient-derived hematopoietic stem and progenitor cells (HSPCs) using CRISPR-Cas9 can rescue neutrophil differentiation. ELANE KO removed the deleterious effects of misfolded NE and restored mature neutrophil formation in vitro.25 However, while an ELANE KO strategy provides a potentially promising treatment for G-CSF poor responders, it seems to favor the generation, selection, or outgrowth of edited cells that lack NE expression altogether.25 This causes concern for human patients, since ELANE KO models in mice have established a non-redundant role for NE in the innate immune defense against certain microbial infections.26, 27, 28, 29, 30 It therefore seems warranted to explore additional gene-repair approaches that can fully restore NE and neutrophil function.
In the CRISPR-Cas9 system, a single guide RNA (sgRNA)-directed Cas9 nuclease introduces specific double-stranded breaks (DSBs) at the targeted sequence. DSBs are predominantly repaired by the non-homologous end joining (NHEJ) pathway, causing micro-deletions/insertions (indels). However, if a DNA donor template is provided, the homology-directed repair (HDR) pathway may be used to precisely repair DSBs.31, 32, 33, 34 Pre-assembled ribonucleoprotein (RNP) complexes of Cas9 protein and synthetic sgRNA, in combination with adeno-associated virus (AAV) serotype 6 for donor template delivery, have led to high HDR efficiencies in human CD34+ HSPCs.35, 36, 37, 38 This approach has been used successfully to repair mutations causing several monogenic blood disorders in patient-derived HSPCs.39, 40, 41 In this study, we describe a CRISPR-Cas9-AAV6-based ELANE gene repair system that achieves high rates of HDR and restores NE function in the edited neutrophils. We demonstrate the feasibility of this approach by repairing mutations in exon 4, which represent ∼34% of the known autosomal dominant ELANE mutations. Additionally, we selectively repaired an individual patient-derived missense mutation at amino acid position 172.
Results
Selection of sgRNAs Targeting ELANE and ELANEL172P
A 3-year-old boy was hospitalized and showed typical symptoms of SCN, including severe neutropenia and infections. A genetic screen and sequencing analysis identified a heterozygous missense c.515T>C mutation in the ELANE gene, converting lysine at position 172 into proline (L172P) (Figure 1A). We developed a “universal” exon 4-based and an L172P-specific CRISPR-Cas9 system to correct ELANE mutations in exon 4 of the ELANE gene in human HSPCs. Using CrispRGold,42,43 we designed a universal sgRNA to target a region of ELANE exon 4 with 60% genome copy (GC) content, termed sgELANE-ex4 hereafter. Since the c.515T>C mutation created a new protospacer adjacent motif (PAM) signal on the minus strand, we also designed an sgRNA specifically targeting the mutant allele that we term sgELANE-L172P hereafter (Figure 1B). To assess the editing efficiencies of these sgRNAs, human HSPCs from healthy donors (HD-HSPCs) or the SCN patient (SCN-HSPCs) were activated and electroporated with sgELANE-ex4- or sgELANE-L172P-containing RNPs, respectively (Figure S1A). At 3 days post-targeting, the sgELANE-ex4 created indels in up to 90% of target alleles in the HD-HSPCs (Figures S1B–S1D). As expected, the sgELANE-L172P specifically targeted the mutant allele with high efficiency (∼77%), leaving the wild-type (WT) allele intact (Figures S2A and S2B). Thus, using these sgRNAs, ∼34% of the known autosomal dominant SCN ELANE mutations (namely mutations in exon 4) may be targeted with high efficiency.
Figure 1.

CRISPR-Cas9-Based ELANE Gene Correction and Off-Target Analysis in SCN Patient-Derived CD34+ HSPCs
(A) Schematic of the human ELANE locus, the ELANE cDNA and NE protein including the L172P (c.515T>C) mutation in exon 4 (top), and Sanger sequencing of the dominant heterozygous ELANEL172P mutation (bottom). (B) Strategy to correct the ELANEL172P mutation using CRISPR-Cas9. PAM sequences are indicated in orange. sgELANE-ex4 targets both the wild-type (WT) and the mutant allele. sgELANE-L172P targets specifically the mutant allele. The donor template includes 0.75 kb of 5′ and 3′ homology arms (HAs), silent mutations (magenta), and a SalI restriction site. External forward primer (Extern For) and reverse primer (Rev) were used to amplify a 1.5-kb fragment of the modified ELANE locus. (C) The on-target and highest risk off-target sites of sgELANE-ex4 and sgELANE-L172P, as predicted by CrispRGold (https://crisprgold.mdc-berlin.de). (D) On-target frequencies were quantified using Sanger sequencing; controls are not shown, as their values are 0. Off-target indel frequencies (on the targeting sequence) were analyzed using amplicon deep sequencing of SCN patient-derived untreated HSPCs (Ctrl) and HSPCs treated with RNPs (∗∗p < 0.01). Data represent mean ± SD for three independent experiments. See also Figures S1 and S2.
Off-Target Analysis of sgELANE-ex4 and sgELANE-L172P
To determine the off-target activity of sgELANE-ex4 and sgELANE-L172P, we used CrispRGold42,43 (https://crisprgold.mdc-berlin.de) to predict the off-target sites with the highest risk, based on their mutation distance to the target site. We selected the top nine and five off-target sites of sgELANE-ex4 and sgELANE-L172P, respectively, for amplicon deep sequencing (Figure 1C). SCN-HSPCs were electroporated with or without sgELANE-ex4- or sgELANE-L172P-containing RNPs, respectively. At 3 days post-editing, genomic DNA (gDNA) was isolated and off-target amplicons were amplified by PCR. The PCR amplicons were pooled and sequenced using MiniSeq. The analysis of these PCR amplicons uncovered no detectable off-target activity for sgELANE-ex4. In the case of sgELANE-L172P, a weak but significant increase in indel events was detected at the top off-target site (OT-11) after treatment with sgELANE-L172P/RNPs. The frequencies of indel events at OT-11, however, were at the level of background sequencing error rates (∼0.2%) (Figure 1D).
Efficient CRISPR-Cas9-Mediated ELANE Correction Systems
Next, to correct ELANE mutations in exon 4 in SCN-HSPCs and to quantify HDR efficiencies, we generated a DNA donor template containing ELANE exon 4 including silent mutations, which create a new SalI restriction site (Figure 1B). We first tested this universal exon 4-based gene correction approach in HD-HSPCs. To this end, HD-HSPCs from two donors were electroporated with sgELANE-ex4 containing RNPs and subsequently infected with AAV6 vectors carrying the ELANE donor template (AAV6-ELANE), at a multiplicity of infection (MOI) of 1 × 105 GC per cell (Figure 2A). The combination of sgELANE-ex4/RNPs and AAV6-ELANE donor vectors resulted in an HDR efficiency of 30% in the HD-HSPCs, as measured by PCR and a SalI-mediated restriction fragment length polymorphism (RFLP) assay at 3 days post-targeting (Figure 2B). By Sanger sequencing, we detected approximately 37%, 37%, and 26% of WT, HDR, and indel sequences, respectively (Figure 2C; Figure S3).
Figure 2.

Efficiency of ELANE Correction in HD- and SCN Patient-Derived CD34+ HSPCs
(A) Schematic of experiment to assess the efficiency of the ELANE correction in either HD-HSPCs or SCN-HSPCs. (B) Left: SalI-RFLP assays showing the HDR events using universal exon 4-based ELANE correction (sgELANE-ex4) in HD-HSPCs from two individuals (HD#1 and HD#2). Stars indicate SalI-cleaved fragments. Right: quantification of three independent duplicate experiments. Data are represented as mean ± SD. (C) Pie chart showing WT, HDR, and indel events of the targeted ELANE locus in HD-HSPCs. (D) SalI-RFLP assays showing efficiency of universal exon 4-based ELANE- (left) or ELANEL172P-specific (right) correction in SCN-HSPCs. Scars indicate SalI-cleaved bands. (E) Quantification of the correction efficiency of universal (sgELANE-ex4) and specific (sgELANE-L172P) systems based on three independent experiments. Data are represented as mean ± SD. (F) Pie chart showing repaired, WT, and mutant (Mut) alleles with or without indel events of the targeted ELANE locus in SCN-HSPCs undergoing universal exon 4-based ELANE gene correction. (G) Pie chart showing repaired and Mut allele with or without indel events of the targeted ELANE locus in SCN-HSPCs undergoing the ELANEL172P-specific correction. See also Figures S3–S5.
Next, we applied this procedure to the SCN-HSPCs harboring the ELANEL172P mutation (Figure 2A). As in the case of the HD-HSPCs, the SalI-cleaved DNA bands were only detectable in the cells that received RNPs and the AAV6-ELANE donor vector (Figure 2D). Quantification of these bands indicated a gene correction efficiency of approximately 30% and 20% using sgELANE-ex4 and sgELANE-L172P, respectively (Figure 2E). Indeed, sequencing of the ELANE locus targeted with sgELANE-ex4/RNPs and AAV6-ELANE donor vectors confirmed that the ELANE allele was repaired in 40% of the cases. As sgELANE-ex4 targets both the WT and mutant alleles, we observed 6% and 8% of indels in the WT and mutant sequences, respectively (Figure 2F; Figure S4). Of note, in the SCN-HSPCs receiving sgELANE-L172P/RNPs and AAV6-ELANE donor vectors, no genetic modifications were detected in the WT allele (Figure S5A). In contrast, the mutant allele was correctly repaired in 56% of the cases or showed indels in 19% of the cases (Figure 2G; Figure S5B). Thus, our approach is suitable to efficiently and specifically repair the ELANEL172P mutation and other mutations in exon 4 of the ELANE gene.
ELANE-Corrected HSPCs Restore Neutrophil Differentiation In Vitro
To confirm the functionality of the corrected ELANE gene, we differentiated the targeted HSPCs into mature neutrophils in vitro. To this end, the targeted HSPCs were expanded with human stem cell factor (hSCF) and human interleukin-3 (hIL-3) cytokines for 3 days and subsequently stimulated with human G-CSF (hG-CSF) and human IL-3 for a further 4–10 days (Figure 3A). To answer whether the corrected ELANE gene restores granulocytic development, we analyzed CD11b−CD15+ promyelocytes by flow cytometry after 3 days of culture in the presence of G-CSF. As expected, the ELANE-corrected HSPCs (with either sgELANE-ex4 or sgELANE-L172P) were able to differentiate into CD11b−CD15+ promyelocytes at a level comparable to HD-HSPC-derived myeloid cultures (∼50%). In contrast, the SCN-HSPC-derived progenitor cells showed strongly impaired promyelocyte differentiation (∼10%) (Figure 3B). Consistent with previous publications,44,45 we also observed an increased percentage of CD14+ monocytes/macrophages in the SCN-HSPC-derived myeloid cultures (∼68% versus ∼22%) (Figure 3B). Using annexin V staining, we detected apoptotic promyelocytes. Fluorescence-activated cell sorting (FACS) analyses showed significantly elevated frequencies of early (annexin V+DAPI−) and late (annexin V+DAPI+) apoptosis in SCN-HSPC-derived promyelocytes compared with HD-HSPC-derived promyelocytes. ELANE gene correction rescued this phenotype and prevented increased apoptosis (Figure 3C). As a result, while SCN-HSPCs showed impaired neutrophil differentiation (< 5%), the ELANE-corrected HSPCs regained the ability to differentiate into CD11bintCD15+CD16+CD66b+ mature neutrophils46 with the same efficiency as the HD-HSPCs (∼40%) (Figure 3D; Figures S6A–S6C). Notably, the ELANE-repaired neutrophils expressed NE protein at a level similar to HD-derived neutrophils (Figure 3E). The mature, edited neutrophils showed the typical morphology with highly lobulated nuclei (Figure 3F). Thus, ELANE correction in SCN-HSPCs restored the ability of these cells to differentiate into mature neutrophils in vitro.
Figure 3.

ELANE-Corrected SCN-HSPCs Restore Neutrophil Differentiation In Vitro
(A) Experimental scheme of in vitro neutrophil differentiation of ELANE-corrected SCN-HSPCs. Two days after AAV infection, the edited HSPCs were cultured in expansion medium supplemented with human SCF and human IL-3 (phase I). 3 days later, human G-CSF was added (phase II). Myeloid cultures were analyzed by flow cytometry at the indicated time points. As a positive control, CD34+ HSPCs from a healthy donor were used (HD-HSPCs). (B) Left: FACS data showing the percentages of CD14+ monocytes/macrophages (Mono/Mϕ) and CD15+ promyelocytes (Promye) in myeloid cultures 10 days after targeting. Right: quantification of two independent experiments. (C) Left: FACS data showing percentages of early (annexin V+DAPI−) and late (annexin V+DAPI+) apoptotic cells among CD15+ promyelocytes gated on (B). Right: quantification of two independent experiments. (D) FACS data showing the percentages of CD11bintCD15+ mature neutrophils at the indicated time points. (E) Western blot data showing expression of NE protein in RNPs/AAV6-ELANE-edited SCN- and in HD-HSPC-derived myeloid cultures at day 18 of neutrophil differentiation. NE protein was detected with both anti-human C-terminal and N-terminal NE antibodies; β-actin was detected as loading control. NE protein was not detectable in myeloid cultures derived from SCN-HSPCs that did not receive both RNPs and AAV6-ELANE donor vectors. (F) Giemsa staining of sorted CD15+CD11bint human neutrophils from the indicated experimental groups. Scale bars, 10 μm. See also Figure S6.
ELANE-Corrected Neutrophils Function Normally
To test whether ELANE-repaired mature neutrophils were equivalent to healthy donor-derived neutrophils, we assessed a variety of functional hallmarks of human neutrophils. First, we measured oxidative burst as detected by oxidation of dihydrorhodamine 123 (DHR) from reactive oxygen species (ROS) produced in response to phorbol myristate acetate (PMA). ELANE-repaired neutrophils (using either sgELANE-ex4 or sgELANE-L172P) produced ROS levels similar to HD-HSPC-derived neutrophils (Figures 4A and 4B). Second, we determined in vitro phagocytic capacity by analyzing the internalization of opsonized, fluorescent zymosan A particles. HD- and ELANE-repaired neutrophils displayed similar levels of phagocytosis as indicated by the median fluorescence intensity and percentage of zymosan+ neutrophils (Figures 4C and 4D). Third, to assess neutrophil extracellular trap (NET) formation, HD and ELANE-repaired neutrophils were stimulated for 4 h with PMA, and subsequently fixed and stained to detect myeloperoxidase (MPO) (present in granules or NETs) and DNA (present in nuclei or NETs). ELANE-repaired and HD-derived neutrophils showed a similar percentage of NETosis (Figures 4E and 4F) and a similar length of NET chromatin spreading (Figures 4G and 4H). Finally, we determined intra-phagosomal killing of Gram-negative bacteria (E. coli). Both HD- and ELANE-repaired neutrophils exhibited efficient bacterial killing as indicated by significantly decreased bacterial colony-forming units (CFU) (Figure 4I). These results confirm that mutant neutrophils upon ELANE gene correction are functionally indistinguishable from healthy donor-derived neutrophils.
Figure 4.

ELANE Correction Restores Neutrophil Functions
ELANE-repaired neutrophils were analyzed for various neutrophil functions and compared to neutrophils from healthy donors (HD neutrophils). (A) FACS data showing the percentages of neutrophils producing reactive oxygen species (ROS) after stimulation with phorbol myristate acetate (PMA) and staining with dihydrorhodamine 123 (DHR). (B) Quantification of two independent experiments. Data are represented as mean ± SD. (C) Histograms showing the percentages and fluorescence intensities of neutrophils after phagosomal uptake of antibody-opsonized Alexa Fluor 488-conjugated zymosan particles. (D) Quantification of two independent experiments. (E) Representative images of the HD- and ELANE-repaired neutrophils stimulated with solvent (DMSO) or PMA and stained for DNA with DAPI (blue). Scale bars, 10 μm. (F) Quantification of NETosis (released NETs as shown in E); each dot represents one captured image from six slides in two independent experiments. (G) Fluorescent images of the HD- and ELANE-repaired neutrophils stimulated with DMSO or PMA and stained with anti-myeloperoxidase (MPO; red) and DAPI (blue). Scale bars, 10 μm. (H) Quantification of NET chromatin spreading as shown in (G); each dot represents one captured image from six slides in two independent experiments. (I) Intra-phagosomal bacterial killing of E. coli: quantification of colony-forming units (CFU) after 0 and 60 min incubation at 37°C in the absence (Crtl) or presence of 2 × 105 the HD- or ELANE-repaired neutrophils. Data represent mean ± SD of two technical replicates.
ELANE-Corrected HSPCs Restore Neutrophil Differentiation In Vivo
To assess whether the ELANE-corrected HSPCs restore neutrophil differentiation also in vivo, we transplanted such cells (termed ELANE-repair) into irradiated humanized NOG-EXL mice, which express human IL-3 and GM-CSF (Figure 5A).47 As a negative control, mutant SCN-HSPCs were separately transplanted (termed ELANE-L172P). Prior to transplantation, the percentages of CD34+CD38- cells among the HSPCs in the ELANE-repair and ELANE-L172P groups were similar (∼62%), with an ELANEL172P correction efficiency of about 20% in the ELANE-repair group (Figures S7A and S7B). At 4 weeks post-reconstitution, human CD45+ cells were detected in the peripheral blood of both the ELANE-L172P and ELANE-repair groups. However, human CD66b+SSChi neutrophils were found only in the ELANE-repair group for up to 17 weeks after transplantation (Figures 5B and 5C). Consistent with this finding, at 20 weeks post-reconstitution, human CD45+ cells were equally detected in the bone marrow and spleen of the transplanted animals in both experimental groups. As expected, mature neutrophils were only found in the bone marrow (3.1% ± 1% of total human CD45+ cells) and the spleen (1.4% ± 0.6% of total human CD45+ cells) of the mice reconstituted with the repaired HSPCs (Figures 6A and 6B). Sanger sequencing of the ELANE gene in mature neutrophils indeed confirmed that they derived from the repaired HSPCs (Figures 6C and 6D). In contrast, human T, B, natural killer (NK) cells, and monocytes/macrophages were present at similar frequencies in the peripheral blood, bone marrow, and spleen of the recipient animals reconstituted with repaired or non-repaired HSPCs (Figures S8A–S8E).
Figure 5.

Human Neutrophils in Peripheral Blood of Recipient Mice Reconstituted with ELANE-Corrected HSPCs
(A) Experimental scheme of ELANEL172P correction in SCN-HSPCs and transplantation of these cells into humanized NOG-EXL mice. Human cells were monitored in the peripheral blood of transplanted mice every 4 weeks. (B) FACS data showing the percentage of human CD45+ cells and standard saline citrate (SSC)hiCD66b+ neutrophils in the peripheral blood of recipient animals reconstituted with ELANE-repaired (orange) or non-repaired (ELANE-L172P, black) HSPCs at 8 weeks post-transplantation. (C) Percentages of human CD45+ cells (left) and human SSChiCD66b+ neutrophils (right) in the peripheral blood of reconstituted mice at the indicated time points after transplantation. Data represent mean ± SD for three mice. See also Figures S7 and S8.
Figure 6.

ELANE-Corrected HSPCs Restore Neutrophil Differentiation In Vivo
(A) FACS data showing the frequencies of human CD45+ cells (top) or CD33+CD15+CD66b+ mature neutrophils (Neu) (bottom) in the bone marrow (left) or spleen (right) of recipients at 20 weeks post-transplantation with non-repaired (ELANE-L172P) or repaired (ELANE-repair) HSPCs. (B) Absolute numbers of human CD45+ cells or neutrophils in the bone marrow (left) or spleen (right) of transplanted animals 20 weeks after reconstitution. Data are represented as mean ± SD for three mice. ∗p < 0.05 (two-tailed Mann-Whitney test). (C) Representative sequences of the ELANE WT, Mut, and repaired (Re) alleles in human mature neutrophils isolated from recipients 20 weeks after transplantation. The WT T nucleotide is indicated in red, the Mut C nucleotide in is indicated in blue, and silent mutations are indicated in magenta. (D) Pie chart summarizing the frequencies of the ELANE WT and repaired alleles in mature neutrophils. (E) Left: FACS data showing percentages of human CD33hiCD15+ Promye and CD33loCD15+ Neu in human CD45+ cells in the bone marrow of recipients 20 weeks after transplantation. Right: quantification for three mice; data are represented as mean ± SD. (F) Left: histograms showing annexin V+ apoptotic cells in human CD33hiCD15+ promyelocytes from recipients 20 weeks after reconstitution. Right: quantification for three mice; data are represented as mean ± SD. See also Figures S7 and S8.
To determine whether the ELANE mutation causes an arrest of neutrophil differentiation at the promyelocyte stage in vivo, we analyzed the frequencies of human CD33hiCD15+ promyelocytes and CD33loCD15+ neutrophils in the bone marrow of recipient animals reconstituted with ELANE-L172P and ELANE-repair HSPCs (Figure 6E). Human promyelocytes and neutrophils were present at frequencies of 2.0% ± 0.12% and 2.6% ± 0.4%, respectively, in the bone marrow of mice reconstituted with repaired HSPCs. In contrast, significantly decreased frequencies of human promyelocytes and neutrophils were detected in the bone marrow of mice transplanted with non-repaired HSPCs (Figure 6E). Most of the SCN-HSPC-derived promyelocytes underwent apoptosis, whereas the corrected ELANE gene rescued the repaired promyelocytes by significantly decreasing the fraction of apoptotic cells (∼50% versus ∼10%) (Figure 6F). Thus, the ELANE-repaired SCN-HSPCs had lost their propensity to undergo apoptosis at the promyelocyte stage in vivo and thereby acquired the potential to differentiate into mature neutrophils in vivo.
Discussion
Approximately 60% of SCN patients harbor autosomal dominant monoallelic ELANE mutations that cause an arrest in neutrophil development, resulting in a profound deficiency of mature neutrophils.20,22 As a result, SCN patients are prone to recurrent life-threatening infections. Although administration of supraphysiological concentrations of G-CSF can override the developmental defect by compensatory mechanisms19 and helps ∼80% of SCN patients, it fails to correct the immune defense function of the mutated neutrophils48 and increases the risk of MDS/AML through mutations in the CSF3R gene (encoding the G-CSF receptor), especially in G-CSF poor responders (∼20%).4,7,11 Thus, nuclease-mediated gene correction in autologous HSPCs with subsequent transplantation represents an attractive alternative, in particular for patients who respond poorly to G-CSF.
In principle, neutrophil differentiation in SCN patients can be rescued by precise gene correction of ELANE mutations in SCN-derived HSPCs (as shown here) or by KO of the mutant ELANE allele.25 Neutrophil counts in humans carrying a genetic deletion of one ELANE allele are normal,49 and NE KO mice produce normal numbers of mature neutrophils with the ability to migrate, phagocytose, and form NETs in a model of deep vein thrombosis.26,27,50 Accordingly, a recent report indicated that CRISPR-Cas9-mediated ELANE KO in SCN patient-derived HSPCs can rescue neutrophil differentiation in vitro.25 However, this study did not address the allelic status of the edited HSPCs. The KO strategy produced neutrophils that were elastase-negative,25 suggesting that selectively removing mutated alleles while preserving intact WT alleles in the same cell may be difficult to achieve. Indeed, complete KO of ELANE in human neutrophils may prove problematic with respect to certain infections, given that NE has important and non-redundant roles in the intracellular and extracellular defense against bacterial and fungal pathogens in mice26, 27, 28, 29, 30 and humans.28,51,52
To more precisely correct autosomal dominant ELANE mutations in SCN patient-derived HSPCs and thereby cure SCN, we developed a CRISPR-Cas9-AAV6-based gene repair approach. We achieved efficient ELANE gene correction (∼40%) in SCN patient-derived HSPCs that maintained engraftment capacity upon transplantation into humanized mice. Furthermore, ELANE correction fully restored neutrophil differentiation, both in vitro and in vivo. Mature, repaired neutrophils showed normal NE expression and neutrophil functions such as ROS generation, phagocytosis, anti-bacterial activity, and NET formation.
We demonstrated the feasibility of our approach with an efficient whole-exon-based ELANE gene correction strategy as well as a gene correction strategy for single mutations that can be tailored to individual patients. In case of the whole exon-sgRNA-based strategy, which targets exon 4 and covers ∼34% of the known autosomal dominant ELANE mutations, we achieved gene correction efficiencies of up to 40%. One allele may be lost by KO, but the repaired allele is sufficient to allow mature neutrophil differentiation. In case of the ELANEL172P-specific correction, ∼56% of HSPCs can be repaired without editing the WT allele.
Regarding safety, off-target mutation frequencies at the top nine off-target sites of sgELANE-ex4 were below detection levels based on amplicon deep sequencing. In the case of sgELANE-L172P, indels were detected at the top off-target site at very low frequencies (<0.2%) and intronic. Consistent with previous publications, our study supports the notion that off-target activity can be largely avoided by the use of specific sgRNAs.53
Our experiments in NOG-EXL humanized mice indicate that the transplanted ELANE-corrected HSPCs maintain engraftment and lineage differentiation capacity.47 In the humanized mice engrafted with the repaired HSPCs, human B cells, NK cells, neutrophils, and myeloid cells were observed up to 20 weeks after transplantation, while human T cells were hardly detectable. Apart from the low T cell numbers, no abnormalities of hematopoietic development were seen in the transplanted animals until the end of the experiments. Thus, given its efficiency, safety, and functionality, ELANE gene correction as presented herein provides an alternative treatment option for SCN.
Materials and Methods
Humanized Mice
NOG-EXL mice, expressing human GM-CSF and IL-3 cytokines, were purchased from Taconic Biosciences (New York, NY, USA). All animal experiments were approved by the Institutional Animal Care and Use Committee (Berlin LaGeSo) and performed by EPO (Berlin, Germany).
Human HSPC Isolation and Culture
Human CD34+ HSPCs were isolated from mobilized peripheral blood of healthy donors or bone marrow of the SCN patient using Ficoll-Paque Plus (GE Healthcare, Chicago, IL, USA) and a human CD34 microbead kit according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). 2 × 105 CD34+ HSPCs were cultured in 1 mL of serum-free StemSpan serum-free expansion medium (SFEM) II (STEMCELL Technologies, Cologne, Germany) supplied with human SCF (100 ng/mL), human thyroperoxidase (TPO) (100 ng/mL), human fms-related tyrosine kinase 3 ligand (FLT3L) (100 ng/mL), human IL-6 (100 ng/mL) (PeproTech, Hamburg, Germany), UM171 (35 nM, STEMCELL Technologies, Cologne, Germany), and SR1 (0.75 μM, STEMCELL Technologies, Cologne, Germany).
AAV-ELANE Repair Template Cloning and Production
To generate the pAAV-ELANE donor vector, a 1.6-kb ELANE gene fragment containing exon 4 and the left and right homology arms were amplified from human genomic DNA and cloned into XhoI/KpnI sites of the pTV vector. Silent mutations and the SalI recognition site were added by overlapping PCR. The NotI-flanked ELANE fragment was cloned into the pAAV-DJ backbone vector (Cell Biolabs, San Diego, CA, USA). To produce recombinant AAV6 (rAAV6) viruses, HEK293T cells were co-transfected with pAAV-ELANE, pAAV6-Rep/Cap, and pAAV-Helper plasmids using the polyethylenimine (PEI) transfection protocol. 12 h later, the medium was replaced by DMEM+/+ supplemented with 10% fetal calf serum (FCS), 25 mM HEPES (Gibco, Carlsbad, CA, USA), and 10 μg/mL gentamycin (Lonza, Basel, Switzerland). Three days later, the cell pellet was collected and lysed by three cycles of thaw-freeze in a dry ice/ethanol bath (10 min per cycle). Then, the cell lysate was cleared by spinning at 3,500 rpm for 15 min. The cleared supernatant was transferred into new Falcon tubes and treated with DNA endonuclease Benzonase (Millipore, Darmstadt, Germany) for 1 h at 37°C. The cell lysate was spun down and the supernatant was loaded into iodixanol gradient tubes (Beckman, Krefeld, Germany) and fractionated by ultra-centrifugation at 58,000 rpm for 130 min at 18°C, using a type 70Ti rotor (Beckman, Krefeld, Germany). The 40% iodixanol layer was collected using an 18G needle and syringe. The supernatant was filtered through a 0.2-μm polyethersulfone (PES) filter and dialyzed overnight with PBS at 4°C in dialysis cassettes (Thermo Scientific, Waltham, MA, USA). Finally, the rAAV6 supernatant was concentrated using Amicon Ultra-15 centrifugal filter units (Millipore, Darmstadt, Germany). The concentration of rAAV6 viruses was measured by real-time PCR using TaqMan probes specific for the AAV inverted terminal repeat (ITR) sequence (Thermo Scientific, Waltham, MA, USA).
sgRNA, Cas9 Protein, and Antibodies
To design a universal sgRNA targeting ELANE exon 4, the coding sequence of ELANE exon 4 was mapped to the human genome (Homo sapiens, hg38) using the latest version of CrispRGold (https://crisprgold.mdc-berlin.de) to identify potential off-target sites, GC content, and folding energy between the targeting sequence and the scaffold RNA. sgRNA with the lowest off-target risk score >11, melting temperature (Tm) ≤60°C, and a scaffold-folding energy ≤20 kcal/mol was selected.42,43 Synthetic sgELANE-ex4 and sgELANE-L172P were purchased from Synthego (Menlo Park, CA, USA). Cas9 protein was purchased from Integrated DNA Technologies (Leuven, Belgium). The following antibodies were purchased from BioLegend (Koblenz, Germany): anti-human CD45-Brilliant Violet (BV)605, anti-mouse CD45-phycoerythrin (PE)/Cy7, anti-human CD19-fluorescein isothiocyanate (FITC), anti-human CD3e-allophycocyanin (APC), anti-human CD15-APC, anti-human CD11b-PE/Cy7, anti-human CD16-BV711, anti-human CD66b-FITC, anti-human CD33-BV785, anti-human CD56-BV421, anti-human CD34-APC, anti-human CD38-PE/Cy7.
RNP Electroporation and AAV6 Infection
To generate the RNP complexes, Cas9 proteins (50 pmol) were mixed with synthetic sgRNAs (100 pmol) at a 1:2 molarity ratio and incubated at 25°C for 10 min. Human CD34+ HSPCs were cultured for 3 days and then washed once with PBS. 2 × 105 CD34+ cells were suspended into 20 μL of P3 electroporation buffer (Lonza, Basel, Switzerland) containing gRNA-Cas9 RNPs. After electroporation using the DZ-100 program (4D, Lonza, Basel, Switzerland), the cells were transferred to pre-warmed SFEM II medium supplied with cytokines and placed into an incubator at 37°C and 5% CO2. 30 min later, the rAAV6 particles were added to electroporated cells at a an MOI of 1 × 105 GC/cell. The medium was changed the next day. At 3 days post-targeting, the dead cells were removed by using a dead cell removal kit according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). The living CD34+ HSPCs were analyzed or harvested for genomic DNA extraction for further analysis.
PCR, T7EI, and RFLP Assays
Genomic DNA from the targeted human CD34+ HSPCs was extracted using the QuickExtract DNA extraction kit (Epicenter, Hessisch Oldendorf, Germany) following the manufacturer’s protocol. A T7EI assay was performed as described previously.34 For the RFLP assay, the targeted fragments were amplified using the Thermococcus kodakarensis (KOD) hot start DNA polymerase (Millipore, Darmstadt, Germany) with gene-specific primers (Table S1). PCR products were purified using AMPure XP beads (Beckman Coulter, Krefeld, Germany) and digested with SalI restriction enzyme for 1 h at 37°C. Cleaved DNA fragments were separated on 1.2% agarose gel, and the DNA concentration of each band was quantified using ImageJ software (NIH, Bethesda, MD, USA). Percentages of indels and HDR were calculated as described.31
DNA Sequencing
PCR products were cloned into the pJET plasmids (Thermo Scientific, Waltham, MA, USA) following the manufacturer’s protocol. Plasmids were isolated using the NucleoSpin plasmid (Macherey-Nagel, Dueren, Germany). Plasmids were sequenced using gene-specific primers (Table S1) by the the Sanger method (LGC Genomics, Berlin, Germany).
Analysis of Off-Targets by PCR Amplicon Deep Sequencing
To perform amplicon deep sequencing for off-target analysis, nine and five of the highest risk off-target sites for sgELANE-ex4 and sgELANE-L172P were selected using CrisprGold, respectively. All off-target sites analyzed were first amplified from genomic DNA by PCR using KAPA HiFi HotStart Mix (Kapa Biosystems, Basel, Switzerland) with gene-specific primers, including overhang adaptor sequences that are comparable to IIlumina Nextera XT index adapters (Table S1), and the following PCR conditions: 95°C for 3 min, 20 cycles (98°C for 20 s, 60°C for 15 s, 72°C for 20 s), and 72°C for 1 min. PCR products were purified using AMPure XP beads, quantified using a Qubit double-stranded DNA (dsDNA) high-sensitivity (HS) assay kit (Invitrogen, Waltham, MA, USA) and normalized to 1 ng/μL. For multiplexing sequencing libraries these PCR products were indexed through a second PCR with a Nextera XT DNA library preparation kit v2 set A (Illumina, Berlin, Germany) using KAPA HiFi HotStart mix and following PCR conditions: 95°C for 3 min, 8 cycles (98°C for 20 s, 60°C for 15 s, 72°C for 20 s), and 72°C for 1 min. Indexed PCR products were cleaned using AMPure XP beads, quantified using a Qubit dsDNA HS assay kit, normalized to 10 ng/μL, and pooled. The amplicon libraries were loaded onto an Illumina MiniSeq for deep sequencing. The deep sequencing reads were filtered for quality and mapped to the amplicons using an in-house-developed tool. The indel events on the off-target site (targeting sequence) were quantified and normalized to the number of reads covering the locus.
CD34+ Cell Transplantation
At 3 days post-targeting, corrected CD34+ HSPCs were collected and washed twice with PBS. Dead cells were removed using a dead cell removal kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The living CD34+ cells were washed once with PBS. 2 × 105 CD34+ cells were intravenously injected into irradiated (1 Gy), 4-week-old NOG-EXL mice. 4, 8, 12, or 17 weeks after reconstitution, the human cells in the peripheral blood of the transplanted mice were analyzed by flow cytometry. Human cells were analyzed in the bone marrow and spleen of the recipient animals after 20 weeks of reconstitution.
In Vitro Neutrophil Differentiation
HD-HSPCs, SCN-HSPCs, or corrected SCN-HSPCs were cultured in Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS, 2 mM l-glutamine, human SCF (100 ng/mL), and human IL-3 (100 ng/mL) for 3 days. At day 4, G-CSF (20 ng/mL) was added to the previous medium. Cells were cultured for another 10 days. Mature neutrophils were determined by FACS as CD11bintCD15+CD16+CD66b+ cells and Giemsa staining to observe the lobulated nuclear morphology.
Analysis of ROS Production Using DHR Flow Cytometric Assay
To measure ROS production, 5x104 the ELANE-corrected and HD-derived neutrophils were first stained with surface makers and subsequently suspended in 200 μL of Hanks’ balanced salt solution (HBSS) (without CaCl2 and MgCl2) containing 100 μM DHR (Invitrogen, Waltham, MA, USA) and 500 U of catalase (Sigma, Munich, Germany) and incubated at 37°C for 5 min. PMA (Sigma, Munich, Germany) was added to 1 μM final concentration and followed by incubation for 15 min. The cells were immediately analyzed by flow cytometry.
Analysis of Phagocytic Capacity Using Zymosan Uptake Assay
To measure phagocytic capacity, 1 × 105 differentiated neutrophils were resuspended in 100 μL of HBSS (without CaCl2 and MgCl2) containing 1 × 106 human serum-opsonized Alexa Fluor 488-conjugated zymosan A BioParticles (Invitrogen, Waltham, MA, USA) and were incubated at 37°C for 30 min. Cells were then washed three times with PBS and resuspended in 1 mL of lyticase (100 U/mL) (Sigma, Munich, Germany) and were incubated at 37°C for 20 min to remove extracellular zymosan. To this end, these cells were washed with PBS, stained with surface markers, and analyzed by flow cytometry.
NET Formation
To quantify NETosis and measure the length of NET chromatin spreading, 2 × 104 ELANE-corrected and HD-derived neutrophils were seeded in a poly-l-lysine-coated chamber slide at 37°C for 30 min. These cells were then treated with 100 nM PMA for 4 h at 37°C. PMA-treated neutrophils were fixed by using 4% paraformaldehyde (Sigma, Munich, Germany) for 15 min at 37°C followed by permeabilizing with PBS/0.25% Triton X-100 for 15 min at room temperature (RT). The slides were stained with primary antibody rabbit anti-human MPO (1:400, Dako/Agilent Technologies, Waldbronn, Germany) at RT for 1 h. The slides were washed three times with PBS/0.05% Tween 20 and developed with secondary antibody goat anti-rabbit immunoglobulin G (IgG) Alexa Fluor 546 (1:600, Invitrogen, Waltham, MA, USA) for 1 h at RT. The slides were washed and mounted using Fluoromount aqueous mounting medium (Sigma, Munich, Germany). Nuclei and DNA were counterstained with DAPI. The stained neutrophils were captured and analyzed for NETosis and NET chromatin spreading using a Keyence BZ-X800 microscope system.
Anti-Bacterial Activity of Neutrophils
Two strains of E. coli ATCC 8739 (ATCC, Manassas, VA, USA) and DH10B (Thermo Scientific, Waltham, MA, USA) were used for neutrophil-mediated bacterial killing assays. To this end, 2 × 105 differentiated neutrophils were mixed with 1 × 106 bacterial cells in a final volume of 200 μL of HBSS (with CaCl2 and MgCl2). Human serum (Sigma, Munich, Germany) was added to give a 1% final concentration for opsonization. At start (0) and 60 min of incubation, 10 μL of reaction mixture was removed and added to 1 mL of endotoxin-free distilled water, adjusted to pH 11, vortexed vigorously for 5 min, and diluted in PBS and 100-μL samples were plated on Luria-Bertani (LB) agar for duplicate viable counts after 18 h at 37°C. The numbers of CFU were determined at 0 and 60 min of incubation by plating serial 10-fold dilutions on LB agar.
FACS Analysis
To analyze human cells in the peripheral blood of the transplanted mice, red blood cells were lysed twice with red blood cell buffer. Single-cell suspensions were prepared from bone marrow and spleen of the recipient animals. These cells were blocked with FcγR antibodies (BioLegend, Koblenz, Germany) for 10 min at 4°C. Subsequently, these cells were stained with fluorescent-conjugated antibodies for 15 min at 4°C. The cells were finally washed with FACS buffer (PBS/1% BSA) and analyzed by BD LSRFortessa. The data were analyzed using FlowJo. In vitro- and in vivo-differentiated human promyelocytes were analyzed for apoptosis using an annexin V staining kit according to the manufacturer’s instructions (BD Pharmingen, Heidelberg, Germany). By gating on CD11b−CD15+ population in vitro, early and late apoptosis levels were determined as annexin V DAPI− and annexin V+DAPI+, respectively.
Western Blotting
To detect NE protein in the differentiated neutrophils, SCN, edited (RNPs only or RNPs/AAV6-ELANE), and HD-HSPC-derived myeloid cells were harvested and lysed at the end of in vitro neutrophil differentiation. The lysates were then run on SDS-PAGE gel and transferred into a polyvinylidene fluoride (PVDF) membrane. The NE protein was developed with anti-human C-terminal (R&D Systems, Minneapolis, MN, USA) and anti-human N-terminal (Sigma, Munich, Germany) NE antibodies; β-actin (Sigma, Munich, Germany) was used for loading controls.
Statistical Analysis
Statistical tests were performed using Prism 7.0 (GraphPad, San Diego, CA, USA) using a two-tailed Mann-Whitney test or two-way ANOVA (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Author Contributions
V.T.C. and K.R. initiated and designed the project. N.T.T., A.W.-G., M.S., and V.T.C. performed experiments. N.T.T. and V.T.C. analyzed and interpreted the experimental data. R.G. analyzed the off-target data. R.K. and G.S. provided materials and reagents. V.T.C., N.T.T., R.G., and K.R. wrote the paper. V.T.C., N.T.T., C.K., and K.R. carried out the revision.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank the families participating in this study and their involvement. We thank H.P. Rahn (FACS core facility, Max-Delbrück-Center for Molecular Medicine, Berlin, Germany) for excellent FACS-related support and K. Petsch for excellent technical support. This work was supported by the European Research Council (ERC Advanced Grant 268921 to K.R.) and by the Helmholtz-Gemeinschaft, Zukunftsthema “Immunology and Inflammation” (ZT-0027).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.08.004.
Contributor Information
Klaus Rajewsky, Email: klaus.rajewsky@mdc-berlin.de.
Van Trung Chu, Email: vantrung.chu@mdc-berlin.de.
Supplemental Information
References
- 1.Bellanné-Chantelot C., Clauin S., Leblanc T., Cassinat B., Rodrigues-Lima F., Beaufils S., Vaury C., Barkaoui M., Fenneteau O., Maier-Redelsperger M. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004;103:4119–4125. doi: 10.1182/blood-2003-10-3518. [DOI] [PubMed] [Google Scholar]
- 2.Lebel A., Yacobovich J., Krasnov T., Koren A., Levin C., Kaplinsky C., Ravel-Vilk S., Laor R., Attias D., Ben Barak A. Genetic analysis and clinical picture of severe congenital neutropenia in Israel. Pediatr. Blood Cancer. 2015;62:103–108. doi: 10.1002/pbc.25251. [DOI] [PubMed] [Google Scholar]
- 3.Grann V.R., Ziv E., Joseph C.K., Neugut A.I., Wei Y., Jacobson J.S., Horwitz M.S., Bowman N., Beckmann K., Hershman D.L. Duffy (Fy), DARC, and neutropenia among women from the United States, Europe and the Caribbean. Br. J. Haematol. 2008;143:288–293. doi: 10.1111/j.1365-2141.2008.07335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg P.S., Alter B.P., Bolyard A.A., Bonilla M.A., Boxer L.A., Cham B., Fier C., Freedman M., Kannourakis G., Kinsey S., Severe Chronic Neutropenia International Registry The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107:4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welte K., Zeidler C., Dale D.C. Severe congenital neutropenia. Semin. Hematol. 2006;43:189–195. doi: 10.1053/j.seminhematol.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Skokowa J., Germeshausen M., Zeidler C., Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr. Opin. Hematol. 2007;14:22–28. doi: 10.1097/00062752-200701000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Donadieu J., Beaupain B., Mahlaoui N., Bellanné-Chantelot C. Epidemiology of congenital neutropenia. Hematol. Oncol. Clin. North Am. 2013;27:1–17. doi: 10.1016/j.hoc.2012.11.003. vii. [DOI] [PubMed] [Google Scholar]
- 8.Germeshausen M., Ballmaier M., Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: results of a long-term survey. Blood. 2007;109:93–99. doi: 10.1182/blood-2006-02-004275. [DOI] [PubMed] [Google Scholar]
- 9.Tidow N., Pilz C., Teichmann B., Müller-Brechlin A., Germeshausen M., Kasper B., Rauprich P., Sykora K.W., Welte K. Clinical relevance of point mutations in the cytoplasmic domain of the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Blood. 1997;89:2369–2375. [PubMed] [Google Scholar]
- 10.Skokowa J., Steinemann D., Katsman-Kuipers J.E., Zeidler C., Klimenkova O., Klimiankou M., Unalan M., Kandabarau S., Makaryan V., Beekman R. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood. 2014;123:2229–2237. doi: 10.1182/blood-2013-11-538025. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg P.S., Zeidler C., Bolyard A.A., Alter B.P., Bonilla M.A., Boxer L.A., Dror Y., Kinsey S., Link D.C., Newburger P.E. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br. J. Haematol. 2010;150:196–199. doi: 10.1111/j.1365-2141.2010.08216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeidler C., Welte K., Barak Y., Barriga F., Bolyard A.A., Boxer L., Cornu G., Cowan M.J., Dale D.C., Flood T. Stem cell transplantation in patients with severe congenital neutropenia without evidence of leukemic transformation. Blood. 2000;95:1195–1198. [PubMed] [Google Scholar]
- 13.Fioredda F., Iacobelli S., van Biezen A., Gaspar B., Ancliff P., Donadieu J., Aljurf M., Peters C., Calvillo M., Matthes-Martin S., Severe Aplastic Anemia the Inborn Error, and the Pediatric Disease Working Parties of the European Society for Blood and Bone Marrow Transplantation (EBMT) and Stem Cell Transplant for Immunodeficiencies in Europe (SCETIDE) Stem cell transplantation in severe congenital neutropenia: an analysis from the European Society for Blood and Marrow Transplantation. Blood. 2015;126:1885–1892, quiz 1970. doi: 10.1182/blood-2015-02-628859. [DOI] [PubMed] [Google Scholar]
- 14.Germeshausen M., Deerberg S., Peter Y., Reimer C., Kratz C.P., Ballmaier M. The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum. Mutat. 2013;34:905–914. doi: 10.1002/humu.22308. [DOI] [PubMed] [Google Scholar]
- 15.Newburger P.E., Pindyck T.N., Zhu Z., Bolyard A.A., Aprikyan A.A., Dale D.C., Smith G.D., Boxer L.A. Cyclic neutropenia and severe congenital neutropenia in patients with a shared ELANE mutation and paternal haplotype: evidence for phenotype determination by modifying genes. Pediatr. Blood Cancer. 2010;55:314–317. doi: 10.1002/pbc.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boxer L.A., Stein S., Buckley D., Bolyard A.A., Dale D.C. Strong evidence for autosomal dominant inheritance of severe congenital neutropenia associated with ELA2 mutations. J. Pediatr. 2006;148:633–636. doi: 10.1016/j.jpeds.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 17.Xia J., Bolyard A.A., Rodger E., Stein S., Aprikyan A.A., Dale D.C., Link D.C. Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br. J. Haematol. 2009;147:535–542. doi: 10.1111/j.1365-2141.2009.07888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horwitz M.S., Duan Z., Korkmaz B., Lee H.H., Mealiffe M.E., Salipante S.J. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109:1817–1824. doi: 10.1182/blood-2006-08-019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skokowa J., Dale D.C., Touw I.P., Zeidler C., Welte K. Severe congenital neutropenias. Nat. Rev. Dis. Primers. 2017;3:17032. doi: 10.1038/nrdp.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grenda D.S., Murakami M., Ghatak J., Xia J., Boxer L.A., Dale D., Dinauer M.C., Link D.C. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110:4179–4187. doi: 10.1182/blood-2006-11-057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köllner I., Sodeik B., Schreek S., Heyn H., von Neuhoff N., Germeshausen M., Zeidler C., Krüger M., Schlegelberger B., Welte K., Beger C. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006;108:493–500. doi: 10.1182/blood-2005-11-4689. [DOI] [PubMed] [Google Scholar]
- 22.Xia J., Link D.C. Severe congenital neutropenia and the unfolded protein response. Curr. Opin. Hematol. 2008;15:1–7. doi: 10.1097/MOH.0b013e3282f13cd2. [DOI] [PubMed] [Google Scholar]
- 23.Nanua S., Murakami M., Xia J., Grenda D.S., Woloszynek J., Strand M., Link D.C. Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood. 2011;117:3539–3547. doi: 10.1182/blood-2010-10-311704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garg B., Mehta H.M., Wang B., Kamel R., Horwitz M.S., Corey S.J. Inducible expression of a disease-associated ELANE mutation impairs granulocytic differentiation, without eliciting an unfolded protein response. J. Biol. Chem. 2020;295:7492–7500. doi: 10.1074/jbc.RA120.012366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nasri M., Ritter M., Mir P., Dannenmann B., Aghaallaei N., Amend D., Makaryan V., Xu Y., Fletcher B., Bernhard R. CRISPR/Cas9-mediated ELANE knockout enables neutrophilic maturation of primary hematopoietic stem and progenitor cells and induced pluripotent stem cells of severe congenital neutropenia patients. Haematologica. 2020;105:598–609. doi: 10.3324/haematol.2019.221804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belaaouaj A., McCarthy R., Baumann M., Gao Z., Ley T.J., Abraham S.N., Shapiro S.D. Mice lacking neutrophil elastase reveal impaired host defense against Gram negative bacterial sepsis. Nat. Med. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- 27.Tkalcevic J., Novelli M., Phylactides M., Iredale J.P., Segal A.W., Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–210. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- 28.Reeves E.P., Lu H., Jacobs H.L., Messina C.G., Bolsover S., Gabella G., Potma E.O., Warley A., Roes J., Segal A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 29.Hirche T.O., Benabid R., Deslee G., Gangloff S., Achilefu S., Guenounou M., Lebargy F., Hancock R.E., Belaaouaj A. Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J. Immunol. 2008;181:4945–4954. doi: 10.4049/jimmunol.181.7.4945. [DOI] [PubMed] [Google Scholar]
- 30.Benabid R., Wartelle J., Malleret L., Guyot N., Gangloff S., Lebargy F., Belaaouaj A. Neutrophil elastase modulates cytokine expression: contribution to host defense against Pseudomonas aeruginosa-induced pneumonia. J. Biol. Chem. 2012;287:34883–34894. doi: 10.1074/jbc.M112.361352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 35.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gundry M.C., Brunetti L., Lin A., Mayle A.E., Kitano A., Wagner D., Hsu J.I., Hoegenauer K.A., Rooney C.M., Goodell M.A., Nakada D. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Rep. 2016;17:1453–1461. doi: 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bak R.O., Dever D.P., Reinisch A., Cruz Hernandez D., Majeti R., Porteus M.H. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. eLife. 2017;6 doi: 10.7554/eLife.27873. e27873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.J., Mitros T., Muñoz D.P., Boffelli D. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuo C.Y., Long J.D., Campo-Fernandez B., de Oliveira S., Cooper A.R., Romero Z., Hoban M.D., Joglekar A.V., Lill G.R., Kaufman M.L. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 2018;23:2606–2616. doi: 10.1016/j.celrep.2018.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavel-Dinu M., Wiebking V., Dejene B.T., Srifa W., Mantri S., Nicolas C.E., Lee C., Bao G., Kildebeck E.J., Punjya N. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019;10:1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu V.T., Graf R., Wirtz T., Weber T., Favret J., Li X., Petsch K., Tran N.T., Sieweke M.H., Berek C. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc. Natl. Acad. Sci. USA. 2016;113:12514–12519. doi: 10.1073/pnas.1613884113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graf R., Li X., Chu V.T., Rajewsky K. sgRNA Sequence Motifs Blocking Efficient CRISPR/Cas9-Mediated Gene Editing. Cell Rep. 2019;26:1098–1103.e3. doi: 10.1016/j.celrep.2019.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nayak R.C., Trump L.R., Aronow B.J., Myers K., Mehta P., Kalfa T., Wellendorf A.M., Valencia C.A., Paddison P.J., Horwitz M.S. Pathogenesis of ELANE-mutant severe neutropenia revealed by induced pluripotent stem cells. J. Clin. Invest. 2015;125:3103–3116. doi: 10.1172/JCI80924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.L’Esperance P., Brunning R., Good R.A. Congenital neutropenia: in vitro growth of colonies mimicking the disease. Proc. Natl. Acad. Sci. USA. 1973;70:669–672. doi: 10.1073/pnas.70.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lakschevitz F.S., Hassanpour S., Rubin A., Fine N., Sun C., Glogauer M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp. Cell Res. 2016;342:200–209. doi: 10.1016/j.yexcr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 47.Ito R., Takahashi T., Katano I., Kawai K., Kamisako T., Ogura T., Ida-Tanaka M., Suemizu H., Nunomura S., Ra C. Establishment of a human allergy model using human IL-3/GM-CSF-transgenic NOG mice. J. Immunol. 2013;191:2890–2899. doi: 10.4049/jimmunol.1203543. [DOI] [PubMed] [Google Scholar]
- 48.Donini M., Fontana S., Savoldi G., Vermi W., Tassone L., Gentili F., Zenaro E., Ferrari D., Notarangelo L.D., Porta F. G-CSF treatment of severe congenital neutropenia reverses neutropenia but does not correct the underlying functional deficiency of the neutrophil in defending against microorganisms. Blood. 2007;109:4716–4723. doi: 10.1182/blood-2006-09-045427. [DOI] [PubMed] [Google Scholar]
- 49.Horwitz M.S., Laurino M.Y., Keel S.B. Normal peripheral blood neutrophil numbers accompanying ELANE whole gene deletion mutation. Blood Adv. 2019;3:2470–2473. doi: 10.1182/bloodadvances.2019000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinod K., Witsch T., Farley K., Gallant M., Remold-O’Donnell E., Wagner D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016;14:551–558. doi: 10.1111/jth.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S., Weinrauch Y., Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 52.Weinrauch Y., Drujan D., Shapiro S.D., Weiss J., Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 2002;417:91–94. doi: 10.1038/417091a. [DOI] [PubMed] [Google Scholar]
- 53.Akcakaya P., Bobbin M.L., Guo J.A., Malagon-Lopez J., Clement K., Garcia S.P., Fellows M.D., Porritt M.J., Firth M.A., Carreras A. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. doi: 10.1038/s41586-018-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.