

In this study, Bloom and Schimenti examine the response of primordial germ cells to DNA damage. Using both environmental and genetic stresses, the authors reveal the importance of the G1 checkpoint in preventing accumulation of complex mutations in the germline, and the differentiation of the DNA damage response during germ cell development.
Keywords: primordial germ cells, DNA damage, FancM, genome maintenance, fertility, transposons, cell cycle checkpoint, single-cell DNA sequencing
Abstract
Germ cells specified during fetal development form the foundation of the mammalian germline. These primordial germ cells (PGCs) undergo rapid proliferation, yet the germline is highly refractory to mutation accumulation compared with somatic cells. Importantly, while the presence of endogenous or exogenous DNA damage has the potential to impact PGCs, there is little known about how these cells respond to stressors. To better understand the DNA damage response (DDR) in these cells, we exposed pregnant mice to ionizing radiation (IR) at specific gestational time points and assessed the DDR in PGCs. Our results show that PGCs prior to sex determination lack a G1 cell cycle checkpoint. Additionally, the response to IR-induced DNA damage differs between female and male PGCs post-sex determination. IR of female PGCs caused uncoupling of germ cell differentiation and meiotic initiation, while male PGCs exhibited repression of piRNA metabolism and transposon derepression. We also used whole-genome single-cell DNA sequencing to reveal that genetic rescue of DNA repair-deficient germ cells (Fancm−/−) leads to increased mutation incidence and biases. Importantly, our work uncovers novel insights into how PGCs exposed to DNA damage can become developmentally defective, leaving only those genetically fit cells to establish the adult germline.
Specification of the germline in humans and mice occurs during embryonic development (Ginsburg et al. 1990), during which PGCs undergo a rapid expansion to populate the fetal gonad (Gomperts et al. 1994). Early in mouse embryogenesis, PGCs are specified as a group of ∼45 cells in the epiblast of 6- to 6.5-d postfertilization embryos (E6–E6.5) (Ewen and Koopman 2010). After specification, PGCs both proliferate and migrate to the location of the future gonads where they undergo roughly nine population doublings over the span of 7 d to reach a peak population of ∼25,000 cells (Nikolic et al. 2016). These PGCs form the founding germ cell population from which the entire adult germline in both females and males is established (Tam and Snow 1981).
Perturbations to PGC development, especially those that cause accumulation of mutations, can profoundly impact the function and quality of the germline at all subsequent stages of development. In particular, early mutational events in PGCs would be expanded clonally, thus pervading the adult germ cell population. Remarkably, the spontaneous mutation rate in gametes is ∼100 times lower than that of somatic cells (Milholland et al. 2017), suggesting that PGCs, and subsequent stages of gametogenesis, have a highly effective DDR. The ability to suppress mutation transmission in germ cells is essential for maintenance of the germline's genome integrity, and thus genetic stability of species and avoidance of birth defects. However, how this suppression is achieved is still incompletely understood.
Studies examining the impact of exogenous genotoxic stressors on germ cells have largely focused on postnatal germ cell development (Russell et al. 1981; Favor 1999; Rinaldi et al. 2017; Singh et al. 2018; Enguita-Marruedo et al. 2019). Fetal germ cells comprise a small population of cells that are difficult to access, but there are some reports in the literature indicating that PGCs are hypersensitive to DNA damage (Hamer and de Rooij 2018). These studies show that mutations in several DNA repair genes impact PGC development, but have subtle effects on other embryonic and postnatal cell types (Nadler and Braun 2000; Agoulnik et al. 2002; Luo et al. 2014; Luo and Schimenti 2015). Furthermore, germ cells in gastrulating mouse embryos readily undergo apoptosis in response to low-dose IR, leading to depletion of the PGC pool; however, the underlying mechanisms were not delineated (Heyer et al. 2000). During normal PGC development, some cells are lost through BAX-mediated apoptosis, implying that there are robust quality control mechanisms present and engaged in these germ cells even under physiological conditions (Rucker et al. 2000; Stallock et al. 2003). Exposure to environmental genotoxic agents during fetal development has the potential to impact not only the fetus, but also the future offspring of the fetus through its developing germline. In addition to genetic effects, intrinsic and extrinsic stressors may evoke epigenetic changes to fetal germ cells, possibly impacting fertility and causing adverse health outcomes in subsequent generations. However, our understanding of how PGCs respond to stressors remains underexplored.
DNA damage in the form of double-strand breaks (DSBs), which can arise spontaneously (for example, during DNA replication) or induced by extrinsic exposures, are particularly dangerous to the genome because they can cause gross chromosomal rearrangements and insertions/deletions (indels). Consequently, the cellular responses to DSBs, commonly induced experimentally by IR, have been studied in many contexts (Featherstone and Jackson 1999; Ciccia and Elledge 2010). While DSBs can be damaging to any cell type in the body, the potential consequences are greater for stem cell populations that would propagate mutations to all progeny cells. Therefore, the DDR in these cells is of particular importance. A tractable system for studying stem cells in mammals are mouse embryonic stem cells (mESCs), and they are highly sensitive to IR exposure compared with other well-studied cell types such as mouse embryonic fibroblasts (MEFs) (Hong and Stambrook 2004; Chuykin et al. 2008; Tichy and Stambrook 2008; Suvorova et al. 2016). Interestingly, PGCs have several properties resembling mESCs, including rapid proliferation, low mutation rate, and similar transcriptomes (Cervantes et al. 2002; Grskovic et al. 2007; Hong et al. 2007). Under proper culture conditions, mESCs can even be differentiated into PGC-like cells (PGCLCs) in just a few days (Hayashi et al. 2011). This raises the possibility that PGCs and mESCs have similar DDRs that are distinct from terminally differentiated cells, and that ESCs, which are very easily cultured, can be used as a guide for studying DDRs of other stem cells including PGCs.
In this study, we examined the PGC response to IR-induced DNA damage at two distinct stages of development: (1) when PGCs are bipotent prior to sex determination at E11.5, and (2) subsequent to the initiation of sex determination at E13.5 (Endo et al. 2019). At E11.5, female and male fetal gonads are morphologically indistinguishable from one another, but by E13.5 the gonads are morphologically distinct (Koubova et al. 2006). Additionally, the developmental trajectories of male and female PGCs begin to diverge at this time. Female germ cells begin to undergo meiotic initiation at E13.5, while male germ cells continue on a mitotic cell cycle program before becoming quiescent ∼2 d later (Anderson et al. 2008). By examining the IR-induced DDR in PGCs both before and after sex determination, we uncovered novel developmental context-dependent responses to DNA damage. We show that before sex determination, irradiated PGCs lack a G1 cell cycle checkpoint similar to mESCs. After sex determination, we show that male PGCs gain G1 checkpoint activity while female PGCs instead prematurely initiate an abortive oogonial differentiation program. We also assessed mutational burden and the role of cell cycle checkpoint in mutation prevention in an intrinsic DNA damage model, Fancm-deficient mice that exhibit p53- and p21-dependent PGC depletion in males (Luo et al. 2014). Overall, our studies reveal the importance of checkpoints in preventing accumulation of complex mutations in the germline, and the differentiation of the DDR during germ cell development.
Results
Mouse PGCs lack a G1 cell cycle checkpoint
In a proliferating population of cells, acute DNA damage can activate cell cycle checkpoints at a number of different cell cycle stages (Shaltiel et al. 2015). These checkpoints give cells a chance to respond to the damage and can lead to a shift in the population's cell cycle distribution compared with control, undamaged cells. The canonical DDR in many cell types involves activation of a checkpoint at the G1 stage of the cell cycle, but, notably, this checkpoint is absent in mouse and primate ESCs, which have a very short G1 phase (Hong and Stambrook 2004; Fluckiger et al. 2006; Hong et al. 2007). The speculated reason for this strategy is that rather than attempting repair of a mutational load sufficient to stop the cell cycle at G1 as do most somatic cells, ESCs sustaining substantial DNA damage of a cell-deleterious nature get culled subsequently by other mechanisms. Mouse neural stem and progenitor cells (NSPCs) and hematopoietic stem cells (HSCs) also do not activate a G1 cell cycle block in response to IR (Roque et al. 2012; Brown et al. 2015).
To determine whether PGCs lack a G1 DNA damage checkpoint, we exposed pregnant mice at E11.5 to 5 Gy IR, then 8 h later dissected fetal gonads for flow cytometric cell cycle analysis. The mice expressed GFP in germ cells (Szabó et al. 2002), enabling us to distinguish PGCs from gonadal somatic cells. The IR dose was chosen based upon previous studies examining IR-induced cell cycle checkpoint activation and DDRs in stem cells and PGCs (Hong and Stambrook 2004; Chuykin et al. 2008; Roque et al. 2012; Wang et al. 2014; Brown et al. 2015; Bloom and Schimenti 2020). As reported previously (Hong and Stambrook 2004), we confirmed that this level of IR exposure causes most MEFs (mouse embryonic fibroblasts) to accumulate at G1, whereas ESC cultures shift dramatically from G1 and S to G2 (Supplemental Fig. S1). Similarly, irradiated E11.5 PGCs accumulated overwhelmingly in G2 rather than G1, indicative of a functional G2/M checkpoint but an absent or weaker G1/S cell cycle checkpoint (Fig. 1A). GFP-negative gonadal somatic cells in the same samples were predominantly in G1, with or without IR exposure (Supplemental Fig. S2).
Figure 1.

PGCs lack a G1 DNA damage-responsive cell cycle checkpoint at E11.5, but E13.5 male germ cells acquire it at E13.5. (A) Representative cell cycle profiles of control and treated primordial germ cells. Percentage of cells in the indicated cell cycle stages are graphed, representing three experimental replicates. (B) Significantly enriched gene ontology terms among up-regulated genes in response to irradiation at E11.5. (C) Representative cell cycle profiles of E13.5 male germ cells. (IR) Ionizing radiation.
Irradiation leads to decreased piRNA metabolism and transposon derepression in the fetal male germline
To better understand the molecular nature of the DDR in bipotent PGCs, we performed RNA-seq on PGCs purified from irradiated embryos compared with control PGCs at the same time point in development (E11.5). Comparisons between irradiated and unirradiated samples 4 h after treatment revealed 282 differentially expressed genes with 124 genes up-regulated twofold or more and 18 genes down-regulated twofold or more (Supplemental Table S1). Only 6% of the significantly differentially expressed genes are annotated as being “responsive to DNA damage stimulus” (Supplemental Tables S1, S2), but gene ontology (GO) analysis (Mi et al. 2019) highlighted a number of significant terms among the IR-induced genes (Fig. 1B), most notably cell-to-cell communication, factors involved in stem cell population maintenance, and activation of apoptotic processes through cysteine endopeptidase activity (Earnshaw et al. 1999). Quantification of PGC numbers indicated that ∼40% of PGCs were lost in response to IR compared with unirradiated controls (Supplemental Fig. S3).
Having found that E11.5 PGCs have an impaired or absent G1 cell cycle checkpoint, we next asked whether and when a G1 checkpoint response gets established in this lineage. This would indicate that the lineage gained a more canonical DDR, typical of most differentiated cells, which can activate checkpoints in both G1 and G2. Strikingly, cell cycle profiles of male embryonic germ cells at E13.5 was indicative of functional checkpoints at both of these cell cycle stages (Fig. 1C). The presence of a robust accumulation of cells in G1 in response to irradiation (in contrast to the marked decrease in E11.5 PGCs), plus the up-regulation of genes associated with G1 checkpoint activity (Supplemental Fig. S4), indicates most of the male germ cells at this stage (but possibly not all, since there was a slight decrease of G1 cells after IR) acquired a DNA damage-responsive G1 checkpoint for the first time in their development.
To gain insight into the molecular consequences of DNA damage in these cells, we examined the transcriptomes of irradiated E13.5 male PGCs compared with unirradiated controls using RNA-seq. This revealed an up-regulation of 1987 genes and down-regulation of 537 genes (Supplemental Fig. S5; Supplemental Table S3). Whereas mRNA levels of genes associated with G1 checkpoint activity were increased (Supplemental Fig. S4), surprisingly, only 4% of up-regulated genes were annotated as being associated with the broad-based GO category “cellular response to DNA damage stimulus” (Supplemental Fig. S6; Supplemental Table S2). This finding could indicate that cells with a robust transcriptional DDR underwent apoptosis prior to collection and were therefore precluded from being sampled and/or that the transcriptional signature of the DDR is activated more immediately after the initial dose of IR.
During normal male PGC development, there is widespread demethylation of the genome (Ernst et al. 2017). This global DNA demethylation leads to derepression of silenced transposons, but also coincides with an up-regulation of piRNA signaling to repress transposon expression (Aravin et al. 2008; Rojas-Ríos and Simonelig 2018). Activation of piRNA signaling is necessary for the progression of developmentally competent fetal male germ cells in order to preserve genome integrity (Kuramochi-Miyagawa et al. 2008; Nguyen and Laird 2019). GO analysis highlighted a number of gene categories down-regulated in response to IR, revealing perturbations to critical male germ cell-specific developmental pathways related to gene silencing, cell differentiation, and piRNA signaling (Fig. 2A). We next examined the expression of genes associated with piRNA metabolism in our data set. The vast majority (all except one) were expressed at lower levels in response to IR (Fig. 2B). We then asked whether the down-regulation of piRNA metabolism led to transposon activation in the irradiated male germ cells. We observed that the majority of expressed transposons fall into families that are capable of transposition (Supplemental Fig. S7; Deniz et al. 2019). Additionally, when we compared the expression of all differentially expressed transposons with an adjusted P value < 0.05, all but one were derepressed in irradiated germ cells (Fig. 2C; Supplemental Table S4).
Figure 2.

Radiation exposure at E13.5 causes down-regulation of piRNA metabolism and derepression of transposons in male germ cells. (A) Significantly enriched GO terms among genes down-regulated in response to irradiation of E13.5 male germ cells. (B) Expression of piRNA metabolic process genes in control and irradiated E13.5 male germ cells. (C) Heat map of differentially expressed transposons between control and irradiated samples with an adjusted P-value <0.05. (D) Percentage of total mapped reads in control and irradiated samples that belong to piRNAs. (E) Classification of transposon-derived small RNAs. (F) Size distribution of small RNAs. (G) Fold change in piRNA expression in response to IR from small-RNA sequencing. The majority of the piRNAs (shown in teal) are not significantly up-regulated in response to IR. (MC) Male no IR, (MIR) male IR-treated).
Finally, we conducted small RNA sequencing to examine the expression of piRNAs. Overall, the percentage of small RNA sequencing reads mapping to piRNAs was reduced in response to IR (Fig. 2D). Consistent with prior studies on fetal piRNA signaling, the majority of piRNAs detected (∼93%) were transposon-derived in origin and associated with LINE-, LTR-, and SINE-families (Fig. 2E; Aravin et al. 2007, 2008). Examination of the size distribution of small RNAs revealed a peak in those small RNAs matching the size bound by Piwi family member, MILI (Fig. 2F; Aravin et al. 2008; Kuramochi-Miyagawa et al. 2008). Overall, the majority of differentially expressed piRNAs were down-regulated in response to IR (Fig. 2G). Therefore, induction of DNA damage in mouse fetal germ cells suppresses piRNA activity by down-regulating both piRNA biogenesis factors and the piRNAs themselves.
Although most piRNAs were less abundantly expressed, the nontransposable element (TE)-derived piRNAs were ∼2.5 times more likely to be up-regulated than down-regulated. This finding may be connected to the up-regulation of Mybl1 (A-Myb) in response to IR. Normally, Mybl1 is absent in fetal germ cells, but present in male germ cells undergoing pachytene piRNA biogenesis (Li 2013). Relevant to results presented in the following section, the enrichment of Mybl1 in irradiated fetal germ cells coincides with an increase in retinoic acid signaling associated with meiosis (Supplemental Fig. S8), and may help contextualize the up-regulation of some pachytene piRNAs in response to IR.
Irradiation of female germ cells leads to an uncoupling of oocyte differentiation and meiosis
As described above, post-sex determination male PGCs (E13.5) acquire a G1 checkpoint response as they come to the end of their highly proliferative phase. With respect to female PGCs, IR caused a marked shift toward cells with 4C DNA content (Fig. 3). However, the fate of normal female germ cells differs at E13.5 as they begin transitioning into a meiotically competent cell cycle program. We hypothesized that rather than representing a lack of a mitotic G1 checkpoint as in E11.5, that these E13.5 female germ cells might actually represent prematurely arising oocytes resulting from IR induction. Consistent with this possibility, we noted that this cell cycle profile was remarkably similar to that reported for normal E14.5 female germ cells (Miles et al. 2010).
Figure 3.
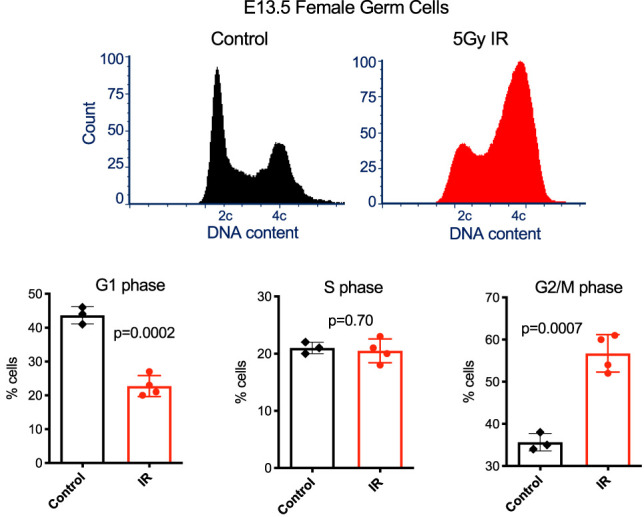
Enrichment of G2/M-phase cells in IR exposed E13.5 female germ cells. Representative cell cycle profiles of control and treated E13.5 female germ cells are presented at the top, and percentages of cells in the indicated cell cycle stages are plotted at the bottom. Data are from three to four biological replicates, as indicated in the plots.
To test this hypothesis, we performed RNA-seq on untreated or IR-exposed E13.5 female germ cells, and observed that IR caused an up-regulation of 1455 genes versus down-regulation of 152 genes (Supplemental Fig. S5; Supplemental Table S3). As with the E13.5 male germ cells, we examined the expression of genes associated with DNA damage and saw little overlap between up-regulation and DNA damage-responsive genes (∼5%) (Supplemental Fig. S6; Supplemental Table S2). While many genes were differentially expressed, enriched GO categories shared among commonly expressed up-regulated and down-regulated genes between E13.5 male and female germ cells highlighted an increase in sex differentiation and apoptotic signaling and a decrease in stem cell division (Supplemental Fig. S5C,D). We then examined the expression of pluripotency-related genes (Lesch et al. 2013; Sangrithi et al. 2017) in irradiated female germ cells. Several genes (23) associated with pluripotency were down-regulated in response to IR (Fig. 4A), consistent with either IR exposure causing premature differentiation of female PGCs, or a biased survival of more differentiated germ cells. Additionally, comparisons between irradiated and unirradiated E13.5 female germ cells 8 h after treatment also revealed an increase in retinoic acid (RA)-responsive genes (Fig. 4B). Entry into meiosis requires the completion of premeiotic DNA replication and an extended prophase I stage (Speed 1982; Soh et al. 2017). This, along with the observation that the amount of IR-induced cell death in female germ cells (∼40%, Supplemental Fig. S3) is greater than the reduction in G1-phase cells (∼25%, Fig. 4), supports the hypothesis that IR leads to premature differentiation of female germ cells rather than merely differential survival of a subset of germ cells that may have entered meiosis. Moreover, initiation of the meiotic program in female germ cells occurs in both a spatial and temporal anterior to posterior wave of RA signaling in the fetal gonad (Koubova et al. 2006). Therefore, based on the increased expression of RA-responsive genes and the dramatic upsurge in G2/M-phase cells (Fig. 3D), we hypothesized that IR stimulates RA-associated gene expression, which in turn stimulates meiotic entry. To explore this possibility, we took advantage of a published data set where the embryonic ovary was dissected into thirds at E14.5 and RNA-seq performed on the anterior and posterior portions (Soh et al. 2015). With the caveat that timing of the RA wave can vary between different strains of mice (Borum 1961; Kolas et al. 2005), comparison with the published data set revealed that gene expression in irradiated fetal germ cells is more similar to the portion of the embryonic gonad that has been exposed to RA and initiated meiotic entry (Fig. 4C; Supplemental Table S7).
Figure 4.

Irradiation exposure at E13.5 leads to premature retinoic acid (RA) signaling and meiotic entry disruption in female germ cells. (A) Expression of pluripotency-associated genes in control and irradiated E13.5 female germ cells. (B) Expression of RA-responsive genes in control and irradiated E13.5 female germ cells. (C) Expression of genes associated with spatial development of the fetal ovary in control and irradiated samples. The heat map is comprised of genes up-regulated in response to RA exposure; pink indicates higher expression, and green indicates lower expression (reference data set used for comparison from Soh et al. 2015). (D) t-SNE plot representing the similarity between the control and irradiated samples compared with Stra8−/− from Soh et al. (2015). (FC) Female no IR, (FIR) female irradiated.
Upon entry into meiosis, a set of meiosis-specific genes, primarily, become highly expressed (Lesch et al. 2013; Sangrithi et al. 2017). Surprisingly, these genes in our data set are not more highly expressed in female germ cells exposed to IR (Supplemental Fig. S9; Supplemental Table S6). This result, which seemingly conflicts with the IR-induced enrichment of gene expression associated with RA signaling, led us to conclude that IR causes an uncoupling of oogonial differentiation from meiotic entry. Previous work in mice has demonstrated that oogonial differentiation and meiosis are dissociable from one another (Dokshin et al. 2013), but, importantly, our work suggests that DNA damage might trigger this dissociation in an otherwise wild-type context.
Why does IR-induced stimulation of RA signaling not lead to premature meiotic entry in E13.5 female germ cells? One possibility relates to one of the master regulators of meiotic entry, Stra8 (Koubova et al. 2014). Stra8 stands for “stimulated by retinoic acid 8” and, as the gene name implies, its activity is dependent on RA (Koubova et al. 2006). While RA activates transcription of Stra8, expression of Stra8 leads to the formation of a negative feedback loop and self-repression (Soh et al. 2015). This mechanism ensures that meiotic entry initiates only once. We hypothesized that IR-induced DNA damage stimulates Stra8 prematurely, leading to an inappropriate and irreversible repression of meiosis. If this is indeed the case, then we would predict that the irradiated germ cell samples would have a similar gene expression profile to Stra8-deficient female germ cells. Using an RNA sequencing data set of female Stra8 mutant germ cells (Soh et al. 2015), this conjecture was borne out (Fig. 4D).
Rescue of Fancm-deficient germ cells via p21-mediated checkpoint loss leads to an enrichment of complex mutations
To assess how germline mutational burden is impacted when DNA damage checkpoints are abrogated, we sought to examine mutation incidence in a PGC proliferation-defective mouse model where DNA damage arises in an endogenous setting. We chose to examine Fancm-deficient mice because this model exhibits intrinsic DNA damage and germ cell reduction could be partially rescued by deletion of p53 or the cyclin-dependent kinase inhibitor, p21 (Luo et al. 2014). Prior work examining the naturally occurring mitotic arrest of male fetal germ cells identified that up-regulation of p21 contributes to germ cell arrest (Western et al. 2008). Beginning at E12.5, Fancm mutants exhibit a cell cycle that is longer/slower compared with controls, indicating that p21-mediated partial rescue of germ cells contributes to a quickening of the cell cycle (Luo et al. 2014). Interestingly, this p21-mediated partial rescue only occurs in male germ cells (Luo et al. 2014), consistent with our findings that there is a sexually dimorphic checkpoint response after sex determination in fetal germ cells.
Fancm is the largest subunit of the Fanconi Anemia (FA) core complex, which is named after a chromosomal instability syndrome that leads to cancer predisposition, bone marrow failure, congenital abnormalities. and infertility (Joenje and Patel 2001). Importantly, a number of additional mouse models containing mutations in FA genes have also been reported to lead to PGC loss, which indicates that this pathway is critical for responding to intrinsic DNA damage in these cells (Pellas et al. 1991; Nadler and Braun 2000; Agoulnik et al. 2002; Wong et al. 2003; Kato et al. 2015; Hill and Crossan 2019). Studies in cell culture systems have shown that Fancm, and other FA genes, facilitate intra-S checkpoint activation at sites of arrested DNA replication forks, particularly in the contexts of interstrand cross-links (ICL) (Sala-Trepat et al. 2000; Deans and West 2009). Fancm has also been reported to mediate fork reversal via translocase activity when the lagging strand template is partially single-stranded and bound by the single-stranded DNA-binding protein RPA (Gari et al. 2008). Additionally, single molecule studies have indicated that Fancm may even be capable of mediating transversal of ICLs by replication forks (Huang et al. 2013).
Using CRISPR/Cas9 genome editing, we simultaneously generated Fancm and p21 null mutations on an isogenic strain background. Characterization of the Fancm mutant revealed phenotypic similarities to the previously published mutant including a partial, but significant, rescue of male germ cells in Fancm−/− p21−/− double mutants (Supplemental Fig. S10; Luo et al. 2014). To examine whether rescuing germ cell quantity through checkpoint bypass led to a decrease in germline genome quality, we compared germline mutation incidence between the single and double mutants by collecting spermatids from wild-type, p21−/−, Fancm−/−, and Fancm−/− p21−/− animals and performing whole-genome single-cell DNA sequencing on them. An increase in mutation incidence in the double mutants would indicate that removing the p21-mediated checkpoint response has a negative impact on germline genome integrity. While p21 is important primarily for the G1 cell cycle checkpoint, it also acts in conjunction with p53 at G2/M (Bunz et al. 1998). A mutational burden similar to controls would suggest that p21 loss facilitates germ cell rescue without impacting germ cell quality. Either outcome will lead to important insights regarding the relationship between cell cycle checkpoint response and germline genome maintenance.
While whole-genome single-cell sequencing can be subject to a relatively high rate of technical error (Wang and Navin 2015; Milholland et al. 2017), initial analysis of all detected variants in the cells revealed an overrepresentation of mutations in double-mutant germ cells (Fig. 5A; Supplemental Table S9). Further analysis comparing the types of mutations per genotype relative to the distribution of detected variants in each cell indicated that the point mutation frequency, while enriched in Fancm−/− p21−/− double-mutant spermatids, did not reach statistical significance when compared with other genotypes (Fig. 5B). Breakdown of point mutation type (Supplemental Fig. S11) also did not show distinctions between any of the genotypes.
Figure 5.
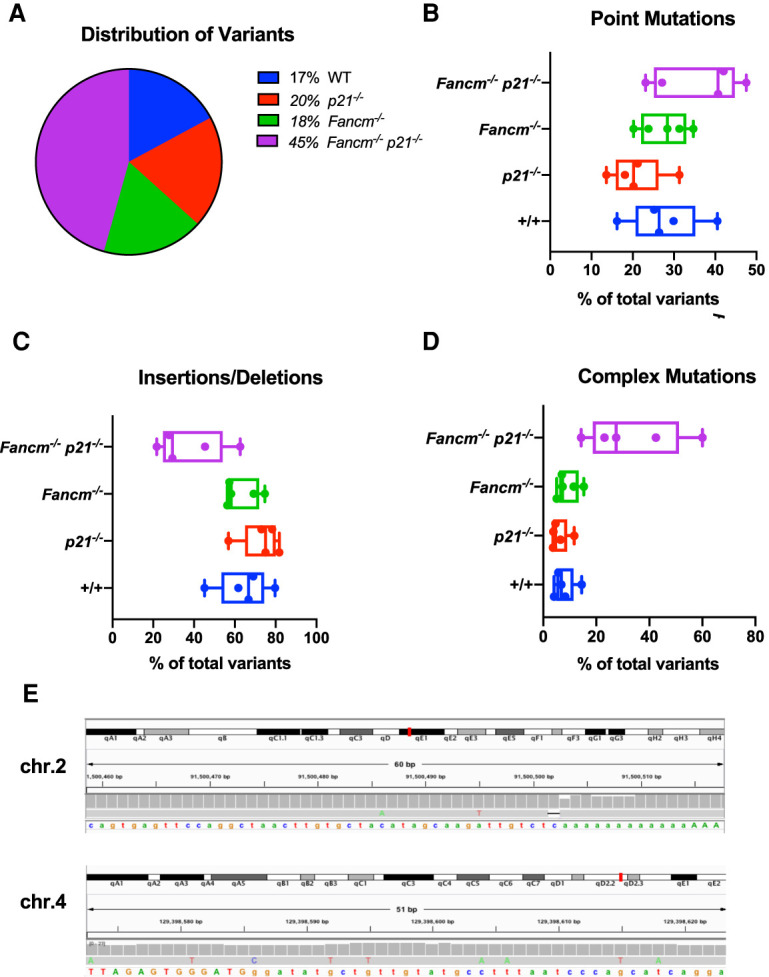
Variant detection in genetically rescued germ cells reveals an alteration mutational profile compared with controls. (A) Distribution of total variants with respect to genotype. (B) Point mutation frequency shown as a percentage of the total number of variants identified in each cell. (Kruskal-Wallis test, q = 0.09 between p21−/− and Fancm−/− p21−/−). (C) Insertion and deletion (InDel) mutation frequency shown as a percentage of the total number of variants identified in each cell. (Kruskal-Wallis test, q = 0.02 between p21−/− and Fancm−/− p21−/−) (D) Complex variant frequency shown as a percentage of the total number of variants identified in each cell. (Kruskal-Wallis test, q = 0.04 between wild type and Fancm−/− p21−/−). (E) Examples of two complex mutations from Fancm−/− p21−/− spermatids. Shown are IGV screen shots.
Next, we examined insertion/deletion (Indel) frequency and, while depleted in Fancm−/− p21−/− mutant cells compared with the other genotypes, the differences only reached the threshold for statistical significance when compared with that of p21−/− cells (Fig. 5C). Furthermore, predicted frameshift-causing variants (Supplemental Fig. S12) did not differ statistically between double-mutant cells and those from wild-type or single mutants. However, clusters of mutations in close proximity to one another were increased in Fancm−/− p21−/− spermatids compared with wild type (Fig. 5D). These complex mutation events were often comprised of Indels along with one or more base substitutions. We defined variants as “complex” if two or more mutations were within 200 nt of one another. Examples of these clustered mutations from individual Fancm−/− p21−/− germ cells are shown in Figure 5E. The enrichment of this mutation cluster signature in Fancm−/− p21−/− germ cells indicates that p21's cell cycle checkpoint function(s) is important for suppressing propagation of germ cells bearing high levels of complex mutations, or which are experiencing a high number of defective replication forks that would lead to such mutational events. It is important to note that since spermatids were sequenced, the mutations could have accumulated in any cell type in the germ lineage.
Discussion
In this study, we examined DNA damage checkpoint activity in mouse PGCs and identified developmental context-dependent responses before and after sex determination in these cells. We found similarities between the DDR of mESCs (Hong and Stambrook 2004), NSPCs (Roque et al. 2012), HSCs (Brown et al. 2015), and E11.5 PGCs. These similarities have biological and experimental implications. Regarding the former, the results suggest that key features of the DDR are similar amongst distinct stem cell types, from highly pluripotent cells (ESCs) to those dedicated to different lineages (PGCs, NSPCs, and HSCs). Experimentally, the data suggest that mESCs, which can be cultured indefinitely and are easily manipulated genetically, could serve as a model for DNA damage responses in PGCs and possibly other cell types that are not easily cultured.
Interestingly, the majority of differentially expressed genes were not associated with canonical DNA damage signaling, and elucidation of the IR-induced DDR in PGCs following sex determination highlights how perturbations to cell differentiation can be used as a highly sensitive response to preserve the genomic integrity of the final surviving germ cell pool. Derepression of transposons and down-regulation of piRNA signaling in the male germline illustrates the deleterious downstream consequences of DNA damage beyond the direct consequences of the IR itself. Our findings are consistent with a study that showed that in wild-type E13.5 male PGCs, there is a subpopulation of cells exhibiting co-occurrence of piRNA pathway down-regulation and up-regulation of apoptotic pathway-associated genes (Nguyen and Laird 2019).
Evidence for RA pathway stimulation in response to DNA damage has also been found in ESCs. In this system, DNA damage-induced RA pathway activation promoted cell differentiation through Stra6. Stra6, like Stra8, is an RA-responsive gene (Carrera et al. 2013; Serio et al. 2019). We also observed a DNA damage-induced increase in RA signaling and differentiation in female PGCs, but based on the specifics of RA-induced meiotic entry, cannot exclude the possibility that there may be at least a partial enrichment of more differentiated cells among the surviving irradiated cells. This scenario implies that differentiated female PGCs are more resistant to DNA damage than their less-differentiated RA naïve counterparts. It is possible that our timing of exogenous DNA damage with cells primed to tolerate hundreds of programmed meiotic DSBs may distinguish these cells from their less differentiated counterparts, but, importantly, we show that the irradiated cells exhibit inappropriate expression of meiosis-associated genes. Therefore, regardless of whether these germ cells represent a bias toward RA-exposed cells prior to IR along with a population induced to differentiate, they do represent a group of cells no longer developmentally competent to establish the germline.
Finally, we assessed the quality of individual haploid germ cells in a DNA replication-repair defective mutant (Fancm−/−) where the source of DNA damage is endogenous. With whole-genome single-cell DNA sequencing, we were able to highlight the potential ramifications of manipulating DNA damage checkpoints to facilitate increased germ cell survival. This is likely relevant to cells affected by extrinsic damage as well. Our results indicate that increasing cell survival in a model of germline DNA repair deficiency leads to germ cells with an increased mutational burden. Repair of Fancm-associated DNA lesions in other cell types has been shown to involve exposure of single-stranded DNA and repair by low fidelity translesion synthesis (TLS) DNA polymerases (Grompe and D'Andrea 2001). In PGCs, Rev7 (a subunit of the DNA polymerase ζ that synthesizes DNA after TLS) has been shown to be essential for cell survival, and its deficiency causes complete loss of PGCs by E13.5 (Watanabe et al. 2013). Taken together, these findings provide additional independent support for the idea that enriched variants identified in the double-mutant spermatids likely arose from TLS events (Harfe and Jinks-Robertson 2000; Stone et al. 2012).
Notably, there are many genes that lead to germ cell depletion, some via apoptosis and some by slowing DNA replication. Importantly, the spectrum of mutations in FA cells that are otherwise not predisposed or exposed to DNA damage has not been previously characterized. In this context, we found that the absence of p21 enables the increased survival of cells with complex mutations, presumably via checkpoint bypass. Although we do not know at what point(s) during male germline development the mutations arose predominantly, our prior genetic experiments showing that PGC depletion (partially rescuable by p21 deletion) was primarily responsible for germ cell loss in Fancm−/− mice (Luo et al. 2014), we believe that PGCs are the significant source of the mutation accumulation. Double-mutant male PGCs cells prior to E13.5 (and thus lacking the G1 checkpoint) would be particularly vulnerable, since FA-deficient cells are defective in the intra-S checkpoint (Dutrillaux et al. 1982; Sabatier and Dutrillaux 1988; Sala-Trepat et al. 2000), and p21 deficiency would compromise the G2 checkpoint as well (Bunz et al. 1998), allowing mutations to go unrepaired without consequence. It will be important to continue exploring the impact of these various contexts on de novo germline variation throughout germ cell development. Overall, our findings provide novel insight into how the germline minimizes mutation transmission to future generations when exposed to DNA damage and replication stress in utero.
Materials and methods
Mouse models
The use of mice in this study was approved by Cornell's Institutional Animal Care and Use Committee. B6;CBA-Tg(Pou5f1-EGFP)2Mnn/J transgenic mouse strain, commonly referred to as Oct4ΔPE-GFP, (Jackson Laboratory 004654) were used to purify PGCs. For the irradiation experiments, mice were placed in a cesium 137 irradiator with a rotating turntable and exposed to the dose of radiation specified.
Generation of Fancmem1/Jcs and p21em1/Jcs mice
p21em1/Jcs was generated using CRISPR/Cas9-mediated genome editing. The sgRNA was in vitro transcribed as described previously (Singh et al. 2014) from a DNA template ordered from Integrated DNA Technologies (IDT). See Supplemental Table S8 for the DNA template primers. Embryo microinjection in C57BL/6J zygotes was performed as described previously using 50 ng/uL sgRNA and 50 ng/uL Cas9 mRNA (TriLink Biotechnologies). The resulting 11-bp deletion was identified with Sanger sequencing of genomic DNA. Editing of the allele generated a novel BstUI restriction site that was used to distinguish between wild-type and mutant alleles after PCR amplification (see Supplemental Table S8 for genotyping primers). Generation and genotyping of Fancmem1/Jcs animals was described previously (McNairn et al. 2019).
Cell lines (related to Supplemental Fig. S1)
V6.4 mESCs (You et al. 1998) were maintained under traditional mESC culture conditions (Tremml et al. 2008). Primary C57BL/6J MEFs were isolated from E13.5 embryos in which organs were removed and the remainder of the embryo was trypsinized to make a cell suspension. Cells were cultured in media comprised of DMEM with 10% FBS, 1× nonessential amino acids, and 100 U/mL penicillin–streptomycin.
Cell cycle analysis
Fetal gonads from embryos whose mothers were either treated or untreated with radiation were dissected and pooled according to treatment condition and sex. Fetal gonads were disaggregated and dissociated into a single cell suspension using 0.25% trypsin-EDTA and 20 μg/mL DNase I for 15 min at 37°C. Trypsin was deactivated with 10% FBS. Suspensions were stained for DNA content with Hoechst 33342 (ThermoFisher Scientific 622495) and propidium iodide (PI) (Life Technologies P3566) for dead cell exclusion. Single-cell suspensions were labeled for 30 min with 100 μg of Hoechst 33342 in a 33°C water bath, shaking at 150 rpm. Prior to cell cycle analysis samples were strained through a prewetted 40-μm filter and labeled with propidium iodide (0.25 μg/mL). Cell cycle analysis was performed using FCS Express 6 software with live PGCs defined as GFP+, PI− and apoptotic PGCs defined as GFP+, PI+. Statistical comparisons of cell cycle graphs were performed using GraphPad Prism8 using unpaired nonparametric Mann–Whitney tests.
RNA-seq sample preparation and gene expression analysis
GFP+ PGCs were purified via FACS and total RNA was isolated using Trizol-LS (ThermoFisher Scientific) according to the manufacturer's instructions. RNA quality was assessed by spectrophotometry (Nanodrop) to determine concentration and chemical purity (A260/230 and A260/280 ratios) and with a fragment analyzer (Advanced Analytical) to determine RNA integrity. Prior to ribosomal RNA extraction, ERCC spike-in RNAs were added according to the manufacturer's directions (Thermo Fisher Scientific 4456740) (Jiang et al. 2011). Ribosomal RNA was subtracted by hybridization from total RNA samples using the RiboZero Magnetic Gold H/M/R Kit (Illumina) and the rRNA-subtracted samples were quantified with a Qubit 2.0 (RNA HS kit, Thermo Fisher Scientific). TruSeq-barcoded RNA-seq libraries were generated with the NEBNext Ultra II RNA library preparation kit (New England Biolabs) and each library was quantified via Qubit 2.0 (dsDNA HS kit, Thermo Fisher Scientific) prior to pooling. For analysis, reads were trimmed to remove adaptor sequences and low quality reads using Cutadapt v1.8 with parameters: -m 50 –q 20 –a AGATCGGAAGAGCACACGTCTGAACTCCAG –match-readwildcards. Reads were then mapped to the mm10 mouse reference genome/transcriptome using Tophat v2.1. For gene expression analysis, Cufflinks v2.2 (cuffnorm/cuffdiff) was used to generate FPKM values and statistical analysis of differential gene expression (Trapnell et al. 2010)
Gene ontology analyses were conducted using PANTHER classification system (Mi et al. 2019) and heat maps were generated using heatmapper.ca (Babicki et al. 2016). The t-SNE plot in Figure 5D was generated using iDEP (Ge et al. 2018).
Transposable element expression analysis
To conduct transposable element differential expression analysis, the software package TEtranscripts (Jin et al. 2015) was used with the default settings and the associated mm10 TE annotation GTF file.
Small RNA-seq sample preparation and analysis
Total RNA was isolated as described above and the presence of small RNAs (<200-nt fragments) was detected with a fragment analyzer (Advanced Analytical). TrueSeq-barcoded RNA-seq libraries were generated with the NEBNext Small RNA library preparation kit (New England Biolabs) and size selected for insert sizes ∼18–50 bp. Each library was quantified with a Qubit 2.0 (dsDNA HS kit, Thermo Fisher Scientific). For piRNA analysis, reads were trimmed using Trim Galore! and then run through TEsmall small RNA-seq pipeline (O'Neill et al. 2018) using the default settings.
Fertility tests, sperm counts, and testis histology
Methods were conducted (as described in Bloom and Schimenti 2020). Statistical comparisons were performed using GraphPad Prism8 using unpaired nonparametric Mann–Whitney tests.
Single-cell DNA sequencing and analysis
Round spermatids were isolated from mice of the following genotypes: wild-type, p21−/−, Fancm−/−, and Fancm−/− p21−/− at postnatal day 26 using fluorescence-activated cell sorting (FACS). To FACS spermatids, Vybrant DyeCycle Violet Stain was used to label cellular DNA according to the manufacturer's instructions (Thermo Fisher Scientific V35003) and PI was used to exclude dead cells as described in “Cell Cycle Analysis.” Spermatids were individually sorted into PCR tubes and flash frozen prior to DNA amplification.
Single cells were subjected to AccuSomatic single-cell multiple displacement amplification (Dong et al. 2017; Milholland et al. 2017) for whole-genome sequencing by Singulomics. Sequencing libraries were also prepared by Singulomics. Amplicons were prepared using the NEBNext DNA library preparation kit following manufacturer's recommendations. The libraries were analyzed for size distribution by an Agilent 2100 Bioanalyzer and quantified using real-time PCR. The libraries were pooled according to their effective concentrations and sequenced on Illumina NovaSeq6000 sequencer with 150-bp paired-end model using the NovaSeq6000 SP reagent kit. Approximately 1 Gb of sequencing data was generated per cell. Parental genomic DNA was isolated from spleens and subjected to 100-bp paired-end whole-genome sequencing using BGI's DNBseq (BGISEQ-500) platform.
Samples were aligned to the mm10 reference genome using BWA-MEM 0.7.17 (Li 2013) and variants were called from sorted BAM files using Platypus 0.8.1 (Rimmer et al. 2014). The following Platypus settings were applied to all samples: –assemble = 1 –assemblyRegionSize = 5000 –maxSize = 5000. BCFtools 1.9 was used to filter out variants present in both individual germ cell genomes and parental genomes in order to identify germ cell-specific variants. Statistical significance was assessed using the Kruskal–Wallis test controlling for multiple comparisons using a Benjamini false discovery rate (FDR) correction. Genome browser images were generated using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011). Supplemental Figure S13 shows the percentage of the genome sequenced at specified minimum depths according to genotype.
Data availability
Raw data files from the RNA-seq, small RNA-seq, and scDNA-seq experiments have been deposited in the GEO database with accession numbers GSE153177, GSE153194, and PRJNA638122.
Supplementary Material
Acknowledgments
We thank J. Grenier and Cornell's Transcriptional Regulation and Expression Facility for assistance with RNA sequencing and small RNA sequencing experiments (P50-HD076210), and J. Glaubitz for helpful discussions regarding single-cell DNA sequencing analysis. R. Munroe and C. Abratte of Cornell's Stem Cell and Transgenic Core Facility generated the Fancmem1/Jcs and p21em1/Jcs mouse lines with partial support from the Empire State Stem Cell Fund (contract no. C024174). This work was supported by National Institutes of Health grant T32HD057854 to J.C.B., and R01HD082568 to J.C.S.
Author contributions: J.C.B. conducted the experiments described and performed data analysis. J.C.S. supervised all aspects of the work. J.C.B. and J.C.S. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.341602.120.
References
- Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, Arango N, Chada KK, Bishop CE. 2002. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet 11: 3047–3053. 10.1093/hmg/11.24.3047 [DOI] [PubMed] [Google Scholar]
- Anderson EL, Baltus AE, Roepers-Gajadien HL, Hassold TJ, de Rooij DG, van Pelt AMM, Page DC. 2008. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc Natl Acad Sci 105: 14976–14980. 10.1073/pnas.0807297105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. 2007. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316: 744–747. 10.1126/science.1142612 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31: 785–799. 10.1016/j.molcel.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. 2016. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44: W147–W153. 10.1093/nar/gkw419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JC, Schimenti JC. 2020. A reporter mouse for in vivo detection of DNA damage in embryonic germ cells. Genesis 58: e23368 10.1002/dvg.23368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borum K. 1961. Oogenesis in the mouse. A study of the meiotic prophase. Exp Cell Res 24: 495–507. 10.1016/0014-4827(61)90449-9 [DOI] [PubMed] [Google Scholar]
- Brown A, Pospiech J, Moehrle B, Martin N, Geiger H. 2015. Assessing the roles of cell cycle checkpoints in HSC. Exp Hematol 43: S54 10.1016/j.exphem.2015.06.088 [DOI] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501. 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- Carrera S, Cuadrado-Castano S, Samuel J, Jones GDD, Villar E, Lee SW, Macip S. 2013. Stra6, a retinoic acid-responsive gene, participates in p53-induced apoptosis after DNA damage. Cell Death Differ 20: 910–919. 10.1038/cdd.2013.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes RB, Stringer JR, Shao C, Tischfield JA, Stambrook PJ. 2002. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc Natl Acad Sci 99: 3586–3590. 10.1073/pnas.062527199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuykin IA, Lianguzova MS, Pospelova TV, Pospelov VA. 2008. Activation of DNA damage response signaling in mouse embryonic stem cells. Cell Cycle 7: 2922–2928. 10.4161/cc.7.18.6699 [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans AJ, West SC. 2009. FANCM connects the genome instability disorders Bloom's syndrome and Fanconi anemia. Mol Cell 36: 943–953. 10.1016/j.molcel.2009.12.006 [DOI] [PubMed] [Google Scholar]
- Deniz Ö, Frost JM, Branco MR. 2019. Regulation of transposable elements by DNA modifications. Nat Rev Genet 20: 417–431. 10.1038/s41576-019-0117-3 [DOI] [PubMed] [Google Scholar]
- Dokshin GA, Baltus AE, Eppig JJ, Page DC. 2013. Oocyte differentiation is genetically dissociable from meiosis in mice. Nat Genet 45: 877–883. 10.1038/ng.2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, Vijg J. 2017. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods 14: 491–493. 10.1038/nmeth.4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrillaux B, Aurias A, Dutrillaux AM, Buriot D, Prieur M. 1982. The cell cycle of lymphocytes in Fanconi anemia. Hum Genet 62: 327–332. 10.1007/BF00304549 [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. 1999. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 68: 383–424. 10.1146/annurev.biochem.68.1.383 [DOI] [PubMed] [Google Scholar]
- Endo T, Mikedis MM, Nicholls PK, Page DC, de Rooij DG. 2019. Retinoic acid and germ cell development in the ovary and testis. Biomolecules 9: 775 10.3390/biom9120775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enguita-Marruedo A, Martín-Ruiz M, García E, Gil-Fernández A, Parra MT, Viera A, Rufas JS, Page J. 2019. Transition from a meiotic to a somatic-like DNA damage response during the pachytene stage in mouse meiosis. PLoS Genet 15: e1007439 10.1371/journal.pgen.1007439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C, Odom DT, Kutter C. 2017. The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat Commun 8: 1411 10.1038/s41467-017-01049-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewen KA, Koopman P. 2010. Mouse germ cell development: from specification to sex determination. Mol Cell Endocrinol 323: 76–93. 10.1016/j.mce.2009.12.013 [DOI] [PubMed] [Google Scholar]
- Favor J. 1999. Mechanisms of mutation induction in germ cells of the mouse as assessed by the specific locus test. Mutat Res 428: 227–236. 10.1016/S1383-5742(99)00050-2 [DOI] [PubMed] [Google Scholar]
- Featherstone C, Jackson SP. 1999. DNA double-strand break repair. Curr Biol 9: R759–R761. 10.1016/S0960-9822(00)80005-6 [DOI] [PubMed] [Google Scholar]
- Fluckiger A-C, Marcy G, Marchand M, Négre D, Cosset F-L, Mitalipov S, Wolf D, Savatier P, Dehay C. 2006. Cell cycle features of primate embryonic stem cells. Stem Cells 24: 547–556. 10.1634/stemcells.2005-0194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Delannoy M, Wu L, Constantinou A. 2008. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci 105: 16107–16112. 10.1073/pnas.0804777105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Son EW, Yao R. 2018. iDEP: an integrated web application for differential expression and pathway analysis of RNA-seq data. BMC Bioinformatics 19: 534 10.1186/s12859-018-2486-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg M, Snow MH, McLaren A. 1990. Primordial germ cells in the mouse embryo during gastrulation. Development 110: 521–528. [DOI] [PubMed] [Google Scholar]
- Gomperts M, Wylie C, Heasman J. 1994. Primordial germ cell migration. Ciba Found Symp 182: 121–134. [DOI] [PubMed] [Google Scholar]
- Grompe M, D'Andrea A. 2001. Fanconi anemia and DNA repair. Hum Mol Genet 10: 2253–2259. 10.1093/hmg/10.20.2253 [DOI] [PubMed] [Google Scholar]
- Grskovic M, Chaivorapol C, Gaspar-Maia A, Li H, Ramalho-Santos M. 2007. Systematic identification of cis-regulatory sequences active in mouse and human embryonic stem cells. PLoS Genet 3: e145 10.1371/journal.pgen.0030145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer G, de Rooij DG. 2018. Mutations causing specific arrests in the development of mouse primordial germ cells and gonocytes. Biol Reprod 99: 75–86. 10.1093/biolre/ioy075 [DOI] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S. 2000. DNA polymerase ζ introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol Cell 6: 1491–1499. 10.1016/S1097-2765(00)00145-3 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M. 2011. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146: 519–532. 10.1016/j.cell.2011.06.052 [DOI] [PubMed] [Google Scholar]
- Heyer BS, MacAuley A, Behrendtsen O, Werb Z. 2000. Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev 14: 2072–2084. [PMC free article] [PubMed] [Google Scholar]
- Hill RJ, Crossan GP. 2019. DNA cross-link repair safeguards genomic stability during premeiotic germ cell development. Nat Genet 51: 1283–1294. 10.1038/s41588-019-0471-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Stambrook PJ. 2004. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci 101: 14443–14448. 10.1073/pnas.0401346101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Cervantes RB, Tichy E, Tischfield JA, Stambrook PJ. 2007. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat Res 614: 48–55. 10.1016/j.mrfmmm.2006.06.006 [DOI] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. 2013. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52: 434–446. 10.1016/j.molcel.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B. 2011. Synthetic spike-in standards for RNA-seq experiments. Genome Res 21: 1543–1551. 10.1101/gr.121095.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Tam OH, Paniagua E, Hammell M. 2015. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31: 3593–3599. 10.1093/bioinformatics/btv422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joenje H, Patel KJ. 2001. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2: 446–458. 10.1038/35076590 [DOI] [PubMed] [Google Scholar]
- Kato Y, Alavattam KG, Sin H-S, Meetei AR, Pang Q, Andreassen PR, Namekawa SH. 2015. FANCB is essential in the male germline and regulates H3K9 methylation on the sex chromosomes during meiosis. Hum Mol Genet 24: 5234–5249. 10.1093/hmg/ddv244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Marcon E, Crackower MA, Höög C, Penninger JM, Spyropoulos B, Moens PB. 2005. Mutant meiotic chromosome core components in mice can cause apparent sexual dimorphic endpoints at prophase or X–Y defective male-specific sterility. Chromosoma 114: 92–102. 10.1007/s00412-005-0334-8 [DOI] [PubMed] [Google Scholar]
- Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC. 2006. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc Natl Acad Sci 103: 2474–2479. 10.1073/pnas.0510813103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koubova J, Hu Y-C, Bhattacharyya T, Soh YQS, Gill ME, Goodheart ML, Hogarth CA, Griswold MD, Page DC. 2014. Retinoic acid activates two pathways required for meiosis in mice. PLoS Genet 10: e1004541 10.1371/journal.pgen.1004541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, et al. 2008. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22: 908–917. 10.1101/gad.1640708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch BJ, Dokshin GA, Young RA, McCarrey JR, Page DC. 2013. A set of genes critical to development is epigenetically poised in mouse germ cells from fetal stages through completion of meiosis. Proc Natl Acad Sci 110: 16061–16066. 10.1073/pnas.1315204110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv arXiv:1303.3997 [Google Scholar]
- Luo Y, Schimenti JC. 2015. MCM9 deficiency delays primordial germ cell proliferation independent of the ATM pathway. Genesis 53: 678–684. 10.1002/dvg.22901 [DOI] [PubMed] [Google Scholar]
- Luo Y, Hartford SA, Zeng R, Southard TL, Shima N, Schimenti JC. 2014. Hypersensitivity of primordial germ cells to compromised replication-associated DNA repair involves ATM–p53–p21 signaling. PLoS Genet 10: e1004471 10.1371/journal.pgen.1004471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNairn AJ, Chuang C-H, Bloom JC, Wallace MD, Schimenti JC. 2019. Female-biased embryonic death from inflammation induced by genomic instability. Nature 567: 105–108. 10.1038/s41586-019-0936-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD. 2019. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14: 703–721. 10.1038/s41596-019-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles DC, van den Bergen JA, Sinclair AH, Western PS. 2010. Regulation of the female mouse germ cell cycle during entry into meiosis. Cell Cycle 9: 408–418. 10.4161/cc.9.2.10691 [DOI] [PubMed] [Google Scholar]
- Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J. 2017. Differences between germline and somatic mutation rates in humans and mice. Nat Commun 8: 15183 10.1038/ncomms15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JJ, Braun RE. 2000. Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. Genesis 27: 117–123. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Laird DJ. 2019. Apoptosis in the fetal testis eliminates developmentally defective germ cell clones. bioRxiv 10.1101/601013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic A, Volarevic V, Armstrong L, Lako M, Stojkovic M. 2016. Primordial germ cells: current knowledge and perspectives. Stem Cells Int 2016: 1741072 10.1155/2016/1741072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill K, Liao W-W, Patel A, Hammell MG. 2018. TEsmall identifies small RNAs associated with targeted inhibitor resistance in melanoma. Front Genet 9: 461 10.3389/fgene.2018.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellas TC, Ramachandran B, Duncan M, Pan SS, Marone M, Chada K. 1991. Germ-cell deficient (gcd), an insertional mutation manifested as infertility in transgenic mice. Proc Natl Acad Sci 88: 8787–8791. 10.1073/pnas.88.19.8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, WGS500 Consortium, Wilkie AOM, McVean G, Lunter G. 2014. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 46: 912–918. 10.1038/ng.3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi VD, Hsieh K, Munroe R, Bolcun-Filas E, Schimenti JC. 2017. Pharmacological inhibition of the DNA damage checkpoint prevents radiation-induced oocyte death. Genetics 206: 1823–1828. 10.1534/genetics.117.203455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Ríos P, Simonelig M. 2018. piRNAs and PIWI proteins: regulators of gene expression in development and stem cells. Development 145: dev161786 10.1242/dev.161786 [DOI] [PubMed] [Google Scholar]
- Roque T, Haton C, Etienne O, Chicheportiche A, Rousseau L, Martin L, Mouthon M-A, Boussin FD. 2012. Lack of a p21waf1/cip-dependent G1/S checkpoint in neural stem and progenitor cells after DNA damage in vivo. Stem Cells 30: 537–547. 10.1002/stem.1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker EB, Dierisseau P, Wagner KU, Garrett L, Wynshaw-Boris A, Flaws JA, Hennighausen L. 2000. Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol 14: 1038–1052. 10.1210/mend.14.7.0465 [DOI] [PubMed] [Google Scholar]
- Russell LB, Selby PB, von Halle E, Sheridan W, Valcovic L. 1981. The mouse specific-locus test with agents other than radiations: interpretation of data and recommendations for future work. Mutat Res 86: 329–354. 10.1016/0165-1110(81)90010-5 [DOI] [PubMed] [Google Scholar]
- Sabatier L, Dutrillaux B. 1988. Effect of caffeine in Fanconi anemia. I. Restoration of a normal duration of G2 phase. Hum Genet 79: 242–244. 10.1007/BF00366244 [DOI] [PubMed] [Google Scholar]
- Sala-Trepat M, Rouillard D, Escarceller M, Laquerbe A, Moustacchi E, Papadopoulo D. 2000. Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp Cell Res 260: 208–215. 10.1006/excr.2000.4994 [DOI] [PubMed] [Google Scholar]
- Sangrithi MN, Royo H, Mahadevaiah SK, Ojarikre O, Bhaw L, Sesay A, Peters AHFM, Stadler M, Turner JMA. 2017. Non-canonical and sexually dimorphic X dosage compensation states in the mouse and human germline. Dev Cell 40: 289–301.e3. 10.1016/j.devcel.2016.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio RN, Laursen KB, Urvalek AM, Gross SS, Gudas LJ. 2019. Ethanol promotes differentiation of embryonic stem cells through retinoic acid receptor-γ. J Biol Chem 294: 5536–5548. 10.1074/jbc.RA118.007153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel IA, Krenning L, Bruinsma W, Medema RH. 2015. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci 128: 607–620. 10.1242/jcs.163766 [DOI] [PubMed] [Google Scholar]
- Singh P, Schimenti JC, Bolcun-filas E. 2014. A mouse geneticist's practical guide to CRISPR applications. Genetics 199: 1–15. 10.1534/genetics.114.169771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Aggarwal LM, Parry SA, Raman MJ. 2018. Radiation dosimetry and repair kinetics of DNA damage foci in mouse pachytene spermatocyte and round spermatid stages. Mutagenesis 33: 231–239. 10.1093/mutage/gey007 [DOI] [PubMed] [Google Scholar]
- Soh YQS, Junker JP, Gill ME, Mueller JL, van Oudenaarden A, Page DC. 2015. A gene regulatory program for meiotic prophase in the fetal ovary. PLoS Genet 11: e1005531 10.1371/journal.pgen.1005531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh YQS, Mikedis MM, Kojima M, Godfrey AK, de Rooij DG, Page DC. 2017. Meioc maintains an extended meiotic prophase I in mice. PLoS Genet 13: e1006704 10.1371/journal.pgen.1006704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed RM. 1982. Meiosis in the foetal mouse ovary. I. An analysis at the light microscope level using surface-spreading. Chromosoma 85: 427–437. 10.1007/BF00330366 [DOI] [PubMed] [Google Scholar]
- Stallock J, Molyneaux K, Schaible K, Knudson CM, Wylie C. 2003. The pro-apoptotic gene Bax is required for the death of ectopic primordial germ cells during their migration in the mouse embryo. Development 130: 6589–6597. 10.1242/dev.00898 [DOI] [PubMed] [Google Scholar]
- Stone JE, Lujan SA, Kunkel TA, Kunkel TA. 2012. DNA polymerase zeta generates clustered mutations during bypass of endogenous DNA lesions in Saccharomyces cerevisiae. Environ Mol Mutagen 53: 777–786. 10.1002/em.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorova II, Grigorash BB, Chuykin IA, Pospelova TV, Pospelov VA. 2016. G1 checkpoint is compromised in mouse ESCs due to functional uncoupling of p53–p21Waf1 signaling. Cell Cycle 15: 52–63. 10.1080/15384101.2015.1120927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó PE, Hübner K, Schöler H, Mann JR. 2002. Allele-specic expression of imprinted genes in mouse migratory primordial germ cells. Mech Dev 115: 157–160. 10.1016/S0925-4773(02)00087-4 [DOI] [PubMed] [Google Scholar]
- Tam PP, Snow MH. 1981. Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. J Embryol Exp Morphol 64: 133–147. [PubMed] [Google Scholar]
- Tichy ED, Stambrook PJ. 2008. DNA repair in murine embryonic stem cells and differentiated cells. Exp Cell Res 314: 1929–1936. 10.1016/j.yexcr.2008.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremml G, Singer M, Malavarca R. 2008. Culture of mouse embryonic stem cells. Curr Protoc Stem Cell Biol 5: 1C.4.1–1C.4.19. 10.1002/9780470151808.sc01c04s5 [DOI] [PubMed] [Google Scholar]
- Wang Y, Navin NE. 2015. Advances and applications of single-cell sequencing technologies. Mol Cell 58: 598–609. 10.1016/j.molcel.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Chan KK, Ming X, Lui VCH, Poon RYC, Lo CM, Norbury C, Poon RTP. 2014. G1 checkpoint establishment in vivo during embryonic liver development. BMC Dev Biol 14: 23 10.1186/1471-213X-14-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Mii S, Asai N, Asai M, Niimi K, Ushida K, Kato T, Enomoto A, Ishii H, Takahashi M, et al. 2013. The REV7 subunit of DNA polymerase ζ is essential for primordial germ cell maintenance in the mouse. J Biol Chem 288: 10459–10471. 10.1074/jbc.M112.421966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Western PS, Miles DC, van den Bergen JA, Burton M, Sinclair AH. 2008. Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells 26: 339–347. 10.1634/stemcells.2007-0622 [DOI] [PubMed] [Google Scholar]
- Wong JCY, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. 2003. Targeted disruption of exons 1 to 6 of the Fanconi anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 12: 2063–2076. 10.1093/hmg/ddg219 [DOI] [PubMed] [Google Scholar]
- You Y, Bersgtram R, Klemm M, Nelson H, Jaenisch R, Schimenti J. 1998. Utility of C57BL/6J × 129/SvJae embryonic stem cells for generating chromosomal deletions: tolerance to γ radiation and microsatellite polymorphism. Mamm Genome 9: 232–234. 10.1007/s003359900731 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data files from the RNA-seq, small RNA-seq, and scDNA-seq experiments have been deposited in the GEO database with accession numbers GSE153177, GSE153194, and PRJNA638122.