

Abstract
SNAI2 overexpression appears to be associated with poor prognosis in breast cancer, yet it remains unclear in which breast cancer subtypes this occurs. Here we show that excess SNAI2 is associated with a poor prognosis of luminal B HER2+ breast cancers in which SNAI2 expression in the stroma but not the epithelium correlates with tumor proliferation. To determine how stromal SNAI2 might influence HER2+ tumor behavior, Snai2-deficient mice were crossed with a mouse line carrying the ErbB2/Neu protooncogene to generate HER2+ breast cancer. Tumors generated in this model expressed SNAI2 in the stroma but not the epithelium, allowing for the role of stromal SNAI2 to be studied without interference from the epithelial compartment. The absence of SNAI2 in the stroma of HER2/ERBB2+ tumors is associated with: (i) lower levels of CYCLIN D1 and reduced tumor epithelium proliferation; (ii) higher levels of AKT and a lower incidence of metastasis; (iii) lower levels of ANGIOPOIETIN-2 and more necrosis. Together, these results indicate that the loss of SNAI2 in cancer-associated fibroblasts limits the production of some cytokines, which influences AKT/ERK tumor signaling and subsequent proliferative and metastatic capacity of ERBB2+ breast cancer cells. Accordingly, SNAI2 expression in the stroma enhanced the tumorigenicity of luminal B ERBB2+ breast cancers. This work emphasizes the importance of stromal SNAI2 in breast cancer progression and patients' prognosis.
Keywords: Stromal SNAI2, Luminal B HER2+ breast cancer, Cancer-associated fibroblasts
Graphical Abstract
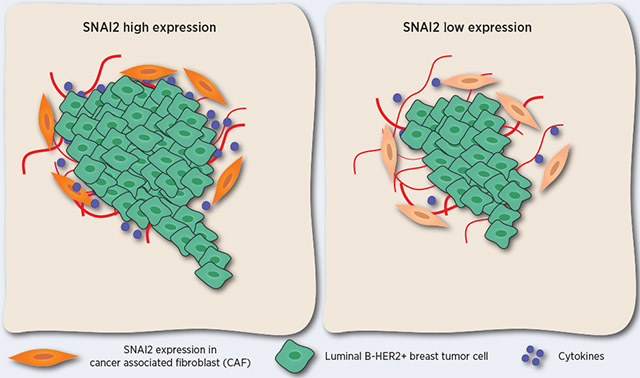
SNAI2 overexpression in the stroma and not in the epithelium is associated with a poor prognosis in luminal B-HER2+ breast cancer patients.
Introduction
Breast cancer is one of the major causes of death among women worldwide (1), with ERBB2/NEU/HER2-positive breast cancers (hereafter ERBB2+) constituting 20-30% of all breast tumors (2). Breast cancer develops in complex tissue environments, with cancer cells depending on these milieus for their sustained growth, invasion and metastasis. The tumor microenvironment contains both cellular and non-cellular components, with different cell types contributing to the cellular compartment: immune cells, endothelial cells, and fibroblasts. Fibroblasts are the most abundant cell type in the tumor-associated stroma (also known as cancer-associated fibroblast or CAFs) where they fulfill multiple roles, influencing the epithelial-mesenchymal transition (EMT) of tumor cells and their ability to acquire stem cell traits (3). CAFs are a heterogeneous population of activated fibroblasts whose activity in the stroma is associated with tumor progression and malignancy. CAFs produce different cytokines, including stromal-derived growth factor (SDF1), and their activity influences the biomechanical homeostasis of the tumor microenvironment and tumor evolution (4).
SNAI2/SLUG is a transcription factor of the SNAIL family with repressor activity (5), dampening the expression of target genes like E-CADHERIN (6), PUMA (7) and BRCA2 (8). However, SNAI2 can also increase the levels of CYCLIN D1 (9) and activate the transcription of other genes, such as VEGF (10), VIMENTIN (11) or MMP-9 (12). SNAI2 is involved in initiating the EMT by repressing E-CADHERIN expression (the CDH gene) (6, 13), and it has been associated with tumor invasion and metastasis, and with the poor prognosis of different tumors like breast cancer (14). Furthermore, SNAI2 fulfills other roles in cells, inhibiting apoptosis (7) or regulating cell movement, adhesion, proliferation (15), and the acquisition of stem cell properties (16), all critical aspects of tumor development and progression. SNAI2 has also been associated with differentiation and morphogenesis in several tissues (13). Indeed, the constitutive deletion of Snai2 in mice results in small body size (17), defects in hematopoiesis, the variable penetrance of pigmentation defects (18), cleft palate (19) and defects in the post-lactation involution of the mammary gland (20).
SNAI2 is expressed in the basal compartment of the mammary gland, and it is involved in programming the basal phenotype of breast cancer (21). We showed previously that SNAI2 is important in the development of luminal breast cancer (20). Indeed, we crossed the Snai2-deficient mouse (17) with a mouse model that overexpresses the ErbB2/Neu protooncogene under the MMTV promoter, mice in which breast cancer develops within a median of 7 months (22). In these MMTV-ErbB2 mice, SNAI2 deficiency leads to longer tumor latency and fewer breast tumors, surprisingly, without any effect on tumor dissemination. Interestingly, these effects on tumor latency and tumor load were compensated by increasing the ERBB2 oncogenic activity after pregnancy, although the incidence of metastases was reduced (20).
Here, we show that overexpression of SNAI2 in Luminal B HER2+ breast tumors can identify a subset of patients with poor prognosis. Interestingly, overexpression of SNAI2 in the stroma and not in the epithelium was associated with increased proliferation of Luminal B HER2+ human breast cancers, evident through the expression of Ki-67, a marker of poor prognosis used in the clinic (23, 24). Besides, the breast tumors that develop in MMTV-Erbb2 mice express SNAI2 in the stroma but not in the epithelium. The defects observed in the mouse model where SNAI2 is ablated in the stroma suggest that SNAI2 in the tumour microenvironment influences distinct aspects of epithelial cell behavior and evolution in breast tumors.
Materials and Methods
Clinical data analyses
The Kaplan-Meier (KM) plotter free database was used to evaluate how SNAI2 overexpression is associated with the evolution of several intrinsic subtypes of breast cancer patients (25). In addition, the compartmentalization of SNAI2 expression in HER2+ breast tumors was studied by immunohistochemistry in 50 breast tumor samples from patients (Supplementary Table S2). According to the Declaration of Helsinki, studies were performed after approval of The Bioethics Committee at the Instituto Biosanitario de Salamanca (IBSAL). Written informed consent was obtained from each patient. The Gene expression profiles from human breast tumor stroma microarrays and their associated clinical data were downloaded from the NCBI gene expression omnibus (GSE36295, GSE9014).
Mice
The mice used in this work have been described elsewhere (20) and they were housed at the Animal Research Core Facility at the University of Salamanca, maintained in ventilated filter cages under SPF conditions with food and water available ad libitum. In brief, FVB/N-Tg(MMTVneu)202Mul/J mice carrying the ErbB2 protooncogene (22) were obtained from the Jackson Laboratory and B6;129S1-Snai2tm2Grid/J Snai2-deficient mice were generated by Dr. Gridley (26). Snai2 WT ErbB2+ and Snai2 KOErbB2+ female mice were bred twice to generate the parous cohort, and after the second lactation, the cohort was followed for mammary tumor formation. All the experimental protocols were previously approved by the Institutional Animal Care and Bioethics board at the University of Salamanca. The ages of the mice that participate in different experiments are shown in Table S9. After embedding in paraffin, tumors sections were stained with hematoxylin-eosin (H/E) to evaluate their pathology under the microscope. PCR of the MMTV 3’LTR promoter was used to detect the ErbB2 transgene and the Snai2 gene in the mice, as described previously (20).
Immunohistochemistry
SNAI2 expression was evaluated in 50 HER2+ human tumor samples by immunohistochemistry (IHC) (Table S2). IHC staining was performed automatically using the Bond Polymer Refine Detection kit (BOND III: Leica, Biosystems, Leica Microsystems). The sections were probed for 60 minutes with an antibody against SNAI2 (clone: 1A6. Novus Biologicals) diluted 1:100 and then mildly counterstained with hematoxylin. Appropriate positive and negative controls were used, and SNAI2 expression was considered positive when the staining was nuclear. The expression of SNAI2 in the tumor epithelium and stroma was analyzed as described in the supplementary methods.
Immunohistochemistry in mouse tissues is described in supplementary methods
Cell cultures
Mouse Embryonic Fibroblasts (MEFs), CAFs, MDA-MB-231, and MCF7 cells were grown in complete DMEM containing glucose (4.5 g/L) and L-glutamine (SIGMA), and supplemented with penicillin (56 IU/ml), streptomycin (56 mg/L, Invitrogen) and 10% FBS (Fetal Bovine Serum: LINUS). BT474 cancer cells were grown in RPMI medium. The cells were maintained at 37 °C in a humid atmosphere containing 5% C02 and where indicated, they were treated with TGFβ1 (transforming growth factor β1, 15 ng/mL: R&D) or SDF1 (10 ng/ml: SIGMA). MEF and CAF generation is described in the supplementary methods. The hCAFs and hNFs were isolated from breast tumors (27), they were grown for three days in the absence of serum, and the culture supernatants were collected and used as conditioned medium (CM) in migration experiments. All cells were routinely tested for mycoplasma contamination. Cell lines were analyzed for authentication by the Genomics Core Facility (Instituto de Investigaciones Biomédicas “Alberto Sols” CSIC-UAM. Madrid, Spain) using the STR PROFILE DATA. STR amplification kit: GenePrintR 10 System (Promega); STR profile analysis software GeneMapper® v3.7 (Life Technologies); Genomic Analyzer System ABI 3130 XL (Applied Biosystems).
Transfection experiments
For RNAi transfection experiments, a pool of 3 constructs specifically targeting SNAI2 (Santa Cruz Biotechnology, sc-38393) and an RNAi Control (#sc-37007) were used. A second pool of RNAi anti-SNAI2 (RNAi #2) was obtained to validate the results (Sigma, #EHU048191). In general, 1×106 cells were transfected with DharmaFECT 1 Transfection Reagent (Dharmacon catalog number T-2001) and incubated for up to 72 hours before starting the experiment. In co-culture experiments, 1×106 hCAFs were transfected with the RNAi indicated and incubated for up to 72 hours. Subsequently, 5×105 cells were seeded in the lower chamber, and 5×105 MCF7, BT474, or MDA-MB-231 breast cancer cells were seeded on the inside of the transwell insert (4 μm pore size: Millipore). The transwell was incubated for 48 hours before breast cancer cells were harvested from the insert and analyzed in western blots.
Protein analyses
The proteins in the tumor samples were analyzed in western blots, as described previously (20). The antibodies used are described in the supplementary methods section.
Xenografts
The mMEF were mixed with MCF-7-ras human breast carcinoma cells expressing an activated RAS oncogene, and the mixtures were injected subcutaneously into immunodeficient nude mice. As a control, normal GFP-labeled, puromycin-resistant, immortalized human mammary stromal fibroblasts were injected subcutaneously into nude mice as pure cultures without MCF-7-RAS cells. Tumors were induced in nude mice by subcutaneous injection of 1x106 MCF7 cells and 3x106 Snai2 WTErbB2+ or Snai2 KOErbB2+ MEFs in PBS supplemented with 0.1% glucose. Tumor growth was determined using digital calipers and the tumor volume was estimated daily using the formula: Tumor volume = Length × Width2 × 0.5.
Gene expression profiling and analysis
The quality and quantity of the total RNA isolated from the cells were determined using an Agilent 2100 Bioanalyzer and NanoDrop ND-1000. Affymetrix GeneChip mouse gene 1.0 ST arrays were used according to the manufacturer’s protocol. Briefly, the robust microarray analysis (RMA) algorithm was used for background correction, intra and inter-microarray normalization, and to calculate the signal for the gene expression analysis. Once the absolute expression of each gene was calculated in each microarray (i.e., the value for each probe set), the significance analysis of microarray (SAM) method (28) was used to calculate the differential expression. This method uses permutations to provide a robust statistical inference of the most significant genes, and it includes P-values adjusted to multiple testing using a false discovery rate (FDR) (29). Gene Set Enrichment Analyses were performed using GSEA ver. 2.2.2 (30) and the hallmark collection of gene sets (31). Gene expression data for mouse breast tumors are available through Gene Expression Omnibus GSE112052.
Statistical analyses
SNAI2 overexpression was associated with patient survival using Kaplan-Meier curves, evaluating the degree of significance with a Log-Rank test. The expression of SNAI2 in the stroma was evaluated as a discrete and dichotomous variable. Thus, the presence of SNAI2 expression (positive and focal) was compared to the absence of expression (negative staining). The association of SNAI2 expression with different histopathological and clinical parameters in patients was achieved using Chi-squared and Fisher’s tests. The number of tumors, the number of lung metastases and other variables were evaluated using a Mann-Whitney U test for two groups, and with the Kruskal-Wallis test for more than two groups. Pearson’s or Spearman’s correlation tests were performed according to the distribution of the data. All these analyses were performed using the SPSS and JMP/SAS statistical packages, and P values ≤ 0.05 were considered statistically significant. We used the “glm” function of the package “stats” of R to fit generalized linear models.
Results
Snai2 overexpression in the stroma of HER2+ breast cancer is associated with poor evolution in patients
Overexpression of SNAI2 is associated with poor prognosis in breast cancer (32, 33), yet it remains unclear which breast tumor subtypes are influenced by the overexpression of SNAI2. Therefore, we studied the association of SNAI2 overexpression with tumor evolution in different breast cancer subtypes in a freely available public database (25) (Fig. 1A-F). There were no differences in tumor evolution between patients with low and high SNAI2 levels without considering any tumor subtype (Fig. 1A and Table S1). Importantly, when we examined intrinsic and Pietenpol tumor subtypes (34), we only found clear differences in the Luminal B group that were ER+ and HER2+ (P=0.06) (Fig. 1D, E and Table S1). Indeed, SNAI2 expression was not associated with prognosis in HER2-enriched (ER-) tumors (Fig. 1F and Table S1). These results indicate an association between the signaling pathways of the HER2 oncogene and the SNAI2 transcription factor in Luminal B breast tumors.
Fig. 1. Association of tumor Snai2 overexpression with evolution in different subtypes of breast cancer.

Survival curves (Kaplan-Meier) were obtained from the Kaplan-Meier plotter database (30). (A) Survival of patients with breast cancer in the whole cohort regardless of tumor subtype. (B) Survival of patients with the Luminal A subtype of tumors. (C) Patients with basal tumors. (D) Patients with luminal B tumors. (E) Patients with luminal B HER2+/ER+ tumors. (F) Patients with HER2+ enriched ER- tumors. The statistical significance of the Kaplan-Meier curves (A-F panels) was evaluated using the Log-Rank test, according to the public database website, Kaplan-Meier plotter. The number of patients (N) in each cohort is shown in Table S1. (G) Bioinformatics analysis of the SNAI2 mRNA levels of normal (N=5) and breast cancer samples (N=45). The data were collected from a public data set of breast cancer (GSE36295). (H, I) Bioinformatics analysis of SNAI2 mRNA levels in the stroma of normal (N=12) and breast cancer samples (N=111). The data were collected from a public data set of stromal gene expression in human breast cancer (GSE9014). (J) SNAI2 protein expression assessed in western blots of normal human fibroblast (hNFs) and CAFs obtained from human breast cancers (hCAFs). (K) Breast tumors with SNAI2 expression in the stroma and the epithelium assessed by immunohistochemistry (L) or only in the epithelium of the tumor. (M) There was no correlation between the immunohistochemical detection of SNAI2 in the epithelium and stroma of HER2+ tumors. (N) Expression of SNAI2 in the tumor stroma but not in the epithelium was associated with the proliferation of HER2+ tumors. Statistical significance in panels M and N was evaluated using Fisher's exact test. (O) Scheme showing in which HER2-positive tumor subtypes there is an association between the stromal expression of SNAI2 and tumor proliferation (measured as Ki-67 expression by immunohistochemistry), evaluated by generalized linear models. Scale bar 100 μm.
Bioinformatics analysis of a public human breast cancer data set (GSE36295) revealed that SNAI2 expression in breast tumors was similar to that in normal breast tissue (Fig. 1G). However, analysis of a human breast cancer data set of stromal gene expression (GSE9014) revealed that SNAI2 expression in the stroma of tumors was significantly stronger than in the normal stroma (Fig. 1H). Moreover, the differences were greater when we compared normal breast tissue with the stroma of HER2+ tumors (Fig. 1I). Furthermore, we analyzed the expression of SNAI2 in human breast cancer fibroblasts (hCAFs) (27), with SNAI2 expressed more strongly in hCAFs than in normal human fibroblasts (hNFs) (Fig. 1J), suggesting a role for SNAI2 in breast cancer CAFs.
SNAI2 expression in different tumors has been associated with poor prognosis (35), yet recently, SNAI2 overexpression in the stroma of ovarian cancer was also associated with poor prognosis (36). Therefore, in a cohort of patients with HER2+ breast cancer, we evaluated the expression of SNAI2 in the epithelial and stromal compartments of the tumor by immunohistochemistry (Fig. 1K and L, and Fig. S1), as well as its association with clinical and pathological variables of the disease (Table S2 and S3). Significantly, there was no association between epithelial and stromal SNAI2 expression (Fig. 1M). For many years, overexpression of Ki-67 has been recognized as a marker of tumor proliferation and poor prognosis in breast cancer (37). Interestingly, we noted that SNAI2 expression in the stroma but not in the epithelium was positively associated with Ki-67 expression (P = 0.03) and hence, tumor proliferation (Fig. 1N). Moreover, we only detected a positive association with SNAI2 expression in the stroma and cell proliferation in the ER+ and progesterone negative (PR-) tumor subtype (Fig. 1O and Table S3), which is the Luminal B HER2+ intrinsic subtype according to the 2013 St Gallen criteria (38). These results are in agreement with the results obtained when the prognosis was evaluated in the freely available Kaplan-Meier plotter database (25). We did not observe any other associations with either clinical or histopathological variables in our cohort.
Collectively, these data suggest that SNAI2 expression in tumor stroma can identify a subgroup of breast cancer patients with poor prognosis into the luminal B HER2+ subtype.
The absence of SNAI2 in the stroma disrupts the changes in gene expression observed in tumors when oncogenic ERBB2 activity was enhanced
The overexpression of SNAI2 in the stroma of HER2+ tumors was associated with enhanced proliferation and poor prognosis of a subset of patients (Fig. 1). Thus, to study how stromal SNAI2 influences the evolution of HER2+ tumors, we took advantage of a transgenic mouse model that overexpresses the protooncogene ErbB2/Neu under the MMTV promoter (22). MMTV-ErbB2/Neu mice develop tumors with a predominantly epithelial component and a minor stromal one, like human ductal carcinoma in situ (DCIS). In the stromal compartment there are fibroblasts and to a lesser extent, cells like endothelial cells and intratumoral leukocytes (39). The tumors that develop in these mice do not express SNAI2 in the predominant epithelial component but rather in the mammary gland (20), as also shown here (Fig. 2A and Fig. S2A). We already demonstrated that the MMTV-ErbB2 mice deficient in Snai2 displayed defects in the development of breast cancer (20). To justify the differences in tumor behavior in the absence of SNAI2 (which is not expressed in the epithelium), we assessed whether SNAI2 was present in another tumor compartment, such as the tumor stroma as seen in human tumors (Fig. 1 and Fig. S1). Indeed, SNAI2 was expressed in the stroma of these tumors when assessed by qPCR and by western blots (Fig. 2B, C), similar to hCAFs (Fig. 1J).
Fig. 2. The profile of breast tumor gene expression is affected by SNAI2 deficiency in a non-cell-autonomous manner.

(A) Detection of SNAI2 in Snai2 WTErbB2 and Snai2 KOErbB2 tumors by western blot. Mouse embryonic fibroblasts (MEFs) were used as a positive control for SNAI2 expression. (B) QPCR quantification of Snai2 in the epithelial and stromal components from Snai2 WTErbB2+ and Snai2 KOErbB2+ tumors. Snai2 RNA is not detected in Snai2 WTErbB2+ mice by QPCR after EpCAM+ cell sorting of the epithelial component, yet it is found in the stromal CD140+ cells. (C) SNAI2 protein in western blots of CAFs from mouse breast tumors. (D) Scheme representing the study design. Tumors from Snai2 WTErbB2 and Snai2 KOErbB2 mice were analyzed, both from nulliparous (ERBB2+ expression: WT N=41 and KO N=33) and parous (ERBB2++ expression) females in which the tumors developed after two pregnancies (WT, N=33, and KO, N=22). (E) The 602 gene expression signature allows unsupervised classification of the tumors from Snai2 WTErbB2 nulliparous and parous mice. (F) The signature does not permit the clustering of Snai2 KOErbB2 nulliparous and parous mice. (G) GSEA analysis: representation of the enrichment of the EMT pathway in tumors from Snai2 WTErbB2+ relative to the tumors from Snai2 KOErbB2+ nulliparous mice (above), and in tumors Snai2 KOErbB2++ relative to the tumors from Snai2 WTErbB2++ parous mice (below). (H) Conditioned medium (CM) from cultured human normal fibroblasts (hNFs) or human cancer-associated fibroblasts (hCAFs) was used to induce MDA-MB-231 cell migration (N=3). (I) Representative western blot of normal hCAFs and hCAFs in which SNAI2 was depleted by specific siRNAs, probed for SNAI2. (J) Migration of MDA-MB-231 cells in the presence of CM from control or SNAI2 depleted hCAFs (N=3). (K) Migration of BT474 cells using CM from control or SNAI2 depleted hCAFs (N=3). Statistical significance in panels I, K was evaluated using an unpaired t-test. In E and F red and blue color represent over-expressed and down-expressed genes, respectively.
SNAI2 deficiency in nulliparous mice (Snai2 KOErbB2+, hereafter) was previously seen to delay the development of ErbB2 -positive breast cancer, leading to longer tumor latency, survival and fewer tumors, yet surprisingly, no effect on metastasis was observed (19). Given the hormone-responsive nature of the MMTV promoter, we used pregnancy and lactation as a strategy to transiently induce the expression of ErbB2 and enhance its oncogenic activity (Fig. 2D) (40). Under these conditions (Snai2 KOErbB2++, hereafter), there was a reduction in the incidence (20) and the number of lung metastases in Snai2 KO ErbB2++ parous mice relative to their WT ErbB2++ counterparts (Fig. S2B, C). However, we did not observe differences in the number of metastases between Snai2 WT ErbB2++ and Snai2 KO ErbB2++ parous mice, considering only mice with lung metastases (Fig. S2D, E). Hence, although Snai2 KO ErbB2++ parous mice develop fewer tumors with the capability to metastasize, those tumors that do metastasize do so as efficiently as tumors that develop in Snai2 WT ErbB2++ parous mice.
Thus, we wondered if the absence of SNAI2 in the stroma might modify the metastatic capacity of ErbB2 tumors leading to differences in incidence. Hence, we studied the molecular mechanisms underlying the different effects of ErbB2 induction on tumor behavior in Snai2 WTErbB2++ and Snai2 KOErbB2++ mice. Microarray analyses of whole-tumor RNA from nulliparous and parous Snai2 WTErbB2 and Snai2 KOErbB2 mice were carried out. We identified 602 transcripts differentially expressed between Snai2 WTErbB2+ tumors from nulliparous mice and those with even stronger ErbB2 induction from parous mice (FDR = 0.05). This signature permits the unsupervised clustering of tumors from Snai2 WTErbB2 nulliparous and parous mice (Fig. 2E and Table S4). However, when these 602 transcripts were compared between tumors from nulliparous and parous Snai2 KOErbB2 mice, the signature was disrupted and it was not possible to classify the tumors from Snai2 KOErbB2 mice in an unsupervised manner (Fig. 2F). Indeed, Snai2 KOErbB2 tumors from nulliparous or parous mice did not cluster together, nor did metastatic and non-metastatic tumors.
Furthermore, we already published that there were no differences in the hierarchy of epithelial cell subpopulations between tumors from nulliparous and parous Snai2 WTErbB2 or Snai2 KOErbB2 mice (19). Hence, the epithelial cell composition of the tumor did not appear to determine the differences in tumor gene expression observed. Moreover, the levels of ERBB2 expression and activation were similar in Snai2 WTErbB2++ and Snai2 KOErbB2++ tumors from parous mice, excluding this as a possible explanation for the differential tumor expression (Fig. S2F and G). Thus, these results suggest that the absence of SNAI2 in the stromal microenvironment affects gene expression in these tumors.
We assessed the differences in gene expression between Snai2 WTErbB2 and Snai2 KOErbB2 mice using gene set enrichment analysis (GSEA) (30). Interestingly, we found that relative to Snai2 KOErbB2+ tumors, Snai2 WTErbB2+ nulliparous tumors were enriched in EMT-related signatures (Fig. 2G, upper graph). Surprisingly, an enrichment of the EMT signature was also detected in tumors from parous Snai2 KOErbB2++ mice despite their lower incidence of metastasis (Fig. 2G, the graphic below). However, this activation seems to be insufficient to promote metastasis in this mouse model. Therefore, the influence of stromal SNAI2 expression on the metastatic capacity of breast cancer cells was analyzed by assessing the impact of luminal hCAFs deficient for SNAI2 on breast cancer cell migration. When the effect of conditioned medium (CM) from hNFs (hNF-CM) and hCAFs (hCAF-CM) was assessed on tumor cell migration, hCAF-CM enhanced tumor cell migration significantly more than hNFs-CM (Fig. 2H). However, the depletion of SNAI2 in hCAFs using siRNAs produced CM that did not stimulate such strong migration of MDA-MB-231 and BT474 tumor cells (Fig. 2I-K). Similar results were obtained when using a different pool of two SNAI2-selective siRNAs (Fig. S2H, I).
All these results suggest that SNAI2 in the stroma could affect gene expression in ERBB2 tumors, thereby influencing the migratory capacity of breast cancer cells in a non-cell-autonomous manner.
SNAI2 deficiency in the stroma alters the proliferation, cell death, migration and the ANGIOPOIETIN levels of ERBB2 tumors
Gene ontology (GO) analysis revealed a significant enrichment of the differentially expressed genes involved in biological processes other than EMT in nulliparous Snai2 WTErbB2+ mice relative to their parous counterparts, including genes involved in the cell cycle, inflammation, apoptosis, metabolism and necrosis. Also, tumors from Snai2 KOErbB2+ mice were enriched in gene sets associated with metabolic pathways and other processes (Fig. 3A). Thus, we evaluated the effect of the absence of SNAI2 in the stroma of tumors from nulliparous and parous mice. Regarding tumor growth, no apparent differences were observed between tumors from nulliparous and parous Snai2 deficient mice (Fig. S3A). However, Ki-67 staining indicated that tumors from Snai2 WTErbB2++ parous mice had a higher proportion of proliferative cells than their Snai2 WTErbB2+ nulliparous counterparts (P = 0.05) (Fig. 3B, C). These differences were not observed in Snai2 KOErbB2 tumors indicating that cell proliferation is dampened in tumors from Snai2 KOErbB2++ parous mice (Fig. 3B, D).
Fig. 3. Histopathological features of tumors from Snai2 WTErbB2 and Snai2 KOErbB2 mice.
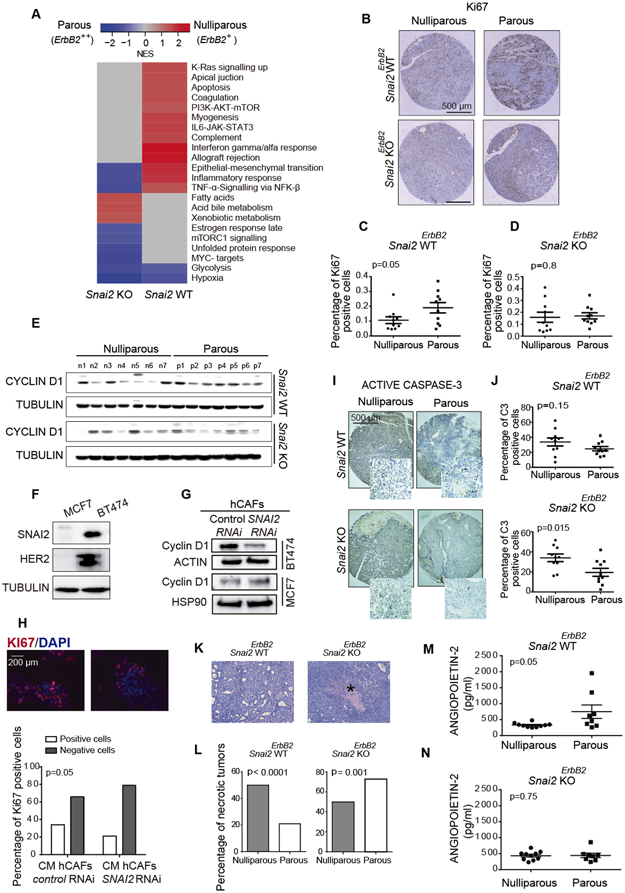
(A) GSEA analysis showed different functional groups of transcripts enriched in tumors from nulliparous and parous Snai2 WTErbB2 and Snai2 KOErbB2 mice. (B) Representative images of tumor proliferation assessed by Ki-67 immunohistochemistry in a tissue-array (N = 10 mice per group). (C, D) The proportion of Ki67-positive cells was quantified in the tissue-array (Mann-Whitney U test). (E) Western blot showing CYCLIN D1 in the different groups of mice studied (N = 7 mice per group): n, nulliparous; p, parous. (F) SNAI2 and HER2 expression in MCF7 and BT474 human breast cancer cell lines assessed by immunoblots. (G) The siRNA SNAI2-depleted hCAFs were incubated with MCF7 and BT474 cells for two days, and the CYCLIN D1 expressed by the MCF7 and BT474 cells was analyzed in western blots. (H) Conditioned medium from hCAFs was used to analyze BT474 cell proliferation through the expression of the Ki67 marker (Chi-squared test). (I) Evaluation of apoptosis through active CASPASE 3 immunohistochemistry in a tissue-array (N = 10 mice per group, Mann-Whitney U test). (J) Quantification of the active CASPASE-3 labeled cells in the tissue-array shown in panel I. (K) Detail of a micronecrotic focus in a tumor from a Snai2 KOErbB2+ parous mouse indicated with an asterisk. (L) The percentage of tumors with micronecrotic foci in the different groups of mice studied (Chi-squared test). (M, N) ANGIOPOIETIN-2 expression analyzed by multiplex bead array (Luminex), 10 tumors per group were studied. Statistical significance in panels C, D, J, M, and N was evaluated using an unpaired t-test.
Since CYCLIN D1 is essential for breast cancer cell proliferation (9), we also evaluated its expression in these tumors. While there were no differences in CYCLIN D1 expression in tumor cells from Snai2 KOErbB2++ parous and Snai2 KOErbB2+ nulliparous mice, stronger CYCLIN D1 expression was observed in tumors from Snai2 WTErbB2++ parous mice than in tumors from Snai2 WTErbB2+ nulliparous mice (Fig. 3E), consistent with their higher rate of proliferation. Since SNAI2 is not expressed in the epithelial component of these tumors (Fig. 2B, C) (20), the weaker proliferation and CYCLIN D1 expression of tumors from Snai2 KOErbB2++ parous mice could be the consequence of the Snai2 deficiency in the stromal compartment. To address this possibility, we analyzed how SNAI2 expression in hCAFs influenced the proliferation of tumor epithelial cells by co-culturing the human breast cancer cell line MCF7, which does not express SNAI2, and BT474, which express SNAI2 and the oncogene HER2 (Fig. 3F), with SNAI2-depleted or control hCAF cells (Fig. S3B). Interestingly, the reduction of SNAI2 expression in the stroma did not affect CYCLIN D1 expression in MCF7 cells but it did affect its expression in the luminal HER2+ breast cancer cell line, BT474 (Fig. 3G). Moreover, transient exposure of BT474 cells to hCAF-CM affected the proliferation of BT474 breast cancer cells (Fig. 3H).
Despite the differences in proliferation, no differences were detected in the rate of tumor growth between nulliparous and parous Snai2 WTErbB2 and Snai2 KOErbB2 mice (Fig. S3A) (20). However, while there was less apoptosis in the tumors from Snai2 KOErbB2++ parous mice than in those from their Snai2 KOErbB2+ nulliparous counterparts (P = 0.015), no such differences were evident in tumors from Snai2 WTErbB2++ parous and Snai2 WTErbB2+ nulliparous mice (Fig. 3I, J). Accordingly, the less intense proliferation and lower apoptosis in tumors from parous Snai2 KOErbB2++ mice could partly explain the similarities in the rates of tumor growth.
Foci of micronecrosis were less frequent in tumors from Snai2 KOErbB2+ nulliparous mice (52%) than in their parous counterparts (70%, P = 0.001). By contrast, they were more frequent in tumors from Snai2 WTErbB2+ nulliparous mice (55%) than in their parous counterparts (20%, P < 0.0001) (Fig. 3K, L). The higher frequency of micronecrosis in tumors from Snai2 KOErbB2++ parous mice might suggest a relative deficiency in blood flow. Indeed, as expected, there was an increase in CD31+ cells in Snai2 WTErbB2++ parous mice relative to their nulliparous counterparts (P = 0.035) (Fig. S3C). Unexpectedly, the same was observed in Snai2 KOErbB2++ parous mice relative to their nulliparous counterparts, with an even stronger statistical significance (P = 0.009) (Fig. S3D).
Increased necrosis in the context of lower tissue proliferation, coupled with an increase in CD31+ cells, might suggest a functional deficit of blood vessels in the tumors of Snai2 KOErbB2++ parous mice. Therefore, we evaluated a battery of angiogenic factors using a multiplex bead array (Luminex assay) (Table S5) and interestingly, we found that Snai2 WTErbB2++ parous tumors had more ANGIOPOIETIN-2 than their nulliparous counterparts, a factor known to be related to vessel physiology and pathophysiology (41). However, this increase was not evident in Snai2 KOErbB2 tumors (Fig. 3M, N, and Table S5). ANGIOPOIETIN-2 and other angiogenic factors can be produced by macrophages and other white blood cells (42) but paradoxically, we also observed a small decrease in keratinocyte chemoattractant (KC) levels in the tumors of Snai2 WTErbB2++ parous mice compared to their nulliparous counterparts. This difference was not found in the tumors of the Snai2 KOErbB2++ parous mice, so it did not contribute to the increased necrosis observed in these animals (Fig. S3E). .
Infiltrating white blood cells in the tumors produce angiogenic factors (43). Differences in F4/80+ macrophages and Gr1+ myeloid cell infiltration between breast tumors from Snai2 WTErbB2 and KOErbB2 nulliparous and parous mice had already been excluded (20). Nevertheless, lymphocyte infiltration has been implicated in the angiogenic capacity of tumors (43) and mice deficient in SNAI2 are defective in peripheral blood lymphocytes (44). However, when we evaluated lymphocyte infiltration in these tumors, there were no differences in tumor lymphocyte infiltration between nulliparous and parous mice (Fig. S3F).
Since SNAI1/SNAIL can partially compensate for the loss of SNAI2 activity in various contexts (19, 44, 45), we quantified SNAI1 levels in the tumors from Snai2 WTErbB2 and Snai2 KOErbB2 mice, both nulliparous and parous. SNAI1 was expressed in both the epithelium and the stroma of tumors (Fig. S3G). Interestingly, although there were no significant differences in epithelial expression between the different cohorts of mice, differences were observed at the stromal level (Table S6A). However, no associations were found between SNAI1/SNAIL expression and the evolution of breast cancer (Table S6B, C).
Together, all these results indicate that SNAI2 expression in the stroma influences the behavior of ERBB2-positive breast tumors at the histopathological level.
SNAI2 deficiency in tumor stroma modifies cell signaling in ERBB2 breast cancer
Given the histopathological differences detected in the tumors, we evaluated the role of SNAI2 at the molecular level. Since we did not previously observe differences in the total and phosphorylated ERBB2 protein in tumors from nulliparous or parous Snai2 WTErbB2 and Snai2 KOErbB2 mice (20) (Fig. S2F, G), the histopathological differences might be driven by signals downstream of ERBB2 (22). When assessed by ELISA, the expression of some primary ERBB2 targets, like pAKT1(S473) and pERK1/2, was elevated in the tumor relative to the normal mammary gland, both in Snai2 WTErbB2 and Snai2 KOErbB2 mice (Fig. 4A, B). These results are consistent with the stronger ERBB2 expression in mammary tumors relative to the mammary gland (22). More total and pERK1/2 was detected in developed tumors from parous mice than in tumors from nulliparous ones, both in the presence and absence of SNAI2 (Fig. 4C, D, and Table S7). These differences between nulliparous and parous mice were much more evident in tumors from Snai2 KOErbB2 mice (P < 0.0001 for total ERK1/2 and P = 0.0003 for pERK1/2) than in tumors from Snai2 WTErbB2 mice (P = 0.028 for total ERK1/2 and P = 0.035 for pERK1/2). Moreover, tumors from Snai2 KOErbB2++ parous mice had more total and pERK1/2 than tumors from Snai2 WTErbB2++ parous mice, a difference not evident in the nulliparous mice (Fig. S4A-D and Table S7).
Fig. 4. Signaling in tumors derived from Snai2 WTErbB2+ and Snai2 KOErbB2+ mice.
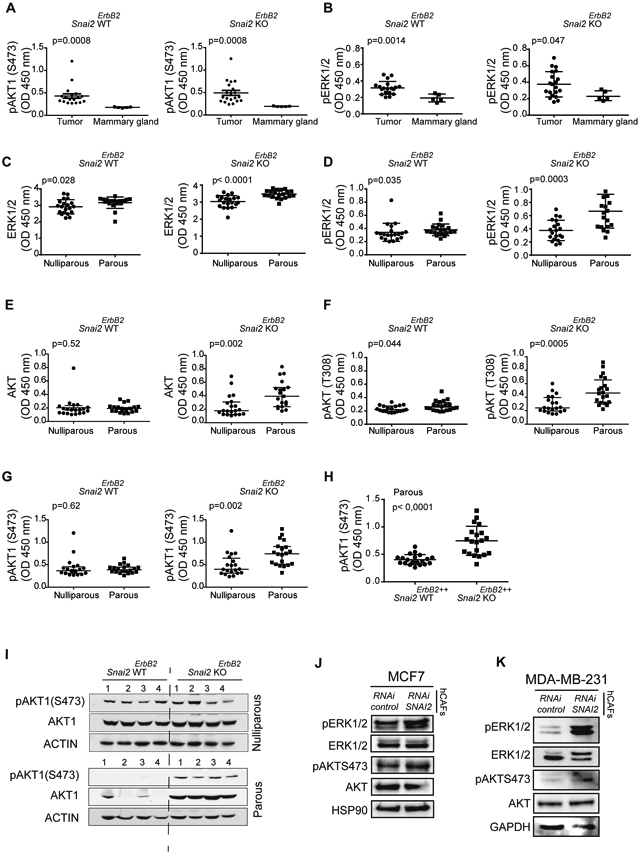
(A and B) Comparison of the signaling in mammary glands and tumors derived from Snai2 WTErbB2 and Snai2 KOErbB2 mice, quantified by ELISA (Mann-Whitney U test); (A) pAKT1 (S473) and (B) ERK1/2(T185/Y187) (pERK1/2, after that). In panels A and B, N = 20 tumors per group and N = 5 mammary glands were studied per group. (C-G) Comparison of the signaling in tumors derived from Snai2 WTErbB2 and Snai2 KOErbB2 mice: (C) Total ERK1/2; (D) pERK1/2; (E) total AKT; (F) pAKT1 (T308); and (G) pAKT1 (S473). (H) Comparison of the signaling in tumors derived from Snai2 WTErbB2++ and Snai2 KOErbB2++ parous mice. In panels C-H, the levels of signaling molecules were quantified by ELISA and compared in N = 20 tumors per group by the Mann-Whitney U test. See also Table S7. (I) Detection of total AKT1 and pAKT1 (S473) in western blots of selected tumors (lower and higher expression) to confirm the sensitivity of the technique (N = 4 mice per group). (J) hCAFs were treated with control or SNAI2 siRNA for two days and then co-cultured with MCF7 cells or (K) MDA-MB-231 in transwell inserts, for two days before analyzing the ERK and AKT signaling pathways in the tumor cells by western blots.
Regarding AKT, tumors from Snai2 KOErbB2++ parous mice had more total AKT and pAKT1(Ser473) than Snai2 WTErbB2++ parous mice (20). Here we confirmed that not only pAKT1(Ser473) but also total and pAKT(T308) were elevated in tumors from Snai2 KOErbB2++ parous mice (Fig. 4E-G and Table S7). Moreover, tumors from Snai2 KOErbB2++ parous mice had more pAKT1 than tumors from Snai2 WTErbB2++ parous mice (Fig. 4H). These results obtained by ELISA were confirmed by western blots (Fig. 4I and Fig. S4E).
To confirm the influence of SNAI2 in the stroma over the AKT/ERK signaling pathway, we co-cultured in transwell inserts MCF7 luminal human breast cancer cells that do not express SNAI2, and MDA-MB-231 and cell lines that express SNAI2, in the presence of SNAI2 siRNAs silenced hCAFs (Fig. S4F), analyzing the signaling downstream of ERBB2. We observed similar results to those obtained in the Snai2 KO ERBB2 mouse model and as such, the absence of SNAI2 in hCAFs was associated with higher levels of phosphorylated ERK1/2 and AKT in breast cancer cells (Fig. 4J, K). Similar results were obtained using a different pool of two SNAI2-selective siRNAs (Fig. S4G).
In conclusion, tumors from Snai2 KOErbB2++ parous mice had more phosphorylated ERK1/2 and AKT than those from Snai2 WTErbB2++ parous mice (Table S7), implicating some activation of the central pathways downstream of ERBB2 (22). Since tumor epithelial cells do not express SNAI2 (Fig. 2A-C and Fig. S2A), these differences must be a consequence of the absence of SNAI2 expression in the tumor microenvironment, as also indicated by our in vitro experiments (Fig. 4J, K).
SNAI2 in the stroma influences the initiation and dissemination of ERBB2 tumors
We assessed whether differences in cell signaling in tumors were associated with breast cancer behavior in the different cohorts of mice studied. Interestingly, we only observed correlations between the tumor initiation time (latency) and most of these molecules in parous Snai2 KOErbB2++ mice (Table S8). Indeed, the pERK1/2 but not the total ERK1/2 levels were positively correlated with tumor latency in parous Snai2 KOErbB2++ mice (P = 0.009) (Fig. 5A, B), whereas total and pERK1/2 levels were not correlated with tumor latency in Snai2 WTErbB2 parous and nulliparous mice, and Snai2 KOErbB2+ nulliparous mice (Table S8). Similar results were obtained when survival was used as the endpoint (Table S8 and Fig. S5A, B), as expected because latency but not the disease duration influences the survival time (19). Also, total and pAKT(T308) levels were only correlated with tumor latency in parous Snai2 KOErbB2++ mice (Fig. 5C, D, and Table S8). When we analyzed this correlation between latency and the different AKT isoforms, the pAKT1(S473) and pAKT2(S474) but not pAKT3(S472) levels were correlated with tumor latency in tumors developed in parous Snai2 KOErbB2++ mice (Fig. 5E-G).
Fig. 5. Correlation between tumor levels of total and phosphorylated AKT and ERK and tumor behavior in Snai2 KOErbB2++ parous mice.
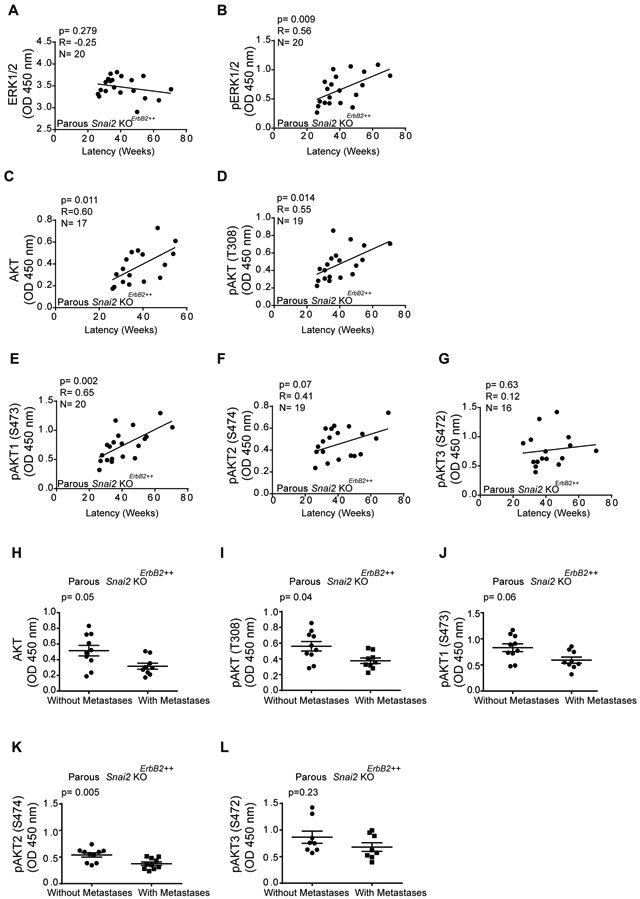
(A-G) Association between tumor latency and signaling elements in the tumor: (a) Total ERK; (B) pERK; (C) total AKT; (D) Pan-pAKT (T308); (E) pAKT1 (S473); (F) pAKT2 (S474); and (G) pAKT3 (S472). All protein isoforms were assessed by ELISA (Spearman correlation, N = 20 mice per group). See also Table S8. (H-L) Levels of pAKT isoforms in tumors from Snai2 KOErbB2++ parous mice with and without dissemination quantified by ELISA (Mann-Whitney U test, N = 10 mice per group). (H) Total AKT; (I) Pan-pAKT (T308); (J) pAKT1 (S473); (K) pAKT2 (S474); and (L) pAKT3 (S472).
The capacity of breast cancer to metastasize has been associated with limited AKT in both humans and mice (46-48). Interestingly, the highest levels of total and phospho-AKT were observed in tumors from Snai2 KOErbb2++ parous mice (Table S7). Moreover, tumors from Snai2 KOErbB2++ parous mice with elevated total AKT and pAKT(T308) did not spread to the lung, unlike the tumors with less total and pAKT (Fig. 5H, I). When we assessed which AKT isoform might be related to the metastatic behavior of the tumors, high levels of pAKT2(S474), and to a lesser extent pAKT1(S473) but not pAKT3(S472), were associated with tumor dissemination to the lung (Fig. 5J-L). These results suggest that the absence of SNAI2 in the stroma affects the ERBB2-dependent activation of AKT1/2 and the ability of tumor cells to metastasize.
SNAI2 directs cytokine production in CAFs
Since differences in the dissemination of tumors derived from Snai2 WTErbB2++ and Snai2 KOErbB2++ parous mice were observed (20), and CAFs have been implicated in tumor dissemination (49), we evaluated the activity of mouse CAFs (mCAFs) derived from tumors developed in mice deficient in Snai2. Deficiencies in cytokine production were evident in the mCAFs isolated from Snai2-lacking tumors relative to those from their WT counterparts using an antibody array (Fig. 6A). Accordingly, there was less SDF1 (CXC12) and CXCL1 in the supernatant (Fig. 6B), and minor changes to the CXCL2, IFN-γ and IL16 (Fig. S6A), all cytokines that support the growth and progression of carcinomas (3, 50). Similar results were obtained when Cxcl1 and Sdf1 mRNAs expression were analyzed in the stromal cells (CD140+/CD31−/EpCAM−) isolated by FACS of tumors developed in Snai2 WTErbB2++ or Snai2 KOErbB2++ parous mice, indicating that the cytokine deficiency arises at the transcriptional level (Fig. 6C).
Fig. 6. SNAI2 in the stroma modified the microenvironment required to support cancer behavior.
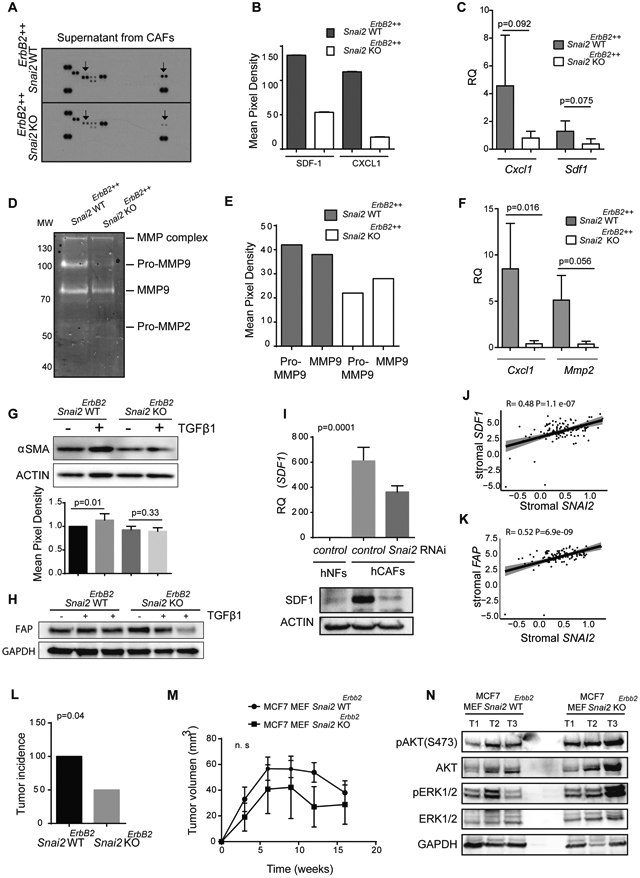
(A) Mouse CAFs (mCAFs) obtained from tumors developed in Snai2 WTErbB2++, and Snai2 KOErbB2++ parous mice were maintained for three days in the absence of serum. The supernatant was analyzed using a mouse cytokine array. Arrows point to differences in CXCL-1 (left) and SDF-1(right) protein expression. (B) Quantification of the cytokine array showed in panel A with the ImageJ software. (C) Relative Cxcl1 and Sdf1 mRNA expression in sorted CD140+/CD31−/EpCAM− cells from tumors developed in WT (N=8) or KO (N=9) parous mice, determined by real-time QPCR (Mann-Whitney U test). (D) Gelatin zymography assay on the supernatant from cultured mCAFs. Supernatants were collected and separated by electrophoresis, and gelatinase activity was visualized by standard staining. (E) Gelatin zymography bands were quantified using the ImageJ software. (F) Relative Cxcl1 and Mmp2 mRNA expression in sorted CD140+/CD31−/EpCAM− cells from metastatic tumors developed in WT and KO parous mice, as determined by real-time QPCR (Mann-Whitney U test). (G) Primary WT or KO mouse embryonic fibroblasts (MEFs) were treated with TGFβ1 (10 ng/mL) for 48 hours in the absence of serum, and αSMA (Student's t-test, N=3), or FAP (H) expression was determined by western blots. (I) Relative SDF1 mRNA expression in WT or SNAI2 depleted human CAFs (hCAFs) determined by real-time QPCR (one-way ANOVA, upper graph) and the SDF1 protein levels were assessed by western blot (lower image). (J, K) A positive correlation between the levels of SNAI2 and (J) SDF1 or (K) FAP was found in the stroma of breast tumors (Pearson's correlation). (L) Incidence of tumors generated by xenografts of WT or Snai2-deficient MEFs mixed with MCF7 cells (Chi-squared test, N=6). (M) Volume of tumors generated by MCF7 cells co-injected with WT or Snai2-deficient primary MEFs in subcutaneous xenografts (tumor volumes were compared using t-tests, N=6). (N) The tumors shown in panel M were analyzed for total and phosphorylated ERK and AKT levels by western blots (N=3).
These differences in cytokine production suggest that SNAI2-deficient mCAFs have a less active pro-tumorigenic phenotype, an activity that includes metalloprotease (MMP) production (51). When the MMP-2 and MMP-9 (gelatinase) activity in supernatants from primary mCAF cultures was examined, there was less MMP-9 activity in the supernatant of SNAI2-deficient mCAFs (Fig. 6D, E). In addition, there appeared to be less Mmp2 mRNA in the mCAFs of tumors from KO ErbB2++ parous mice than from WT ErbB2++ parous mice, yet these differences were not statistically significant (Fig. S6B). Interestingly, Cxcl1 and Mmp2 were expressed more weakly in mCAFs obtained from Snai2 KOErbB2++ metastatic tumors (Fig. 6F), suggesting that tumors require enhanced cytokine expression from CAFs to disseminate efficiently. Moreover, we only observed significant differences in the levels of Cxcl1 but not in those of Sdf1 or Mmp2 and Mmp9 in the mCAFs isolated from nulliparous Snai2 WTErbB2+ or Snai2 KOErbB2+ mice (Fig. S6C, D). These differences could help explain why tumors from parous mice are more malignant than those from nulliparous ones.
Fibroblasts are transformed into CAFs by TGFβ, and CAFs are permanently activated by an autocrine loop involving TGFβ itself (27, 52). Thus, we assessed whether the deficient cytokine production by mCAFs was associated with a defect in MEF activation in response to TGFβ. The absence of SNAI2 in fibroblasts was related to the failure of TGFβ1 to efficiently induce αSMA (α-Smooth Muscle Actin), FAP (Fibroblast Activating Protein) or S100A4 production (Fig. 6G, H and Fig. S6E). However, the TGFβ signaling did not seem to be affected (Fig. S6F).
While the oncogenic activity of ERBB2 increases by pregnancy, the incidence of lung metastases was reduced in Snai2 KOErbB2++ parous mice (20) (Fig. S2B, C). Interestingly, SDF1, which plays an essential role in tumor metastasis (53), was weakly expressed in Snai2-deficient mCAFs (Fig. 6A-C). Indeed, we confirmed that SDF1 potentiated human MDA-MB-231 breast cancer cell migration (Fig. S6G) and significantly, hCAFs produced more SDF1 than hNFs. Indeed, depletion of SNAI2 in hCAFs using specific siRNAs significantly dampened SDF1 RNA and protein expression (Fig. 6I). Furthermore, incubating human breast cancer cells with CM from SNAI2-depleted hCAFs confirmed the reduction in cell migration (Fig. 2I-K and S2I). Also, the addition of SDF1 to the CM obtained from SNAI2 depleted hCAFs partially rescued the defect observed in cell migration (Fig. S6H). Interestingly, further bioinformatics studies of a public human breast cancer data set (GSE9014) of stromal gene expression, revealed a statistically significant correlation between the transcript levels of SNAI2 and the hCAFs activating proteins SDF1 and FAP in the tumor stroma (Fig. 6J, K), yet not with CXCL1 (Fig. S6I).
Normal fibroblasts can inhibit cancer cell growth via direct cell-cell interactions and secreted paracrine factors (54). SNAI2-depleted fibroblasts were transformed into less functional CAFs than WT fibroblasts, although we did not previously observe differences in mouse tumor growth rate (Fig. S3A). Thus, we wondered if co-injection of MCF7 breast cancer cells, which does not express SNAI2, with primary MEFs of WT or KO Snai2 origin, would modify tumor cell growth in xenografts. Interestingly, the Snai2-deficient fibroblasts reduced the incidence of tumor generation (Fig. 6L) but not tumor growth (Fig. 6M), as seen in the MMTV-ErbB2 mice. Moreover, there was more total and phosphorylated ERK and AKT in MCF7 tumors when Snai2 was absent from the stroma (Fig. 6N). Hence, SNA2 in the stroma appears to be required to generate a tumor niche that supports tumor development.
Together, these results suggest that SNAI2 is required for the efficient transformation of fibroblasts to CAFs and normal CAFs cytokine production (e.g., SDF1). These defects could in part explain how SNAI2 in the stroma compartment influences breast cancer evolution.
DISCUSSION
SNAI2 participates in tumor pathogenesis through its anti-apoptotic activity and its prometastatic influence (15, 55).
Importantly, the influence of SNAI2 on cell behavior has, in most cases, been related to the direct activity of SNAI2 in the cells that express it. Here we assessed the non-autonomous cell effects of SNAI2 deficiency that might underlie the reduced proliferation and apoptosis in tumors, their enhanced necrosis, and the altered signaling and transcription in such tissue. All these data support the idea that the influence of SNAI2 in the extra-epithelial compartment has a significant impact on the epithelium itself. In this study, we observed an association between tumor Snai2 overexpression and the poor evolution of breast cancer patients, specifically with the luminal B ERBB2 tumor subtype. Also, we showed an association of SNAI2 stromal expression with ERBB2 tumor epithelial cell proliferation. All this suggests an indirect functional interaction between these two molecular pathways, from two different compartments, ERBB2 expressed in the tumor epithelium and SNAI2 expressed in the tumor stroma. Thus, we highlight the clinical relevance of the interaction between ERBB2 in the tumor epithelium and SNAI2 in the stroma, contributing to tumor proliferation and poor prognosis in patients with ERBB2 breast cancer. Indeed, the ZEB1/p53 axis was recently identified as a stroma-specific signaling pathway that promotes breast tumor development (56), emphasizing the importance of the levels of stromal transcriptional factors for breast cancer progression.
Fibroblasts constitute a major component of the tumor microenvironment and there is a large amount of data demonstrating their capacity to promote cancer through paracrine effects, escorting tumor cells through all the steps of carcinogenesis (3). Here we illustrate the complex interactions in the tumor-host microenvironment. The expression of SNAI2 in CAFs stimulates tumor cell migration thought the release of different cytokines into the medium. At the same time, SNAI2 depleted CAFs produce less SDF1, CXCL1 and MMPs, all of which are important in the tumorigenic process (57). In prostate cancer, SNAI2 is required for SDF1 and MMP-9 expression (58), and here we found that SNAI2 is required for SDF1 production by CAFs in both mice and humans, as well as for the heterocellular signaling loop established between cancer cells and fibroblasts. Primary fibroblasts deficient in Snai2 cannot be transformed into CAFs as efficiently as Snai2 WT fibroblasts. Thus, our results suggest that SNAI2 is not only crucial for stromal activation by cancer cells but also to maintain the secreted signals that enhance the invasive behavior of tumor cells, thereby favoring tumor progression.
While SNAI2 has been primarily implicated in the EMT, the data presented here is the first in vivo evidence supporting the idea that SNAI2 in the tumor stromal compartment plays an essential role in metastasis. However, we were only able to detect a role for SNAI2 in tumor dissemination when ERBB2 levels were increased.
Although there was no defect in tumor spreading in nulliparous mouse tumors, we observed a tumor spreading defect in parous mice, in which there were fewer tumors with the ability to metastasize. However, the tumors from Snai2 KO mice that metastasized seem to do so just as efficiently as the tumors developed by Snai2 WT mice. These results suggest that there is no defect in metastasis homing but rather, in the primary tumor. Moreover, the metastasis deficit is associated with higher levels of AKT1 in the primary tumor, consistent with data from other models (59-61).
All the data obtained in this study suggest that the decreased expression of SNAI2 by CAFs reduces cytokine production and influences the tumor-host microenvironment by modifying the ERK/AKT pathways. Other unknown elements of the EMT program are also likely to be affected (62). Thus, more studies will be necessary to clarify the mechanism by which SNAI2 expression in CAFs affects metastasis in the luminal breast tumors.
In summary, the absence of SNAI2 in the stroma dramatically affects tumor behavior, gene expression, the signaling pathways activated in response to the ERBB2 oncogene, and tumor malignancy due to the effects on tumor latency, proliferation and metastasis. Indeed, SNAI2 overexpression in the stroma is associated with poor prognosis in patients with breast cancer of the luminal B HER2+ subtype. These data enhance the potential of SNAI2 as a therapeutic target since inhibiting its activity in different compartments could have a more potent antitumor effect than initially expected due to its non-autonomous cell effects in the epithelium.
Supplementary Material
Significance.
Stromal SNAI2 expression enhances the tumorigenicity of luminal B HER2+ breast cancers and can identify a subset of patients with poor prognosis, making SNAI2 a potential therapeutic target for this disease.
Acknowledgments
We thank Dr. García Macías and the Comparative Pathology Core Facility for support with the pathology, and Isabel Ramos for her technical assistance. We would also like to thank Dr. Sánchez-García for many useful comments. We thank Carmen Rodríguez for her help in performing the immunohistochemistry to evaluate the SNAI2 expression, Paloma Bragado for the gift of the anti-S100A4 antibody and Ana García Casas and Rocío Puras Pardo for graphic design.
Financial Support
SCLL was the recipient of a Ramón y Cajal research contract from the Spanish Ministry of Economy and Competitiveness (Ministerio de Economía y Competitividad; MINECO), and the work was supported by a MINECO/FEDER research grant (SAF2015-64499-R; RTI2018-094130-B-100). N.S. was partially supported by the RTI2018-094130-B-100 budget. JPL was partially supported by FEDER funds, the Carlos III Health Institute (PIE14/00066), MINECO (SAF2014-56989-R, SAF2017-88854R), Castile and León (CSI234P18), the Sandra Ibarra Foundation for Solidarity against Cancer and the “We can be heroes” Foundation. NGS is a recipient of an FPU fellowship (MINECO/FEDER). RCC, MMSF and ABG were funded by fellowships from the Spanish Regional Government of Castile and León. The development of the Snai2 KO mice was supported by NIH grant R01HD034883 to TG.
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
Availability of data and materials
The data sets obtained and/or analyzed in the current study are available from the corresponding author upon reasonable request.
References
- 1.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. [DOI] [PubMed] [Google Scholar]
- 2.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–12. [DOI] [PubMed] [Google Scholar]
- 3.Mezawa Y, Orimo A. The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res. 2016;365(3):675–89. [DOI] [PubMed] [Google Scholar]
- 4.Kalluri R The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98. [DOI] [PubMed] [Google Scholar]
- 5.Nieto MA, Sargent MG, Wilkinson DG, Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264(5160):835–9. [DOI] [PubMed] [Google Scholar]
- 6.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116(Pt 3):499–511. [DOI] [PubMed] [Google Scholar]
- 7.Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123(4):641–53. [DOI] [PubMed] [Google Scholar]
- 8.Tripathi MK, Misra S, Khedkar SV, Hamilton N, Irvin-Wilson C, Sharan C, et al. Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J Biol Chem. 2005;280(17):17163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mittal MK, Singh K, Misra S, Chaudhuri G. SLUG-induced elevation of D1 cyclin in breast cancer cells through the inhibition of its ubiquitination. J Biol Chem. 2011;286(1):469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang HW, Menon LG, Black PM, Carroll RS, Johnson MD. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer. 2010;10:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30(12):1436–48. [DOI] [PubMed] [Google Scholar]
- 12.Olmeda D, Montes A, Moreno-Bueno G, Flores JM, Portillo F, Cano A. Snai1 and Snai2 collaborate on tumor growth and metastasis properties of mouse skin carcinoma cell lines. Oncogene. 2008;27(34):4690–701. [DOI] [PubMed] [Google Scholar]
- 13.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132(14):3151–61. [DOI] [PubMed] [Google Scholar]
- 14.Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62(6):1613–8. [PubMed] [Google Scholar]
- 15.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3(3):155–66. [DOI] [PubMed] [Google Scholar]
- 16.Nassour M, Idoux-Gillet Y, Selmi A, Come C, Faraldo ML, Deugnier MA, et al. Slug controls stem/progenitor cell growth dynamics during mammary gland morphogenesis. PLoS One. 2012;7(12):e53498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang R, Lan Y, Norton CR, Sundberg JP, Gridley T. The Slug gene is not essential for mesoderm or neural crest development in mice. Dev Biol. 1998;198(2):277–85. [PubMed] [Google Scholar]
- 18.Perez-Losada J, Sanchez-Martin M, Rodriguez-Garcia A, Sanchez ML, Orfao A, Flores T, et al. Zinc-finger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood. 2002;100(4):1274–86. [PubMed] [Google Scholar]
- 19.Murray SA, Oram KF, Gridley T. Multiple functions of Snail family genes during palate development in mice. Development. 2007;134(9):1789–97. [DOI] [PubMed] [Google Scholar]
- 20.Castillo-Lluva S, Hontecillas-Prieto L, Blanco-Gomez A, Saez-Freire MD, Garcia-Cenador B, Garcia-Criado J, et al. A new role of SNAI2 in postlactational involution of the mammary gland links it to luminal breast cancer development (vol 34, pg 4777, 2015). Oncogene. 2015;34(36):4797–8. [DOI] [PubMed] [Google Scholar]
- 21.Storci G, Sansone P, Trere D, Tavolari S, Taffurelli M, Ceccarelli C, et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol. 2008;214(1):25–37. [DOI] [PubMed] [Google Scholar]
- 22.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89(22):10578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soliman NA, Yussif SM. Ki-67 as a prognostic marker according to breast cancer molecular subtype. Cancer Biol Med. 2016;13(4):496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jurikova M, Danihel L, Polak S, Varga I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016;118(5):544–52. [DOI] [PubMed] [Google Scholar]
- 25.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–31. [DOI] [PubMed] [Google Scholar]
- 26.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. P Natl Acad Sci USA. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alves CL, Elias D, Lyng MB, Bak M, Ditzel HJ. SNAI2 upregulation is associated with an aggressive phenotype in fulvestrant-resistant breast cancer cells and is an indicator of poor response to endocrine therapy in estrogen receptor-positive metastatic breast cancer. Breast Cancer Res. 2018;20(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou S, Sun X, Yu L, Zhou R, Li A, Li M, et al. Differential expression and clinical significance of epithelial-mesenchymal transition markers among different histological types of triple-negative breast cancer. J Cancer. 2018;9(3):604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front Biosci (Landmark Ed). 2009;14:3035–50. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Yang X, Xu S, Jin P, Li X, Wei X, et al. Reprogramming of stromal fibroblasts by SNAI2 contributes to tumor desmoplasia and ovarian cancer progression. Mol Cancer. 2017;16(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penault-Llorca F, Radosevic-Robin N. Ki67 assessment in breast cancer: an update. Pathology. 2017;49(2):166–71. [DOI] [PubMed] [Google Scholar]
- 38.Untch M, Gerber B, Harbeck N, Jackisch C, Marschner N, Mobus V, et al. 13th st. Gallen international breast cancer conference 2013: primary therapy of early breast cancer evidence, controversies, consensus - opinion of a german team of experts (zurich 2013). Breast Care (Basel). 2013;8(3):221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cardiff RD, Wellings SR. The comparative pathology of human and mouse mammary glands. J Mammary Gland Biol Neoplasia. 1999;4(1):105–22. [DOI] [PubMed] [Google Scholar]
- 40.Taneja P, Frazier DP, Kendig RD, Maglic D, Sugiyama T, Kai F, et al. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn. 2009;9(5):423–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells. 2019;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubbard NE, Lim D, Mukutmoni M, Cai A, Erickson KL. Expression and regulation of murine macrophage angiopoietin-2. Cell Immunol. 2005;234(2):102–9. [DOI] [PubMed] [Google Scholar]
- 43.Quigley DA, Kristensen V. Predicting prognosis and therapeutic response from interactions between lymphocytes and tumor cells. Mol Oncol. 2015;9(10):2054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pioli PD, Dahlem TJ, Weis JJ, Weis JH. Deletion of Snai2 and Snai3 Results in Impaired Physical Development Compounded by Lymphocyte Deficiency. Plos One. 2013;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Gridley T. Compensatory regulation of the Snai1 and Snai2 genes during chondrogenesis. J Bone Miner Res. 2013;28(6):1412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004;64(9):3171–8. [DOI] [PubMed] [Google Scholar]
- 47.Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res. 2010;70(11):4260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castellanos-Martin A, Castillo-Lluva S, Saez-Freire Mdel M, Blanco-Gomez A, Hontecillas-Prieto L, Patino-Alonso C, et al. Unraveling heterogeneous susceptibility and the evolution of breast cancer using a systems biology approach. Genome Biol. 2015;16:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugai T, Uesugi N, Kitada Y, Yamada N, Osakabe M, Eizuka M, et al. Analysis of the expression of cancer-associated fibroblast- and EMT-related proteins in submucosal invasive colorectal cancer. J Cancer. 2018;9(15):2702–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48. [DOI] [PubMed] [Google Scholar]
- 51.Taguchi A, Kawana K, Tomio K, Yamashita A, Isobe Y, Nagasaka K, et al. Matrix metalloproteinase (MMP)-9 in cancer-associated fibroblasts (CAFs) is suppressed by omega-3 polyunsaturated fatty acids in vitro and in vivo. PLoS One. 2014;9(2):e89605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158(3):564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang H, Watkins G, Parr C, Douglas-Jones A, Mansel RE, Jiang WG. Stromal cell derived factor-1: its influence on invasiveness and migration of breast cancer cells in vitro, and its association with prognosis and survival in human breast cancer. Breast Cancer Res. 2005;7(4):R402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alkasalias T, Flaberg E, Kashuba V, Alexeyenko A, Pavlova T, Savchenko A, et al. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc Natl Acad Sci U S A. 2014;111(48):17188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inukai T, Inoue A, Kurosawa H, Goi K, Shinjyo T, Ozawa K, et al. SLUG, a ces-1-related zinc finger transcription factor gene with antiapoptotic activity, is a downstream target of the E2A-HLF oncoprotein. Mol Cell. 1999;4(3):343–52. [DOI] [PubMed] [Google Scholar]
- 56.Fu R, Han CF, Ni T, Di L, Liu LJ, Lv WC, et al. A ZEB1/p53 signaling axis in stromal fibroblasts promotes mammary epithelial tumours. Nat Commun. 2019;10(1):3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barcellos-Hoff MH, Lyden D, Wang TC. The evolution of the cancer niche during multistage carcinogenesis. Nat Rev Cancer. 2013;13(7):511–8. [DOI] [PubMed] [Google Scholar]
- 58.Uygur B, Wu WS. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol Cancer. 2011;10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoeli-Lerner M, Chin YR, Hansen CK, Toker A. Akt/protein kinase b and glycogen synthase kinase-3beta signaling pathway regulates cell migration through the NFAT1 transcription factor. Mol Cancer Res. 2009;7(3):425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu H, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, et al. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc Natl Acad Sci U S A. 2006;103(11):4134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chin YR, Toker A. The Actin-Bundling Protein Palladin Is an Akt1-Specific Substrate that Regulates Breast Cancer Cell Migration. Molecular Cell. 2010;38(3):333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castillo-Lluva S, Tatham MH, Jones RC, Jaffray EG, Edmondson RD, Hay RT, et al. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat Cell Biol. 2010;12(11):1078–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.