

Abstract
Aims/hypothesis
Melanocortin 4 receptor (MC4R) mutation is the most common cause of known monogenic obesity in humans. Unexpectedly, humans and rodents with MC4R deficiency do not develop hyperglycaemia despite chronic obesity and insulin resistance. To explain the underlying mechanisms for this phenotype, we determined the role of MC4R in glucose homeostasis in the presence and absence of obesity in mice.
Methods
We used global and hypothalamus-specific MC4R-deficient mice to investigate the brain regions that contribute to glucose homeostasis via MC4R. We performed oral, intraperitoneal and intravenous glucose tolerance tests in MC4R-deficient mice that were either obese or weight-matched to their littermate controls to define the role of MC4R in glucose regulation independently of changes in body weight. To identify the integrative pathways through which MC4R regulates glucose homeostasis, we measured renal and adrenal sympathetic nerve activity. We also evaluated glucose homeostasis in adrenaline (epinephrine)-deficient mice to investigate the role of adrenaline in mediating the effects of MC4R in glucose homeostasis. We employed a graded [13C6]glucose infusion procedure to quantify renal glucose reabsorption in MC4R-deficient mice. Finally, we measured the levels of renal glucose transporters in hypothalamus-specific MC4R-deficient mice and adrenaline-deficient mice using western blotting to ascertain the molecular mechanisms underlying MC4R control of glucose homeostasis.
Results
We found that obese and weight-matched MC4R-deficient mice exhibited improved glucose tolerance due to elevated glucosuria, not enhanced beta cell function. Moreover, MC4R deficiency selectively in the paraventricular nucleus of the hypothalamus (PVH) is responsible for reducing the renal threshold for glucose as measured by graded [13C6]glucose infusion technique. The MC4R deficiency suppressed renal sympathetic nerve activity by 50% in addition to decreasing circulating adrenaline and renal GLUT2 levels in mice, which contributed to the elevated glucosuria. We further report that adrenaline-deficient mice recapitulated the increased excretion of glucose in urine observed in the MC4R-deficient mice. Restoration of circulating adrenaline in both the MC4R- and adrenaline-deficient mice reversed their phenotype of improved glucose tolerance and elevated glucosuria, demonstrating the role of adrenaline in mediating the effects of MC4R on glucose reabsorption.
Conclusions/interpretation
These findings define a previously unrecognised function of hypothalamic MC4R in glucose reabsorption mediated by adrenaline and renal GLUT2. Taken together, our findings indicate that elevated glucosuria due to low sympathetic tone explains why MC4R deficiency does not cause hyperglycaemia despite inducing obesity and insulin resistance.
Keywords: Diabetes, Endocrinology, Hypothalamus, Melanocortin 4 receptor, Mouse model, Obesity
Graphical Abstract
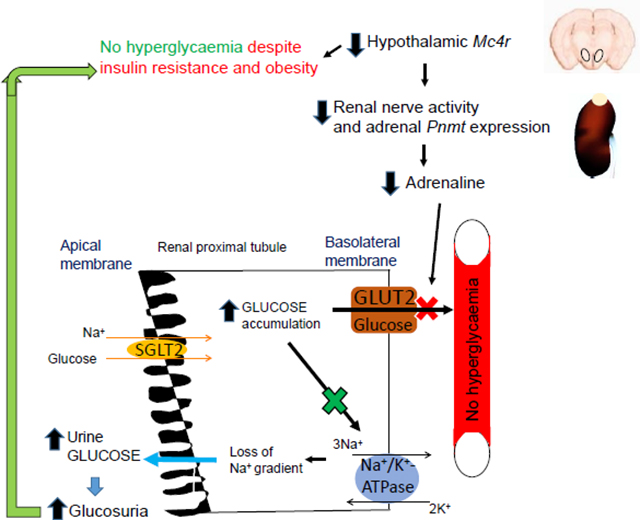
Decreased adrenaline due to hypothalamic MC4R deficiency reduces kidney GLUT2 levels and glucose reabsorption. This phenomenon increases glucosuria and improves glucose tolerance. SGLT2, sodium-glucose cotransporter 2
Introduction
The melanocortin 4 receptor (MC4R) is a primary effector of the central melanocortin system in regulating energy balance [1–5]. Mutations in the MC4R gene are the most common cause of known monogenic obesity in humans [1–3]. Moreover, MC4R deficiency causes insulin resistance in humans [2, 6] and rodents [1, 5, 7]. MC4R is an established therapeutic target for treating some types of obesity [8–10]. MC4R agonists have shown promising results in treating obesity in individuals that exhibit mutations in the leptin receptor or in the melanocortin signalling pathway upstream of MC4R [8–10]. In the general population, obesity and insulin resistance increase the risk of diabetes [11–15]. However, humans and rodents with MC4R deficiency either do not develop [2, 5, 16–20] or have a relatively lower risk [21] of developing hyperglycaemia despite obesity and insulin resistance. Identifying molecular mechanisms that protect MC4R-deficient individuals from developing hyperglycaemia will advance our understanding of the function of the central melanocortin system in glucose homeostasis and may facilitate the development of new therapeutics to control diabetes in the general population. Therefore, we aimed to elucidate why MC4R deficiency does not cause hyperglycaemia despite inducing insulin resistance and obesity.
Based on our previous observations in hypothalamus-specific proopiomelanocortin (POMC)-deficient mice [22, 23], we determined the role of MC4R in glucose reabsorption to ascertain whether elevated glucosuria protects from hyperglycaemia during MC4R deficiency. We used MC4R-deficient mice that were either overweight or weight-matched compared with their wild-type (WT) littermates to investigate the role of MC4R in glucose homeostasis. In addition to global MC4R-deficient mice, we employed a Cre-Lox recombination strategy to rescue, knockout or knockdown Mc4r in specific nuclei of the brain to further identify the sites that control renal glucose reabsorption via the melanocortin system.
Methods
For detailed Methods, please refer to the Electronic supplementary material (ESM).
Animal care and generation of mice
All mouse procedures were approved by the Institutional Animal Care and Use Committee at the University of Rochester or the University of Iowa and were performed according to the Public Health Service guidelines for the humane care and use of experimental animals. We housed mice in ventilated cages under controlled temperature (~23°C) and photoperiod (12 h light/dark cycle, lights on from 06:00 hours to 18:00 hours) conditions and fed them Hydropac water (Lab Products, USA) and regular laboratory chow (catalogue no. 5010; LabDiet, USA). We purchased Mc4rloxTB/+ mice from the Jackson Laboratory, USA (C57BL/6J genetic background, catalogue no. 032518) and generated Mc4rloxTB/loxTB mice and their littermate controls (representative genotypes, ESM Fig. 1). We obtained Mc4rloxP/loxP mice [24] from D. P. Olson (University of Michigan, USA) with permission from B. B. Lowell (Harvard University, USA). We crossed these mice with Sim1Cre (006395, the Jackson Laboratory) to knockout Mc4r predominantly in the PVH [24] (including some other regions that express Sim1 gene). S. Ebert provided adrenaline-deficient mice (Pnmt knockout [KO], phenylethanolamine N-methyltransferase,) that were generated by his team as described previously [25]. Experimenters were not blinded to either group assignment or to outcome assessment. See ESM Methods.
Glucose/insulin tolerance tests and glucose-stimulated insulin secretion assay
For glucose tolerance tests, we fasted mice for 6 h (08:00–14:00 hours) or overnight (18:00–08:00 hours) and administered glucose by oral gavage or i.p. injections. For ITTs, we fasted mice for 6 h (08:00– 14:00 hours) and administered insulin by i.p. injections. Before the administration of glucose (100 mg/mouse in 300 μl 0.9% (wt/vol.) saline [154 mmol/l NaCl]; catalogue no. G8270; Sigma, USA) or insulin (15 mU/mouse; Humulin R, Eli Lilly, USA) in different cohorts of mice, we measured their baseline blood glucose levels (tail-vein blood, AlphaTRAK 2 glucometer, USA) at 0 min. Post glucose or insulin administration, we measured blood glucose levels at different times as noted in Fig. 1. We used the trapezoidal rule to calculate total AUC for further integrated analyses of the measured blood glucose levels at different times during the tests.
Fig. 1.

Improved glucose tolerance despite insulin resistance in 6h fasted obese male Mc4r null 24-week-old C57Bl/6 mice. (a) Body weight, (b) OGTT, (c) fasting plasma insulin levels, (d) ITT in obese Mc4r null 24-week-old C57Bl/6 mice. Bar graphs in (b) and (d) represent the corresponding AUC. Two-tailed unpaired Student’s t test or repeated measures two-way ANOVA followed by Bonferroni’s multiple comparison test were used for comparisons. **p<0.01, ***p<0.001;. Error bars are mean ± SEM. Mc4r−/−, Mc4rloxTB/loxTB
For glucose-stimulated insulin secretion assay in freely moving awake mice, we cannulated the carotid artery and jugular vein, and allowed the mice to recover from surgery for 3–5 days [22, 26]. On the day of the experiment, we fasted mice for 6 h (08:00–14:00 hours) and administered an i.v. bolus of 60 mg glucose then measured blood glucose and insulin levels at different times as indicated in Fig. 2a and b, respectively.
Fig. 2.

Elevated glucosuria but no enhancement in beta cell function post glucose administration in obese male Mc4r null 24-week-old C57Bl/6 mice. (a) Blood glucose levels during the IVGTT (mice were fasted for 6 h before the test); n=7 WT and n=6 Mc4r−/−, (b) plasma insulin levels during IVGTT; n=7 (c) 24 h urine glucose concentration at baseline, (d) 24 h urine glucose concentration after administering 250 mg glucose by oral gavage, (e) 24 h urine glucose concentration after administering 200 mg glucose by i.p. injection. Two-tailed unpaired Student’s t test or repeated measures two-way ANOVA followed by Bonferroni’s multiple comparison test were used for comparisons. **p<0.01, ***p<0.001 vs WT; †p<0.01 vs 0 min, WT. Error bars are mean ± SEM. Mc4r−/−, Mc4rloxTB/loxTB
Plasma insulin and adrenaline/noradrenaline measurements
We fasted mice for 6 h (08:00–14:00 hours) and collected tail blood for insulin measurements. We centrifuged the blood at 2,000g at 4°C for 20 min to separate plasma from whole blood. We measured plasma insulin levels using an ELISA (mouse insulin assay kit, catalogue no. 90080; Crystal Chem, USA). Similarly, we used ELISAs to measure plasma adrenaline (epinephrine) and noradrenaline (norepinephrine) (catalogue nos ADU39-K01 and NOU39-K01, respectively; Eagle Bioscience, USA). We did not fast mice before adrenaline or noradrenaline assays. We euthanised mice and collected trunk blood for the assay between 14:00 hours and 15:00 hours on the day of the experiment for all the groups.
Urine glucose, sodium, protein and creatinine assays
We housed mice individually in metabolic cages (Tecniplast, USA) and let them acclimate to the cages for 1 week before using them for experiments. We collected 24 h urine samples at baseline (before glucose administration) and after glucose challenge (250 mg oral, or 100–200 mg i.p. injections in 300 μl 0.9% saline). We used a glucose colorimetric test (catalogue no. 10009582; Cayman Chemical, USA) to measure urine glucose levels. Urine sodium, creatinine and protein levels were also measured by colorimetric tests according to the manufacturers’ instructions (sodium, catalogue no. 211096, Abcam USA; creatinine, catalogue no. 500701, Cayman Chemical USA; protein, catalogue no. 23227, Pierce USA). The absorbance was measured using a spectrophotometer as per manufacturers’ instructions (Epoch, BioTek, USA).
Restoration of Mc4r in different brain regions
Mc4rloxTB/loxTB mice were anaesthetised with isoflurane, placed in a stereotaxic frame (Model 1900, Kopf Instruments) and the skull was exposed for intracranial injections of AAV-Cre or AAV-GFP as described in the ESM Methods.
Quantitative real-time PCR
To assess expression of Mc4r and Pnmt, quantitative real-time PCR (qPCR) was performed as described previously [27]. See ESM Methods for further details.
Intracranial surgery in Mc4rloxP/loxP mice
We used the SomnoSuite system (Kent Scientific, USA) to deliver isoflurane (2–5%) to anaesthetise mice before placing them in a stereotaxic frame (Model 1900; Kopf Instruments, USA). With a continuous flow of isoflurane (2%) in mouse nose mask, the mouse skull was exposed for intracranial injections. We injected AAV-Cre-GFP or AAV-GFP (~50 nl, , titre 8×1012 vg/ml; University of North Carolina Vector Core, USA) bilaterally into the paraventricular nucleus of the hypothalamus (PVH) of 7-week-old Mc4rloxP/loxP mice (coordinates from bregma: anteroposterior, ~0.70 mm; mediolateral, ~0.22 mm; dorsoventral, ~4.80 mm) using a 33 G Hamilton syringe attached to UltraMicroPump (UMP3–1; WPI, USA). Four weeks after the administration of the viral vectors, we measured glucose tolerance and urine glucose levels in these mice.
In situ hybridisation
Fluorescence in situ hybridisation was performed on 14 μm thick slices of flash frozen brains using RNAscope fluorescent multiplex reagents (ACD, USA). RNA probes for Mc4r or GFP (catalogue numbers 319181-C2, 409011; ACD) were incubated with the brain slices and signal amplification was achieved using the multiplex reagents as described previously [26, 28].
Liver glycogen levels and pyruvate tolerance test
Hepatic glycogen levels were measured using colorimetric reagents from Sigma (catalogue no. MAK016, USA). For pyruvate tolerance tests, mice were fasted for 6 h and blood glucose levels were measured at baseline and at 15, 30, 60 and 120 min after the administration of sodium pyruvate (60 mg in 0.9% saline per mouse, ip; catalogue no. P5280; Sigma, USA).
Renal and adrenal sympathetic nerve activity
Sympathetic nerve activity was assessed using direct multifibre recording as described in the ESM Methods.
Graded [13C6]glucose infusion procedure
Escalated doses of [13C6]glucose were infused into the jugular vein and blood and urine samples were collected at different times as described in ESM Methods.
Isolation and culture of mouse primary renal tubular cells
We isolated mouse primary renal proximal tubular epithelial cells from 8-week-old WT mice using a published protocol [29]. For the glucose transport assay, we incubated the proximal tubule cells with Hanks’ balanced salt solution (HBSS) containing [13C6]glucose (5 mmol/l) in the presence or absence of adrenaline/noradrenaline (10 nmol/l in HBSS). The cells were grown in cell culture inserts (catalogue no. MCRP24H48; Millipore, USA) that are used for studying small molecule transport. On the day of the experiment, the labelled glucose was added to the apical (top) side of the cells in cell culture inserts (treated with 10 nmol/l adrenaline or noradrenaline in HBSS) and glucose was measured at the basolateral (bottom) side of the cells at different times as shown in Fig. 5b. [13C6]Glucose was measured by LC-MS assay using Shodex (USA) HILICpak VG-50 2D column by the URMC Mass Spectrometry Resource Laboratory.
Fig. 5.
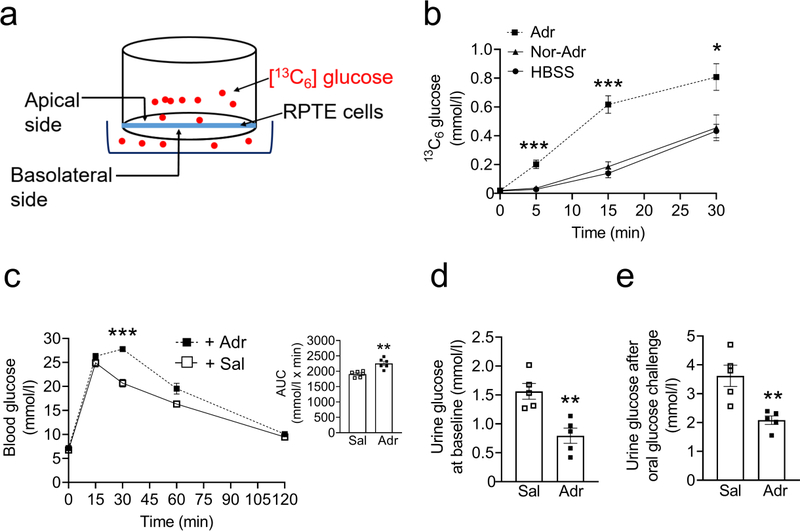
Adrenaline increases glucose transport in the mouse primary renal tubular epithelial (RPTE) cells and reverses improved glucose tolerance and elevated glucosuria in hypothalamus-specific MC4R-deficient mice. (a) Schematic of the experiment setup and cell culture inserts. (b) [13C6]Glucose levels after treatment with adrenaline or noradrenaline in the RPTE cells; n=5 (c) OGTT, (d) 24 h urine glucose concentration at baseline, (e) 24 h urine glucose concentration after administering 250 mg glucose by oral gavage in 18- to 24-week-old Mc4rloxP/loxP+AAV-Cre mice whose plasma adrenaline levels are restored. Two-tailed unpaired Student’s t test or repeated measures two-way ANOVA followed by Bonferroni’s multiple comparison test were used for comparisons. *p<0.05, **p<0.01, ***p<0.001 for adrenaline vs noradrenaline and HBSS groups in part (b) and for adrenaline group vs saline group in (c–e). Adr, adrenaline; Nor-Adr, noradrenaline; Sal, saline
Restoration of adrenaline in MC4RPVH- or adrenaline-deficient mice
To restore the plasma adrenaline levels in mice lacking MC4R in the PVH (MC4RPVH-deficient mice), we injected adrenaline (5 μg in 300 μl 0.9% saline, per mouse, i.p.), or 0.9% saline in the control group, 1 h prior to either glucose tolerance tests or placing the mice in metabolic cages for 24 h urine collections. For adrenaline-deficient mice, we infused adrenaline (1 μg/day) or 0.9% saline using Alzet 2002 osmotic pumps as described previously [27].
Western blotting
We performed western blotting to measure renal GLUT1, GLUT2, sodium–glucose cotransporter (SGLT)1, SGLT2 and Na+/K+-ATPase as per our published protocol [22]. For full details, see ESM Methods.
Statistical analyses
All data are presented as mean ± SEM. Two-tailed Student’s unpaired t test was used to compare results between two independent groups. Comparisons between two independent groups involving one dependent variable with repeated measures were made by repeated measures two-way ANOVA followed by a Bonferroni post hoc multiple comparison test. All analyses were performed using Prism 8.0 (GraphPad, USA) and a p value <0.05 was considered significant.
Results
MC4R-deficient mice have improved glucose tolerance despite obesity and insulin resistance
We used Mc4rloxTB/loxTB mice, in which MC4R deficiency can be reversed by Cre recombinase that removes the loxP-flanked transcriptional blocking sequences and rescues Mc4r gene expression [3]. As shown previously [1, 3], MC4R-deficient mice exhibit obesity due to hyperphagia and reduced energy expenditure. We administered equal amounts of glucose to different groups of mice, regardless of changes in their body weights, for glucose tolerance tests. We found that the MC4R-deficient mice had normal fasting blood glucose levels coupled with improved glucose tolerance despite obesity and insulin resistance (male: Fig. 1; female: ESM Fig. 2). Moreover, IPGTTs also showed that MC4R-deficient mice have improved glucose tolerance compared with their littermate controls (Male: AUC, 2,184 ±158 vs 2,852±72, female: AUC, 2,421 ±190 vs 2,946 ±128 mmol/l × min, MC4R-deficient vs WT, respectively, p<0.05).
MC4R-deficient mice exhibit elevated glucosuria but no enhancement in beta cell function
We performed a glucose-stimulated insulin secretion assay to determine if an increase in insulin secretion to compensate for the insulin resistance can explain the improved glucose tolerance in MC4R-deficient mice. Supporting the results obtained from the OGTT and IPGTT, the IVGTT also demonstrated improved glucose tolerance in MC4R-deficient mice (Fig. 2a). WT mice showed a significantly higher increase in glucose-stimulated insulin secretion compared with baseline (time 0, Fig. 2b). In contrast, there was no such increase in insulin secretion in response to glucose challenge in MC4R-deficient mice (Fig. 2b), which already have elevated baseline plasma insulin levels due to insulin resistance. These data indicate that enhanced glucose-induced insulin secretion is not the underlying cause of improved glucose tolerance in MC4R-deficient mice.
Based on our observation of elevated glucosuria in hypothalamus-specific
POMC-deficient mice [22, 23], we measured urine glucose levels in MC4R-deficient mice. Indeed, the MC4R-deficient mice exhibited elevated glucosuria at baseline and after glucose administration compared with their WT littermates (Fig. 2c–e). Moreover, the MC4R-deficient mice had increased 24 h urine volume and exhibited natriuresis without any changes in creatinine and total protein levels (ESM Table 1 and ESM Fig. 3). Taking into account 24 h urine volume, the MC4R-deficient mice exhibited an increase of total glucose excretion of different magnitude and intensity, depending on the route and amount of glucose administration, compared with their control littermates, as shown in ESM Table 1.
MC4R in the PVH controls glucose reabsorption
Mc4r is expressed in the PVH, medial amygdala and lateral hypothalamus, in addition to hindbrain regions, such as the nucleus tractus solitarius (NTS) and rostral ventrolateral medulla (RVLM) [30]. To identify the areas that mediate the effects of MC4R on kidney glucose reabsorption and glucose homeostasis, we restored Mc4r in different brain regions in reversible Mc4rloxTB/loxTB using the Cre-Lox strategy [3]. We injected AAV-Cre or AAV-GFP into the above-mentioned Mc4r expressing areas in different cohorts of Mc4rloxTB/loxTB mice. We confirmed the accuracy of the injections by measuring Mc4r expression in each of the regions using qPCR at the end of the study (ESM Fig. 4a). Mice that did not show significant restoration of Mc4r expression after the injections were excluded from the experiments or data analyses. We found that restoration of Mc4r in the PVH reversed the phenotype of improved glucose tolerance and elevated glucosuria observed in otherwise global MC4R-deficient mice (ESM Fig. 4b, c). These data indicated that PVH-specific MC4R (MC4RPVH) is sufficient to maintain glucose homeostasis through renal glucose reabsorption.
To investigate whether MC4R in the PVH is necessary for glucose reabsorption, we used two mouse models: Mc4rloxP/loxP;Sim1Cre and Mc4rloxP/loxP with AAV-Cre administration selectively into the PVH in adult mice.
Obese Mc4rloxP/loxP;Sim1Cre mice had improved glucose tolerance and elevated glucosuria like that observed in global MC4R-deficient mice (Fig. 3a–c), demonstrating the role of hypothalamic MC4R in glucose regulation. Results from weight-matched mice also supported this phenotype (ESM Fig. 5). It is important to note that the weight-matched Mc4rloxP/loxP;Sim1Cre mice did not exhibit elevated glucosuria at baseline (ESM Fig. 5d) because they were not fed ad libitum and, therefore, their blood glucose levels at baseline remained below the renal threshold for glucose excretion. However, the mice did show elevated glucosuria (ESM Fig. 5e) after we administered 250 mg glucose by oral gavage, further validating the role of MC4R in glucose reabsorption.
Fig. 3.

Improved glucose tolerance and elevated glucosuria in male 18- to 24-week-old hypothalamus-specific Mc4r knockout mice. (a) Body weight, (b) OGTT (mice were fasted for 6 h before the test), (c) 24 h urine glucose concentration in Mc4rloxP/loxP;Sim1Cre mice. (d) Representative images of fluorescence in situ hybridisation showing reduced Mc4r in the PVH; n=5, four sections per mouse and four areas of interest per section were analysed. Scale bar, 100 μm. (e) Body weight, (f) OGTT, (g) urine [13C6]glucose levels in Mc4rloxP/loxP+AAV-Cre mice during the graded glucose infusion procedure; n=5. Bar graphs in (b) and (f) represent the corresponding AUC. Two-tailed unpaired Student’s t test or repeated measures two-way ANOVA followed by Bonferroni’s multiple comparison test were used for comparisons. *p<0.05, **p<0.01, ***p<0.001. Error bars are mean ± SEM. 3V, third ventricle
To account for any developmental or compensatory changes, or a possible non-specific knockout of Mc4r outside the PVH, which may have impacted the results obtained from Mc4rloxP/loxP;Sim1Cre mice, we also determined glucose homeostasis in adult Mc4rloxP/loxP mice that were made MC4RPVH deficient by administration of AAV-Cre selectively in the PVH, as described previously [3, 26]. In line with the findings in Mc4rloxP/loxP;Sim1Cre mice, the obese MC4RPVH-deficient mice (Fig. 3d) also showed improved glucose tolerance and elevated glucosuria (Fig. 3e–g). The number of cells expressing Mc4r in the PVH was reduced by 70% without any difference in the total number of cells (DAPI staining) after AAV-Cre administration compared with the mice that received AAV-GFP/control vector (AAV-Cre: 217 ±21 vs AAV-GFP: 721 ±51, n = 5, four sections per mouse and four areas of interest per section were analysed, number of cells expressing Mc4r, p<0.01). Accuracy of intra-PVH injections was confirmed after the study by observing the expression of GFP in the PVH. Data from only those mice that had GFP confined to the PVH were used for analyses (ESM Fig. 6a).
We did not observe any differences in the hepatic glycogen levels and gluconeogenesis between WT and the MC4R-deficient (Mc4rloxTB/loxTB) mice (ESM Fig. 6b, c). Moreover, renal Mc4r expression was negligible compared with that in the hypothalamus in WT mice (Mc4r: 0.6 ±0.2% vs 100 ±12%, kidney vs hypothalamus, respectively, p<0.001). Altogether, these data demonstrate that MC4R in the PVH regulates glucose homeostasis via renal glucose reabsorption.
MC4R deficiency reduces renal sympathetic nerve activity and circulating adrenaline levels
To determine the integrative pathways that link the hypothalamic MC4R and kidney glucose reabsorption, we measured circulating adrenaline levels, renal and adrenal sympathetic nerve activities in Mc4rloxP/loxP;Sim1Cre mice. We observed that Mc4rloxP/loxP;Sim1Cre mice had reduced plasma adrenaline and noradrenaline levels (Fig. 4a, b). Moreover, the MC4R deficiency in Sim1 neurons suppressed the renal sympathetic nerve activity by 50% compared with their control littermates without affecting blood pressure (Fig. 4c, d).
Fig. 4.

Reduced renal sympathetic nerve activity and adrenal Pnmt expression in male 8- to 16-week-old Mc4rloxP/loxP;Sim1Cre mice vs Mc4rloxP/loxP mice. (a) Plasma adrenaline, (b) plasma noradrenaline. (c) Representative traces of blood pressure, renal sympathetic nerve activity (RSNA) and adrenal sympathetic nerve activity (ADSNA). (d) RSNA, (e) ADSNA and (f) adrenal Pnmt expression in male 8- to 16-week-old Mc4rloxP/loxP;Sim1Cre mice vs Mc4rloxP/loxP mice. Two-tailed unpaired Student’s t test was used for comparisons. *p<0.05, **p<0.01, ***p<0.001;. Error bars are mean ± SEM
Unexpectedly, the Mc4rloxP/loxP;Sim1Cre mice had increased adrenal sympathetic nerve activity (Fig. 4e) despite the reduced circulating adrenaline levels. The increased sympathetic tone was probably a compensatory response to the reduced Pnmt expression (Fig. 4f) and consequent decrease in adrenaline synthesis. Taken together, the results indicate the essential role of the hypothalamic MC4R in regulating renal and adrenal sympathetic nerve activity that likely affect circulating adrenaline and noradrenaline levels.
Adrenaline increases glucose transport in the mouse primary renal tubular cells
We determined the direct effects of adrenaline and noradrenaline (both 10 nmol/l in HBSS) on glucose transport in the mouse primary renal tubular cells isolated from 8-week-old WT mice using [13C6]glucose (5 mmol/l) to identify the hormone that integrates the effects of the hypothalamic MC4R on glucose reabsorption. We found that glucose transport was increased across the cell culture inserts (Fig. 5a), representing glucose reabsorption, in the adrenaline- but not the noradrenaline-treated group compared with the control group (Fig. 5b). These data indicate the direct role of adrenaline in glucose reabsorption.
Restoration of adrenaline reverses the improved glucose tolerance and elevated glucosuria in MC4RPVH-deficient mice
Because we observed that adrenaline increased glucose transport in primary renal tubular cells, we hypothesised that adrenaline would reverse elevated glucosuria in MC4RPVH-deficient mice. To test this hypothesis, we injected adrenaline (5 μg in 300μl 0.9% saline per mouse) or saline into MC4RPVH-deficient mice (Mc4rloxP/loxP+AAV-Cre mice) to restore their plasma adrenaline levels and measured glucose tolerance in addition to urine glucose levels. Circulating adrenaline in MC4RPVH-deficient mice was reduced (6,461 ±1,179 vs 18,227 ±3,918 pmol/l, p<0.05) compared with their littermate controls. The restoration of adrenaline reversed the improved glucose tolerance and elevated glucosuria in the MC4RPVH-deficient mice (Fig. 5c–e). These data demonstrate the role of adrenaline in integrating the effects of the hypothalamic MC4R in glucose homeostasis via the reabsorption of glucose.
Adrenaline-deficient mice recapitulate the phenotype of elevated glucosuria that was observed in MC4R-deficient mice
We found that Pnmt-KO (adrenaline-deficient) mice (Fig. 6a, b) had improved glucose tolerance and elevated glucosuria compared with their WT littermates (Fig. 6c, d, e). There was no change in 24 h urine volume between the adrenaline-deficient mice and the controls (1,006 ±277 vs 1,058 ±107 μl, Pnmt-KO vs WT, respectively). Moreover, there was no difference in the hepatic glycogen levels between WT and adrenaline-deficient mice (ESM Fig. 6d). Adrenaline-deficient mice had hyperinsulinaemia, but not reduced insulin sensitivity as measured by ITT (Fig. 6f, g).
Fig. 6.

Blood and urine glucose levels in male 6- to 9-week-old-mice adrenaline-deficient (Pnmt-KO) mice before (a–g) and after (h–j) restoration of their plasma adrenaline levels. (a) Plasma adrenaline, (b) plasma noradrenaline, (c) OGTT, (d) 24 h urine glucose concentration at baseline, (e) 24 h urine glucose concentration after administering 250 mg glucose by oral gavage in adrenaline-deficient mice, (f) ITT, (g) plasma insulin levels in adrenaline-deficient mice, (h) plasma adrenaline (to validate its restoration), (i) OGTT, (j) 24 h urine glucose concentration at baseline and (k) 24 h urine glucose concentration after administering 100 mg glucose by i.p. injection, following restoration of plasma adrenaline in otherwise adrenaline-deficient mice. Bar graphs in (c), (f) and (i) represent the corresponding AUC. Two-tailed unpaired Student’s t test or repeated measures two-way ANOVA followed by Bonferroni’s multiple comparison test were used for comparisons. *p<0.05, **p<0.01, ***p<0.001.. Error bars are mean ± SEM. Adr, adrenaline; Sal, saline
We implanted osmotic minipumps (1 μg/day adrenaline or saline, s.c.) to restore plasma adrenaline (Fig. 6h) in otherwise adrenaline-deficient mice to validate the essential role of adrenaline in glucose homeostasis via glucose reabsorption. We evaluated glucose tolerance on the 4th day and measured urine glucose levels on the 12th day of the infusion. Adrenaline infusion reversed the improved glucose tolerance and the elevated glucosuria observed in the adrenaline-deficient mice (Fig. 6i, j, k) without affecting liver glycogen levels (ESM Fig. 6f). Together, these data point to the direct role of adrenaline in systemic glucose homeostasis via renal glucose reabsorption independently of changes in insulin sensitivity.
Decreased renal GLUT2 is responsible for elevated glucosuria in MC4R- and adrenaline-deficient mice
Renal GLUT2 was decreased by 20–25% in both MC4RPVH- and adrenaline-deficient mice compared with their littermate controls (Fig. 7a, b). In contrast, the levels of SGLT2, SGLT1, and GLUT1 were not different between the groups (ESM Fig.7). Adrenaline treatment restored renal GLUT2 levels in MC4RPVH-deficient and adrenaline-deficient mice (Fig. 7c, d). These results indicate the role of renal GLUT2 in mediating the effects of hypothalamic MC4R and adrenaline on glucose reabsorption.
Fig. 7.

Decreased renal GLUT2 and Na+/K+-ATPase levels/activity in hypothalamus-specific MC4R- and adrenaline-deficient mice. (a, b) Decreased renal cortical GLUT2 levels in hypothalamus-specific MC4R-deficient (Mc4rPVH) (a) and adrenaline-deficient (Pnmt-KO) (b) mice. (c, d) Increased renal cortical GLUT2 levels after treatment with adrenaline in hypothalamus-specific MC4R-deficient (c) and Pnmt-KO (d) mice. (e, f) Decreased renal Na+/K+-ATPase levels in hypothalamus-specific MC4R-deficient (e) and Pnmt-KO (f) mice. (g, h) Decreased renal Na+/K+-ATPase activity in hypothalamus-specific MC4R-deficient (g) and Pnmt-KO (h) mice. Proteins of interest and internal control were probed on the same membrane as described in the Methods. Two-tailed unpaired Student’s t test was used for comparisons. *p<0.05, **p<0.01, ***p<0.001. Error bars are mean ± SEM. Adr, adrenaline; Sal, saline; Mc4rPVH, Mc4rloxP/loxP with AAV-Cre; Ctrl, Mc4rloxP/loxP with AAV-GFP
To explain how the reduced renal GLUT2 increases glucosuria in MC4RPVH- and adrenaline-deficient mice, we measured the levels and activity of Na+/K+-ATPase in the plasma membrane fractions of the kidneys. The Na+ gradient maintained by Na+/K+-ATPase in the renal proximal tubular cells facilitates glucose reabsorption [31]. However, a decrease in GLUT2 causes accumulation of glucose in the proximal tubular cells (as depicted in the Graphical Abstract), which in turn is known to inhibit Na+/K+-ATPase [32, 33]. Consistent with these findings, both the MC4RPVH- and adrenaline-deficient mice had reduced levels and activity of Na+/K+-ATPase (Fig. 7e–h). The reduced Na+/K+-ATPase activity also probably accounts for natriuresis in MC4RPVH-deficient mice. Taken together, these results indicate that MC4RPVH- and adrenaline-deficient mice exhibit elevated glucosuria because of reduced renal GLUT2 and probably due to consequent decrease in renal Na+/K+-ATPase activity.
Discussion
Statement of principal findings
We have identified elevated glucosuria as a mechanism by which MC4R-deficient mice remain protected from hyperglycaemia despite obesity and insulin resistance. We report an essential role of the hypothalamic MC4R in renal glucose reabsorption through the effects of adrenaline on renal GLUT2.
Strengths and weaknesses of the study
A major strength of this study is that we have used both global and hypothalamus-specific MC4R-deficient obese or lean mice (male and female) to precisely define the brain regions that contribute to glucose homeostasis via MC4R. Moreover, MC4R deficiency was induced either since birth or in adult mice to account for the confounding effects of developmental or compensatory changes on glucose homeostasis. We employed a state-of-the-art graded [13C6]glucose infusion procedure and primary mouse kidney cells to further validate the effects of MC4R/adrenaline on glucose reabsorption.
A major weakness of this study is that we did not determine whether acute renal sympathetic nerve stimulation is enough to increase glucose reabsorption via renal GLUT2, which is beyond the scope of this study. We observed a 20–25% reduction in renal GLUT2 levels in MC4R-deficient mice. Although this moderate reduction in GLUT2 levels is sufficient to cause elevated glucosuria and maintain normal blood glucose levels in MC4R-deficient mice, we speculate that kidney-specific Glut2 knockout mice will exhibit significantly higher glucosuria than that observed in MC4R-deficient mice, and further establish the function of renal GLUT2 in regulating systemic glucose homeostasis.
Strengths and weaknesses in relation to other studies
Both weight-matched and obese MC4R-deficient mice showed hyperinsulinaemia and reduced insulin sensitivity to decrease blood glucose levels compared with their WT littermates. These data support previous observations that MC4R directly regulates plasma insulin levels and insulin sensitivity independently of obesity [7, 16]. Few previous studies had reported hyperglycaemia in Mc4r null mice fed ad libitum [1, 34, 35]. Mc4r null mice exhibit hyperphagia, which confounds blood glucose levels without fasting because the mice may consume food just before blood sample collection. Moreover, in most of the previous studies that evaluated glucose tolerance tests in Mc4r null mice [36–38], the amount of glucose was administered based on the mouse body weight; thereby disproportionately increasing the amount of glucose delivered in obese Mc4r null mice relative to their lean controls. Such a practice may misdiagnose obese mice as having impaired glucose tolerance. In clinical research or practice, a fixed amount of glucose (75 g) is administered orally to measure glucose tolerance in humans, regardless of their body weight [39]. Similarly, a fixed dose of glucose is also recommended for glucose tolerance tests in animal studies to avoid confounding effects of obesity on glucose clearance [40–42]. The bedding material of the mouse cages is another factor that could have confounded blood glucose levels in the reports that demonstrate either unchanged [20] or impaired [4] glucose tolerance in Mc4r null mice. It is critical to use aspen wood chip bedding (we use this material) in mouse cages for experiments that involve fasting because mice eat standard bedding materials such as corncob, as reported by the Jackson Laboratory [43] and supported empirically by our previous reports [22, 23].
Human MC4R deficiency decreases sympathetic tone as measured by adrenaline and noradrenaline levels in urine [44]. Consistent with our previous report [26], MC4RPVH-deficient mice exhibited reduced circulating adrenaline and noradrenaline levels. We further show in the current study that MC4R deficiency selectively in the Sim1 neurons decreases renal sympathetic nerve activity. The low sympathetic tone also protects MC4R-deficient mice and humans from hypertension [17, 44–46]. Moreover, MC4R-deficient mice have normal bowman’s space, glomerular area, mesangial matrix, albumin excretion, renal TGF-β and collagen expression [17], indicating that the elevated excretion of glucose observed in our present study is not the outcome of any known renal injury in these mice.
GLUT2 mutations cause glucosuria in mice [47, 48] and humans [49, 50]. Renal GLUT2 is increased in rodent models of diabetes [51, 52], causing enhanced glucose reabsorption and worsening hyperglycaemia. GLUT2 is also present in the liver and may be affected by adrenaline [53]. However, it is known that hepatocytes from Glut2 knockout mice do not exhibit any changes in glucose transport because of redundant glucose regulating pathways in the liver [54]. Moreover, liver-specific GLUT2-deficient mice [55] have normal glucose tolerance until the loss of beta cells, after which the mice start to show impaired glucose tolerance. In contrast, we report that reduced renal GLUT2 improved glucose tolerance in MC4RPVH- and adrenaline-deficient mice by elevating glucosuria, demonstrating the function of kidney-specific GLUT2 in systemic glucose homeostasis.
Unanswered questions and future research
The decrease in circulating adrenaline levels in the MC4R-deficient mice was likely caused by the combination of reduced renal sympathetic nerve activity and reduced adrenal Pnmt expression. Renal sympathetic nerve activity [56] and enzymes [57, 58] in the kidneys other than phenylethanolamine N-methyltransferase (PNMT) are reported to contribute to circulating or urine adrenaline levels. However, the mechanisms underlying the hypothalamic MC4R regulation of Pnmt expression and subsequently, secretion or synthesis of adrenaline are as yet unclear. Moreover, it is unknown whether MC4R signalling regulates renal Na+/K+-ATPase directly or secondary to renal GLUT2. Future studies are necessary to answer these questions.
Conclusion
In summary, this study demonstrates that MC4R selectively in the PVH contributes to glucose homeostasis by regulating glucose reabsorption via circulating adrenaline and renal GLUT2. These findings indicate that MC4R deficiency does not cause hyperglycaemia, even in the face of insulin resistance and obesity, because of elevated glucosuria mediated by low sympathetic tone.
Supplementary Material
Research in context.
What is already known about this subject?
Melanocortin 4 receptor (MC4R)-deficient humans and rodents do not develop hyperglycaemia despite obesity and Insulin resistance
MC4R mutations are the most common cause of known monogenic obesity
Mc4r knockout mice recapitulate the phenotype of MC4R-deflclent humans
What Is the key question?
What are the molecular and physiological bases of MC4R control of glucose homeostasis?
What are the new findings?
MC4R In the hypothalamus contributes to glucose homeostasis via regulation of renal glucose reabsorption
Circulating adrenaline mediates the effects of hypothalamic MC4R on glucose reabsorption
Renal glucose transporter GLUT2 mediates the effects of MC4R and adrenaline on glucosuria
How might this Impact on clinical practice In the foreseeable future?
Elevated glucosuria may explain why Individuals with MC4R deficiency do not develop hyperglycaemia despite obesity and Insulin resistance. Thus, It Is possible that blocking kidney-specific GLUT2 might protect against diabetes by Increasing glucose excretion
Acknowledgements
We thank D. P. Olson (University of Michigan, Ann Arbor, MI, USA), B. B. Lowell (Harvard University, Cambridge, MA, USA) for providing Mc4rloxP/loxP mice; V. Vallon (University of California, San Diego, CA, USA), L. Harrison-Bernard (LSUHSC, New Orleans, LA, USA) for discussion and critical comments on the research design of, and results obtained from, this study; G. Pryhuber, C. Poole and S. Mack (URMC Pediatric Histology Service, Rochester, NY, USA) for their help with cryostat and brain sections; K. Welle (University of Rochester Mass Spectrometry Resource Laboratory, Rochester, NY, USA) for help with the [13C6]glucose assay; V. K. Thomas and J. Zhang (URMC Center for Advanced Light Microscopy and Nanoscopy, Rochester, NY, USA) for help with microscopy. Some of the findings of this study were reported in abstract form at ENDO 2020, Endocrine Society Meeting.
Funding
This study was supported by funding from an Endocrine Fellows Foundation grant to AE. NIDDK/NIH DK113115 and DK122190 grants, in addition to Pilot Research Award and Startup funds from the Department of Medicine, URMC, to KHC. NIH/NHLBI HL084207, Department of Veterans Affairs BX004249, funding from the University of Iowa Fraternal Order of Eagles Diabetes Research Center and the Iowa Neuroscience Institute to KR. NIH instrument grant S10OD025242 to University of Rochester Mass Spectrometry Resource Laboratory. The URMC Pediatric Histology Service is supported by the Department of Pediatrics at the University of Rochester and NHLBI/NIH HL122700 and HL148861 grants to G. H. Deutsch, T. J. Mariani, and G. S. Pryhuber.
Abbreviations
- MC4R
Melanocortin 4 receptor
- qPCR
Quantitative real-time PCR
- POMC
Proopiomelanocortin
- SGLT
Sodium–glucose cotransporter
- WT
Wild type
Footnotes
Data availability
Data related to this manuscript are available on request from the authors.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- [1].Huszar D, Lynch CA, Fairchild-Huntress V, et al. (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88(1): 131–141 [DOI] [PubMed] [Google Scholar]
- [2].Farooqi IS, Yeo GS, Keogh JM, et al. (2000) Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. The Journal of clinical investigation 106(2): 271–279. 10.1172/JCI9397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Balthasar N, Dalgaard LT, Lee CE, et al. (2005) Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123(3): 493–505. 10.1016/j.cell.2005.08.035 [DOI] [PubMed] [Google Scholar]
- [4].Berglund ED, Liu T, Kong X, et al. (2014) Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nature neuroscience 17(7): 911–913. 10.1038/nn.3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tan HY, Steyn FJ, Huang L, Cowley M, Veldhuis JD, Chen C (2016) Hyperphagia in male melanocortin 4 receptor deficient mice promotes growth independently of growth hormone. The Journal of physiology 594(24): 7309–7326. 10.1113/jp272770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ramachandrappa S, Farooqi IS (2011) Genetic approaches to understanding human obesity. The Journal of Clinical Investigation 121(6): 2080–2086. 10.1172/JCI46044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L (2001) Central melanocortin receptors regulate insulin action. J Clin Invest 108(7): 1079–1085. 10.1172/jci12954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kuhnen P, Clement K, Wiegand S, et al. (2016) Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med 375(3): 240–246. 10.1056/NEJMoa1512693 [DOI] [PubMed] [Google Scholar]
- [9].Collet TH, Dubern B, Mokrosinski J, et al. (2017) Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol Metab 6(10): 1321–1329. 10.1016/j.molmet.2017.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Clement K, Biebermann H, Farooqi IS, et al. (2018) MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med 24(5): 551–555. 10.1038/s41591-018-0015-9 [DOI] [PubMed] [Google Scholar]
- [11].Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106(4): 473–481. 10.1172/jci10842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444(7121): 840–846. 10.1038/nature05482 [DOI] [PubMed] [Google Scholar]
- [13].Ganz ML, Wintfeld N, Li Q, Alas V, Langer J, Hammer M (2014) The association of body mass index with the risk of type 2 diabetes: a case–control study nested in an electronic health records system in the United States. Diabetology & Metabolic Syndrome 6(1): 50 10.1186/1758-5996-6-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wainberg M, Mahajan A, Kundaje A, et al. (2019) Homogeneity in the association of body mass index with type 2 diabetes across the UK Biobank: a Mendelian randomization study. PLOS Medicine 16(12): e1002982 10.1371/journal.pmed.1002982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schnurr TM, Jakupović H, Carrasquilla GD, et al. (2020) Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: a case-cohort study. Diabetologia 63:1324–1334. 10.1007/s00125-020-05140-5 [DOI] [PubMed] [Google Scholar]
- [16].Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD (2000) The central melanocortin system can directly regulate serum insulin levels. Endocrinology 141(9): 3072–3079. 10.1210/endo.141.9.7665 [DOI] [PubMed] [Google Scholar]
- [17].do Carmo JM, Tallam LS, Roberts JV, et al. (2009) Impact of obesity on renal structure and function in the presence and absence of hypertension: evidence from melanocortin-4 receptor-deficient mice. American journal of physiology Regulatory, integrative and comparative physiology 297(3): R803–R812. 10.1152/ajpregu.00187.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Iepsen EW, Zhang J, Thomsen HS, et al. (2018) Patients with obesity caused by melanocortin-4 receptor mutations can be treated with a glucagon-like peptide-1 receptor agonist. Cell Metab 28(1): 23–32.e23. 10.1016/j.cmet.2018.05.008 [DOI] [PubMed] [Google Scholar]
- [19].Stepp DW, Osakwe CC, Belin de Chantemele EJ, Mintz JD (2013) Vascular effects of deletion of melanocortin-4 receptors in rats. Physiol Rep 1(6): e00146–e00146. 10.1002/phy2.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Litt MJ, Okoye GD, Lark D, et al. (2017) Loss of the melanocortin-4 receptor in mice causes dilated cardiomyopathy. Elife 6: e28118 10.7554/eLife.28118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baron M, Maillet J, Huyvaert M, et al. (2019) Loss-of-function mutations in MRAP2 are pathogenic in hyperphagic obesity with hyperglycemia and hypertension. Nat Med 25(11): 1733–1738. 10.1038/s41591-019-0622-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chhabra KH, Adams JM, Fagel B, et al. (2016) Hypothalamic POMC deficiency improves glucose tolerance despite insulin resistance by increasing glycosuria. Diabetes 65(3): 660–672. 10.2337/db15-0804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chhabra KH, Morgan DA, Tooke BP, Adams JM, Rahmouni K, Low MJ (2017) Reduced renal sympathetic nerve activity contributes to elevated glycosuria and improved glucose tolerance in hypothalamus-specific Pomc knockout mice. Mol Metab 6(10): 1274–1285. 10.1016/j.molmet.2017.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shah BP, Vong L, Olson DP, et al. (2014) MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proceedings of the National Academy of Sciences 111(36): 13193 10.1073/pnas.1407843111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ebert SN, Rong Q, Boe S, Pfeifer K (2008) Catecholamine-synthesizing cells in the embryonic mouse heart. Annals of the New York Academy of Sciences 1148: 317–324. 10.1196/annals.1410.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tooke BP, Yu H, Adams JM, et al. (2019) Hypothalamic POMC or MC4R deficiency impairs counterregulatory responses to hypoglycemia in mice. Mol Metab 20: 194–204. 10.1016/j.molmet.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chhabra KH, Adams JM, Jones GL, et al. (2016) Reprogramming the body weight set point by a reciprocal interaction of hypothalamic leptin sensitivity and Pomc gene expression reverts extreme obesity. Mol Metab 5(10): 869–881. 10.1016/j.molmet.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang F, Flanagan J, Su N, et al. (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. The Journal of molecular diagnostics : JMD 14(1): 22–29. 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ding W, Yousefi K, Shehadeh LA (2018) Isolation, characterization, and high throughput extracellular flux analysis of mouse primary renal tubular epithelial cells. Journal of visualized experiments : JoVE(136): 57718 10.3791/57718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu H, Kishi T, Roseberry AG, et al. (2003) Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. The Journal of Neuroscience 23(18): 7143–7154. 10.1523/jneurosci.23-18-07143.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ghezzi C, Loo DDF, Wright EM (2018) Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61(10): 2087–2097. 10.1007/s00125-018-4656-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rivelli JF, Amaiden MR, Monesterolo NE, et al. (2012) High glucose levels induce inhibition of Na,K-ATPase via stimulation of aldose reductase, formation of microtubules and formation of an acetylated tubulin/Na,K-ATPase complex. Int J Biochem Cell Biol 44(8): 1203–1213. 10.1016/j.biocel.2012.04.011 [DOI] [PubMed] [Google Scholar]
- [33].Owada S, Larsson O, Arkhammar P, et al. (1999) Glucose decreases Na+,K+-ATPase activity in pancreatic beta-cells. An effect mediated via Ca2+-independent phospholipase A2 and protein kinase C-dependent phosphorylation of the α-subunit. J Biol Chem 274(4): 2000–2008. 10.1074/jbc.274.4.2000 [DOI] [PubMed] [Google Scholar]
- [34].Itoh M, Suganami T, Nakagawa N, et al. (2011) Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol 179(5): 2454–2463. 10.1016/j.ajpath.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ste Marie L, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD (2000) A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc Natl Acad Sci U S A 97(22): 12339–12344. 10.1073/pnas.220409497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Morgan DA, McDaniel LN, Yin T, et al. (2015) Regulation of glucose tolerance and sympathetic activity by MC4R signaling in the lateral hypothalamus. Diabetes 64(6): 1976–1987. 10.2337/db14-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].You P, Hu H, Chen Y, et al. (2016) Effects of melanocortin 3 and 4 receptor deficiency on energy homeostasis in rats. Scientific reports 6: 34938–34938. 10.1038/srep34938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mosialou I, Shikhel S, Liu J-M, et al. (2017) MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543(7645): 385–390. 10.1038/nature21697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].ADA (2018) 2. Classification and Diagnosis of Diabetes: Standards of Medical care in diabetes—2018. Diabetes Care 41(Suppl 1): S13–S27. 10.2337/dc18-S002%J [DOI] [PubMed] [Google Scholar]
- [40].McGuinness OP, Ayala JE, Laughlin MR, Wasserman DH (2009) NIH experiment in centralized mouse phenotyping: the Vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am J Physiol Endocrinol Metab 297(4): E849–855. 10.1152/ajpendo.90996.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J (2008) Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab 295(6): E1323–E1332. 10.1152/ajpendo.90617.2008 [DOI] [PubMed] [Google Scholar]
- [42].Ayala JE, Samuel VT, Morton GJ, et al. (2010) Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 3(9–10): 525–534. 10.1242/dmm.006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Garcia-Arocena D (2014) 7 Tips every dio mice user should know. Available from https://wwwjaxorg/news-and-insights/jax-blog/2014/may/seven-tips-every-dio-mice-user-should-know Accessed 05 November 2019
- [44].Greenfield JR, Miller JW, Keogh JM, et al. (2009) Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med 360(1): 44–52. 10.1056/NEJMoa0803085 [DOI] [PubMed] [Google Scholar]
- [45].Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE (2005) Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension 46(2): 326–332. 10.1161/01.HYP.0000175474.99326.bf [DOI] [PubMed] [Google Scholar]
- [46].Samuelsson A-MS, Mullier A, Maicas N, et al. (2016) Central role for melanocortin-4 receptors in offspring hypertension arising from maternal obesity. Proc Natl Acad Sci U S A 113(43): 12298–12303. 10.1073/pnas.1607464113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Guillam M-T, Hümmler E, Schaerer E, et al. (1997) Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nature Genetics 17(3): 327–330. 10.1038/ng1197-327 [DOI] [PubMed] [Google Scholar]
- [48].Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M (2000) Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem 275(31): 23751–23758. 10.1074/jbc.M002908200 [DOI] [PubMed] [Google Scholar]
- [49].Sakamoto O, Ogawa E, Ohura T, et al. (2000) Mutation analysis of the GLUT2 gene in patients with Fanconi–Bickel syndrome. Pediatr Res 48(5): 586–589. 10.1203/00006450-200011000-00005 [DOI] [PubMed] [Google Scholar]
- [50].Grunert SC, Schwab KO, Pohl M, Sass JO, Santer R (2012) Fanconi–Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol Genet Metab 105(3): 433–437. 10.1016/j.ymgme.2011.11.200 [DOI] [PubMed] [Google Scholar]
- [51].Kamran M, Peterson RG, Dominguez JH (1997) Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. Journal of the American Society of Nephrology 8(6): 943–948 [DOI] [PubMed] [Google Scholar]
- [52].Chin E, Zamah AM, Landau D, et al. (1997) Changes in facilitative glucose transporter messenger ribonucleic acid levels in the diabetic rat kidney. Endocrinology 138(3): 1267–1275. 10.1210/endo.138.3.5015 [DOI] [PubMed] [Google Scholar]
- [53].Miura T, Kato M, Iwamoto N, et al. (2000) Effect of epinephrine on GLUT2 protein content in mouse liver. Biol Pharm Bull 23(11): 1374–1376. 10.1248/bpb.23.1374 [DOI] [PubMed] [Google Scholar]
- [54].Guillam MT, Burcelin R, Thorens B (1998) Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proc Natl Acad Sci U S A 95(21): 12317–12321. 10.1073/pnas.95.21.12317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Seyer P, Vallois D, Poitry-Yamate C, et al. (2013) Hepatic glucose sensing is required to preserve β cell glucose competence. The Journal of Clinical Investigation 123(4): 1662–1676. 10.1172/JCI65538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang WH, Zhou QN, Lu YM, et al. (2018) Renal denervation reduced ventricular arrhythmia after myocardial infarction by inhibiting sympathetic activity and remodeling. J Am Heart Assoc 7(20): e009938 10.1161/jaha.118.009938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ziegler MG, Kennedy B, Elayan H (1989) Rat renal epinephrine synthesis. The Journal of Clinical Investigation 84(4): 1130–1133. 10.1172/JCI114276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ziegler MG, Aung M, Kennedy B (1997) Sources of human urinary epinephrine. Kidney International 51(1): 324–327. 10.1038/ki.1997.40 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.