

Abstract
The identification of major histocompatibility complex (MHC)-binding peptides in mass spectrometry (MS)-based immunopeptideomics relies largely on database search engines developed for proteomics data analysis. However, because immunopeptidomics experiments do not involve enzymatic digestion at specific residues, an inflated search space leads to a high false positive rate and low sensitivity in peptide identification. In order to improve the sensitivity and reliability of peptide identification, we developed DeepRescore, a post-processing tool that combines peptide features derived from deep learning predictions, namely accurate retention time and MS/MS spectra predictions, with previously used features to rescore peptide-spectrum matches. Using two public immunopeptidomics datasets, we showed that rescoring by DeepRescore increased both the sensitivity and reliability of MHC-binding peptide and neoantigen identifications compared to existing methods. We also showed that the performance improvement was, to a large extent, driven by the deep learning-derived features. DeepRescore was developed using NextFlow and Docker and is available at https://github.com/bzhanglab/DeepRescore.
Keywords: Bioinformatics, Retention time, Proteomics, Deep learning, Immunopeptidomics
1. Introduction
The major histocompatibility complex (MHC), also called the human leukocyte antigen (HLA) complex in humans, can bind to peptide antigens and display them on the cell surface for recognition by T-cells. The rich repertoire of peptides presented by MHC class I (MHC-I) and MHC class II (MHC-II) complexes, referred to as the immunopeptidome, reflects the health state of a cell. MHC-binding peptides derived from mutated and other cancer-specific proteins, pathogens, and self-peptides, in the case of autoimmunity, serve as leading targets for T-cell recognition. Liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS) is one of the most commonly utilized approaches to comprehensively interrogate the naturally presented MHC-binding peptide repertoire [1].
The most widely used method for peptide identification from LC-MS/MS-based immunopeptidomics data is database searching, in which MS/MS spectra are searched against a reference protein sequence database or a customized, sample-specific protein sequence database. This method was originally developed for the analysis of MS/MS spectra generated in bottom-up shotgun proteomics studies [2]. Database search tools developed for the analysis of shotgun proteomics data, such as Comet [3], MS-GF+ [4], X!Tandem [5], MaxQuant [6], and Mascot [7], can be and have been used for the analysis of immunopeptidomics data [8, 9]. However, this comes with a major risk. In shotgun proteomics experiments, proteins are digested into peptides by enzymes such as trypsin before LC-MS/MS analysis [10]. Due to the sequence specificity of enzyme cleavage, the database search space can be significantly reduced through an enzyme-specific search. In contrast, immunopeptidomics experiments do not utilize enzymatic digestion, and thus the database search space is much larger. This inflated search space can lead to a high false positive rate and low sensitivity in the identification of MHC-binding peptides [2, 11].
Several tools, such as MS-Rescue [12] and MHCquant [13], have been developed to address this limitation. To improve the sensitivity of MHC class I peptide identification, MS-Rescue uses high confidence peptide identifications, i.e., the peptides passing 1% false discovery rate (FDR) threshold, to train a model in order to rescore peptide identifications of lower confidence. Similarly, MHCquant uses Percolator [14] to rescore peptide-spectrum matches (PSMs) identified by Comet. Percolator uses semi-supervised machine learning to build an SVM-based classifier that can better discriminate between target and decoy PSMs. This is achieved by incorporating additional features not initially used in PSM scoring in the classifier. Classifier training begins with parsing the initial database searching results to form a group of high-scoring PSMs from the target proteins (positives) and another group of PSMs from decoy proteins (negatives). Assuming there are false positives and false negatives in the initial results, the learned classifier is applied to the entire set and computes a new score for each PSM. The procedure is repeated multiple times until convergence. This new score routinely increases the number of confident identifications because original search engine scores typically fail to address the specific characteristics of individual experiments. Compared to database searching without rescoring, rescoring using Percolator in MHCquant significantly improves the sensitivity of peptide identification in immunopeptidomics data analysis [13].
The performance of Percolator is tightly associated with the features used for semi-supervised learning. Recent advancements in deep learning have enabled accurate peptide retention time (RT) prediction [15–17] and MS/MS spectrum prediction [15, 18, 19] for a given peptide sequence. We reasoned that adding these new, peptide-specific features to Percolator could further improve the confidence and sensitivity of peptide identification in immunopeptidomics data analysis. Here, we present DeepRescore, a novel immunopeptidomics data analysis tool that leverages deep learning-derived peptide features to rescore PSMs. We demonstrate the performance of DeepRescore using two public immunopeptidomics datasets and experimental validation with synthetic peptides.
2. Materials and methods
2.1. Datasets and processing
Two public immunopeptidomics datasets were used in our study. The first dataset was from an in-depth immunopeptidomics analysis of native melanoma tissue samples from 25 melanoma patients, which was generated by a Q Exactive HF mass spectrometer [9]. Raw MS/MS data files were downloaded from PRIDE with accession number PXD004894. The data for HLA class I binding peptides from the sample Mel15, which included 938,131 MS/MS spectra, were used in our study and were denoted as dataset D1. The second dataset was from a large immunopeptidomics analysis of 95 mono-allelic cell lines, which was generated by a Fusion Lumos (Thermo Scientific) [20]. Raw MS/MS data files were downloaded from MassIVE with accession number MSV000084442. The data files from the A*11:01 mono-allelic cell line, which included 279,700 MS/MS spectra, were used in this study and were denoted as dataset D2. Raw MS/MS data files from both datasets were converted to MGF files using ProteoWizard (v3.0.19014) [21].
We performed database searching for both datasets using four widely used search engines: MS-GF+ (v2018.10.15), Comet (v2018.01 rev. 4), X!Tandem (v2017.2.1.2), and MaxQuant (v1.6.5.0). The first dataset (D1) was searched against a customized database. Specifically, somatic mutations for the tumor sample were downloaded from the original study and annotated using ANNOVAR [22]. Then Customprodbj (https://github.com/bzhanglab/customprodbj) [17] was used to build a customized database, which includes reference proteins in the human RefSeq protein database (56,659 sequences), variant proteins containing somatic mutations, and 245 contaminant protein sequences. These processes were automated using NeoFlow (https://github.com/bzhanglab/neoflow) [17]. For the second dataset (D2), the human protein database (63,691 sequences) downloaded from UCSC Genome Browser was used, with 245 contaminant protein sequences appended. For both datasets, the MS/MS data were searched against the protein databases with decoy sequences, and parameters for database searching were set as follows: enzyme specificity, unspecific cleavage; variable modification, oxidation of methionine; fixed modification, carbamidomethyl of cysteine; precursor ion mass tolerance, 10 ppm; MS/MS mass tolerance, 0.02 Da. Only peptides with length between 8 and 25 were considered. Peptide identifications were filtered using the target-decoy strategy [23] implemented in PGA [24] based on the PSM scores reported by each search engine (i.e., Evalue from MS-GF+, Comet, and X!Tandem, and posterior error probability from MaxQuant), and FDR was controlled at 1% at both PSM and peptide levels.
2.2. Deep learning-derived PSM quality-indicating features
For each PSM, we considered two PSM quality-indicating features: (1) the absolute difference between the predicted retention time (RT) of the associated peptide and the observed RT associated with the spectrum, and (2) the similarity between the predicted MS/MS spectrum of the associated peptide and the experimental MS/MS spectrum.
For RT prediction, we used AutoRT (https://github.com/bzhanglab/AutoRT), a peptide sequence-based RT prediction tool that uses deep learning and transfer learning to achieve high prediction accuracy [17]. Specifically, deep neural network models we previously trained on the basis of a large public dataset containing 136,791 peptides [25] were employed as the base models for transfer learning in this study. To train LC-MS/MS run-specific models, the base models were fine-tuned using high-quality peptides identified in each run and their RTs using transfer learning. In Section 3.2, high-quality peptides were defined as the peptides passing 1% FDR filtering at both PSM and peptide levels according to at least three of the four search engines to ensure high quality of the training samples for all search engines. In Section 3.3, high-quality peptides were defined as the peptides passing 1% FDR for each search engine separately for usability in real applications. In order to avoid overfitting, dropout and early stop were used in AutoRT. The run-specific models were used to predict RTs for all PSMs, and delta RT, denoted as RTdelta, was calculated for each PSM as follows:
GPTime, a traditional machine learning based RT prediction tool [26], was also included in this study for comparison.
For MS/MS spectrum prediion, we used pDeep2 and the pre-trained model from the original study [19]. For each PSM, the normalized spectral angle (SA) between the predicted MS/MS spectrum and the experimental MS/MS spectrum was calculated as previously described [27]:
where S1 and S2 are the normalized spectra for the experimental MS/MS spectrum and the predicted MS/MS spectrum, respectively.
2.3. Implementation of DeepRescore
DeepRescore was implemented using Docker and NextFlow. The workflow of DeepRescore is shown in Figure 1. The current implementation supports four search engines: MS-GF+, Comet, X!Tandem, and MaxQuant. DeepRescore takes PSM identification results from a search engine and MS/MS data in MGF format as input, although the latter is not required for MaxQuant. For each PSM, three sets of features are extracted or calculated. The first set is the set of search engine specific features, which include multiple scores for each search engine (e.g., Score, PEP, and Delta Score for MaxQuant). The second set is the set of search engine independent features that are commonly reported for all search engines. This set of features was adopted from our previous studies [28]. The third set includes the deep learning derived features described above, including delta RT and SA. All features used in DeepRescore are summarized in Supplementary Table S1. These features were integrated by Percolator (v3.4) [29] to generate a new score for each PSM. These new scores were used to filter the PSMs by controlling FDR at 1% at both PSM and peptide levels. The results of DeepRescore can be imported into PDV [30] for visualization. The source code of DeepRescore is available at https://github.com/bzhanglab/DeepRescore.
Figure 1. Overview of the study design and the DeepRescore workflow.
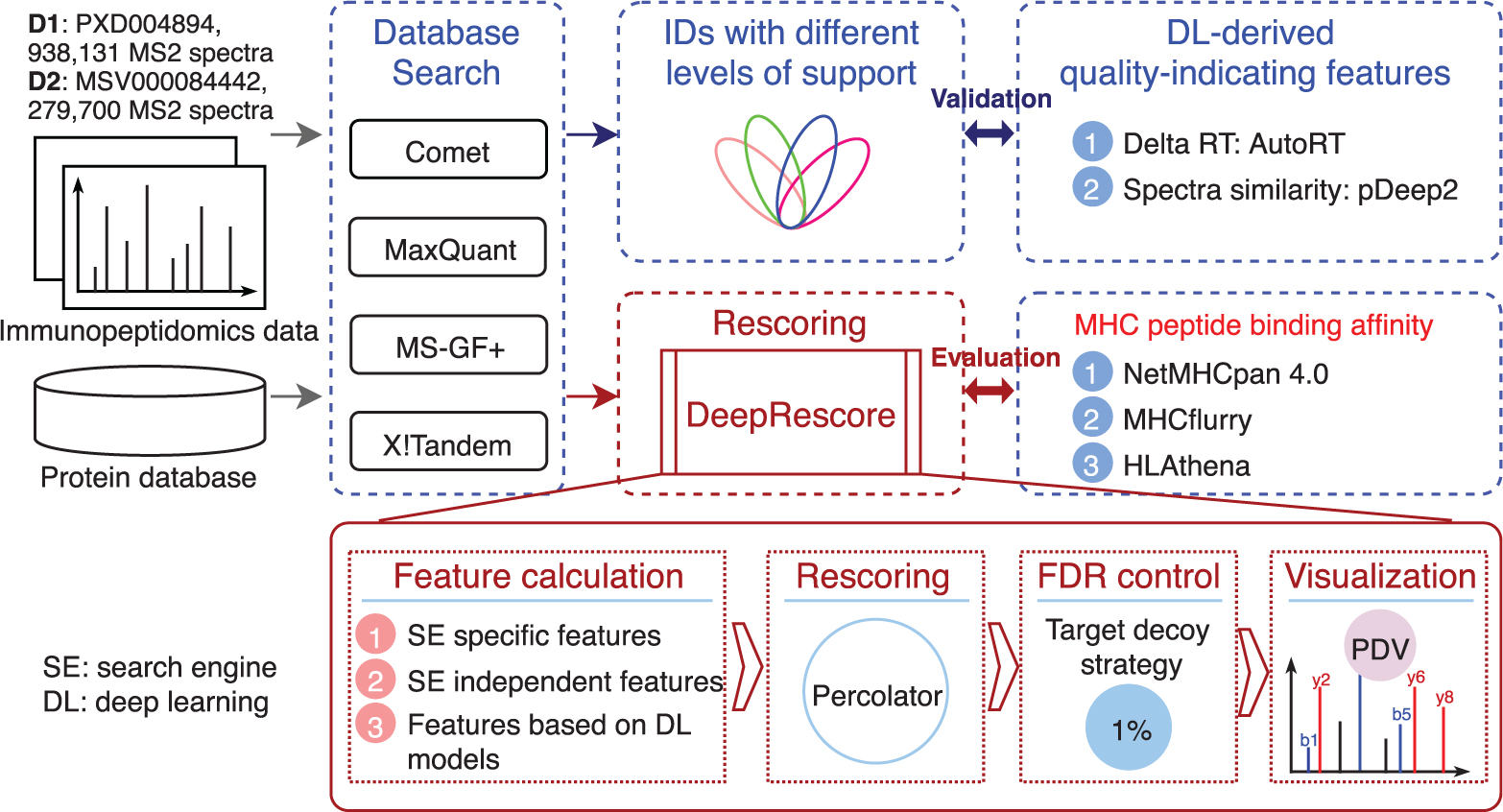
2.4. Evaluation
To evaluate the quality of peptides identified by DeepRescore, we calculated MHC-peptide binding affinity using three tools: NetMHCpan (v4.0) [31], MHCflurry (v1.6.0) [32], and HLAthena (http://hlathena.tools/) [20], respectively. Only peptides with length between 8 and 11 were considered unless otherwise noted. For each HLA allele, binding affinities were predicted for all identified peptides, and percentile rank scores were calculated for each peptide. The tumor sample in D1 has multiple HLA alleles. For a given peptide, the best percentile rank score across all HLA alleles of the sample was used. Following previous studies [31, 33], we defined MHC binders as peptides with a percentile rank score of 2% or less.
2.5. Peptide synthesis and LC-MS/MS analysis
Reference peptides were synthesized by Biomatik (Biomatik, Canada) and analyzed using a nano-LC 1000 system (Thermo Fisher Scientific, San Jose, CA) coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Briefly, peptide at 1μM concentration were pooled together, and the 18 peptide mixture was loaded onto a 2cm × 100μm ID pre-column trap packed with Reprosil-Pur Basic C18 1.9 μm beads (Dr.Maisch GmbH, Germany). The peptides were resolved on a 5cm × 150μm ID analytical column packed with the same stationary phase. A 90-minute gradient of 2–30% acetonitrile/0.1% formic acid at a flow rate of 800nl/min was used. The peptides were directly electrosprayed into the mass spectrometer operated in a data-dependent mode with ‘Top 10’ method. The full MS scan was performed in Orbitrap in the range of 300–1400m/z at 120,000 resolution followed by HCD fragmentation (CE32%) and MS/MS acquisition (15,000 resolution). Precursor isolation width was set at 2m/z, with AGC of 1X105, and maximum ion accumulation time of 120ms. The dynamic exclusion was set to 2s. The raw MS/MS data was converted to MGF file using ProteoWizard (v3.0.19014). Then the MS/MS data was searched against a database which contains the selected synthetic peptides using Comet (v2018.01 rev. 4). The parameters for database searching were set as follows: enzyme specificity, unspecific cleavage; variable modification, oxidation of methionine; fixed modification, carbamidomethyl of cysteine; precursor ion mass tolerance, 10 ppm; MS/MS mass tolerance, 0.02 Da. The identification result was loaded into PDV for visualization.
3. Results and Discussion
3.1. Different search engines give vastly different peptide identifications
We applied four widely used search engines, namely Comet, MS-GF+, X!Tandem, and MaxQuant, to the two immunopeptidomics datasets (D1 and D2). For D1, Comet, MS-GF+, X!Tandem, and MaxQuant identified 23400, 22789, 20727, and 17122 unique peptide sequences, respectively (Figure 2A, left). Comet identified the most peptides, 3%, 13% and 37% more peptides than MS-GF+, X!Tandem, and MaxQuant, respectively. Together, the four search engines identified a total of 31447 unique peptides. Among these peptides, only 33% were commonly identified by all four search engines, whereas 24% were uniquely identified by only one of them.
Figure 2. Search engine comparison and validation of two deep learning-derived quality-indicating features.
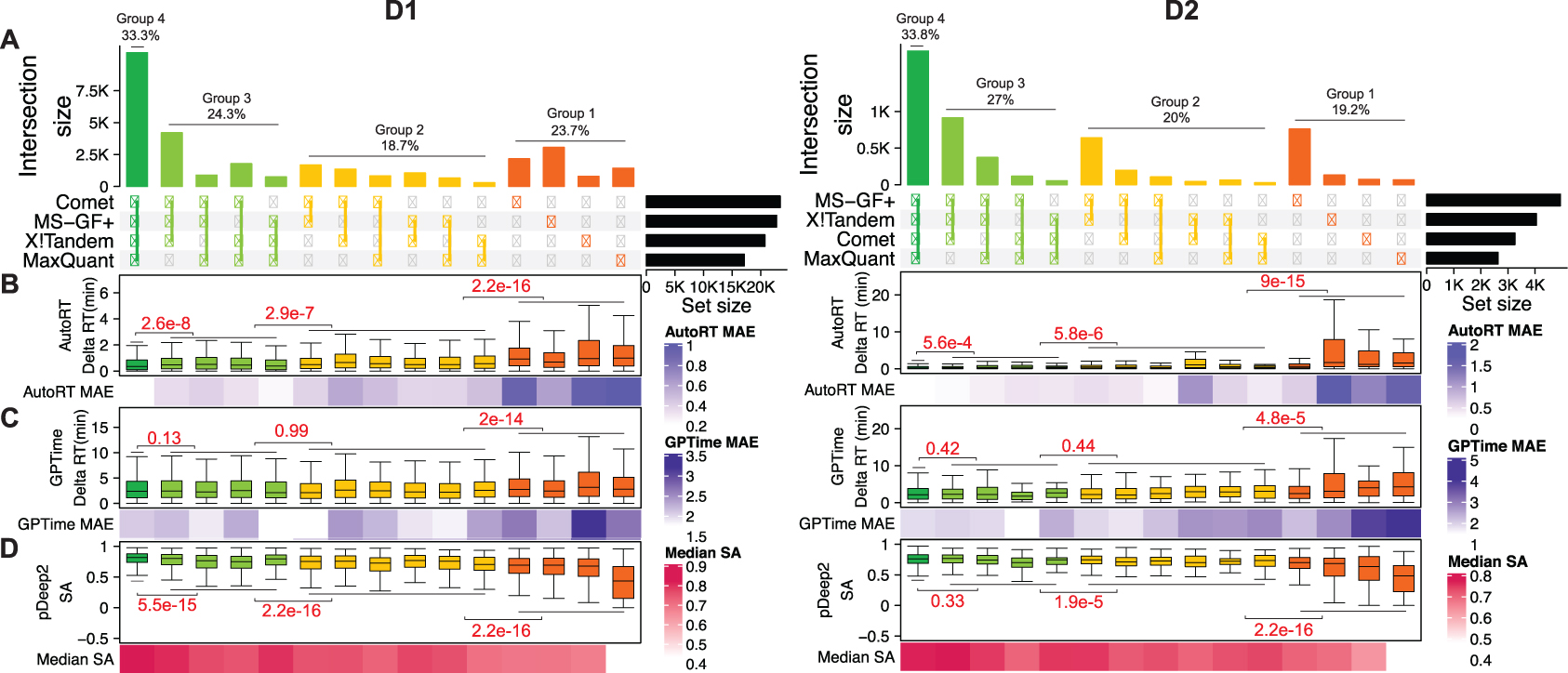
(A) Comparison of the peptides identified by four search engines in two immunopeptidomics datasets. (B) Distribution of AutoRT-derived delta RT for different peptide groups shown in A. (C) Distribution of GPTime-derived delta RT for different peptide groups. (D) SA (spectra angle) distribution for different peptide groups. The red numbers showing above the horizontal lines are p values from Kolmogorov–Smirnov test of the neighboring peptide groups. MAE: median absolute error. The boxplots representing the delta RT distribution of AutoRT and GPTime for the peptides identified by four search engines and three search engines were from the testing data (20% of the peptides in each run identified by at least three search engines) which were not used during training.
For D2, Comet, MS-GF+, X!Tandem, and MaxQuant identified 3255, 4935, 4048, and 2635 unique peptide sequences, respectively (Figure 2A, right). MS-GF+ identified the most peptides, 52%, 22%, and 87% more peptides than Comet, X!Tandem, and MaxQuant, respectively. The four search engines identified a total of 5,397 unique peptides. Among these peptides, only 34% were commonly identified by all the four search engines, whereas 19% were uniquely identified by only one of them.
These results showed that applying different search engines to the same immunopeptidomics dataset gives vastly different peptide identification results. Such inconsistency has been previously reported [13, 34]. Low overlap between results from different search engines suggests that considerable room for improvement remains for individual search engines.
3.2. Validity of deep learning-derived PSM quality-indicating features
One effective approach to improve search engine results is to rescore PSMs by considering additional features [14]. We reasoned that features indicating PSM quality that are independent of search engine scoring would be strong features for such rescoring. Here, we considered two such features: (1) delta RT: the absolute difference between predicted RT and the experimentally observed RT of a peptide, and (2) SA: the similarity between predicted MS/MS spectrum of a peptide and corresponding experimental MS/MS spectrum. Delta RT and SA were computed based on two deep learning tools, AutoRT and pDeep2, respectively. In both datasets, target PSMs showed considerably lower delta RTs and higher SAs compared to decoy PSMs across all four search engines (Figure S1), supporting the utility of delta RT and SA as PSM quality-indicating features.
To further evaluate the validity of these features as PSM quality-indicating features, we classified the union of peptides identified by any of the four search engines (1% FDR) into four groups based on the number of search engines identifying each peptide. Specifically, groups 1, 2, 3 and 4 represent peptides that were identified by 1, 2, 3, and 4 search engines, respectively. In general, peptides identified by more search engines are more reliable than those identified by fewer search engines. Thus, a good PSM quality-indicating feature should be able to distinguish these four groups.
For comparison, we calculated delta RTs based on both AutoRT prediction and GPTime prediction. The RTs predicted by AutoRT were in general much closer to experimentally observed RTs in both datasets than the RTs predicted by GPTime, as indicated by much lower delta RTs (Figure 2B–C). According to both calculations, group 4 peptides showed the lowest delta RTs, whereas group 1 showed the highest delta RTs in both datasets. We further compared the delta RTs between all neighboring groups (4 vs. 3, 3 vs. 2, and 2 vs. 1). In both datasets, all three comparisons showed significant differences (p<0.01, Kolmogorov–Smirnov test) according to delta RTs calculated based on AutoRT prediction (Figure 2B), whereas only one out of the three comparisons showed a significant difference according to delta RTs calculated based on GPTime prediction (Figure 2C).
For SA scores calculated by pDeep2, group 4 peptides showed the highest SA scores, whereas group 1 showed the lowest SA scores in both datasets (Figure 2D). In the comparison between neighboring groups, all three comparisons showed significant difference in D1. In D2, two comparisons were statistically significant, and one was not significant, which may be explained by the small sample size of D2.
Together, these results showed that both delta RT derived from AutoRT and SA derived from pDeep2 are useful PSM quality indicating features. Moreover, we also showed that AutoRT outperforms GPTime in RT prediction, and more accurate RT prediction provides a better indication of the quality of peptide identifications.
3.3. DeepRescore increases the sensitivity of peptide identification
To improve search engine results through PSM rescoring, we developed DeepRescore, which considers three types of feature sets for rescoring: (1) search engine specific features; (2) search engine independent features, and (3) deep learning-derived features. Based on these features, Percolator was used to rescore all PSMs, including both target and decoy PSMs, and these new scores were used to filter the PSMs.
In D1, when used in combination with DeepRescore, Comet, MaxQuant, MS-GF+, and X!Tandem identified 35683, 34204, 28104 and 32776 unique peptide sequences (1% FDR, Supplementary Table S2), respectively, which were 56%, 109%, 27% and 61% higher, respectively, than the numbers originally reported for each search engine at the same FDR level (Figure 3A). Such improvements were reproducible across different FDR thresholds from 0.5% to 10% (Figure S2). In D2, DeepRescore increased peptide identification by 61%, 109%, 10%, and 34% for Comet, MaxQuant, MS-GF+, and X!Tandem, respectively, at 1% FDR (Figure 3A, Supplementary Table S3), and these improvements were also reproducible for different FDR levels (Figure S2).
Figure 3. Sensitivity of DeepRescore and feature contributions.
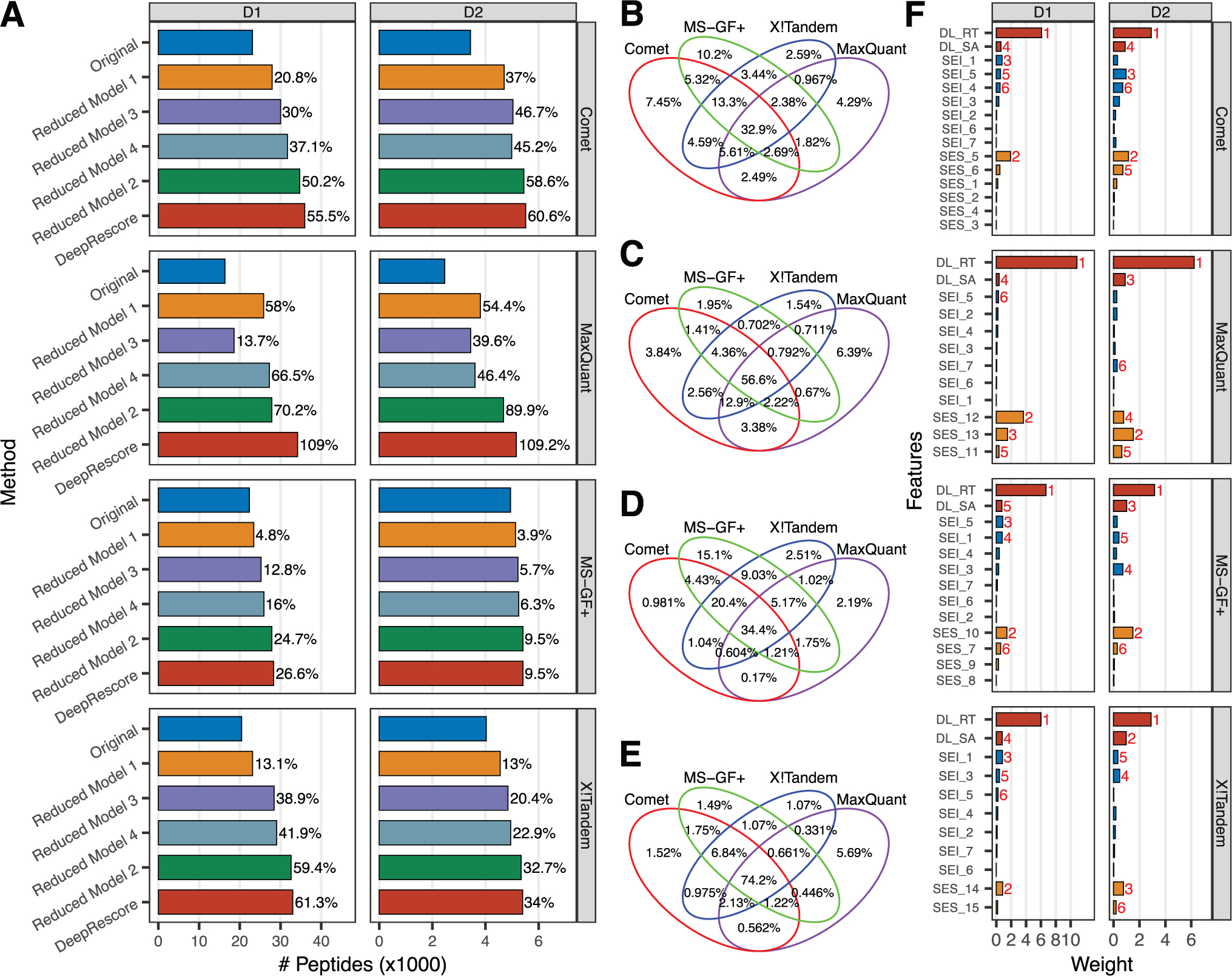
(A) The numbers of unique peptide sequences identified by different methods in datasets D1 and D2, respectively. Original: no rescoring; DeepRescore: rescoring using all the features available in DeepRescore; Reduced Model 1: dropping delta RT and SA from the full DeepRescore model; Reduced Model 2: dropping all other features from DeepRescore while keeping only delta RT, SA, and the search engine’s original PSM score; Reduced Model 3: further dropping delta RT from Reduced Model 2; Reduced Model 4: further dropping SA from Reduced Model 2. (B-E) Comparisons of peptides identified from datasets D1 (B-C) or D2 (D-E) by the four search engines either with or without DeepRescore. (F) Feature weights extracted from the iteratively trained SVM as part of the Percolator algorithm for each search engine on dataset D1 and D2.
Without PSM rescoring, MaxQuant identified the fewest peptides among the four search engines in both datasets. However, after PSM rescoring by DeepRescore, the numbers of identified peptides were comparable across different search engines (Figure 3A). Importantly, after rescoring by DeepRescore, the proportions of peptides commonly identified by all four search engines increased from 33% to 57% in D1 and from 34% to 74% in D2 (Figure 3B–E).
We also compared the peptides identified by DeepRescore with those reported in the original studies [9, 20]. Four and three search engines identified more peptides than the original reports for D1 (Figure S3) and D2 (Figure S4A), respectively. For example, Comet with DeepRescore identified 55% more peptides in D1 compared to the original report (Figure S3). We observed this increase despite using a 1% peptide FDR in this study, which is more stringent than the thresholds used in the original studies. Specifically, 1% PSM FDR was used in the original studies for D1 and D2.
In summary, these results clearly demonstrate the power of DeepRescore for increasing the sensitivity of peptide identification in immunopeptidomics.
3.4. Dominant contribution of the deep learning-derived features in DeepRescore
To investigate the contributions of the deep learning-derived features to performance improvement, we performed rescoring using a series of reduced models (Figure 3A). Compared to the full DeepRescore models, dropping delta RT and SA (Reduced Model 1) led to greatly reduced peptide identifications for all search engines in both datasets. In contrast, dropping all other features from DeepRescore while keeping only delta RT, SA, and the search engine’s original PSM score (Reduced Model 2) led to only very minor reductions in peptide identifications for three out of the four search engines in both datasets. These results demonstrated dominant contributions of the deep learning-derived features in DeepRescore. Compared to Reduced Model 2, further dropping delta RT (Reduced Model 3) or SA (Reduced Model 4) led to evident reductions in peptide identifications, suggesting complementary contributions from these two deep-learning derived features.
We also investigated the contribution of individual features by looking at the weights of features in rescoring, which were extracted from the iteratively trained SVM as part of the Percolator algorithm. Remarkably, delta RT showed the highest weight among all features across all search engines in both datasets (Figure 3F). SA was also among the top five features in all comparisons.
Of note, delta RT was used as a feature for PSM rescoring in immunopeptidomics data analysis in a recently published study [13], in which the data did not support delta RT as an effective feature for improving peptide identification. In that study, delta RT was computed based on OpenMS RTModel, which was based on oligo-kernel support vector regression. One possible explanation for these apparently contradictory conclusions is that AutoRT might be more accurate for RT prediction, which is critical for accurate delta RT estimation and effective PSM rescoring. Consistent with this hypothesis, we replaced AutoRT derived delta RT with GPTime derived delta RT and observed obvious performance reductions across all four search engines in both datasets (Figure S5).
Results in Figure 3F showed that delta RT is a stronger feature than SA. We note that the model we used for MS/MS spectrum prediction was trained on the basis of proteomics data from trypsin digested proteome rather than immunopeptidomics data. Thus, the model can be potentially improved by using a large, high quality immunopeptidomics data for training when it becomes available [2].
Taken together, these results demonstrated that DeepRescore performance relies on accurate RT and MS/MS spectrum predictions and that these deep learning-derived features are the dominant contributors to the improvements provided by DeepRescore.
3.5. Peptides identified by DeepRescore are of high quality
To further evaluate the quality of the peptides identified by DeepRescore, we used MHC-peptide binding affinity as a systematic and unbiased quantitative metric and three MHC-peptide binding prediction tools (NetMHCpan, MHCflurry and HLAthena) were used in this study. Prediction models underlying these three tools use different algorithms and training data. Although all three tools have been shown to provide high prediction accuracy, they have unique strengths and weaknesses and thus provide independent data for our evaluation. We compared peptide identification results from (1) original search engine scores without PSM rescoring, (2) rescoring using Reduced Model 1, in which delta RT and SA were dropped from the full DeepRescore model, and (3) rescoring using the full DeepRescore model. Only peptides with length between 8 and 11 were considered in this evaluation, which covered at least 75% of the identified peptides across different search engines in the two datasets (Figure S6). For both datasets, the vast majority of the peptides identified by methods (1) and (2) were also identified by method (3) (Figure 4A). Among all peptides with length between 8 and 11 identified by DeepRescore, 84%−95% were considered MHC binders by the three prediction tools across all search engines in both datasets, which was comparable to the proportions resulted from method (1) and (2) (Figure S7). The ratios of MHC binders among peptides uniquely identified by DeepRescore were 86% – 93% for D1 and 71% – 86% for D2, and these ratios were obviously higher than those among peptides not identified by DeepRescore (Figure 4C). In the results above, MHC binders were defined as peptides with a percentile rank score of 2% or less for predicted MHC-peptide binding affinity [31, 33]. Similar patterns were observed with a wide range of cutoffs (Figure S8). These results suggest a higher proportion of false positives among peptides not identified by DeepRescore compared to those uniquely identified by DeepRescore. Remarkably, in HLAthena-based evaluation, peptides uniquely identified by DeepRescore in D2 also showed obviously higher ratios of binders than those uniquely reported in the original study even though HLAthena was trained using the peptides identified in D2 in the original study (Figure S4B). Thus, DeepRescore not only increases the sensitivity of peptide identification but also effectively reduces false positives.
Figure 4. Quality evaluation of DeepRescore identifications.
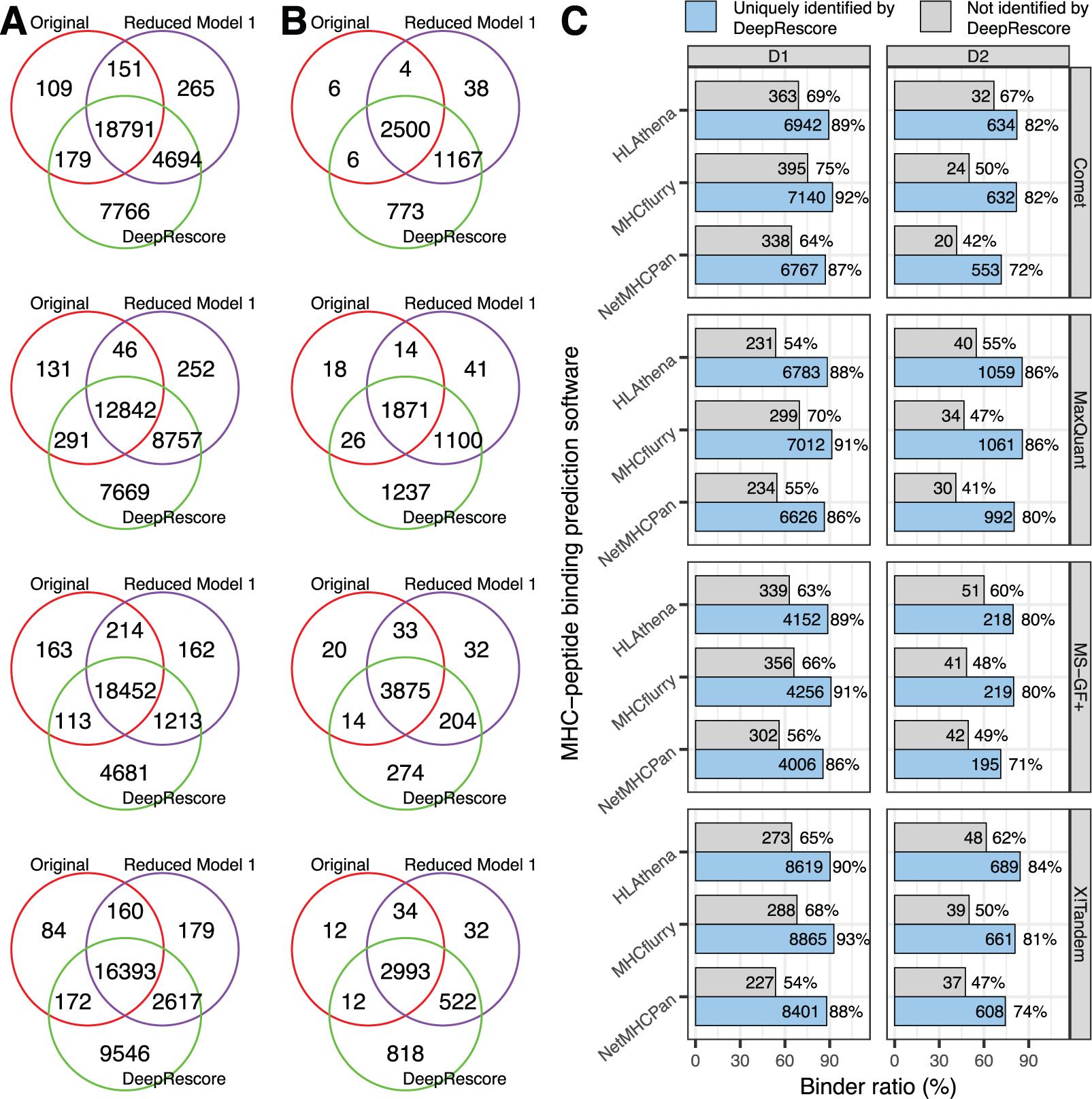
(A) Venn diagrams comparing peptides identified by different methods in dataset D1. (B) Venn diagrams comparing peptides identified by different methods in dataset D2. (C) Ratios of MHC binders predicted by different tools among peptides uniquely identified by DeepResore or among peptides not identified by DeepRescore in the two datasets.
3.6. Neoantigen identification using DeepRescore
Neoantigens encoded by tumor-specific somatic mutations are ideal targets for T cell-based immunotherapy, and they could be identified by an immunoproteogenomics approach, in which immunopeptidomics data are searched against sample-specific, customized protein sequence databases derived from paired DNA sequencing data. We built a customized database for D1 based on the somatic variants detected in the same sample using DNA sequencing data and then searched the MS/MS data against the customized database using Comet followed by PSM rescoring using DeepRescore. Our analysis identified 12 unique putative neoantigens with length between 8 and 12 (Supplementary Table S4). For the same sample, the original study reported 8 putative neoantigens, and a recent reanalysis of the sample by Bichmann et al. [13] reported 11 putative neoantigens. All but two neoantigens reported in those studies were identified by DeepRescore (Figure 5A). The two neoantigens not identified by DeepRescore but reported in the previous studies had high RT errors and low SAs (Tables S4) and are likely to be false positives. Taken together, the results demonstrated the benefit of using DeepRescore for neoantigen identification from immunopeptidomics data.
Figure 5. Comparison of neoantigen identification using different methods.
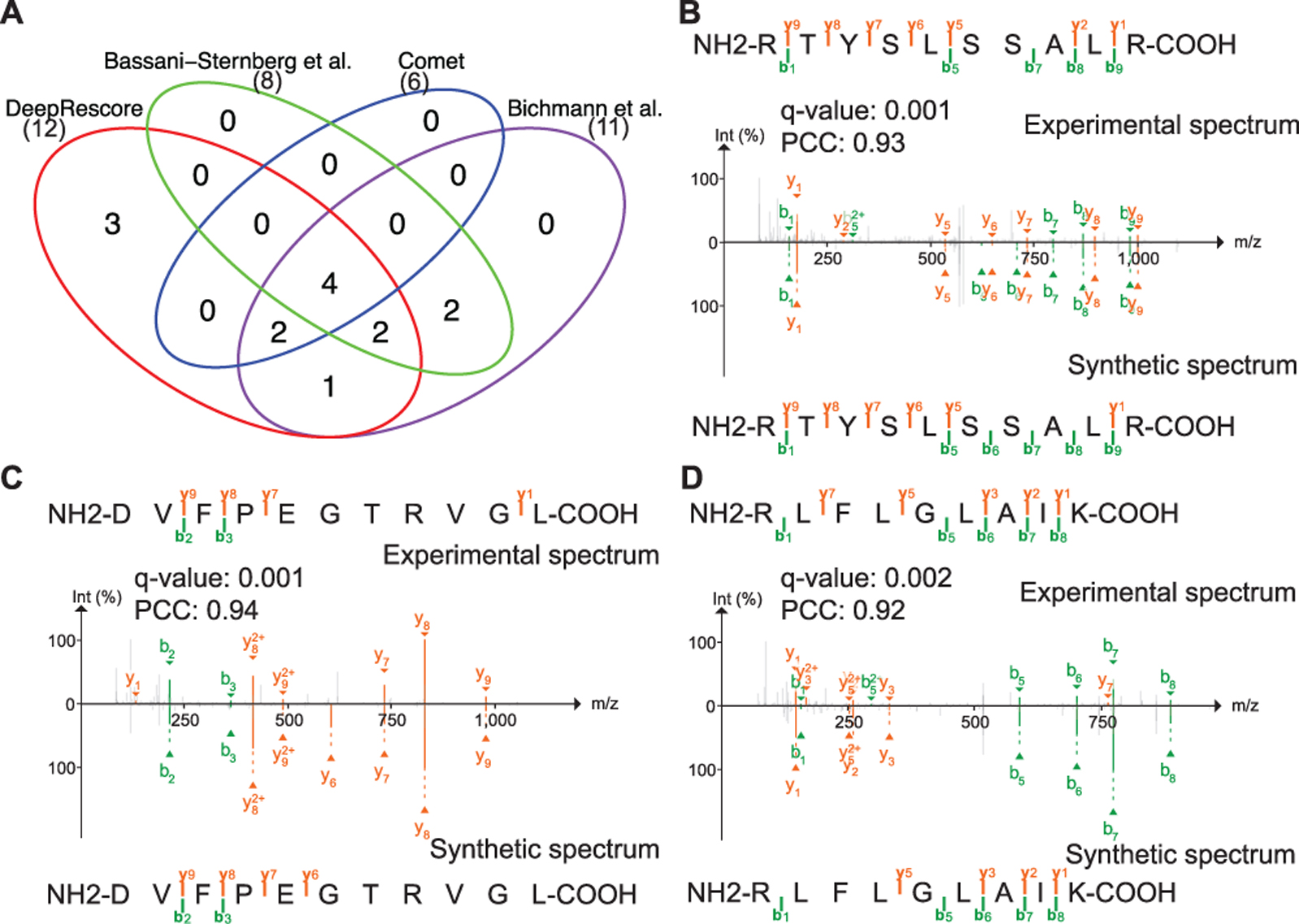
(A) The venn diagram comparing neoantigens identified by four different methods. (B-D) Annotated spectra for neoantigens uniquely identified by DeepRescore. The precursor peak for spectrum showing in (B) was removed and the raw spectrum was shown in Figure S9.
3.7. Experimental validation of peptides identified by DeepRescore
In order to further validate the quality of the peptides uniquely identified by DeepRescore, we selected 18 peptides (q-value <= 0.01) from D1 for experimental validation using synthetic peptides (Supplementary Table S5). The 18 peptides included 3 neoantigens uniquely identified by DeepRescore and 15 peptides from reference proteins. Among the 15 peptides, 12 were uniquely identified by DeepRescore and were randomly selected from three different confidence groups (low, medium and high), with 4 peptides for each group. The other 3 were randomly selected from peptides identified by both DeepRescore and other methods, with 1 peptide for each confidence group. These overlapping peptides were used as controls. For all the three neoantigens, we observed a high similarity (Pearson’s correlation coefficient (PCC) > 0.9)) between the original experimental spectra used to identify the peptides and the spectra from corresponding synthetic peptides (Figure 5B–D, Figure S9). Among the 12 reference protein-derived peptides uniquely identified by DeepRescore, a high similarity (PCC > 0.9) between experimental and synthetic spectra were found for 11 (Figure S10). The PCCs observed for these peptides were comparable to those for the three control peptides identified by both DeepRescore and other methods (Figure S10). Taken together, the results clearly demonstrated the high quality of peptides uniquely identified by DeepRescore.
Although the original experimental spectra are highly similar to the corresponding spectra from the synthetic peptides, some of the peaks in the original experimental spectra were not matched to any peaks in the spectra from corresponding synthetic peptides. These peaks are likely from background noise especially when their intensities are low or from the co-isolated peptides due to the complexity of the immunopeptidomics samples. We observed more unmatched peaks for neoantigen peptides than for peptides from reference proteins. This may result from the low abundance of the neoantigen peptides. Thus, validation using synthetic peptides is recommended when considering neoantigens identified by DeepRescore for clinical applications.
4. Concluding remarks
MS-based immunopeptidomics is the primary analytical methodology for high-throughput and direct identification of MHC binding peptides in vivo. However, applying different search engines to the same immunopeptidomics dataset gives vastly different peptide identification results. To improve peptide identification in immunopeptidomics, we validated delta RT and SA, two deep learning-derived features as indicators of PSM quality and then combined them with other previously used features for PSM rescoring in DeepRescore. We showed that DeepRescore increased both the sensitivity and quality of MHC-binding peptide and neoantigen identifications compared to existing methods, and the performance improvement was, to a large extent, driven by the deep learning-derived features. DeepRescore is freely available to the scientific community to utilize for sensitive and reproducible MHC-binding peptide and neoantigen identification from immunopeptidomics data.
Supplementary Material
Figure S1. Distributions of Delta RT and spectral angle for target and decoy peptides. (A-B) Delta RT distribution for dataset D1 (A) and D2 (B). (C-D) Spectral angle distribution for datasets D1 (C) and D2 (D).
Figure S2. The numbers of peptide sequences identified by different methods at different FDR levels for datasets D1 (A) and D2 (B).
Figure S3. Comparison of DeepRescore identifications with the report from the original study producing dataset D1.
Figure S4. Comparison of DeepRescore identifications with the report from the original study producing dataset D2.
Figure S5. Comparison of peptide identifications in D1 (A) and D2 (B) using rescoring models based on delta RT derived from AutoRT prediction (green) or GPTime prediction (blue) and Reduced Model 1 (red).
Figure S6. Length distribution of identified peptides. (A) D1 without DeepRescore. (B) D1 with DeepRescore. (C) D2 without DeepRescore. (D) D2 with Deep Rescore.
Figure S7. Ratios of predicted MHC binders among all peptides identified by each method in the two datasets.
Figure S8. Ratios of predicted MHC binders among peptides uniquely identified by DeepResore or among peptides not identified by DeepRescore in the two datasets across different percentile rank score cutoffs.
Figure S9. Annotated spectrum for peptide RTYSLSSALR.
Figure S10. Annotated spectra of the peptides selected for experimental validation.
Table S1. All features used in DeepRescore.
Table S2. Peptides identified from dataset D1 using different search engines in combination with DeepRescore.
Table S3. Peptides identified from dataset D2 using different search engines in combination with DeepRescore.
Table S4. Neoantigens identified using different methods.
Table S5. The list of peptides selected for experimental validation.
Statement of significance of the study:
Comprehensive characterization of the human immunopeptidome plays a critical role in understanding the immune system and guiding vaccine development and immunotherapies. Mass spectrometry (MS)-based immunopeptidomics is the most commonly used approach to comprehensively interrogate human immunopeptidome. However, sensitive and reliable peptide identification from immunopeptidomics data remains a major challenge. To address this challenge, we leveraged the power of deep learning and developed a new computational tool named DeepRescore. Using two public immunopeptidomics datasets, we showed that DeepRescore increased both the sensitivity and reliability of peptide and neoantigen identifications compared to existing methods. DeepRescore is freely available to the scientific community to allow sensitive and reproducible peptide and neoantigen identification from immunopeptidomics data.
Acknowledgements:
we thank Eric Jaehnig for proofreading the manuscript. We gratefully acknowledge the support of NVIDIA Corporation with the donation of the Titan Xp GPU used for this research.
Funding: This study was supported by the National Cancer Institute (NCI) CPTAC award U24 CA210954, the Cancer Prevention & Research Institutes of Texas (CPRIT) award RR160027, and funding from the McNair Medical Institute at The Robert and Janice McNair Foundation. B.Z. is a CPRIT Scholar in Cancer Research and a McNair Scholar. BCM Mass Spectrometry Proteomics Core is supported by the Dan L. Duncan Comprehensive Cancer Center NIH award (P30 CA125123) and CPRIT Core Facility Award (RP170005).
Abbreviations:
- RT
retention time
- PSM
peptide spectrum match
- SA
spectral angle
- FDR
false discovery rate
- MHC
major histocompatibility complex
- HLA
human leukocyte antigen
- PCC
Pearson correlation coefficient
Footnotes
The authors have declared no conflicts of interest.
REFERENCES
- [1].Bassani-Sternberg M, Coukos G, Curr Opin Immunol 2016, 41, 9. [DOI] [PubMed] [Google Scholar]
- [2].Vizcaino JA, Kubiniok P, Kovalchik KA, Ma Q, Duquette JD, Mongrain I, Deutsch EW, Peters B, Sette A, Sirois I, Caron E, Mol Cell Proteomics 2020, 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Eng JK, Jahan TA, Hoopmann MR, Proteomics 2013, 13, 22. [DOI] [PubMed] [Google Scholar]
- [4].Kim S, Pevzner PA, Nat Commun 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Craig R, Beavis RC, Bioinformatics 2004, 20, 1466. [DOI] [PubMed] [Google Scholar]
- [6].Cox J, Mann M, Nat Biotechnol 2008, 26, 1367. [DOI] [PubMed] [Google Scholar]
- [7].Perkins DN, Pappin DJ, Creasy DM, Cottrell JS, Electrophoresis 1999, 20, 3551. [DOI] [PubMed] [Google Scholar]
- [8].Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, Kloetzel PM, Stumpf MP, Heck AJ, Mishto M, Science 2016, 354, 354; [DOI] [PubMed] [Google Scholar]; Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, Busby M, Duke F, Yang A, Young L, Ojo NC, Caldwell K, Abhyankar J, Boucher T, Hart MG, Makarov V, Montpreville VT, Mercier O, Chan TA, Scagliotti G, Bironzo P, Novello S, Karachaliou N, Rosell R, Anderson I, Gabrail N, Hrom J, Limvarapuss C, Choquette K, Spira A, Rousseau R, Voong C, Rizvi NA, Fadel E, Frattini M, Jooss K, Skoberne M, Francis J, Yelensky R, Nat Biotechnol 2018; [DOI] [PubMed] [Google Scholar]; Caron E, Espona L, Kowalewski DJ, Schuster H, Ternette N, Alpizar A, Schittenhelm RB, Ramarathinam SH, Lindestam Arlehamn CS, Chiek Koh C, Gillet LC, Rabsteyn A, Navarro P, Kim S, Lam H, Sturm T, Marcilla M, Sette A, Campbell DS, Deutsch EW, Moritz RL, Purcell AW, Rammensee HG, Stevanovic S, Aebersold R, Elife 2015, 4; [DOI] [PMC free article] [PubMed] [Google Scholar]; Freudenmann LK, Marcu A, Stevanovic S, Immunology 2018, 154, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, Martignoni ME, Werner A, Hein R, C. Peschel HBD, Rad R, Cox J, Mann M, Krackhardt AM, Nat Commun 2016, 7, 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aebersold R, Mann M, Nature 2003, 422, 198. [DOI] [PubMed] [Google Scholar]
- [11].Faridi P, Purcell AW, Croft NP, Proteomics 2018, 18, e1700464. [DOI] [PubMed] [Google Scholar]
- [12].Andreatta M, Nicastri A, Peng X, Hancock G, Dorrell L, Ternette N, Nielsen M, Proteomics 2019, 19, e1800357. [DOI] [PubMed] [Google Scholar]
- [13].Bichmann L, Nelde A, Ghosh M, Heumos L, Mohr C, Peltzer A, Kuchenbecker L, Sachsenberg T, Walz JS, Stevanovic S, Rammensee HG, Kohlbacher O, J Proteome Res 2019, 18, 3876. [DOI] [PubMed] [Google Scholar]
- [14].Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ, Nat Methods 2007, 4, 923. [DOI] [PubMed] [Google Scholar]
- [15].Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, Knaute T, Rechenberger J, Delanghe B, Huhmer A, Reimer U, Ehrlich HC, Aiche S, Kuster B, Wilhelm M, Nat Methods 2019, 16, 509. [DOI] [PubMed] [Google Scholar]
- [16].Guan S, Moran MF, Ma B, Mol Cell Proteomics 2019, 18, 2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wen B, Li K, Zhang Y, Zhang B, Nat Commun 2020, 11, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou XX, Zeng WF, Chi H, Luo C, Liu C, Zhan J, He SM, Zhang Z, Anal Chem 2017, 89, 12690; [DOI] [PubMed] [Google Scholar]; Tiwary S, Levy R, Gutenbrunner P, Salinas Soto F, Palaniappan KK, Deming L, Berndl M, Brant A, Cimermancic P, Cox J, Nat Methods 2019, 16, 519. [DOI] [PubMed] [Google Scholar]
- [19].Zeng WF, Zhou XX, Zhou WJ, Chi H, Zhan J, He SM, Anal Chem 2019, 91, 9724. [DOI] [PubMed] [Google Scholar]
- [20].Sarkizova S, Klaeger S, Le PM, Li LW, Oliveira G, Keshishian H, Hartigan CR, Zhang W, Braun DA, Ligon KL, Bachireddy P, Zervantonakis IK, Rosenbluth JM, Ouspenskaia T, Law T, Justesen S, Stevens J, Lane WJ, Eisenhaure T, Lan Zhang G, Clauser KR, Hacohen N, Carr SA, Wu CJ, Keskin DB, Nat Biotechnol 2020, 38, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P, Nat Biotechnol 2012, 30, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang K, Li M, Hakonarson H, Nucleic Acids Res 2010, 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Elias JE, Gygi SP, Nat Methods 2007, 4, 207. [DOI] [PubMed] [Google Scholar]
- [24].Wen B, Xu S, Zhou R, Zhang B, Wang X, Liu X, Xu X, Liu S, BMC Bioinformatics 2016, 17, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meier F, Geyer PE, Virreira Winter S, Cox J, Mann M, Nat Methods 2018, 15, 440. [DOI] [PubMed] [Google Scholar]
- [26].Maboudi Afkham H, Qiu X, The M, Kall L, Bioinformatics 2017, 33, 508. [DOI] [PubMed] [Google Scholar]
- [27].Toprak UH, MCP 2014, 2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wen B, Du C, Li G, Ghali F, Jones AR, Kall L, Xu S, Zhou R, Ren Z, Feng Q, Xu X, Wang J, Proteomics 2015, 15, 2916; [DOI] [PubMed] [Google Scholar]; Wen B, Li G, Wright JC, Du C, Feng Q, Xu X, Choudhary JS, Wang J, Proteomics 2014, 14, 1011. [DOI] [PubMed] [Google Scholar]
- [29].The M, MacCoss MJ, Noble WS, Kall L, J Am Soc Mass Spectrom 2016, 27, 1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li K, Vaudel M, Zhang B, Ren Y, Wen B, Bioinformatics 2019, 35, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M, J Immunol 2017, 199, 3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J, Cell Syst 2018, 7, 129. [DOI] [PubMed] [Google Scholar]
- [33].Andreatta M, Alvarez B, Nielsen M, Nucleic Acids Res 2017, 45, W458; [DOI] [PMC free article] [PubMed] [Google Scholar]; Alvarez B, Barra C, Nielsen M, Andreatta M, Proteomics 2018, 18, e1700252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chong C, Muller M, Pak H, Harnett D, Huber F, Grun D, Leleu M, Auger A, Arnaud M, Stevenson BJ, Michaux J, Bilic I, Hirsekorn A, Calviello L, Simo-Riudalbas L, Planet E, Lubinski J, Bryskiewicz M, Wiznerowicz M, Xenarios I, Zhang L, Trono D, Harari A, Ohler U, Coukos G, Bassani-Sternberg M, Nat Commun 2020, 11, 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distributions of Delta RT and spectral angle for target and decoy peptides. (A-B) Delta RT distribution for dataset D1 (A) and D2 (B). (C-D) Spectral angle distribution for datasets D1 (C) and D2 (D).
Figure S2. The numbers of peptide sequences identified by different methods at different FDR levels for datasets D1 (A) and D2 (B).
Figure S3. Comparison of DeepRescore identifications with the report from the original study producing dataset D1.
Figure S4. Comparison of DeepRescore identifications with the report from the original study producing dataset D2.
Figure S5. Comparison of peptide identifications in D1 (A) and D2 (B) using rescoring models based on delta RT derived from AutoRT prediction (green) or GPTime prediction (blue) and Reduced Model 1 (red).
Figure S6. Length distribution of identified peptides. (A) D1 without DeepRescore. (B) D1 with DeepRescore. (C) D2 without DeepRescore. (D) D2 with Deep Rescore.
Figure S7. Ratios of predicted MHC binders among all peptides identified by each method in the two datasets.
Figure S8. Ratios of predicted MHC binders among peptides uniquely identified by DeepResore or among peptides not identified by DeepRescore in the two datasets across different percentile rank score cutoffs.
Figure S9. Annotated spectrum for peptide RTYSLSSALR.
Figure S10. Annotated spectra of the peptides selected for experimental validation.
Table S1. All features used in DeepRescore.
Table S2. Peptides identified from dataset D1 using different search engines in combination with DeepRescore.
Table S3. Peptides identified from dataset D2 using different search engines in combination with DeepRescore.
Table S4. Neoantigens identified using different methods.
Table S5. The list of peptides selected for experimental validation.