

Abstract
The maintenance of skeletal muscle mass depends on the overall balance between the rates of protein synthesis and degradation. Thus, age-related muscle atrophy and function, commonly known as sarcopenia, may result from decreased protein synthesis, increased proteolysis, or simultaneous changes in both processes governed by complex multifactorial mechanisms. Growing evidence implicates oxidative stress and reactive oxygen species (ROS) as an essential regulator of proteolysis. Our previous studies have shown that genetic deletion of CuZn superoxide dismutase (CuZnSOD, Sod1) in mice leads to elevated oxidative stress, muscle atrophy and weakness, and an acceleration in age-related phenotypes associated with sarcopenia. The goal of this study is to determine whether oxidative stress directly influences the acceleration of proteolysis in skeletal muscle of Sod1−/− mice as a function of age. Compared to control, Sod1−/− muscle showed a significant elevation in protein carbonyls and 3-nitrotyrosine levels, suggesting high oxidative and nitrosative protein modifications were present. In addition, age-dependent muscle atrophy in Sod1−/− muscle was accompanied by an upregulation of the cysteine proteases, calpain, and caspase-3, which are known to play a key role in the initial breakdown of sarcomeres during atrophic conditions. Furthermore, an increase in oxidative stress-induced muscle atrophy was also strongly coupled with simultaneous activation of two major proteolytic systems, the ubiquitin-proteasome and lysosomal autophagy pathways. Collectively, our data suggest that chronic oxidative stress in Sod1−/− mice accelerates age-dependent muscle atrophy by enhancing coordinated activation of the proteolytic systems, thereby resulting in overall protein degradation.
Electronic supplementary material
The online version of this article (10.1007/s11357-020-00200-5) contains supplementary material, which is available to authorized users.
Keywords: Protein oxidation, Proteolysis, Sarcopenia, Autophagy, Ubiquitin proteasome, Muscle atrophy
Introduction
Aging is associated with a progressive loss of muscle mass, a phenotype that is known as sarcopenia. Although sarcopenia is well documented in experimental animal models and in the clinic, the etiology and exact underlying mechanisms of this condition remain poorly understood. The maintenance of skeletal muscle mass depends on the overall balance between the rates of protein synthesis and degradation. Thus, age-related muscle atrophy may result from decreased protein synthesis, increased proteolysis, or simultaneous changes in both processes, which are governed by complex multifactorial mechanisms (Ferrington et al. 2005; Fry and Rasmussen 2011; Gordon et al. 2013; Husom et al. 2004).
Growing evidence implicates oxidative stress and ROS as an important regulator of proteolysis in catabolic muscle atrophy (Ott and Grune 2014; Powers et al. 2005; Tang et al. 2017). ROS can directly or indirectly damage proteins to alter their function and increase their turnover rate. Prochniewicz et al. demonstrated that permeabilized fibers exposed to H2O2 in vitro significantly reduced the contractility of myosin (Prochniewicz et al. 2008). In addition, Tiago and colleagues also reported that peroxynitrite (ONOO−) can react with F-actin to induce depolymerization in skeletal muscle. Moreover, peroxynitrite has been shown to inhibit myosin ATPase function, thus reducing muscle contractility (Tiago et al. 2006).
It is widely accepted that the ubiquitin proteasome pathway is the main route by which proteins are degraded during muscle atrophy. This pathway involves the targeted degradation of proteins via modification by ubiquitin and subsequent proteolysis by the 26S proteasome (Li et al. 2005). Although the 20S core proteasome can selectively degrade oxidatively modified proteins in a ubiquitin-independent manner, ROS have been demonstrated to activate the ubiquitin proteasome pathway in vivo and in vitro (Jung et al. 2006; Ott and Grune 2014). C2C12 cells treated with H2O2 results in an increased expression of the ubiquitin-activating enzyme, E214k, and the E3 ligases, atrogin-1 and MuRF1. These increases correlate with increased ubiquitin-conjugating activity, increased proteasome activity, and decreased myosin protein (Gomes-Marcondes and Tisdale 2002; Li et al. 2003). Although several studies demonstrated an increased ROS generation and oxidative damage in aged skeletal muscle (Mansouri et al. 2006; Navarro et al. 2001; Powers et al. 1992; Powers et al. 2012), there has been a paucity of in vivo evidence that directly links ROS and oxidative damage to activation of proteolytic pathways. Therefore, the purpose of this study is to determine the cause-and-effect relationship of chronic oxidative stress in vivo and the activation of proteolytic pathways in skeletal muscle as a function of age. To test our hypothesis, we utilized a mouse model that lacks the important antioxidant enzyme, CuZnSOD (Sod1−/−). Sod1−/− mice are characterized by very high levels of oxidative stress and elevated levels of oxidative damage to lipid, protein, and DNA in several tissues, including skeletal muscle (Jang et al. 2012; Jang et al. 2010; Muller et al. 2005). Moreover, Sod1−/− mice exhibit dramatic age-related muscle atrophy that is accelerated compared to age-matched wild-type (Jang et al. 2012; Jang et al. 2010; Muller et al. 2005). Here, we report that oxidative stress in vivo activates proteolysis by concurrent upregulation of cysteine proteases, the ubiquitin proteasome, and lysosomal autophagy. Acceleration of muscle wasting seen in Sod1−/− mice is in part due to increased oxidative damage and simultaneous upregulation of protein degradation pathways.
Material and methods
Animals
The Sod1−/− mice used in this study have been described previously (Jang et al. 2012; Jang et al. 2010). Briefly, the mice were maintained under specific pathogen-free conditions in the heterozygous state (Sod1+/-) and backcrossed with C57BL/6J females (Jackson Laboratory) for more than 20 generations. For tissue harvesting, mice were anesthetized using ketamine (30 mg/kg) and xylazine (4 mg/kg) and euthanized by cervical dislocation. Visceral organs were collected and weighed; any gross pathological observations, such as tumors, cysts, and organ deformities, were recorded. All of the measurements were done in female animals. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio and Georgia Institute of Technology.
Immunohistochemistry and immunofluorescence
Muscles frozen in isopentane were sectioned (10 μm) from the mid-belly and fixed in 4% paraformaldehyde. For measurement of fiber size, sections were incubated in blocking buffer for 1 h and then incubated in anti-laminin (L9393; Sigma-Aldrich) overnight at 4 °C. Alexa Fluor 488 (Invitrogen) fluorescent dye conjugated to an anti-rabbit secondary antibody was used for visualization. Images were visualized with a fluorescent microscope (Nikon Inc.) and captured with a SPOT RT camera. Fiber cross-sectional area measurements were calculated using the Nikon Element software.
Transmission electron microscopy
We fixed muscle block (1 mm3 in size) with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, post-fixed with 1% osmium tetroxide followed by 1% uranyl acetate, dehydrated them through a graded series of ethanol washes and embedded them in resin. Blocks were cut in ultrathin (80 nm) sections on Reichert Ultracut UCT, stained the sections with uranyl acetate followed by lead citrate, and viewed on a JEOL 1230 EX transmission electron microscope at 80 kV.
Protein carbonyls
Protein carbonyls were measured using the method described by Chaudhuri et al (Chaudhuri et al. 2006). Briefly, the cytosolic extracts from hindlimb muscles (gastrocnemius, plantaris, tibialis anterior, extensor digitorum longus, and quadriceps) were diluted to 1 mg/mL, mixed with FTC (1 mM), and incubated at 37 °C for 150 min in the dark. The proteins were precipitated with an equal volume of 20% chilled TCA (v/v) and centrifuged at 16,000×g for 5 min at 25 °C. The pellets were then re-suspended and washed five times, with 100% ethanol/ethyl acetate (1:1) to remove the unbound FTC. The final pellets were then dissolved in phosphate buffer pH 8.0 containing 0.5 mM MgCl2, 1 mM EDTA, and 8 M urea. The concentration of the protein in each sample was measured by Bradford assay, and approximately 15–25 μg of protein were subjected to 12% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). It is to be noted that all the free FTC may not be quantitatively removed from the pellets by extensive washing with ethanol/ethyl acetate. In that case, the free residual FTC will be removed from the gel during electrophoresis. After electrophoresis, the image of the fluorescent protein on the gel was captured with the Typhoon 9400 using an excitation wavelength of 488 nm and an emission filter at 520 nm with a 40-nm bandpass. The intensity of fluorescence for each lane (from top to bottom of the lane) was calculated using ImageQuant 5.0 (Amersham, GE Health) software.
Caspase-3 and Calpain activity
Caspase-3 activity was measured in the cytosolic extracts from hindlimb muscles (gastrocnemius, plantaris, tibialis anterior, extensor digitorum longus, and quadriceps) using the synthetic peptide n-acetyl-DEVD-AMC (BD Biosciences, San Diego, CA). Active caspases cleave the AMC from the peptide and the free AMC fluoresces. Briefly, 1 mL of assay buffer (20 mM HEPES, 10% glycerol, 1 M DTT, and 14 μL of Ac-DEVD-AMC/mL of buffer) and 50 μL of sample and standards were added to a microcentrifuge tube and protected from light. Samples were incubated at 37 °C for 60 min, after which fluorescence was measured on a spectrofluorometer at 380 -nm excitation and 440-nm emission. All samples were measured in triplicate. Results are reported as pmol/min/mg protein. Calpain activity was measured in the muscle cytosolic fraction using a fluorometric assay kit (Oncogene Research Products, San Diego, CA). This assay utilizes the unique ability of calpain to digest synthetic substrate Suc-LLVY AMC in the presence of the Ca2+ ion and the reducing agent TCEP. Released free AMC was measured fluorometrically at an excitation wavelength of 380 nm and an emission wavelength of 440 nm. All samples were measured in triplicate.
Proteasome activity
The fluoropeptides LLE-AMC, AAF-AMC, and LSTR-AMC were used to measure the caspase-like, chymotrypsin-like, and trypsin-like activities, respectively, as previously described (Kisselev and Goldberg 2005). Experiments were performed at 37 °C in microtiter plates using a Fluoroskan-FL Ascent type 374 microplate reader (excitation = 360, emission = 460) to monitor fluorescence every 5 min for 2 h. In the proteasome-enriched fractions, activity was measured in the absence and the presence of 200 μM Ada-(Ahx)3-(Leu)3-vinyl sulfone (Biomol, Enzo Life Sciences, Plymouth, PA) in the buffer containing 50 mM Tris (pH 7.5), 5 mM MgCl2, 20 mM KCl, 2.5 μg protein and peptides with concentrations from 12 to 200 μM for AAF and LLE, and 3–400 μM for LSTR. The rate of proteolysis was determined for each peptide concentration by comparing the linear response of fluorescence with a standard curve of AMC. The rate of proteolysis as a function of peptide concentration was plotted, and the maximal activity (Vmax) and the concentration of substrate at the half-maximal activity (Km) were determined by fitting a hyperbolic model defined by the Michaelis–Menton equation to the experimental data.
Native gel electrophoresis
Native gel electrophoresis has been used to determine if the proteasome remains intact in a higher molecular weight form (i.e., 26S) or disassembles during the assay (20S). This could reflect the physiological properties of the tissue tested or whether or not the extraction process leads to disassembly. About 50 μg of fractionated muscle lysates from WT and Sod1−/− were run on a 3–12% non-denaturing, gradient polyacrylamide gel (Invitrogen, Carlsbad, CA). The gel was run at 30 V for 30 min in 4 °C, followed by 35 V for 1 h, 50 V for 1 h, and further increased to 75 V for 3 more hours. Peptidolytic activity of proteasomes was detected after incubating the gels in a Suc-LLVY-MCA substrate dissolved in 50 mM Tris pH 8.0, 5 mM MgCl2, 1 mM DTT, 1 mM ATP, and 0.02% SDS for 15, 30, and 60 min at 37 °C. Proteasome bands were identified by the release of highly fluorescent, free AMC. Following the in-gel assay, the protein from the gel was transferred to PVDF via a wet transfer procedure and subjected to Western blotting analyses to identify the various proteasome subunits and whether the proteasome remained intact or was disassembled into 20S, 19S, or other complexes.
Western blot analysis
Western blot analysis was performed using a Bio-Rad Criterion Gel system. The immunoblots were detected with an enhanced chemiluminescence (ECL) system (Amersham: Piscataway, NJ) and then visualized and scanned on a Typhoon 9400 (Amersham: Piscataway, NJ). Quantification of the immunoblots was performed with ImageQuant Software (Sunnyvale, CA). The following antibodies against proteasome subunits were used: α4 (mouse mAb, 1:1,000, PW8120), α7 (mouse mAb, 1:1,000, PW8110), β2 (mouse mAb, 1:1,000, PW9300), MECL1 (β2i) (rabbit pAb, 1:1,000, PW8350), LMP7 (β5i) (mouse mAb, 1:1,000, PW8845), β4 (rabbit pAb, 1:1,000, PW8890), Rpn7(rabbit pAb, 1:1,000, PW8225), Rpn10 (mouse mAb, 1:1,000, PW9250), Rpt5 (mouse mAb, 1:1,000, PW8770) (Enzo Life Sciences, Plymouth Meeting, PA, USA), and PA28α (goat pAb, 1:500, sc-21267) (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Statistical analyses
All statistical analyses were performed on GraphPad Prism 8, and data are presented as mean ± standard error (SEM). The student t test was used to compare differences between Sod1 null and control WT group. For autophagic flux analyses, 2-way ANOVA with Bonferroni’s post hoc test was used. A p-value of less than 0.05 was considered statistically significant.
Results
Accelerated muscle atrophy in Sod1−/− mice
In a comparison of different hind-limb muscles between WT and Sod1−/− mice, the overall loss of wet weight of the muscle normalized to bodyweight clearly showed muscle atrophy in Sod1−/− mice (Supplemental Fig. 1A). To test whether the loss of muscle mass was the consequence of reduced myofiber cross sectional area, gastrocnemius muscle sections from 20 months old WT and Sod1−/− mice were stained with hematoxylin and eosin as well as immunofluorescence imaging with anti-laminin (Fig. 1a (a and b)), and these sections were used to measure fiber cross-sectional area (Fig. 1a (c and d)). Next, to examine muscle morphology, we analyzed ultrathin longitudinal sections from WT and Sod1−/− muscle using electron microscopy (EM). EM analyses revealed that in Sod1−/−, the z-lines on the myofibrils were disoriented, and the myofibril diameters were considerably smaller compared to WT (Fig. 1a (e and f)). In an agreement with the visual analysis, in Sod1−/− muscle fibers, the average cross-sectional area was ~40% less than the age-matched WT mice (Fig. 1c). As shown previously by our laboratory (Jang et al. 2012; Jang et al. 2010), denervation and alterations in neuromuscular junctions were present in Sod1−/− muscle (Supplemental Fig. 1B). The postsynaptic endplates of Sod1−/− were extensively fragmented and distributed compared to the pretzel-like form of WT endplates. The pre-terminal side of motor axons in Sod1−/− mice demonstrates regions of sprouting and abnormal thinning as compared to those in WT. Collectively, these data support the notion that the significant muscle atrophy seen in Sod1−/− muscle is due to denervation of NMJ (Jang et al. 2010).
Fig. 1.

Muscle atrophy in Sod1−/− mice is correlated with protein oxidation. a Morphological and fiber cross-sectional area comparison at 20 months in hematoxylin and eosin (a, b), laminin-stained sections (c, d), and transmission electron micrograph (e, f) of gastrocnemius muscle. Scale bar: 100 μm (in a–d) and 2 μm (in e and f). b Ratio of gastrocnemius wet-weight normalized to body weight. Values were compared using student t test. ***p < 0.001. c Quantification of mean cross-sectional area in a. Values were compared using student t test; ***p < 0.001. d Protein carbonyl levels in cytosolic fraction of WT and Sod1−/− muscle at 5 months and 20 months. Values were compared using two-way ANOVA with Bonferroni post hoc test; *p < 0.05 (WT vs Sod1−/−) and #p < 0.05 (5 months vs 20 months). e A representative Western blot analysis of 3-nitrotyrosin residues from total muscle lysates at 5 months and 20 months
Increased oxidized proteins in Sod1−/−
Oxidatively damaged proteins accumulate in aged skeletal muscle, and oxidative modification of contractile proteins impairs muscle function (Ott and Grune 2014; Prochniewicz et al. 2008). Therefore, we assessed the amount of oxidized proteins in muscle from wild-type and Sod1−/− mice by measuring protein carbonyl and nitrotyrosylated proteins. As shown in Fig. 1d, protein carbonyls were increased by 45% in the soluble cytosolic fraction from young Sod1−/− mice compared to young WT mice (5 months). Similarly, protein carbonyls were elevated by nearly 40% in 20-month-old WT mice. However, in 20-month-old Sod1−/− mice, carbonyl levels remained elevated at the level of young Sod1−/− mice and did not increase further with age. Peroxynitrite (ONOO−), which is formed in vivo by the diffusion-limited reaction of superoxide anion (O ×-) and nitric oxide (NO), which are both produced in abundance in skeletal muscle (Tidball 2005). Nitration of the amino acid tyrosine, to produce 3-nitrotyrosine, is reportedly a reliable index of damage by peroxynitrite (Duncan 2003). Therefore, we measured the level of total proteins containing 3-nitrotyrosine using Western blot analysis (Fig. 1e). Nitrated proteins showed a trend of increase in Sod1−/− muscle homogenate at both 5 months and 20 months, but it did not reach statistical significance.
Increased cysteine protease calpain and caspase-3 levels in Sod1−/−
The ubiquitin proteasome system is the major protein degradation pathway during muscle atrophy conditions (Bonaldo and Sandri 2013). However, the proteasome cannot degrade intact myofibrillar proteins. Thus, several investigators suggested that disassembled sarcomere by the Ca2+-dependent protease calpain is a necessary first step in initiating the muscle atrophy process (Chopard et al. 2009; Huang and Forsberg 1998; Kandarian and Jackman 2006; Williams et al. 1999). Previous work from Du et al. revealed that activation of caspase-3 may also present a similar function in triggering initiation of muscle proteolysis (Du et al. 2004). Therefore, we determined whether the cysteine proteases, calpain and caspase- 3, are activated in WT and Sod1−/− muscle. As demonstrated in Fig. 2b, m-calpain, measured at middle age (11–16 months), showed a trend toward an increase in Sod1−/− muscle homogenate but did not reach statistical significance. On the other hand, caspase-3 activity was significantly elevated in Sod1−/− muscle compared to age-matched WT (Fig. 2a). Additionally, we measured α-spectrin, which is a substrate for both calpain and caspase-3, to further confirm their activation. Using Western blot analysis, we found that 150-kDa cleavage product was significantly higher in Sod1−/− (Fig. 2c). In all, our data suggest that its conjunction with increased oxidative damage, activation of the cysteine proteases, calpain and caspase-3, may trigger the proteolytic process by cleaving contractile proteins in the actomyosin complex.
Fig. 2.
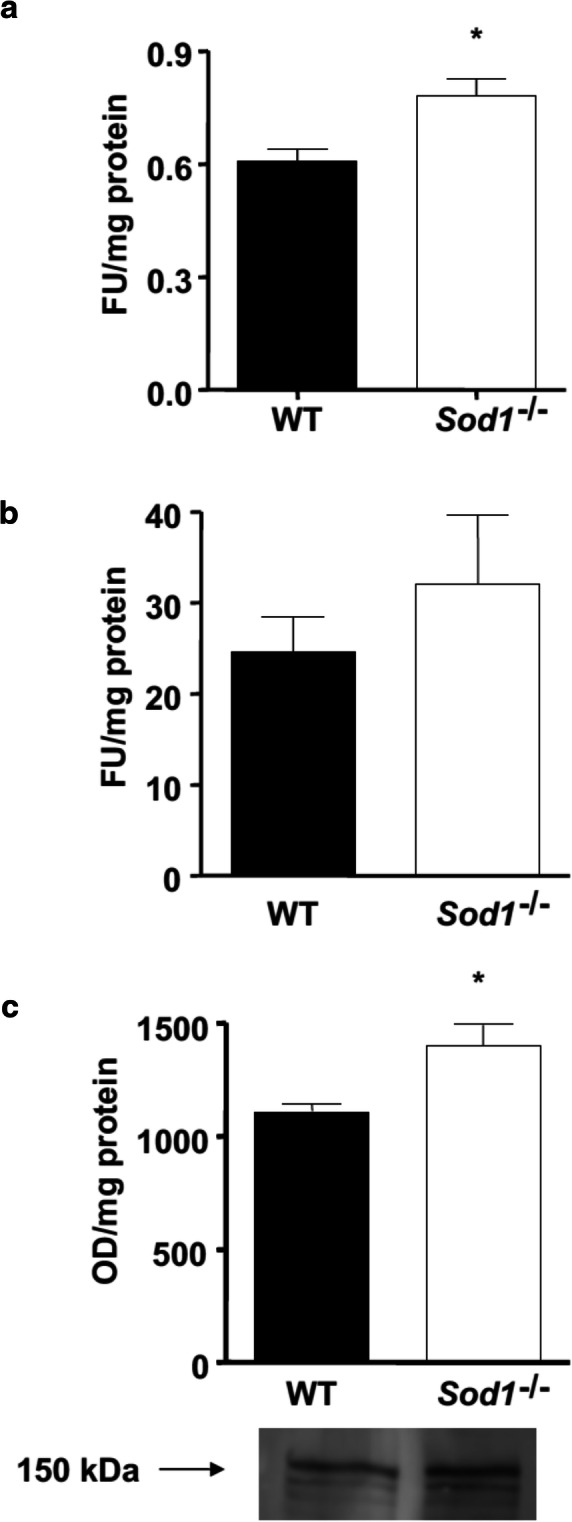
Elevated levels of cysteine proteases activity in Sod1−/− mice. a Caspase-3 activity measured at 11–16 months from hind-limb muscle (b) m-Calpain activity measured at 11–16 months from hind-limb muscle (c) Western blot quantification and representative image of calpain substrate, α-spectrin cleavage product (150 kDa)
Increased ubiquitinylated protein in Sod1−/−
Once myofibrillar proteins are cleaved and released from the actomyosin complex, they are tagged with poly-ubiquitin for further degradation by the 26S proteasome through the actions of E1 ubiquitin-activating enzyme, E2 conjugating enzyme, and E3 ligases (Bonaldo and Sandri 2013; Du et al. 2004; Jackman and Kandarian 2004). The level of ubiquitin-bound proteins from the cytosolic fraction was elevated in middle-aged Sod1−/− mice (Fig. 3a). As illustrated in Fig. 3b, only the chymotrypsin-like peptidase activity was increased in Sod1−/− muscle at both 5 months and 20 months, and no changes were seen in either caspase-like or trypsin-like peptidase activity (data not shown). On the other hand, we did not see any changes in MAFbx1 and MuRF-1, two common E3 ligases that are known to play a pivotal role in multiple conditions of muscle wasting (Bonaldo and Sandri 2013), suggesting different E3 ligase may be involved (data not shown). Furthermore, in an agreement with the increased proteasome activity in Sod1 null muscle, a significant increase in the protein levels of various proteasome assembly was seen in Sod1−/− homogenates with the exception of 20S proteasome. However, the protein levels of individual 20S subunits were assessed and β5 and Rpt5 as illustrated in Fig. 3e and f, respectively. Interestingly, subunit PA28α, in contrary to the other two, exhibited a significant decrease in Sod1−/− mice as compared to their age-matched WT (Fig. 3g).
Fig. 3.

A loss of Sod1 induces muscle atrophy through ubiquitin protein systems. a A representative Western blot of total ubiquitin-bound protein bound and free ubiquitin from cytosolic fraction from 11 to 16-month-old muscle. b 20S Chymotrypsin-like proteasome activity from 5 month (top) and 20 month (bottom) old muscle. Values were compared using student t test; *p < 0.05. c A quantification of native gel analyses of different proteasome assembly muscle homogenates at 15 months of age. Values were compared using student t test; *p < 0.05. Protein levels of 20S proteasome subunit α7 (d) and β5 (e), 19S subunit Rpt5 (F), and 11S proteasome activator PA28α (g). Values were compared using student t test; *p < 0.05, **p < 0.01, and ***p < 0.001
Increased macro-autophagy in Sod1−/− muscle
Autophagy is one of the cellular pathways that deal with protein degradation and has been affected in response to stress (Kroemer et al. 2010). To assess the significance of this pathway, we examined the autophagosome formation and autophagic proteins (ATG) in response to the chronic oxidative stress condition in Sod1−/− mice. As shown in Fig. 4a, autophagosomes were formed in the gastrocnemius muscle fibers of 15-month-old Sod1−/− mice along with increased accumulation of lipid vacuoles. Intriguingly, mitochondrial damage and swollen mitochondria co-localized with autophagosome and lipid vacuoles, suggesting that the ROS and oxidative damage to mitochondria that is present in Sod1−/− mice may induce mitochondrial autophagy (Fig. 4a (b and c)). Immunoblotting of the microtubule-associated protein light chain 3 (LC3) in the absence and presence of autophagosome–lysosome fusion inhibitor (Chloroquine) as well as the autophagy-related protein Beclin-1 was used to monitor the autophagic flux in Sod1−/− mice (Fig. 4b). During autophagy, LC3-I (the cytosolic form of LC3) is lipidated to form LC3-II, which is specifically recruited to the phagophore to form autophagosomes. In the presence of a lysosomal protease inhibitor that prevents the fusion between autophagosomes and lysosomes, the amount of LC3-II accumulates, generating an autophagic flux (Mizushima et al. 2010). Beclin-1 is a protein that plays an important role in autophagy by being involved in the engagement of membranes to form autophagosomes (Mizushima et al. 2010). Quantification of the Western blot analysis demonstrates a significant increase in Beclin-1 protein, but the LC3-I/LC3-II ratio was not significantly different (Fig. 4b and d). However, when autophagic flux was assessed after the treatment with Chloroquine to block lysosomal fusion of autophagosome (Fig. 4d), a significant decrease in LC3-I/LC3-II was observed in Sod1−/− muscle (Fig. 4e), suggesting more accumulation of autophagosome in Sod1 null myofibers. In addition, the protein level of the Atg5-Atg12 conjugate, which facilitates the lipidation of LC3, was elevated in Sod1−/− muscle, which further supports an increase in the autophagic flux of Sod1 null fibers (Fig. 4e).
Fig. 4.

Elevated levels of macro-autophagy in muscle of Sod1−/− mice. a Transmission electron microscope (TEM) images of muscle from 15-month-old gastrocnemius muscle from WT (a) and Sod1−/− (b, c, d) mice. Black rectangle represents autophagosome formation in Sod1−/− muscle and higher magnification of shown in (d). Increased accumulation of lysosomal vacuoles shown in (c). Scale bar: 2 μm (in a–c) and 500 nm (in d). b Representative Western blot of autophagic proteins Beclin-1 (top) and LC3-I and II (bottom). c and d A quantification of Western blot analyses shown in (b). Values were compared using student t test; **p < 0.01. e Autophagic flux was measured 24 h after Chloroquine treatment. Autophagosome formation as measurement by ratio between LC3-I and LC3-II (top) and Atg5–12 (bottom). Actin was used as loading control. f and g A quantification of Western blot analyses shown in (d). Values were compared using two-way ANOVA with Bonferroni post hoc test; *p < 0.05 (WT vs Sod1−/−) and #p < 0.05 (5 months vs 20 months)
Discussion
A growing amount of evidence suggests that the aging of a multi-cellular organism is strongly associated with the loss of protein homeostasis, commonly termed as proteostasis. The maintenance of proteostasis is particularly important for skeletal muscle fibers due to its high content of proteins (Egerman and Glass 2014). The accumulation of abnormal proteins during aging is widely believed to result from defects in protein breakdown (Davies and Shringarpure 2006; Ferrington et al. 2005). However, very few experimental data support these hypotheses. Rates of protein turnover regulate the levels of specific proteins (Bonaldo and Sandri 2013; Egerman and Glass 2014; Ferrington et al. 2005). In addition, changes in skeletal muscle protein mass depend on the overall balance between rates of protein synthesis and breakdown. Muscle wasting may result from decreased protein synthesis, increased proteolysis, or simultaneous changes in both processes. These alterations are presumably multi-factorial. Major factors implicated in the etiology of sarcopenia include decreased physical activity, declines in α-motor neurons, malnutrition, increased cytokine activity, oxidative stress, mitochondrial dysfunction, and abnormalities in growth hormones and sex steroid hormones (Argiles et al. 2005; Fulle et al. 2004; Giresi et al. 2005). The causative role of oxidative stress in sarcopenia has been demonstrated in previous studies (Fulle et al. 2004; Roubenoff and Hughes 2000; Sakellariou et al. 2018; Vasilaki et al. 2017). The direct relation between chronic oxidative stress and protein degradation via the ubiquitin proteasome system and the autophagy–lysosome system in vivo, however, has not been thoroughly addressed. In this present study, we utilized an aged Sod1−/− mouse model, which is characterized by elevated levels of oxidative stress and oxidative damage to lipid, protein, and DNA in different types of tissue, including skeletal muscle (Jang et al. 2010; Muller et al. 2006). In an agreement with previous findings, we detected an increase in oxidized protein levels as verified by the measurement of protein carbonyls and nitrotyrosylated proteins in Sod1−/− mice as compared to those in age-matched WT mice (Sakellariou et al. 2018). In conjunction with the increased oxidative damage, the cysteine proteases, calpain and caspase-3, were upregulated in Sod1−/− mice, with the latter being significantly elevated. Our data suggest that an increase in protein modifications by oxidative stress activates calpain and caspase-3 pathways, which may subsequently trigger the proteolytic process by cleaving the contractile proteins in the actomyosin complex (Du et al. 2005; Du et al. 2004). Intriguingly, we observed several morphological alterations in myofibrils near interfibrillar mitochondria (Figs. 1a and 4a). Previously, we demonstrated that Sod1 null muscle display an age-dependent increase in mitochondrial ROS emission (Jang et al. 2010). Thus, future studies elucidating the link between mitochondrial ROS and proteostasis, as well as signaling pathways that govern this process, will be needed.
The ubiquitin proteasome is the main proteolytic system for degrading misfolded proteins, as well as oxidized proteins (Ott and Grune 2014; Sakellariou et al. 2018). In skeletal muscle fibers, proteasome requires the disassembly of sarcomere by Ca2+-dependent proteases, calpain and caspase-3, to initiate the proteolytic process as it cannot directly degrade intact myofibrillar proteins (Du et al. 2005; Du et al. 2004; Tang et al. 2017). Thus, the initial cleavage and release of the myofibrillar proteins from the actomyosin complex are crucial steps for poly-ubiquitination and degradation by the 26S proteasome (Du et al. 2005; Du et al. 2004; Tang et al. 2017).
While our data, in part, support the basic premise of the oxidative stress theory of aging in skeletal muscle, due to the deletion of the Sod1 gene in the whole body, we cannot rule out the contribution of oxidative stress coming from tissue other than skeletal muscle. In support of this notion, muscle-specific deletion of Sod1 and global deletion of Sod1 show different changes in ubiquitin proteasome activity, suggesting cell-autonomous and non-cell autonomous effects of oxidative stress potentially regulate this process (Sakellariou et al. 2018). Furthermore, it has been well documented that proteostasis of muscle is regulated by humoral factors such as growth hormone, IGF-1, and more (Bassel-Duby and Olson 2006; Bodine et al. 2001; Egerman and Glass 2014). Thus, oxidative damage in the other endocrine organ or oxidative modification of the secretome may also influence the acceleration of age-dependent muscle wasting of the Sod1 knockout mice. Moreover, one of the emerging concepts in aging research is the organ to organ crosstalk. It will be interesting to test the effects of systemic oxidative stress using the parabiosis model or tissue-specific deletion of Sod1 in endocrine organs.
The autophagy–lysosome system is another essential proteolytic pathway that is involved in the selective turnover of cytoplasmic proteins and organelles (Masiero et al. 2009; Masiero and Sandri 2010;
Sandri 2010). Recent studies demonstrated that autophagy is required for the maintenance of muscle mass, and inhibition of autophagy accelerates atrophy and myopathy in adult skeletal muscle (Masiero et al. 2009; Masiero and Sandri 2010). Similar to these studies, we demonstrated that levels of a key autophagic protein, Beclin-1, were significantly elevated in response to the chronic oxidative stress in Sod1−/− mice. Moreover, the autophagic flux was notably altered in Sod1 null muscle following the treatment of muscle fibers with Chloroquine with a decrease in LC3-I/LC3-II ratio. Several recent studies indicated that crosstalk between ubiquitin proteasome and autophagy is a crucial event in intracellular quality control and recycling of essential proteins (Kocaturk and Gozuacik 2018; Nam et al. 2017). Both ubiquitin proteasome and selective autophagy recognize their target by ubiquitin tags via p62/sequestosome-1. Moreover, reciprocal regulation mechanisms are thought to be present between these conserved degradation pathways. However, whether oxidative stress or an accumulation of oxidized proteins disrupts this crosstalk merits further investigation. Emerging evidence also suggests that a controlled level of ROS plays a central role in redox regulation and directs intracellular signaling pathways in skeletal muscle (Jackson 2008; Jackson 2015; Sakellariou et al. 2018). Therefore, we cannot completely rule out the role of ROS in proteostasis (Bonet-Costa et al. 2019). Nevertheless, we can postulate that in the Sod1 knockout model and during aging, high levels of ROS/RNS generation overwhelm the antioxidant defense and lead to chronic oxidative stress, which in turn, disrupts the redox balance and alters regulatory or quality control mechanism of proteolytic systems.
In summary, our results establish a cause-and-effect relationship between oxidative stress in vivo and reciprocal activation of a major protein degradation pathways in age-associated muscle wasting. Thus, a better understanding of these proteolytic pathways may provide a druggable therapeutic target against sarcopenia and frailty.
Electronic supplementary material
(DOCX 469 kb)
Acknowledgments
We thank the Physiological Research Laboratory and core facilities at the UTHSCSA/Sam and Ann Barshop Institute for Aging and Longevity Studies and Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology for the use of shared equipment and services.
Author contributions
YCJ, KR, MSL, AB, FLM, AP, AC, JJC, and NL performed all experiments and analyzed the data. YCJ, AGR, and HVR designed the studies, co-wrote, and edited the manuscript.
Funding information
Research reported in this publication was supported by the National Institutes of Health under award numbers R01AG020591 (A.G.R.), P01AG051442 (A.G.R. and H.V.R.), and R21AR072287 (Y.C.J.), S&R Foundation (Y.C.J.), and Merit Review Grant (H.V.R) from the US Department of Veterans Affairs. Dr. Van Remmen is the recipient of a Senior Research Career Scientist award (1IK6BX005234) from the Department of Veterans Affairs.
Compliance with ethical standards
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest
The authors declare that they have no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Young C. Jang, Email: young.jang@gatech.edu
Holly Van Remmen, Email: holly-vanremmen@omrf.org.
References
- Argiles JM, Busquets S, Felipe A, Lopez-Soriano FJ. Molecular mechanisms involved in muscle wasting in cancer and ageing: cachexia versus sarcopenia. Int J Biochem Cell Biol. 2005;37:1084–1104. doi: 10.1016/j.biocel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006. [DOI] [PubMed]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–9. [DOI] [PubMed]
- Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonet-Costa V, Sun PY, Davies KJA. Measuring redox effects on the activities of intracellular proteases such as the 20S proteasome and the immuno-proteasome with fluorogenic peptides. Free Radic Biol Med. 2019;143:16–24. doi: 10.1016/j.freeradbiomed.2019.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AR, de Waal EM, Pierce A, Van Remmen H, Ward WF, Richardson A. Detection of protein carbonyls in aging liver tissue: a fluorescence-based proteomic approach. Mech Ageing Dev. 2006;127:849–861. doi: 10.1016/j.mad.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Chopard A, Hillock S, Jasmin BJ. Molecular events and signalling pathways involved in skeletal muscle disuse-induced atrophy and the impact of countermeasures. J Cell Mol Med. 2009;13:3032–3050. doi: 10.1111/j.1582-4934.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KJ, Shringarpure R. Preferential degradation of oxidized proteins by the 20S proteasome may be inhibited in aging and in inflammatory neuromuscular diseases. Neurology. 2006;66:S93–S96. doi: 10.1212/01.wnl.0000192308.43151.63. [DOI] [PubMed] [Google Scholar]
- Du J, et al. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–123. doi: 10.1172/JCI18330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Hu Z, Mitch WE. Cellular signals activating muscle proteolysis in chronic kidney disease: a two-stage process. Int J Biochem Cell Biol. 2005;37:2147–2155. doi: 10.1016/j.biocel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Duncan MW. A review of approaches to the analysis of 3-nitrotyrosine. Amino Acids. 2003;25:351–361. doi: 10.1007/s00726-003-0022-z. [DOI] [PubMed] [Google Scholar]
- Egerman MA, Glass DJ. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol. 2014;49:59–68. doi: 10.3109/10409238.2013.857291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Husom AD, Thompson LV. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- Fry CS, Rasmussen BB. Skeletal muscle protein balance and metabolism in the elderly. Curr Aging Sci. 2011;4:260–268. doi: 10.2174/1874609811104030260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulle S, Protasi F, di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, et al. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp Gerontol. 2004;39:17–24. [DOI] [PubMed]
- Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, et al. Identification of a molecular signature of sarcopenia. Physiol Genomics. 2005;21:253–63. [DOI] [PubMed]
- Gomes-Marcondes MC, Tisdale MJ. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002;180:69–74. doi: 10.1016/S0304-3835(02)00006-X. [DOI] [PubMed] [Google Scholar]
- Gordon BS, Kelleher AR, Kimball SR. Regulation of muscle protein synthesis and the effects of catabolic states. Int J Biochem Cell Biol. 2013;45:2147–2157. doi: 10.1016/j.biocel.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Forsberg NE. Role of calpain in skeletal-muscle protein degradation. Proc Natl Acad Sci U S A. 1998;95:12100–12105. doi: 10.1073/pnas.95.21.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, Ferrington DA. Altered proteasome function and subunit composition in aged muscle. Arch Biochem Biophys. 2004;421:67–76. doi: 10.1016/j.abb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–C843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- Jackson MJ. Redox regulation of skeletal muscle. IUBMB Life. 2008;60:497–501. doi: 10.1002/iub.72. [DOI] [PubMed] [Google Scholar]
- Jackson MJ. Redox regulation of muscle adaptations to contractile activity and aging. J Appl Physiol (1985) 2015;119:163–171. doi: 10.1152/japplphysiol.00760.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, et al. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell. 2012;11:770–82. 10.1111/j.1474-9726.2012.00843.x. [DOI] [PMC free article] [PubMed]
- Jung T, Engels M, Kaiser B, Poppek D, Grune T. Intracellular distribution of oxidized proteins and proteasome in HT22 cells during oxidative stress. Free Radic Biol Med. 2006;40:1303–1312. doi: 10.1016/j.freeradbiomed.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 2006;33:155–165. doi: 10.1002/mus.20442. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005;398:364–378. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system front. Cell Dev Biol. 2018;6:128. doi: 10.3389/fcell.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol. 2003;285:C806–C812. doi: 10.1152/ajpcell.00129.2003. [DOI] [PubMed] [Google Scholar]
- Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–70. [DOI] [PMC free article] [PubMed]
- Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, et al. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev. 2006;127:298–306. [DOI] [PubMed]
- Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010;6:307–309. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- Masiero E, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, et al. Absence of Cu,Zn-Sod causes severe oxidative stress and acceleration of age- dependent skeletal muscle atrophy. Free Radical Biology and Medicine. 2005;39:S127. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Muller FL, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Nam T, Han JH, Devkota S, Lee HW. Emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome. Syst Mol Cells. 2017;40:897–905. doi: 10.14348/molcells.2017.0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Lopez-Cepero JM, Sanchez del Pino MJ. Skeletal muscle and aging. Front Biosci. 2001;6:D26–D44. doi: 10.2741/A591. [DOI] [PubMed] [Google Scholar]
- Ott C, Grune T. Protein oxidation and proteolytic signalling in aging. Curr Pharm Des. 2014;20:3040–3051. doi: 10.2174/13816128113196660709. [DOI] [PubMed] [Google Scholar]
- Powers SK, Lawler J, Criswell D, Lieu FK, Dodd S. Alterations in diaphragmatic oxidative and antioxidant enzymes in the senescent Fischer 344 rat. J Appl Physiol (1985) 1992;72:2317–2321. doi: 10.1152/jappl.1992.72.6.2317. [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, DeRuisseau KC. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288:R337–R344. doi: 10.1152/ajpregu.00469.2004. [DOI] [PubMed] [Google Scholar]
- Powers SK, Smuder AJ, Judge AR. Oxidative stress and disuse muscle atrophy: cause or consequence? Curr Opin Clin Nutr Metab Care. 2012;15:240–245. doi: 10.1097/MCO.0b013e328352b4c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochniewicz E, et al. Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am J Physiol Cell Physiol. 2008;294:C613–C626. doi: 10.1152/ajpcell.00232.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubenoff R, Hughes VA. Sarcopenia: current concepts. J Gerontol A Biol Sci Med Sci. 2000;55:M716–M724. doi: 10.1093/gerona/55.12.M716. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, et al. Comparison of whole body SOD1 knockout with muscle-specific SOD1 knockout mice reveals a role for nerve redox signaling in regulation of degenerative pathways in skeletal muscle. Antioxid Redox Signal. 2018;28:275–295. doi: 10.1089/ars.2017.7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Tang H, et al. Smad3 initiates oxidative stress and proteolysis that underlies diaphragm dysfunction during mechanical ventilation. Sci Rep. 2017;7:14530. doi: 10.1038/s41598-017-11978-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiago T, Ramos S, Aureliano M, Gutierrez-Merino C. Peroxynitrite induces F-actin depolymerization and blockade of myosin ATPase stimulation. Biochem Biophys Res Commun. 2006;342:44–49. doi: 10.1016/j.bbrc.2006.01.112. [DOI] [PubMed] [Google Scholar]
- Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, Richardson A, Van Remmen H, Brooks SV, Larkin L, McArdle A, et al. Role of nerve-muscle interactions and reactive oxygen species in regulation of muscle proteostasis with ageing. J Physiol. 2017;595:6409–15. 10.1113/JP274336. [DOI] [PMC free article] [PubMed]
- Williams AB, Decourten-Myers GM, Fischer JE, Luo G, Sun X, Hasselgren PO. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J. 1999;13:1435–1443. doi: 10.1096/fasebj.13.11.1435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 469 kb)