

Abstract
A chiral synthesis of a series of hexahydroisobenzofuran (HIBF) nucleosides has been accomplished via glycosylation of a stereo-defined (syn-isomer) sugar motif 16 with the appropriate silylated bases. All nucleoside analogs were obtained in 52–71% yield as a mixture of α- and β-anomeric products increasing the breadth of the novel nucleosides available for screening. The structure of the novels bicyclic HIBF nucleosides was established by a single crystal X-ray structure of the β-HIBF thymine analog 22b. Furthermore, the sugar conformation for these nucleosides was established as N-type. Among the novel HIBF nucleosides synthesized, twenty-five compounds were tested as inhibitor of HIV-1 in human peripheral blood mononuclear (PBM) cells and seven were found to be active (EC50 = 12.3–36.2 μM). Six of these compounds were purine analogs with β-HIBF inosine analog 22o being the most potent (EC50 = 12.3 μM) among all compounds tested. The striking resemblance between didanosine (ddI) and 22o may explain the potent anti-HIV activity.
Introduction
Despite prodigious efforts in combating the HIV infection, it remains the leading cause of death worldwide.1 Among the various therapeutic treatments available for HIV-1, nucleoside reverse transcriptase inhibitors (NRTIs) has proven to be an important class of drugs with a variety of nucleoside analogs at the forefront. For example, zidovudine (AZT), zalcitabine (ddC), didanosine (ddI), stavudine (d4T, 1 in Fig. 1), lamivudine (3TC), abacavir (ABC), viread (TDF) and emtricitabine (FTC) have been approved by the FDA as anti-HIV drugs.2 However, the therapeutic potential of these compounds is often compromised by drug resistance and toxicity.3 In order to overcome these limitations and design improved inhibitors of HIV-1, several investigators4 have modified the carbohydrate moiety of the nucleosides resulting in enhanced understanding of the structure–activity relationship (SAR).
Fig. 1.
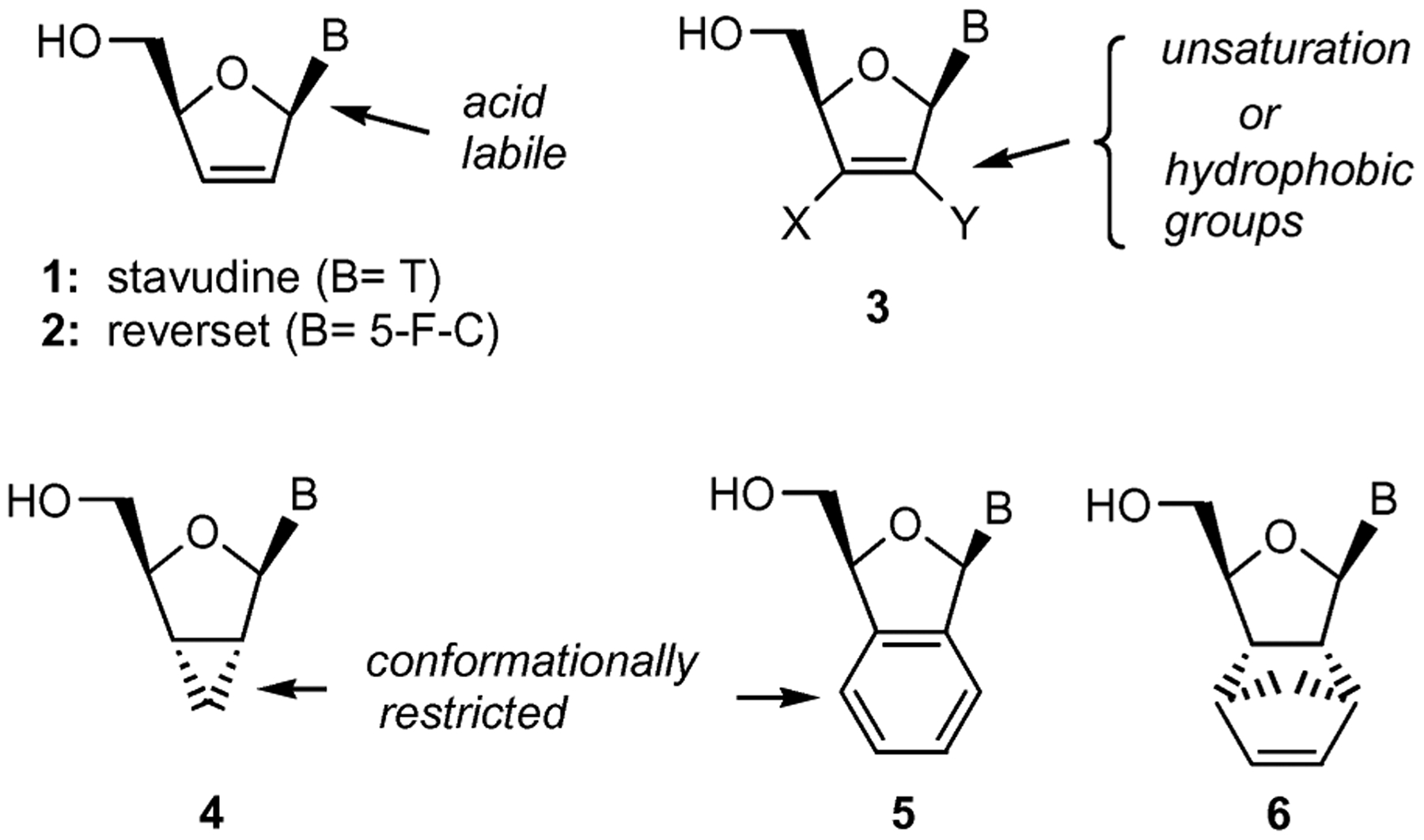
Designed features of modified nucleosides.
The anti-HIV studies with various carbohydrate-modified nucleosides have established three salient SAR features that contribute to the observed activity. First, the placement of a 2′,3′-double bond in the structures of d4T (1), abacavir, reverset (D-d4FC, 2) and elvucitabine (L-d4TC) is essential to their anti-HIV-1 activity.5 For instance, molecular modeling studies with d4T-triphosphate indicate that the double bond is positioned in close proximity to the aromatic side chain of Tyr 115 in the active site of HIV-RT, thus contributing to desirable π–π interactions.6 Second, the acid labile nature of the 2′,3′-dideoxypurine analogs can be overcome by introducing an electronegative fluorine atom or other hydrophobic groups at the 2′ or 3′-positions (structure 3) of the carbohydrate moiety.7 Such groups improve the stability of these compounds at a lower pH and increase their lipophilicity for uptake, both essential features for oral delivery of drugs. Third, nucleoside analogs with a variety of conformational restrictions imposed on the sugar ring (4, 5 and 6 in Fig. 1) have resulted in modulation of the enzyme–substrate recognition.8 In particular, planar pseudo-sugar ring systems are usually preferred. This concept has led to the synthesis of several bicyclic nucleoside analogs with some exhibiting N-type (2′-exo/3′-endo) sugar conformation and antiviral activity.9
Based on our ongoing interest10 in the design of conformationally restricted nucleoside analogs for various drug-discovery approaches, we decided to explore the possibility of introducing these three attributes to one molecule and ultimately discover new compounds with antiviral activity. More specifically, we envisioned that a hexahydroisobenzofuran (HIBF) nucleoside, represented by structure 7 (Fig. 2), could contain all three characteristics discussed above. Earlier, it was reported that 3-hydroxymethyl-1,3-dihydrobenzo[c]furan nucleosides 5 were found to be inactive as inhibitors of HIV.11 This lack of activity was attributed to the steric effect of the rigid benzene ring, which reduces the ability of the molecule to access the enzyme-binding site. Replacement of the benzene ring with an HIBF system may offer multiple advantages, such as: (i) possible interactions with Tyr 115 in the active site of HIV-RT due to the 7′–8′ double bond; (ii) stabilization of the glyocosidic linkage at a lower pH due to the C–C bond at the 2′-position; (iii) adoption of the preferred N-type sugar conformation for improved recognition by cellular enzymes, due to the rigid bicyclic motif of HIBF; and (iv) improved lipophilicity for enhanced cellular uptake. With this in mind, we embarked on the synthesis of HIBF type nucleosides.
Fig. 2.
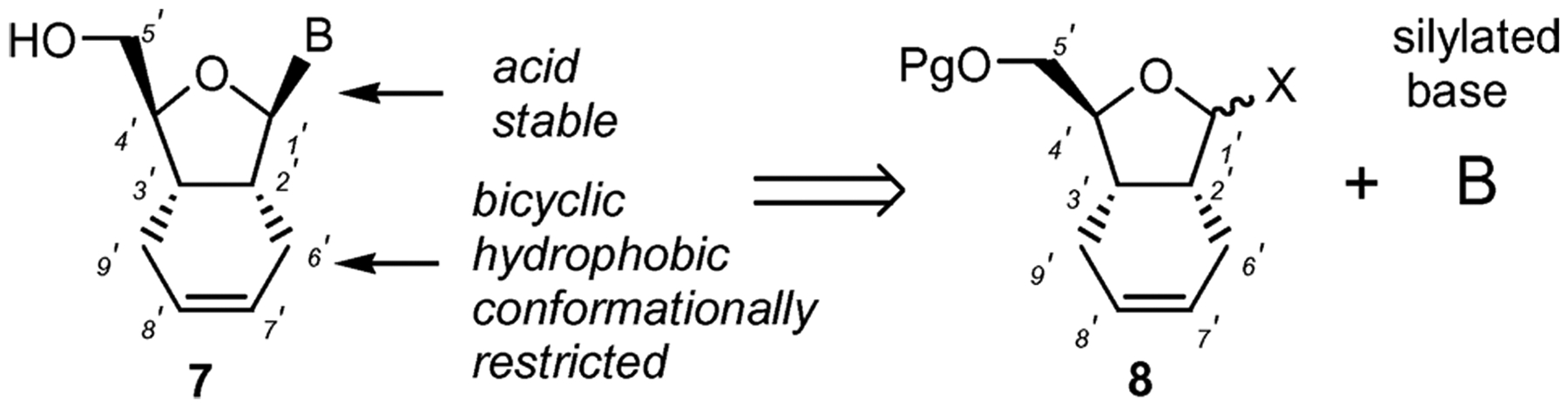
Concept for the design and synthesis of HIBF nucleosides.
Although many variants of the bicyclic nucleosides have been studied, HIBF nucleosides have not received much attention. As illustrated in Fig. 2, our synthetic approach is based on glycosidation of an appropriately protected HIBF motif 8 with a variety of nucleobases. This strategy is advantageous for a number of reasons. The chirally defined starting material 8 could be synthesized in few steps from inexpensive d-mannitol. The chemically inert HIBF moiety is not expected to interfere in the glycosylation step. The glycosylation reaction would most likely result in the formation of both α- and β-anomers offering both varieties of nucleosides in one step. Appropriately protected 8 could be synthesized in bulk and a large number of nucleoside analogs could be synthesized in parallel mode in a relatively short time. As such, the overall approach is convergent and amenable to scale-up, should any of these nucleosides be required in larger amounts for further studies. Moreover, the HIBF nucleoside family of compounds offers a unique SAR link between the 2′,3′-dideoxynucleosides and the structural requirements for appending a second ring without abolishing the antiviral activity. Herein we report in detail the synthesis of several HIBF nucleosides and their antiviral and cytotoxicity profiles.
It should be noted that while this work was in progress, a communication appeared in the literature12 describing the synthesis of the uridine analog 7 (B = Uracil). The reported synthesis departs from uridine and employs a ring-closure metathesis reaction to form the cyclohexene ring. As such, this route is not convergent and suffers from a multi-step process that results in the formation of cis- and trans-isomeric products. Moreover, the biological activities of 7 have not been reported.
Results and discussion
Synthesis of HIBF nucleosides
The bicyclic core of the designed nucleosides was synthesized as shown in Scheme 1. Commercially available d-mannitol was converted to butenolide 10 via a sequence of steps that includes: (a) acetonide protection of the 1,2 and 5,6 diol; (b) oxidative cleavage of the 3,4 diol; (c) Wittig olefination of the resulting aldehyde and (d) acid-catalyzed acetonide deprotection and lactonization of the resulting hydroxy ester.13,14 Initial attempts to silylate butenolide 10 under basic conditions (imidazole, DMAP) led to complete racemization of the starting material, due to the relative acidity of the 4′ hydrogen. However, silylation under acidic conditions (NH4NO3, TBDPSCl, DMF)15 produced the desired compound 11 as a single isomer in excellent yield. The Diels–Alder cycloaddition between 11 and butadiene proceeded exclusively from the opposite face of the bulky TBDPS group and afforded 12 as a single enantiomer in 85% yield.16 DIBAL-H reduction of lactone 12 proceeded exclusively from the less sterically hindered α-face forming β-lactol 13 in 92% yield.
Scheme 1.
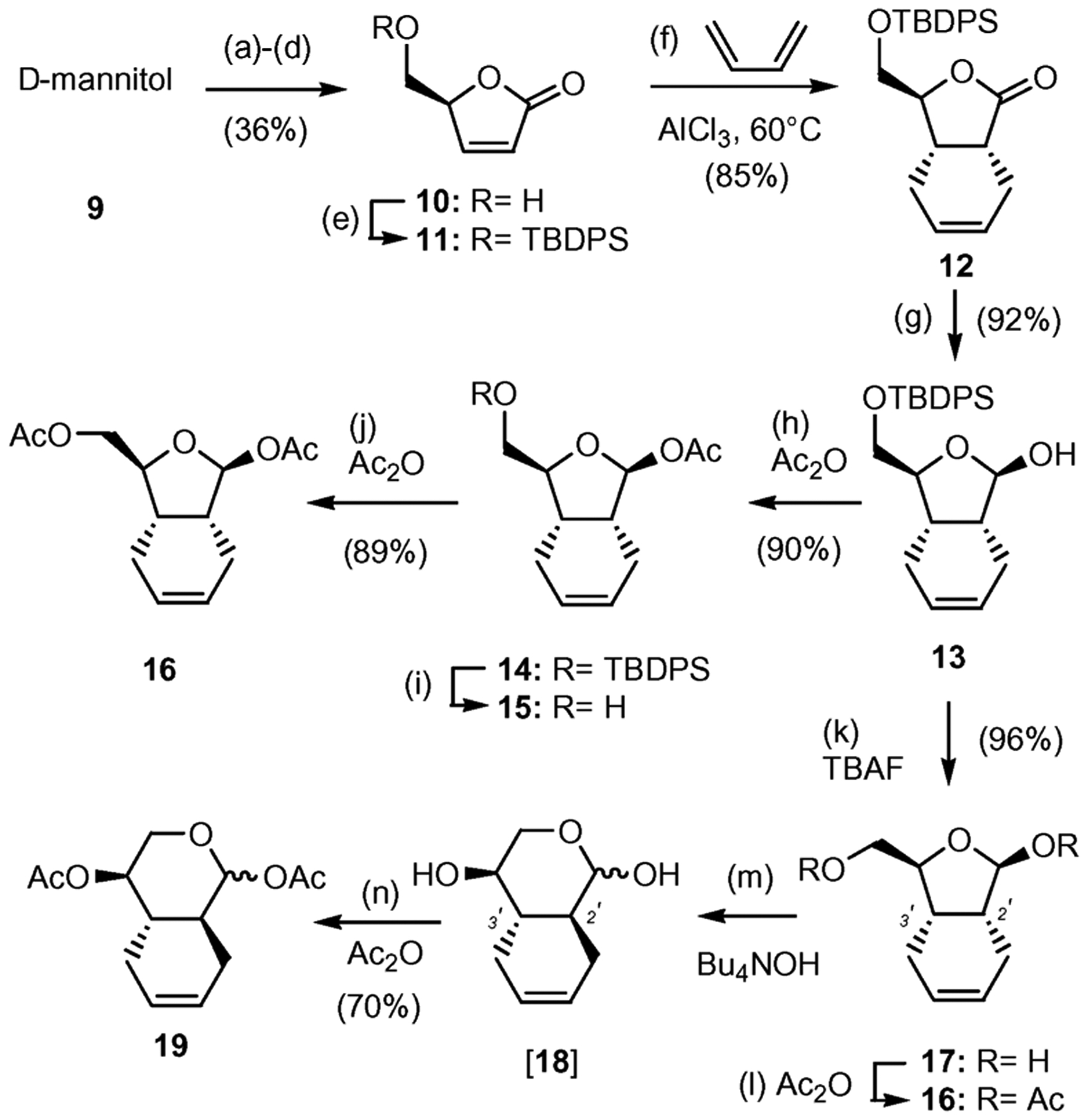
Synthesis of bicyclic scaffold 16. Reagents and conditions: (a) 0.01 equiv SnCl2, 2,2-dimethoxypropane–DME (2 : 3), reflux, 4 h, 78%; (b) 2.0 equiv NaIO4, sat. aq. NaHCO3–CH2Cl2 (1 : 20), 0 °C, 2 h, 73%; (c) 1.5 equiv Ph3P=CHCO2Me, MeOH, 0 °C, 16 h, 70%; (d) conc. H2SO4–MeOH (1 : 100), 25 °C, 1 h, 90% (e) 1.2 equiv TBDPSCl, 5.0 equiv NH4NO3, DMF, 25 ° (f) 0.33 equiv AlCl3, CH2Cl2, 60 °C, 6 d, 85%; (g) 1.1 equiv DIBAL-H (1 M), CH2Cl2, −78 °C 1 h, 92%; (h) 3.0 equiv Ac2O, pyridine, 25 °C, 12 h, 90%; (i) 1.2 equiv TBAF (1 M), THF, 25 °C, 30 min, 87%; (j) 3.0 equiv Ac2O, pyridine, 25 °C, 12 h, 89%; (k) 1.2 equiv TBAF (1 M), THF, 25 °C, 25 min, 96%; (l) 4.0 equiv Ac2O, pyridine, 25 °C, 12 h, 92%; (m) 1.3 equiv Bu4NOH (33% in H2O), THF, 25 °C, 6 h; (n) 4.0 equiv of Ac2O, pyridine, 25 °C, 12 h, 70% (over two steps).
Initially we targeted lactol 13 as the glycosyl donor for coupling with appropriately protected nucleobases. These efforts proved to be unsuccessful, presumably due to the steric hindrance of the bulky TBDPS group. To overcome this problem we decided to change the protecting group at the 5′ center to an acetate functionality. To this end, compound 13 was first acetylated at the C1′ center and then underwent a TBAF-induced desilylation to form alcohol 15 in 78% combined yield. Acetylation of 15 then formed bicyclic scaffold 16 in 89% yield. Alternatively, desilylation of 13 formed diol 17 that, without further purification, was converted to compound 16 (2 steps, 88% combined yield). It is worth noting that, upon standing, diol 17 can undergo conversion to lactol 18, in which the C2′ stereocenter was isomerized to form a more stable 6,6 trans bicyclic motif. In fact, treatment of 17 under basic conditions (Bu4NOH, H2O) followed by acetylation of the reaction mixture led to isolation of diacetate 19 in 70% combined yield.
The glycosidation of diacetate 16 proceeded under Hilbert–Johnson conditions17 (silylated base, DBU, TMSOTf) to produce a mixture of α- and β-nucleosides in 52–71% overall yield (Scheme 2). The ratio of α/β-anomers was determined by LCESI-MS studies used in combination with 1H NMR on the crude reaction mixture. In addition, spectroscopic studies including NOESY measurements confirmed that the β-anomer was the major compound. Unambiguous confirmation of the structure of the synthesized compounds was obtained by a single crystal X-ray analysis of 22b, the fully deprotected product of 20b (see Fig. 3).
Scheme 2.
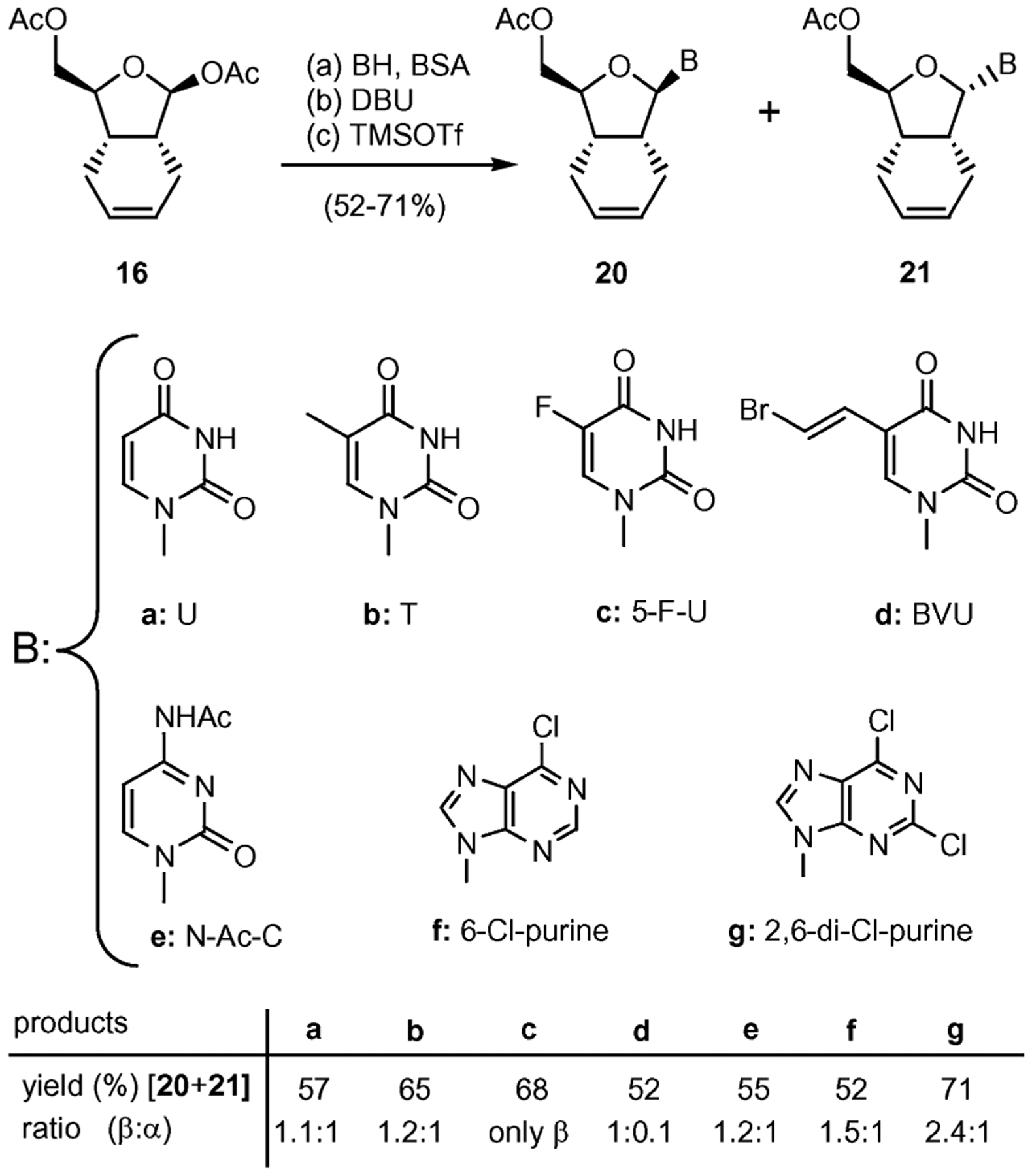
Glycosylation of 16 with pyrimidines and purines. Reagents and conditions: (a) 0.5 equiv 16, 1.0 equiv base, 3.5 equiv BSA, MeCN, 75 °C, 1 h; (b) 1.6 equiv DBU, 25 °C; (c) 3.0 equiv TMSOTf, 0–25 °C, 2–3 h, 52–71% (over three steps).
Fig. 3.
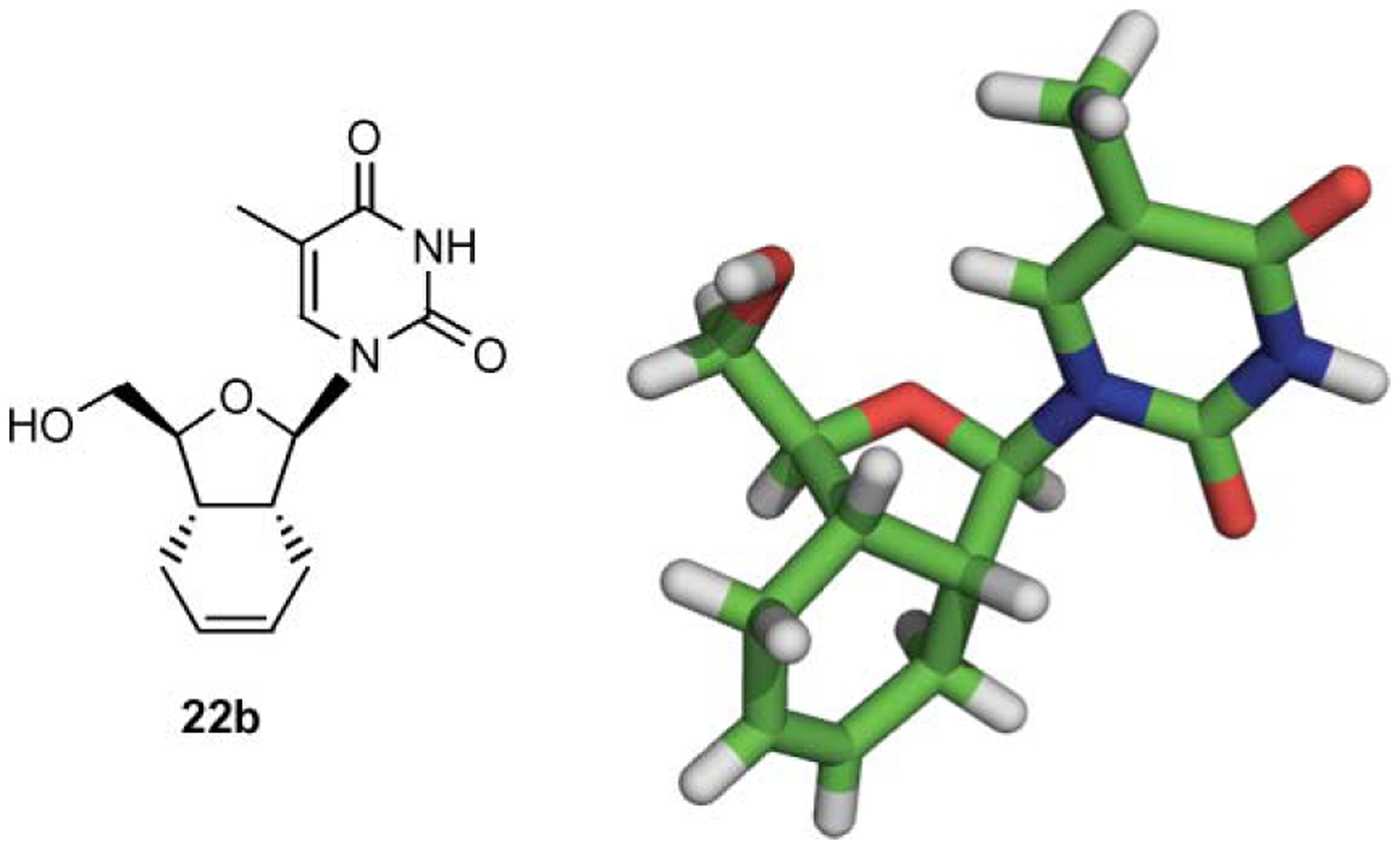
Single X-ray structure of compound 22b.
The coupled nucleosides were deprotected under NH3–MeOH conditions as shown in Scheme 3.18 We observed that the 5′-acetate group was easily removed in all cases at room temperature. Under these conditions all pyrimidine nucleosides (20a–e, 21a–b and 21d–e) formed the corresponding 5′-deprotected HIBF nucleosides in excellent yields. These conditions were also successfully used for the selective deprotection of 6-Cl-purine 20f leading to compound 22f. However, treatment of 20f with saturated NH3–MeOH at 110 °C formed the 6-aminopurine analog 22i, while under less drastic conditions produced the 6-methoxypurine 22k.
Scheme 3.
Deprotection of HIBF nucleosides. Reagents and conditions: (a) 2 M NH3–MeOH, rt for obtaining a–d, f, h; (b) NH3 sat–MeOH, 110 °C for obtaining i, j, m; (c) 2 M NH3–MeOH, 90–100 °C for obtaining k, l. Yields are reported in the Experimental section.
Treatment of 2,6-dichloropurine 21g under ammonia led to a mixture of monoamino (23j) and diaminopurine derivative (23m) in quantitative combined yield. The mixture of compounds 23j and 23m was purified by silica gel column chromatography (gradient 50–100% EtOAc–hexane). This transformation is reproducible with both anomeric nucleosides (see Scheme 4).
Scheme 4.
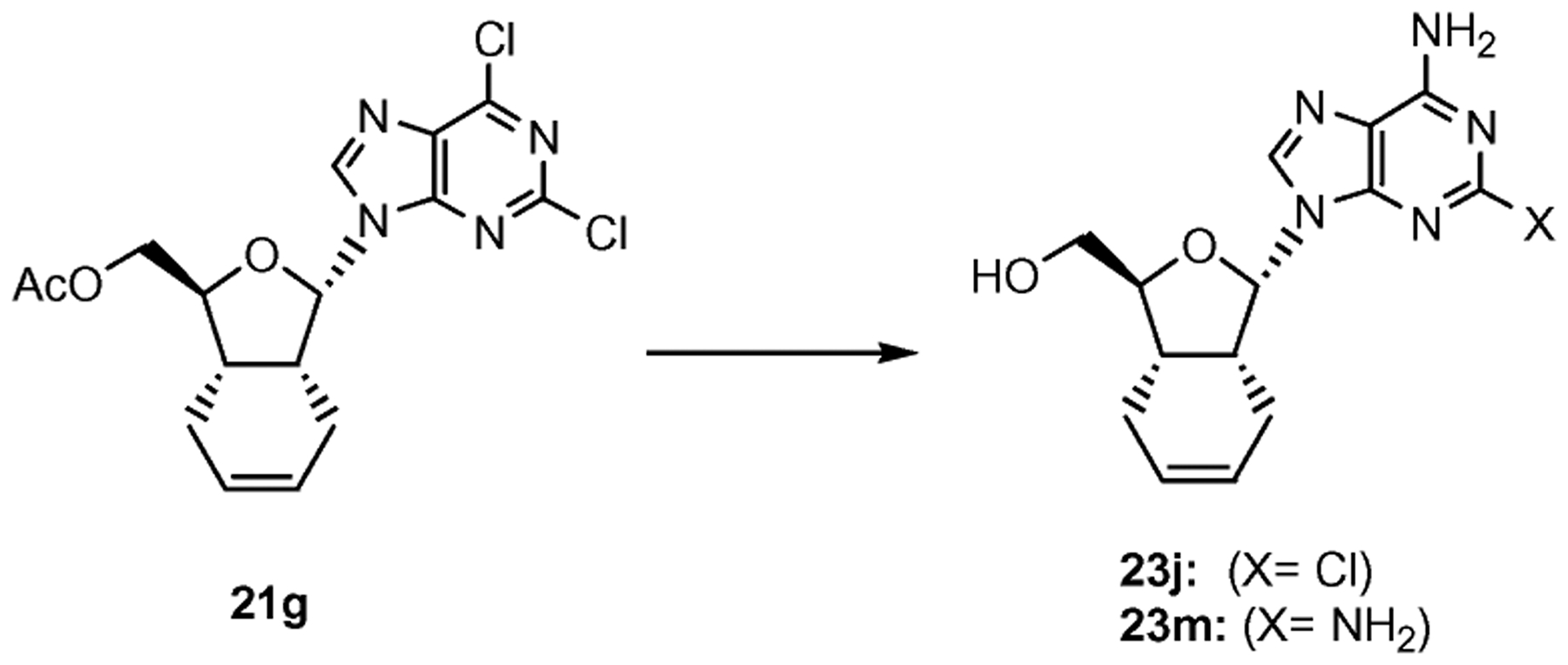
Synthesis of α-diamino purine bicyclic nucleoside 23. Reagents and conditions: NH3 sat–MeOH, 110 °C, 36 h, 40% of 23j and 60% of 23m.
Alternatively, the diaminopurine 22m could be obtained by reaction of 20g with NaN3 and Me3P (Scheme 5).19 Under these conditions the double bond remained unchanged.
Scheme 5.
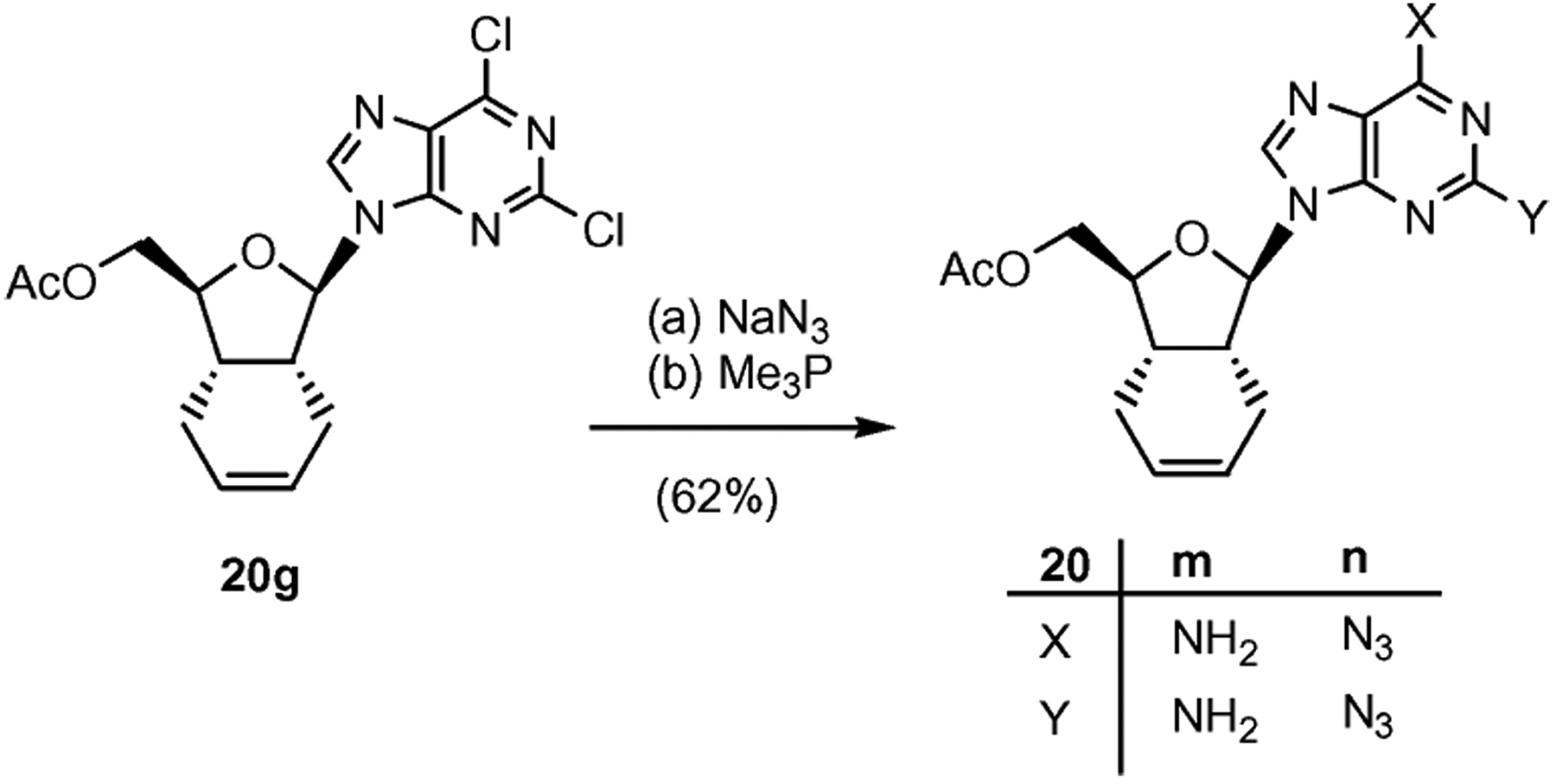
Purine transformations in β-nucleoside 20g. Reagents and conditions: (a) 4 equiv NaN3, DMF, 70 °C, 4 h, 80%; (b) 2.2 equiv Me3P, THF–H2O (1 : 1), 24 h, 77%.
The inosine compound 22o was obtained via enzymatic deamination of adenosine analogue 22i (Scheme 6).20 This reaction was slower than the reported deamination of natural adenosine and only afforded a 50% yield of the desired product after 72 h. We attribute this sluggish reactivity to the lipophilic nature of the HIBF nucleoside 22i. Nonetheless, it is remarkable to observe that the presence of the cyclohexene ring is not incompatible to the recognition by the enzyme. The recognition of 22i as a substrate for adenosine deaminase further confirms that the 5′-hydroxyl group was accessible to the active site in the enzyme for its transformation to 22o.21
Scheme 6.
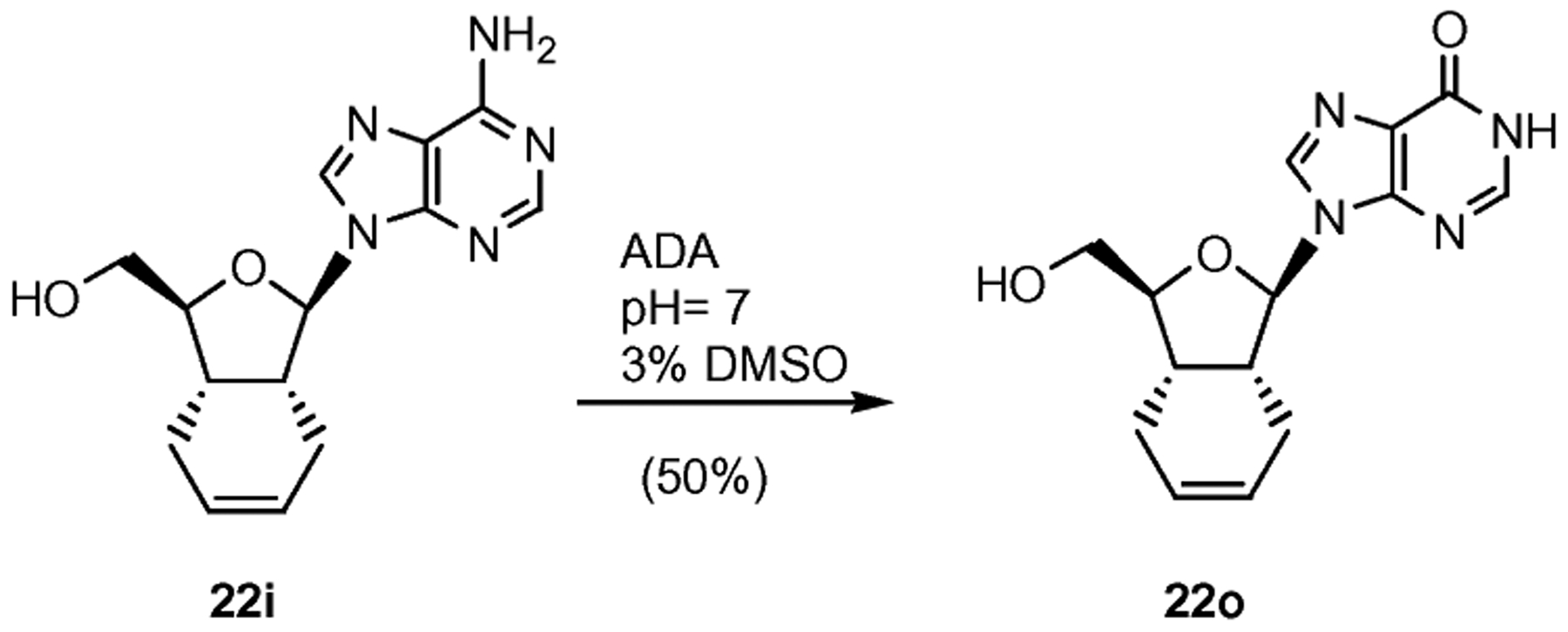
Synthesis of inosine 22o through enzymatic deamination. Reagents and conditions: 1% ADA, phosphate buffer pH = 7, 3% DMSO, 35 °C, 72 h, 50%.
Single crystal X-ray data for compound 22b is given in Table 1. HIBF nucleoside analog 22b crystallized in the form of colorless needles.
Table 1.
Crystal data for HIBF nucleoside analog 22b
| Formula | C14H18N2O4 |
|---|---|
| Molecular weight | 278.30 |
| Crystal system | Tetragonal |
| Space group | P42/n |
| a, Å | 22.892 (12) |
| b, Å | 22.892 (12) |
| c, Å | 5.224 (4) |
| Z | 8 |
| Final R | 0.074 |
| Reflections | 7109 |
Fig. 3 is a PyMOL 0.9922 drawing, which corresponds to single-crystal X-ray diffraction studies of compound 22b.23 Note that the sugar conformation is North type with the 2′-exo and 3′-endo configurations.
Evaluation of HIBF nucleosides
Acid stability studies.
The acid stability of the glycosidic linkage in 22i (HIBF-ddA) was studied by a direct comparison with 2′,3′-didehydro-2′,3′-dideoxynucleoside (d4A, Fig. 4). The cleavage of the glycosidic bond was studied at pH 2 buffered solution and 37 °C.24 The degradation of the nucleoside to the free adenine was followed by high performance liquid chromatography (HPLC), where samples were collected at different time points. The data indicated that under experimental conditions d4A was degraded at a significantly faster rate compared to the HIBF nucleoside 22i ( d4A is about 20 min while of 22i is about 6 h). The data confirmed that placement of a C–C bond at the 2′-position enhances significantly the stability of HIBF nucleosides towards lower pH.
Fig. 4.
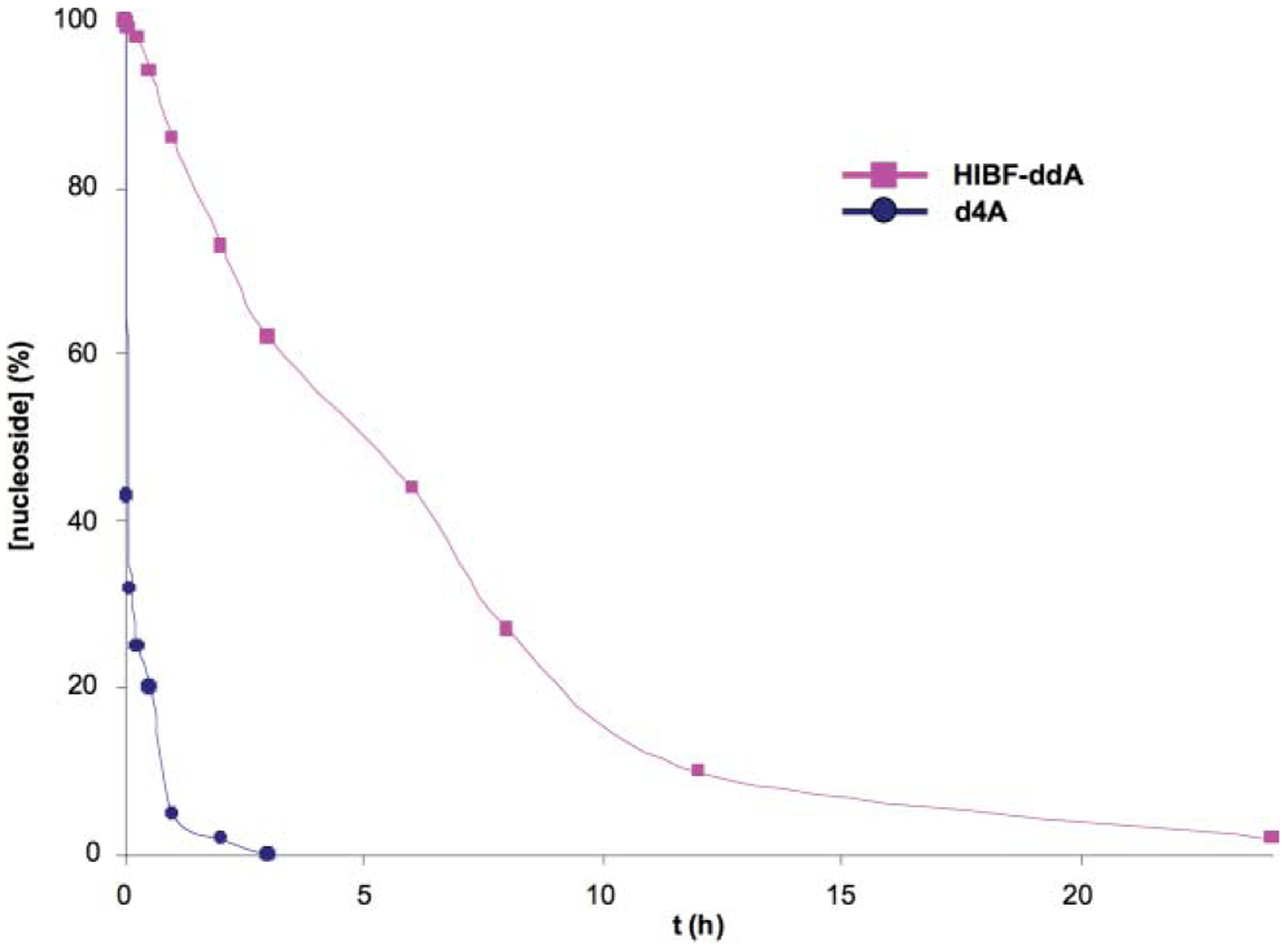
Percentage versus time curves for d4A and HIBF-ddA (22i) in pH = 2 buffer at 37 °C.
Biological evaluation
Antiviral assays.
A set of twenty-five compounds were tested against HIV-1 (strain LAI) and compared to those of 3′-azido-3′-deoxythymidine (AZT, zidovudine) in an assay with human peripheral blood mononuclear (PBM) cells. Some of the 5′-O-acetyl HIBF nucleosides were also included in the testing as potential prodrugs because acyl groups are known to hydrolyze in the presence of cellular esterases generating the free nucleosides. Furthermore, some nucleosides were tested as mixtures of α/β-anomers because these were obtained as an inseparable mixture. A summary of the data, expressed as the effective concentration required to inhibit viral replication by 50% (EC50) and 90% (EC90), is presented in Table 2. HIBF nucleoside analogs 20g/21g, 22j, 22l, 22o, 23d, 23f, and 23j demonstrate modest activity when compared with the AZT as a control. The potencies (EC50) of the compounds were in the order of 22o > 22j > 23d > 22l > 20g/21g > 23f > 23j. In general, the purine nucleosides were found to be more potent than the corresponding pyrimidine nucleosides, with the exception of β−5-(2-bromovinyl)uridine 23d. Among various HIBF purine nucleoside derivatives tested for anti-HIV-1, two α-anomers (6-Cl-purine 23f, 2-Cl-adenosine 23j) and a mixture of α/β-anomers (2,6-di-Cl-purines 20g/21g) also exhibited moderate activities. The HIBF nucleoside 22o containing the inosine base is the most potent nucleoside analog in this series of compounds (EC50 = 12.3 μM). Interestingly, the 2-Cl-adenosine derivative 22j showed closely related antiviral activity as inosine analog 22o (EC50 16.5 μM vs. 12.3 μM). The similar anti-HIV-1 activities of these two nucleosides could be attributed to possible deamination of the 22j furnishing a 2-chloro-inosine analog with a close structural resemblance to 22o. This observation is in agreement with the transformation of the adenosine analog 22i to 22o in the presence of adenosine deaminase (Scheme 6). In fact, the masked activity of the inosine analog is also evident in 2-Cl-6-OMe-purine 22l and 6-Cl-purine 23f. Both nucleosides could potentially be hydrolyzed under physiological conditions exposing the anti-HIV motif of the 2-chloro-inosine analog and its activity. It is noteworthy that the natural configuration of β−2-Cl-adenine derivative 22j exhibited 2-fold greater potency than its corresponding αanomer 23j (EC50 16.5 μM vs. 36.2 μM). Therefore, the observed activity of the mixture α/β−2,6-di-Cl-purine 20g/21g could be due to the presence of β-anomer in the mixture tested. Clearly, there is a correlation between the six active purine nucleoside analogs with the inosine analog being most active. The structural resemblance of the ddI and inosine analog 22o is remarkable and warrants further studies with the HIBF series of purine nucleosides.
Table 2.
Effect of analogs against HIV-1 (strain LA1) in human peripheral blood mononuclear (PBM) cells
| Anti-HIV-1 activity in PBM cellsa | Cytotoxicity (IC50, μM)b | |||||
|---|---|---|---|---|---|---|
| Analog | Base | EC50, μM | EC90, μM | PBM cells | CEM cells | VERO cells |
| AZT | β-T0 | 0.0015 | 0.006 | > 100 | 14.3 | 50.6 |
| 20a+21a | α/β-U | 60.7 | > 100 | > 100 | > 100 | > 100 |
| 20b+21b | α/β-T | 82.02 | > 100 | > 100 | > 100 | > 100 |
| 20d+21d | α/β-BVU | > 100 | > 100 | > 100 | > 100 | > 100 |
| 20e+21e | α/β-N-Ac-C | > 100 | > 100 | > 100 | > 100 | > 100 |
| 20g+21g | α/β-2,6-di-Cl-purine | 24.6 | 62.4 | 43.1 | 5.9 | 18.9 |
| 22a | β-U | 90.5 | > 100 | > 100 | 67.0 | > 100 |
| 22b | β-T | > 100 | > 100 | > 100 | > 100 | > 100 |
| 22c | β-5-F-U | > 100 | > 100 | > 100 | > 100 | > 100 |
| 22d | β-BVU | 77.8 | > 100 | > 100 | > 100 | > 100 |
| 22f | β-6-Cl-purine | 70.3 | > 100 | > 100 | 95.6 | > 100 |
| 22i | β-A | > 100 | > 100 | > 100 | > 100 | > 100 |
| 22j | β-2-Cl-A | 16.5 | 39.1 | 27.2 | > 100 | 34.7 |
| 22l | β-2-Cl-6-OMe-purine | 20.2 | 95.6 | > 100 | > 100 | 76.2 |
| 22m | β-2,6-di-NH2-purine | > 100 | > 100 | > 100 | > 100 | > 100 |
| 22o | β-I | 12.3 | 44.3 | 41.4 | 20.1 | 50.3 |
| 23b | α-T | > 100 | > 100 | > 100 | > 100 | > 100 |
| 23d | α-BVU | 19.0 | 74.2 | 31.9 | 34.2 | 19.6 |
| 23f | α-6-Cl-purine | 28.5 | 46.9 | 54.2 | 21.9 | 98.3 |
| 23i | α-A | 90.6 | > 100 | > 100 | ≥ 100 | > 100 |
| 23j | α-2-Cl-A | 36.2 | 67.1 | 30.6 | > 100 | 61.9 |
| 23m | α-2,6-di-NH2-purine | > 100 | > 100 | ≥ 100 | > 100 | > 100 |
| 24b | α/β-T | > 100 | > 100 | > 100 | > 100 | > 100 |
| 24h | α/β-C | > 100 | > 100 | > 100 | > 100 | > 100 |
| 24i | α/β-A | > 100 | > 100 | > 100 | > 100 | > 100 |
| 24j | α/β-2-Cl-A | 76.2 | > 100 | > 100 | > 100 | > 100 |
Cytotoxicity assays.
The compounds were evaluated for their potential toxic effects on uninfected phytohemagglutinin-stimulated human PBM cells, and also in CEM and Vero (African green monkey kidney) cells. Primary human PBM cells were selected, because these lymphocytes represent slow-growing cells, and were also used in the antiviral assay as host cells. The lymphocytic CEM cells were selected, as they are a continuous nontransformed cell line that replicates quickly and is commonly used for anti-HIV-1 assays. Vero cells were used, as they not only grow rapidly but are also anchored on the surface. The toxicities in these cells may translate into the observed anti-HIV-1 activity. It is noteworthy that the observed cytotoxicity in PBM and CEM cells with inosine analog 22o could be eliminated by introduction of suitable functionality on the purine ring. For example, β−2-Cl-6-OMe-purine 22l is not cytotoxic to the PBM and CEM cells. No significant toxicity was detected for most of the compounds devoid of the anti-HIV activity.
In summary, results of both antiviral and cytotoxicity studies indicate that these compounds could offer valuable leads into the development of novel NRTIs with improved therapy for HIV-1 treatment.
Conclusions
In summary, the present study provides for the first time a direct access to a variety of bicyclic nucleoside analogs starting with d-mannitol. The designed HIBF nucleosides contain the three essential SAR elements, which are: (i) the presence of a double bond in the carbohydrate portion of the structure; (ii) the stabilization of the glycosidic linkage at lower pH and (iii) the adoption of a preferred N-conformation. The NMR and X-ray studies confirmed that the sugar adopts an N-conformation that may be responsible for the observed anti-HIV-1 activity with some of the compounds. The anti-HIV-1 activity exhibited by purine analogs 22o, 22j and 22l is of particular interest because of the close resemblance with the US FDA approved anti-HIV-1 drug ddI. Despite the modified bicyclic carbohydrate motif, the adenosine analog 22i was deaminated with ADA indicating its ability to mimic the natural adenosine. Therefore, it is reasonable to postulate that the observed anti-HIV-1 activity with purine analogs is perhaps dependent on the recognition by cellular ADA. Interestingly, the antiviral activity and cytotoxicity was modulated by the nature of the substituent on the purine ring. The intracellular metabolism of these nucleosides deserves further attention with particular emphasis on specificity for the ADA.
The anti-HIV-1 activity of ddI is due to its transformation to dideoxyadenosine triphosphate, which inhibits the activity of HIV-1 RT by competing with natural substrate, 2′-deoxyadenosine triphosphate as well as by its incorporation into viral DNA causing termination of the viral chain elongation. Based on the observed anti-HIV-1 activity with various purine analogs, we propose that the HIBF nucleosides are mimics of ddI and exert their activity via incorporation into viral DNA. Because these compounds are stable at lower pH, we believe that they could offer superior bioavailability and longer half-life in vivo after oral administration. The transformation of some of the lead compounds (e.g. 22o) into their triphosphate analogs is currently under progress for further biological studies. The synthetic methodology described in this study offers a facile and efficient entry into a variety of bicyclic nucleoside analogs with a wider range of therapeutic applications, such as synthesis of novel nucleosides as antiviral compounds and building blocks for the construction of antisense oligonucleotides.
Experimental section
General techniques
All reagents were bought from Aldrich and Acros at highest commercial quality and used without further purification except where noted. Air- and moisture-sensitive liquids and solutions were transferred via syringe or stainless steel cannula. Organic solutions were concentrated by rotary evaporation below 45 °C at approximately 20 mmHg. All non-aqueous reactions were carried out under anhydrous conditions using flame-dried glassware within an argon atmosphere in dry, freshly distilled solvents, unless otherwise noted. THF and CH2Cl2 were purified by passage through a bed of activated alumina. Pyridine (Py) was distilled from CaH2 prior to use. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. Reactions were monitored by TLC carried out on 0.25 mm E. Merck silica gel plates (60F-254) using UV light as visualizing agent and 7% ethanolic phosphomolybdic acid, or p-anisaldehyde solution and heat as developing agents.E. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash chromatography. LC-ESI-MS analyses were carried out in a chromatograph with UV detector at 280 nm using a VYDAC C-18 column, Cat. Number: 218TP54, (4.6 mm × 250 nm); flow 1 mL min−1; r.t.; gradient MeOH–H2O as eluent. NMR spectra were recorded on Varian Mercury 300, 400 and/or Unity 500 MHz or Bruker AV-400 or 600 MHz instruments and calibrated using residual undeuterated solvent as an internal reference. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. IR spectra were recorded on a Nicolet 320 Avatar FT-IR spectrometer and values are reported in cm−1 units. Optical rotations were recorded on a Jasco P-1010 polarimeter and values are reported as follows: (c: g/100 mL, solvent). High resolution mass spectra (HRMS) were recorded on a VG 7070 HS mass spectrometer under electron spray ionization (ESI) conditions. X-Ray data were recorded on a Bruker SMART APEX 3 kW Sealed Tube X-ray diffraction system.
The bicyclic motif of the synthetic hexahydroisobenzofuran nucleosides is abbreviated below as HIBF. The assignment of the NMR data is based on the nucleoside numbering (1′–5′) and the second ring as 6′–9′ positions (Fig. 2).
Synthesis of lactone 12.
It was obtained following a previously described procedure.14 Rf (33% hexane–EtOAc): 0.41; (c 3.40, CH2Cl2); IR (film): v 3070, 3043, 2932, 2857, 1775, 1112, 704 cm−1; MS (ESI+, m/z): 407 [(M + H)+, 100%]; Exact mass found for C25H30O3Si: 406.1964.
Synthesis of lactol 13.
A solution of 12 (12.8 g, 31.5 mmol) in dry CH2Cl2 (150 mL) was cooled to −78 °C under argon and treated with DIBAL-H (35 mL, 35 mmol, 1 M in CH2Cl2) for 1 h. The reaction was quenched with MeOH (10 mL) and the solution was brought to r.t. over a 20 min period. Saturated Rochelle salt was added (stirred for 15 min) and the crude product was extracted with EtOAc (4 × 100 mL). Lactol 13 was isolated in 92% yield as yellow oil. Rf (33% hexane–EtOAc): (c 1.9, CH2Cl2); MS (ESI+, m/z): 409 [(M + H)+, 100%]; Exact mass found for C25H32O3Si: 408.2120.
Synthesis of acetate 14.
Lactol 13 (6.65 mmol) was dissolved in pyridine (40 mL) and Ac2O (1.8 mL, 19.95 mmol) was added dropwise. The reaction was stirred overnight and then quenched with saturated NaHCO3. The solution was extracted with CH2Cl2 (3 × 60 mL). The combined organic extracts were dried (Na2SO4). The residue was purified by flash chromatography (10% hexane–EtOAc) to obtain 14 (90%). Rf (25% hexane–EtOAc): 0.65; (c 0.6, CHCl3); IR (film): v 3069, 3025, 2925, 2849, 1744, 1421, 1226, 1099 cm−1; MS (ESI+, m/z): 468 [(M + NH4)+, 100%], 473 [(M + Na)+, 45], and 489 [(M + K)+, 15].
Synthesis of acetate 15.
Compound 14 (3.34 mmol) was dissolved in 60 mL of dry THF and then treated with TBAF (4.3 mL, 4.3 mmol, 1 M in THF). The reaction was kept under argon at r.t. for 30 min. The solution was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried using Na2SO4 and then filtered. The residue was purified by flash chromatography (30% hexane–EtOAc) to give 15 (89%). Rf (EtOAc): 0.74; (c 0.9, CHCl3); IR (film): v 3019, 2922, 2845, 1728, 1427, 1369, 1233, 932 cm−1; MS (ESI+, m/z): 230 [(M + NH4)+, 50%], 235 [(M + Na)+, 75], and 251 [(M + K)+,20].
Synthesis of diacetate 16.
A solution of 17 (31 mmol) in pyridine (100 mL) was treated with Ac2O (12 mL, 124 mmol). The reaction was stirred overnight and quenched with saturated NaHCO3. The solution was extracted with CH2Cl2 (4 × 200 mL). The combined organic extracts were dried using Na2SO4 and then filtered. The residue was purified by flash chromatography (15% hexane–EtOAc) to afford 16 (93%). Rf (50% EtOAc–hexane): 0.53; (c 1.9, CHCl3); IR (film): v 3030, 2918, 2838, 1737, 1641, 1440, 1239, 933 cm−1; MS (ESI+, m/z): 272 [(M + NH4)+, 20%] and 277 [(M + Na)+, 100].
Synthesis of diol 17.
Lactol 13 (31.5 mmol) was dissolved in THF (100 mL) under argon and then treated with TBAF (37.8 mL, 37.8 mmol, 1 M in THF) at r.t. for 25 min. The solution was quenched with brine and extracted with EtOAc (3 × 100mL). The residue was chromatographed using a gradient of 30–70% EtOAc– hexane to give 17 (96%). R (EtOAc): 0.44; (c 1.9, CH2Cl2); MS (ESI+, m/z): 193 [(M + Na)+, 55%].
Synthesis of diacetate 19.
A solution of 13 (31.5 mmol) in THF (100 mL) was treated with TBAOH (47.3 mmol, 33% in H2O) at r.t. for 6 h. Then, solvents were removed in vacuo. The orange residue was treated in pyridine (100 mL) with Ac2O (12 mL, 124 mmol). The reaction was stirred overnight and then quenched with saturated NaHCO3. The solution was extracted with CH2Cl2 (4 × 200 mL). The combined organic extracts were dried using Na2SO4, and then filtered. The residue was purified by flash chromatography (20% hexane–EtOAc) to give 19 (70%). Rf (50% EtOAc–hexane): 0.55; IR (film): v 3026, 2931, 2839, 1734, 1633, 1233, 936 cm−1; MS (ESI+, m/z): 272 [(M + NH4)+, 80%] and 277 [(M + Na)+, 100].
General procedure for condensation of diacetate 16 with heterocyclic bases
To a stirred solution of the different bases (1.81 mmol) in dry MeCN (5.0 mL), BSA was added (1.5 mL, 6.338 mmol). The reaction mixture was heated at 70 °C for 1.5 h. To the resulting clear mixture was added at r.t. a solution of diacetate 16 (0.9 mmol) in dry MeCN (3 mL), followed by DBU (0.434 mL, 2.899 mmol). This solution was cooled to 0 °C and then TMSOTf was added dropwise (1.05 mL, 5.798 mmol). The resulting mixtures were stirred for 2–3 h. Each mixture was poured into an ice-cool saturated NaHCO3 solution and extracted with CH2Cl2 (3 × 25 mL). Residues were purified by silica gel column chromatography to give 20 and 21 using a gradient of 33–100% EtOAc–hexane.
HIBF-α/β-uracil (20a+21a).
R (EtOAc): 0.55; (c 0.7, CHCl3); IR (KBr): v 3200, 3032, 2922, 2844, 1743, 1686, 1461, 1271, 1237, 1042, 705 cm−1; MS (ESI+, m/z): 307 [(M + H)+, 70%], 324 [(M + NH)+, 80], and 329 [(M + Na)+, 100]; LC-ESI-MS (UV 280 nm): coupling ratio β/α 1.1 : 1 tR(min) = 17.46, 17.91; Exact mass found for C15H18N2O5: 306.1206.
HIBF-α/β-thymine ((20b+21b).
Rf (50% hexane–EtOAc):0.22; (c 1.0, CHCl3); IR (KBr): v 3197, 3032, 2926, 1744, 1684, 1662, 1473, 1271, 1237, 1044 cm−1; MS (ESI+, m/z): 321 [(M + H)+, 100%], 338 [(M + NH)+, 90], and 343 [(M + Na)+, 95]; LC-ESI-MS (UV 280 nm): coupling ratio β/α1.2 : 1 tR(min) = 19.11, 19.29; Exact mass found for C16H20N2O5: 320.1371.
HIBF-β−5-fluorouracil (20c).
Rf (50% hexane–EtOAc): 0.29; (c 1.3, CHCl3); IR (KBr): v 3189, 3075, 3044, 2950, 2857, 1718, 1661, 1469, 1262, 1230, 1093, 1064 cm−1; 19F NMR (CDCl3, 282 MHz): δ 167.1; MS (ESI+, m/z): 325 [(M + H)+, 30%], 342 [(M + NH4)+, 60], and 347 [(M + Na)+, 100]; LC-ESI-MS (UV 280 nm): tR(min) 19.31; Exact mass found for C15H17FN2O5: 324.1122.
HIBF-β−5-bromovinyluracil (20d).
Rf (50% hexane–EtOAc):0.41; (c 0.6, CHCl3); IR (KBr): v 3424, 2926, 2874, 1793, 1747, 1700, 1472, 1368, 1280, 1166, 1109, 1041 cm−1; MS (ESI+, m/z): 433 [(M79Br + Na)+, 100%], 435 [(M81Br + Na)+, 97]; Exact mass found for C17H19BrN2O5: 410.0872.
HIBF-α/β-cytosine (20e+21e).
Rt (EtOAc): 0.24; (c 0.4, MeOH); MS (ESI+, m/z): 370 [(M + Na)+, 100%]; LC-ESI-MS (UV 280 nm): coupling ratio β/α 1.2 : 1 tR(min) = 19.09,19.63; Exact mass found for C17H21N3O5: 347.1480.
HIBF-α/β−6-chloropurine (20f+21f).
Rf (50% hexane– EtOAc): 0.37; (c 1.0, CHCl3); IR (KBr): v 3389, 2942, 2890, 1708, 1633, 1489, 1245, 1193, 1094, 920 cm−1; MS (ESI+, m/z): 371 [(M35Cl + Na)+, 100], 373 [(M37Cl + Na)+, 30]; LC-ESI-MS (UV 280 nm): coupling ratio β/α1.5 : 1 tR(min) = 21.19, 21.43; Exact mass found for C16H17ClN4O3: 348.0982.
HIBF-α/β−2,6-dichloropurine (20g+21g).
The two anomers were separated. 20g: Rf (50% hexane–EtOAc): 0.52; (c 0.3, CHCl3); IR (KBr): v 3370, 2940, 2879, 1728, 1627,=1480, 1239, 1190, 1076, cm−1; MS (ESI+, m/z): 405 [(M35Cl35Cl + Na)+, 100%], 407 [(M35Cl37Cl + Na)+, 80], and 409 [(M37Cl37Cl + Na)+, 10%]; LC-ESI-MS (UV 280 nm): ratio β/α2.4 : 1 tR(min) 23.62, 24.27; Exact mass found for C16H16Cl2N4O3: 382.0600.
21g.
Rf (50% hexane–EtOAc): 0.52; (c 1.3, CHCl3); MS (ESI+, m/z): 405 [(M35Cl35Cl + Na)+, 100%], 407 [(M35Cl37Cl + Na)+, 80], and 409 [(M37Cl37Cl + Na)+, 10]; LC-ESI-MS (UV 280 nm): coupling ratio β/α 2.4 : 1 tR(min) = 23.62, 24.27; Exact mass found for C16H16Cl2N4O3: 382.0651.
General protocols for deprotection of HIBF nucleoside derivatives 20–21
Method A (deprotection of 5′-OAc group). Nucleoside derivatives 20a–f+21a–f (0.1 mmol) were dissolved in a 2 M NH3–MeOH solution (1.5 mL) and stirred at r.t. overnight. Residues were purified by silica gel column chromatography using a gradient of 33–100% EtOAc–hexane. Method B. Nucleoside derivatives 20f+21f, 20g and 21g (0.1 mmol) were treated with a NH3–MeOH saturated solution (1.5 mL) and stirred at 110 °C in a Parr reactor for 36 h. Residues were purified by silica gel column chromatography (gradient 50–100% EtOAc–hexane) obtaining 22i, 23i (95–97%), 22j–22m, 23j–23m (quantitative combined yield). Method C. Nucleoside derivatives 20f and 20g (0.11 mmol) were treated with a 2 M NH3–MeOH solution (1.8 mL) and stirred at 90–100 °C in a Parr reactor for 30 h. Residues were purified by silica gel column chromatography using a gradient 50–100% EtOAc–hexane.
HIBF-β-uracil (22a).
From 20a using Method A. Rf (EtOAc):0.40; (c 0.3, CHCl3); MS (ESI+, m/z): 287 [(M + Na)+, 100%]; Exact mass found for C13H15N2O4: 264.1104.
HIBF-β-thymine (22b).
From 20b using Method A. Rf (50% hexane–EtOAc): 0.16; (c 0.3, CHCl3); MS (ESI+, m/z): 301 [(M + Na)+, 100%]; Exact mass found for C14H18N2O4: 278.1257.
HIBF-β−5-fluorouracil (22c).
From 20c using Method A. Rf (50% hexane–EtOAc): (c 0.8, CHCl3); IR (KBr): v 3508, 3448, 3156, 3040, 2925, 2851, 1682, 1652, 1266, 1093, 1066, 1026 cm−1; 19F NMR (CDCl3, 282 MHz): δ 167.0; MS (ESI+, m/z): 305 [(M + Na)+, 100%]; Exact mass found for C13H15FN2O4: 282.1014.
HIBF-β−5-bromovinyluracil (22d).
From 20d using Method A. Rf (50% hexane–EtOAc): 0.32; (c 0.6, CHCl3); MS (ESI+, m/z): 369 [(M79Br + H)+, 100%] and 371 [(M81Br + H)+, 96]; Exact mass found for C15H17BrN2O4: 368.0395.
HIBF-β−6-chloropurine (22f).
From 20f using Method A. Rf (EtOAc): 0.50; (c 1.3, CHCl3); MS (ESI+, m/z): 329 [(M35Cl + Na)+, 100%] and 331 [(M37Cl + Na)+, 20]; Exact mass found for C14H15ClN4O2: 306.0682.
HIBF-β−6-aminopurine (22i).
From 20f using Method B. Rf (5% CH2Cl2–MeOH): 0.20; (c 0.8, DMSO); MS (ESI+, m/z): 288 [(M + H)+, 100%] and 310 [(M + Na)+, 60]; Exact mass found for C14H17N5O2: 287.0713.
HIBF-β−6-amino-2-chloropurine (22j).
From 20g using Method B. Rf (10% EtOAc–MeOH): 0.44; (c 0.3, DMSO); MS (ESI+, m/z): 322 [(M35Cl + H)+, 100%], 324 [(M37Cl + H)+, 20], 344 [(M35Cl + Na)+, 80], and 346 [(M37Cl + Na)+, 10].
HIBF-β−6-methoxypurine (22k).
From 20f using Method C. Rf (EtOAc): 0.39; MS (ESI+, m/z): 325 [(M + Na)+, 100%]; Exact mass found for C15H18N4O3: 302.0774.
HIBF-β−2-chloro-6-methoxypurine (22l).
From 20g using Method C. Rf (EtOAc): 0.44; MS (ESI+, m/z): 359 [(M35Cl + Na)+, 100%] and 361 [(M37Cl + Na)+, 20]; Exact mass found for C15H17ClN4O3: 336.1102.
HIBF-β−2,6-diaminopurine (22m).
From 20g using Method B. Rf (10% EtOAc–MeOH): 0.17; (c 2.5, DMSO); MS (ESI+, m/z): 303 [(M + H)+, 100%].
HIBF-α-thymine (23b).
From 21b using Method A. Rf (50% hexane–EtOAc): 0.10; (c 0.3, CHCl3); MS (ESI+, m/z): 301 [(M + Na)+, 100%]; Exact mass found for C14H18N2O4: 278.1260.
HIBF-α−5-bromovinyluracil (23d).
From 21d using Method A. Rf(50% hexane–EtOAc): 0.14; MS (ESI+, m/z): 369 [(M79Br + H)+, 100] and 371 [(M81Br + H)+, 96]; Exact mass found for C15H17BrN2O4: 368.0874.
HIBF-α−6-chloropurine (23f).
From 21f using Method A. Rf (EtOAc): 0.40; (c 0.8, CHCl3); MS (ESI+, m/z): 329 [(M35Cl + Na)+, 100%] and 331 [(M37Cl + Na)+, 30]; Exact mass found for C14H15ClN4O2: 306.0548.
HIBF-α−6-aminopurine (23i).
From 21f using Method B. Rf (5% CH2Cl2–MeOH): 0.09; (c 2.5, MeOH); MS (ESI+, m/z): 288 [(M + H)+, 100%] and 310 [(M + Na)+, 60]; Exact mass found for C14H17N5O2: 287.0772.
HIBF-α−6-amino-2-chloropurine (23j).
From 21g using Method B. Rf (6.6% EtOAc–MeOH): 0.37; (c 1.0, DMSO); MS (ESI+, m/z): 322 [(M35Cl + H)+, 100%] and 324 [(M37Cl + H)+, 20].
HIBF-α−2,6-diaminopurine (23m).
From 21g using Method B. Rf (10% EtOAc–MeOH): 0.12; (c 0.8, MeOH); MS (ESI+, m/z): 303 [(M + H)+, 100%].
HIBF-α/β-uracil (24a).
From 20a+21a using Method A. Rf (EtOAc): 0.40β, 0.36α; MS (ESI+, m/z): 287 [(M + Na)+, 100%].
HIBF-α/β-cytosine (24h).
From 20h+21h using Method A. Rf(EtOAc–MeOH 10%): 0.20; (c 0.7, MeOH); MS (ESI+, m/z): 264 [(M + H)+, 70%] and 286 [(M + Na)+, 100]; Exact mass found for C13H17N3O3: 263.1318.
Synthesis of HIBF-β−2,6-diaminopurine (20m).
A solution of 20n (30 mg, 0.076 mmol) in a mixture THF–H2O was treated with Me3P (15.4 μL, 0.174 mmol). Immediately, N2 was given off and the solution became yellowish. The reaction was stirred for 26 h and then solvents were removed. The residue was purified by silica gel chromatography (2–10% MeOH–EtOAc) to give 20m in 73% yield. Rf(20% EtOAc–MeOH): 0.49; MS (ESI+f, m/z): 367 [(M + Na)+, 100%].
Synthesis of HIBF-β−2,6-diazidopurine (20n).
A solution of 20g (0.262 mmol, 100 mg) in DMF (1.8 mL) was treated with NaN3 (1.048 mmol, 68 mg) at 70 °C for 4 h. Solvent was removed and the residue obtained was purified by silica gel flash chromatography (gradient 30–50% EtOAc–hexane) to give 20n in 76% yield. Rf(50% hexane–EtOAc): 0.47; MS (ESI+, m/z): 419 [(M + Na)+, 100%].
Synthesis of HIBF-β-hypoxanthine 22o.
Nucleoside 22i (20 mg, 0.070 mmol) was dissolved in a phosphate buffer pH = 7/3% DMSO (400 μL) and treated with 1 mg of ADA dissolved in that buffer (100 μL). The residue was purified by flash chromatography (gradient 5–20% MeOH–EtOAc) to give 22o (50%). Rf (10% EtOAc–MeOH): 0.10; MS (ESI+, m/z): 289 [(M + H)+, 100%] and 311 [(M + Na)+, 60].
Antiviral assays
The procedures for the antiviral assays in human peripheral blood mononuclear (PBM) cells have been reported previously.25 Briefly, uninfected phytohemagglutinin-stimulated human PBM cells were infected with HIV-1 (strain LA1) (about 63 000 disintegrations of RT activity per minute per 107 cells per 10 ml of medium). The HIBF nuclesoside analogs were then added to duplicate or triplicate cultures. Uninfected and untreated PBM cells were grown in parallel at equivalent cell concentrations as controls. The cultures were maintained in a humidified 5% CO2–95% air incubator at 37 °C for 6 days after infection, at which point all cultures were sampled for supernatant RT activity. The supernatant was clarified, and the viral particles were then pelleted at 40 000 rpm for 30 min by using a rotor and suspended in virus-disrupting buffer. The RT assay was performed in 96-well microdilution plates by using (rA)n·(dT)12–18 as the template primer. The RT results were expressed in disintegrations per minute per millilitre of originally clarified supernatant.
Cytotoxicity assays
The HIBF nucleoside analogs were evaluated for their potential toxic effects on uninfected phytohemagglutinin-stimulated human PBM cells and also in CEM and Vero (African green monkey kidney) cells as described previously.26 PBM cells were obtained from the whole blood of healthy HIV-1 and hepatitis B virus-seronegative volunteers and collected by single-step Ficoll-Hypaque discontinuous gradient centrifugation. CEM (CEM-CCRF) cells were a T-lymphoblastoid cell line that was obtained from the American Type Culture Collection, Rockville, Md. The CEM cells were maintained in RPMI 1640 medium supplemented with 20% heat-inactivated fetal bovine serum, penicillin (100 U ml−1), and streptomycin (100 mg ml−1). The PBM and CEM cells were cultured with and without drug for 6 days, at which time portions were counted for cell proliferation and viability by the trypan blue exclusion method. Only the effects on cell growth are reported, since these correlated well with cell viability. The toxicity of the compounds in Vero cells was assessed after 3 days of treatment with a hemacytometer.
Supplementary Material
Acknowledgements
Financial support of this work by the Spanish Ministerio de Educación y Ciencia (MEC) (Project MEC-07-CTQ-61126) is gratefully acknowledged. A. D.-R. thanks MEC for a predoctoral fellowship. We also thank Dr A. G. DiPasquale and Professor A. L. Rheingold (UCSD X-Ray Facility) for the reported crystallographic studies and Dr Y. Su (UCSD Mass Spectrometry Facility) for mass analysis and the NSF (CHE-0116662, CHE-9709183 and CHE-0741968) for NMR and MS instrumentation grants. R. F. S. is supported in part by NIH grants 5P30-AI-50409 (CFAR), 5R37-AI-041980 and by the Department of Veterans Affairs. We are grateful to Kim L. Rapp and Jason Grier for their excellent technical assistance.
Footnotes
Electronic supplementary information (ESI) available: 1H, 13C NMR spectral data. The level of purity is indicated by the inclusion of copies of 1H, 13C and 19F NMR spectra. In addition, some 2D NMR experiments are shown, which were used to assign the peaks. CCDC reference number 666543. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b818707j
References and notes
- 1.(a) Meadows CD and Gervay-Hague J, ChemMedChem, 2006, 1, 16–29; [DOI] [PubMed] [Google Scholar]; (b) Fauci AS, Science, 2006, 313, 409–409. [DOI] [PubMed] [Google Scholar]
- 2.Franklin D, Roemer AS and Placidi L, The global antiviral therapeutics market, in Frontiers in Nucleosides and Nucleic Acids, ed. Schinazi RF and Liotta DC, IHL Press, 2004, pp. 159–184. [Google Scholar]
- 3.(a) Schinazi RF, Larder BA and Mellors JW, Internatl. Antiviral News, 2000, 8, 129; [Google Scholar]; (b) Ono SK, Nakane H, Herdewijn P, Balzarini J and De Clercq E, Mol. Pharmacol, 1989, 35, 578–583; [PubMed] [Google Scholar]; (c) Moyle GJ, Immunol. Infect. Dis, 1995, 5, 170–182. [Google Scholar]
- 4.(a) Wang J, Jin Y, Rapp KL, Schinazi RF and Chu CK, J. Med. Chem, 2007, 50, 1828–1839; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Casu F, Chiacchio MA, Romeo R and Gumina G, Curr. Org. Chem, 2007, 11, 999–1016; [Google Scholar]; (c) Kim MJ and Chun MW, Aust. J. Chem, 2007, 60, 291–295; [Google Scholar]; (d) Sard H, Nucleosides Nucleotides, 1994, 13, 2321–2328; [Google Scholar]; (e) Okabe M and Sun R-C, Tetrahedron Lett, 1989, 30, 2203–2206; [Google Scholar]; (f) Beard AR, Butler PI,Mann J and Partlet NK, Carbohydr. Res, 1990, 205, 87–91; [DOI] [PubMed] [Google Scholar]; (g) Wu J-C and Chattopadhyaya J, Tetrahedron Lett, 1990, 46, 2587–2592; [Google Scholar]; (h) Ewing DF, Fahmi N-E, Len C, Mackenzie G and Pranzo A,J. Chem. Soc., Perkin Trans 1, 2000, 3561–3565; [Google Scholar]; (i) Hossain N, Plavec J, Thibaudeau C and Chattopadhyaya J, Tetrahedron, 1993, 49, 9079–9088. [Google Scholar]
- 5.Lee K, Choi Y, Gumina G, Zhou W, Schinazi RF and Chu CK,J. Med. Chem, 2002, 45, 1313–1320. [DOI] [PubMed] [Google Scholar]
- 6.Choo H, Chong Y and Chu CK, Bioorg. Med. Chem. Lett, 2003, 13, 1993–1996. [DOI] [PubMed] [Google Scholar]
- 7.(a) Marquez VE, Tseng CK-H, Mitsuya H, Aoki S, Kelley JA,Ford H, Roth JS, Broder S, Johns DG and Driscoll JS, J. Med. Chem, 1990, 33, 978–985; [DOI] [PubMed] [Google Scholar]; (b) Ruxrungtham K, Boone E, Ford H, Drisoll JS, Davey RT and Lane HC, Antimicrob. Agents Chemother, 1996, 40, 2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russ P, Schelling P, Scapozza L, Folkers G, De Clercq E and Marquez VE, J. Med. Chem, 2003, 46, 5045–5054. [DOI] [PubMed] [Google Scholar]
- 9.(a) Choi Y, George C, Comin MJ, Barchi JJ Jr, Kim HS, Jacobson KA, Balzarini J, Mitsuya H, Boyer PL, Huges SH and Marquez VE, J. Med. Chem, 2003, 46, 3292–3299; [DOI] [PubMed] [Google Scholar]; (b) Webb TR, Mitsuya S and Broder S, J. Med. Chem, 1988, 31, 1475–1479. [DOI] [PubMed] [Google Scholar]
- 10.(a) Haly B, Bharadwaj R and Sanghvi YS, Synlett, 1996, 687–689; [Google Scholar]; (b) Sanghvi YS, Cook PD, Carbohydrates: synthetic methods and applications in antisense therapeutics; an overview in Carbohydrates Modifications in Antisense Research, ed. Sanghvi YS and Cook PD, ACS Symp. Ser., 1994, 580, 1–23; [Google Scholar]; (c) Sanghvi YS, Synthesis of Nitrogen Containing Linkages for Antisense Oligonucleotides, in Carbohydrate Mimics: Concepts and Methods, ed. Chapleur Yves, VCH Publication, 1998, pp. 523–36. [Google Scholar]
- 11.Egron D, Perigaud C, Gosselin G, Aubertin A-M, Faraj A, Selouane M, Postel D and Len C, Bioorg. Med. Chem. Lett, 2003, 13, 4473–4475. [DOI] [PubMed] [Google Scholar]
- 12.Drone J, Egorov M, Hatton W, Bertrand M-J, Len C and Lebreton J, Synlett, 2006, 1339–1342. [Google Scholar]
- 13.(a) Schmid CR, Bryant JD, Dowlatzedah M, Phillips JL, Prather DE, Shantz RD, Sear NL and Vianco CS, J. Org. Chem, 1991, 56, 4056–4058; [Google Scholar]; (b) Mann J and Weymouth-Wilson A, Carbohydr. Res, 1991, 216, 511–515; [Google Scholar]; (c) Fazio F and Scheider MP, Tetrahedron: Asymmetry, 2000, 11, 1869–1876. [Google Scholar]
- 14.(a) Brady TP, Kim SH, Wen K and Theodorakis EA, Angew. Chem., Int. Ed, 2004, 43, 739–742; [DOI] [PubMed] [Google Scholar]; (b) Brady TP, Kim SH, Wen K,Kim C and Theodorakis EA, Chem.–Eur. J, 2005, 11, 7175–7190. [DOI] [PubMed] [Google Scholar]
- 15.Hardinger SA and Wijaya N, Tetrahedron Lett, 1993, 34, 3821–3824. [Google Scholar]
- 16.(a) Casas R, Parella T, Branchadell V, Oliva A, Ortuño RM and Guignant A, Tetrahedron, 1992, 48, 2659–2680; [Google Scholar]; (b) Ortuño RM, Ballesteros M, Corbera J, Sanchez-Ferrando F and Font J, Tetrahedron, 1988, 44, 1711–1719; [Google Scholar]; (c) Ortuño RM, Corbera J and Font J, Tetrahedron Lett, 1986, 27, 1081–1084. [Google Scholar]
- 17.(a) Hilbert GE and Johnson TB, J. Am. Chem. Soc, 1930, 52, 4489–4494; [Google Scholar]; (b) Vorbruggen H, Ruh-Pohlenz C, Handbook of nucleoside synthesis, Wiley Interscience, 2001, pp. 10–24. [Google Scholar]
- 18.Sanghvi YS, Bhattacharya BK, Kini GD, Matsumoto SS, Larson SB, Jolley WB, Robins RK and Revankar GR, J. Med. Chem, 1990, 33, 336–344. [DOI] [PubMed] [Google Scholar]
- 19.(a) Staudinger H and Meyer J, Helv. Chim. Acta, 1919, 2, 635–646; [Google Scholar]; (b) Robins MJ and Barr PJ, J. Org. Chem, 1983, 48, 1854–1862. [Google Scholar]
- 20.Santaniello E, Ciuffreda P and Alessandrini L, Synthesis, 2005, 509–526. [Google Scholar]
- 21.Gupta M and Nair V, Collect. Czech. Chem. Commun, 2006, 71, 769–787. [Google Scholar]
- 22.DeLano WL, “The PyMOL Molecular Graphics System”; DeLano Scientific LLC, San Carlos, CA, USA, http://www.pymol.org. [Google Scholar]
- 23.CCDC-666543 contains the supplementary crystallographic data for compound 22b. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre. via www.ccdc.cam.ac.uk/products/csd/request/.
- 24.Jeannot F, Gosselyn G, Strandring D, Bryant M, Sommadossi J-P, Loi AG, La Colla P and Mathé P, Bioorg. Med. Chem, 2002, 10, 3153–3161. [DOI] [PubMed] [Google Scholar]
- 25.Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL,Xie M-W, Hart GC, Smith GA and Hahn EF, Antimicrob. Agents Chemother, 1990, 34, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ and Schinazi RF, Antimicrob. Agents Chemother, 2002, 46, 3854–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.