

Abstract
COVID‐19 is characterized by dysregulated immune responses, metabolic dysfunction and adverse effects on the function of multiple organs. To understand host responses to COVID‐19 pathophysiology, we combined transcriptomics, proteomics, and metabolomics to identify molecular markers in peripheral blood and plasma samples of 66 COVID‐19‐infected patients experiencing a range of disease severities and 17 healthy controls. A large number of expressed genes, proteins, metabolites, and extracellular RNAs (exRNAs) exhibit strong associations with various clinical parameters. Multiple sets of tissue‐specific proteins and exRNAs varied significantly in both mild and severe patients suggesting a potential impact on tissue function. Chronic activation of neutrophils, IFN‐I signaling, and a high level of inflammatory cytokines were observed in patients with severe disease progression. In contrast, COVID‐19‐infected patients experiencing milder disease symptoms showed robust T‐cell responses. Finally, we identified genes, proteins, and exRNAs as potential biomarkers that might assist in predicting the prognosis of SARS‐CoV‐2 infection. These data refine our understanding of the pathophysiology and clinical progress of COVID‐19.
Keywords: COVID‐19 pathophysiology, immunopathogenesis, multi‐omics, multiple organ damage, SARS‐CoV‐2
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Proteomics, metabolomics and RNAseq data map immune responses in COVID‐19 patients with different disease severity, revealing molecular makers associated with disease progression and alterations of tissue‐specific proteins.
Introduction
Coronaviruses (family Coronaviridae) are a diverse group of positive‐sense single‐stranded RNA viruses with enveloped virions (Masters & Perlman, 2013; Cui et al, 2019). Coronaviruses are well known due to the emergence of Severe Acute Respiratory Syndrome (SARS) in 2002–2003 and Middle East Respiratory Syndrome (MERS) in 2012, both of which caused thousands of cases in multiple countries (Ksiazek et al, 2003; Bermingham et al, 2012; Cui et al, 2019). Coronaviruses naturally infect a broad range of vertebrate hosts including mammals and birds (Cui et al, 2019). As coronavirus primarily target epithelial cells, they are generally associated with gastrointestinal and respiratory infections (Masters & Perlman, 2013; Cui et al, 2019). In addition, they cause hepatic and neurological diseases of varying severity (Masters & Perlman, 2013).
The world is currently experiencing a disease pandemic (COVID‐19) caused by a newly identified coronavirus called SARS‐CoV‐2 (Wu et al, 2020a). At the time of writing, there have been more than ~25 million cases of SARS‐CoV‐2 and ~830,000 deaths globally (WHO, 2020). The disease leads to both mild and severe respiratory manifestations, with the latter prominent in the elderly and those with underlying medical conditions such as cardiovascular and chronic respiratory disease, diabetes, and cancer (Guan et al., 2020). In addition to respiratory syndrome, mild gastrointestinal and/or cardiovascular symptoms and neurological manifestations have been documented in hospitalized COVID‐19‐infected patients (Gupta et al, 2020; Mao et al, 2020). These data point to the complexity of COVID‐19 pathogenesis, especially in patients experiencing severe disease.
SARS‐CoV‐2 is able to use angiotensin‐converting enzyme 2 (ACE 2) as a receptor for cell entry (Hoffmann et al, 2020; Zheng et al, 2020a; Zhou et al, 2020b). Aside from lungs, ACE2 is expressed in other organs including heart, liver, kidney, pancreas, and small intestines (Li et al, 2020; Liu et al, 2020; Zou et al, 2020; Chen et al, 2020a). More recently, ACE2 expression has also been found in Leydig cells in the testes (Li et al, 2020; Wang & Xu, 2020) and neurological tissue (Baig et al, 2020; Bullen et al, 2020; Xu & Lazartigues, 2020). As such, it is possible that these organs might also be infected by SARS‐CoV‐2, and recent autopsy studies have also revealed multi‐organ damage including heart, liver, intestine, pancreas, brain, kidney, and spleen in fatal COVID‐19‐infected patients (Lax et al, 2020; Menter et al, 2020; Varga et al, 2020; Wichmann et al, 2020; Wang et al, 2020c). The host immune response to SARS‐CoV‐2 may also impact pathogenicity, resulting in severe tissue damage and, occasionally, death (Tay et al, 2020). Indeed, several studies have reported lymphopenia, exhausted lymphocytes, and cytokine storms in COVID‐19‐infected patients, especially those with severe symptoms (Blanco‐Melo et al, 2020; Cao, 2020; Chua et al, 2020; Liao et al, 2020). Numerous clinical studies have also observed the elevation of lactate dehydrogenase (LDH), IL‐6, troponin I, inflammatory markers, and D‐dimer in COVID‐19‐infected patients (Zhou et al, 2020a; Wang et al, 2020b). However, despite the enormous burden of morbidity and mortality due to COVID‐19, we know little about its pathophysiology, even though this establishes the basis for successful clinical practice, vaccine development, and drug discovery.
Using a multi‐omics approach employing cutting‐edge transcriptomic, proteomic, and metabolomic technologies, we identified significant molecular alterations in patients with COVID‐19 compared with uninfected controls in this study. Our results refine the molecular view of COVID‐19 pathophysiology associated with disease progression and clinical outcome.
Results
Patient cohort and clinical characteristics
We studied 66 clinically diagnosed and laboratory confirmed COVID‐19‐infected patients hospitalized at the Shanghai Public Health Clinical Center, Shanghai, China, between January 31 and April 7, 2020 (Fig 1A, Datasets EV1 and EV2). At the time of writing, 55 (49 mild and 6 severe) of the 66 patients have recovered and been discharged following treatment, while five patients (1 mild and 4 severe) remain in the hospital and are receiving ongoing treatment. Unfortunately, six patients (all severe) died.
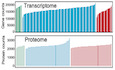
Figure 1. Study design and patient cohort.
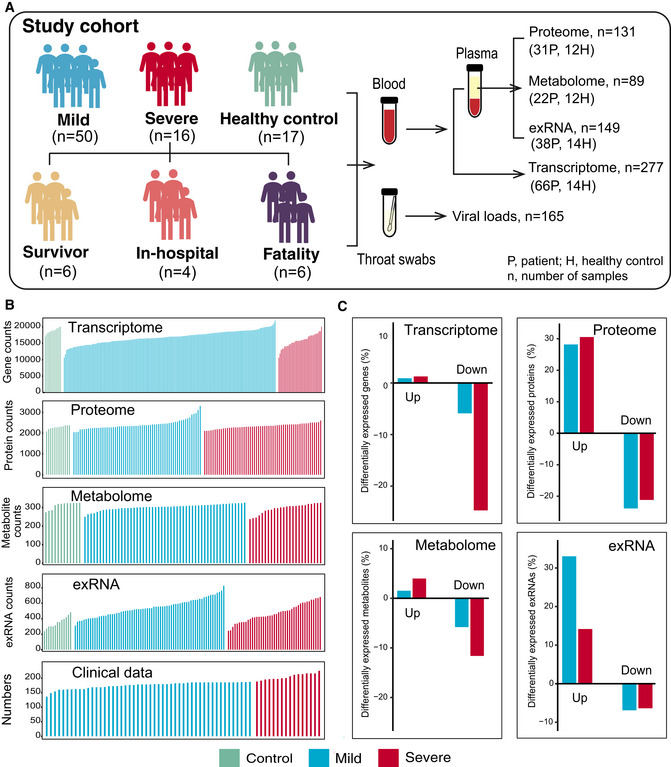
- Schematic summary of the study design and patient cohort. Both blood and throat swabs were collected in different timepoint (1 to 5). Not all samples were analyzed in all omics approach due to different requirements of sample quality and amounts for different approach. The sample numbers for each omics data set were shown, and specific sampling time for each sample in each data set was listed in Dataset EV1.
- The number of expressed genes and detected proteins, metabolites, exRNAs, and clinical parameters in high‐quality patient samples.
- Summary of differentially expressed genes, proteins, metabolites, and exRNAs between uninfected controls and COVID‐19‐infected patients (mild and severe) in the multi‐omics data. Y‐axis showed the proportion of upregulated/downregulated genes, proteins, metabolites, and exRNAs in those that were detected in total.
Molecular variation associated with COVID‐19 pathophysiology
Serial blood and throat swab samples were collected from all patients, as well as from 17 healthy volunteers. To determine whether COVID‐19 pathophysiology was associated with particular molecular changes, a total of 23,373 expressed genes, 9,439 proteins, 327 metabolites, and 769 exRNAs were examined using a multi‐omics approach combining transcriptomics, proteomics, and metabolomics (Fig 1B). Compared with healthy controls, mild and severe patients had significantly different expression patterns (higher or lower) in 6.79 and 26.0% of expressed genes, 52.1 and 51.7% of proteins, 7.34 and 15.6% of metabolites, and 39.9 and 20.5% of exRNAs, respectively (Fig 1C, Datasets [Link], [Link], [Link], [Link]). Significant differences in the principal component 1 (PC1), PC2, and/or PC3 between healthy controls, mild, and severe COVID‐19‐infected patients were observed in multi‐omics data (Figs 2A and EV1), suggesting that the molecular changes identified are likely to reflect the severity of COVID‐19‐infected patients.
Figure 2. Molecular characteristics of COVID‐19‐infected patients.
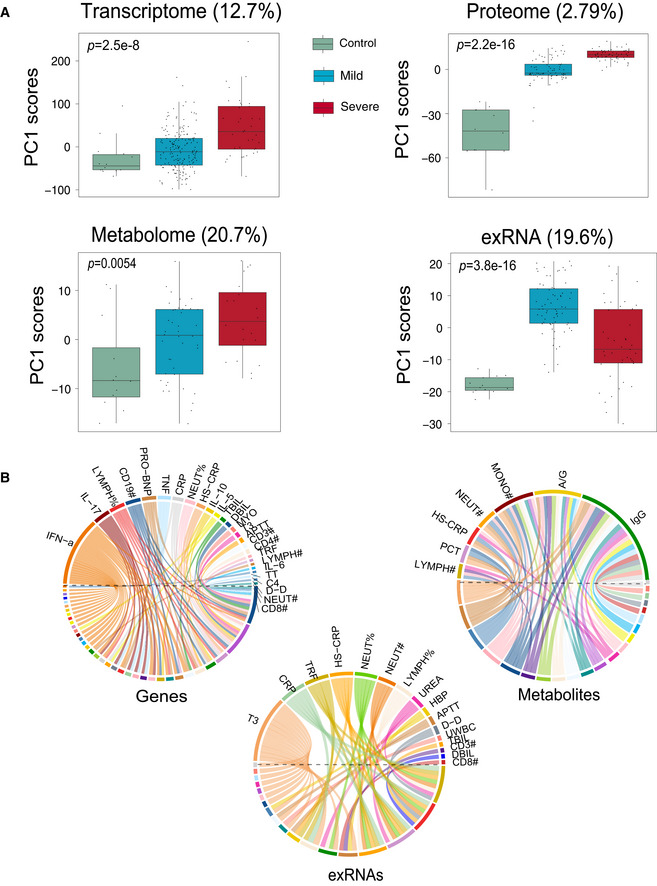
- Scores of principal components 1 (PC1) of each sample from the transcriptome (control, n = 14; mild, n = 179; severe, n = 37), proteome (control, n = 12; mild, n = 80; severe, n = 57), metabolome (control, n = 12; mild, n = 42; severe, n = 20), and exRNA‐seq (control, n = 14; mild, n = 179; severe, n = 37) principal component analyses. Differences between groups were estimated using Kruskal–Wallis test. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
- Circos plots showing the significant correlations between clinical parameters and the multi‐omics data.
Figure EV1. Scores of principal components 2 and 3 (PC2 and PC3) of each sample from the RNA‐seq, proteome, metabolome, and exRNA‐seq principal component analyses.
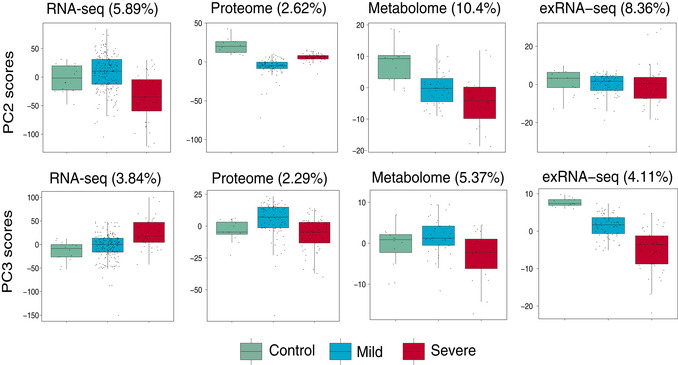
For RNA‐seq: control, n = 14; mild, n = 179; severe, n = 37; For proteome, control, n = 12; mild, n = 80; severe, n = 57; For metabolome, control, n = 12; mild, n = 42; severe, n = 20; For exRNA‐seq: control, n = 14; mild, n = 179; severe, n = 37. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
Remarkably, there were significant correlations between multi‐omics data and classical blood and biochemical parameters (Fig 2B). This was best reflected in the proteomic analysis (Fig 3) where there was a significant downregulation in the tricarboxylic acid cycle (TCA) and glycolytic pathways in both mild and severe patients compared with healthy controls (Figs 3A and EV2A). However, the hypoxia‐inducible factors (HIF‐1) signaling pathways and well‐known host defense pathways (e.g., T‐cell receptor signaling pathway, ISG15 antiviral signaling pathway) were elevated in these patients (Figs 3A and EV2A). Additionally, we applied weighted gene co‐expression network analysis (WGCNA) to separate the proteomic profiles into 33 co‐expression modules (ME0‐ME32) (Fig 3B). Among these, six modules showed significant correlation with clinical parameters (Fig 3B). Module 1, comprising 12 proteins, was strongly associated with activated partial thromboplastin time (APTT), with their downregulated expression likely indicating higher APTT values (Fig 3C). In contrast, levels of plasma IL‐6 and IL‐10 in patients were positively correlated with the expression of proteins in Module 15 (Fig 3D). Notably, correlations between the proteins in these modules were also identified, suggesting that proteins may interact in defining clinical outcome (Fig EV2B and C). In addition to proteins, lipoprotein variation was also significantly correlated with immune changes including IgG, monocytes, and procalcitonin (Fig 2B). Combined, these data suggest the association between specific molecular variations and the pathophysiological changes in COVID‐19‐infected patients.
Figure 3. Protein variation associated with COVID‐19 pathophysiology.
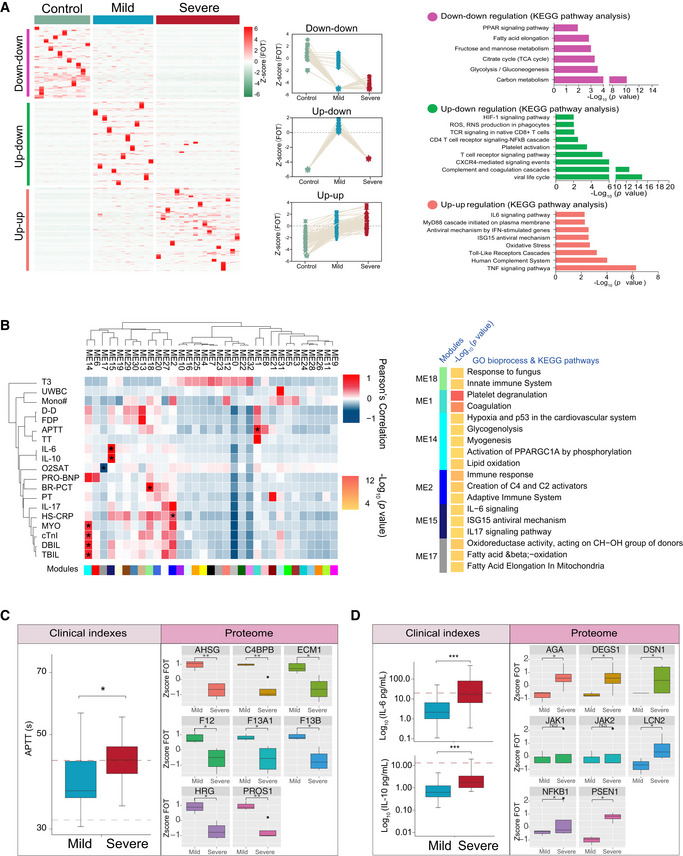
- Different expression of proteins in samples collected at the first timepoint. The heatmap (left panel) indicated expression patterns of proteins overrepresented (fold change > 2) in healthy control (1,656 proteins), mild (1,547 proteins), and severe (2,362 proteins) patient groups. The top categories enriched for clusters are shown. Values for each protein in each sample (columns) are color‐coded based on expression levels: low (blue) and high (red) z‐scored FOT. The line plots (middle panel) indicated selected gene sets with up‐up (upregulation in both mild and severe patients compared with healthy controls) or up‐down or down‐down regulation trend during disease progression. The bar plot (right panel) indicated Gene Ontology annotations with up‐up or up‐down or down‐downregulation trend during disease progression (Fisher’s exact test, P < 0.05).
- The Weighted Gene Co‐Expression Network Analysis (WGCNA) of 31 COVID‐19 samples shows modules that are highly correlated with clinical features (left heatmap). Among the 33 modules (ME0‐ME32), six of them showed significant correlation with clinical parameters (asterisk in the heatmap). Gene Ontology (GO) enrichment analysis were performed on these six modules, and the enriched pathways were presented on the right panel (Fisher’s exact test, P < 0.05).
- Boxplots showing differences of the APTT time (left) and ME1‐enriched protein expressions (right) between mild (n = 13) and severe (n = 18) COVID‐19‐infected patients. Differences between groups were estimated using Mann–Whitney–Wilcoxon test. *P < 0.05, **P < 0.01, ***P < 0.001. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
- Boxplots showing differences of IL‐6 level, IL‐10 level (left), and ME15‐enriched protein expressions (right) between mild (n = 13) and severe (n = 18) COVID‐19‐infected patients. Differences between groups were estimated using Mann–Whitney–Wilcoxon test. *P < 0.05, **P < 0.01, ***P < 0.001. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
Figure EV2. Molecular variation associated with COVID‐19 pathophysiology.
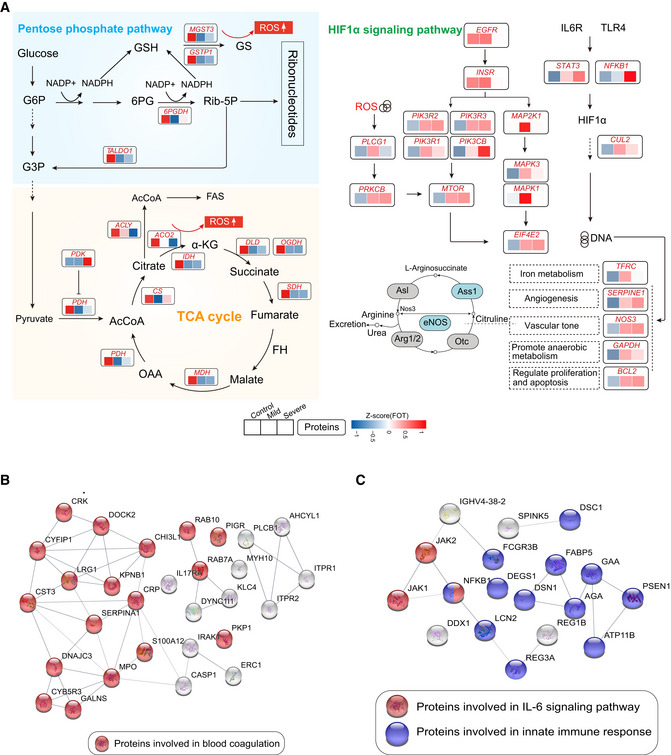
- Systematic summary of the proteins and signaling cascades significantly altered in healthy control patients (TCA, PEP) and in mild or severe patients (HIF‐1α). Values for each protein in all samples analyzed (columns) are color‐coded based on expression levels: low (blue) and high (red) z‐scored FOT. Blue and gray circles present proteins that could and could not be detected in this study, respectively; eNOS, endothelial nitric oxide synthase; OTC, Ornithine carbamoyltransferase, mitochondrial; Asl, argininosuccinate lyase; Ass1, argininosuccinate synthase; Arg1/2, arginase‐1/‐2.
- Network indicating protein–protein interactions among module 1 enriched proteins.
- Network indicating protein–protein interactions among module 2 enriched proteins.
Tissue damage caused by SARS‐CoV‐2
Recent data suggests that SARS‐CoV‐2 infection is associated with multi‐tissue injury and organ damage (Gupta et al, 2020; Zheng et al, 2020a; Wu et al, 2020b). Compared with healthy controls, alteration of tissue‐enhanced proteins (see Methods) was observed in COVID‐19‐infected patients, suggesting that COVID‐19 has a potential impact on multiple organs including the lung, liver, brain, testis, and intestine (Figs 4A and 5C). Notably, the majority of tissue‐enhanced proteins related to fundamental functions of the tissue in question (identified by GO analysis) were significantly downregulated in COVID‐19‐infected patients (Fig 4B). As expected, lung‐enhanced proteins varied significantly in the plasma of both mild and severe patients. The activation of the HIF‐1 signaling pathway and reactive oxygen species metabolic processes was seen in all patients (Fig 4D). Liver‐ and brain‐enhanced proteins also varied significantly, followed by those from the testis, intestine, and other organs (Fig 4A). Meanwhile, we have also observed a significant decline of brain‐enhanced proteins regulating neurotransmitter synthesis (GLS, OGDH, DLD, etc.), neurotransmitter transport (GLUL, GLUD2, GLUD1), and the numbers of neurotransmitter receptors (HTRA3, GRIK3, and GRIA3), as well as a significant decrease in proteins including ENO1, MBP, and NEFM (Fig 4C and F). Liver‐enhanced proteins, that regulate the transportation of sterol and cholesterol, were downregulated, while those involved in acute inflammatory response were elevated in both mild and severe patients (Fig 4E). Testis‐enhanced proteins involved in the cell cycle and cell proliferation were upregulated in all male patients, although proteins (e.g. YBX2) associated with reproduction were significantly downregulated. Heart‐specific proteins related to cardiac muscle contraction and oxidative reduction were reduced in COVID‐19‐infected patients (Fig 5C).
Figure 4. Variation of tissue‐enhanced proteins in COVID‐19‐infected patients.
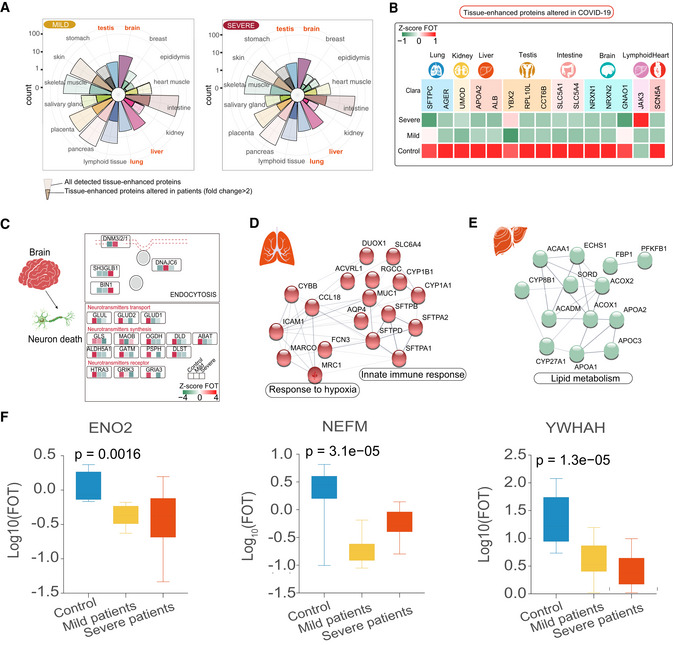
- Rose plots indicating the number of all detected and differently expressed tissues‐enhanced proteins in mild (left) and severe patients (right) compared with healthy controls. Tissues labeled in orange represented tissues with the largest proportions (100% of all detected tissue‐enhanced proteins) of altered tissues‐enhanced proteins.
- Heatmap indicating expression patterns of proteins related to organ function among control, mild, and severe patient groups. Values for each protein in three groups (rows) are color‐coded based on expression levels: low (green) and high (red) z‐scored FOT.
- Systematic summary of brain‐enhanced expressed proteins and signaling cascades significantly altered in COVID‐19‐infected patients (neurotransmitters transport, synthesis). Values for each protein at all analyzed samples (columns) are color‐coded based on the expression levels: low (green) and high (red) z‐scored FOT.
- Network summarizing lung‐enhanced expressed proteins and signaling cascades significantly altered in COVID‐19‐infected patients (HIF‐1α signaling pathway).
- Network summarizing liver‐enhanced expressed proteins and signaling cascades significantly altered in COVID‐19‐infected patients (Lipid metabolism).
- Boxplots indicating the expression level of known brain dysfunctional biomarkers in control (n = 12), mild (n = 13), and severe (n = 18) patients. Differences between groups were estimated using Kruskal–Wallis test. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
Figure 5. Patterns of tissue damage associated with COVID‐19.

- Bar plots showing the X‐Cell scores of specific cell types across control, mild, and severe COVID‐19‐infected patients.
- Boxplot showing the selected cell type specific proteins among control (n = 12), mild (n = 13), and severe (n = 18) COVID‐19‐infected patients. Differences between groups were estimated using Kruskal–Wallis test. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
- Systematic summary of the GO pathways enriched by tissue‐enhanced proteins that exhibited altered expression among control, mild, and severe patient groups. The heatmap of each panel indicates expression patterns of tissues‐enhanced proteins among control, mild, and severe patient groups. The top panel on the heatmap indicated the age and gender information of patients. The fold changes of tissue‐enhanced proteins between mild/severe patient samples and control samples are shown on the right panel of heatmap.
We also looked at the relative proportion of different cell populations and the expression of tissue injury‐related exRNAs. We identified 16 cell types based on proteomes whose abundance changed significantly following virus infection (Fig 5A and B). For example, the set of proteins expressed by alveolar type 1/2 epithelial cells (AT1 and AT2) were significantly downregulated in all patients (Fig 5B). In addition, the majority of tissue injury‐related exRNAs across all tissues showed differential expression, including lung (55 in 92 pre‐identified, P < 0.0001), kidney (14 in 22, P < 0.0001), liver (17 in 22, P < 0.0001), brain (8 in 16, P = 0.0016), and heart (5 in 6, P < 0.0001) (Fig EV3C). Finally, variation in some tissue‐enhanced proteins and tissue injury‐related exRNAs was also associated with COVID‐19 severity. For example, brain‐enhanced proteins enriched in tubulin accumulation were upregulated in mild disease patients. However, proteins significantly upregulated in severe patients were enriched in liver steatosis AOP and in multi‐drug resistance factors (Fig EV3A and B). A large proportion of tissue injury‐related exRNAs were expressed differently in mild and severe patients in most tissues analyzed (30/92 in lung, 10/22 in kidney, 8/22 in liver, and 3/6 in heart), except brain (1/16) (Fig EV3C). Together, these data suggest that COVID‐19 potentially impacts multiple organs.
Figure EV3. Comparative analysis of tissue injury in mild and severe COVID‐19‐infected patients.
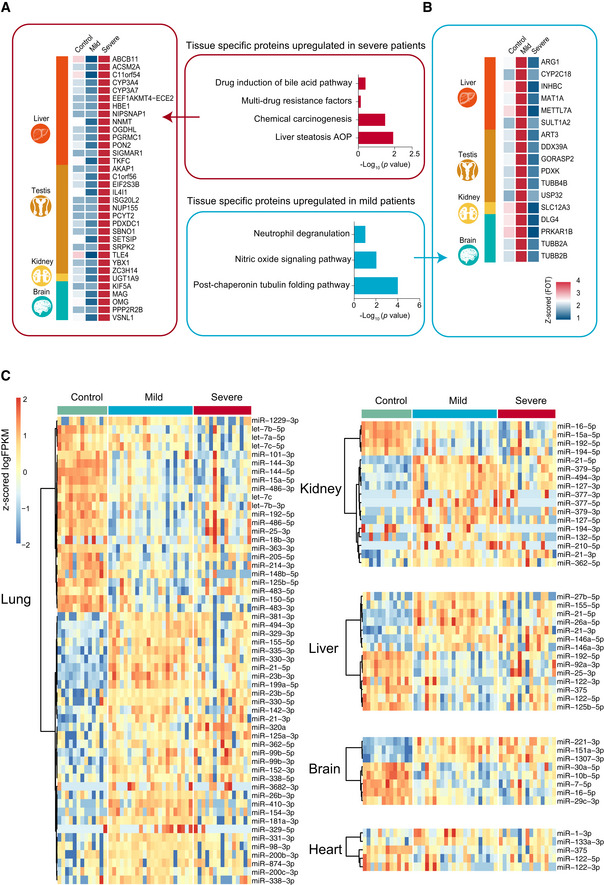
-
A, BThe heatmap indicates expression patterns of tissue‐enhanced biomarkers among the healthy control, mild, and severe patient groups. (A), tissue‐enhanced proteins upregulated in severe patients; (B), tissue‐enhanced proteins upregulated in mild patients. Values for each protein in all samples analyzed (columns) are color‐coded based on expression levels: low (blue) and high (red) z‐scored FOT. The bar plots indicate GO processes and pathways enriched by tissue‐specific proteins upregulated in mild patients (A) and severe patients (B) (Fisher’s exact test, P < 0.05).
-
CexRNA data related to tissue injury were collected from publications. The majority of tissue injury‐related exRNAs across all tissues analyzed showed differential expression. Differentially expressed exRNAs were identified using a t‐test < 0.05 and fold change> 2 or < 0.5.
Immunopathological changes in COVID‐19‐infected patients
Immune responses can cause severe damage to the cells or tissues that defend hosts against viral infection (Newton et al, 2016; Baseler et al, 2017; Cicchese et al, 2018; Chua et al, 2020; Gupta et al, 2020; Tay et al, 2020). Analysis of whole blood transcriptomic data revealed that gene sets, including an antiviral IFN signature (M75 module), were enriched at the first sampling timepoint (Fig 6A, Dataset EV7). Notably, IFN signaling was continuously activated in severe patients during the entire period of hospitalization (Fig 6A), while negative regulators of innate immune signaling (e.g. TRIM59, USP21, and NLRC3) were downregulated (Fig EV4A). Additionally, clinical data showed significant increases of IL‐6, IL‐8, and IL‐10 levels in severe patients compared with mild patients (Figs 2F and EV4B). Combined, these data suggest that the continuous activation of IFN‐I signaling and a high level of inflammatory cytokines likely impact COVID‐19 immunopathology.
Figure 6. Differences in immune responses among COVID‐19‐infected patients.
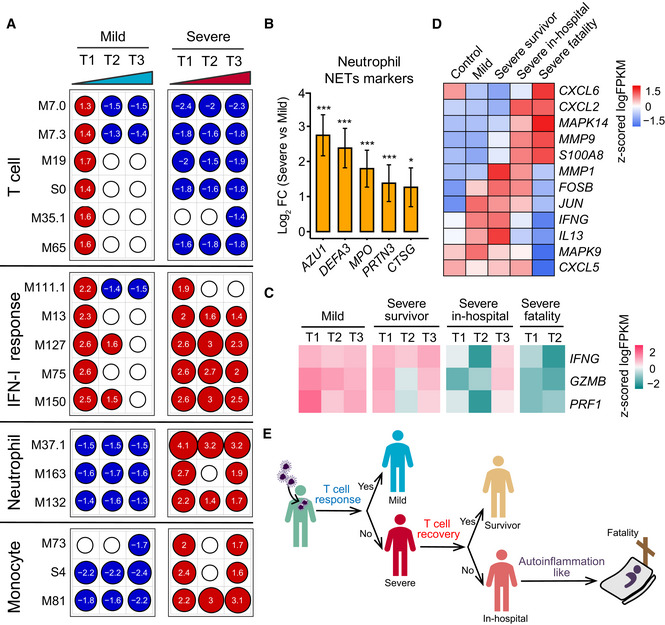
- Transcriptional profiles reflect the dynamic immune response in COVID‐19. GSEA (FDR < 0.25; 1,000 permutations) was used to identify positive (red), negative (blue), or no (white) enrichment of BTMs (gene sets). The graph shows the normalized enrichment score (NES) of each selected BTM in the different timepoints (T1, T2, and T3) for patients with mild or severe COVID‐19 illness, in comparison with healthy controls. The timepoints 1, 2, 4 or 1, 3, 5 were used as T1, T2, and T3 for patients with four or five sampling timepoints, respectively.
- Expression levels of NETs’ markers for individual transcripts in severe (n = 37) versus mild (n = 179) comparisons. Data were represented as means ± SEM, and differences between groups were estimated using Student’s t‐test. *P < 0.05; **P < 0.01; ***P < 0.001.
- Heatmap of IFNG, GZMB, and PRF1 gene expression in COVID‐19‐infected patients. Average expression values were centered and scaled. Red indicates a higher expression, and blue indicates a lower expression.
- Heatmap of the expression of genes enriched in IL‐17 signaling pathway between healthy control and COVID‐19‐infected patients. Average expression values were centered and scaled. Red indicates a higher expression and blue indicates a lower expression.
- T‐cell and innate immune response elucidate immunopathology of COVID‐19.
Figure EV4. Immune characteristics in COVID‐19‐infected patients.
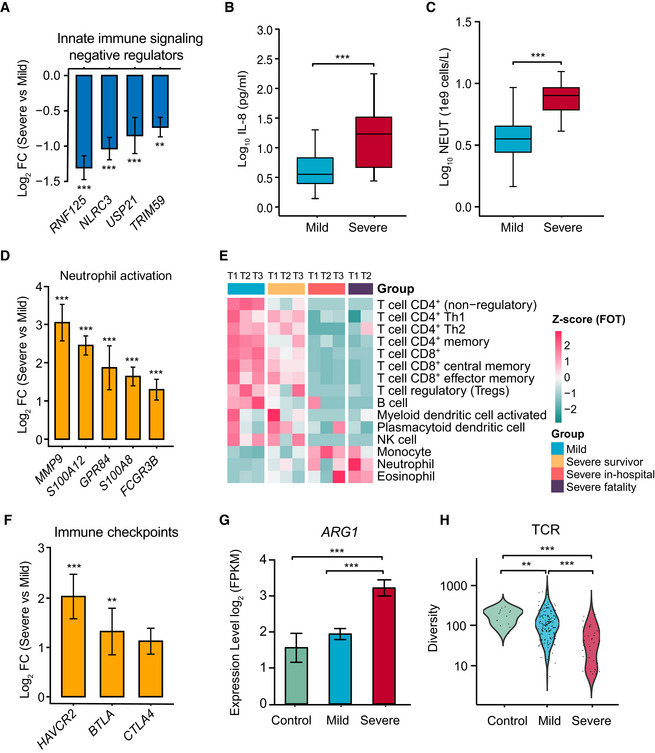
- Normalized gene expression of innate immune signaling negative regulators in mild versus severe COVID‐19‐infected patient comparisons. Mild, n = 179; severe, n = 37. Differences between groups were estimated using Student’s t‐test. **P < 0.01; ***P < 0.001.
- Level of plasma cytokines IL‐8 between mild and severe patient groups. Mild, n = 35; severe, n = 16. Differences between groups were estimated using Student’s t‐test. ***P < 0.001. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
- Absolute neutrophil count (NEUT) between mild and severe patient groups. Mild, n = 50; severe, n = 16. Differences between groups were estimated using Student’s t‐test. ***P < 0.001. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
- Normalized expression of neutrophil activation genes in mild versus severe COVID‐19‐infected patient comparisons. Mild, n = 179; severe, n = 37. Data were represented as means ± SEM. Differences between groups were estimated using Student’s t‐test. ***P < 0.001.
- Cell type enrichment analysis of the RNA sequencing data using the X‐Cell tool among COVID‐19‐infected patient subgroups from longitudinal samples. The colors of the heatmap represent the Z‐scaled values of fractions of each cell type from different stages and subgroups. The red color stands for a higher proportion whereas the blue color denotes a lower cell population.
- Expression levels of immune checkpoints, normalized to CD3G mild versus severe patient comparisons. Mild, n = 179; severe, n = 37. Data were represented as means ± SEM. Differences between groups were estimated using Student’s t‐test. **P < 0.01; ***P < 0.001.
- Expression levels of ARG1 between control and COVID‐19‐infected patient subgroups. Control, n = 14; mild, n = 179; severe, n = 37. Data were represented as means ± SEM. Differences between groups were estimated using Student’s t‐test. ***P < 0.001.
- Comparison of TCR diversity between healthy controls and COVID‐19‐infected patient subgroups. Control, n = 14; mild, n = 179; severe, n = 37. Differences between groups were estimated using Student’s t‐test. **P < 0.01; ***P < 0.001.
Higher neutrophil counts were observed clinically in severe patients but not in mild patients during hospitalization (Fig EV4C). Examination of the neutrophil transcriptomic signatures revealed that excessive neutrophil activation was associated with severe rather than mild disease, consistent with results of recently published studies (Chua et al, 2020; Zhang et al, 2020). These markers involved those utilized in neutrophil chemotaxis, activation, and migration (Fig EV4D). Notably, genes encoding molecules associated with neutrophil extracellular traps (NETs) were significantly upregulated in severe disease patients (Fig 6B). As excess NETs formation can lead to tissue damage (Kruger et al, 2015), our data imply that the excessive activation of neutrophils may contribute to COVID‐19 pathogenesis.
As in the case of influenza viruses that can be cleared by strong T‐cell responses (van de Sandt et al, 2014; Wang et al, 2015), SARS‐CoV‐2 immunity in mild patients was characterized by a robust T‐cell response, reflected in T‐cell signaling activation (M7.3 module, M35.1 module) and T‐cell differentiation (M19 module) on admission, followed by subsequent rapid reduction (Fig 6A). However, a negative T‐cell signaling was continuously observed in severe patients along time. In agreement with transcriptomic data, clinical data showed that severe patients lost ~59.1% of their total T‐cell population (CD3, severe versus mild: 383.38 versus 937.24 cells per ml), 62.3% of their CD4 T cells (208.63 versus 554.08 cells per ml), and 52.8% of their CD8 T cells (162.81 versus 344.96 cells per ml). Importantly, the CD4 T cells in the severe‐survivor groups maintained certain level of T‐cell activation, while those in the severe‐fatality group did not (Fig EV4E). Additionally, T cells in the survivors were primed by dendritic cells and expressed high levels of IFNG and GZMB (Fig 6C) as other studies reported (Braun et al, 2020; Sekine et al, 2020). T‐cell dysfunction was observed in the severe group, which could in part be due to an inhibitory status based on the expression levels of multiple exhaustion markers (Fig EV4F). Strikingly, the severe disease group had a greater abundance of ARG1 (Fig EV4G). Finally, the mild group had higher TCR diversity than the severe group (Fig EV4H). In sum, our data suggest that the T‐cell response is indispensable to successful host defense against SARS‐CoV‐2.
Finally, we investigated the immune signatures associated with poor COVID‐19 prognosis. Notably, KEGG functional analysis revealed that gene sets of the “IL‐17 signaling pathway” were significantly enriched in the severe‐fatal group. Further analysis of the signature components revealed that p38 MAPK activation was dominant in fatal cases, while higher levels of IL13 and IFNG were present in survivors (Fig 6D). These gene signatures might contribute to greater neutrophil influx (CXCL2 and CXCL6) and inflammation (S100A8), and could be detrimental in the severe disease group (Fig 6E).
Comprehensive changes in lipoprotein metabolism in COVID‐19‐infected patients
To reveal metabolic changes in COVID‐19‐infected patients, we quantified 348 metabolite parameters in small metabolites, lipoprotein subclasses, and their compositional components. The PCA scores plot revealed an obvious metabolomic trajectory from mild to severe COVID‐19, and gradually away from healthy controls (Fig 7A). Such group‐clustering patterns were independently confirmed by PCA scores plots from all NMR‐detectable metabolite signals, all MS‐detectable signals for lipids, and hydrophilic molecules in plasma samples (Figs EV5A–D). Our data therefore indicate that a concentration of changes in plasma metabolites is associated with COVID‐19 severity.
Figure 7. COVID‐19‐associated metabolomic changes in blood plasma.
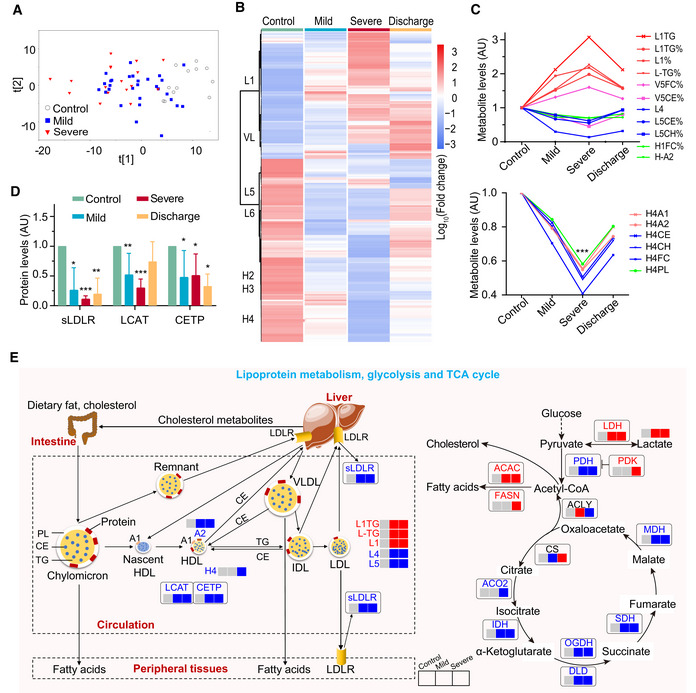
- Plasma metabolomic changes revealed a trajectory in COVID‐19 severity, from healthy control (n = 12), mild (n = 29), to severe (n = 17).
- Changes in the concentration of plasma metabolites are associated with COVID‐19 severity. The discharge group consist of all patients (mild and severe) that were recovered and discharged. L1, low‐density lipoprotein subclass‐1 (LDL1); VL, very low‐ density lipoprotein (VLDL); L5, LDL5; L6, LDL6; H2: high‐density lipoprotein subclass‐2 (HDL2); H3: HDL3; H4: HDL4.
- COVID‐19 severity is associated with significant changes in lipoprotein subclasses including high‐density lipoprotein subclass‐1 (HDL1), HDL4, low‐density lipoprotein subclasses (LDL1, LDL4, LDL5), very low‐density lipoprotein subclass‐5 (VLDL5), and their compositional components (ApoA1, triglycerides, cholesterol). TG: triglycerides; FC: free cholesterol; CE: cholesteryl esters; CH: total cholesterol (i.e., FC + CE); PL: total phospholipids; A1: ApoA1; A2: ApoA2; L1TG: TG in LDL1; L1TG%: percentages of L1TG in total lipids of LDL1; L1%: percentage of LDL1 in all LDL; L‐TG%: percentages of L‐TG(TG in LDL) in total lipids of LDL; V5FC%, V5CE%: percentages of V5FC(FC in VLDL5) and V5CE(CE in VLDL5) in total lipids of VLDL5; L5CE%, L5CH%: percentages of L5CE(CE in LDL5) and L5CH(CH in LDL5) in total lipids of LDL5; H1FC%: percentages of H1FC (FC in HDL1) in total lipids of HDL1; H‐A2: ApoA2 in both HDL and nascent HDL; H4A1, H4A2, H4CE, H4CH, H4FC, H4PL: ApoA1, ApoA2, CE, CH, FC, and PL in HDL4.
- Plasma levels of key enzymes and proteins directly involving lipoprotein metabolism are indicators for COVID‐19 severity. Data were represented as means ± SD and differences between groups were estimated using Student’s t‐test. *P < 0.05; **P < 0.01; ***P < 0.001. Control, n = 12; mild, n = 29; severe, n = 17; discharge, n = 16; sLDLR: soluble low‐density lipoprotein receptor; LCAT: lecithin‐cholesterol acyltransferase; CEPT: cholesteryl‐ester transfer protein.
- COVID‐19 caused dysregulation in lipoprotein metabolism, glycolysis, and TCA cycle. The three boxes from left to right are control, mild, and severe, in which gray means normal, blue means decrease, red means increase. CS: Citrate synthase; IDH: Isocitrate dehydrogenase; ACO2: Aconitase; OGDH: α‐ketoglutarate dehydrogenase; DLD: Dihydrolipoyl dehydrogenase; SDH: Succinic dehydrogenase; MDH: Malate dehydrogenase; PDH: Pyruvate dehydrogenase; PDK: Pyruvate dehydrogenase kinase; ACLY: ATP citrate lyase; ACAC: Acetyl coenzyme A carboxylase; FASN: Fatty acid synthetase; LDH: Lactate dehydrogenase.
Figure EV5. COVID‐19‐caused changes in plasma metabolomics and clinical biochemistry associated with disease severity.
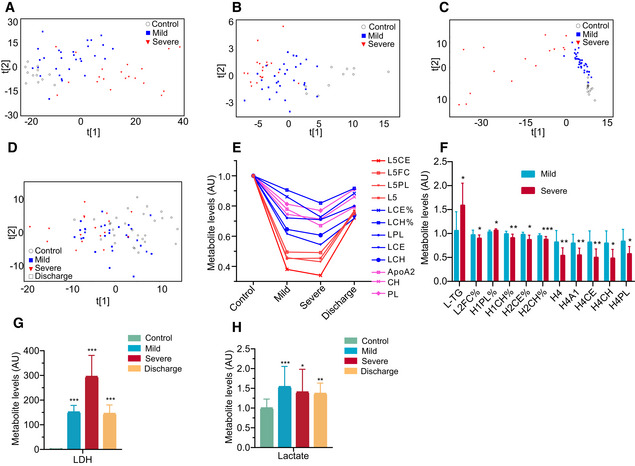
-
A–CCOVID‐19 severity was associated with plasma metabolomic phenotypes defined by all MS‐detectable lipidomic compounds (A), hydrophilic metabolites (B), and all NMR‐detectable metabolite signals (C).
-
DPCA scores revealed a variation in plasma metabolomic trajectory among healthy controls, patients with mild and severe COVID‐19, and upon discharge. The discharge group comprised all patients (mild and severe) that were recovered and discharged.
-
E–HCOVID‐19 severities are associated with changes in levels of the compositional components of lipoprotein subclasses (E and F), lactate (H), lactate dehydrogenase (G) in blood plasma. Data in bar plots are represented as means ± SD. Control, n = 12; mild, n = 29; severe, n = 17; discharge, n = 16; Differences between groups were estimated using Student’s t‐test. *P < 0.05; **P < 0.01; ***P < 0.001. HDL: high‐density lipoprotein; IDL: intermediate‐density lipoprotein; LDL: low‐density lipoprotein; VLDL: very low‐density lipoprotein; TG: triglycerides; FC: free cholesterol; CE: cholesteryl esters; CH: total cholesterol (i.e., FC + CE); PL: total phospholipids; L5CE, L5FC, L5PL:CE, FC and PL in LDL5; L5:LDL5; LCE, LCH: CE and CH in LDL; LCE%, LCH%: percentages of LCE and LCH in total lipids of LDL; LPL: PL in LDL; L‐TG: TG in LDL; L2FC%: percentages of L2FC (FC in LDL2) in total lipids of LDL2; H1PL%: percentages of H1PL (PL in HDL1) in total lipids of HDL1; H1CH%: percentages of H1CH (CH in HDL1) in total lipids of HDL1; H2CE%: percentages of H2CE (CE in HDL2) in total lipids of HDL2; H2CH%: percentages of H2CH (CH in HDL2) in total lipids of HDL2; H4A1, H4CE, H4CH, H4PL: ApoA1, CE, CH and PL in HDL4; AU: The metabolite concentration of each sample was normalized using the average of the control group; LDH: lactate dehydrogenase.
Further statistical analyses highlighted the major changes in the levels of lipoprotein subclasses and their compositional components including LDL1 (L1TG), LDL4, VLDL5, HDL1, and HDL4 (Figs 7C and EV5E). Compared with healthy controls, the level of triglycerides (TG) in LDL1 and free cholesterol (FC) in all VLDL5 lipids were significantly elevated in both mild and severe patients, while there were significant decreases in LDL4 and LDL5, cholesterol in LDL, cholesterol esters in VLDL5, Apo‐A2 in both HDL and nascent HDL, FC in HDL1 together with total cholesterol and phospholipids (PL). Interestingly, HDL4 and its components had significant lower levels in severe patients. Compared with mild patients, L1TG and PL in HDL1 were increased in severe patients, while cholesterol in HDL1, HDL2, HDL4, and its components decreased (Fig EV5F). Fortunately, most of these lipoproteins recovered following patients discharge (Figs 7Band C, and EV5D).
The levels of some key proteins involved in lipoprotein metabolism, including the soluble low‐density lipoprotein receptor (sLDLR), lecithin‐cholesterol acyltransferase (LCAT), and the cholesteryl ester transfer protein (CETP), were significantly reduced in mild and severe COVID‐19‐infected patients than those in healthy controls (Fig 7D and E). Additionally, enzymes such as ACO2, IDH, OGDH, DLD, SDH, and MDH in the TCA cycle were lower in COVID‐19‐infected patients compared with healthy controls, while the enzymes central to fatty acid synthesis (Acetyl coenzyme A carboxylase [ACAC] and Fatty acid synthetase [FASN]) were elevated. Finally, significant concurrent elevations in plasma lactate and LDH were observable in patients compared with healthy controls (Figs 7E and EV5G and H). In sum, these data reveal the dysregulation in lipoprotein metabolism, glycolysis, and TCA cycle during SARS‐CoV‐2 infection.
Viral load is associated with disease prognosis of severe COVID‐19‐infected patients
The severity and clinical outcome of COVID‐19 were also associated with viral load. Overall, SARS‐CoV‐2 RNA loads on admission were significantly higher in the throat swabs of the five fatal cases compared with those who survived (mean, 1.26 × 105 versus 3.98 × 103 copies/ml, respectively; P = 0.04) (Fig 8A and Dataset EV8). Although viral load declined during the period of hospitalization in both survival and fatal cases, it remained elevated in fatal cases compared with survivors.
Figure 8. Comparative analysis of severe‐survival patients and severe‐fatal patients.
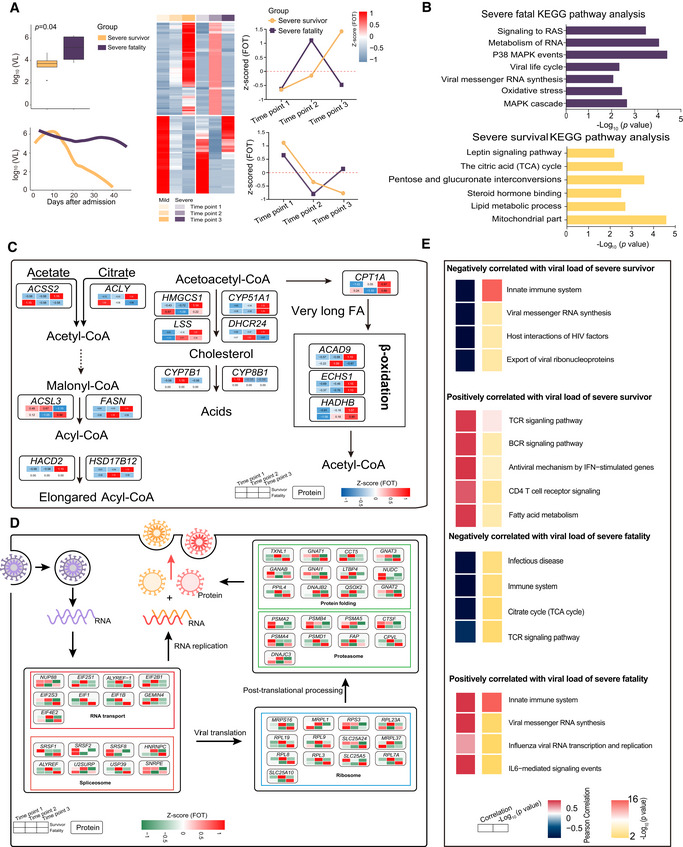
-
ABoxplot comparison of viral loads between severe‐survival patients (yellow, n = 8) and severe‐fatal patients (purple, n = 5). Line graph represents the temporal changes in viral load in time (days) after hospital admission of the patients. The cluster heatmap represents expression patterns of 1,541 proteins that exhibited temporal changes across timepoints in severe‐survival (left) and severe‐fatal (right) patients. The line plots indicate the expression trends of 1,541 selected proteins. VL, viral loads; FOT, the fraction of total. Differences between groups were estimated using Mann–Whitney–Wilcoxon test. The horizontal box lines in the boxplots represent the first quartile, the median, and the third quartile. Whiskers denote the range of points within the first quartile − 1.5 × the interquartile range and the third quartile + 1.5 × the interquartile range.
-
BEnriched annotations for corresponding clusters showed in Fig 6A (Fisher’s exact test, P < 0.05).
-
C, DSystematic summary of proteins and signaling cascades significantly altered in severe‐survival patients (lipid metabolism; (C)) and severe‐fatal (viral life cycle; (D)). Values for each protein in all samples analyzed (columns) are color‐coded based on the expression levels: low (blue) and high (red) z‐scored FOT.
-
EFor each of the four panels, the heatmaps on the left indicate the Pearson correlation of proteins with viral load in severe survivors and severe fatalities, the heatmap on the right indicate the significant pathways enriched by proteins positively or negatively correlated with viral load in severe survivors and severe fatalities (Fisher’s exact test, P < 0.05).
Estimation of the correlation coefficient between viral load and protein expression revealed that proteins participating in antiviral processes, including the TCR and BCR signaling pathway, were positively associated with viral load changes in severe survivors. Additionally, proteins participating in viral life cycle processes, including viral messenger RNA synthesis and innate immune responses, were only positively associated with viral load changes in the severe‐fatal group (Fig 8E). Notably, proteins (e.g., FASN, ACSS2, CPT1A, HADHB) involved in pathways including mitochondrial function, lipid metabolic process, steroid hormone process, and TCA cycle were continuously upregulated in the severe‐survivor compared with the severe‐fatal group (Fig 8B and C). However, this upregulation was only observed during the early stage following admission in the severe‐fatal group. Surprisingly, proteins related to viral life cycle, viral RNA synthesis, and oxidative stress (e.g., EIF, EIFB, RPL19, SLCA24) were downregulated in the several‐survivor cases following admission, but maintained high levels in the severe‐fatal patients (Fig 8B and D). Hence, SARS‐CoV‐2 may exploit host resources over the duration of its infection.
Biomarkers predictive of clinical outcomes of COVID‐19‐infected patients
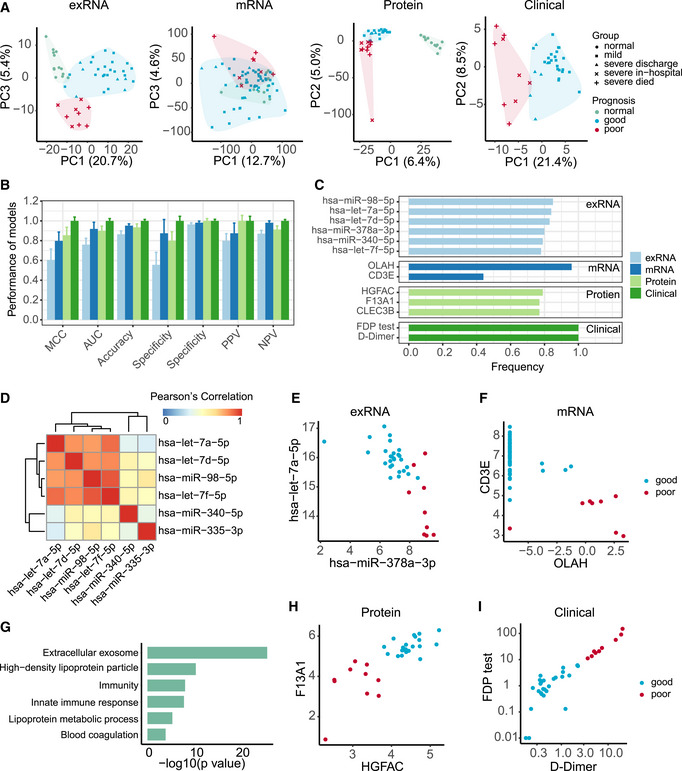
As many molecules associated with COVID‐19 pathophysiology were identified, we investigated whether particular molecular changes could be used as biomarkers to predict clinical outcomes. Using an unsupervised PCA, the exRNA, mRNA, proteomics and the corresponding clinical covariate data sets across all timepoints, or a subset from the first timepoint, clustered into three clinical phenotypes: (i) samples from healthy controls; (ii) samples from COVID‐19‐infected patients with a good prognosis; and (iii) samples from COVID‐19‐infected patients with a poor prognosis (Methods; Fig 9A). Given this, prognostic classification models were constructed. Predictive models based on all four types of data worked well, especially those utilizing the clinical covariates and the proteomic data (Fig EV6), suggesting that all four types of data collected at admission contain key prognostic information.
Figure 9. Biomarkers predictive of clinical outcomes of COVID‐19‐infected patients.
-
APrincipal component analysis of exRNA, transcriptome, proteome, and clinical covariate data from samples collected at the first timepoint. The first two components were used to describe the distribution of samples based on expressed genes, proteins, and clinical data, respectively, whereas the first and third components were used for samples based on the exRNA data.
-
BPerformance of prognostic models based on exRNA, transcriptome, proteome, and the corresponding clinical covariate data sets. Model performance of the fivefold cross‐validation was assessed using the Matthews correlation coefficient (MCC), AUC, accuracy, sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). Data were represented as means ± SD.
-
CThe most frequently selected features of exRNA‐, transcriptome‐, proteome‐, and clinical‐based models. Features were simultaneously identified from each of the four data sets and for each of the four machine learning algorithms based on the frequency of variables used by AI models during 50 runs of the fivefold cross‐validation.
-
DCorrelation heatmap among the most frequently selected features (frequency> 0.78) used in the exRNA‐based model. Members in the let‐7 family selected for the exRNA‐based predictors (hsa‐miR‐98‐5p, hsa‐let‐7a‐5p, hsa‐let‐7d‐5p, hsa‐let‐7f‐5p) were highly correlated with each other.
-
E–IBiomarkers identified from exRNA (E), transcriptome (F), proteome (H), and clinical data (I)‐based models exhibited a clear separation between those patients with either good or poor prognosis. (G) Functional enrichment of 110 protein features selected from random forest modeling (Fisher’s extract test, P < 0.05).
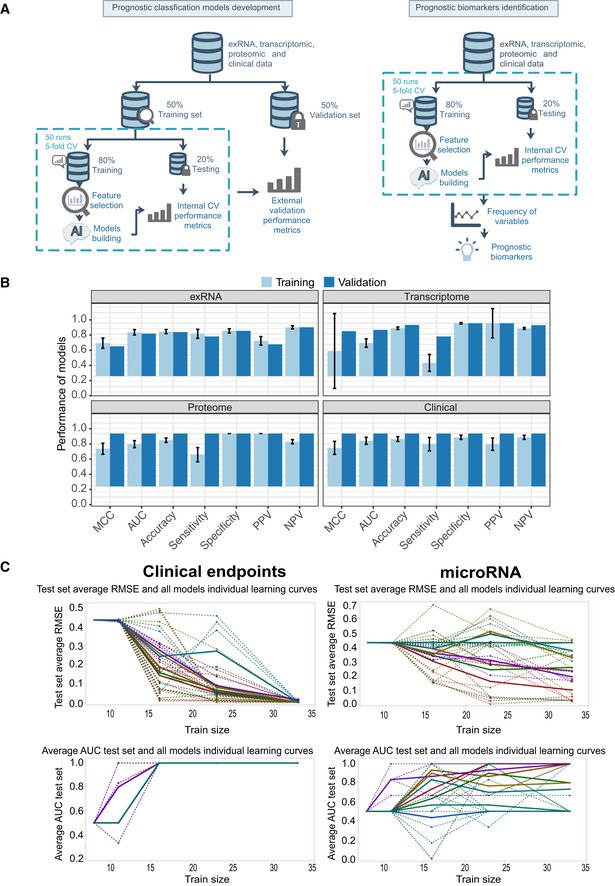
Figure EV6. Training and validation set performance in exRNA‐, transcriptome‐, proteome‐, and clinical‐ models.
- Workflow of prediction model construction.
- Performance of AI models in the training and validation set based on exRNA (n = 37), transcriptome (n = 63), proteome (n = 31), and the corresponding clinical covariate data sets (n = 37). Data are represented as means ± SD.
- Learning curve model comparison (LCMC) revealing sample size effects on the accuracy and variability of the predictive models using cross‐validation. Each individual root means square error (RMSE) learning curve and the average for each of 8 models is shown. Each color represents a model. A solid line represents the average for each model, whereas a dashed line represents one random iteration for each model.
In addition, we identified robust predictive models and prognostic biomarkers from each of the four types of data using a previously described approach (Shi et al, 2010) (Fig 9B and C). One or two features (expressed genes, proteins, exRNAs, and biochemical parameters) in each data set were able to clearly separate patients into two groups characterized by different prognoses (Fig 9D–I). Poor prognosis was associated with increased levels of D‐dimer (P = 0.004) and fibrinogen degradation products (FDP; P = 0.02), and with a decrease in F13A1 expression (P < 0.002; Fig 9H and I), suggesting that blood clotting status may be one of the key factors to monitor in COVID‐19 progression. For the mRNA‐based model, poor prognosis was associated with lower levels of CD3E and higher levels of OLAH, and hence highly concordant with immune responses in COVID‐19‐infected patients (Fig 9E). Additionally, exRNA‐based predictors included members of the let‐7 family (Fig 9D and E). Finally, the protein‐based models highlighted features enriched in extracellular exosomes, lipoprotein metabolic processes, innate immune responses, and blood coagulation (Fig 9G and H).
Discussion
The COVID‐19 pandemic has had a profound impact on a global scale (WHO, 2020). Despite the enormous burden or morbidity and mortality due to COVID‐19, we know little about its pathophysiology, even though this establishes the basis for successful clinical practice, vaccine development, and drug discovery. Current clinical practice may be unable to provide a precision supportive therapy when a novel disease like COVID‐19 emerges, in part explaining the high case fatality rates often observed at the beginning of outbreaks (Alonso et al, 2019). We used a multi‐omics approach to identify numerous expressed genes, proteins, metabolites, and exRNAs from COVID‐19‐infected patients with a range of disease severities and that were significantly correlated with key clinical features as well as to classic blood and biochemical parameters (Fig 2). These data therefore provide a comprehensive molecular view of the pathophysiology of COVID‐19. Finally, based on our multi‐omics data (Appendix Fig S1), mild and severe COVID‐19 cases may need different therapeutic strategies.
COVID‐19 severity and clinical outcome were significantly associated with multi‐organ damage. Due to the widespread presence of ACE2 in humans (preprint: Chai et al, 2020; preprint: Chen et al, 2020c; Hamming et al, 2004; preprint: Fan et al, 2020; Zou et al, 2020), SARS‐CoV‐2 is able to infect many organs (Puelles et al, 2020; Tay et al, 2020; Wadman et al, 2020). In addition to pneumonia (Wu et al, 2020a; Zhou et al, 2020b), several clinical studies have reported mild gastrointestinal, cardiovascular symptoms, and neurological manifestations in hospitalized COVID‐19‐infected patients (Baig, 2020; Chen et al, 2020b; Guan et al., 2020; Mao et al, 2020; Xu et al, 2020). Histopathologic investigation and autopsies have also documented damage in other organs in addition to the lung (Barton et al, 2020; Fox et al, 2020; Lax et al, 2020; Menter et al, 2020; Su et al, 2020; Wadman et al, 2020; Wang et al, 2020d). The molecular data generated here not only supported the occurrence of damage in multiple organs including the lung, liver, heart, intestine, and pancreas in COVID‐19‐infected patients as reported previously (Cai et al, 2020; Varga et al, 2020; Zheng et al, 2020b; Wang et al, 2020c), but also highlighted the impact of SARS‐CoV‐2 infection on less studied organs such as the brain and testis (Puelles et al, 2020; Yang et al, 2020). In the case of brain and testis, a key issue is how SARS‐CoV‐2 is able to cross the blood–brain or the blood–testis barriers? One possibility might be that heparin was prescribed for coaggregation problems commonly observed in some COVID‐19, even though it increases permeability (Gautam et al, 2001; Oschatz et al, 2011; Lin et al, 2020). It was also recently reported that SARS‐CoV‐2 could enter the nervous system via trespassing the neuromucosal interface in the olfactory mucosa by exploiting the close vicinity of olfactory mucosal and nervous tissue (preprint: Meinhardt et al, 2020). More importantly, the alteration (upregulated or downregulated) of tissue‐enhanced proteins and tissue damage‐related exRNAs are significantly correlated with clinical severity and outcome.
SARS‐CoV‐2 infection results in acute lung injury (ALI) in patients, with ground‐glass opacity in most computed tomography (CT) reports from our facility and in other hospitals (Zhu et al, 2020; Chen et al, 2020b; Zhou et al, 2020b). Autopsy disclosed histologic changes in lungs included edema, fibrinous/proteinaceous exudates, hyperplastic pneumocytes, patchy inflammation, multinucleated giant cells, and diffuse alveolar damage (Barton et al., 2020; Tian et al., 2020). The data generated here revealed that the number of AT1 and AT2 cells reduced significantly in severe patients, suggesting destruction of the alveolar epithelium (Fig 5A), which in turn will lead to the accumulation alveolar fluid and hence cause hypoxia (Vadász & Sznajder, 2017). In addition, we noted that HIF1a signaling was modified which may further worsen ALI (Dada et al, 2003). Thus, our molecular data suggest that removal of excess alveolar fluid and the restoration of alveolar structure will be of major clinical importance.
Our data also identified immune pathophysiology a factor that greatly impacted COVID‐19 clinical outcome. The innate immune response against viruses begins immediately after a host acquires a viral infection, whereas there is a delay before the onset of adaptive immunity (Murphy & Weaver, 2016). Unlike SARS‐CoV and influenza virus, SARS‐CoV‐2 may be present in patients for longer time periods, especially those with severe syndrome (Du et al, 2020; Wang et al, 2020a). Several studies have reported that severe COVID‐19‐infected patients experience lymphopenia, impaired adaptive immunity, uncontrolled inflammatory innate responses, and cytokine storms (Guan et al., 2020; Huang et al, 2020; Qin et al, 2020; Tan et al, 2020; Wang et al, 2020b). While it is believed that T cells play an important role in fighting the infection in the case of Ebola virus, influenza virus, SARS‐CoV (Zhao et al, 2010; Sridhar et al, 2013; Channappanavar et al, 2014; van de Sandt et al, 2014; Ruibal et al, 2016), and also in SARS‐CoV‐2 (Braun et al, 2020; Sekine et al, 2020; Zhou et al, 2020c; Wang et al, 2020e), more data are needed to confirmed the function of T‐cell response in SARS‐CoV‐2‐infected individuals.
Our longitudinal analyses provided evidence that patients with mild or severe symptoms who succeeded in T‐cell mobilization promptly controlled SARS‐CoV‐2 infection and symptoms (Figs 2C and 6A and C). In contrast, those (especially severe‐fatal) patients that failed to mount a sound T‐cell response maintained a continuous pro‐inflammatory response and suffered from cytokine storms as well as excess NETs (Figs 6A and B), both of which are known to cause systematic tissue damages (Akiyama et al, 2019; Bohmwald et al, 2019). In sum, our data indicate that T cells play a key role in controlling SARS‐CoV‐2 infection.
It is believed that p38 signaling, collagenase (MMP9), neutrophil chemo‐attractants (CXCL2 and CXCL6) and S100A8 are autoinflammation‐like signatures (Mattos et al, 2002; Halayko & Ghavami, 2009; Chung, 2011; Cheng et al, 2019). Remarkably, these molecules were significantly upregulated in severe‐fatal in comparison with mild and severe‐survival patients (Fig 6D), the former of which also exhibited a persistent elevation of type I interferon responses (Fig 6A). Together, the data generated here indicate that autoinflammation may amplify disease in very severe cases of COVID‐19. While a lack of controls from other respiratory pathogens might be a limitation of this study to determine whether these findings are unique to COVID‐19, it has been reported recently that SARS‐CoV‐2 infection has differences of tropism, replication, and innate immune response with other respiratory pathogens. SARS‐CoV‐2 was found to replicate better than SARS‐CoV, but not as well as the 2009 pandemic influenza H1N1 virus in bronchial epithelium (Hui et al, 2020). In addition, SARS‐CoV‐2 has a less potent induced effect on pro‐inflammatory cytokines compared with H5N1, H1N1, and MERS‐CoV (Hui et al, 2020). Examination of lungs from patients who died from COVID‐19 and H1N1 infection found that vascular angiogenesis was able to distinguish the pulmonary pathobiology of these two infections. Moreover, significant differences of CD4 T cell, CD8 T cell, and neutrophil counts, as well as the expression of inflammation‐related genes, were observed between patients with COVID‐19 and those with influenza (Ackermann et al, 2020). Based on these findings, we suggest that at least some of the changes we observed between mild and severe COVID‐19‐infected patients are likely to be specific to COVID‐19. Clearly, the pathology and pathophysiology of SARS‐CoV‐2 and its differences to other respiratory pathogens merits additional study.
Patients will receive better and more precise therapy if we are able to identify molecular biomarkers associated with prognosis at the beginning of disease presentation. For example, a 21‐gene expression assay, which can predict clinical outcome, is used in the case of breast cancer (Sparano et al, 2018). To date, however, almost no biomarkers have been used to accurately predict prognosis in the case of emerging infectious diseases (Wynants et al, 2020). In this study, some of molecules identified at the beginning phase of COVID‐19 were significantly correlated to both classical blood and biochemical parameters, and more importantly to disease severity. Based on our previous work (Shi et al, 2010; Su et al, 2014a; Zhang et al, 2015), we established classification models based on each of four data types: exRNAs, mRNA, proteins, and biochemical parameters. Notably, COVID‐19 clinical outcomes could be accurately predicted using just one or two biomarkers in each data type. In addition, these biomarkers may have biological functions directly relevant to COVID‐19 pathophysiology. For example, biomarkers let‐7 family from exRNAs, OLAH, and CD3E from mRNAs, and C4A and C4B from proteomes concordantly revealed the importance of T‐cell activation and the suppression inflammatory responses. However, because of the relatively small patient sample size utilized here, it is clear that more work is needed to confirm the reliability and practicality these biomarkers, and particularly to validate the predictive power of these biomarkers in larger cohorts.
In sum, we have identified a large number of molecules associated with COVID‐19 pathophysiology, some of which may also be effective predictive biomarkers of clinical outcome at the onset of disease. Taken together, these data suggest that distinct immune responses and multi‐organ damage have a major impact on COVID‐19 severity and disease prognosis.
Materials and Methods
Study design and patient cohort
According to arrangements made by the Chinese Government, all adult patients in Shanghai diagnosed with COVID‐19 were admitted to the Shanghai Public Health Clinical Center. We enrolled 66 COVID‐19‐infected patients who were treated at the Shanghai Public Health Clinical Center between January 31 and April 7, 2020. While these patients consisted of four clinical types according to WHO classification, we divided them into two groups based on clinical signs and the need for oxygen because of the limited sample size: (i) mild (50/66, 75.8%)—with clinical signs of pneumonia but without oxygen support, and (ii) severe (16/66, 24.2%)—with oxygen support using non‐invasive ventilation, tracheal tube, tracheotomy assist ventilation, or extracorporeal membrane oxygenation (ECMO) (Fig 1A and Dataset EV1). The mild patient group included both mild and moderate patients based on WHO classification, while the severe group comprised severe and critical COVID‐19‐infected patients. All human samples included in the present study were obtained after approval of the research by the Shanghai Public Health Clinical Center Ethics Committee (YJ‐2020‐S018‐02), together with the written informed consent from each patient.
Sample collection and processing
A total of 248 blood samples, comprising 1–2 ml each, were collected from 66 COVID‐19‐infected patients by professional healthcare workers over a 5‐week period, with 65 patients having more than one sampling timepoints (Dataset EV1). In addition, 17 blood samples were collected from 17 uninfected volunteers and utilized as healthy controls. Samples were transported to the research laboratory within 2 h of collection. For RNA extraction, 200 μl of whole blood was mixed with 1 ml TRIzol reagent (Qiagen), followed by 15‐min incubation at room temperature and subsequent freezing at −80°C before total RNA extraction. The remaining whole blood samples (800–1,800 μl) were processed immediately to separate plasma and subsequently stored at −80°C until use.
All clinical data were recorded by the clinicians. SARS‐CoV‐2 infection was first determined by quantitative real‐time RT–PCR using the Takara One Step PrimeScript RT–PCR kit (Takara RR064A) as previously described (Wu et al, 2020a). For positive samples, digital RT–PCR was then used to obtain the absolute viral loads (copies per ml) following the manufacturer’s instructions (BioDigital General dPCR kit, Jiangsu Saint Genomics, Cat no. CSJ‐3‐0018). Specifically, 15 μl of RNA solution was mixed with 20 μl reaction buffer. The reaction buffer comprised 7 μl 5 × RT–PCR buffer, 3 μl Taq polymerase and 10 μl SARS‐CoV‐2 detection mix provided in the kit. Subsequently, 30 μl of these dRT–PCR mixture and 25 μl oil mixture (provided by the kit) was added to the chip, followed by a amplification program of 37°C, 10 min; 55°C, 10 min; 95°C, 10 min; 45 × (95°C, 20 s; 60°C, 40 s); 25°C, hold. Nuclease‐free water was used as negative control and performed RNA extraction together with samples. Positive controls were provided by the kit itself.
RNA and exRNA extraction and library construction
Total RNA from whole blood samples was extracted using the RNeasy Plus Universal Mini Kit (Qiagen) following the manufacturer’s instructions. The quantity and quality of RNA solution were assessed using a Qubit Flex fluorometer (Invitrogen) and an Agilent Bioanalyzer (Agilent Technologies) before library construction and sequencing. RNA library construction was performed as described using the VAHTS Universal V6 RNA‐seq Library Prep Kit for Illumina (Vazyme, China). Ribosomal, globin and RN7S RNAs were depleted using specially designed probes (Vazyme, China).
Plasma samples were divided into aliquots and used for extracellular RNA (exRNA) extraction and library construction, protein extraction, and metabolomic analyses. For exRNA library preparation, total RNA including exRNA was extracted using the miRNeasy Serum/Plasma Advanced Kit (Qiagen). The exRNA library was prepared using the NEXTflex Small RNA‐seq Kit v3 (PerkinElmer). RNA quantity and quality were determined as mentioned above. After final library quantification using a Qubit Flex fluorometer (Invitrogen) and quality control using the Bioptic Qsep100 to confirm the expected size distributions, all libraries (RNA and exRNA) were pair‐end (150‐bp reads) sequenced on the Illumina NovaSeq6000 platform (Illumina).
RNA‐seq data analysis
Data processing and filtering criteria
Preliminary processing of raw reads was performed using FASTP v0.19.6 to remove adapter sequences and obtain trimmed reads (Chen et al, 2018). The sequence AGATCGGAAGAGCACACGTCTGAACTCCAGTCA was used as the R1 adapter sequence while AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT was used as the R2 adapter sequence. HISAT2 v2.1 (Pertea et al, 2016) was used for read alignment to the human genome, build 38. Samtools v1.3.1 was used to generate intermediate result files for quality assessment of the aligned reads by BamQC v2.0.0 (https://github.com/s‐andrews/BamQC) and insert size distribution analysis. The assembly of aligned reads and assessment of expression levels were processed through StringTie v1.3.4. Gene counts were determined with preDE.py (http://ccb.jhu.edu/software/stringtie/) based on results derived from Ballgown (https://github.com/alyssafrazee/ballgown). Ensembl transcript annotation (version: Homo_sapiens.GRCh38.93.gtf) with 58,395 genes was used.
A QC analysis and library filtering were performed before downstream biological analysis. Libraries that passed the following criteria were retained: (i) more than five million reads; (ii) more than 90% of reads aligned to the human reference genome; and (iii) over 10,000 genes were expressed (a gene with FPKM > 0.5 was identified as an expressed gene). In addition, to monitor data quality across batches, libraries of some healthy control samples were constructed and sequenced 2–3 times. The average expression profile of the multiple libraries from each healthy control sample was calculated for follow‐up analyses.
Immunoassay
Immune repertoires were extracted with MiXCR, a software tool that extracts T‐cell receptor (TCR) and immunoglobulin (IG) repertoires from RNA‐seq data (Bolotin et al, 2015; Bolotin et al, 2017). The number of clonotypes was then calculated using VDJtools, using the output from MiXCR (Shugay et al, 2015).
Differentially expressed genes (DEGs)
To identify DEGs, Student’s t‐test was applied to the expression matrix. Genes with P‐values < 0.05 as well as a fold change > 2 or < 1/2 were labeled as upregulated and downregulated genes, respectively (Su et al, 2014b). This straightforward approach of combining a fold change cutoff with a non‐stringent P‐value threshold has been demonstrated to yield reproducible and robust lists of DEGs for both microarray and RNA‐seq‐based gene expression analyses (Shi et al, 2006; Su et al, 2014b).
Functional and cell type enrichment analyses
Functional analyses were conducted based on genes differentially expressed between several subgroups of COVID‐19‐infected patients compared with healthy control samples. GSEA (Gene Set Enrichment Analyses) was performed to identify significantly enriched functional classes of gene sets correlated with blood transcription modules (BTM) described by Li et al. (2014), KEGG pathways, and Gene Ontology (GO) terms. A default FDR (false discovery rate) value of q < 0.25 was considered statistically significant. The Normalized Enrichment Score (NES) of significant immune modules from BTMs was used to denote enrichment levels.
The fraction of the cell subsets was calculated using the enrichment score‐based algorithm X‐Cell from the RNA‐seq data (Aran et al, 2017). Briefly, the expression profile (FPKM) of all 230 samples was employed as raw signatures. The R package immunedeconv was applied to obtain enrichment scores of 35 immune cell types, estimating immune cell fractions including T cell, monocyte, and neutrophil by summation of the scores in each sample (Sturm et al, 2019).
exRNA‐seq data analysis
Alignment, quantification, and quality control
Libraries were sequenced in two batches, with an average sequencing depth of 15.7M raw reads per library. All FASTQ files were delivered to the ExceRpt small RNA sequencing data analysis pipeline (docker v4.6.3) (Rozowsky et al, 2019). Default parameters were used with exception of (i) the sequence TGGAATTCTCGGGTGCCAAGG was given as the 3’adapter sequence, ignoring the adapter sequences guessed by the pipeline; (ii) the random barcode length was set to 4; and (iii) the priority of the reference libraries during read assignment was set to miRNA > piRNA> tRNA> GENCODE> circRNA (Godoy et al, 2018). Pre‐compiled genome and transcriptome indices of human genome, build 38 were used. The raw read count matrix was then normalized using count per million (CPM).
A QC analysis was performed prior to biological analysis by removing (i) libraries with low sequencing depths (< 1M raw reads); (ii) libraries with mapping ratio lower than 50%, and (iii) libraries with low transcript‐genome ratios. To minimize the impact of noise due to low expression levels, only 769 miRNAs with at least 1 count per million in no less than 10% of the total number of samples were included in the final analysis.
Differentially expressed exRNAs
To identify differentially expressed exRNAs, Student’s t‐tests were applied to the normalized expression matrix. exRNAs with P‐values < 0.05, as well as fold change > 2 or < 1/2, were labeled as upregulated and downregulated exRNAs, respectively, in a similar manner to the RNA‐seq data.
Tissue damage‐related miRNAs
exRNAs reported to be associated with tissue damage were collected from publications (Wang et al, 2009; Godwin et al, 2010; Wang et al, 2010; Zhou et al, 2016; Atif & Hicks, 2019) (Dataset EV9). Fisher’s exact test was used to determine whether the proportions of differentially expressed (DE) exRNAs in tissue damage‐related exRNAs were significantly higher than the proportions of DE exRNAs in the entire data set of 769 miRNAs.
Proteome analysis
Plasma protein extraction and trypsin digestion
Plasma samples used for protein extraction were first removed the tip 14 highest abundance plasma proteins using an immunodepleting kit (Thermo Fisher) according to the manufacturer’s instructions, and then inactivated at 85°C for 10 min. The depleted plasma was digested by trypsin at an enzyme to protein mass ratio of 1:25 overnight at 37°C, and the peptides were then extracted and dried (SpeedVac, Eppendorf).
LC‐MS/MS acquisition of plasma samples
Samples were measured using LC‐MS instrumentation consisting of an EASY‐ nLC 1200 ultra‐high‐pressure system (Thermo Fisher Scientific) coupled via a nano‐electrospray ion source (Thermo Fisher Scientific) to a Fusion Lumos Orbitrap (Thermo Fisher Scientific). The peptides were dissolved with 12 μl loading buffer (0.1% formic acid in water), and 5 μl was loaded onto a 100 μm I.D. × 2.5 cm, C18 trap column at a maximum pressure 280 bar with 14 μl solvent A (0.1% formic acid in water). Peptides were separated on 150 μm I.D. × 15 cm column (C18, 1.9μm, 120 Å, Dr. Maisch GmbH) with a linear 15–30% Mobile Phase B (ACN and 0.1% formic acid) at 600 nl/min for 75 min. The MS analysis was performed in a data‐independent manner (DIA). The DIA method consisted of MS1 scan from 300–1,400 m/z at 60k resolution (AGC target 4e5 or 50 ms). Then, 30 DIA segments were acquired at 15k resolution with an AGC target 5e4 or 22 ms for maximal injection time. The setting “inject ions for all available parallelizable time” was enabled. HCD fragmentation was set to normalized collision energy of 30%. The spectra were recorded in profile mode. The default charge state for the MS2 was set to 3.
Peptide identification and protein quantification
All data were processed using Firmiana (Feng et al, 2017).The DIA data were search against UniProt human protein database (updated on 2019.12.17, 20406 entries) using FragPipe (v12.1) with MSFragger (2.2) (Kong et al, 2017). The mass tolerances were 20 ppm for precursor and 50 mmu for product ions. Up to two missed cleavages were allowed. The search engine set cysteine carbamidomethylation as a fixed modification and N‐acetylation and oxidation of methionine as variable modifications. Precursor ion score charges were limited to +2, +3, and +4. The data were also searched against a decoy database so that protein identifications were accepted at a false discovery rate (FDR) of 1%. The results of DIA data were combined into spectra libraries using SpectraST software. A total of 327 libraries were used as reference spectra libraries.
DIA data was analyzed using DIA‐NN (v1.7.0) (Demichev et al, 2020). The default settings were used for DIA‐NN (Precursor FDR: 5%, Log lev: 1, Mass accuracy: 20 ppm, MS1 accuracy: 10 ppm, Scan window: 30, Implicit protein group: genes, Quantification strategy: robust LC (high accuracy)). Quantification of identified peptides was calculated as the average of chromatographic fragment ion peak areas across all reference spectra libraries. Label‐free protein quantifications were calculated using a label‐free, intensity‐based absolute quantification (iBAQ) approach (Zhang et al, 2012). We calculated the peak area values as parts of corresponding proteins. The fraction of total (FOT) was used to represent the normalized abundance of a particular protein across samples. FOT was defined as a protein’s iBAQ divided by the total iBAQ of all identified proteins within a sample. The FOT values were multiplied by 105 for the ease of presentation and missing values were imputed with 10−5.
To better understand the impact of COVID‐19 on patients, we mapped our plasma proteome data with the Human Protein Atlas (HPA) database and determined different expressions of tissue‐enhanced proteins. Tissue‐enhanced protein was defined as proteins encoded by genes which have an elevated expression (at least fourfold higher mRNA level) in the specific type of tissue compared with the average level in all other tissues (Uhlén et al, 2015). In total, 544 tissue‐enhanced proteins were detected, among which 335 tissue‐enhanced proteins were expressed significantly differently in COVID‐19‐infected patients’ blood samples compared with healthy controls.
Metabolome analysis
NMR spectroscopy
The plasma samples used for NMR analysis were first treated with 56°C for 30 min. Our subsequent quantitative measurements of samples from healthy controls showed that such treatments caused no differences to the quantification results.
NMR analysis was conducted on a 600 MHz NMR spectrometer (Bruker Biospin) as reported previously (Jimenez et al, 2018) with some minor modifications. In brief, 320 μl of each plasma sample was mixed with 320 μl of a phosphate buffer (0.085 M containing 10% D2O) with composition described previously (Jiang et al, 2012), and 600 μl mixture was transferred into a 5mm NMR tube for NMR analysis. 152 parameters of the plasma were then quantified using a server‐based software package (Bruker Biospin), including 112 parameters for lipoproteins (including main fractions, subclasses, and compositional components therein), two acute‐phase glycoproteins together with 41 small metabolites (such as amino acids, ketone bodies, glucose, carboxylic acids, ethanol). We also quantified six ratio parameters for saturated, unsaturated, monounsaturated, and polyunsaturated fatty acids from the diffusion‐edited spectra (Xu et al, 2012). We further calculated 187 more ratio‐parameters (such as the cholesterol‐to‐triglyceride ratio, percentage of triglycerides, and cholesterol in total lipids) from the quantitative data for lipoproteins. A total of 348 quantitative parameters obtained were collectively employed to define the metabolomic phenotypes of each of the human plasma samples.
Development of prognostic models
Model development
Four data sets representing the (i) clinical tests, (ii) exRNA‐seq, (iii) mRNA‐seq, and (iv) proteomics quantification analysis were used to develop prognostic models for the prediction of patient outcomes (i.e. good or poor). Patients with a “good” outcome included those with mild or severe syndrome but who were discharged after treatment, while patients with “poor” outcomes included those who died or remained in ICU for more than 2 months. The numbers of samples used in predictive modeling were as follows: for the exRNA‐seq and the matched clinical testing data sets, a total of 37 patients were used, including 28 patients with good outcomes and 9 patients with poor outcomes. For the RNA‐seq data set, 63 patients were used, including 55 patients with good outcomes and 8 patients with poor outcomes. For the proteomics data set, 31 patients were used, including 21 patients with good outcomes and 10 patients with poor outcomes. Samples from the first sampling timepoint from each patient were used in the analysis.
Prognostic models were developed and validated using a two‐layer validation strategy (Fig EV6A) to prevent information leaking from the training set to the validation set (Shi et al, 2010). Briefly, patients were first divided into training and validation sets with equal size based on outcome and admission date. The training set was then used to select variables and train prognostic models using multiple machine learning algorithms, including nearest mean classification (NMC), k‐nearest neighbors (KNN), support vector machine (SVM), and random forest (RF) through an internal‐layer of 50 runs of fivefold cross‐validation process to resist overfitting. Next, a final model was built using the whole training set with the best performing machine learning algorithm as defined above. The final model was further validated using the validation set as an external‐layer evaluation. Model performance was assessed in terms of the Matthews correlation coefficient (MCC), AUC, accuracy, sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
Prognostic biomarkers were identified based on the frequency of variables selected by machine learning algorithms. Because the sample size was relatively small compared with the large number of variables, it was difficult to identify stable biomarkers as indicated by the low frequencies of the variables used in the prognostic models (Shi et al, 2010) (Fig EV6A and B). To detect more robust prognostic biomarkers, 50 runs of fivefold cross‐validation process were therefore applied to the whole data set. The variables used by the best performing machine learning algorithm were identified as prognostic biomarkers for each data set.
Learning curve model comparison
Learning curve model comparison (LCMC) was performed using Predictive Modeling Review as available in JMP Genomics 10 (https://www.jmp.com/en_us/software/genomics‐data‐analysis‐software.html). LCMC reveals the effects of sample size on the accuracy and variability of the predictive models using 10 runs of fourfold cross‐validation.
We performed LCMC with prognosis (good or poor) as target variables, and the clinical variables, exRNA, mRNA, or proteomics measurements as predictors. Figure EV6C shows each individual (RMSE) and (AUC) learning curve and the average for each of the eight partition tree models for clinical endpoints, as well as exRNA using K‐fold cross‐validation. The LCMC suggested that with up to 15 samples, eight partition tree models reached AUC as 1 for clinical variables. However, more than 23 and 30 samples were needed for one and three models, respectively, to reach AUC of 1 for exRNA‐seq data. The variability of RMSE and AUC for the proteomic and mRNA‐seq data (not shown) were between that observed for clinical variables and the exRNA data.
Statistical analyses
Univariate statistical analysis was performed using Student’s t‐test, Mann–Whitney U tests or ANOVA tests to compare continuous variables. Chi‐square tests and Fisher’s exact tests were used for the comparison of categorical variables. P‐values were adjusted using Bonferroni correction or the Benjamini and Hochberg False Discovery Rate (FDR) in multiple comparisons, with P < 0.05 considered to be statistically significant. Principal components analysis (PCA) was conducted with univariance scaling, with the scores plot showing a distribution of metabolomic phenotypes for healthy participants and patients with moderate or severe COVID‐19 (and upon discharge). Correlations were tested using Pearson correlation coefficients. Locally Weighted Linear Regression (Loess) was used for visualizing the time series data. All analyses were performed using appropriate R packages (version 3.5.1). Circos plots were generated using the circlize package (Gu et al, 2014).
Author contributions
Study conception, design: Y‐ZZ; Design and development of methodology: Y‐ZZ, T‐YZ, CD, LMS, HRT, YL, ZY, LJ; Clinical work and sample collection: T‐YZ, YL, Z‐GS; Experiments: Y‐MC, Y‐TZ, Y‐ZW, Q‐XH, S‐YS, Z‐GS, F‐HD, Y‐PA, Q‐SC, Q‐WL; Data analysis: YY, Y‐ZW, Q‐XH, Y‐MC, FQ, LS, Z‐YC, J‐CY, Z‐QS, J‐WF, P‐CL; Manuscript writing: Y‐ZZ, CD, FQ, LS, YY, Y‐ZW, Y‐MC, Q‐XH, LMS, HRT, ECH.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Review Process File
Acknowledgements
We acknowledge the support from Volvo Car Corporation. This study was supported by the National Natural Science Foundation of China (grants 32041004, 31930001, 81861138003, 81672057, and 91846302), the Special National Project on investigation of Basic Resources of China (grant 2019FY101500), Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01), and the Science and Technology Commission of Shanghai Municipality for International Partnership Project (20490780100). E.C.H. is supported by an ARC Australian Laureate Fellowship (FL170100022).
The EMBO Journal (2020) 39: e105896.
Contributor Information
Chen Ding, Email: chend@fudan.edu.cn.
Tong‐Yu Zhu, Email: zhutongyu@shphc.org.cn.
Yong‐Zhen Zhang, Email: zhangyongzhen@shphc.org.cn.
Data availability
The datasets produced in this study are available in the following databases:
RNA‐Seq and exRNA‐Seq Data: NODE OEP000868 (http://www.biosino.org/node/project/detail/OEP000868)
Raw mass spectrometry data: iProX IPX 0002186001 (https://www.iprox.org/page/subproject.html?id=IPX0002186001)
References
- Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N Engl J Med 383: 120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Zeisbrich M, Ibrahim N, Ohtsuki S, Berry GJ, Hwang PH, Goronzy JJ, Weyand CM (2019) Neutrophil extracellular traps induce tissue‐invasive monocytes in granulomatosis with polyangiitis. Front Immunol 10: 2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso DO, Iglesias A, Coelho R, Periolo N, Bruno A, Córdoba MT, Filomarino N, Quipildor M, Biondo E, Fortunato E (2019) Epidemiological description, case‐fatality rate, and trends of Hantavirus Pulmonary Syndrome: 9 years of surveillance in Argentina. J Med Virol 91: 1173–1181 [DOI] [PubMed] [Google Scholar]
- Aran D, Hu Z, Butte AJ (2017) xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18: 220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atif H, Hicks SD (2019) A review of microRNA biomarkers in rraumatic brain Injury. J Exp Neurosci 13: 1179069519832286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig AM (2020) Neurological manifestations in COVID‐19 caused by SARS‐CoV‐2. CNS Neurosci Ther 26: 499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID‐19 virus targeting the CNS: Tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11: 995–998 [DOI] [PubMed] [Google Scholar]
- Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S (2020) COVID‐19 autopsies, Oklahoma, USA. Am J Clin Pathol 153: 725–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baseler L, Chertow DS, Johnson KM, Feldmann H, Morens DM (2017) The pathogenesis of Ebola virus disease. Annu Rev Pathol 12: 387–418 [DOI] [PubMed] [Google Scholar]
- Bermingham A, Chand M, Brown C, Aarons E, Tong C, Langrish C, Hoschler K, Brown K, Galiano M, Myers R (2012) Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill 17: 20290 [PubMed] [Google Scholar]
- Blanco‐Melo D, Nilsson‐Payant BE, Liu W‐C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D et al (2020) Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell 181: 1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmwald K, Gálvez NM, Canedo‐Marroquín G, Pizarro‐Ortega MS, Andrade‐Parra C, Gómez‐Santander F, Kalergis AM (2019) Contribution of cytokines to tissue damage during human respiratory syncytial virus infection. Front Immunol 10: 452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, Hemmers S, Putintseva EV, Obraztsova AS, Shugay M et al (2017) Antigen receptor repertoire profiling from RNA‐seq data. Nat Biotechnol 35: 908–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM (2015) MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12: 380–381 [DOI] [PubMed] [Google Scholar]
- Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, Hippenstiel S, Dingeldey M, Kruse B, Fauchere F et al (2020) SARS‐CoV‐2‐reactive T cells in healthy donors and patients with COVID‐19. Nature 587: 270–274 [DOI] [PubMed] [Google Scholar]
- Bullen CK, Hogberg HT, Bahadirli‐Talbott A, Bishai WR, Hartung T, Keuthan C, Looney MM, Pekosz A, Romero JC, Sillé FCM et al (2020) Infectability of human BrainSphere neurons suggests neurotropism of SARS‐CoV‐2. Altex 37: 665–671 [DOI] [PubMed] [Google Scholar]
- Cai Q, Huang D, Yu H, Zhu Z, Xia Z, Su Y, Li Z, Zhou G, Gou J, Qu J (2020) Characteristics of liver tests in COVID‐19 patients. J Hepatol 73: 712–713 32348791 [Google Scholar]
- Cao X (2020) COVID‐19: immunopathology and its implications for therapy. Nat Rev Immunol 20: 269–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, Zhou J, Shi G, Fang N, Fan J (2020) Specific ACE2 expression in cholangiocytes may cause liver damage after 2019‐nCoV infection. bioRxiv 10.1101/2020.02.03.931766 [PREPRINT] [DOI] [Google Scholar]
- Channappanavar R, Zhao J, Perlman S (2014) T cell‐mediated immune response to respiratory coronaviruses. Immunol Res 59: 118–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li X, Chen M, Feng Y, Xiong C (2020a) The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS‐CoV‐2. Cardiovasc Res 116: 1097–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y (2020b) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet 395: 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Yu J, Wang K, Chen Z, Wen C, Xu Z (2020c). The spatial and cell‐type distribution of SARS‐CoV‐2 receptor ACE2 in human and mouse brain. bioRxiv 10.1101/2020.04.07.030650 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34: i884–i890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Ma XL, Wei YQ, Wei XW (2019) Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer 1871: 289–312 [DOI] [PubMed] [Google Scholar]
- Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, Debnath O, Thürmann L, Kurth F, Völker MT et al (2020) COVID‐19 severity correlates with airway epithelium‐immune cell interactions identified by single‐cell analysis. Nat biotechnol 38: 970–979 [DOI] [PubMed] [Google Scholar]
- Chung KF (2011) p38 mitogen‐activated protein kinase pathways in asthma and COPD. Chest 139: 1470–1479 [DOI] [PubMed] [Google Scholar]
- Cicchese JM, Evans S, Hult C, Joslyn LR, Wessler T, Millar JA, Marino S, Cilfone NA, Mattila JT, Linderman JJ (2018) Dynamic balance of pro‐and anti‐inflammatory signals controls disease and limits pathology. Immunol Rev 285: 147–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li F, Shi Z‐L (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17: 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI (2003) Hypoxia‐induced endocytosis of Na, K‐ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC‐ζ. J Clin Invest 111: 1057–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M (2020) DIA‐NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods 17: 41–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Yu X, Li Q, Li X, Qin T, Luo Q, Wang M, Jiang M, Bai L, Wang X (2020) Duration for carrying SARS‐CoV‐2 in COVID‐19 patients. J Infect 81:e78–e79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Li K, Ding Y, Lu WL, Wang J (2020) ACE2 expression in kidney and testis may cause kidney and testis damage after 2019‐nCoV infection. MedRxiv 10.1101/2020.02.12.20022418 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Ding C, Qiu N, Ni X, Zhan D, Liu W, Xia X, Li P, Lu B, Zhao Q (2017) Firmiana: towards a one‐stop proteomic cloud platform for data processing and analysis. Nat Biotechnol 35: 409–412 [DOI] [PubMed] [Google Scholar]
- Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS (2020) Pulmonary and cardiac pathology in African American patients with COVID‐19: an autopsy series from New Orleans. Lancet Respir Med 8: 681–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam N, Olofsson AM, Herwald H, Iversen LF, Lundgren‐Åkerlund E, Hedqvist P, Arfors K‐E, Flodgaard H, Lindbom L (2001) Heparin‐binding protein (HBP/CAP37): a missing link in neutrophil‐evoked alteration of vascular permeability. Nat Med 7: 1123–1127 [DOI] [PubMed] [Google Scholar]
- Godoy PM, Bhakta NR, Barczak AJ, Cakmak H, Fisher S, MacKenzie TC, Patel T, Price RW, Smith JF, Woodruff PG et al (2018) Large differences in small RNA composition between human biofluids. Cell Rep 25: 1346–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J (2010) Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci USA 107: 14339–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, Brors B (2014) circlize Implements and enhances circular visualization in R. Bioinformatics 30: 2811–2812 [DOI] [PubMed] [Google Scholar]
- Guan W‐J, Ni Z‐Y, Hu Y, Liang W‐H, Ou C‐Q, He J‐X, Liu L, Shan H, Lei C‐L, Hui DS (2020). Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382: 1708–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, Bikdeli B, Ahluwalia N, Ausiello JC, Wan EY et al (2020) Extrapulmonary manifestations of COVID‐19. Nat Med 26: 1017–1032 [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Ghavami S (2009) S100A8/A9: a mediator of severe asthma pathogenesis and morbidity? Can J Physiol Pharmacol 87: 743–755 [DOI] [PubMed] [Google Scholar]
- Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203: 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N‐H, Nitsche A et al (2020) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet 395: 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KPY, Cheung M‐C, Perera RAPM, Ng K‐C, Bui CHT, Ho JCW, Ng MMT, Kuok DIT, Shih KC, Tsao S‐W et al (2020) Tropism, replication competence, and innate immune responses of the coronavirus SARS‐CoV‐2 in human respiratory tract and conjunctiva: an analysis in ex‐vivo and in‐vitro cultures. Lancet Respir Med 8: 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Huang J, Wang Y, Tang H (2012) Eliminating the dication‐induced intersample chemical‐shift variations for NMR‐based biofluid metabonomic analysis. Analyst 137: 4209–4219 [DOI] [PubMed] [Google Scholar]
- Jimenez B, Holmes E, Heude C, Tolson RF, Harvey N, Lodge SL, Chetwynd AJ, Cannet C, Fang F, Pearce JTM et al (2018) Quantitative lipoprotein subclass and low molecular weight metabolite analysis in human serum and plasma by (1)H NMR spectroscopy in a multilaboratory trial. Anal Chem 90: 11962–11971 [DOI] [PubMed] [Google Scholar]
- Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI (2017) MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat Methods 14: 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D (2015) Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog 11: e1004651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348: 1953–1966 [DOI] [PubMed] [Google Scholar]
- Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, Vander K, Bargfrieder U, Trauner M (2020) Pulmonary arterial thrombosis in COVID‐19 with fatal outcome: results from a prospective, single‐center, clinicopathologic case series. Ann Intern Med 173: 350–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M‐Y, Li L, Zhang Y, Wang X‐S (2020) Expression of the SARS‐CoV‐2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty 9: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Rouphael N, Duraisingham S, Romero‐Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G et al (2014) Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 15: 195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, Wang F et al (2020) Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med 26: 842–844 [DOI] [PubMed] [Google Scholar]
- Lin L, Lu L, Cao W, Li T (2020) Hypothesis for potential pathogenesis of SARS‐CoV‐2 infection——a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect 9: 727–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Long X, Zhang B, Zhang W, Chen X, Zhang Z (2020) ACE2 expression in pncreas may cause pancreatic damage ffter SARS‐CoV‐2 infection. Clin Gastroenterol Hepatol 18: 2128–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Wang M, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Hu Y (2020) Neurologic manifestations of hospitalized patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol 77: 683–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P, Perlman S (2013) Coronaviridae In Fields virology, Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed) pp825–858. Philadelphia, PA: Lippincott Williams & Wilkins; [Google Scholar]
- Mattos W, Lim S, Russell R, Jatakanon A, Chung KF, Barnes PJ (2002) Matrix metalloproteinase‐9 expression in asthma: effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest 122: 1543–1552 [DOI] [PubMed] [Google Scholar]
- Meinhardt J, Radke J, Dittmayer C, Mothes R, Franz J, Laue M, Schneider J, Brünink S, Hassan O, Stenzel W et al (2020). Olfactory transmucosal SARS‐CoV‐2 invasion as port of central nervous system entry in COVID‐19 patients. bioRxiv 10.1101/2020.06.04.135012 [PREPRINT] [DOI] [PubMed] [Google Scholar]
- Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, Frank S, Turek D, Willi N, Pargger H et al (2020) Postmortem examination of COVID‐19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 77: 198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K, Weaver C (2016) Janeway's immunobiology. Garland Science, New York, USA. [Google Scholar]
- Newton AH, Cardani A, Braciale TJ (2016) The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol 38: 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oschatz C, Maas C, Lecher B, Jansen T, Björkqvist J, Tradler T, Sedlmeier R, Burfeind P, Cichon S, Hammerschmidt S (2011) Mast cells increase vascular permeability by heparin‐initiated bradykinin formation in vivo. Immunity 34: 258–268 [DOI] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016) Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, Chilla S, Heinemann A, Wanner N, Liu S et al (2020) Multiorgan and renal tropism of SARS‐CoV‐2. N Engl J Med 383: 590–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, Xie C, Ma K, Shang K, Wang W (2020) Dysregulation of immune response in patients with COVID‐19 in Wuhan, China. Clin Infect Dis 71: 762–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Kitchen RR, Park JJ, Galeev TR, Diao J, Warrell J, Thistlethwaite W, Subramanian SL, Milosavljevic A, Gerstein M (2019) exceRpt: a comprehensive analytic platform for extracellular RNA profiling. Cell Syst 8: 352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruibal P, Oestereich L, Ludtke A, Becker‐Ziaja B, Wozniak DM, Kerber R, Korva M, Cabeza‐Cabrerizo M, Bore JA, Koundouno FR et al (2016) Unique human immune signature of Ebola virus disease in Guinea. Nature 533: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T, Perez‐Potti A, Rivera‐Ballesteros O, Straling K, Gorin J‐B, Olsson A, Llewellyn‐Lacey S, Kamal H, Bogdanovic G, Muschiol S (2020) Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID‐19. Cell 183: 158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu T‐M, Goodsaid FM, Pusztai L (2010) The MicroArray Quality Control (MAQC)‐II study of common practices for the development and validation of microarray‐based predictive models. Nat Biotechnol 28: 827–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY et al (2006) The MicroArray Quality Control (MAQC) project shows inter‐ and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24: 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI et al (2015) VDJtools: Unifying Post‐analysis of T Cell Receptor Repertoires. PLoS Comput Biol 11: e1004503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, Geyer CE Jr, Dees EC, Goetz MP, Olson JA Jr (2018) Adjuvant chemotherapy guided by a 21‐gene expression assay in breast cancer. N Engl J Med 379: 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, Bean T, Barclay W, Deeks JJ, Lalvani A (2013) Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 19: 1305–1312 [DOI] [PubMed] [Google Scholar]
- Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, List M, Aneichyk T (2019) Comprehensive evaluation of transcriptome‐based cell‐type quantification methods for immuno‐oncology. Bioinformatics 35: i436–i445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Yang M, Wan C, Yi L‐X, Tang F, Zhu H‐Y, Yi F, Yang H‐C, Fogo AB, Nie X et al (2020) Renal histopathological analysis of 26 postmortem findings of patients with COVID‐19 in China. Kidney Int 98: 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Fang H, Hong H, Shi L, Zhang W, Zhang W, Zhang Y, Dong Z, Lancashire LJ, Bessarabova M et al (2014a) An investigation of biomarkers derived from legacy microarray data for their utility in the RNA‐seq era. Genome Biol 15: 523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Łabaj PP, Li S, Thierry‐Mieg J, Thierry‐Mieg D, Shi W, Wang C, Schroth GP, Setterquist RA, Thompson JF et al (2014b) A comprehensive assessment of RNA‐seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 32: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Liu Y, Zhou R, Deng X, Li F, Liang K, Shi Y (2020) Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. Immunology 160: 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP (2020) The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol 20: 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Hu W, Niu L, Liu H, Xu H, Xiao S‐Y (2020) Pulmonary pathology of early phase 2019 novel coronavirus (COVID‐19) pneumonia in two patients with lung cancer. J Thorac Oncol 15: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Vadász I, Sznajder JI (2017) Gas exchange disturbances regulate alveolar fluid clearance during acute lung injury. Front Immunol 8: 757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sandt CE, Kreijtz JH, de Mutsert G, Geelhoed‐Mieras MM, Hillaire ML, Vogelzang‐van Trierum SE, Osterhaus AD, Fouchier RA, Rimmelzwaan GF (2014) Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross‐react with the newly emerging H7N9 virus. J Virol 88: 1684–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H (2020) Endothelial cell infection and endotheliitis in COVID‐19. Lancet 395: 1417–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadman M, Couzin‐Frankel J, Kaiser J, Matacic C (2020). How does coronavirus kill? Clinicians trace a ferocious rampage through the body, from brain to toes. Science https://wwwsciencemagorg/news/2020/04/how‐does‐coronavirus‐kill‐clinicians‐trace‐ferocious‐rampage‐through‐body‐brain‐toes [DOI] [PubMed] [Google Scholar]
- Wang B, Wang L, Kong X, Geng J, Xiao D, Ma C, Jiang X, Wang P‐H (2020a). Long‐term coexistence of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2) with antibody response in Coronavirus Disease 2019 (COVID‐19) Patients. J Med Virol 92: 1684–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y et al (2020b) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 323: 1061–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang H, Fan J, Zhang Y, Wang H, Zhao Q (2020c) Pancreatic injury patterns in patients with Coronavirus Disease 19 Pneumonia. Gastroenterology 159: 367–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Zhu JQ, Zhang JT, Li Q, Li Y, He J, Qin YW, Jing Q (2010) Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J 31: 659–666 [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang S, Marzolf B, Troisch P, Brightman A, Hu Z, Hood LE, Galas DJ (2009) Circulating microRNAs, potential biomarkers for drug‐induced liver injury. Proc Natl Acad Sci USA 106: 4402–4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fang X, Cai Z, Wu X, Gao X, Min J, Wang F (2020d) Comorbid chronic diseases and acute organ injuries are strongly correlated with disease severity and mortality among COVID‐19 patients: a systemic review and meta‐analysis. Research 2020: 2402961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu W, Hu G, Xia S, Sun Z, Liu Z, Xie Y, Zhang R, Jiang S, Lu L (2020e) SARS‐CoV‐2 infects T lymphocytes through its spike protein‐mediated membrane fusion. Cell Mol Immunol 10.1038/s41423-020-0424-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wan Y, Qiu C, Quinones‐Parra S, Zhu Z, Loh L, Tian D, Ren Y, Hu Y, Zhang X et al (2015) Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8(+) T cells. Nat Commun 6: 6833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu X (2020) scRNA‐seq profiling of human testes reveals the presence of the ACE2 receptor, A target for SARS‐CoV‐2 infection in spermatogonia, leydig and sertoli cells. Cells 9: 940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2020). Weekly Operational Update on COVID‐19‐28 August 2020.
- Wichmann D, Sperhake J‐P, Lütgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Schröder AS et al (2020) Autopsy findings and venous thromboembolism in patients with COVID‐19. Ann Intern Med 173: 268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zhao S, Yu B, Chen Y‐M, Wang W, Song Z‐G, Hu Y, Tao Z‐W, Tian J‐H, Pei Y‐Y et al (2020a) A new coronavirus associated with human respiratory disease in China. Nature 579: 265–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, Liu C, Yang C (2020b) Nervous system involvement after infection with COVID‐19 and other coronaviruses. Brain Behav Immun 87: 18–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynants L, Van Calster B, Bonten MM, Collins GS, Debray TP, De Vos M, Haller MC, Heinze G, Moons KG, Riley RD (2020) Prediction models for diagnosis and prognosis of covid‐19 infection: systematic review and critical appraisal. BMJ 369: m1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lazartigues E (2020) Expression of ACE2 in human neurons supports the neuro‐invasive potential of COVID‐19 virus. Cell Mol Neurobiol 10.1007/s10571-020-00915-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wu J, An Y, Xiao C, Hao F, Liu H, Wang Y, Tang H (2012) Streptozotocin‐induced dynamic metabonomic changes in rat biofluids. J Proteome Res 11: 3423–3435 [DOI] [PubMed] [Google Scholar]
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L (2020) Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med 8: 420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Chen S, Huang B, Zhong J‐M, Su H, Chen Y‐J, Cao Q, Ma L, He J, Li X‐F et al (2020) Pathological findings in the testes of COVID‐19 patients: clinical implications. Eur Urol Focus 6: 1124–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhou X, Qiu Y, Song Y, Feng F, Feng J, Song Q, Jia Q, Wang J (2020) Clinical characteristics of 82 cases of death from COVID‐19. PLoS One 15: e0235458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yu Y, Hertwig F, Thierry‐Mieg J, Zhang W, Thierry‐Mieg D, Wang J, Furlanello C, Devanarayan V, Cheng J (2015) Comparison of RNA‐seq and microarray‐based models for clinical endpoint prediction. Genome Biol 16: 133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang J, Xu C, Li N, Liu H, Ma J, Zhu Y, Xie H (2012) LFQ uant: a label‐free fast quantitative analysis tool for high‐resolution LC‐MS/MS proteomics data. Proteomics 12: 3475–3484 [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhao J, Perlman S (2010) T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus‐infected mice. J Virol 84: 9318–9325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y‐Y, Ma Y‐T, Zhang J‐Y, Xie X (2020a) COVID‐19 and the cardiovascular system. Nat Rev Cardiol 17: 259–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X (2020a) Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. The Lancet 395: 1054–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Hara H, Dai Y, Mou L, Cooper DK, Wu C, Cai Z (2016) Circulating organ‐specific MicroRNAs serve as biomarkers in organ‐specific diseases: implications for organ allo‐ and xeno‐transplantation. Int J Mol Sci 17: 1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL et al (2020b) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, Sun R, Tian Z, Xu X, Wei H (2020c). Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID‐19 patients. Natl Sci Rev 7: 998–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382: 727–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Chen K, Zou J, Han P, Hao J, Han Z (2020) Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med 14: 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Review Process File
Data Availability Statement
The datasets produced in this study are available in the following databases:
RNA‐Seq and exRNA‐Seq Data: NODE OEP000868 (http://www.biosino.org/node/project/detail/OEP000868)
Raw mass spectrometry data: iProX IPX 0002186001 (https://www.iprox.org/page/subproject.html?id=IPX0002186001)