

Summary
Oxytosis was first described over 30 years ago in nerve cells as a non-excitotoxic pathway for glutamate-induced cell death. The key steps of oxytosis, including glutathione depletion, lipoxygenase activation, reactive oxygen species accumulation and calcium influx, were identified using a combination of chemical and genetic tools. A pathway with the same characteristics as oxytosis was identified in transformed fibroblasts in 2012 and named ferroptosis. Importantly, the pathophysiological changes seen in oxytosis and ferroptosis are also observed in multiple neurodegenerative diseases as well as in the aging brain. This led to the hypothesis that this pathway could be used as a screening tool to identify novel drug candidates for the treatment of multiple age-associated neurological disorders, including Alzheimer’s disease (AD). Using this approach, we have identified several AD drug candidates, one of which is now in clinical trials, as well as new target pathways for AD.
Keywords: glutamate toxicity, oxidative stress, glutathione, lipoxygenases, calcium, glutathione peroxidase 4, mitochondria
Graphical Abstract

TOC:
Oxytosis was first described over 30 years ago in nerve cells and its key steps were subsequently identified. Ferroptosis shares these key steps. The pathophysiological changes seen in oxytosis/ferroptosis are also observed in neurodegenerative diseases. Thus, this pathway can be used to identify new therapies to treat neurodegenerative diseases.
Introduction
Cells die from a number of causes Over the last few decades, it has become clear that while the process of dying can be initiated by a large number of conditions, from aging to acute toxicities, the process itself frequently requires a series of biochemical and often transcriptional changes. The execution of this sequence is called programmed or regulated cell death, and multiple iterations of these pathways have been identified using genetics, small molecule inhibitors or a combination of both (Tang et al., 2019). After the description of apoptosis in 1972 (Kerr et al., 1972), one of the next regulated cell death pathways to be defined was oxytosis (Tan et al., 2001a). The identification of the components of this pathway was largely based on the use of small molecule inhibitors of enzymes and cellular processes, an early application of a chemical biology approach to understanding a natural process.
If the goal is to use a regulated cell death pathway as the basis for drug discovery, it is then necessary to show that it is involved in the disease. If it is, then components of the pathway can be used as the basis for drug discovery to treat that disease. In this review, we will first outline the history of oxytosis and its relationship to another regulated cell death pathway called ferroptosis, followed by a more detailed review of each of the major components in this pathway. We will then describe how each component is related to neurodegenerative disease, followed by an outline of the AD drug discovery process based upon the oxytosis pathway that we have used to get an AD drug candidate into clinical trials and another completing the investigational new drug (IND) process required to initiate a clinical trial. Finally, we will summarize how the chemical biology approach based upon these drug candidates is being used to understand the molecular contribution of aging to AD.
The History of Oxytosis & Ferroptosis
In order to understand why oxytosis is highly relevant to neurological disorders, it is important to look at the history behind the discovery of this regulated cell death pathway. A timeline of this work is shown in Figure 1. In the late 1980’s there was a burgeoning interest in understanding how glutamate killed nerve cells. Glutamate had been implicated in the nerve cell death associated with several neurodegenerative diseases as well as that following acute insults to the central nervous system (CNS). Therefore, it was reasoned that understanding how glutamate killed nerve cells could lead to new treatments for multiple neurological disorders (Choi, 1988). At this time, it was thought that glutamate exclusively killed nerve cells by an excitotoxic pathway mediated by ionotropic glutamate receptors (Choi, 1988). Thus, the reports from Coyle’s group on a glutamate-induced, calcium-dependent form of cell death which appeared to be distinct from excitotoxicity were very intriguing. Initially, using the N18-RE-105 neuronal hybridoma cell line, these investigators showed that millimolar concentrations of glutamate or certain glutamate analogues induced cell death that was independent of excitotoxic glutamate receptors. Cell death was blocked by removal of extracellular calcium or inhibition of calcium uptake (Murphy et al., 1988) as well as by multiple antioxidants including vitamin E and idebenone (Miyamoto et al., 1989). However, the key observation was that the first step in this form of glutamate toxicity was the glutamate-mediated inhibition of cystine uptake through the cystine/glutamate antiporter thereby resulting in the depletion of glutathione (GSH), the major endogenous intracellular antioxidant, and thus subsequent oxidative stress (Murphy et al., 1989). Within the cell, cystine is reduced to cysteine, the rate limiting amino acid for GSH synthesis. Indeed, glutamate-induced cell death could be mimicked by cystine deprivation (Murphy et al., 1989). Importantly, immature primary neurons that lack excitotoxic glutamate receptors, but not glial cells, were highly sensitive to inhibition of cystine uptake by glutamate or to cystine deprivation (Murphy et al., 1990). Similarly, mature primary hippocampal neurons but not the underlying glial cells were also highly sensitive to cystine deprivation (Murphy et al., 1989).
Figure 1:

Time line of key discoveries in oxytosis/ferroptosis.
At about the time, we had also developed an interest in how glutamate killed nerve cells and based on some early results (Schubert et al., 1992) decided to explore further the oxidative glutamate toxicity pathway. Starting with HT-4 cells, a mouse hippocampal nerve cell line that was immortalized with SV-40 T antigen (Morimoto and Koshland, 1990), we identified a subclone, HT22, that was particularly sensitive to glutamate but was killed exclusively via the oxidative pathway (Davis and Maher, 1994). HT22 cells lack ionotropic glutamate receptors (Maher and Davis, 1996), but have many neuronal characteristics including an electrically excitable membrane, GABA receptors, and functional cholinergic properties (Liu et al., 2009; Schriau et al., 2009). Using these cells as our primary research tool, but supporting our results with experiments in immature rat cortical primary neurons, we went on to investigate many aspects of this new regulated cell death pathway including the roles of lipoxygenases (LOXs), mitochondria, cyclic guanosine monophosphate (cGMP), iron and calcium influx, each of which is detailed below. In 2001, we published a comprehensive review of this work that also gave the name oxytosis to this new regulated cell death pathway which reflected both its dependence on oxidative stress and the fact that it was distinct from apoptosis (Tan et al., 2001a). For these studies we employed a variety of techniques to elucidate the oxytosis pathway including the generation and characterization of resistant cell lines (Sagara et al., 1998) and the use of gene expression libraries to identify genes that promote or inhibit cell death (Tan et al., 2001b).
Since 2001 a number of other laboratories have provided additional key insights into the oxytosis pathway including those of Axel Methner (Albrecht et al., 2010), Marcus Conrad (Seiler et al., 2008), Carsten Culmsee (Dolga et al., 2018) and Rajiv Ratan (Ratan, 2020) as will be described below.
A pathway with these same characteristics was described in transformed fibroblasts in a 2012 paper from Brent Stockwell’s laboratory as an iron dependent, non-apoptotic form of cell death and named ferroptosis (Dixon et al., 2012). In this paper, cell death was induced by a small molecule called erastin that was developed in the Stockwell laboratory and found to be an inhibitor of the cystine/glutamate transporter. Curiously, erastin had previously been shown by the Stockwell laboratory to be both selective for oncogenic Ras transformed cells (Dolma et al., 2003) and a voltage-dependent anion channel 3 (VDAC3) inhibitor (Yagoda et al., 2007). Despite the fact that erastin induced cell death by inhibiting the cystine/glutamate antiporter, the same target as glutamate, the likelihood that oxytosis and ferroptosis were the same pathway was not considered. In 2014, the Stockwell laboratory showed that another compound, RSL3, that they had previously described as inducing an iron dependent, VDAC3 independent cell death pathway (Yang and Stockwell, 2008), inhibited glutathione peroxidase 4 (GPx4) (Yang et al., 2014). Loss of GPx4 had previously been shown by the Conrad laboratory to induce a form of cell death with all of the characteristics of oxytosis including a requirement for LOX activity and inhibition by vitamin E (Seiler et al., 2008). Since GPx4 activity is dependent on adequate GSH levels, it makes sense that GSH loss due to cystine depletion would be upstream of GPx4 inhibition as part of the same cell death pathway. However, it should be recognized that RSL3 inhibits selenocysteine-containing proteins and so is not entirely specific for GPx4 (Gao et al., 2018). Since most of these proteins play anti-oxidant roles, it is likely that some of the effects of RSL3 could be due to inhibition of these other proteins as well.
Although oxytosis was first described in nerve cells, we have shown in multiple studies that mouse embryonic fibroblasts (MEFs) are also sensitive to glutamate toxicity (Lewerenz et al., 2014; Lewerenz and Maher, 2009) consistent with the original characterization of ferroptosis in transformed fibroblasts (Dixon et al., 2012). Thus, all of the evidence indicates that these are the same cell death pathways (Lewerenz et al., 2018) (Table 1) and we believe a more appropriate name for this pathway is oxytosis/ferroptosis (Lewerenz et al., 2018). However, since there are multiple points at which this pathway can be initiated (GSH loss, GPX4 loss) and multiple readouts used by different laboratories, further investigation of each of the steps activated by distinct inducers is certainly warranted.
Table 1:
Markers of Oxytosis and Ferroptosis
| Glutamate, -Cys, HCA | Erastin | RSL3 | |
|---|---|---|---|
| ⇓GSH | + | + | − |
| Iron requirement | + | + | + |
| ⇑ROS | + | + | + |
| ⇑Lipid peroxides | + | + | + |
| ETC | + | + | − |
| Mitochondrial alterations | + | + | + |
| ⇑ LOX activity | + | + | + |
| Calcium dysregulation | + | + | + |
| Protein synthesis | + | + | − |
| Caspase activity | − | − | − |
| Nuclear condensation | − | − | − |
The biological role of oxytosis/ferroptosis is an interesting question that remains to be elucidated. It has been proposed that oxytosis/ferroptosis may act as a tumor suppressor pathway (Dixon et al., 2012; Stockwell et al., 2020). As discussed here, there is now growing evidence that it may also be a key driver of pathology in (neuro)degenerative diseases. Because its activation can be detrimental to normal cells, it could be a cancer control mechanism that has unintended, negative consequences. Interestingly, oxytosis/ferroptosis appears to be quite highly conserved evolutionarily suggesting that it may have a more fundamental role in cellular physiology (Conrad et al., 2018; Stockwell et al., 2017; Stockwell et al., 2020). Clearly, this is an area for further investigation.
The Basic Pathway
An outline of the oxtosis/ferroptosis pathway is shown in Figure 2 and briefly described below, followed by a more detailed analysis of each component of the pathway. The initiating step in the classical experimental paradigm for the induction of oxytosis/ferroptosis is the inhibition of cystine uptake into the cells. Although cystine can be imported into cells via four transport systems, the glutamate/cystine antiporter, system xc− (Lewerenz et al., 2013) is the critical player in oxytosis/ferroptosis. System xc− is a heterodimeric amino acid transporter comprised of xCT (SLC7A11) and 4F2hc (SLC3A2) as the heavy chain, that specifically transports cystine, glutamate and the non-proteinogenic amino acid cystathionine (Kobayashi et al., 2015; Lewerenz et al., 2013). Importantly, other substrate inhibitors of system xc− like homocysteate and quisqualate inhibit cystine uptake and cause cell death in cultured cells (Maher and Davis, 1996; Murphy and Baraban, 1990; Murphy et al., 1989). Furthermore, cells derived from xCT knock-out mice do not survive in cell culture unless cystine in the culture medium is replaced by β-mercaptoethanol (β–ME) as an alternative source of reducing equivalents (Sato et al., 2005). Over the years, additional inhibitors of system xc− including erastin (see above) and sulfasalazine (Gout et al., 2001) have been described. In addition to cystine starvation or inhibition of cystine import, inhibition of GSH synthesis by buthionine sulfoximine (BSO), an inhibitor of glutamate cysteine ligase (GCL), the rate-limiting enzyme in GSH biosynthesis, can also induce oxytosis (Ishige et al., 2001b; Lewerenz et al., 2003; Li et al., 1997b). This observation further supports the relevance of GSH depletion for the initiation of oxytosis/ferroptosis.
Figure 2:
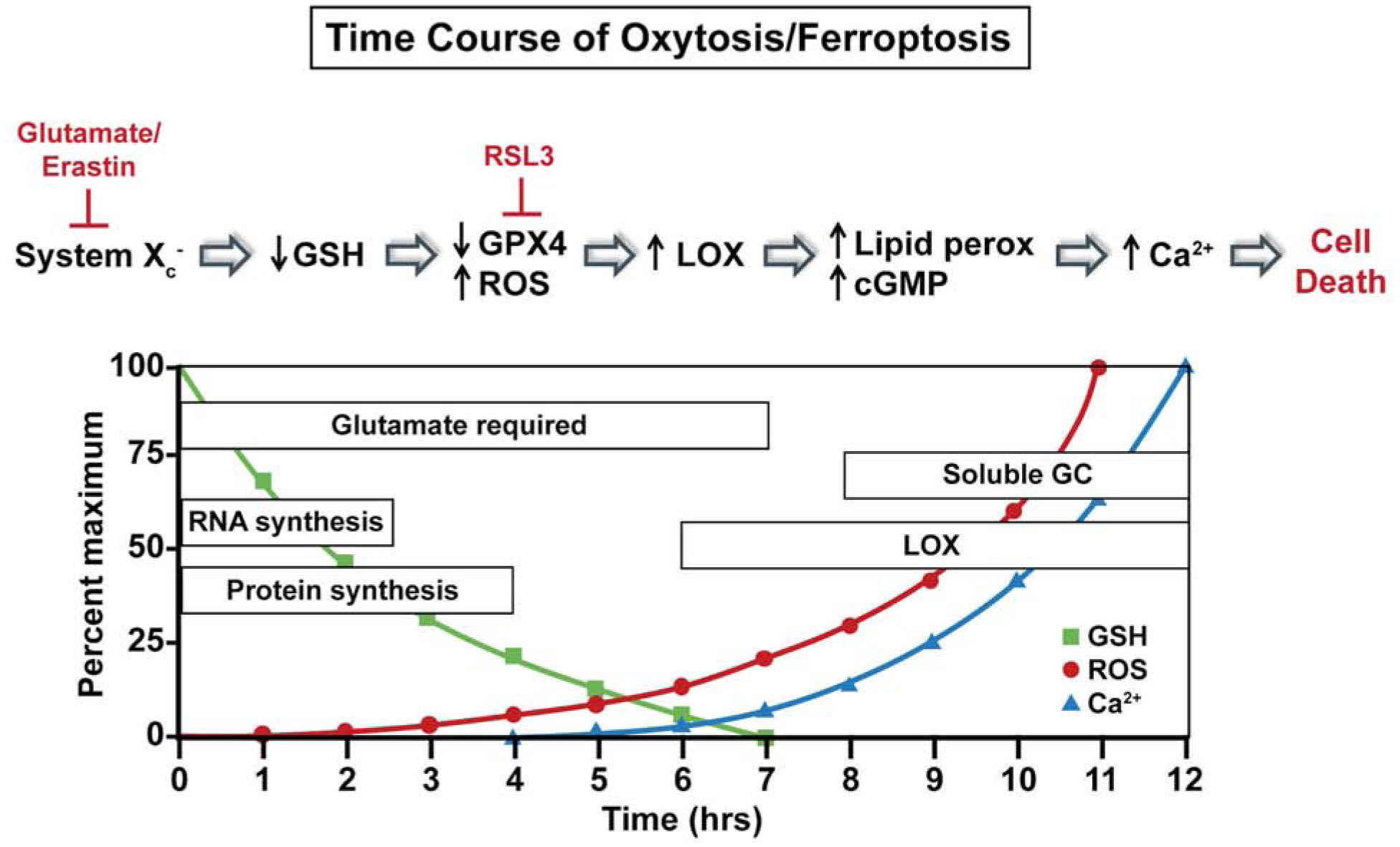
Time course of major pathophysiological changes that occur during oxytosis/ferroptosis.
Following the inhibition of system xc− by glutamate, GSH levels drop in a time-dependent manner. When the GSH levels fall below ~20% (~6–8 hours after glutamate treatment), reactive oxygen species (ROS) levels start to increase exponentially (Tan et al., 1998a). It is important to note that the ROS levels themselves do not kill the cells directly but rather give rise to a number of downstream effectors that culminate in cell death. Thus, the accumulation of large amounts of intracellular ROS is necessary but not sufficient to cause death. Indeed, compounds that block pathways downstream of ROS accumulation can be protective even in the presence of elevated levels of ROS (e.g. Maher and Schubert, 2000). The major initial source of these ROS appears to be the mitochondrial electron transport chain (ETC) (Tan et al., 1998a). Other sources of ROS, including the NADPH oxidase Nox4 (Ha et al., 2010) and lysosomes (Kubota et al., 2010), may also contribute to the increase in ROS in response to GSH depletion.
There is also a requirement for RNA and protein synthesis during the course of GSH depletion for the subsequent production of ROS. Macromolecule synthesis inhibitors block cell death induced by glutamate or erastin treatment in multiple cell types (Dixon et al., 2012; Tan et al., 1998b) but only when added within a few hours of treatment, suggesting an early requirement for these proteins. In contrast, the protein synthesis inhibitor cycloheximide does not protect from RSL3 toxicity in either transformed fibroblasts (Yang and Stockwell, 2008) or HT22 cells (Lewerenz et al., 2018). This observation is consistent with GPx4 inhibition being downstream of GSH depletion (Seiler et al., 2008). Both GPx4 inhibition and GSH depletion lead to the activation of 12/15-lipoxygenase (12/15LOX also known as 15-LOX-1) (Li et al., 1997b; Seiler et al., 2008), likely because high concentrations of GSH inhibit LOX activity (Li et al., 1997b). GPx4 is unique in its ability to reduce lipid hydroperoxides, the products of 12/15LOX, embedded in membranes (Imai and Nakagawa, 2003). GPx4 inhibition can also directly lead to an increase in lipid peroxidation because without its activity, any lipid peroxidation that is initiated will continue to propagate (Zimniak, 2011). However, the precise role of lipid peroxidation in oxytosis/ferroptosis and whether it is the direct cause of cell death or rather activates a pathway that eventually leads to cell death is still unclear (Feng and Stockwell, 2018; Lei et al., 2019; Stoyanovsky et al., 2019). It is likely that there are multiple roles including direct disruption of membrane dynamics, alterations in signaling pathways and modulation of protein function, both of the latter mediated by secondary products of lipid peroxidation such as hydroxyeicosatetraenoic acids (HETEs), the reactive carbonyls 4-hydroxynon-2-enal (4-HNE) and malondialdehyde (MDA) (Ayala et al., 2014; Feng and Stockwell, 2018; Zimniak, 2011) and cyclopentenone isoprostanes (Musiek et al., 2006). All of these molecules have been shown to accumulate in various models of oxytosis/ferroptosis (eg Ates et al., 2020; Doll et al., 2017; Li et al., 1997b; Musiek et al., 2006; Zou et al., 2020).
The ROS generated downstream of GSH depletion can also act directly as initiators of lipid peroxidation (Ayala et al., 2014; Zimniak, 2011). In this case, the key ROS is the hydroxyl radical. This is formed through redox cycling by the Fenton reaction whereby free ferrous iron (Fe+2) reacts with hydrogen peroxide, Fe+2 is generated in the Haber Weiss reaction by the interaction of superoxide with ferric iron (Fe+3) (Ayala et al., 2014). Other transition metals, including copper, can also contribute to hydroxyl radical formation in cells (Ayala et al., 2014). Both iron and copper contribute to oxytosis/ferroptosis (Maher, 2017), while only the contribution of iron to this pathway is reflected in the name ferroptosis (Dixon et al., 2012). In addition, it is important to note that this name is at least partly based on the protective activity of iron chelators against ferroptosis (Dixon et al., 2012), but these compounds also chelate copper and zinc (Crisponi et al., 2015). Interestingly, one of the molecules that is thought to limit the redox cycling activity of Fe+2 is GSH (Stoyanovsky et al., 2019) suggesting that GSH depletion can increase the levels of both of the substrates of the Fenton reaction.
Further evidence for the role of lipid peroxides in ferroptosis comes from several recent studies. Two groups (Bersuker et al., 2019; Doll et al., 2019) demonstrated a protective role against ferroptosis for apoptosis inducing factor mitochondria-associated 2 (AIFM2) which they renamed ferroptosis suppressor protein 1 (FSP1). This protein works through ubiquinone (CoQ10) to trap lipid peroxyl radicals. Another study showed that nitric oxide (NO) can act as a negative regulator of ferroptosis by both scavenging lipid peroxides and their by-products and by interacting with reactive intermediates of 12/15-LOX (Kapralov et al., 2020). Importantly, inducible nitric oxide synthase (iNOS)/NO was able to substitute for GPx4 as a protective mechanism in cells treated with RSL3. This pathway is likely of particular relevance in immune cells which have high levels of iNOS activity and NO production when they express a pro-inflammatory phenotype.
As noted earlier, the requirement for calcium influx for the execution of cell death in the oxytosis pathway was seen in the earliest studies (Murphy et al., 1988). This influx is downstream of ROS production and LOX activation since 12/15LOX inhibitors prevent the rise in intracellular calcium following GSH depletion (Li et al., 1997b). More on the specifics of calcium influx will be discussed below. However, it should be noted that while inhibitors of calcium influx clearly protect from multiple inhibitors of system xc− (Maher et al., 2018) as well as RSL3 (Lewerenz et al., 2018), this step in the cell death pathway has not been as thoroughly investigated following treatment with RSL3.
About 10–12 hours after the induction of oxytosis/ferroptosis by glutamate, erastin or RSL3, when ROS, lipid peroxidation and intracellular calcium levels have reached their maximum, the Culmsee lab showed that the pro-apoptotic Bcl-2 family member Bid (BH3-interacting domain death agonist) translocates to the mitochondria and Bid-loaded mitochondria accumulate around the nucleus and lose their membrane integrity (Jelinek et al., 2018; Landshamer et al., 2008). This leads to the translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus where it promotes cell death (Landshamer et al., 2008). Importantly, calpains, calcium-dependent proteases, can activate Bid (Cabon et al., 2012; Chen et al., 2001) and AIF (Norberg et al., 2010). Some calpain inhibitors have been reported to protect from oxytosis (Elphick et al., 2008) suggesting that this might be one way that calcium influx contributes to cell death.
Further Insight into Key Elements of the Oxytosis/Ferroptosis Pathway
Lipoxygenases (LOXs)
Lipid peroxides can be formed both non-enzymatically through the action of ROS as described above and enzymatically. Several enzymes can generate lipid peroxides including cyclooxygenases (COXs), LOXs and cytochrome P450 oxidoreductases (Ayala et al., 2014). All three enzymes require iron for activity. However, the COX inhibitor indomethacin does not protect against either oxytosis (Li et al., 1997b) or ferroptosis (Yang et al., 2014). In contrast, a very recent study in cancer cells showed that knockdown of cytochrome P450 oxidoreductase protected cells against multiple inducers of oxytosis/ferroptosis by reducing lipid peroxidation and its secondary products (Zou et al., 2020). The group of enzymes that has received the most attention in the context of oxytosis/ferroptosis is the LOXs which are non-heme iron containing enzymes that can act on both free polyunsaturated fatty acids (PUFAs) (mainly arachidonic acid and linoleic acid) as well as PUFAs that are esterified in phospholipids or cholesterol esters (for review Kuhn et al., 2015). The products of LOXs include a wide variety of bioactive lipid mediators. LOX naming is complicated and has changed over the years (Kuhn et al., 2015). For example, what was called 12/15LOX or 12LOX in humans and mice, respectively, is now called ALOX15 or 15-LOX-1. In 1997 we identified 12-lipoxygenase activity (12LOX) and 12LOX-mediated peroxidation of arachidonic acid as an important link between GSH depletion and ROS accumulation in HT22 cells (Li et al., 1997b). During the induction of oxytosis/ferroptosis by glutamate or BSO, the cellular uptake of arachidonic acid was enhanced, 12LOX activity (measured as the production of 3H-12-HETE from 3H-arachidonic acid in cell lysates) was increased and LOX proteins were translocated to the membrane fraction. In addition, exogenous arachidonic acid potentiated cell death. Most importantly, we showed that there was a direct link between GSH depletion and LOX activation in agreement with earlier reports showing that GSH inhibits LOX activity (Hagmann et al., 1993; Li et al., 1997b). Consistent with these observations, MEF deficient in ALOX15 were protected against BSO-induced cell death (Seiler et al., 2008).
However, whether specific LOXs are more likely to contribute to oxytosis/ferroptosis is still not clear. HT22 cells express both ALOX15 and ALOX15B based on both protein and biochemical analyses (generation of 12-HETE) (Li et al., 1997b; Rai et al., 2014; Wenzel et al., 2017; Yigitkanli et al., 2013). Inhibition of LOX activity in HT22 cells by multiple inhibitors with different reported specificities blocked calcium influx and cell death induced by GSH depletion (Li et al., 1997b; Pallast et al., 2009). Surprisingly, the ALOX5 inhibitor zileuton also protected HT22 cells against cell death induced by either glutamate or erastin (Liu et al., 2015).
A very similar pharmacological profile was reported for genetically engineered MEF in which cell death associated with lipid peroxidation could be induced by GPx4 inactivation (Seiler et al., 2008). In these cells, both linoleic and arachidonic acid exacerbated cell death, while multiple LOX inhibitors protected against GPx4 deficiency (Seiler et al., 2008). Using redox phospholipidomics, Kagan et al. showed that double- and triple-oxygenated arachidonic and adrenic acid-containing phosphatidylethanolamine species with C18 fatty acids (C18:0 or C18:1) at the sn-1 position and C20:4 or C22:4 fatty acids at the sn-2 position are the preferential substrates for ALOX15 in ferroptosis induced by RSL3 or GPx4 deficiency in MEF (Kagan et al., 2017). This observation that PUFAs incorporated into phospholipids are key targets of LOXs in ferroptosis is consistent with the finding that the activity of acyl-CoA synthetase long-chain member 4 (ACSL4) is required for the induction of cell death in GPx4-deficient MEF (Doll et al., 2017). ACSL4 preferentially esterifies CoA to long chain free PUFAs in an ATP dependent manner thereby activating them for phospholipid synthesis.
As indicated above, we showed that GSH depletion causes the translocation of LOX from the cytoplasm to the cell membrane in HT22 cells (Li et al., 1997b). Recently, another mechanism for altering the substrate specificity of LOXs was described (Wenzel et al., 2017). It was found that when ALOX15 or ALOX15B form complexes with the promiscuous small scaffolding protein, phosphatidylethanolamine-binding protein 1 (PEBP1), their ability to oxidize PUFAs shifts from free PUFAs to esterified PUFAs thereby inducing the generation of phosphatidylethanolamine lipid hydroperoxides (Wenzel et al., 2017). In addition, ALOX15 has been reported to be involved in programmed organelle degradation by binding to intracellular membranes of various organelles while sparing the plasma membrane (van Leyen et al., 1998). ALOX15 was further shown to bind to mitochondria in vitro causing membrane disintegration and ROS generation (Pallast et al., 2009). This is consistent with the disintegration of intracellular organelles observed in oxytosis/ferroptosis (Tan et al., 1998b; Tirosh et al., 2000).
Despite the biochemical, pharmacological and genetic evidence for a role for LOXs in oxytosis/ferroptosis, their precise role is still under debate. Indeed, a recent study suggested that several commonly used LOX inhibitors, including NDGA, zileuton and PD146176, protect from oxytosis/ferroptosis by virtue of their radical trapping antioxidant activity (Shah et al., 2018) rather than via their effects on LOX activity. Thus, it will be important to incorporate genetic manipulations into future work on the role of LOXs in oxytosis/ferroptosis. In addition, based on the published data, it is likely that LOXs may play more or less important roles in oxytosis/ferroptosis depending on both the cell type and the initiating stimulus.
Mitochondria
The production of ROS is central to many forms of regulated cell death (Tang et al., 2019). There are multiple sources of ROS in cells, including the mitochondrial electron transport chain (ETC), and a wide array of enzymes including the monoamine oxidases, tyrosine hydroxylase, amino acid oxidases, various dehydrogenases, NADPH oxidases (NOXs) and xanthine oxidase (Maher and Schubert, 2000). Among these monoamine oxidases, NOX4 (Case et al., 2013), α-ketoglutarate dehydrogenase and α-glycerophosphate dehydrogenase (Adam-Vizi and Tretter, 2013) are found in mitochondria and therefore could contribute to mitochondrial ROS generation. In the case of oxytosis/ferroptosis induced by GSH depletion, the mitochondrial ETC is essential for ROS production and subsequent cell death. We showed in 1998 that ETC inhibitors, as well as the mitochondrial electron transport uncoupler FCCP which dissipates the mitochondrial membrane potential (Voet and Voet, 1990), block ROS production and prevent cell death in HT22 cells following treatment with glutamate or cystine deprivation (Tan et al., 1998a). These results were recently reproduced by another lab using cystine deprivation as the initiating insult (Gao et al., 2019). Reverse electron transport (RET) from Complex II to Complex I appears to be the major source of the ROS (Liu and Schubert, 2009) consistent with the effects of FCCP since a high mitochondrial membrane potential is needed for RET (Onukwufor et al., 2019).
In contrast to compounds that induce oxytosis/ferroptosis via GSH depletion, neither ETC inhibitors (Gao et al., 2019) nor FCCP (unpublished results) protect from RSL3 toxicity. However, RSL3 along with erastin and glutamate all increase the levels of the mitochondrial specific ROS probe mitoSOX (Jelinek et al., 2018; Neitemeier et al., 2017). Similar results were obtained with GPx4 knockdown in human Caco-2 cells (Cole-Ezea et al., 2012). Furthermore, this study (Cole-Ezea et al., 2012) supports the idea that while ETC-produced ROS do not play a role in RSL3 toxicity, other mitochondrial sources of ROS potentially do such as monoamine oxidases, NOX4 (Case et al., 2013) and/or mitochondrial dehydrogenases (Adam-Vizi and Tretter, 2013). In addition, ruthenium red, an inhibitor of mitochondrial calcium uptake, also blocks high rates of ROS formation and cell death in response to glutamate, suggesting that mitochondrial calcium contributes to ROS production (Tan et al., 1998a). Interestingly, ruthenium red also protects from erastin and RSL3 toxicity in HT22 cells (unpublished).
Whereas we have observed a transient hyperpolarization of the mitochondria during the exponential increases in ROS and intracellular calcium (Liu and Schubert, 2009; Tan et al., 1998a) in HT22 cells treated with glutamate, others reported depolarization upon the release of AIF from the mitochondria in the final phase of the cell death process (Landshamer et al., 2008). However, a recent study (Kang et al., 2014) provides further support for the idea that hyperpolarization actually precedes the final depolarization which occurs near to the time of cell death and would be more consistent with the protective effects of the mitochondrial uncoupler FCCP against oxytosis/ferroptosis induced by GSH depletion and the role of RET in ROS production.
Calcium
Among the targets of LOX metabolites such as 12- and 15-HETE is soluble guanylate cyclase (sGC) (Brune and Ullrich, 1991; Li et al., 1997a). In 1997, we showed that the sGC inhibitor LY83583 inhibits the death of HT22 cells but not GSH loss or ROS accumulation following glutamate or BSO treatment (Li et al., 1997a). Importantly, LY83583 also protects against erastin and RSL3 toxicity (Lewerenz et al., 2018). Moreover, cGMP levels increase after 6 hr of glutamate treatment when GSH is depleted and ROS begin to exponentially accumulate (Li et al., 1997a). The cell permeable and phosphodiesterase resistant cGMP analogue, CPT-cGMP, can potentiate glutamate toxicity, even when added up to 8 hr after treatment with glutamate (Henke et al., 2013; Li et al., 1997a). Moreover, cell death similar to oxytosis/ferroptosis can be induced directly by CPT-cGMP (Henke et al., 2013; Ishige et al., 2001a). Mechanistically, we found that elevated cGMP levels contribute to calcium influx which is required for oxytotic/ferroptotic cell death (Li et al., 1997a; Murphy et al., 1988; Murphy et al., 1989).
During the exponential phase of ROS accumulation in oxytosis, there is a sharp increase in cellular calcium that immediately precedes cell death (Tan et al., 1998a). Indeed, the protective activity of the general calcium channel inhibitor CoCl2 against glutamate-induced oxytosis is retained until the very late stage of the cell death pathway indicating the functional relevance of the late calcium accumulation for cell death execution (Davis and Maher, 1994; Li et al., 1997a; Tan et al., 1998a). CoCl2 as well as other inhibitors of calcium influx in HT22 cells such as apomorphine (Ishige et al., 2001a) also protect against erastin and RSL3 toxicity (Lewerenz et al., 2018; Maher et al., 2018), further supporting a role for late stage calcium influx in oxytosis/ferroptosis. Moreover, similar to glutamate, erastin was recently shown to induce calcium influx in HT22 cells (Nagase et al., 2020).
In addition, an inhibitor of store-operated calcium entry (SOCE), 2-aminoethoxydiphenyl borate, strongly inhibited glutamate-induced oxytosis in HT22 cells with the same time profile as CoCl2 (Henke et al., 2013), indicating that SOCE might contribute to the final rise in cytosolic calcium during oxytosis. SOCE is triggered when endoplasmic reticulum (ER) calcium depletion leads to the translocation of the ER membrane-resident stromal interaction molecule 1/2 (Stim1/2) to the plasma membrane where recruitment of calcium release-activated calcium modulator (Orai) Ca2+ channel proteins promotes extracellular-derived Ca2+ influx into the cytosol to replenish ER stores (Maher et al., 2018). Indeed, siRNA mediated knock-down of Orai1, but not of other components of SOCE protected against glutamate-induced oxytosis and decreased the peak of intracellular calcium immediately before cell rupture (Henke et al., 2013). Very recently, we showed that Orai3 knockdown also protects against glutamate-induced oxytosis in HT22 cells and that both Orai1 and Orai3 knockdown also protect against RSL3-induced ferroptosis (Goldberg et al., 2020) thereby providing further support for calcium influx as part of the cell death mechanism underlying both glutamate and RSL3 toxicity. The observation that Orai1 knock-down also protects against CPT-cGMP-induced cell death connects these two pathways. However, the exact mechanisms underlying the modulation of SOCE by cGMP in this cell death paradigm remains to be fully characterized.
Relevance of the Oxytosis/Ferroptosis Pathway to Neurodegeneration and Aging
GSH Depletion
If oxytosis/ferroptosis is involved in old age-associated neurodegenerative diseases and other neurological disorders, then there should be evidence that the key steps in this cell death pathway are altered in these conditions (Table 2). The first step in oxytosis/ferroptosis is GSH depletion. GSH plays a central role in maintaining cellular redox homeostasis (Schafer and Buettner, 2001). A fairly large number of studies have shown age-dependent decreases in total GSH and/or reduced GSH levels in the brain (for reviews see Ballatori et al., 2009; Currais and Maher, 2013; Mandal et al., 2019). Importantly, a study using double-edited proton (1H) magnetic resonance spectroscopy (MRS) to look at GSH levels in the occipital cortex of healthy young (20 yr) and elderly (77 yr) human subjects found that GSH levels were decreased by ~30% in the elderly subjects (Emir et al., 2011). Moreover, multiple studies have shown decreases in plasma and/or red blood cell GSH levels with age in humans (Droge et al., 2006; Giustarini et al., 2006; Maher, 2005; Sekhar et al., 2011), which is exacerbated in mild cognitive impairment and AD (Bermejo et al., 2008; Rae and Williams, 2017). However, measurements of GSH levels in the brains of AD patients have produced conflicting results. Although some studies reported a decrease (Ansari and Scheff, 2010; Gu et al., 1998), other papers either reported no change (Karelson et al., 2001; Makar et al., 1995; Perry et al., 1987) or an increase (Adams et al., 1991) in GSH levels in the brains of AD patients relative to age-matched controls. Since these studies all used autopsied tissue, variables such as varied techniques as well as post-mortem interval and the quality of the samples could account for these differences. However, a more recent study that used live subjects in combination with 1H MRS to look at GSH levels in the hippocampi and frontal cortices of control, MCI and AD subjects found an AD-dependent decrease in GSH in both regions that correlated with declines in cognitive function (Mandal et al., 2015). Moreover, the levels of hippocampal GSH could discriminate between healthy controls and MCI subjects while cortical GSH levels could discriminate between MCI and AD patients. Further analysis showed that the decreases in GSH were not secondary to tissue atrophy. Interestingly, two recent studies in mice that used knockdown of GCL in all neurons (Fernandez-Fernandez et al., 2018) or only forebrain neurons (Feng et al., 2017), to specifically reduce neuronal GSH levels, showed an enhanced, age-dependent development of cognitive deficits relative to control animals, supporting the idea that GSH loss contributes to cognitive impairments in AD. Importantly, in the former study (Fernandez-Fernandez et al., 2018), the decrease in GSH levels in the hippocampus was modest but comparable to that seen in MCI and AD subjects (Mandal et al., 2015).
Table 2:
Neurological Disorders and Markers of Oxytosis/Ferroptosis
| AD | PD | HD | ALS | stroke | aging | |
|---|---|---|---|---|---|---|
| ⇓GSH | + | + | + | + | + | + |
| ⇑LOX | + | + | ? | + | + | + |
| ⇑Lipid peroxides | + | + | + | + | + | + |
| Mitochondrial Dysfunction | + | + | + | + | + | + |
| Ca+2 Dysregulation | + | + | + | + | + | + |
There is also evidence for GSH loss playing a key role in Parkinson’s disease (PD). A number of studies have found that total GSH levels are specifically decreased in the substantia nigra (SNc) of PD patients and that there is a positive correlation between the severity of the disease and the extent of GSH loss (Schulz et al., 2000). There is also a positive correlation between decreases in total blood GSH and disease severity that might provide a biomarker for PD (Mischley et al., 2016). The decrease in GSH precedes other PD-associated changes in the SNc (Martin and Teisman, 2009; Zeevalk et al., 2008) and is not due to nerve cell death since it can be observed in surviving dopaminergic neurons (Pearce et al., 1997). Indeed, it has been argued that GSH loss may play an active role in PD development and progression and precipitate all of the other pathological changes associated with the disease (Martin and Teisman, 2009). Animal studies have shown that treatment with GSH analogues can reduce the neurodegeneration seen in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD in mice (Chinta et al., 2006). More importantly, GSH administration to a small group of PD patients resulted in significant improvements in disability (Sechi et al., 1996). GSH loss has also been noted in the motor cortex in ALS patients (Rae and Williams, 2017).
Together these and other results (Currais and Maher, 2013) suggest that GSH loss contributes to neuronal damage and death in neurodegenerative diseases which is consistent with a role for oxytosis/ferroptosis in these diseases.
Lipid peroxidation and LOX activity
Lipid peroxidation and the production of lipid peroxides from LOXs are also associated with a number of neurological disorders, including neurodegenerative diseases (Ayala et al., 2014; Kuhn et al., 2015; Reed, 2011; Singh and Rao, 2018; Zimniak, 2011). Since the brain is particularly rich in PUFAs, the cells of this tissue are very susceptible to lipid peroxidation. Lipid peroxidation is considered an important hallmark of AD cellular pathology (Bradley-Whitman and Lovell, 2015; Gaschler and Stockwell, 2017; Sultana et al., 2013) and is also seen in PD and Huntington’s disease (HD) (Reed, 2011). Not only is the peroxidation of membrane lipids detrimental to lipid bilayer function and cellular integrity, the by-products of lipid peroxidation can act as electrophilic aldehydes to crosslink with DNA or covalently bind to amino acids, thus interfering with nucleic acid and protein structure and function. Additionally, they can act as signaling molecules able to induce inflammation (Breitzig et al., 2016; Uchida, 2017; Yadav and Ramana, 2013). One of the major products of lipid peroxidation is the electrophilic aldehyde 4-HNE which is formed from n-6 PUFAs (Zimniak, 2011). Increases in 4-HNE-modified proteins have been detected in AD, PD, HD and ALS (Reed, 2011) as well as during aging (Zimniak, 2011). Many of these proteins play important roles in energy metabolism and mitochondrial function (Reed, 2011). These observations suggest that lipid peroxidation could provide a feed forward mechanism to drive oxytotic/ferroptotic regulated cell death rather than simply cause membrane damage.
LOXs, particularly 12/15LOX, have also been implicated in some neurological disorders (Kuhn et al., 2015; Singh and Rao, 2018). While much of the work has been done using LOX inhibitors which may in some cases also have radical trapping activity and therefore not be as specific as has been assumed (Shah et al., 2018), several studies have used LOX gene knockouts to demonstrate a role for these enzymes in neurodegeneration. These include studies on ischemic stroke in mice where ALOX15 knockdown protects from damage (van Leyen et al., 2006) and AD where 12/15LOX knockdown reduces memory loss and amyloid accumulation in the Tg2576 transgenic mouse model (Joshi et al., 2015). Consistent with these observations, overexpression of 12/15LOX in the same mouse strain led to increased memory loss and amyloid accumulation (Joshi et al., 2015). Interestingly, 12/15LOX is upregulated in the brain regions that are most vulnerable to AD early in the course of the disease, suggesting a role for this enzyme in disease development (Joshi et al., 2015).
5LOX has also been implicated in AD (Joshi and Practico, 2015). 5LOX activity requires an activating protein called FLAP (Joshi and Practico, 2015) and there are FLAP inhibitors that may not suffer from the same concerns as LOX inhibitors. 5LOX expression increases with age in the brain in both mice and men and this is exacerbated in AD (Joshi and Practico, 2015). Similar to the results with the genetic manipulations of 12/15LOX, knockout of either 5LOX or FLAP in both Tg2576 and 3xFAD transgenic AD mice improves the overall AD phenotype while overexpression of 5LOX is associated with an exacerbation of the phenotype (Joshi and Practico, 2015). Thus, based on the results with gene knockout mice, both 12/15LOX and 5LOX appear to play a role in AD and stroke, further supporting a role for oxytosis/ferroptosis in the nerve cell damage and death seen in these diseases.
Mitochondrial Dysfunction
Evolutionarily, the acquisition of mitochondria played a key energetic role in the establishment of biological complexity, expanding life from unicellular to multicellular organisms (Lane and Martin, 2010). Mitochondria play a key role in cell differentiation and function by mediating numerous aspects of metabolism (Agathocleous and Harris, 2013; Folmes et al., 2012; Harris et al., 2012). As such, detrimental changes in mitochondrial homeostasis can lead to a variety of human diseases (Chan, 2006; Lin and Beal, 2006; Navarro and Boveris, 2010; Wallace, 2005).
The brain is one of the most energy demanding tissues in the body (Belanger et al., 2011). Therefore, it comes as no surprise that of the several features that characterize the aging process in the brain, a decline in cerebral energy metabolism precedes the pathology and symptoms of AD and is more severe than that observed in normal aging (Costantini et al., 2008; Cunnane et al., 2011; Currais, 2015; Yin et al., 2014). In fact, several lines of evidence indicate that neurons try to shift metabolism towards glycolysis during AD as an adaptive response to mitochondrial dysfunction (reviewed in Atlante et al., 2017). Thus, given that mitochondrial energy production from glucose metabolism supports the majority of brain activity and the maintenance of neuronal homeostasis, it is likely that a failure to supply cells with adequate energy contributes to the neuropathological cascade in AD. Most of the energy derived from glucose oxidation is produced in mitochondria, and multiple mitochondrial-dependent functions are impaired during aging and AD. Evidence that mitochondrial dysfunction occurs early and acts causally in AD pathogenesis includes declines in ETC activity and respiration, increases in mitochondrial-derived oxidative stress, significant changes in the mitochondrial metabolite landscape, oxidative damage to mitochondrial DNA, RNA, lipid and protein species, and an accumulation of mitochondrial DNA (mtDNA) mutations (Currais, 2015; Lin and Beal, 2006; Onyango et al., 2016; Swerdlow and Khan, 2004; Yin et al., 2016).
Although the role of ROS as the origin of age-dependent mitochondrial decline is still controversial, evidence for deficits in mitochondrial function extends to other neurodegenerative diseases, including PD, HD and ALS (Boumezbeur et al., 2010; Boveris and Navarro, 2008; Chan, 2006; Lin and Beal, 2006; Sorbi et al., 1983; Wallace, 2005).
Calcium Dysregulation
The movement of calcium both across the plasma membrane as well as between intracellular organelles, including the ER and mitochondria, plays a critical role in many of the fundamental functions of neurons. Thus, it is not surprising that dysregulation of calcium homeostasis is associated with multiple neurological disorders (Zundorf and Reiser, 2011). These changes include alterations in calcium buffering capacity, deregulation of calcium channel activities and alterations of other calcium regulated proteins (Zundorf and Reiser, 2011). Interestingly, dysregulation of SOCE is observed in aging (Chandran et al., 2019) and de-regulation of SOCE components may contribute to AD pathogenesis (Tong et al., 2018). Furthermore, drugs that block calcium influx have shown positive effects in preclinical models of some neurological disorders (Zundorf and Reiser, 2011). In C. elegans models of human neurodegenerative diseases there is evidence that a prolonged increase in intracellular cytoplasmic calcium is the final common step required for a form of non-apoptotic cell death (Alvarez et al., 2020).
Together, these results support the idea that dysregulation of calcium homeostasis contributes to neuronal damage and death in neurodegenerative diseases and is consistent with a role for oxytosis/ferroptosis in these diseases.
Using a drug discovery platform based upon oxytosis/ferroptosis to identify novel neuroprotective compounds
Because all of the detrimental cellular changes described above that occur during the process of oxytosis/ferroptosis are associated with aging and age-associated neurodegenerative diseases, we have used it as our primary screen to identify potential AD drug candidates (Prior et al., 2014). Although the in vivo relevance of system xc- inhibition to neurodegeneration is not clear, what is clear is that the perturbation of the cellular redox balance is a central event in this process and can be initiated by a variety of pathophysiologically relevant insults. For our screening platform (Figure 3), which has yielded two AD drug candidates in and near clinical trials, chemical libraries or compounds synthesized in our laboratories are initially screened in the oxytosis/ferroptosis assay using glutamate and HT22 cells. This assay is robust, highly reproducible, and can easily be automated for robotic screening. Since the discovery of oxytosis, a number of other labs have used this assay to screen curated groups of compounds to identify those that might have the best potential to work in vivo. Examples of these screens include herbal extracts (Kobayashi et al., 2000), cannabinoids (Marsicano et al., 2002), analogues of estradiol (Prokai et al., 2001), flavonoid derivatives (Schramm et al., 2018), oxindole derivatives (Hirata et al., 2018) and LOXs (Armstrong et al., 2016).
Figure 3:

AD drug discovery pipeline using oxytosis/ferroptosis as the primary screen.
Because neurodegenerative diseases are usually initiated and/or executed by multiple toxicities associated with the aging brain, we have incorporated several secondary phenotypic screening assays into the platform and require that a drug candidate be effective in all of them before moving the compound into pharmacological and animal studies. These include assays for proteotoxicity, trophic factor loss, inflammation, and energy deprivation (Prior et al., 2014; Schubert et al., 2018). Once a compound is identified with a low EC50, high stability and good brain penetrance it is then put into animal disease models and its molecular target identified (Goldberg et al., 2018). Once the target is known, it is then used in a chemical biology approach to identify related aging and disease pathways (Currais et al., 2019). Below we discuss four compounds discovered through initial screens based upon the HT22 oxytosis/ferroptosis pathway (Figure 4, Table 3).
Figure 4:
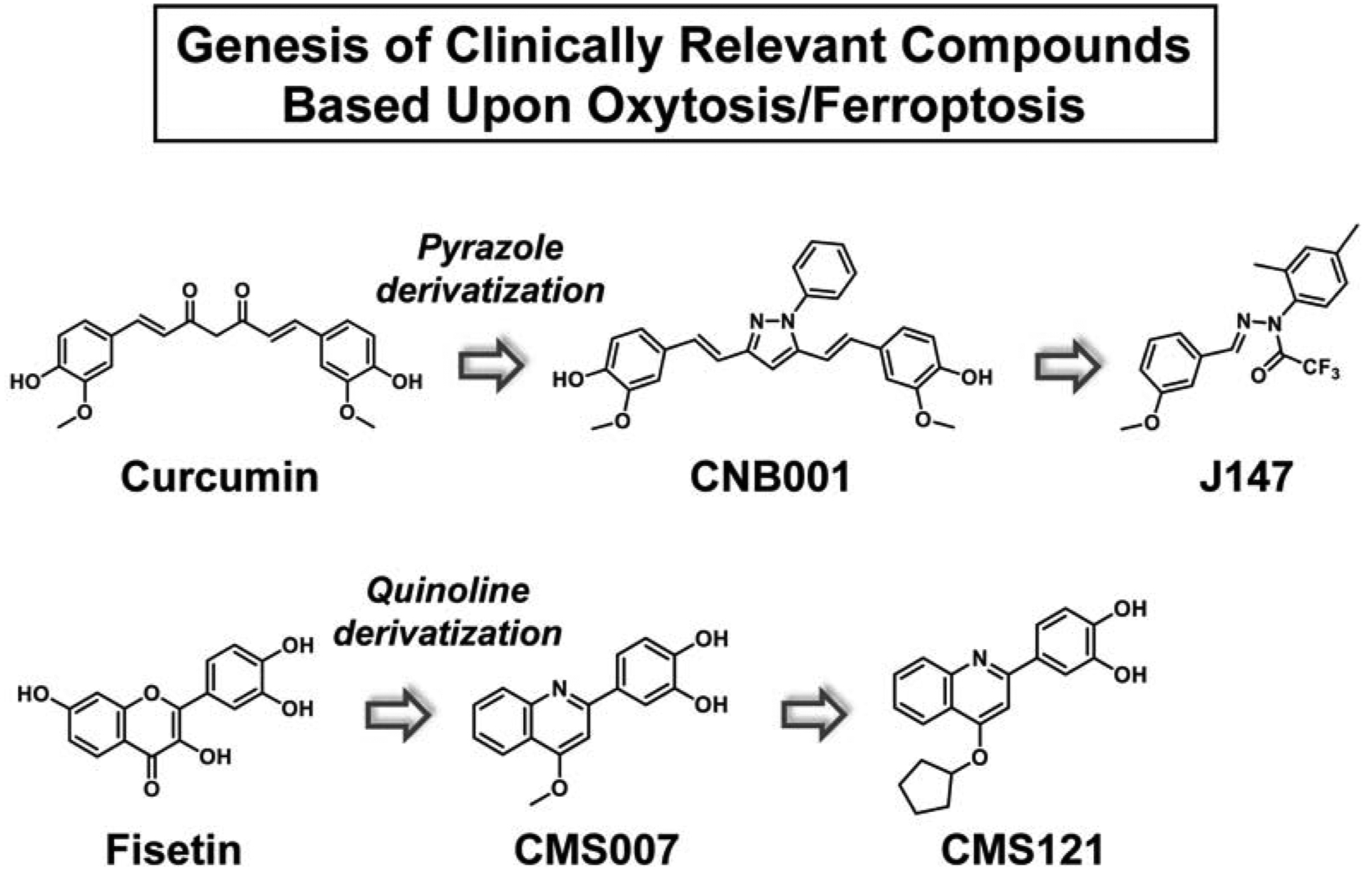
Genesis of AD drug candidates based on oxytosis/ferroptosis as the primary screen.
Table 3:
EC50s of Clinically Relevant Compounds for Protection Against Inducers of Oxytosis/Ferroptosis
| Compound | Glutamate EC50 (μM) | Erastin EC50 (μM) | RSL3 EC50 (μM) |
|---|---|---|---|
| Fisetin | 3 | 2 | 3 |
| CMS121 | 0.2 | 0.3 | 0.2 |
| Curcumin | 10 | 15 | 15 |
| CNB001 | 1 | 2 | 0.2 |
| J147 | 0.04 | 0.06 | 0.15 |
It should be noted that while cultured cells die rapidly following the induction of oxytosis/ferroptosis, this might not necessarily be the case in vivo. As described above, oxytosis/ferroptosis follows a series of well-defined steps so that once the pathway is initiated in vivo, cells could traverse these steps over an extended time period thereby leading to the neuronal dysfunction, synaptic loss, neuroinflammation and eventually cell death that is characteristic of these diseases. Thus, if oxytosis/ferroptosis plays an important role in the nerve cell damage and death that is characteristic of AD as well as other neurodegenerative diseases, then it should be considered that inhibition of this pathway in the brain in vivo could be relevant throughout the course of disease progression and not only at its point of initiation.
Fisetin and CMS121
In our first attempt to use the oxytosis/ferroptosis pathway to identify new neuroprotective compounds, we screened a group of ~30 flavonoids using HT22 cells and glutamate to initiate the cell death pathway (Ishige et al., 2001b). We identified the mechanisms underlying their protective effects and found that only two, fisetin and quercetin, were also able to maintain GSH levels in the presence of oxidative stress, indicating that this is not a common property of flavonoids. Since GSH loss is the critical step in the classical oxytosis/ferroptosis pathway, we decided to pursue further studies on fisetin. These studies demonstrated that fisetin has both direct antioxidant activity and maintains the levels of GSH under conditions of stress by inducing the transcription factors, Nrf2 and ATF4 (Ehren and Maher, 2013). Moreover, fisetin was shown to facilitate long term potentiation (LTP) both in hippocampal slices (Maher et al., 2006) and in mouse brains (He et al., 2018) and oral administration of fisetin promoted learning and memory in mice using the object recognition test (Maher et al., 2006). Fisetin is also effective in two different models of stroke (Gelderblom et al., 2012; Maher et al., 2007). Using three different models of HD (mutant huntingtin-expressing PC12 cells, mutant huntingtin-expressing Drosophila and the R6/2 mouse) we found that fisetin was able to reduce the impact of mutant huntingtin in each of these disease models (Maher et al., 2011). In addition, fisetin prevents learning and memory deficits in APPswe/PS1dE9 (huAPP/PS1) double transgenic AD mice (Currais et al., 2014) and rapidly aging SAMP8 mice (Currais et al., 2018). Fisetin is an effective inhibitor of all three LOXs with IC50 values as good or better than those of known LOX inhibitors (Maher, 2015). Importantly, in the mouse model of AD, fisetin was found to significantly reduce the levels of pro-inflammatory 5-HETE and 12-HETE, the primary metabolites of 5LOX and 12/15LOX, respectively, in the brains of the AD mice (Currais et al., 2014). These results clearly indicate that fisetin is able to inhibit both 5LOX and 12LOX not only in vitro but in vivo as well, at least in the context of AD. Thus, fisetin targets two of the major steps in oxytosis/ferroptosis both in cell culture and in vivo. Fisetin is currently in clinical trials for age-associated disorders including frailty, osteoporosis and chronic kidney disease (NCT03325322, 03430037, 03675724, 04210986 and 04313634).
Fisetin’s relatively high EC50 in cell based assays (2–5 μM) as well as its low lipophilicity (cLogP 1.24), high tPSA (107Å) and high number of hydrogen bond donors (HBD = 5) suggested that there was room for medicinal chemical improvement. Using structure-activity relationship (SAR)-driven iterative chemistry, we synthesized more than 160 derivatives of fisetin based on several different chemical scaffolds (Chiruta et al., 2012). We used a multi-tiered approach to screening that included protection against oxytosis/ferroptosis as our primary assay to identify fisetin derivatives with significantly enhanced neuroprotective activity while at the same time maintaining other key activities (Chiruta et al., 2012). While all of the fisetin derivatives had improved medicinal chemical properties more consistent with those of known CNS drugs, only about 20 had greatly enhanced neuroprotective activity against multiple insults including oxytosis/ferroptosis (Chiruta et al., 2012). Absorption, distribution, metabolism and excretion (ADME) and pharmacokinetic studies on the six most promising derivatives showed that one, CMS121, had peak brain levels following a single oral dose of 20 mg/kg that greatly exceeded its average EC50 in the in vitro neuroprotection assays as well as good oral bioavailability. Based on these results, CMS121 was tested in rapidly aging SAMP8 mice (Currais et al., 2019). We believe that SAMP8 mice are the very best animal model for the most prevalent, sporadic form (>97% of cases) of AD because this is the only mouse AD model that is not biased toward the genetic form of the disease and that manifests the pathology (Aβ, tau phosphorylation, memory loss, vascular damage) and sequence of disease progression similar to human AD (Currais et al., 2015; Pallas, 2012). We used a rigorous paradigm wherein two groups of mice were aged to 9 months and then fed with control or CMS121 diet for an additional 4 months (13 month old mice) while another group of 9 month old mice was used as a control group (Currais et al., 2019). We found that CMS121 reduced metabolic and gene transcription markers of aging in SAMP8 brains while preserving cognition when administered at advanced stages of the aging process, demonstrating that it delays molecular aspects of aging and cognitive loss in this model. Very recently, we reported on the effects of CM121 in the APPswe/PS1ΔE9 transgenic mouse model of AD (Ates et al., 2020). Similar to the SAMP8 study, the mice were aged to 9 months, a time when they already show cognitive impairment (Currais et al., 2014), and then treated for 3 months with CMS121. CMS121 treatment reduced lipid peroxidation and neuroinflammation in the brains of the mice while alleviating cognitive decline as assessed in multiple behavioral tests. We further showed that the beneficial effects of CMS121 in this model were associated with its ability to partially inhibit the lipid biosynthetic enzyme, fatty acid synthase (FASN) thereby reducing the levels of PUFAs, the targets of lipid peroxidation. In cells, knockdown of FASN phenocopied the protective effects of CMS121 against both oxytosis/ferroptosis and inflammation. CMS121 is currently in the IND process.
CNB001 and J147
Another sequence of drug discovery based upon the oxytosis/ferroptosis pathway started with the natural product curcumin. This Indian spice was being studied as an AD therapeutic by Greg Cole and Sally Frautschy (Yang et al., 2005) and became of interest to us when it was shown to have an EC50 of 5 micromolar in the HT22 oxytosis assay but lacked neurotrophic activity. We had identified another compound, cyclohexyl bisphenol A, that had neurotrophic activity but minimal anti-oxytotic activity. Because of the structures of these molecules, it was possible to make combinatorial libraries incorporating features of both, and based upon both the oxytosis and neurotrophic activity assays, a pyrazole derivative of curcumin was selected that had both activities, resulting in CNB001 (Liu et al., 2008). Because of its broad neuroprotective activity, this compound was tested for therapeutic efficacy in multiple animal disease models and its molecular target identified. CNB001 was shown to reduce disease pathology in mouse models of traumatic brain injury (Wu et al., 2011), lung inflammation (Narumoto et al., 2012), PD (Jayaraj et al., 2014) and AD (Valera et al., 2013). In addition, it dramatically improved both pathological and behavioral outcomes in ischemic stroke models in both rabbits and monkeys (Lapchak et al., 2019; Lapchak et al., 2011). However, because of its structure, CNB001 proved difficult to move further toward the clinic and additional chemistry and screening was initiated.
SAR-driven iterative chemistry initially based exclusively on the HT22 oxytosis assay for screening was used to develop a 250 compound library with structural relationships to CNB001 but lacking hydroxyl groups. These results suggested that only two aromatic rings with methoxy and methyl groups in defined positions and a nitrogen-nitrogen containing bridge related to the pyrazole in CNB001 were necessary for activity. A single pot library of these compounds was made and screened initially in the HT22 oxytosis assay. The most active compound was then isolated and its structure determined by NMR (Chen et al., 2011). This compound, J147, has a low nanomolar EC50 in the oxytosis/ferroptosis assay as well as in the other assays in our screening platform. It was then put into multiple rodent models of memory and AD (Chen et al., 2011; Currais et al., 2015; Prior et al., 2013), tested for safety in animals for IND approval and is currently in Phase 1 clinical trials (NCT03838185). The target was identified as ATP5A, a catalytic subunit of the mitochondrial ATP synthase complex (Goldberg et al., 2018).
Using oxytosis/ferroptosis-based drug candidates in a chemical biology approach to uncover aging and disease pathways
Having identified a set of compounds that inhibit oxytosis/ferroptosis and are protective in animal models of AD, our focus has evolved towards understanding how these compounds can be used in a chemical biology approach to identify specific age-related molecular pathways with therapeutic potential for AD (Figure 5).
Figure 5:
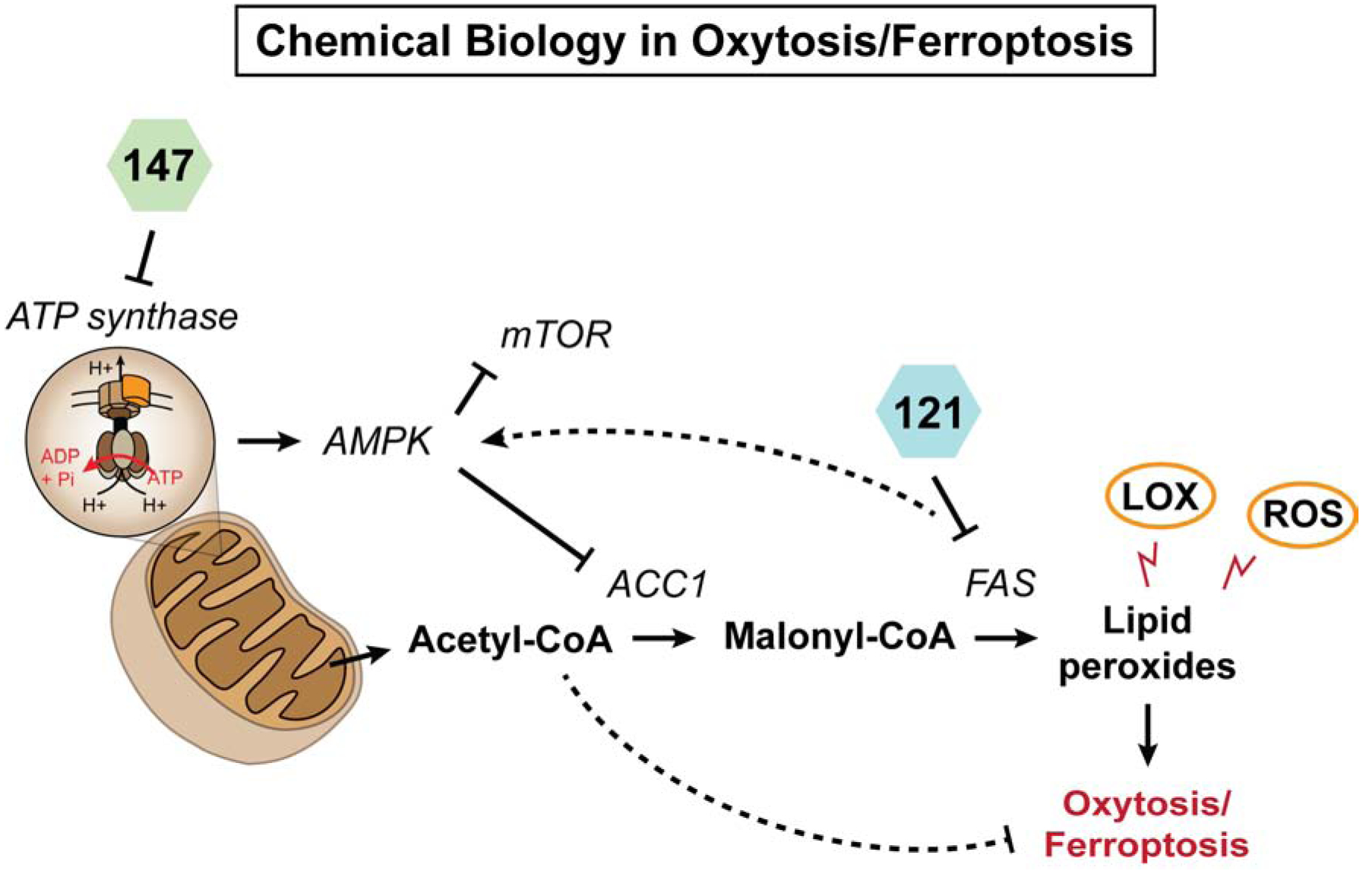
Our chemical biology approach targeting oxytosis/ferroptosis.
To address this, large data omic analyses were carried out and a set of bioinformatics tools was developed to identify the relevant pathways as defined by their modulation by compounds that have therapeutic efficacy in the disease model (Currais et al., 2015; Currais et al., 2019). By integrating different types of molecular parameters (genes, metabolites and proteins), we identified mitochondrial acetyl-CoA as a central metabolite underlying the neuroprotective actions of CMS121 and J147. The regulation of acetyl-CoA metabolism by the compounds was associated with a preservation of mitochondrial homeostasis and histone acetylation in two distinct symptomatic mouse models of AD at an epitope that is required for memory and is downregulated in the brains of AD patients (Currais et al., 2019).
Most importantly, the effects on acetyl-CoA metabolism were a result of the inhibition of acetyl-CoA carboxylase 1 (ACC1), an enzyme responsible for the conversion of acetyl-CoA into malonyl-CoA, via activation of 5’ AMP-activated protein kinase (AMPK) (Currais et al., 2019). A decrease in free PUFAs was also observed (Currais et al., 2019). Direct inhibition or knockdown of ACC1 also led to increased acetyl-CoA levels and was itself very neuroprotective against oxytosis/ferroptosis (Currais et al., 2019). The identification of the AMPK/ACC1 axis as a new molecular regulator of oxytosis/ferroptosis, whether by directly decreasing the levels of free PUFAs or increasing the levels of acetyl-CoA, warrants further investigation. While these experiments are required to clarify the value of ACC1/acetyl-CoA as a therapeutic target for aging and dementia, activation of AMPK has largely been described in the literature as a canonical target of a diverse range of anti-aging approaches (Burkewitz et al., 2014; Wang et al., 2019). In the case of J147, AMPK activation was shown to occur via its action on the mitochondrial ATP synthase (Goldberg et al., 2018). In the case of CMS121, it is likely through its action on FASN (Zhou et al., 2007).
Interestingly, a recent study from Lee et al (Lee et al., 2020) confirmed these findings showing that engagement of the AMPK/ACC1 pathway is protective against oxytosis/ferroptosis in renal ischemia-reperfusion injury. Thus, using oxytosis/ferroptosis as our primary screen not only did we identify potential new AD drug candidates but we are also finding new targets for protection against oxytosis/ferroptosis.
Conclusions
Oxytosis was first described in nerve cells over 30 years ago as a regulated cell death pathway involving GSH depletion, LOX activation, ROS accumulation and mitochondrial and calcium dysregulation. It was subsequently re-described in transformed fibroblasts and re-named ferroptosis. All of the pathophysiological changes that occur in cells undergoing oxytosis/ferroptosis are also seen in the brain in multiple neurodegenerative diseases. Thus, it has proven to be an invaluable model both for identifying novel drug candidates for the treatment of these diseases and for characterizing novel therapeutic pathways. However, despite these results, conclusive evidence for the role of oxytosis/ferroptosis in these diseases is still needed. The ongoing human clinical studies with our compounds should provide further clarification. Importantly, the role of oxytosis/ferroptosis appears to go beyond the brain, and its value for the understanding of other diseases of old age and improvement of human health is a challenge that lies ahead.
Highlights:
The oxytosis and ferroptosis cell death pathways share many characteristics.
The adverse effects of oxytosis/ferroptosis overlap with neurodegenerative diseases.
Oxytosis/ferroptosis can be used to develop therapies for neurodegenerative diseases.
Such therapies can also be used to identify novel protective molecular pathways.
Acknowledgements:
This review is dedicated to the memory of Dave Schubert. He taught us the value of persistence. This work was supported by grants from NIH (RO1 AG046153 and RF1 AG054714 to PM and DS, and R41 AI104034 to PM), the Edward N. & Della Thome Memorial Foundation (PM), the Shiley-Marcos Alzheimer’s Disease Research Center at University of California San Diego (AC) and an Innovation Award from the Salk Institute (AC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: DS is an unpaid advisor for Abrexa Pharmaceuticals, a company working on the development of J147 for AD therapy. The Salk Institute holds the patents for CMS121 (US9744164) and J147 (US8779002).
References
- Adam-Vizi V, and Tretter L (2013). The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem Int 62, 757–763. [DOI] [PubMed] [Google Scholar]
- Adams JD, Klaidman LK, Odunze IN, Shen HC, and Miller CA (1991). Alzheimer’s and Parkinson’s disease. Brain levels of glutathione, glutathione disulfide and vitamin E. Mol Chem Neuropathol 14, 213–226. [DOI] [PubMed] [Google Scholar]
- Agathocleous M, and Harris WA (2013). Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol 23, 484–492. [DOI] [PubMed] [Google Scholar]
- Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, and Methner A (2010). Mechanisms of oxidative glutamate toxicity: The glutamate/cystine antiporter system x(c)- as a neuroprotective drug target. CNS Neurol Disord Drug Targets 9, 373–382. [DOI] [PubMed] [Google Scholar]
- Alvarez J, Alvarez-Illera P, Garcia-Casas P, Fonteriz RI, and Montero M (2020). The role of calcium signaling in aging and neurodegeneration: Insights from Caenorhabditis elegans models. Cells 9, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, and Scheff SW (2010). Oxidative stress in the progression of Alzheimer’s disease in the frontal cortex. J Neuropathol Exp Neurol 69, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong MM, Freedman CJ, Jung JE, Zheng YL, Kalyanaraman C, Jacodson MP, Simeonov A, Maloney DJ, van Leyen K, Jadhav A, et al. (2016). A potent and selective inhibitor targeting human and murine 12/15 LOX. Bioorg Med Chem 24, 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ates G, Goldberg J, Currais A, and Maher P (2020). CMS121, a fatty acid synthase inhibitor, protects against excess lipi peroxidation and inflammation and alleviates cognitive loss in a transgenic model of Alzheimer’s disease. Redox Biol in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlante A, de Bari L, Bobba A, and Amadoro G (2017). A disease with a sweet tooth: exploring the Warburg effect in Alzheimer’s disease. Biogerontol 18, 301–319. [DOI] [PubMed] [Google Scholar]
- Ayala A, Munoz MF, and Arguelles S (2014). Lipid peroxidation: Production, metabolism and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014, 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, and Hammond CL (2009). Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem 390, 191–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Allaman I, and Magistretti PJ (2011). Brain energy metabolism:focus on astrocyte-neuron metabolic copperation. Cell Metab 14, 724–738. [DOI] [PubMed] [Google Scholar]
- Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, and Villar AM (2008). Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from mild cognitive impairment. Free Rad Res 42, 162–170. [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong BCK, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumezbeur F, Mason GF, de Graaf RA, Behar KL, Cline GW, Shulman GI, Rothman DL, and Petersen KF (2010). Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab 30, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, and Navarro A (2008). Brain mitochondrial dysfunction in aging IUBMB Life 60, 308–314. [DOI] [PubMed] [Google Scholar]
- Bradley-Whitman MA, and Lovell MA (2015). Biomarkers of lipid peroxidation in Alzheimer’s disease (AD): an update. Arch Toxicol 89, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitzig M, Bhimineni C, Lockey N, and Kolliputi N (2016). 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Amer J Physiol Cell Physiol 311, C537–C543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune B, and Ullrich V (1991). 12-Hydroperoxyeicosatetraenoic acid inhibits main platelet functions by activation of soluble guanylate cyclase. Mol Pharmacol 39, 671–678. [PubMed] [Google Scholar]
- Burkewitz K, Zhang Y, and Mair WB (2014). AMPK at the nexus of energetics and aging. Cell Metab 20, 10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabon L, Galan-Maol P, Bouharrour A, Delavallee L, Brunelle-Navas M-N, Lorenzo HK, Gross A, and Susin SA (2012). BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death Differ 19, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, Li S, Basu U, Tian J, and Zimmerman MC (2013). Mitochondrial-localized NADPH oxidase 4 is a soure of superoxide in angiotensin II-stimulated neurons. Amer J Physiol Heart Circ Physiol 305, H19–H28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2006). Mitochondria: dynamic organelles in disease, aging and development. Cell 125, 1241–1252. [DOI] [PubMed] [Google Scholar]
- Chandran R, Kumar M, Kesavan L, Jacob RS, Gunasekaran S, Lakshmi S, Sadasivan C, and Omkumar RV (2019). Cellular calcium signaling in the aging brain. J Chem Neuroanat 95, 95–114. [DOI] [PubMed] [Google Scholar]
- Chen M, He H, Zhan S, Krajewski S, Reed JC, and Gottlieb RA (2001). Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem 276, 30724–30728. [DOI] [PubMed] [Google Scholar]
- Chen Q, Prior M, Dargusch R, Roberts A, Riek R, Eichmann C, Chiruta C, Akaishi T, Abe K, Maher P, et al. (2011). A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease. PLoS ONE 6, e27865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Rajagopalan S, Butterfield DA, and Anderson JK (2006). In vitro and in vivo neuroprotection by g-glutamylcysteine ethyl ester against MPTP: Relevance to the role of glutathione in Parkinson’s disease. Neurosci Lett 402, 137–141. [DOI] [PubMed] [Google Scholar]
- Chiruta C, Schubert D, Dargusch R, and Maher P (2012). Chemical modification of the multi-target neuroprotective compound fisetin. J Med Chem 55, 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. [DOI] [PubMed] [Google Scholar]
- Cole-Ezea P, Swan D, Shanley D, and Hesketh J (2012). Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining phosphorylation complexes in gut epithelial cells. Free Rad Biol Med 53, 488–497. [DOI] [PubMed] [Google Scholar]
- Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, and Stockwell BR (2018). Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev 32, 602–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini LC, Barr LJ, Vogel JL, and Henderson ST (2008). Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci 9, S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisponi G, Nurchi VM, Crespo-Alonso M, Sanna G, Zoroddu MA, Alberti G, and Biesuz R (2015). A speciation stuy on the perturbing effects of iron chelators on the homeostasis of essential metal ions. PLoS ONE 10, e0133050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, Castellano A, Pifferi F, Bocti C, Paquet N, et al. (2011). Brain fuel matabolism, aging and Alzheimer’s disease. Nutrition 27, 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A (2015). Ageing and inflammation-A central role for mitochondria in brain health and disease. Ageing Res Rev 21, 30–42. [DOI] [PubMed] [Google Scholar]
- Currais A, Farrokhi C, Dargusch R, Armando A, Quehenberger O, Schubert D, and Maher P (2018). Fisetin reduces the impact of aging on behavior and physiology in the rapidly aging SAMP8 mouse. J Gerentol A Biol Sci Med Sci 73, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A, Goldberg J, Farrokhi C, Chang M, Prior M, Dargusch R, Daugherty D, Armando A, Quehenberger O, Maher P, et al. (2015). A comprehensive multiomics approach toward understanding the relationship between aging and dementia. Aging 7, 937–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A, Huang L, Goldberg J, Petrascheck M, Ates G, Pinto-Duarte A, Shokhirev M, Schubert D, and Maher P (2019). Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 8, e47866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A, and Maher P (2013). Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid Redox Signal 19, 813–822. [DOI] [PubMed] [Google Scholar]
- Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, and Maher P (2014). Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 13, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JB, and Maher P (1994). Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell lines. Brain Res 652, 169–173. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: An iron-dependent form of non-apoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolga AM, POppermann S, Richter M, Honrath B, Neitemeier S, Jelinek A, Ganjam G, and Culmsee C (2018). Molecular mechanisms underlying oxytosis In Apoptosis and Beyond: The Many Ways Cells Die, Radosevich J, ed. (Hoboken, NJ: Wiley; ). [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, Costa da Silva M, Ingold I, Grocin AG, da Silva TNX, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, and Stockwell BR (2003). Identification of genotyps-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. [DOI] [PubMed] [Google Scholar]
- Droge W, Kinscherf R, Hildebrandt W, and Schmitt T (2006). The deficit in low molecular weight thiols as a target for antiageing therapy. Curr Drug Targets 7, 1505–1512. [DOI] [PubMed] [Google Scholar]
- Ehren JL, and Maher P (2013). Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem Pharmacol 85, 1816–1826. [DOI] [PubMed] [Google Scholar]
- Elphick LM, Hawat M, Toms NJ, Meinander A, Mikhailov A, Eriksson JE, and Kass GEN (2008). Opposing roles for caspase and calpain proteases in L-glutamate-induced oxidative neurotoxicity. Toxicol Appl Pharmacol 232, 258–267. [DOI] [PubMed] [Google Scholar]
- Emir UE, Raatz S, McPherson S, Hodges JS, Torkelson C, Tawfik P, White T, and Terpstra M (2011). Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed 24, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, and Stockwell BR (2018). Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 16, e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Rosca M, fan Y, Hu Y, Feng P, Lee H-G, Monnier VM, and Fan X (2017). Gclc deficiency in mouse CNS causes mitochondrial damage and neurodegeneration. Hum Mol Gen 26, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Fernandez S, Bobo-Jimenez V, Requejo-Aguilar R, SGonzalez-Fernandez S, Resch M, Carabias-Carrasco M, Ros J, Almeida A, and Bolanos JP (2018). Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol 38, 5415–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, and Terzic A (2012). Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Yang F, Che J, Han Y, Chen N, Bak DW, Lai S, Xie X, Weerapana E, and Wang CX (2018). Selenium-encoded isotopic signature target profiling. ACS Cent Sci 4, 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Yi JJ, Zhu JW, Minikes AM, Monian P, Thompson CB, and Jiang X (2019). Role of mitochondria in ferroptosis. Mol Cell 73, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, and Stockwell BR (2017). Lipid peroxidation in cell death. Biochem Biophys Res Commun 482, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Lewerenz J, Birkenmayer G, Orozco D, Ludewig P, Thundyil J, Arumugam TV, Gerloff C, Tolosa E, et al. (2012). The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J Cereb Blood Flow Metab 32, 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustarini D, Dalle-Donne I, Lorenzini S, Milzani A, and Rossi R (2006). Age-related influence on thiol, disulfide and protein-mixed disulfide levels in human plasma. J Gerentol A Biol Sci Med Sci 61, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Goldberg J, Currais A, Ates G, Huang L, Shokhirev M, Maher P, and Schubert D (2020). Targeting of intracellular Ca+2 stores as a therapeutic strategy against age-related neurotoxicities. Aging Mech Dis in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J, Currais A, Prior M, Fischer WH, Chiruta C, Ratliff E, Daughtery D, Dargusch R, Finley D, Esparza-Molto PB, et al. (2018). The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 17, 12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout PW, Buckley AR, Simms CR, and Bruchovsky N (2001). Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the Xc- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640. [DOI] [PubMed] [Google Scholar]
- Gu M, D. OA, Toffa SE, Cooper JM, Dexter DT, Jenner P, Marsden CD, and Schapira AH (1998). Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J Neurol Sci 158, 24–29. [DOI] [PubMed] [Google Scholar]
- Ha JS, Lim HM, and Park SS (2010). Extracellular hydrogen peroxide contributes to oxidative glutamate toxicity. Brain Res 1359, 291–297. [DOI] [PubMed] [Google Scholar]
- Hagmann W, Kagawa D, Renaud C, and Honn KV (1993). Activation and protein distribution of 12-lipoxygenase in HEL cells: induction of membrane-association by phorbol ester TPA, modulation of activity by glutathione and 13-HPODE and Ca+2-dependent translocation to membranes. Prostaglandins 46, 471–477. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, and Attwell D (2012). Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- He WB, Abe K, and Akaishi T (2018). Oral administration of fisetin promotes the induction of hippocampal lonr-term potentiation in vivo. J Pharmacol Sci 136, 42–45. [DOI] [PubMed] [Google Scholar]
- Henke N, Albrecht P, Bouchachia I, Ryazantseva M, Knoll K, Lewerenz J, Kaznacheyeva E, Maher P, and Methner A (2013). The plasma membrane channel ORA1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis 4, e470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y, Yamada C, Ito Y, Yamamoto S, Nagase H, Oh-hashi K, Kiuchi K, Suzuki H, Sawada M, and Furuta K (2018). Novel oxindole derivatives prevent oxidative stress-induced cell death in mouse hippocampal HT22 cells. Neuropharmacol 135, 242–252. [DOI] [PubMed] [Google Scholar]
- Imai H, and Nakagawa Y (2003). Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Rad Biol Med 34, 145–169. [DOI] [PubMed] [Google Scholar]
- Ishige K, Chen Q, Sagara Y, and Schubert D (2001a). The activation of dopamine D4 receptors inhibits oxidative stress-induced nerve cell death. J Neurosci 21, 6069–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishige K, Schubert D, and Sagara Y (2001b). Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med 30, 433–446. [DOI] [PubMed] [Google Scholar]
- Jayaraj RL, Elangovan N, Dhanalakshmi C, Manivasagam T, and Essa MM (2014). CNB-001, a novel pyrozole derivative mitigates motor impairments associated with neurodegeneration via suppression of neuroinflammatory and apoptotic response in experimental Parkinson’s disease mice. Chem Biol Interact 220, 149–157. [DOI] [PubMed] [Google Scholar]
- Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, Diederich WE, and Culmsee C (2018). Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Rad Biol Med 117, 45–57. [DOI] [PubMed] [Google Scholar]
- Joshi YB, Giannopoulos PF, and Practico D (2015). The 12/15-lipoxygenase as an emerging therapeutic target for Alzheimer’s disease. Trends Pharmacol Sci 36, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi YB, and Practico D (2015). The 5-lipoxygenase pathay: oxidative and inflammatory contributions to the Alzheimer’s disease phenotype. Front Cell Neurosci 8, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, P. AJ, Doll S, St. Croix CM, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Tiziani S, Park G, Kaul M, and Paternostro G (2014). Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nature Commun 5, 3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, St. Croix CM, Mikulska-Ruminiska K, Liu B, Shrivastava I, et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nature Cell Biol 16, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelson E, Bogdanovic N, Garlind A, Winblad B, Zilmer K, Kullisaar T, Vihalemm T, Kairane C, and Zilmer M (2001). The cerebrocortical areas in normal brain aging and in Alzheimer’s disease: noticeable differences in the lipid peroxidation level and in antioxidant defense. Neurochem Res 26, 353–361. [DOI] [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AH, and Currie AR (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi MS, Han D, and Packer L (2000). Antioxidants and herbal extracts protect HT-4 neuronal cells against glutamate-induced cytotoxicity. Free Rad Res 32, 115–124. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Sato M, Kasakoshi T, Tusutsui T, Sugimoto M, Osaki M, Okada F, Igarashi K, Hiratake J, Homma T, et al. (2015). Cystathionine is a novel substrate of cystine/glutamate transporter: implciations for immune function. J Biol Chem 290, 8778–8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, and Takeuchi T (2010). Constitutive reactive oxygen species generation from autophagosomes/lysosomes in neuronal oxidative toxicity. J Biol Chem 285, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Banthiya S, and van Leyen K (2015). Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta 1851, 308–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landshamer S, Hoehn M, Barth N, Duvezin-Caubet S, Schwake G, Tobaben S, Kazhdan I, Becattini B, Zahler S, Vollmar A, et al. (2008). Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ 15, 1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, and Martin W (2010). The energetics of genome complexity. Nature 467, 929–934. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Boitano PD, Bombien R, Cook DJ, Doyan S, Lara JM, and Schubert D (2019). CNB-001, a pleiotropic drug is efficacious in embolized agyrencephalic New Zealand white rabbits and ischemic gyrencephalic cynomolgus monkeys. Exp Neurol 313, 98–108. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Schubert DR, and Maher PA (2011). Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke. J Neurochem 116, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al. (2020). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 22, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Bai T, and Sun Y (2019). Mechanisms of ferroptosis and relations with regulated cell death: A review. Front Physiol 10, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Ates G, Methner A, Conrad M, and Maher P (2018). Oxytosis/ferroptosis-(Re-)emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases on the central nervous system. Front Neurosci 12, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Baxter P, Kassubek R, Albrecht P, Van Liefferinge J, Westhoff MA, Halatsch ME, Karpel-Massler G, Meakin PJ, Hayes JD, et al. (2014). Phosphoinositide 3-kinases upregulate system xc- via eukaryotic transcription factor 2α and activating transcription factor 4-A pathway active in glioblastoma and epilepsy. Antioxid Redox Signal 20, 2907–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas P, Massie A, Smolders I, Methner A, Pergande M, et al. (2013). The cystine/glutamate antiporter system x(c)- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18, 522–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Letz J, and Methner A (2003). Activation of stimulatory heteromeric G proteins increases glutathione and protects neuronal cells against oxidative stress. J Neurochem 87, 522–531. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, and Maher P (2009). Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem 284, 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, and Schubert D (1997a). Requirement for cGMP in nerve cell death caused by glutathione depletion. J Cell Biol 139, 1317–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, and Schubert D (1997b). A role of 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Lin MT, and Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Liu J, Li L, and Suo WZ (2009). HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci 84, 267–271. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dargusch R, Maher P, and Schubert D (2008). A broadly neuroprotective derivative of curcumin. J Neurochem 105, 1336–1345. [DOI] [PubMed] [Google Scholar]
- Liu Y, and Schubert DR (2009). The specificity of neuroprotection by antioxidants. J Biomed Res 16, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang W, Li Y, Xiao Y, Cheng JL, and Jia J (2015). The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull 38, 1234–1239. [DOI] [PubMed] [Google Scholar]
- Maher P (2005). The effects of stress and aging on glutathione metabolism. Ageing Res Rev 4, 288–314. [DOI] [PubMed] [Google Scholar]
- Maher P (2015). How fisetin reduces the impact of age and disease on CNS function. Front Biosci 7, 58–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P (2017). Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: implications for age-related neurodegenerative diseases. Free Rad Biol Med 115, 92–104. [DOI] [PubMed] [Google Scholar]
- Maher P, Akaishi T, and Abe K (2006). Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc Natl Acad Sci USA 103, 16568–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, Dargusch R, Bodai L, Gerard P, Purcell JM, and Marsh JL (2011). ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Gen 20, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, and Davis JB (1996). The role of monoamine metabolism in oxidative glutamate toxicity. J Neurosci 16, 6394–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, Salgado KF, Zivin JA, and Lapchak PA (2007). A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res 1173, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, and Schubert D (2000). Signaling by reactive oxygen species in the nervous system. Cell Mol Life Sci 27, 1287–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, van Leyen K, Dey PN, Honrath B, Dolga A, and Methner A (2018). The role of Ca+2 in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 70, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makar TK, Cooper AJ, Tofel-Grehl B, Thaler HT, and Blass JP (1995). Carnitine, carnitine acetyltransferase, and glutathione in Alzheimer brain. Neurochem Res 20, 705–711. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Saharan S, Tripathi M, and Murari G (2015). Brain glutathione levels-a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol Psychiatry 78, 702–710. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Shukla D, Tripathi M, and Ersland L (2019). Cognitive improvement with glutathione supplement in Alzheimer’s disease: a way forward. J Alzheimer’s Dis 68, 531–535. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Moosmann B, Hermann H, Lutz B, and Behl C (2002). Neuroprotective properties of cannibinoids agaisnt oxidative stress: role of the cannabinoid receptor CB1. J Neurochem 80, 448–456. [DOI] [PubMed] [Google Scholar]
- Martin HL, and Teisman P (2009). Glutathione-a review on its role and significance in Parkinson’s disease. FASEB J 23, 3263–3272. [DOI] [PubMed] [Google Scholar]
- Mischley LK, Standish LJ, Weiss NS, Padowski JM, Kavanagh TJ, White CC, and Rosenfeld ME (2016). Glutathione as a biomarker in Parkinson’s disease: Associations with aging and disease severity. Oxid Med Cell Longev 2016, 9409363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto M, Murphy TH, Schnaar RL, and Coyle JT (1989). Antioxidants protect against glutamate-induced cytotoxicity in a neuronal cell line. J Pharmacol Exper Ther 250, 1132–1140. [PubMed] [Google Scholar]
- Morimoto BH, and Koshland DE (1990). Induction and expression of long and short term neurosecretory potentiation in a neural cell line. Neuron 5, 875–880. [DOI] [PubMed] [Google Scholar]
- Murphy TH, and Baraban JM (1990). Glutamate toxicity in immature cortical neurons precedes development of glutamate receptor currents. Brain Res 57, 146–150. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Malouf AT, Sastre A, Schnaar RL, and Coyle JT (1988). Calcium-dependent glutamate cytotoxicity in a neuronal cell line. Brain Res 444, 325–332. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, and Coyle JT (1989). Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, and Coyle JT (1990). Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J 4, 1624–1633. [PubMed] [Google Scholar]
- Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, and McLaughlin B (2006). Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem 97, 1301–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Katagiri Y, Oh-hashi K, Geller HM, and Hirata Y (2020). Reduced sulfation enhanced oxytosis and ferroptosis in mouse hippocampal HT22 cells. Biomolecules 10, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumoto O, Matsuo Y, Sakaguchi M, Shoji S, Yamashita N, Schubert D, Abe K, Horiguchi K, Nagase T, and Yamashita N (2012). Suppressive effects of a pyrozole of curcumin on airway inflammation and remodeling. Exp Mol Pathol 93, 18–25. [DOI] [PubMed] [Google Scholar]
- Navarro A, and Boveris A (2010). Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci 2012, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, Ganjam G, Dolga AM, Oppermann S, and Culmsee C (2017). BID links ferroptosis to mitochondrial cell death pathways. Redox Biol 12, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg E, Karlsson M, Korenkovska O, Szydlowski S, Silberberg G, Uhlen P, Orrenius S, and Zhivotovsky B (2010). Critical role for hyperpolarization-activated cyclic nucleotide-gated channel 2 in AIF-mediated apoptosis. EMBO J 29, 3869–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onukwufor JO, Berry BJ, and Wojtovich AP (2019). Phsyiologic implications of reactive oxygen species production by mitochondrial complex I reverse electorn transport. Antioxid 8, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango IG, Dennis J, and Khan SM (2016). Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies. Aging Dis 7, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas M (2012). Senescence-accelerated mice P8: A tool to study brain aging and Alzheimer’s disease in a mouse model. ISRN Cell Biol 2012, 917167. [Google Scholar]
- Pallast S, Arai K, Wang X, Lo EH, and van Leyen K (2009). 12/15-Lipoxygenase targets neuronal mitochondria under stress. J Neurochem 111, 882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RK, Owen A, Daniel S, Jenner P, and Marsden CD (1997). Alterations in the disttribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm 104, 661–677. [DOI] [PubMed] [Google Scholar]
- Perry TL, Yong VW, Bergeron C, GHansem S, and Jones K (1987). Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer’s disease. Ann Neurol 21, 331–336. [DOI] [PubMed] [Google Scholar]
- Prior M, Chiruta C, Currais A, Goldberg J, Ramsey J, Dargusch R, Maher PA, and Schubert D (2014). Back to the future with phenotypic screening ACS Chem Neurosci 5, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior M, Dargusch R, Ehren J, Chiruta C, and Schubert D (2013). The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimer’s Res Ther 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai L, Oon S-M, Prokai-tatrai K, Abboud KA, and Simpkins JW (2001). Synthesis and biological evaluation of 17beta-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants against oxidative stress. J Med Chem 44, 110–114. [DOI] [PubMed] [Google Scholar]
- Rae CD, and Williams SR (2017). Glutathione in the human brain: review of its roles and measurement by magnetic resonance spectroscopy. Anal Biochem 529, 127–143. [DOI] [PubMed] [Google Scholar]
- Rai G, Joshi N, Jung JE, Liu Y, Schultz L, Yasgar A, Perry S, Diaz G, Zhang Q, Kenyon V, et al. (2014). Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J Med Chem 57, 4035–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR (2020). The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol 27, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed TT (2011). Lipid peroxidation and neurodegenerative disease. Free Rad Biol Med 51, 1302–1319. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Dargusch R, Chambers D, Davis J, Schubert D, and Maher P (1998). Cellular mechanisms of resistance to chronic oxidative stress. Free Radic Biol Med 24, 1375–1389. [DOI] [PubMed] [Google Scholar]
- Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K, Moriguchi T, et al. (2005). Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem 280, 37423–37429. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, and Buettner GR (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad Biol Med 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- Schramm S, Huang B, Gunesch S, Lang F, Roa J, Hogger P, Sabate R, Maher P, and Decker M (2018). Regioselective synthesis of 7-O-esters of the flavonolignan silibinin and SARs lead to compounds with overadditive neuroprotective effects. Eur J Med Chem 146, 93–107. [DOI] [PubMed] [Google Scholar]
- Schriau MG, Dun NJ, and Bau HH (2009). Cell electrophysiology with carbon nanopipettes. ACS Nano 3, 563–568. [DOI] [PubMed] [Google Scholar]
- Schubert D, Currais A, Goldberg J, Finley K, Petrascheck M, and Maher P (2018). Geroneuroprotectors: Effective geroprotectors for the brain. Trends Pharmacol Sci 39, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Kimura H, and Maher P (1992). Growth factors and vitamin E modify neuronal glutamate toxicity. ProcNatlAcadSciUSA 89, 8264–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, and Dichgans J (2000). Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 267, 4904–4911. [DOI] [PubMed] [Google Scholar]
- Sechi G, Deledda MG, Bua G, Satta WM, Deiana GA, Pes GM, and Rosati G (1996). Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog Neuro-Psychopharmacol Biol Psychiat 20, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metabolism 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Sekhar RV, Patel SG, Guthikonda AP, Reid M, Balasubramanyam A, Taffet GE, and Jahoor F (2011). Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Amer J Clin Nutr 94, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, and Pratt DA (2018). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci 4, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, and Rao GN (2018). Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res 73, 28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbi S, Bird ED, and Blass JP (1983). Decreased pyrivate dehydrogenase complex activity in Huntington’s and Alzheimer’s brain. Ann Neurol 13, 72–78. [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Jiang X, and Gu W (2020). Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol 30, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV, Vladimirov YA, et al. (2019). Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Rad Biol Med 133, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Perluigi DA, and Butterfield DA (2013). Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer’s disease brain. Free Rad Biol Med 62, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, and Khan SM (2004). A mitochondrial cascade hypothesis for sporadic Alzheimer’s disease. Med Hypotheses 63, 8–20. [DOI] [PubMed] [Google Scholar]
- Tan S, Sagara Y, Liu Y, Maher P, and Schubert D (1998a). The regulation of reactive oxygen species production during programmed cell death. J Cell Biol 141, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Schubert D, and Maher P (2001a). Oxytosis: a novel form of programmed cell death. Curr Top Med Chem 1, 497–506. [DOI] [PubMed] [Google Scholar]
- Tan S, Somia N, Maher P, and Schubert P (2001b). Regulation of antioxidant metabolism by translation initiation factor 2a. J Cell Biol 152, 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Wood M, and Maher P (1998b). Oxidative stress in nerve cells induces a form of cell death with characteristics of both apoptosis and necrosis. J Neurochem 71, 95–105. [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Vanden Berghe T, Vandenbeele P, and Kroemer G (2019). The molecular machinery of regulated cell death. Cell Res 29, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh O, Sen CK, Roy S, and Packer L (2000). Cellular and mitochondrial changes in glutamate-induced HT4 neuronal cell death. Neurosci 97, 531–541. [DOI] [PubMed] [Google Scholar]
- Tong BCK, Wu AJ, Li M, and Cheung KH (2018). Calcium signaling in Alzheimer’s disease and therapies. Biochim Biophys Acta 1865. [DOI] [PubMed] [Google Scholar]
- Uchida K (2017). HNE as an inducer of COX2. Free Rad Biol Med 111, 169–172. [DOI] [PubMed] [Google Scholar]
- Valera E, Dargusch R, Maher PA, and Schubert D (2013). Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J Neurosci 33, 10512–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Duvoisin RM, Engelhardt H, and Wiedmann M (1998). A function for lipoxygenase in programmed organelle degradation. Nature 395. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee S-R, Jin G, Arai K, and Lo EH (2006). Baicalein and 12/15 lipoxygenase in the ischemic brain. Stroke 37, 3014–3018. [DOI] [PubMed] [Google Scholar]
- Voet D, and Voet JG (1990). Biochemistry (New York: John Wiley and Sons; ). [DOI] [PubMed] [Google Scholar]
- Wallace DC (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: q dawn for evolutionary medicine. Annu Rev Genomics Hum Genet 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zimmerman HR, and Ma TP (2019). Therapeutic potential of AMP-activated protein kinase in Alzheimer’s disease. J Alzheimer’s Dis 68, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel SE, Tyurina YY, Zhao J, St. Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. (2017). PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Ying Z, Schubert D, and Gomez-Pinilla F (2011). Brain and spinal cord interaction: A dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabil Neural Repair 25, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav UCS, and Ramana KV (2013). Regulation of NF-kB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid Med Cell Longev 2013, 690545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, et al. (2005). Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques and reduces amyloid in vivo. J Biol Chem 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem & Biol 15, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigitkanli K, Pekcec A, Karatas H, Pallast S, Mandeville E, Joshi N, Smirnova N, Gazaryan I, Ratan RR, Witztum JL, et al. (2013). Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann Neurol 73, 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Boveris A, and Cadenas E (2014). Mitochondrial energy metabolism and redox signlaing in brain aging and neurodegeneration. Antioxid Redox Signal 20, 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Sancheti H, Liu Z, and Cadenas E (2016). Mitochondrial function in ageing: coordination with signalling and transcriptional pathways. J Physiol 594, 2025–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Razmpour R, and Bernard LP (2008). Glutathione and Parkinson’s Disease: Is this the elephant in the room? Biomed Pharmacother 62, 236–249. [DOI] [PubMed] [Google Scholar]
- Zhou W, Han WF, Landree LE, Thupari JN, Pinn ML, Bililign T, Kim EK, Vadlamudi A, Medghalchi SM, Meskini RE, et al. (2007). Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res 67, 2964–2971. [DOI] [PubMed] [Google Scholar]
- Zimniak P (2011). Relationship of electrophilic stress to aging. Free Rad Biol Med 51, 1087–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, H. L, Graham ET, Delk AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, and Schreiber SL (2020). Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundorf G, and Reiser G (2011). Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal 14, 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]