

Abstract
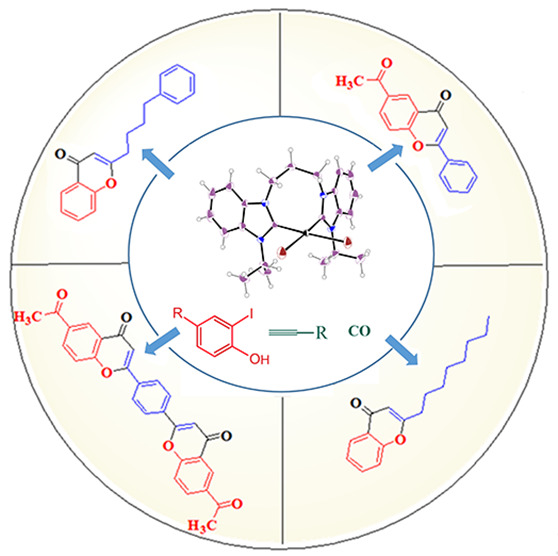
The one-pot regioselective and catalytic synthesis of bioactive chromones and flavones was achieved via phosphine-free cyclocarbonylative Sonogashira coupling reactions of 2-iodophenols with aryl alkynes, alkyl alkynes, and dialkynes. The reactions are catalyzed by new dibromidobis(NHC)palladium(II) complexes. The new bridged N,N′-substituted benzimidazolium salts (L1, L2, and L3) and their palladium complexes C1, C2, and C3 were designed, prepared, and fully characterized using different physical and spectroscopic techniques. The molecular structures of complexes C1 and C3 were determined by single-crystal X-ray diffraction analysis. They showed a distorted square planar geometry, where the Pd(II) ion is bonded to the carbon atoms of two cis NHC carbene ligands and two cis bromido anions. These complexes displayed a high catalytic activity in cyclocarbonylative Sonogashira coupling reactions with low catalyst loadings. The regioselectivity of these reactions was controlled by using diethylamine as the base and DMF as the solvent.
Introduction
Chromones are valuable compounds that have attracted the attention of researchers because of their multiple uses and their utmost importance in the medical and pharmaceutical fields.1−3 These bioactive compounds comprise important structural moieties that showed activity in the treatment of various diseases such as anticancer,3−5 anti-HI,6 asthma,7 anti-inflammatory,8 antianalgesic,3,8 antimicrobial,9 antidiabetic,10 and antioxidant11 agents (Figure 1).
Figure 1.
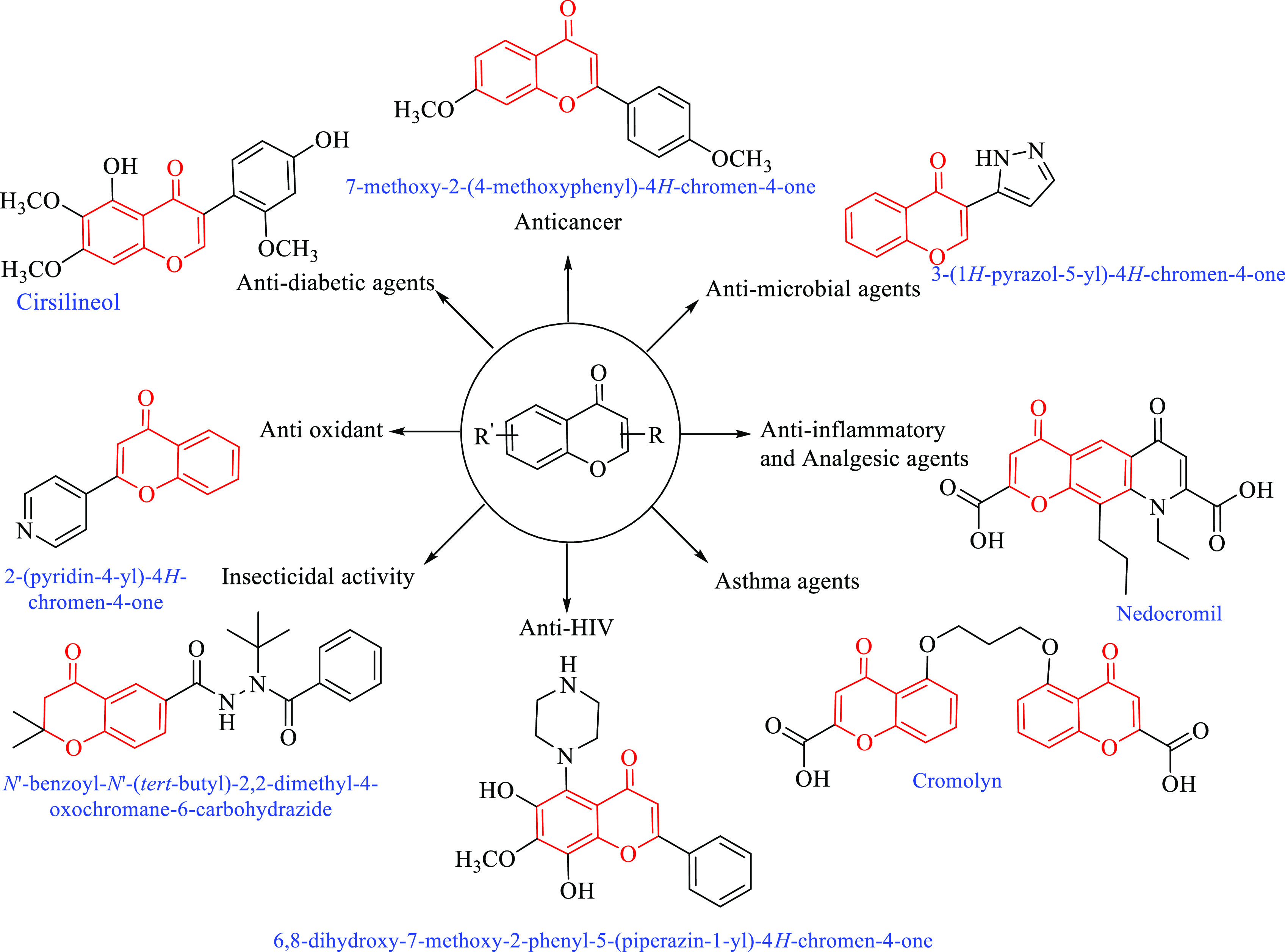
High scope of the pharmacological profile of chromone derivatives in the current literature.
Moreover, the chromones and flavones are interesting moieties in agrochemicals, insecticides, and natural products.1−5,12,13 They were extracted from different plant sources such as citrus lemon, sweet oranges, mandarin, grapefruit, ginger, Casimiroa edulis, and others.14−18
The synthesis of chromones and flavones by traditional methods usually requires several step reactions.2,13,19,20 For example, alkyl lithium reagents were used for ortho-directed metalation of methoxymethyl aryl ethers followed by the reaction with a conjugated unsaturated aldehyde; then, the allylic alcohol intermediate was oxidized with “periodinane”. The obtained ortho-allylic ketone product was heated with acetic acid to yield the corresponding chromanone that was dehydrogenated with pyrrolidone hydrotribromide in dimethyl sulfoxide.2 This method and other similar methods have drawbacks including abundant side reactions, low yields, and difficulty in separation.19−21 Friedel–Crafts acylation of suitably substituted benzoyl chlorides with alkynes was reported for chromone and flavone synthesis,22,23 but this method is associated with the use of large quantities of hazardous soluble Lewis acids (e.g., AlCl3, FeCl3, and TiCl4), which made this method as one of the most challenges for green chemistry.24,25 Copper(II)-catalyzed cyclization of 1-(2-hydroxyaryl)-3-aryl-1,3-propanedione under microwave irradiation was reported for the synthesis of functionalized flavones and chromones.26,27 Other synthetic protocols were developed in recent years such as the tandem acyl transfer-cyclization reactions catalyzed by Lewis base,28 the carbonylative coupling of p-hydroxyacetophenones with aryl bromides29 and the annulation of salicylaldehydes with benzaldehydes or alkynes.30 Recently, a new improved method for the single-step synthesis of chromones and flavones was the palladium-catalyzed cyclocarbonylative Sonogashira coupling reaction of 2-halophenols with terminal alkynes.31−37 Different palladium complexes and protocols were evaluated in the catalytic cyclocarbonylative Sonogashira coupling reactions of different alkynes with 2-iodophenols.31−33 Nevertheless, these methods were associated with some shortcomings such as the high loading of palladium catalysts and the need for the phosphine ligands.32−35,39 Researchers have continued the investigation for the synthesis and development of efficient palladium catalysts for the cyclocarbonylative Sonogashira coupling reactions in order to overcome the problems of low catalyst activity, high loading of catalyst, long reaction time, high temperature, and high CO pressure. A catalytic system including palladium complexes with mixed “phosphines” or “nitrogen”-based ligands such as pyridines, pyrroles, or imidazoles showed high activity in the synthesis of flavones and less efficacy in the production of alkyl-functionalized chromones.38 The difficulty was in the control of the regioselectivity of the reaction between six-membered ring “chromones” and five-membered ring “aurones” products. There are numerous publications that report the control of the selectivity through the type of the base, catalyst, or solvent. However, most of these protocols have considered high loading of both catalysts and phosphine ligands and also led to low yields of the corresponding chromones when terminal alkyl alkynes reacted with 2-iodophenols in the presence of carbon monoxide.33,35,40−42
Recently, the focus on the use of N-heterocyclic carbene (NHC)–metal complexes has increased. This field made a significant revolution in organometallic chemistry because of the catalytic applications in different organic transformations.43−49
The NHC ligands are known as strong σ-electrons donors, which enable them to form strong bonds with metals and to stabilize the metal complexes.50,51 Another important feature of NHCs as ligands is the flexibility to design their structures for different catalytic purposes. For instance, the electronic and steric properties can be tailored for special applications in catalysis.52,53 An interesting approach is the use of chelating bridged bis(N-heterocyclic carbene) ligands. The resulting complexes are expected to have more electron-rich centers and have higher thermodynamic stability by the chelate effect, as compared to the mono NHC counterparts. This would favor the oxidative addition step of the aryl halide substrate to the palladium(0) active species. Additionally, the steric properties can be controlled by varying the N-substituents. These bridged NHC–palladium complexes showed high activity in different applications.54−56 There are limited publications that consider Pd–NHC complexes for the synthesis of chromones and flavones via catalytic cyclocarbonylation of ortho-functionalized aryl halides with terminal alkynes. Nevertheless, these palladium complexes were active with aryl alkynes but showed lower catalytic activity with terminal alkyl alkynes.38
Previously, we have reported the synthesis and characterization of new mixed ligand dibromido and diiodido-palladium(II)–NHCs–pyridine (Br2–Pd–NHC–Py) and (I2–Pd–NHC–Py) complexes and their catalytic applications in the carbonylative Sonogashira coupling reactions and carbonylative Suzuki–Miyaura coupling reactions of aryl iodides with different alkynes or boronic acids in the presence of CO.57−59
In this work, we report the results of the regioselective synthesis of chromones via the one-pot cyclocarbonylative Sonogashira coupling reactions of 2-iodophenols with aryl alkynes, alkyl alkynes, and dialkynes catalyzed by the active-bridged bis(NHC)–palladium(II) catalysts C1, C2, and C3. We have succeeded to crystallize the new palladium complexes C1 and C3 and characterize them by single-crystal X-ray diffraction analysis. Also, the study of the steric and electronic effects was conducted using various benzimidazole N-substituents. In addition, we report the catalytic synthesis of the new compound, 2,2′-(1,4-phenylene)-bis(6-acetyl-4H-chromen-4-one) (8c).
Results and Discussion
Synthesis
N-alkylation reactions of benzimidazole with alkyl bromides (2-bromopropane, benzyl bromides) using potassium hydroxide as the base were conducted efficiently to produce 1-alkyl benzimidazoles (S1 and S2) in very good yields (Scheme 1). Further, bridged bis-benzimidazolium bromide salts (L1, L2, and L3) were prepared in good to very good yields by direct alkylation of the 1-alkyl benzimidazoles (S1 and S2) with dibromoalkanes (1,3-dibromopropane and 1,4-dibromobutane) (Scheme 2). The proton NMR spectra showed downfield singlet peaks at 9.83 ppm (L1), 10.05 ppm (L2), and 10.15 ppm (L3), which are assigned to C-2 protons of the benzimidazole rings. These singlet peaks confirm the formation of the benzimidazolium bromides.
Scheme 1. Synthesis of Alkyl-1H-benzo[d]imidazoles.

Scheme 2. Synthesis of Bridged NHC Salt Ligand Precursors.

The bridged N-heterocyclic biscarbene palladium(II) complex-bridged-bis(NHC)PdBr2 (C1, C2, and C3) were synthesized in very good yields by reacting palladium acetate with 1.0 equiv of the appropriate ligand precursor (L1, L2, or L3) (Scheme 3). The absence of the singlet peaks for the acidic C-2 protons in 1H NMR spectra confirmed the formation of the new palladium complexes. Furthermore, 13C NMR spectra for the palladium–NHC complexes showed new signals assigned to C–Pd at 181.7, 181.1, and 176.3 ppm for C1, C2, and C3, respectively. The ESI mass spectra of the three complexes showed peaks at 546.8 (C1), 560.8 (C2), and 642.8 (C3) that confirm the formation of the Pd–carbene complexes.
Scheme 3. Synthesis of Bridged-Bis(NHC)PdBr2 Complexes (C1, C2 and C3).

The molecular structures of complexes C1 and C3 are depicted in (Figures 2 and 3), respectively. Selected bond distances and bond angles are given in (Table 7). C1 crystallized with one molecule, located on a mirror plane, in the asymmetric unit, as a dimethylformamide solvate, while C3 crystallized with one molecule in the asymmetric unit as mixed hydrate/dichloromethane solvate. In both complexes, the Pd(II) ion is coordinated by the chelating-bridged bis-carbene ligand and two bromide ions at cis positions, in a distorted square planar geometry. The cis bond angles are in the ranges 88.66(17)–92.97(2) and 87.4(3)–97.01(4)° in C1 and C3, respectively. The Pd–C and Pd–Br bond distances are within the range reported in the literature.55,60 The former is 1.997(3) Å in C1 and in the range [1.964(8)–1.985(8) Å] in C3, while the latter is 2.4906(4) Å in C1 and in the range [2.4659(11)–2.4704(11) Å] in C3. The chelate C–Pd–C bite angle values are 88.66(17) and [87.4(3), 87.7(3)°] in C1 and C3, respectively. The larger bond distances in C1 are consistent with the larger steric hindrance of the isopropyl group opposing the formation of the chelate complex.
Figure 2.
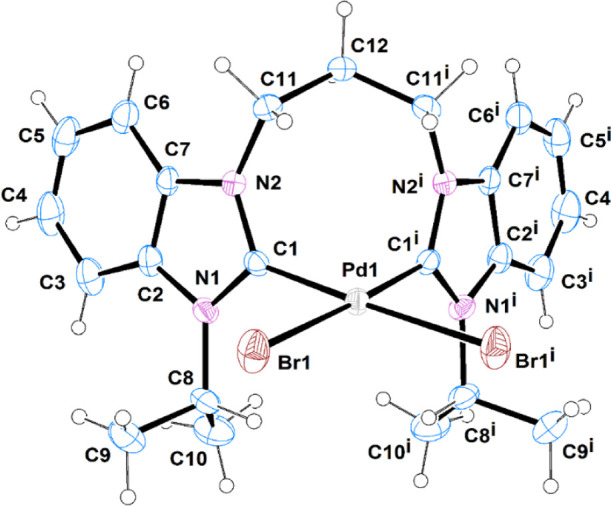
ORTEP diagrams of C1 showing the atomic labeling scheme. Thermal ellipsoids are drawn at the 30% probability level. Symmetry code i = x, −y + 1/2, z.
Figure 3.
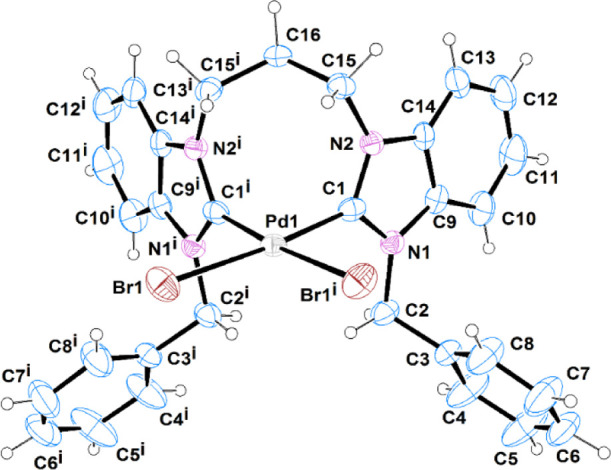
ORTEP diagrams of C3 showing the atomic labeling scheme. Thermal ellipsoids are drawn at the 30% probability level. Symmetry code: i = x, −y + 3/2, z.
Table 7. Selected Bond Lengths [Å] and Bond Angles [deg] for Compounds C1 and C3a.
| C1 | C3 | ||
|---|---|---|---|
| Pd(1)–C(1)#2 | 1.997(3) | Pd(1)–C(1) | 1.964(8) |
| Pd(1)–C(1) | 1.997(3) | Pd(1)–Br(1) | 2.4659(11) |
| Pd(1)–Br(1) | 2.4906(4) | Pd(2)–C(17) | 1.985(8) |
| Pd(1)–Br(1)#2 | 2.4906(4) | Pd(2)–Br(2) | 2.4704(11) |
| C(1)–N(2) | 1.344(4) | N(1)–C(1) | 1.352(10) |
| C(1)–N(1) | 1.350(4) | N(1)–C(9) | 1.395(10) |
| C(2)–N(1) | 1.397(4) | N(2)–C(1) | 1.355(9) |
| C(7)–N(2) | 1.388(4) | N(2)–C(14) | 1.372(9) |
| C(8)–N(1) | 1.475(4) | N(2)–C(15) | 1.458(9) |
| C(11)–N(2) | 1.469(4) | N(3)–C(17) | 1.339(10) |
| N(3)–C(25) | 1.373(9) | ||
| N(3)–C(18) | 1.463(9) | ||
| N(4)–C(17) | 1.339(8) | ||
| N(4)–C(30) | 1.401(9) | ||
| N(4)–C(31) | 1.452(9) | ||
| C(1)#2–Pd(1)–C(1) | 88.66(17) | C(1)–Pd1–C(1)#1 | 87.4(3) |
| C(1)#2–Pd(1)–Br(1) | 177.48(8) | C(1)–Pd(1)–Br(1) | 175.0(2) |
| C(1)–Pd(1)–Br(1) | 89.17(8) | C(1)#1–Pd(1)–Br(1) | 88.5(2) |
| C(1)#2–Pd(1)–Br(1)#2 | 89.17(8) | Br1–Pd1–Br1#1 | 95.49(4) |
| C(1)–Pd(1)–Br(1)#2 | 177.48(8) | C(17)#2–Pd(2)–C(17) | 87.7(3) |
| Br(1)–Pd(1)–Br(1)#2 | 92.97(2) | Br2#2–Pd(2)–Br(2) | 97.01(4) |
| C(17)–Pd(2)–Br(2) | 175.2(2) | ||
| C(17)#2–Pd(2)–Br(2) | 87.6(2) | ||
Symmetry codes: #1 = x, −y + 3/2, z #2 = x, −y + 1/2, z.
There are various catalytic systems used for the synthesis of chromones and flavones reported in the literature. However, they suffer from significant shortcomings such as high catalyst loading, long reaction time, the use of phosphines, and the low selectivity.32−37 Recently, NHC–Pd(II) complexes were evaluated in various cross coupling reactions53−56 but their applications in cyclocarbonylative Sonogashira coupling reactions of 2-iodophenols with terminal alkynes are still limited, where the catalytic system is prepared in situ by mixing the palladium(II) precursors with the NHC ligands.38
In this work, we have examined the application of the newly synthesized bridged-bis(NHC)PdBr2 complexes (C1, C2, and C3) in the cyclocarbonylative Sonogashira coupling reactions of various 2-iodophenols with aryl and alkyl alkynes. The optimization of the reactions was conducted by using the bridged-bis(NHC)PdBr2 complex C1. For this purpose, a model reaction of 2-iodophenol and phenylacetylene under pressurized CO in the presence of a catalytic amount of C1 was adopted (eq 1). The results of the optimization of the reactions are summarized in Table 1. Only traces of 2-phenyl-4H-chromen-4-one (3aa) were obtained when a neat reaction was conducted using 1.0% mol of C1 as the catalyst and diethylamine as the base at room temperature for 16 h (Table 1, entry 1). However, a conversion of 59.5% was obtained at 80 °C, and a higher conversion (85.5%) was achieved at 100 °C. These reactions produced 2-phenyl-4H-chromen-4-one (3aa) as a major compound. The isolated yield of 3aa gradually increased by increasing the temperature (Table 1, entries 2–3). When diethylamine in the neat reaction was replaced by triethylamine at 80 and 100 °C using C1, the cyclocarbonylative of 2-iodophenols with phenylacetylene via a 6-endo cyclization mode leading to six-membered ring product “flavone” (3aa) and 5-exo cyclized five-membered ring product “aurone” (4aa) was formed. Moreover, the increase of the temperature from 80 to 100 °C increased the conversion from 85 to 100% and favors the formation of the aurone product 4aa (57 and 66%) (Table 1, entries 4–5). However, when tetrahydrofuran (THF) was used as a solvent at temperatures (80 and 100 °C) with triethylamine as the solvent, the conversions dropped to 69 and 93%, respectively, with no significant change in the regioselectivity (Table 1, entries 6–7). Nevertheless, the use of Et2NH as the base with THF as the solvent at 80 and 100 °C led to higher conversions (68 and 84.5%) (Table 1, entries 8–9). Consequently, under the same experimental conditions, triethylamine oriented the cyclocarbonylative Sonogashira reactions toward the production of aurone 4aa as the major product. In addition, diethylamine in THF produced flavone 3aa in high isolated yields. When potassium carbonate was used as the base in THF at 100 °C (Table 1, entry 10), a full conversion was observed to produce flavone 3aa and aurone 4aa (38%/62%). In toluene as a solvent, the cyclocarbonylative Sonogashira reaction of 2-iodophenol with phenylacetylene under the optimized conditions (Et2NH/1.0% mol of C1/100 °C/16 h) was converted (97%) which led to the formation of two products flavone “3aa” and aurone “4aa” with a ratio of 93/7 (Table 1, entry 11). We have also observed that in THF as the solvent and Et2NH as the base, the catalyst’s C1 loading can be decreased from 1.0 to 0.5 mol % of C1 leading to high conversion (80%) to produce flavone 3aa as the only product of the reaction (Table 1, entry 12). An excellent isolated yield of 2-phenyl-4H-chromen-4-one (3aa) (96%) was achieved with 98% conversion of 2-iodophenol when the DMF was used as solvent under optimized conditions [Et2NH/C1 (0.5 mol %)/100 °C/16 h] (Table 1, entry 13); small amounts of aurone 4aa were obtained (3aa/4aa = 96:4). A decrease in the temperature from 100 to 80 and 50 °C under the optimized conditions [DMF/Et2NH/C1 (0.5 mol %)/16 h] demonstrated a gradual decline in the conversion of 2-iodophenol (83% at 80 °C and 38% at 50 °C) and the isolated yields of the flavone 3aa were 81% at 80 °C and 35% at 50 °C (Table 1, entries 14, 15). Therefore, 100 °C was considered as the optimized temperature for the subsequent reactions. The effect of the reaction time was also studied. After 16, 12, and 6 h, the isolated yields of the flavone 3aa decreased from 92 to 72 and 46%, respectively (Table 1, entries 13, 16, 17). When DMF was replaced by other solvents such as THF, toluene, and in the neat Et2NH under the optimized conditions [Et2NH/C1 (0.5 mol %)/100 °C/16 h], a significant decrease in the conversions and isolated yields in flavone was observed (Table 1, entries 18–20).
Table 1. Cyclocarbonylative Sonogashira Coupling Reactions of 2-Iodophenol (1a) with Phenylacetylene (2a) by C1a,b.
| selectivity
(%)c |
||||||||
|---|---|---|---|---|---|---|---|---|
| No | C1 mol % | base | solvent | T (°C) | time (h) | conv. (%)c | 3aa (%)d | 4aa (%)d |
| 1 | 1.0 | Et2NH | Et2NH | RT | 16 | traces | traces | traces |
| 2 | 1.0 | Et2NH | Et2NH | 80 | 16 | 59.5 | 100 (55) | |
| 3 | 1.0 | Et2NH | Et2NH | 100 | 16 | 85.5 | 100 (82) | |
| 4 | 1.0 | Et3N | Et3N | 80 | 16 | 85 | 43 (34) | 57 (45) |
| 5 | 1.0 | Et3N | Et3N | 100 | 16 | 100 | 34 (30) | 66 (63) |
| 6 | 1.0 | Et3N | THF | 80 | 16 | 69 | 40 (25) | 60 (38) |
| 7 | 1.0 | Et3N | THF | 100 | 16 | 93 | 36 (30) | 64 (56) |
| 8 | 1.0 | Et2NH | THF | 80 | 16 | 68 | 100 (64) | |
| 9 | 1.0 | Et2NH | THF | 100 | 16 | 84.5 | 100 (81) | |
| 10 | 1.0 | K2CO3 | THF | 100 | 16 | 96 | 38 (33) | 62 (57) |
| 11 | 1.0 | Et2NH | Toluene | 100 | 16 | 97 | 93 (88) | 7 |
| 12 | 0.5 | Et2NH | THF | 100 | 16 | 80 | 100 (77) | |
| 13 | 0.5 | Et2NH | DMF | 100 | 16 | 98 | 96 (92) | 4 |
| 14 | 0.5 | Et2NH | DMF | 80 | 16 | 83 | 100 (81) | |
| 15 | 0.5 | Et2NH | DMF | 50 | 16 | 38 | 100 (35) | |
| 16 | 0.5 | Et2NH | DMF | 100 | 12 | 76 | 100 (72) | |
| 17 | 0.5 | Et2NH | DMF | 100 | 6 | 49 | 100 (46) | |
| 18 | 0.5 | Et2NH | THF | 100 | 16 | 72 | 100 (70) | |
| 19 | 0.5 | Et2NH | Toluene | 100 | 16 | 82 | 95 (76) | 5 |
| 20 | 0.5 | Et2NH | Et2NH | 100 | 16 | 79 | 100 (76) | |
| 21 | 0.5 | K2CO3 | DMF | 100 | 16 | 97 | 70 (65) | 30 (27) |
| 22 | 0.5 | Et3N | DMF | 100 | 16 | 95 | 60 (54) | 40 (35) |
Optimization of reaction conditions.
Reaction conditions: C1 (mol %), 2-iodophenol (0.5 mmol), phenylacetylene (0.6 mmol), base (1.0 mmol), solvent (2.5 mL), and CO (100 psi), 100 °C.
Determined by GC and GC–MS.
Isolated yield.
Additionally, the study of the role of the base on the regioselectivity was also conducted using Et3N and K2CO3 as bases with DMF as solvent under optimized conditions [C1 (0.5 mol %)/100 °C/16 h]. For instance, the use of K2CO3 as the base produced a mixture of flavone 3aa and aurone 4aa (70%/30%) (Table 1, entry 21). Similarly, the use of trimethylamine gave lower regioselectivity producing a mixture of flavone 3aa and aurone 4aa (60%/40%) (Table 1, entry 22). On the other hand, when Et3N was used with THF as a solvent, the isolated yield in flavone 3aa dropped to 30% (Table 1, entry 7) and increased with DMF to 54%. These results showed the importance of Et2NH and DMF as regioselective factors in the production of flavones.
Based on the above results, the subsequent reactions of the scope of substrates were carried out using 0.5 mol % of C1, Et2NH as the base, and DMF as the solvent at 100 °C for 16 h. The catalytic reactions using C1 under the optimized conditions were highly regioselective. The reactions resulted in the formation of the flavone “2-phenyl-4H-chromen-4-one” (3aa) as the major product; only traces of the aurone “2-benzylidenebenzofuran-3(2H)-one” (4aa) were detected. It is important to note that because of the use of an excess of Et2NH as compared to phenylacetylene, small amounts of N,N-diethyl-3-phenylprop-2-ynamide were produced as a side product from the carbonylative coupling of phenylacetylene with Et2NH.
 |
1 |
The catalytic activity of the other synthesized bridged-bis(NHC) PdBr2 catalysts (C2 and C3) were also evaluated under the same optimized conditions (CO/Et2NH/DMF/100 °C/16 h). Their catalytic efficiency in the cyclocarbonylative Sonogashira coupling reactions was slightly less than C1 (Table 2). The conversions with C2 and C3 were 88 and 84%, respectively. The isolated yields of the flavone 3aa were 85 and 79%, respectively (Table 2, entries 2, 3).
Table 2. Cyclocarbonylative Sonogashira Coupling Reactions of 2-Iodophenol (1a) with Phenylacetylene (2a)a,b.
| selectivity
(%)c |
||||
|---|---|---|---|---|
| no. | catalyst (mol %) | conv. (%)c | 3aa (%)d | 4aa (%)d |
| 1 | C1 (0.5%) | 98 | 96 (92) | 4 |
| 2 | C2 (0.5%) | 88 | 96 (85) | 4 |
| 3 | C3 (0.5%) | 84 | 95 (79) | 5 |
| 4e | Pd–NHC–Py1 (0.05%) | 83 | 89 (77) | 11 |
| 5e | Pd–NHC–Py2(0.5%) | 79 | 86 (72) | 14 |
| 6 | Pd(OAc)2 (0.5%) L1 (0.5%) | 81 | 94 (75) | 6 |
| 7 | Pd(OAc)2 (5%) DPPF (10%) | 100 | 88 (85) | 12 |
| 8f | Pd(OAc)2 (5%) DPPF (10%) | 95 | 50 (46) | 50 (45) |
| 9 | Pd(OAc)2 (0.5%) | 38 | 89 (32) | 11 |
| 10 | PdBr2 (0.5%) | 29 | 89 (24) | 11 |
| 11 | PdCl2(PPh3)2 (0.5%) | 60 | 86 (49) | 14 |
| 12 | Pd/C (10%) | 90 | 92 (81) | 8 |
Effect of the type of catalyst.
Reaction conditions: [Pd] (mol %), 2-iodophenol (0.5 mmol), phenylacetylene (0.6 mmol), Et2NH (1.0 mmol), DMF (2.5 mL), CO (100 psi), 100 °C, 16 h.
Determined by GC and GC–MS.
Isolated yield.
Heteroleptic N-heterocyclic carbene palladium(II) complexes (Pd–NHC–Py).59
THF was used as a solvent.
In order to rationalize the relative activity of the three complexes, one can consider the relative sigma donation effects of the ligand precursors L1, L2, and L3 that can be probed by the proton NMR chemical shift of the C-2 protons of the benzimidazole rings, observed at 9.83, 10.05, and 10.15 ppm for L1, L2, and L3 respectively. The most downfield signals were consistent with the highest electron donating effect of the isopropyl group in L1 and L2 relatively to the benzyl group in L3. The results of the comparative catalytic evaluation of the corresponding palladium–NHC complexes C1, C2, and C3 are in agreement with the strong electron donating effect of the ligand in C1 and the stability of the corresponding 8-membred ring chelate.
In addition, the catalytic activity for the mono NHC palladium(II) complexes was also evaluated under the optimized reaction conditions. In fact, the two heteroleptic (NHC)–pyridine palladium(II) complexes, which were previously reported,59 Pd–NHC–Py1 “(1,3-disopropyl-1,3-dihydro-2H-benzo[d]imidazole-2-ylidene)(pyridin-1(2H)-yl) palladium(II) bromide” and Pd–NHC–Py 2 “(1-isopropyl,3-benzyl-1,3-dihydro-2H-benzo[d] imidazole-2-ylidene)(pyridin-1(2H)-yl)palladium(II) bromide” (Figure 4), were evaluated in the cyclocarbonylative Sonogashira [Pd–NHC–Py (0.5 mol %)/Et2NH/DMF/100 psi/100 °C/16 h] (Table 2, entries 4–5). These complexes catalyzed the carbonylation reaction with very good conversions (83 and 79%, respectively) of 2-iodophenol and high selectivity (89 and 86%) toward the flavone. However, the conversion and the isolated yield of flavone are lower than those obtained with the bridged-bis(NHC)PdBr2 complexes.
Figure 4.

NHC–palladium(II)–pyridine complexes (Pd–NHC–Py1 and Pd–NHC–Py2).
Furthermore, Pd(OAc)2 was used with an equal amount of the ligand L1 in the cyclocarbonylative Sonogashira coupling reaction under the optimized conditions. The conversion was lower (81%) with high selectivity (94%) for the flavone 3aa (Table 2, entry 6). This result confirms the superiority of the catalytic activity of C1 as compared to the “in situ”-formed palladium complex.
For comparison, commercially available palladium complexes were also considered (Table 2, entries 7–12) under different protocols in the presence of other ligands, such as 1,1′-bis(diphenylphosphino)ferrocene (DPPF), and using a higher catalyst loading. The results of the catalytic evaluation confirmed again the high catalytic efficiency and selectivity of the bridged-bis(NHC)PdBr2 catalysts C1, C2, and C3.
The model cyclocarbonylative Sonogashira coupling reactions under optimized conditions showed high catalytic activity and selectivity with the newly synthesized bridged-bis(NHC)PdBr2 complexes (C1, C2, and C3). The catalyst C1 was considered in the study of the scope of substrates. The reactions were conducted under the optimized experimental conditions [C1 (0.5 mol %), 2.0 equiv of Et2NH, 3 mL of DMF, 100 psi CO, 100 °C, 16 h]. Different 2-iodophenols were reacted with various aryl alkynes (eq 2, Table 3). Numerous chromones and flavones were produced in excellent yields via cyclocarbonylative Sonogashira coupling reactions of various aryl alkynes with different electron-withdrawing-substituted 2-iodophenols (4′-hydroxy-3′-iodoaryls). The carbonylation reaction of 2-iodophenol with alkynes that have activating electron releasing groups afforded the corresponding flavones in excellent to very good isolated yields (86–91%) (Table 3, entry 1–3). In addition, excellent yield (93%) of flavone was also obtained when the electron-withdrawing-substituted aryl alkyne (1-ethynyl-4-nitrobenzene) reacted with 2-iodophenol (Table 3, entry 4). The cyclocarbonylative Sonogashira coupling reactions of phenylacetylene and aryl alkynes having electron-donating substituents with deactivated 2-iodophenols were also very successful with excellent isolated yields (91–98%) (Table 3, entries 5–8). On the other hand, a decrease in the isolated yield of the corresponding chromone to 74% was obtained when the cyclocarbonylative Sonogashira coupling reaction was carried out with 2-iodophenol and 2-ethynylanisole (Table 3, entry 9), probably because of the steric hindrance of the methoxy substituent in the ortho position of an alkyne substrate.
 |
2 |
Table 3. Cyclocarbonylative Sonogashira Coupling Reactions of 4′-Hydroxy-3′-iodoaryls (1a–d) with Aryl Alkynes (2a–e) Catalyzed by the C1a.

Reaction conditions: C1 (0.50 mol %), 4′-hydroxy, 3′-iodoaryl (0.50 mmol), aryl alkyne (0.60 mmol), Et2NH (1.0 mmol), DMF (2.5 mL), CO (100 psi), 100 °C, 16 h.
Isolated yield.
Cyclocarbonylative Sonogashira Coupling Reactions of 2-Iodophenol (1a–b) with Alkyl Alkynes (5a–e) Catalyzed by the Bridged-Bis(NHC)PdBr2 Complex C1
The research studies of the cyclocarbonylative Sonogashira coupling reactions of 2-iodophenols with alkyl alkynes are still very limited, where low yields of chromones were produced in the presence of palladium–phosphine or palladium-mixed ligand complexes. Interestingly, various chromones were produced in good yields via the cyclocarbonylative coupling reactions of different 2-iodophenols with various alkyl alkynes catalyzed efficiently by the complex C1 under the optimized conditions (Et2NH/DMF/110 °C/24 h) (eq 3) (Table 4). For instance, the cyclocarbonylative coupling reaction of 2-iodophenol with 1-heptyne (Table 4, entry 1), 1-decyne (Table 4, entry 2), 6-phenyl-1-hexyne (Table 4, entry 3), and 3-cyclohexyl-1-propyne (Table 4, entry 4) was accomplished successfully to produce the corresponding chromones 6 in good to excellent yields (53–90%). These results showed a relation between the chain lengths on the alkyl alkynes and the yields of corresponding chromones. Alkynes with shorter alkyl chain, such as 1-heptyne (Table 4, entry 1), were more reactive than the alkyl alkynes having longer alkyl chains such as 1-decyne (Table 4, entry 2). The cyclocarbonylative Sonogashira coupling reactions of 2-iodophenol with either 6-phenyl-1-hexyne or 3-cyclohexyl-1-propyne led to good and very good yields (53 and 71%) of their corresponding chromones (Table 4, entries 3–4). The reactivity 2-iodophenols were improved with 4′-hydroxy-3′-iodoacetophenone (1b) having an electron-withdrawing substituent. The reaction of 1b with 3-cyclohexyl-1-propyne and 1-heptyne catalyzed by C1 gave very good to excellent isolated yields of the corresponding chromones (77–90%) (Table 4, entries 5–6). In conclusion, useful chromones were produced in high yields by the cyclocarbonylative coupling of different 2-iodophenols with various alkyl alkynes in the presence of bridged-bis(NHC)PdBr2 as the catalyst.
 |
3 |
Table 4. Cyclocarbonylative Sonogashira Coupling Reactions of 4′-Hydroxy-3′-iodoaryls (1a–d) with Alkyl Alkynes (5a–e) Catalyzed by C1a.

Reaction conditions: C1 (1.0 mol %), 4′-hydroxy, 3′-iodoaryl (0.50 mmol), alkyl alkyne (0.60 mmol), Et2NH (1.0 mmol), DMF (2.5 mL), CO (100 psi), 110 °C, 24 h.
Isolated yield.
Cyclocarbonylative Sonogashira Coupling Reaction of 2-Iodophenol with Dialkynes Catalyzed by Complex C1
Considering the high efficiency of the newly synthesized complexes C1, C2, and C3, we have investigated the cyclocarbonylative Sonogashira coupling reaction of 2-iodophenol with dialkynes. Remarkably, these reactions successfully proceeded to obtain symmetrical diflavones in very good to excellent isolated yields. For instance, the cyclocarbonylative coupling reactions of 1,4-diethynylbenzene and 1,3-diethynylbenzene with 2 equiv of 2-iodophenol or 4-hydroxy-3-iodoacetophenone proceeded smoothly to yield the corresponding para-diflavones (8a) and (8c) (eq 4) and (eq 4) in 86 and 92%, respectively. Furthermore, meta-diflavone was obtained in 74% (8b) (eq 4).
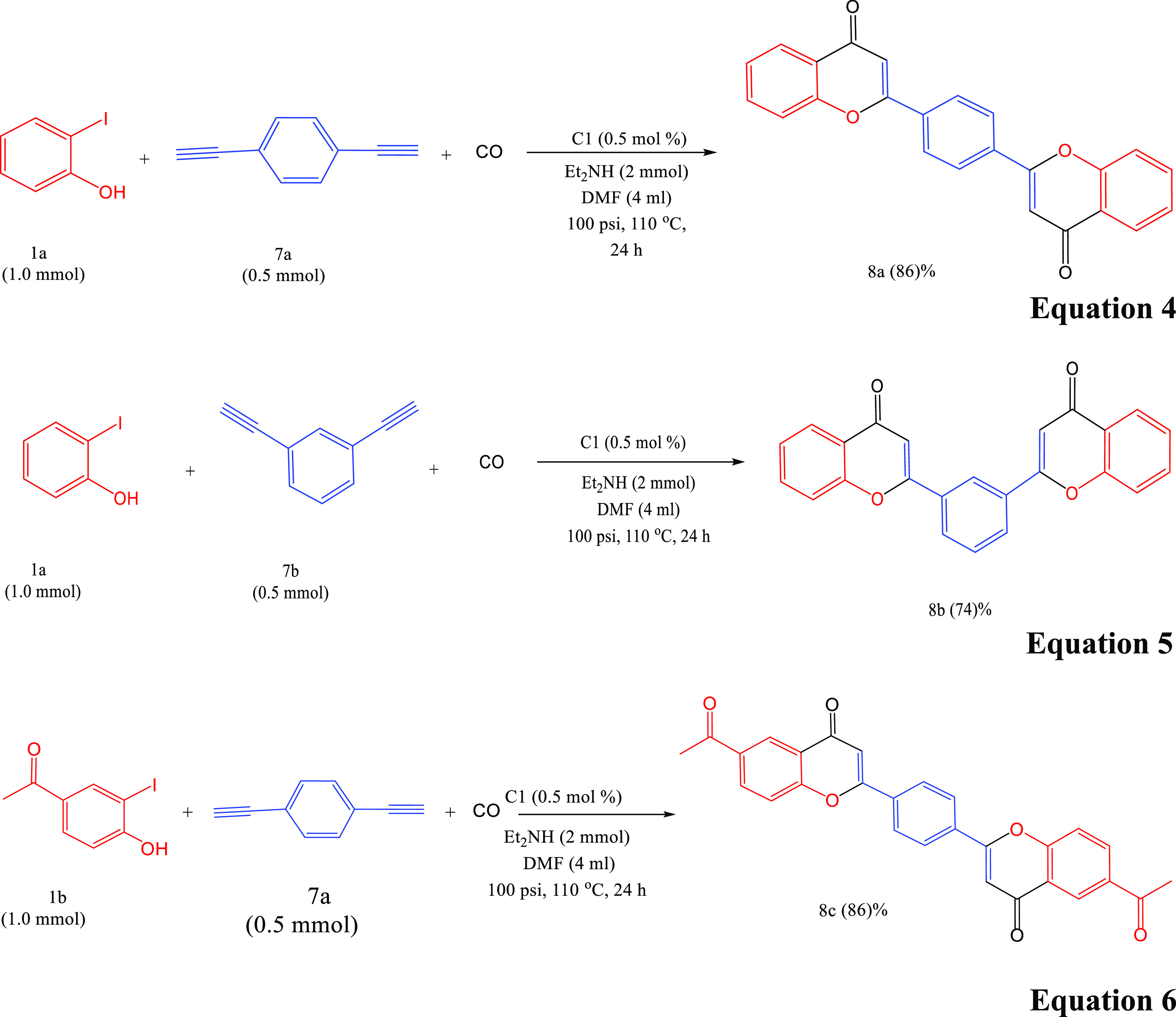 |
4 |
The excellent results obtained with the catalytic cyclocarbonylative coupling of 2-iodophenols with alkynes and diarylalkynes showed the high catalytic activity of the newly synthesized bridged-N-heterocyclic carbene palladium complexes C1, C2, and C3. The complex C1 exhibited superior catalytic efficiency with much lower loading in the absence of additional phosphine ligands as compared to other reported catalytic systems in the literature (Table 5).
Table 5. Comparison of the Activity of the New Catalytic System Including Bridged-Bis(NHC)PdBr2 (C1) in Cyclocarbonylative Sonogashira Coupling Reactions of 2-Iodophenol and Alkynes with Literature Data.
| refs | catalyst | co-catalyst/ligand/additive | base | time (h) | yield (%) |
|---|---|---|---|---|---|
| (33) | PdCL2 (5 mol %) | (H13C6)3P+C14H29Br– (1.5 g) | Et3N | 24 | 64–95 |
| (31) | PdCL2 (5 mol %) | PPh3 (10 mol %) | Et3N | 24 | 35–95 |
| (35) | Pd(OAc)2 (5 mol %) | DPPF (5 mol %) | piperazine | 24 | 30–95 |
| (32) | Pd2(dba)3 (1.5 mol %) | 1,3,5,7-tetramethyl-2,4,8-trioxa-6-phenyl-6-phosphaadamantane (3 mol %) | DBU | 0.5 MW | 62–95 |
| (40) | Pd(Pph3)4 (3 mol %) | Ac2O (6 mol %) | Et3N | 51–82 | |
| (38) | Pd–NHC–Py (0.5 mol %) | imidazole (0.5 mol %) pyridine (0.5 mol %) | Et2NH | 24 | 25–98 |
| (49) | Pd/C (1 mol %) | Et2NH | 20 | 74–98 | |
| this work | bridged-bis(NHC)PdBr2 (0.5 mol %) | Et2NH | 16 | 53–98 |
Plausible Mechanisms for the Cyclocarbonylative Sonogashira Coupling Reactions
Chromones and aurones were produced via carbonylative Sonogashira coupling reactions followed by the cyclization reactions. Initially, in the presence of the base, alkynes are deprotonated and the palladium(II) precatalyst (II) undergoes a substitution of the bromides by the acetylides to produce the palladium(II) intermediate (III), which undergoes reductive elimination of the dialkynes to generate the Pd(0) (IV) as the active catalytic species (Scheme 4, pathway A). The palladium(II) intermediate (Ar–Pd–I) (V) is then formed via the oxidative addition of 2-iodophenol to the Pd(0). The acyl palladium intermediate (VI) is formed by the insertion of carbon monoxide (CO) into the Ar–Pd bond. Then, the iodide is substituted by the acetylide to produce the palladium intermediate VII. The reductive elimination in the presence of the base yields the carbonylative Sonogashira product VIII with the subsequent regeneration of the Pd(0) catalyst.58,61
Scheme 4. Plausible Mechanism for the Cyclocarbonylative Sonogashira Coupling Reaction Catalyzed by Bridged-Bis(NHC)PdBr2 Complexes; Production of Chromone [Pathway B or (C,D)], and Aurone (Pathway C–E).

The production of aurones or chromones depends on the type of base used in the reaction.
In the presence of diethylamine, the favored Michael addition to the intermediate VIII forms the Michael adduct IX (Scheme 4, pathway B), which is usually stabilized in DMF as an ideal solvent in these reactions.62,63 This Michael adduct (IX) was detected and identified in the reaction mixture by gas chromatography–mass spectrometry (GC–MS) [m/z = 296.5; C19H22NO2 (MH+)]. The diethylamine is then eliminated to produce the cyclic enolate X, which is converted into the corresponding chromone.
When triethylamine was used as a base, no reaction was observed in the absence of the palladium catalyst. Furthermore, the palladium catalyzed reaction in the presence of triethylamine led to the formation of a mixture of aurones and chromones (Table 3, entries 4–7). In this case, it is suggested that the oxidative addition of O–H to the Pd(0) species generates the palladium intermediate XI (Scheme 4, pathway C). The subsequent insertion of the alkyne into Pd–H proceeds following two pathways to generate either the seven-membered metallacycle XII (pathway D) or the six-membered metallacycle XIII (pathway E) as intermediates. These undergo further reductive elimination to produce chromones and aurones, respectively.
Conclusions
High regioselectivity of phosphine-free cyclocarbonylative Sonogashira coupling reactions of 2-iodophenols with alkynes toward chromones was achieved using new bridged-bis(NHC)PdBr2 chelate catalysts. The preligands and the corresponding complexes C1, C2, and C3 were synthesized and fully characterized using various physical, analytical, and spectroscopic techniques including single-crystal X-ray diffraction. The catalytic efficiency was evaluated in the cyclocarbonylative Sonogashira coupling reactions of various 2-iodophenols with aryl and alkyl alkynes including the dialkynes under CO pressure and in the absence of any additional phosphine ligands. The palladium complex C1 was more active as compared to C2 and C3 complexes. In general, C1, C2, and C3 showed high catalytic activity with lower catalyst loading as compared to other known catalytic systems. Excellent isolated yields of the expected chromones and flavones were obtained for the cyclocarbonylative Sonogashira coupling reactions of 2-iodophenols with the aryl alkynes and good to very good yields with alkyl alkynes or aryl dialkynes. The regioselectivity of these reactions toward the chromones was favored with diethylamine as a base and in DMF as a solvent. It is worth noting the synthesis of a new compound, 2,2′-(1,4-phenylene) bis(6-acetyl-4H-chromen-4-one) (8c), by the one-pot cyclocarbonylative Sonogashira coupling reaction of 4-hydroxy-3-iodoacetophenone with 1,4-diethynylbenzene catalyzed by C1.
Experimental Section
Materials and Instrumentation
All precursors used for the synthesis of NHCs, palladium complexes, 2-iodophenols, and alkynes were purchased from Sigma-Aldrich and used directly as received unless specified otherwise. Flash column chromatography (packed with 60 F Silica gel from Fluka Chemie AG, Buchs, Switzerland) was used to purify the products.
1H and 13C NMR spectra were recorded on a Bruker EQUIN55 (400 MHz for 1H; 101 MHz for 13C) and JOEL 1500 MODEL (500 MHz for 1H; 125 MHz for 13C) NMR spectrometer in different deuterated solvents like CDCl3, CD2Cl2, DMSO-d6, and DMF-d7. Chemical shifts were reported in ppm downfield of tetramethylsilane, used as the internal standard. The collected data were reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet and m = multiplet), and coupling constant in Hz. 13C NMR spectra were obtained with complete proton decoupling. The products were analyzed by GC–MS (Agilent GC–MS; GC 6890N and MS 5975B; 30 m HP-5 capillary column). A separate GC (Agilent 6890) was used to monitor the reactions and analyze the products. Mass spectra were obtained using the Fisher ESI-TOF/MS spectrometer with the ESI scan (0.104–0.499 min, 17 scans) Frag = 180.0 V. Elemental analyses for the ligand precursors, corresponding complexes, and products were performed on PerkinElmer Series 11 (CHNS/O) Analyzer 2400. Merck 60 F254 silica gel plates (250 μm layer thickness) were used for thin-layer chromatography (TLC) analyses. Single-crystal X-ray data collection for complexes C1 and C3 was performed at 298 K on a Bruker D8 Quest diffractometer (Mo Kα radiation λ = 0.71073 Å). Data were collected and integrated using the Bruker APEX3 software package.64 Multiscan absorption correction was performed using SADABS.65 The structures were solved by direct methods with SHELXS using the SHELXTL package and refined using full-matrix least squares procedures on F2 via the program SHELXL-2014.66 ORTEP3 was used for molecular graphics.67 All hydrogen atoms were included at calculated positions using a riding model. The crystal data and refinement details for C1 and C3 are given in (Table 6). Selected bond lengths and bond angles are given in (Table 7).
Table 6. Crystal and Structure Refinement Data of Compounds C1 and C3.
| C1 | C2 | |
|---|---|---|
| CCDC deposition # | 1961539 | 1950067 |
| empirical formula | C26H35Br2N5OPd | C32H32Br2CL2N4OPd |
| formula weight | 699.81 | 825.73 |
| temperature (K) | 298(2) | 298(2) |
| wavelength (Å) | 0.71073 | 0.71073 |
| crystal system | orthorhombic | monoclinic |
| space group | Pnma | P21/m |
| unit cell dimensions | ||
| a (Å) | 16.2158(6) | 10.5561(8) |
| b (Å) | 18.1879(7) | 23.972(2) |
| c (Å) | 9.2992(4) | 13.3418(11) |
| α (deg) | 90 | 90 |
| β (deg) | 90 | 92.033(2) |
| γ (deg) | 90 | 90 |
| volume (Å3) | 2742.63(19) | 3374.0(5) |
| Z | 4 | 4 |
| density (calculated, g/cm3) | 1.695 | 1.626 |
| absorption coefficient (mm–1) | 3.619 | 3.108 |
| F(000) | 1400 | 1640 |
| θ range data collect. (deg) | 2.512–28.371 | 2.564–28.367 |
| index ranges | –21 ≤ h ≤ 21, –24 ≤ k ≤ 24, 12 ≤ l ≤ 12 | –14 ≤ h ≤ 14, –31 ≤ k ≤ 32, –17 ≤ l ≤ 17 |
| reflections collected | 76,298 | 123,299 |
| independent reflections | 3528 [R(int) = 0.0387] | 8604 [R(int) = 0.1448] |
| absorption correction | semiempirical from equivalents | semiempirical from equivalents |
| refinement method | full-matrix least-squares on F2 | full-matrix least-squares on F2 |
| data/restraints/parameters | 3528/18/173 | 8604/6/399 |
| goodness-of-fit on F2 | 0.989 | 1.112 |
| ginal R indices [I > 2σ(I)] | R1 = 0.0342, wR2 = 0.1080 | R1 = 0.0716, wR2 = 0.1905 |
| R indices (all data) | R1 = 0.0473, wR2 = 0.1299 | R1 = 0.1308, wR2 = 0.2298 |
| largest diff. peak and hole (e Å3) | 1.039 and −0.978 | 1.673 and −1.585 |
Synthesis of 1-Alkyl Benzimidazole (S1 and S2)
A dry and clean round-bottom flask was charged with benzimidazole (10.0 mmol), excess amount of alkyl bromide (12.2 mmol), 2-bromopropane or benzyl bromide, and an appropriate base, potassium hydroxide (20.0 mmol) with 2-bromopropane or cesium carbonate (20.0 mmol) with benzyl bromide, and 1.00 mmol of tetrabutylammonium bromide. 100 mL of acetonitrile was used to dissolve the prepared mixture with continuous stirring at 80 °C for 24 h. TLC (1/1 = hexane/ethyl acetate) was used for monitoring the reaction until no free benzimidazole was observed. After the completion of the reactions, the solvents were removed under vacuum by the rotary evaporator. The oily products were collected as residues; the purification of the products was conducted by extraction two times with 30 mL ethyl acetate and 20 mL distilled water. The aqueous layers were separated and then washed with ethyl acetate. The separated organic layers were dried and washed several times by n-hexane.
1-Isopropyl-1H-benzo[d]midazole (S1)
Yield = 77%. Sticky brown oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.85 (s, 1H, NCHN), 7.64–7.62 (1H, m, Ar-H), 7.25–7.22 (1H, m, Ar-H), 7.11–7.08 (2H, m, Ar-H), 4.42 (1H, sep, 3J = 6.76 Hz, NCH), 1.40 (6H, d, 3J = 6.76 Hz, NC(CH3)2). 13C{1H} NMR (500 MHz, CDCl3): δ (ppm) 143.5 (NCN), 139.8, 132.7, 122.1, 121.5, 119.8, 109.7, (Ar-H), 47.2 (NCH), 22.02 [NC(CH3)2]. Anal. Calcd for C10H12N2, (160): C, 74.97%; H, 7.55%; N, 17.48%. Found: C, 74.84%; H, 7.23%; N, 17.23%.
1-Benzyl-1H-benzo[d]imidazole (S2)
Yield = 87%. Light yellow solid. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.98 (1H, s, NCHN), 7.83 (1H, d, 3J = 7.63 Hz, Ar-H), 7.34–7.24 (6H, m, Ar-H), 7.18 (2H, d, 3J = 7.02 Hz, Ar-H), 5.36 (2H, m, NCH2-Ph); 13C{1H} NMR (500 MHz, CDCl3): δ (ppm); 143.1 (NCN), 135.4, 129, 128.3, 127.06, 123.1, 122.3, 120.3, 110.04 (Ar-H), 48.8 (NCH2); Anal. Calcd for C14H12N2, (208): C, 80.74%; H, 5.81%; N, 13.45%. Found: C, 80.89%; H, 6.03%; N, 13.73%.
Synthesis of the Precursors for Alkylene-Bridged N-Heterocyclic Dicarbene Ligands (L1, L2, and L3)
1-Alkyl benzimidazole (5.0 mmol) and 2.5 mmol of dibromoalkane (1,3-dibromopropane or 1,4-dibromobutane) were introduced into a dried 100-mL round bottom flask. The mixture was refluxed in 35 mL of 1,4-dioxane under stirring at 103 °C for 12 h. The products appeared as white precipitates. They were collected by filtration and washed three times with 15 mL of 1,4-dioxane and then by 15 mL of toluene to remove any traces of the starting materials. The products were dried under vacuum and then collected as a white precipitate. Characterization of the alkylene-bridged N-heterocyclic dicarbene salts was conducted with different spectroscopic techniques including 1H NMR, 13C NMR, elemental analysis, and ESI.
3,3′-(Propane-1,3-diyl)-bis(1-isopropyl-1H-benzo[d]24midazole-3-ium) Bromide (L1)
Yield = 91%. White solid. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 9.83 (2H, s, NCHN), 8.15–8.11 (4H, m, Ar-H), 7.71–7.69 (2H, m, Ar-H), 5.05 (2H, sep, 3J = 6.71 Hz, NCH), 4.67 (4H, t, 3J = 7.01 Hz, CH2), 2.67 (2H, qui, 3J = 7.02 Hz, CH2), 1.61 (12H, d, 3J = 6.7 Hz, NC(CH3)2); 13C{1H} NMR (125 MHz, DMSO): δ (ppm) 140.7 (NCN), 131.3, 130.5, 126.7, 126.6, 114.1, 113.7, (Ar-H), 50.7 (NCH), 44.1 (NCH2), 28.0 (CH2), 21.6 (NC(CH3)2). Anal. Calcd for C23H30N4Br2, (522.3): C, 52.89%; H, 5.79%; N, 10.73%. Found: C: 52.37%, H: 5.8, %, N, 10.97%. ESI m/z: 442 [M – Br–]+, m/z 441 [M – Br–(−H+)].
3,3′-(Butane-1,4-diyl)-bis(1-isopropyll-1H-benzo[d]24midazole-3-ium) Bromide (L2)
Yield = 76%. White solid. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 10.05 (2H, s, NCHN), 8.13–8.11 (4H, m, Ar-H), 7.68 (4H, dd, 3J = 6.1 Hz, 3J2 = 2.75 Hz, Ar-H), 5.05 (2H, sept, 3J = 6.71 Hz, NCH), 4.58 (4H, m, NCH2), 2.04–1.99 (4H, m, CH2), 1.62 [12H, d, 3J = 6.71 Hz (CH3)2]. 13C{1H} NMR (125 MHz, DMSO): δ (ppm) 140.7 (NCN), 131.3, 130.6, 126.7, 126.6, 114.1, 113.8, (Ar-H), 50.7 (NCH), 46.3 (NCH2), 25.6 (CH2), 21.64 [NC(CH3)2]. Anal. Calcd for C24H32N4Br2, (536.3): C, 53.74%; H, 6.01%; N: 10.45%. Found: C: 52.12%, H: 5.91%, N: 10.79%. ESI m/z: 456 [M – Br–]+.
3,3′-(Propane-1,3-diyl)-bis(1-benzyl-1H-benzo[d]25midazole-3-ium) Bromide (L3)
Yield = 65%. White solid. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 10.15 (2H, s, NCHN), 8.14 (2H, d, 3J1 = 7.93 Hz, Ar-H), 7.96 (2H, d, 3J1 = 7.32 Hz, Ar-H), 7.66–7.62 (4H, m, Ar-H), 7.53 (4H, d, 3J1 = 7.02 Hz, Ar-H), 7.39–7.36 (4H, m, Ar-H), 5.80 (4H, s, NCH2Ph), 4.62 (4H, m, NCH2CH2), 2.06 (2H, m, CH2). 13C{1H} NMR (125 MHz, CDCl3): δ (ppm); 141.31, 132.99, 131.51, 130.77, 129.27, 129.10, 128.52, 127.26, 127.14, 114.08, 113.64 (C-arom), 51.95 (NCH2), 51.37 (NCH2), 22.44 2(CH2CH). Anal. Calcd for C31H30N4Br2, (618.4): C, 60.61%; H, 4.89%; N: 9.06%. Found: C: 60.83%, H: 5.11%, N: 9.79%. ESI m/z: 538.5 [M – Br–]+.
Synthesis of Palladium Bridged N-Heterocyclic Dicarbene Complexes (C1, C2, and C3)
A round-bottom flask (50 mL) was charged with bridged N-heterocyclic dicarbene ligand precursors (L1, L2, or L3) (0.50 mmol), palladium(II) acetate (0.50 mmol), and 15 mL of dimethyl sulfoxide as a solvent. The orange solutions were heated at 70 °C for 24 h under stirring. The products were obtained as white precipitates. They were collected by filtration and washed twice with 15 mL of distilled water and then 15 mL of hexane. The residues were purified by extraction three times with dichloromethane/H2O (5 mL/5 mL). Finally, the dichloromethane extracts were evaporated in vacuum and the products were obtained as white crystals. Single crystals for complex C1 were obtained by the slow crystallization procedure using a saturated solution of dichloromethane/acetonitrile (10:1) (v/v). Full characterization of the three complexes was accomplished by different physical and spectroscopic techniques such as 1H and 13C NMR, ESI-MS, elemental analysis, and single-crystal X-ray diffraction analysis.
Dibromido (1,1′-Diisopropyl-3,3′-propylenedibenzimidazoline-2,2-diylidene)palladium(II) (C1)
Yield = 92%. White solid. 1H NMR (500 MHz, DMF-d7): δ (ppm) 8.08 (2H, d, 3J = 8.24 Hz, Ar-H, (the signals of aromatic protons overlap with the solvent signal), 7.97 (2H, d, 3J = 8.24 Hz, Ar-H), 7.45–7.37 (4H, m, Ar-H), 5.99 (2H, m, NCH), 5.43 (2H, m, CH2), 5.11 (2H, m, CH2), 2.19 (2H, m, CH2CH2CH2), 1.94 [6H, d, 3J = 6.71 Hz, NC(CH3)2], 1.78 [6H, d, 3J = 5.80 Hz, NC(CH3)2]. 13C{1H} NMR (125 MHz, CD2CL2): δ (ppm); 181 (Pd–C)[carbene signal (NCbinimN)], 135.2, 133.3, 132.3, 123.8, 123.7, 123.4, 123.1, 122.9, 113.2, 113, 110.7, 110.5 (Ar-H), 55.4 (NCH), 53.4 (NCH), 49.5 (NCH2), 47.8 (NCH2), 30.0 (CH2), 28.5 (CH2), 22.1, 21.7, 21.3 [NC(CH3)2]; Anal. Calcd for C23H28N4Br2Pd, (626.74): C, 44.08%; H, 4.50%; N, 8.94%; Found: C, 43.86%; H, 4.25%; N, 8.63%. ESI m/z: 546.82 [M – Br]+.
Dibromido (1,1′-Diisopropyl-3,3′-butylenedibenzimidazoline-2,2-diylidene)palladium(II) (C2)
Yield = 68%. Orange solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.49 (2H, d, 3J = 7.28 Hz, Ar-H), 7.30 (2H, d, 3J = 7.44 Hz Ar-H), 7.17–7.14 (4H, m, Ar-H), (the signals of aromatic protons overlap with the solvent signal), 6.00–5.96 (2H, m, NCH), 5.75–5.71 (2H, m, NCH2), 4.45–4.41 (2H, m, NCH2), 1.80–1.45 (4H, m, NCH2CH2). The signals of methylene protons were not assigned clearly because of the overlap with the isopropyl methyl signals, 1.74 [6H, d, 3J = 6.56 Hz (CH3)2], 1.69 [6H, d, 3J = 7.16 Hz (CH3)2]. 13C{1H} NMR (125 MHz, CD2CL2): δ (ppm); 181.1 (Pd–C)[carbene signal (NCbinimN)], 135.9, 132.9, 122.9, 122.8, 112.9, 111.0, (Ar-H), 48.2 (NCH), 30.1 (NCH2), 27.7 (CH2), 21.2 [NC(CH3)2]. Anal. Calcd for C24H30N4Br2Pd, (640.7): C, 44.99%; H, 4.72%; N, 8.74%; Found, C, 44.78%; H, 4.89%; N, 8.53%. ESI m/z: 560.00 [M – Br]+.
Dibromido (1,1′-Dibenzyl-3,3′-propylenedibenzimidazoline-2,2-diylidene)palladium(II) (C3)
Yield = 73%. White crystals. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.71–7.69 (m, 2H, C–H-pyr), 7.48 (m, 4H, C–H-arom), 7.45–7.41 (m, 6H, C–H arom), 7.35–7.29 [m, 6H, C–H-(phenyl)], 5.87 (s, 4H, CH2-Ph), 5.25 (m, 4H, NCH2), 1.67 (m, 2H, NC(CH2). 13C NMR (125 MHz, CD2CL2): δ (ppm) 176.39 (Pd–C), 135.14, 134.96, 133.86, 129.08, 128.38, 127.58, 127.42, 127.29, 124.11, 123.86, 112.37, 110.27 (C-arom), 52.39 NCH2, 49.50 NCH2 31.93 NCH2, 29.90 NCH2, 23.00 CH2. Anal. Calcd for C31H28Br2N4Pd, (722.8): C, 51.51%; H, 3.90%; N, 7.75%. Found: C, 50.82; H, 4.03; N, 7.93. ESI m/z: 642.90 [M – Br]+.
Procedure for the Cyclocarbonylative Sonogashira Coupling Reactions
Chromones and flavones were synthesized via cyclocarbonylative Sonogashira coupling reactions that were carried out in a 45 mL stainless steel autoclave equipped with a glass liner, gas inlet valve, and pressure gauge. Palladium–bis(NHC) complex (0.50 mol %), functionalized 2-iodophenol (0.50 mmol), alkyne (0.55 mmol), base (1.00 mmol) and an anhydrous solvent (2 mL) were charged into the glass liner of the autoclave. Then, the autoclave was vented carefully three times with carbon monoxide and then pressurized to 100 psi of carbon monoxide. The mixture was stirred and heated to the required temperature for a specific period of time. At the end of the reaction period, the autoclave was cooled down to room temperature and the excess of CO was discharged carefully under the fume hood. The reaction was extracted three times with 5 mL of distilled water and 10 mL of ethyl acetate. The ethyl acetate extracts were combined and concentrated in a rotary evaporator under reduced pressure. Flash chromatography was used to purify the reaction mixture using silica gel and an eluent (pentane/ethyl acetate = 7:1) to afford the corresponding chromones or flavones. Various physical and spectroscopic techniques such as 1H and 13C NMR, GC, and GC–MS were used to fully characterize the products. The spectral data of the chromones and flavones prepared in this study were in full agreement with those reported in the literature.33,35−37,40,41,49,68
Analytical and Spectroscopic Data of the New Compound [2,2′-(1,4-Phenylene) bis(6-acetyl-4H-chromen-4-one)] (8c)
Yiled = 92%. Yellowish green solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.81 (2H, s, C–H-arom), 8.38 (2H, d 3J = 9.2 Hz, C–H-arom), 8.15 (4H, s, C–H-arom), 7.68 (2H, d, 3J = 8.38 Hz, C–H-arom), 6.96 (2H, s, C–H-arom), 3.89 (6H, s, O=C–CH3). 13C NMR (125 MHz, CDCl3): δ (ppm) 196.58 (C=O), 177.91 (C=O), 163.84, 158.85, 133.99, 132.91, 127.26, 126.39, 123.45, 118.95, 107.89 (C-arom), 26.73 (O=C–CH3). IR (ν cm–1), para aromatic overtone at 1900 (2000–1700), 1766, 1689, 1626, 1591, 1505, 1459, 1253, 1157. GC–MS m/z: 449.44(M+). Anal. Calcd for C28H18O6, (450.45): C, 74.66%; H, 4.04%; Found: C, 75.12; H, 4.11.
Acknowledgments
Saudi Aramco is acknowledged for funding this project under the award number CHEM2432. We also acknowledge the Chemistry department at King Fahd University of Petroleum & Minerals (KFUPM) for all support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04706.
The authors declare no competing financial interest.
Supplementary Material
References
- Harvey R. G.; Hahn J. T.; Bukowska M.; Jackson H. A new chromone and flavone synthesis and its utilization for the synthesis of potentially antitumorigenic polycyclic chromones and flavones. J. Org. Chem. 1990, 55, 6161. 10.1021/jo00312a023. [DOI] [Google Scholar]
- Ferreira A.; Pousinho S.; Fortuna A.; et al. Flavonoid compounds as reversal agents of the P-glycoprotein-mediated multidrug resistance: biology, chemistry and pharmacology. Phytochem. Rev. 2015, 14, 233–272. 10.1007/s11101-014-9358-0. [DOI] [Google Scholar]
- Keri R. S.; Budagumpi S.; Pai R. K.; Balakrishna R. G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340. 10.1016/j.ejmech.2014.03.047. [DOI] [PubMed] [Google Scholar]
- Ismail K. A.; Abd El Aziem T. Synthesis and biological evaluation of some novel 4H-benzopyran-4-one derivatives as nonsteroidal antiestrogens. Eur. J. Med. Chem. 2001, 36, 243. 10.1016/s0223-5234(01)01218-1. [DOI] [PubMed] [Google Scholar]
- Maiti A.; Cuendet M.; Kondratyuk T.; Croy V. L.; Pezzuto J. M.; Cushman M. Synthesis and Biological Evaluation of (+/-)-abyssinone II and Its Analogues as Aromatase Inhibitors for Chemoprevention of Breast Cancer. J. Med. Chem. 2007, 50, 350. 10.1021/jm060915+. [DOI] [PubMed] [Google Scholar]
- Babu T. H.; Subba Rao V. R.; Tiwari A. K.; Suresh Babu K.; Srinivas P. V.; Ali A. Z.; Rao J. M. Synthesis and biological evaluation of novel 8-aminomethylated oroxylin A analogues as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1659. 10.1016/j.bmcl.2008.01.055. [DOI] [PubMed] [Google Scholar]
- Netzer N. C.; Küpper T.; Voss H. W.; Eliasson A. H. The actual role of sodium cromoglycate in the treatment of asthma--a critical review, Sleep Breath. Sleep Breath. 2012, 16, 1027. 10.1007/s11325-011-0639-1. [DOI] [PubMed] [Google Scholar]
- Rowe B. H.; Kelly K.; Spooner C. H. Early Emergency Department Treatment of Acute Asthma With Systemic Corticosteroids. Cochrane Database Syst. Rev. 2000, 4, 2731. 10.1002/14651858.cd002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musthafa T. N. M.; Siddiqui Z. N.; Husain F. M.; Ahmad I. Microwave-assisted solvent-free synthesis of biologically active novel heterocycles from 3-formylchromones. Med. Chem. Res. 2012, 20, 1473. 10.1007/s00044-010-9386-2. [DOI] [Google Scholar]
- Islam M. N.; Jung H. A.; Sohn H. S.; Kim H. M.; Choi J. S. Potent α-glucosidase and protein tyrosine phosphatase 1B inhibitors from Artemisia capillaris. Arch Pharm. Res. 2013, 36, 542. 10.1007/s12272-013-0069-7. [DOI] [PubMed] [Google Scholar]
- Kuroda M.; Uchida S.; Watanabe K.; Mimaki Y. Chromones from the tubers of Eranthis cilicica and their antioxidant activity. Phytochem 2009, 70, 288. 10.1016/j.phytochem.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Zhao P.-L.; Li J.; Yang G.-F. Synthesis and insecticidal activity of chromanone and chromone analogues of diacylhydrazines. Bioorg. Med. Chem. 2007, 15, 1888. 10.1016/j.bmc.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Lei J.; Li Y.; He L.-J.; Luo Y.-F.; Tang D.-Y.; Yan W.; Lin H.-K.; Li H.-Y.; Chen Z.-Z.; Xu Z.-G. Expeditious access of chromone analogues via a Michael addition-driven multicomponent reaction. Org. Chem. Front. 2020, 7, 987. 10.1039/d0qo00145g. [DOI] [Google Scholar]
- Gentili B.; Horowitz R. M. Flavonoids of citrus-VII : Limocitrol and isolimocitrol. Tetrahedron 1964, 20, 2313. 10.1016/s0040-4020(01)97619-7. [DOI] [Google Scholar]
- Ranganna S.; Govindarajan V. S.; Ramana K. V. R.; Kefford J. F. Citrus fruits — Varieties, chemistry, technology, and quality evaluation. Part II. Chemistry, technology, and quality evaluation. A. Chemistry. Crit. Rev. Food Sci. Nutr. 1983, 18, 313. 10.1080/10408398309527366. [DOI] [PubMed] [Google Scholar]
- Govindarajan V. S.; Connell D. W. Ginger-chemistry, Technology, and Quality Evaluation: Part 2. Crit. Rev. Food Sci. Nutr. 1982, 17, 189. 10.1080/10408398209527348. [DOI] [PubMed] [Google Scholar]
- Govindarajan V. S. Ginger — chemistry, technology, and quality evaluation: Part 1. Crit. Rev. Food Sci. Nutr. 1982, 17, 1. 10.1080/10408398209527343. [DOI] [PubMed] [Google Scholar]
- Hostetler G. L.; Ralston R. A.; Schwartz S. J. Flavones: Food Sources, Bioavailability, Metabolism, and Bioactivity. Adv. Nutr. 2017, 8, 423. 10.3945/an.116.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimble M. A.; Gibson J. S.; Sperry J. Pyrans and their Benzo Derivatives: Synthesis. Compr. Heterocycl. Chem. II 2008, 7, 419. 10.1016/b978-008044992-0.00608-8. [DOI] [Google Scholar]
- Zhong Y.-L.; Boruta D. T.; Gauthier D. R.; Askin D. An efficient synthesis of 4-chromanones. Tetrahedron Lett. 2011, 52, 4824. 10.1016/j.tetlet.2011.07.018. [DOI] [Google Scholar]
- Demirayak S.; Yurttas L.; Gundogdu-Karaburun N.; Karaburun A. C.; Kayagil I. New chroman-4-one/thiochroman-4-one derivatives as potential anticancer agents. Saudi Pharm. J. 2017, 25, 1063. 10.1016/j.jsps.2017.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bam R.; Chalifoux W. A. One-Pot Domino Friedel–Crafts Acylation/Annulation between Alkynes and 2-Methoxybenzoyl Chlorides: Synthesis of 2,3-Disubstituted Chromen-4-one Derivatives. J. Org. Chem. 2018, 83, 9929. 10.1021/acs.joc.8b01357. [DOI] [PubMed] [Google Scholar]
- Kim H. Y.; Song E.; Oh K. Unified Approach to (Thio)chromenones via One-Pot Friedel–Crafts Acylation/Cyclization: Distinctive Mechanistic Pathways of β-Chlorovinyl Ketones. Org. Lett. 2017, 19, 312. 10.1021/acs.orglett.6b03348. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Nachtsheim B. J. A review of new developments in the Friedel–Crafts alkylation – From green chemistry to asymmetric catalysis. Beilstein J. Org. Chem. 2010, 6, 6. 10.3762/bjoc.6.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori G.; Maggi R.. Advances in Friedel–Crafts Acylation Reactions: Catalytic and Green Processes; CRC Press: Boca Raton, 2009. [Google Scholar]
- Kabalka G. W.; Mereddy A. R. Microwave-assisted synthesis of functionalized flavones and chromones. Tetrahedron Lett. 2005, 46, 6315. 10.1016/j.tetlet.2005.07.038. [DOI] [Google Scholar]
- Wen L.-R.; Jin X.-J.; Niu X.-D.; Li M. Copper-Catalyzed Tandem Reactions for Synthesis of Pyrazolo[5,1-a]isoquinolines with Heterocyclic Ketene Aminals as Ligands. J. Org. Chem. 2015, 80, 90. 10.1021/jo5020323. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Saito K.; Fujino Y.; Doi T. A concise total synthesis of biologically active frutinones via tributylphosphine-catalyzed tandem acyl transfer-cyclization. Tetrahedron 2014, 70, 3452. 10.1016/j.tet.2014.03.073. [DOI] [Google Scholar]
- Wu X.-F.; Neumann H.; Beller M. Palladium-Catalyzed Carbonylation Reaction of Aryl Bromides with 2-Hydroxyacetophenones to Form Flavones. Chem.—Eur. J. 2012, 18, 12595. 10.1002/chem.201202141. [DOI] [PubMed] [Google Scholar]
- Baruah S.; Kaishap P. P.; Gogoi S. Palladium-Catalyzed Carbonylation Reaction of Aryl Bromides with 2-Hydroxyacetophenones to Form Flavones. Chem. Commun. 2016, 52, 13004. 10.1039/c6cc07204f. [DOI] [PubMed] [Google Scholar]
- Liang B.; Huang M.; You Z.; Xiong Z.; Lu K.; Lu R.; Fathi R.; Chen J.; Yang Z. Pd-Catalyzed Copper-Free Carbonylative Sonogashira Reaction of Aryl Iodides with Alkynes for the Synthesis of Alkynyl Ketones and Flavones by Using Water as a Solvent. J. Org. Chem. 2005, 70, 6097. 10.1021/jo050498t. [DOI] [PubMed] [Google Scholar]
- Awuah E.; Capretta A. Access to Flavones via a Microwave-Assisted, One-Pot Sonogashira–Carbonylation–Annulation Reaction. Org. Lett. 2009, 11, 3210. 10.1021/ol901043q. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Alper H. Synthesis of Chromones via Palladium-Catalyzed Ligand-Free Cyclocarbonylation of o-Iodophenols with Terminal Acetylenes in Phosphonium Salt Ionic Liquids. J. Org. Chem. 2010, 75, 948. 10.1021/jo902210p. [DOI] [PubMed] [Google Scholar]
- Liu J.; Liu M.; Yue Y.; Zhang N.; Zhang Y.; Zhuo K. Construction of the flavones and aurones through regioselective carbonylative annulation of 2-bromophenols and terminal alkynes. Tetrahedron Lett. 2013, 54, 1802. 10.1016/j.tetlet.2013.01.043. [DOI] [Google Scholar]
- Zhou B.; Wu Z.; Ma D.; Ji X.; Zhang Y. Synthesis of indoles through Palladium-catalyzed three-component reaction of aryl iodides, alkynes, and diaziridinone. Org. Lett. 2018, 20, 6440. 10.1021/acs.orglett.8b02750. [DOI] [PubMed] [Google Scholar]
- Guo M.; Wei Z.; Yang J.; Xie Z.; Zhang W. β-Alkynone accelerated PPM level Pd-catalyzed Sonogashira coupling reaction. Catalysts 2020, 10, 302. 10.3390/catal10030302. [DOI] [Google Scholar]
- Yang D.; Wang Z.; Wang X.; Sun H.; Xie Z.; Fan J.; Zhang G.; Zhang W.; Gao Z. Pd catalyzed couplings of “superactive esters” and terminal alkynes: Application to flavones and γ-benzopyranones construction. J. Mol. Catal. A: Chem. 2017, 426, 24. 10.1016/j.molcata.2016.10.030. [DOI] [Google Scholar]
- Xue L.; Shi L.; Han Y.; Xia C.; Huynh H. V.; Li F. Pd–carbene catalyzed carbonylation reactions of aryl iodides. Dalton Trans. 2011, 40, 7632. 10.1039/c1dt10433k. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Wang Z.; Wu X.-F. Transition-Metal-Catalyzed Carbonylative Synthesis and Functionalization of Heterocycles. Chin. J. Org. Chem. 2019, 39, 573. 10.6023/cjoc201809004. [DOI] [Google Scholar]
- Qi X.; Li R.; Wu X.-F. Selective palladium-catalyzed carbonylative synthesis of aurones with formic acid as the CO source. RSC Adv. 2016, 6, 62810. 10.1039/c6ra13615j. [DOI] [Google Scholar]
- Miao H.; Yang Z. Regiospecific carbonylative annulation of iodophenol acetates and Acetylenes To Construct the Flavones by a New Catalyst of Palladium–Thiourea–dppp complex. Org. Lett. 2000, 2, 1765. 10.1021/ol000087t. [DOI] [PubMed] [Google Scholar]
- Zhu F.; Yahui Li L.; Zechao W.; Xiao-Feng W. Highly efficient synthesis of flavones via Pd/C-catalyzed cyclocarbonylation of 2-iodophenol with terminal acetylenes. Catal. Sci. Technol. 2016, 6, 2905. 10.1039/c6cy00613b. [DOI] [Google Scholar]
- Aktaşa A.; Barut D.; Ruya C.; Parham K.; Yetkin T.; Muhittin G.; Gülçinc A. İ. novel morpholine liganded Pd-based N-heterocyclic carbene complexes: Synthesis, characterization, crystal structure, antidiabetic and anticholinergic properties. Polyhedron 2019, 159, 345. 10.1016/j.poly.2018.11.048. [DOI] [Google Scholar]
- Li Y.; Jin G. F.; An Y. Y.; Das R.; Han Y. F. metal-carbene-templated photochemistry in solution: a universal route towards cyclobutane derivatives. Chin. J. Chem. 2019, 37, 1147–1152. 10.1002/cjoc.201900348. [DOI] [Google Scholar]
- Nguyen V. H.; Ibrahim M. B.; Mansour W. W.; El Ali B. M.; Huynh H. V. postmodification approach to charge-tagged 1,2,4-triazole-derived NHC Palladium(II) complexes and their applications. Organometallics 2017, 36, 2345. 10.1021/acs.organomet.7b00329. [DOI] [Google Scholar]
- Kumar A.; Yuan D.; Huynh H. V. stereoelectronic profiling of expanded-ring N-heterocyclic carbenes. Inorg. Chem. 2019, 58, 7545. 10.1021/acs.inorgchem.9b00786. [DOI] [PubMed] [Google Scholar]
- Li Y.; An Y. Y.; Fan X. X.; Liu X.-X.; Li X.; Hahn Y. Y.; Wang Y.-Y.; Han Y.-F. . Strategy for the Construction of Diverse Poly-NHC-Derived Assemblies and Their Photoinduced Transformations. Angew. Chem., Int. Ed. 2020, 59, 10073–10080. 10.1002/anie.201912322. [DOI] [PubMed] [Google Scholar]
- Tao W.; Wang X.; Ito S.; Nozaki K. Palladium complexes bearing an N-heterocyclic carbene–sulfonamide ligand for cooligomerization of ethylene and polar monomers. J. Polym. Sci., Part A: Polym. Chem. 2019, 57, 474. 10.1002/pola.29270. [DOI] [Google Scholar]
- Kostyukovich A. Y.; Tsedilin A. M.; Sushchenko E. D.; Eremin D. B.; Kashin A. S.; Topchiy M. A.; Asachenko A. F.; Nechaev M. S.; Ananikov V. P. In situ transformations of Pd/NHC complexes with N-heterocyclic carbene ligands of different nature into colloidal Pd nanoparticles. Inorg. Chem. Front. 2019, 6, 482. 10.1039/c8qi01095a. [DOI] [Google Scholar]
- Wang T.; Xie H.; Liu L.; Zhao W.-X. N-heterocyclic carbene-palladium(II) complexes with benzoxazole or benzothiazole ligands: Synthesis, characterization, and application to Suzuki–Miyaura cross-coupling reaction. J. Organomet. Chem. 2016, 804, 73. 10.1016/j.jorganchem.2015.12.039. [DOI] [Google Scholar]
- Ma X.; Wang H.; Chen W. N-Heterocyclic carbene-stabilized Palladium complexes as organometallic catalysts for bioorthogonal cross-coupling reactions. J. Org. Chem. 2014, 79, 8652. 10.1021/jo5014228. [DOI] [PubMed] [Google Scholar]
- Hopkinson M. N.; Richter C.; Schedler M.; Glorius F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Nelson D. J.; Nolan S. P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. 10.1039/c3cs60146c. [DOI] [PubMed] [Google Scholar]
- Huynh H. V.; Jothibasu R. Syntheses and catalytic activities of Pd(II) dicarbene and hetero-dicarbene complexes. J. Organomet. Chem. 2011, 696, 3369. 10.1016/j.jorganchem.2011.07.018. [DOI] [Google Scholar]
- Mansour W.; Fettouhi M.; El Ali B. Novel and efficient bridged bis(N -heterocyclic carbene)palladium(II) catalysts for selective carbonylative Suzuki–Miyaura coupling reactions to biaryl ketones and biaryl diketones. Appl. Organomet. Chem. 2020, 34, e5636 10.1002/aoc.5636. [DOI] [Google Scholar]
- Sakaguchi S.; Yoo K. S.; O’Neill J.; Lee J. H.; Stewart T.; Jung K. W. Chiral Palladium(II) complexes possessing a tridentate N-heterocyclic carbene amidate alkoxide ligand: access to oxygen-bridging dimer structures. Angew. Chem., Int. Ed. 2008, 47, 9326. 10.1002/anie.200803793. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.; Malik I.; Mansour W.; Sharif M.; Fettouhi M.; El Ali B. Efficient N-heterocyclic carbene palladium(II) catalysts for carbonylative Suzuki-Miyaura coupling reactions leading to aryl ketones and diketones. J. Organomet. Chem. 2018, 859, 44. 10.1016/j.jorganchem.2018.01.028. [DOI] [Google Scholar]
- Ibrahim M.; Malik I.; Mansour W. W.; Sharif M.; Fettouhi M.; El Ali B. Novel (N-heterocyclic carbene)Pd(pyridine)Br2 complexes for carbonylative Sonogashira coupling reactions: Catalytic efficiency and scope for arylalkynes, alkylalkynes and dialkynes. Appl. Organomet. Chem. 2018, 32, e4280 10.1002/aoc.4280. [DOI] [Google Scholar]
- Mansour W.; Suleiman R.; Fettouhi M.; El Ali B. Soft Heteroleptic N-Heterocyclic Carbene Palladium(II) Species for Efficient Catalytic Routes to Alkynones via Carbonylative Sonogashira Coupling. ACS Omega 2020, 5, 23687–23702. 10.1021/acsomega.0c02413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M. G.; McGuinness D. S.; Vanston C. R. Chelated bis(NHC) complexes of saturated (imidazolin-2-ylidene) NHC ligands: structural authentication and facile ligand fragmentation. Dalton Trans. 2017, 46, 3051. 10.1039/c7dt00327g. [DOI] [PubMed] [Google Scholar]
- Hao W.; Sha J. C.; Sheng S. R.; Cai M. Z. The first heterogeneous carbonylative Sonogashira coupling reaction catalyzed by MCM-41-supported bidentate phosphine palladium(0) complex. J. Mol. Catal. A: Chem. 2009, 298, 94. 10.1016/j.molcata.2008.09.031. [DOI] [Google Scholar]
- Mohapatra S.; Baral N.; Mishra N. P.; Panda P.; Nayak S. Michael Addition of Imidazole to α, β -Unsaturated Carbonyl/Cyano Compound. Open Chem. 2018, 5, 18. 10.2174/1874842201805010018. [DOI] [Google Scholar]
- Boncel S.; Saletra K.; Hefczyc B.; Walczak K. Z. Michael-type addition of azoles of broad-scale acidity to methyl acrylate. Beilstein J. Org. Chem. 2011, 7, 173. 10.3762/bjoc.7.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruker . Apex3 v2017.3-0, SAINT V8.38A; Bruker AXS Inc.: Madison (WI), USA, 2017.
- Krause L.; Herbst-Irmer R.; Sheldrick G. M.; Stalke D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3. 10.1107/s1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. 10.1107/s0021889897003117. [DOI] [Google Scholar]
- Zhao X.; Zhou J.; Lin S.; Jin X.; Liu R. C–H functionalization via remote hydride elimination: Palladium catalyzed dehydrogenation of ortho-acyl phenols to flavonoids. Org. Lett. 2017, 19, 976. 10.1021/acs.orglett.6b03652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.