

Summary
The incorporation of resistance genes into wheat commercial varieties is the ideal strategy to combat stripe or yellow rust (YR). In a search for novel resistance genes, we performed a large‐scale genomic association analysis with high‐density 660K single nucleotide polymorphism (SNP) arrays to determine the genetic components of YR resistance in 411 spring wheat lines. Following quality control, 371 972 SNPs were screened, covering over 50% of the high‐confidence annotated gene space. Nineteen stable genomic regions harbouring 292 significant SNPs were associated with adult‐plant YR resistance across nine environments. Of these, 14 SNPs were localized in the proximity of known loci widely used in breeding. Obvious candidate SNP variants were identified in certain confidence intervals, such as the cloned gene Yr18 and the major locus on chromosome 2BL, despite a large extent of linkage disequilibrium. The number of causal SNP variants was refined using an independent validation panel and consideration of the estimated functional importance of each nucleotide polymorphism. Interestingly, four natural polymorphisms causing amino acid changes in the gene TraesCS2B01G513100 that encodes a serine/threonine protein kinase (STPK) were significantly involved in YR responses. Gene expression and mutation analysis confirmed that STPK played an important role in YR resistance. PCR markers were developed to identify the favourable TraesCS2B01G513100 haplotype for marker‐assisted breeding. These results demonstrate that high‐resolution SNP‐based GWAS enables the rapid identification of putative resistance genes and can be used to improve the efficiency of marker‐assisted selection in wheat disease resistance breeding.
Keywords: common wheat (Triticum aestivum L.), stripe rust, 660K SNP array, GWAS, candidate region association analysis, serine/threonine protein kinase (STPK), marker‐assisted breeding
Introduction
Stripe rust or yellow rust (YR) caused by Puccinia striiformis f. sp. tritici (Pst) is an important foliar disease that has been associated with up to 100% yield losses in wheat (Chen, 2014). The incorporation of resistance genes into commercial varieties is the ideal strategy to combat YR. With our gradually improved understanding of 'durable resistance', greater emphases are being placed on adult‐plant resistance (APR) or high‐temperature adult‐plant resistance (HTAPR), which are affected by actual growth stage and temperature (Niks et al., 2015). Quantitative genetics analyses have shown that APR is usually controlled by multiple inherited loci (Chen, 2013). The best‐known Pst‐APR genes are Yr18, Yr36 and Yr46, which confer a degree of resistance to multiple races and have been cloned (Fu et al., 2009; Krattinger et al., 2009; Moore et al., 2015). Studies on the molecular genetics of APR have indicated that there is more than one model for the mechanism of durable resistance (Brown, 2015). Thus, it is vital for researchers and breeders to understand the genetic basis of stripe rust resistance in current elite breeding populations and continuously search for novel genes.
However, quantitative resistance is based on multiple loci, each with a small effect, thus increasing the difficulty of identification. The large genome size and allopolyploidy of common wheat result in complex quantitative inheritance of APR and cause slow progress in breeding for APR (Sánchez‐Martín and Keller, 2019). With important advances in high‐throughput sequencing and wheat genomic sequencing, large numbers of molecular markers have been developed that facilitate the progress of more efficient mapping techniques (Juliana et al., 2019). In particular, genome‐wide association studies (GWAS) can identify associations between phenotypic variation and nucleotide polymorphisms using a diverse population panel (Bazakos et al., 2017). As numerous natural allelic variations can be simultaneously detected in a single study and a large number of historical chromosomal recombination events occur over multiple generations of natural populations, GWAS is becoming a powerful tool to dissect the genetic basis of complex agronomic traits and identify potential causal genes (Xu et al., 2017).
On the other hand, the identification of causal genes that underlie agronomic traits directly from GWAS results remains difficult. First, population genetic structure can limit the detection of rare allele variants and occasionally generate false associations between phenotype and non‐causal genes (Bazakos et al., 2017). Although several statistically robust models have been built, such as population structure assessment and correction, false positives caused by population structure may not be entirely eliminated (Kang et al., 2008; Yu et al., 2006). To address this problem, another independent population can be reconstructed to validate the resulting marker–trait associations (MTAs) (Lipka et al., 2015). Second, a large extent of linkage disequilibrium (LD) can give rise to a single LD block that displays a remarkable association with the trait of interest but harbours a variety of candidate genes (Schaid et al., 2018). Typically, the extent of LD in self‐pollinating crops spans several hundred kilobases (kb), as documented in rice (Huang et al., 2011), and can occasionally reach megabases (Mb), as in wheat (Cheng et al., 2019; Wu et al., 2020). Thus, with high‐level LD, further investigation is necessary to conclusively identify the causal gene(s). Recently, an efficient GWAS method using whole‐genome sequencing (WGS) was developed in rice for the rapid identification of trait causal genes without the need for additional experiments, based on the estimated functional importance of each nucleotide polymorphism (Yano et al., 2016). Similar practices have been used in wheat. For example, four stem rust resistance genes were rapidly cloned through a combination of association genetics and R gene enrichment sequencing (AgRenSeq) (Arora et al., 2019). Exome association mapping provided another route for the detection of functional SNP variants in wheat leaf rust resistance (Liu et al., 2020). In addition, high‐density SNP array analysis has also become an alternative approach for the refinement of candidate genes. The updated version of the wheat 660K SNP array includes 660 009 SNP sites distributed over all chromosomes and encompassing the majority of genes (Sun et al., 2020). It has been widely used in many genetic studies that are focused on gene fine‐mapping and cloning in China (Li et al., 2019; Rasheed et al., 2017).
Recently, members of the post‐Yr26‐virulent races group (herein referred to as post‐V26) have become the most prevalent forms of Pst that threaten wheat production in China (McIntosh et al., 2018). Unexploited wheat germplasm is a potentially valuable source of genetic diversity that can enhance and enrich breeding germplasm with needed traits for the sustainable improvement of modern cultivars (Hao et al., 2011; Zhuang, 2003). Here, a diversity panel of 411 advanced breeding lines were collected from International Maize and Wheat Improvement Center (CIMMYT) and International Centre for Agricultural Research in the Dry Areas (ICARDA) bread‐wheat breeding programmes, which were expected to have effective and novel resistance genes, thus making them ideal for association mapping. Then, they were evaluated for their responses to post‐V26 races in seedling and in multi‐location field trials with plants either artificially or naturally inoculated over three cropping seasons. Subsequently, we used GWAS to dissect the genetic architecture of these lines and detect QTL associated with variation in stripe rust resistance. Using high‐resolution SNPs from the wheat 660K SNP array, resequencing data and PCR‐based sequencing data, we attempted to refine the number of causal alleles based on analysis of another independent validation panel of over 1000 wheat accessions, as well as analysis of the estimated functional importance of each nucleotide polymorphism. Our study describes promising results that will accelerate marker‐assisted selection for the improvement of stripe rust resistance in Chinese wheat breeding programmes and delineate prospective targets for the cloning of novel resistance genes.
Results
Genotyping by SNP array reveals abundant genetic diversity
After filtering out low‐quality SNP markers, a total of 378,441 SNPs were retained and used for the following analyses. A position could be assigned to 371,972 SNPs, which were distributed over each of the 21 chromosomes (Figure S1a; Table 1; Table S2). Marker density varied among chromosomes with a minimum of 7.19 markers per Mb on chromosome 4D and a maximum of 53.54 markers per Mb on chromosome 3B (Table 1; Figure S1b). In addition, these filtered SNPs were used in a BLAST analysis of the ‘Chinese Spring’ reference genome to analyse and predict their effects on gene structure and function. BLAST analysis revealed that out of the 378 441 SNPs, 41 588 (10.99%) were intron variants and 162 080 (42.82%) mapped within 2 kb upstream or downstream of genic regions. The remaining SNPs were located in gene exons, with 28 728 (7.59%) of these SNPs causing non‐synonymous mutations. Finally, we identified the SNPs associated with 57 833 genes, which accounted for 53.6% of all high‐confidence genes, and found that 36 042 genes (62.32%) possessed at least two SNPs (Table S3; Figure S1c).
Table 1.
Summary of the genetic diversity in the subgenomes and chromosomes of 411 wheat accessions and evaluation of the effective number of independent SNPs, including suggested P‐value thresholds
| Chromosome | No. of markers | Effective number | Suggested P‐value | % markers | Length (Mb) | Marker density | He | PIC | LD (Mb) |
|---|---|---|---|---|---|---|---|---|---|
| 1A | 23,120 | 5,255 | 1.90E‐04 | 6.11 | 594.02 | 38.92 | 0.68 | 0.27 | 2.37 |
| 2A | 26,079 | 5,825 | 1.72E‐04 | 6.89 | 780.76 | 33.40 | 0.75 | 0.30 | 2.38 |
| 3A | 16,040 | 4,234 | 2.36E‐04 | 4.24 | 750.73 | 21.37 | 0.70 | 0.28 | 1.64 |
| 4A | 19,191 | 4,170 | 2.40E‐04 | 5.07 | 744.54 | 25.78 | 0.66 | 0.27 | 4.06 |
| 5A | 20,430 | 4,533 | 2.21E‐04 | 5.40 | 709.76 | 28.78 | 0.74 | 0.30 | 4.14 |
| 6A | 15,935 | 3,554 | 2.81E‐04 | 4.21 | 617.97 | 25.79 | 0.69 | 0.28 | 2.40 |
| 7A | 24,184 | 6,124 | 1.63E‐04 | 6.39 | 736.69 | 32.83 | 0.65 | 0.26 | 2.03 |
| 1B | 19,684 | 4,748 | 2.11E‐04 | 5.20 | 689.38 | 28.55 | 0.68 | 0.27 | 4.15 |
| 2B | 27,097 | 6,988 | 1.43E‐04 | 7.16 | 801.25 | 33.82 | 0.70 | 0.28 | 2.88 |
| 3B | 44,479 | 7,352 | 1.36E‐04 | 11.75 | 830.7 | 53.54 | 0.57 | 0.23 | 7.45 |
| 4B | 12,690 | 2,231 | 4.48E‐04 | 3.35 | 673.47 | 18.84 | 0.71 | 0.28 | 3.09 |
| 5B | 31,251 | 6,135 | 1.63E‐04 | 8.26 | 713.02 | 43.83 | 0.72 | 0.29 | 4.35 |
| 6B | 21,231 | 5,523 | 1.81E‐04 | 5.61 | 720.95 | 29.45 | 0.72 | 0.29 | 2.95 |
| 7B | 16,396 | 3,221 | 3.10E‐04 | 4.33 | 750.61 | 21.84 | 0.68 | 0.27 | 3.29 |
| 1D | 10,210 | 2,561 | 3.90E‐04 | 2.70 | 495.44 | 20.61 | 0.71 | 0.28 | 1.98 |
| 2D | 10,119 | 3,453 | 2.90E‐04 | 2.67 | 651.81 | 15.52 | 0.72 | 0.29 | 3.28 |
| 3D | 6,748 | 6,424 | 1.56E‐04 | 1.78 | 615.48 | 10.96 | 0.62 | 0.25 | 2.29 |
| 4D | 3,664 | 1,024 | 9.77E‐04 | 0.97 | 509.85 | 7.19 | 0.68 | 0.27 | 0.89 |
| 5D | 7,190 | 2,227 | 4.49E‐04 | 1.90 | 566.04 | 12.70 | 0.66 | 0.27 | 0.86 |
| 6D | 7,052 | 2,242 | 4.46E‐04 | 1.86 | 473.56 | 14.89 | 0.69 | 0.27 | 0.62 |
| 7D | 9,182 | 1,010 | 9.90E‐04 | 2.43 | 638.65 | 14.38 | 0.67 | 0.27 | 1.09 |
| A genome | 144,979 | 38.31 | 4934.47 | 29.38 | 0.70 | 0.28 | 2.60 | ||
| B genome | 172,828 | 45.67 | 5179.38 | 33.37 | 0.68 | 0.27 | 5.50 | ||
| D genome | 54,165 | 14.31 | 3950.83 | 13.71 | 0.68 | 0.27 | 1.30 | ||
| Total | 371,972 | 14064.68 | 26.45 | 0.69 | 0.28 | 3.20 | |||
| Average | 3.23E‐04 |
Genetic diversity was analysed using markers with known chromosomal positions. Overall, the spring wheat diversity panel showed relatively high genetic diversity, with He and PIC values of all genomes of 0.69 and 0.28, respectively (Table 1; Figure S1d, e). Although the D genome possessed fewer markers than the A and B genomes, there was little difference in genetic diversity among the three genomes. The mean values of He and PIC for the three subgenomes were 0.68–0.70 and 0.27–0.28, respectively (Table 1). The genetic diversity results of the validation panel of 1,045 accessions with 660K SNP array data are provided in Table S4. Approximately 75% of the markers displayed PIC values exceeding 0.20, demonstrating the informativeness of these markers.
Estimation of population structure and linkage disequilibrium
According to the ΔK method of Bayesian clustering, hierarchical clustering, kinship analysis, phylogenetic tree construction and PCA analyses, geographical origin (Africa and America) and historical era (old landraces to modern elite lines) were the two major factors that determined the classification of diversity in this panel. The population of 411 accessions was first structured into two main subpopulation groups based on geographical distribution (herein referred to as Sp1 and Sp2) (Figure 1a–e). Sp1 was frequently associated with Mediterranean countries such as Morocco and Egypt, whereas Sp2 predominantly included accessions from countries in America, South Asia and Oceania, such as Mexico, the United States, India and Australia. In view of different eras, Sp1 and Sp2 were further subdivided into two and three distinct subgroups (herein referred to as Sp1‐1, Sp1‐2, Sp2‐1, Sp2‐2 and Sp2‐3), respectively (Figure 1c–e). Sp1‐1 consisted of 68 early ICARDA varieties, and Sp1‐2 included a mixture of 176 modern varieties and breeding lines. Sp2‐1, Sp2‐2 and Sp2‐3 contained a mixture of 88 modern varieties and breeding lines, 37 genetic stocks, and a mixture of 42 landraces and earlier varieties. The STRUCTURE membership coefficients revealed a high degree of admixture in a large number of accessions, particularly among cultivars and modern breeding materials, which was mainly observed in a number of lines from other breeding programmes in each cluster. This result is consistent with the shuttle breeding of CIMMYT and ICARDA and the frequent germplasm exchange that characterizes modern wheat breeding worldwide.
Figure 1.
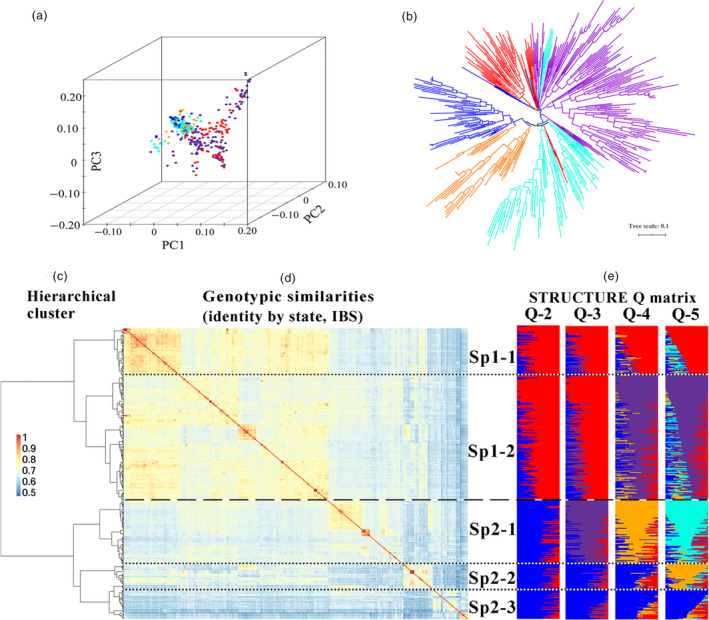
Population structure of the 411 included wheat accessions. (a) Principal components analysis (PCA). (b) Neighbour‐joining tree analysis. (c, d, e) Subgroups inferred by hierarchical clustering, kinship and structure analysis. Vertical lines indicate genetic similarity thresholds used to classify accessions into two main groups (dashed lines) and five subgroups (dotted lines). (d) 411 × 411 kinship matrix based on a simple matching of genetic similarities (IBS, identity by state). Separations among hierarchical‐based groups are shown as horizontal dashed lines for main groups and as dotted‐dashed lines for subgroups. (e) Matrices of membership coefficients of accessions corresponding to 2–5 hypothetical subpopulations derived from the STRUCTURE analysis.
LD analysis was assessed based on 717 701 068 pairwise comparisons of 371,972 SNPs, and pairwise LD was estimated using the squared‐allele frequency correlation (r 2). A plot of the LD estimates (r 2) as a function of physical distance in Mb indicated that there was a clear decay of LD with physical distance (Figure S2). Comparison of LD among subgenomes and chromosomes showed that the LD decay was varied. Overall, the average LD decay distance for the whole genome was approximately 3.2 Mb. LD decayed faster in the D genome (1.3 Mb) than in the A (2.6 Mb) and B (5.5 Mb) genomes (Figure S2b–d). We believe that faster LD decay in the D genome is compatible with wheat evolutionary history (Dubcovsky and Dvorak, 2007). The D genome was the last be incorporated into common wheat and was therefore subjected to less artificial selection than the A and B genomes. Several haplotype blocks harbouring favourable alleles or combinations of alleles tend to be stable, and the LD decay distance is increased through artificial selection‐driven evolution. For example, in this study, the LD decay of all chromosomes ranged from 0.62 Mb to over 7.45 Mb, indicating that different genomic regions have been subjected to various artificial selections and that the haplotype diversity is expansive in this diversity panel (Figure S2).
Phenotypic variation in response to stripe rust
In the stripe rust resistance tests with six Pst races at the seedling stage, the infection type (IT) distributions skewed towards susceptible scores with mean IT values of 7.6–8.1 on a 0–9 scale (Figure S3; Table S5). In the spring wheat diversity panel assessed here, because less than 5% of individuals exhibited resistant reactions (IT 0–3) to each of the tested Pst races, the seedling phenotypes were not used for GWAS analysis to minimize false positive errors. By contrast, greater stripe rust resistance was observed in the field tests at the adult‐plant stage, although the susceptibility checks always indicated high rates of infection. The IT and DS distributions were skewed towards low values in all resistance trials, ranging from 4.4 to 4.9, and 30.0% to 34.7%, respectively (Figure S4a, b; Table S5). IT and DS values were continuous in all environments, indicating that the effects were conferred by APR and the responses were quantitative. Pearson's correlation coefficients were 0.78–0.95 for IT and 0.88–0.96 for DS across all environments (Figure S5a, b). Such significant correlations (P < 0.0001) indicated that stripe rust responses were consistent across the environments and most likely same resistance genes conferred resistance in all environments. As expected, the correlation between IT and DS was highest within the same environment, ranging from 0.80 to 0.91 (Figure S4c). The broad‐sense heritability H 2 was calculated as 0.48 ± 0.07 and 0.55 ± 0.07 for the IT and DS data, respectively (Table S5). The extent to which stripe rust responses of the 411 different accessions were influenced by population structure was analysed, and the accessions in Sp1 generally displayed more resistance than those in Sp2 (Figure S4d, e).
GWAS reveals several significant SNPs associated with known stripe rust resistance loci
The stripe rust responses including IT and DS values of the 411 accessions across nine field environments and the best linear unbiased predictions (BLUPs) were used in association tests based on univariate linear mixed model analysis. Based on the suggested threshold P‐value < 2.90 × 10‐4, 358–801 significant SNP–trait associations were detected. In order to identify stable loci, only QTL associated with APR within at least seven environments including BLUP were considered as high‐confidence QTL, which filtered the common significant SNP–trait associations down to 292 (Table S6). For convenience, tagged SNPs for each QTL were selected based on those exhibiting the strongest association with stripe rust responses alongside the smallest SNP‐associated P‐value, the largest phenotypic variance explained (R 2) and the largest number of environments in which significant trait associations were detected. As a result, a total of 19 QTL regions were identified on chromosome arms 1AL, 1BL, 2AS, 2AL, 2BS, 2BL, 3AL, 3BS, 3BL, 4BS, 4BL, 6BS, 6BL, 7AL, 7BL and 7DS (Figure 2a‐j; Figure S6a‐j; Figure S7). The frequency of SNP marker alleles associated with resistance ranged from 0.06 to 0.93, although marker alleles are not necessarily indicative of functional resistance alleles (Table 2). The phenotypic variation explained (PVE) by individual QTL was 1.5–9.6%, and the total value of PVE contributed by all QTL was 57.8–74.1%. Of the 19 assigned QTL, five loci were potentially novel based on their unique chromosomal locations, determined by referring to the consensus and physical maps, and two of these had been mapped in our previous studies. Among the other 14 QTL, nine were co‐located with characterized Yr genes, namely Yr29 on chromosome arm 1BL, Yr17 on 2AS, Yr32 on 2AL, Yr30 on 3BS, Yr80 on 3BL, Yr78 on 6BS, Yr75 on 7AL, Yr39 or Yr2 on 7BL and Yr18 on 7DS (Figure 2a–j; Figure S7; Table 2). The remaining five QTL identified in this GWAS were in agreement with the candidate regions reported in previous QTL mapping or GWAS studies (Figure S7; Table 2). Moreover, YrNP63‐2BS, YrSnb.1‐2BL, YrRC‐4BL and YrSnb.2‐6BL had been mapped using CIMMYT‐derived bi‐parent populations in our previous studies (Wu et al., 2017, 2018a; Zeng et al., 2019).
Figure 2.
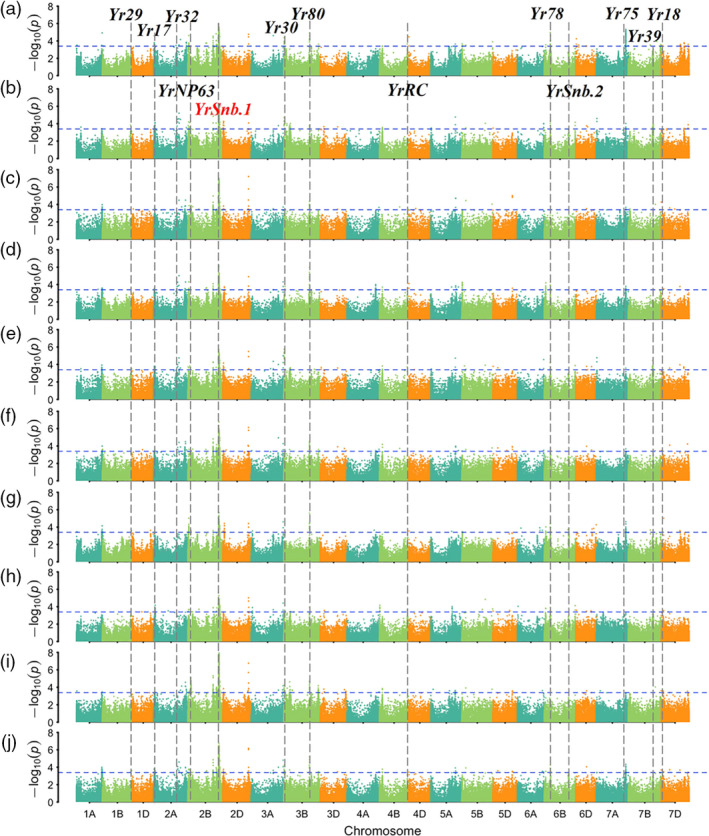
Genome‐wide association analysis results for the severity of stripe rust across ten tested environments. Assessed environments included (a) 2017‐Jiangyou, (b) 2017‐Tianshui, (c) 2017‐Yanlging, (d) 2018‐Jiangyou, (e) 2018‐Tianshui, (f) 2018‐Yanlging, (g) 2019‐Jiangyou, (h) 2019‐Tianshui, (i) 2019‐Yanlging and (j) BLUP (the best linear unbiased predictions). The horizontal line shows the genome‐wide significance threshold –log10 (P) value of 3.4. The A, B and D genomes are in blue‐green, pale green and orange, respectively. The QTL detected in this GWAS panel associated with the known Yr loci are listed in the corresponding chromosome.
Table 2.
Significant quantitative trait loci (QTL) that are associated with adult‐plant stripe rust resistance in multiple environments
| QTL name | Chr. | Tag‐marker | Genetic position (cM) † | Physical position (Mb) | R allele and its ratio ‡ | −Log10 (P) | R 2 (%) | Environments § | Postulated or linked genes |
|---|---|---|---|---|---|---|---|---|---|
| QYr.nwafu‐1AL | 1AL | AX‐108800039 (SNP1) | 278.60 | 587.45 | T/C (0.33) | 3.58–3.99 | 1.9–3.1 | IT and DS: All | IWA3215 |
| QYr.nwafu‐1BL | 1BL | AX‐94947139 (SNP2) | 245.57 | 673.96 | C/T (0.24) | 3.37–3.61 | 1.5–2.7 | DS: All | Yr29 |
| QYr.nwafu‐2AS | 2AS | AX‐109458303 (SNP3) | 3.41 | 24.41 | T/G (0.13) | 3.62–3.82 | 1.7–3.0 | DS: All | Yr17 |
| QYr.nwafu‐2AL.1 | 2AL | AX‐108752496 (SNP4) | 426.3 | 574.70 | T/C (0.63) | 3.81–4.59 | 2.8–4.5 | IT and DS: All | Yr32 |
| QYr.nwafu‐2AL.2 | 2AL | AX‐111630281 (SNP5) | 508.27 | 742.15 | G/A (0.87) | 3.55–4.86 | 3.6–4.9 | IT and DS: All | Novel |
| QYr.nwafu‐2BS | 2BS | AX‐108948038 (SNP6) | 54.636 | 108.67 | G/A (0.06) | 3.40–4.85 | 2.9–4.8 | IT and DS: All | YrNP63 |
| QYr.nwafu‐2BL.1 | 2BL | AX‐109485942 (SNP7) | 86.68 | 576.10 | G/A (0.46) | 3.86–5.00 | 2.9–5.0 | IT and DS: All | Many QTL |
| QYr.nwafu‐2BL.2 | 2BL | AX‐110363517 (SNP8) | 93.34 | 708.24 | G/C (0.61) | 4.90–6.74 | 5.0–9.6 | IT and DS: All | YrSnb.1 (Novel) |
| QYr.nwafu‐3AL | 3AL | AX‐111810295 (SNP9) | 296.22 | 732.09 | C/T (0.93) | 3.50–4.81 | 2.5–4.6 | IT and DS: All | Novel |
| QYr.nwafu‐3BS | 3BS | AX‐94578994 (SNP10) | 2.28 | 2.49 | G/A (0.70) | 3.45–4.32 | 2.5–4.4 | IT and DS: All | Yr30 |
| QYr.nwafu‐3BL.1 | 3BL | AX‐108870372 (SNP11) | 56.892 | 586.15 | G/A (0.86) | 3.90–5.93 | 3.9–6.1 | IT and DS: All | Yr80 |
| QYr.nwafu‐3BL.1 | 3BL | AX‐109534273 (SNP12) | 189.84 | 793.05 | T/C (0.21) | 3.16–3.77 | 2.2–3.8 | IT and DS: All | YrSf |
| QYr.nwafu‐4BS | 4BS | AX‐112287589 (SNP13) | 58.5 | 68.89 | T/C (0.12) | 3.44–5.22 | 2.9–5.1 | IT and DS: All | Novel |
| QYr.nwafu‐4BL | 4BL | AX‐89420204 (SNP14) | 97.83 | 642.70 | G/A (0.80) | 3.40–3.56 | 3.2–3.8 | DS: All | YrRC (Novel) |
| QYr.nwafu‐6BS | 6BS | AX‐110586294 (SNP15) | 46.69 | 149.3 | C/T (0.89) | 3.40–4.52 | 3.4–3.9 | IT and DS: All | Yr78 |
| QYr.nwafu‐6BL | 6BL | AX‐111699663 (SNP16) | 50.1 | 599.08 | C/A (0.18) | 4.28–5.44 | 2.8–4.6 | DS: All | YrSnb |
| QYr.nwafu‐7AL | 7AL | AX‐110983606 (SNP17) | 304.82 | 675.26 | G/A (0.70) | 3.71–4.65 | 3.3–4.8 | IT and DS: All | Yr75 |
| QYr.nwafu‐7BL | 7BL | AX‐110448553 (SNP18) | 119.05 | 553.43 | C/A (0.35) | 3.05‐4.46 | 1.5‐3.8 | DS and IT: ①②③④⑤⑥⑩ | Yr39 + Yr2 |
| QYr.nwafu‐7DS | 7DS | AX‐109857040 (SNP19) | 87.71 | 47.71 | A/G (0.34) | 3.47–3.96 | 3.1–4.2 | DS: All | Yr18 |
Position of each SNP based on 660K wheat consensus map (Cui et al., 2017)
Resistance allele for each QTL is indicated by underlining, along with its ratio
IT: Infection type; DS: Disease severity; Environments, ①: 2017‐Yangling, ②: 2018‐Yangling, ③: 2019‐Yangling, ④: 2017‐Tianshui, ⑤: 2018‐Tianshui, ⑥: 2019‐Tianshui, ⑦: 2017‐Jingyou, ⑧: 2018‐Jianyou, ⑨: 2019‐Jingyou, ⑩: BLUP (the best linear unbiased predictions).
Haplotype and candidate gene analysis for the cloned gene Yr18
To verify the accuracy, reliability and validity of these multi‐environment significant SNPs, the cloned locus Yr18 was selected as an example to demonstrate the a priori experiment of haplotype analysis and revalidation. At the proximal end of the short arm of chromosome 7D, there was a peak close to Yr18. A total of 26 polymorphisms were mapped to a candidate region from 47.379 to 47.711 Mb (332 kb) estimated using pairwise LD correlations (r 2 ≥ 0.6) (Figure 3a‐c). Among these polymorphisms in the candidate region, only one polymorphism was classified as G1 (AX‐94713206) and G3 (AX‐111197303), respectively, no G2 polymorphisms were found, two polymorphisms (AX‐89378255 and AX‐109857040) were classified as G4, and the others were classified as G5. The G1 SNP AX‐94713206 corresponded to a C to T change in the twelfth exon of the TraesCS7D01G080300 ORF, which caused a tyrosine to histidine replacement (Figure 3d; Table S7). TraesCS7D01G080300 encodes an ATP‐binding cassette (ABC) transporter G family protein that is identical to the resistance allele Yr18res (Krattinger et al., 2009). Although the SNP AX‐109857040 is located in an intron, it was significantly associated with resistance, which is consistent with previous studies (Krattinger et al., 2013). Due to the Yr18res 3‐bp deletion in the eleventh exon, the SNP AX‐95209823 (C/G) could not distinguish this site accurately, and thus, it was not a significant MTA. It should be noted that there were two more significant MTAs outside the Yr18 coding region. The accessions carrying haplotype 1 (we herein refer to the haplotype corresponding to Yr18res as ‘1’ and the other as ‘2’) showed more resistance than those carrying haplotype 2 (Figure 3e), which agreed with previous studies concerning the Yr18 locus. The discriminatory effectiveness of these SNPs was validated in the second independent diversity panel of 1045 wheat accessions, and they performed comparably to gene‐specific SNPs (Figure 3e). In addition, a pair of near‐isogenic lines (Hap1: +Yr18, Hap2: −Yr18) in the Avocet S variety background showed different responses to stripe rust in the field (Figure 3f; Table S8). These results indicate that the candidate genes analysis method can identify trait‐associated genes or DNA variants, and, furthermore, that combinations of significant MTAs can help to identify favourable haplotype(s) and improve the efficiency of marker‐assisted selection in wheat breeding.
Figure 3.
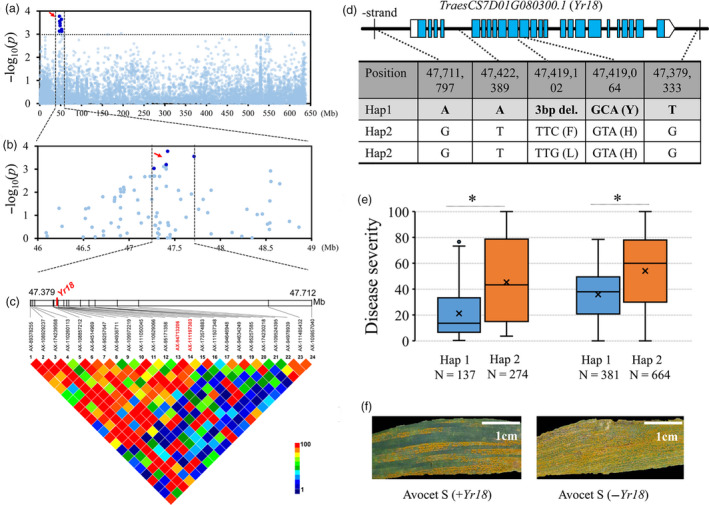
Identification of the causal gene for stripe rust resistance associated with the peak on chromosome 7D. (a) Manhattan plot of single polymorphism‐based association analysis. Dashed line represents a significance threshold (−log10 P = 3.04). Significantly associated single nucleotide polymorphisms (SNPs) are shown as dark blue points, and other SNPs are shown as light blue points. (b, c) Local Manhattan plot of single‐polymorphism‐based association (top) and LD heatmap (bottom) surrounding the peak on chromosome 7D. Arrow indicates the position of nucleotide variations in TraesCS7D01G080300 (Yr18). Dashed lines indicate the candidate region for the peak. (d) The exon‐intron structure of Yr18 and its DNA polymorphisms. del, deletion. (e) Disease severities were based on the haplotypes for Yr18 in different panels of the population. Differences between the haplotypes were statistically analysed using Student's t‐test (*P < 0.05). (f) Stripe rust responses of near‐isogenic lines (±Yr18) in the common wheat cultivar Avocet S background. Scale bar, 1 cm.
Identification of a novel candidate region
Similarly, we analysed the highest peak on chromosome 2B, which was mapped close to YrSnb.1 identified in our QTL mapping. The YrSnb.1 region was previously shown to span an interval of 2.2 cM corresponding to <4 Mb (Zeng et al., 2019). In this GWAS panel, LD analysis within the YrSnb.1 region was initially from 707.418 Mb to 712.236 Mb (4.8 Mb) (Figure 4a). As an experimental control and to more accurately identify candidate regions, GWAS was performed on the independent validation panel, following which the above‐mentioned candidate region was mapped from 707.668 to 708.346 Mb (612.9 kb) (Figure 4b). Association analysis was also performed in the 612.9 kb region using the set of 63 resequencing common wheat genotypes (Figure 5a; Table S9). There were 45 polymorphisms from the SNP array and over 700 polymorphic DNA variants from the resequencing data in this region, covering all 12 candidate high‐confidence (HC) genes (Figure 5b; Table S7). Most of the polymorphisms with significant P‐values surrounded three genes (TraesCS2B01G512900, TraesCS2B01G513000, and TraesCS2B01G513100). Further significant sequence variations were not identified the coding regions of TraesCS2B01G512900 and TraesCS2B01G513000 except TraesCS2B01G513100. Among these, three polymorphisms (AX‐111730867, Rv‐680, and AX‐108806204) that were significantly associated with stripe rust responses (−log10 P ≥ 4.85) were classified as G1, all of which were located within the gene TraesCS2B01G513100 (Figure 5e). These SNPs (AX‐111730867, Rv‐680, and AX‐108806204) changed a G to an A, a C to a T, and a G to an A, causing glutamate to lysine, alanine to valine, and alanine to threonine substitutions, respectively (Table S7). In addition, two polymorphisms were classified as G2 (AX‐110906149 and AX‐109929582) and G3 (AX‐95654572 and AX‐110363517), whereas no G4 polymorphisms were observed and the others were classified as G5 (Figure 5e). The two G2 SNPs AX‐110906149 and AX‐109929582 were both located in the promoter region of TraesCS2B01G513000 and likely affect gene expression regulation (Figure 5d; Table S7). The G3 SNPs AX‐95654572 and AX‐110363517 were located in the 3′ downstream regions of the genes TraesCS2B01G512900 and TraesCS2B01G513100, respectively (Figure 5c, e; Table S7). Interestingly, TraesCS2B01G513100, TraesCS2B01G513000, and TraesCS2B01G512900 all encode serine/threonine protein kinases (STPKs). The amino acid sequences of these STPKs were compared with those in other subgenomes. The allelic variations in different genomes are presented in Figure S8 and show that the STPK sequences in the B genome are obviously different from those in the A and D genomes.
Figure 4.
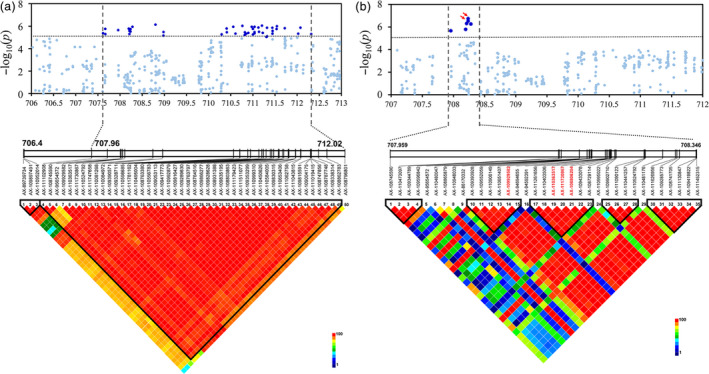
Candidate region associated with stripe rust resistance on chromosome 2B detected in different GWAS panels. Manhattan plots showing the significant SNP associations for the QTL underlying stripe rust resistance detected within (a) an extensive genomic region (707.418–712.236 Mb) using a panel of 411 spring wheat lines and (b) a refined region (707.668 to 708.346) using an independent validation population of 1045 wheat accessions. Grey horizontal dashed line represents a significance threshold (−log10 P = 4.85). Significantly associated SNPs in the two data sets are shown as dark blue points, and other SNPs are shown as light blue points. The upper‐triangular halves of the linkage disequilibrium (LD, as r 2) matrices between SNPs within the candidate region are shown as heat maps below the Manhattan plots. SNP names with red text in LD plots indicate the physical positions of SNPs with significant associations.
Figure 5.
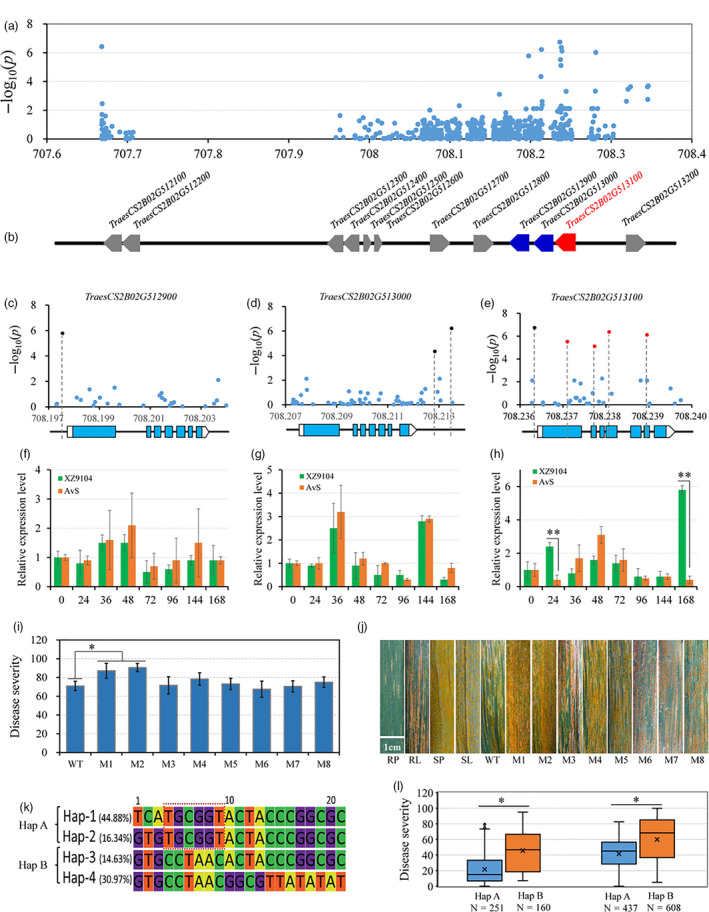
Identification of the causal gene for stripe rust resistance associated with the peak on chromosome 2B. (a) Manhattan plots of polymorphic DNA variants‐based association analysis in the candidate region using resequencing data. (b) The HC genes in the candidate region. (c, d, e) Exon‐intron structures of TraesCS2B01G512900, TraesCS2B01G513000 and TraesCS2B01G513100 and their corresponding DNA polymorphisms with significant associations. (f, g, h) The relative expression levels of TraesCS2B01G512900, TraesCS2B01G513000 and TraesCS2B01G513100 in two cultivars (AvS and XZ9104) with extremely opposite YR phenotypes using qRT‐PCR. Each bar represents the mean ± SD of three biological replicates. (i) Disease severity data based on the functional variants in TraesCS2B01G512900 (M8), TraesCS2B01G513000 (M7) and TraesCS2B01G513100 (M1‐M6) in the EMS mutants. (j) Stripe rust responses for different HIFs and their parents, Snb "S" (resistant parent, RP) and ZM9023 (susceptible parent, SP), and for the durum wheat cultivar Kronos (wild type, WT) and its mutant lines. Scale bar, 1 cm. (k) Haplotype genotype and frequencies in the candidate region and the core of the favourable allele combination are in the red box. (l) Disease severities were based on the haplotypes for YrSnb.1 in different panels of the population. The asterisks indicate significant differences among groups or lines at the P < 0.05 level (Student's t‐test).
Validation of the causal genes
We subsequently analysed the expression levels of all 12 candidate genes using a qRT‐PCR assay in wheat flag leaves at the adult‐plant stage. We found that only the positive allele of TraesCS2B01G513100 in cultivar XZ9104 was up‐regulated six‐fold and fourteen‐fold by Pst inoculation at 24 and 168 h compared with the negative allele in cultivar AvS (Figure 5h). No other genes were differentially expressed between AvS and XZ9104 at any time point (Figure 5f, g and Figure S9). RNA‐seq data indicated that TraesCS2B01G513100 was expressed in flag leaves, spikes and awns, which is consistent with the stripe rust resistance observed at the adult‐plant stage (Figure S10). In mutation analysis, eight mutant lines were selected: Kronos3186 and Kronos3312 with a premature stop codon in TraesCS2B01G512900 and TraesCS2B01G513000, respectively; and Kronos1064, Kronos2338, Kronos3557, Kronos3545, Kronos2969 and Kronos2619, which carry missense mutations in TraesCS2B01G513100. Following the assessment and comparison of these mutants' responses to stripe rust, only Kronos1064 and Kronos2338 exhibited greater susceptibility than wild type (Figure 5i, j; Table S10). These results indicate that TraesCS2B01G513100 is the most likely candidate gene involved in stripe rust resistance.
Association analysis of the STPK gene and identification of allelic variations
To identify more genetic variations, TraesCS2B01G513100 was resequenced in 64 accessions with opposite extreme phenotypes and six NILs derived from the HIFs (Table S8). The sequenced region harboured a 3130‐bp genomic DNA fragment corresponding to the full‐length TraesCS2B01G513100 locus, including its exons, introns and part of primer region and UTRs. In total, 18 SNPs and two insertion/deletions (indels) were identified (Figure 5e and Table S11). An MLM‐based association analysis was performed between all SNPs/indels and the previously assigned stripe rust responses for each accession. One new, non‐synonymous SNP identified (Rv‐686) in the TraesCS2B01G513100 coding region was highly associated with YR (P < 7.69E‐07) (Figure 5e; Table S11).
Based on the LD distance of 612.9 kb, the candidate region was divided into five blocks, which formed four haplotypes (herein referred to as Hap‐1–4). Coupled with the estimation of candidate gene analysis, Hap‐1, Hap‐2, Hap‐3 and Hap‐4 had frequencies of 44.88%, 16.34%, 14.63% and 30.97%, respectively, within the GWAS panel (Figure 5k). Estimation of the contribution of each haplotype towards phenotype variation revealed that Hap‐1 and Hap‐2 had the greatest effect on disease resistance compared with the susceptibility of Hap‐4 and Hap‐3 (Table S1). Taken together, these results indicate that 'TGCGGT' comprises the core of the favourable allele combination and likely underlies the effect on stripe rust resistance. Hap‐1 and Hap‐2 were then combined into Hap‐A, and Hap‐3 and Hap‐4 were combined into Hap‐B. The combination 'TGCGGT' was directly or indirectly related to the gene TraesCS2B01G513100, and the correlating SNPs were developed into derived cleaved amplified polymorphic sequences (dCAPS) and kompetitive allele‐specific PCR (KASP) markers for assisted selection in future resistance breeding (https://galaxy.triticeaetoolbox.org/; Table S12). In addition, the other HIFs from the ZM9023/Snb "S" cross were classified into two major groups using the above‐mentioned PCR markers that distinguish different haplotypes, and the responses of these two groups to stripe rust in the field were assessed (Figure 5j, l). The HIFs carrying the 'TGCGGT' haplotype displayed greater stripe rust resistance, indicating the efficiency of marker‐assisted selection in wheat resistance breeding. Moreover, TraesCS2B01G513100 is a promising target for further functional validation using reverse genetics approaches such as virus‐induced gene silencing, overexpression or transgenic analyses.
Discussion
Multi‐processing environments facilitate the excavation of robust resistance
Many previous studies have shown that multiple loci are involved in complex quantitative resistance (St. Clair, 2010). Wheat stripe rust responses and the resulting phenotypes in the field are consistently affected by host resistance levels, pathogen population structure and weather conditions (Chen and Kang, 2017). Therefore, the ability of resistance‐associated loci to provide protection against disease is dependent on the coevolution between the host and the Pst population in the field. Effective disease resistance also depends on the application of resistance‐associated loci in integrated disease control practices (Nelson et al., 2018). Dissecting the stability of resistance by combining multi‐processing environments with MTAs can provide insights into the long‐term durability of resistance‐associated loci. From this perspective, the loci characterized as imparting environment‐dependent resistance would not be suitable for future marker‐assisted breeding due to the considerable risk of disease (Bazakos et al., 2017). In our study, a diversity panel of wheat accessions was evaluated for stripe rust responses across multiple environments. In order to eliminate environmental interference as much as possible, we also used a linear mixed model to estimate the BLUPs. As strict control measures within the experiment to identify stable loci, only 33.8% (292) of the resistance‐associated loci in this GWAS panel were retained, and some of them were co‐localized at several previously reported Yr genes/QTL regions. As extensively reported in past studies, Yr18, Yr29, Yr30 and Yr78 have been widely used in wheat resistance breeding worldwide due to their durability (Rajpal et al., 2016). It is worth noting that these known Yr genes/QTL were not the most significant loci characterized in this study, despite the fact that they were detected in almost all environments. The MTAs of major effect were located on chromosome 2BL in the proximity of YrSnb.1 identified in our QTL mapping. This novel QTL explained the largest phenotypic variation and warranted further investigation.
The combination of association and haplotype analysis permits refinement of candidate resistance‐associated loci
A common fault of GWAS is the generation of false‐positive associations due to population structure. In this study, an integrated method involving PCA, structure (Q) and a kinship matrix (K) was used to perform population structure adjustment and correction, thereby minimizing the false positive rate (Yu et al., 2006). Meanwhile, another independent diversity panel comprising a natural population was also analysed to validate the MTA results. The extent of LD is determined by the nature of different species and population structure, but it is not invariable. There are differences in LD decay among different segments of the same chromosome, with segments located close to the chromosome telomeres exhibiting lower LD decay than those close to the centromere. This variation in LD decay is highly correlated with the recombination ability of chromosomal segments (Bazakos et al., 2017). Therefore, LD decay impacts the positioning of candidate resistance‐associated regions, depending on whether the significant marker is near the telomeres or the centromere. In this GWAS panel, LD varied across chromosomes and subgenomes, and LD decay ranged from 0.62 Mb to 7.45 Mb. The LD decay of the QTL on chromosome 7DS (Yr18) was 332 kb (r 2 = 0.6), as this region was distant from the centromere. A favourable haplotype carrying gene variants improves the efficiency of marker‐assisted selection in breeding. The LD decay of the major QTL on chromosome 2BL (YrSnb.1) was 4.8 Mb, despite the fact that this region was located at the end of the chromosome. Interestingly, the diversity of SNPs and the pattern of LD in this region was different in the validation diversity panel, and independent validation enabled the candidate region to be narrowed down to 612.9 kb (r 2 = 0.8). This method greatly reduced the computing workload for haplotype analysis. A similar result reported recently detailed how an initial extensive 25‐Mb candidate region on chromosome 3D was ultimately narrowed down to a 1‐Mb region using a validation population (Liu et al., 2019). Here, analysis of different haplotypes and phenotypes of over 1500 wheat accessions, including those from a natural population as well as breeding lines, facilitated identification of favourable allele core sequences. Furthermore, the correlating SNPs were developed into practical PCR markers that can be used to improve the efficiency of marker‐assisted selection in wheat stripe rust resistance breeding.
High‐resolution SNPs enable the prediction of candidate resistance genes
The large extent of LD in wheat makes it difficult to analyse candidate resistance genes. The high‐resolution SNPs identified using the 660K SNP array covered 57,833 high‐confidence genes (53.6% of all genes) and provided insights into the functional causal variant(s) underlying stripe rust resistance. The method of estimating the functional importance of each nucleotide polymorphism serves to predict candidate genes (Yano et al., 2016). First, we identified significant SNPs using GWAS and analysed the LD of the candidate regions containing significant SNPs in different diversity panels. Then, we extracted information for the candidate genes, including the function of polymorphisms, which were validated by their relationships with stripe rust resistance. With this approach, as expected, we delimited the cloned candidate gene Yr18 to within 332 kb and successfully identified Yr18 as a resistance‐associated gene. We similarly analysed a QTL‐based candidate region on 2BL and found several functional associations within the coding and promoter regions of causal genes. The candidate genes TraesCS2B01G512900, TraesCS2B01G513000 and TraesCS2B01G513100 all encoded STPKs, and their amino acid sequences were quite different from that of their respective homologs in the A and D genomes. STPK is known to play a role in plant defence. For instance, STPK‐V, a member of the STPK family in Haynaldia villosa, enhances powdery mildew resistance by decreasing the haustorium index dramatically and mediating H2O2 accumulation (Cao et al., 2011). Although our gene expression and mutation analysis indicated that TraesCS2B01G513100 was the most likely candidate resistance gene, TraesCS2B01G512900 and TraesCS2B01G513000 should not be disregarded. Resistance genes are generally grouped in clusters in plants, and some may play simultaneous central roles in resistance (Kourelis and van der Hoorn, 2018; Zhao et al., 2016). Therefore, the characterization of these genes using molecular biology methods may reveal further molecular mechanisms of stripe rust resistance in wheat.
This study demonstrates the feasibility of predicting causal resistance genes using high‐resolution SNP‐based GWAS in common wheat. In previous studies, allelic variants of Ppd‐D1 (chromosome 2D) and Rht‐D1 (chromosome 4D) loci, which were shown to affect plant growth traits during the stem elongation phase, were precisely identified by wheat 90K SNP array‐based GWAS (Guo et al., 2018). Coincidentally, the SNP AX‐109665328 was found to associate with Rht‐D1 and several candidate genes involved in abiotic stress tolerance, such as those encoding WRKY transcription factors, which were co‐localized in a candidate region identified through wheat 660K SNP array‐based GWAS (Li et al., 2019). The most effective use of wheat 90K SNP‐based GWAS was in the identification of the flour‐colour gene TaRPP13L1 gene that was successfully identified from a 20‐kb candidate region and verified by the functional SNP Excalibur_c5938_1703 (Chen et al., 2019). It should be noted that each of the above‐mentioned traits is controlled by conserved genes. However, there are abundant variations in resistance traits, including their presence/absence in different genomes (Arora et al., 2019). From this point of view, the candidate genes considered in this study were restricted to those annotated in the Chinese Spring reference genome, and thus, other resistance genes absent in the reference genome cannot be ruled out. In addition, an inevitable limitation of SNP array‐based GWAS is that single candidate gene association analysis cannot be performed due to insufficient suitable DNA variants within the gene region. Deep next‐generation sequencing approaches, such as Pan‐genome and 10 × genomics, or whole‐genome resequencing of a diversity panel, can overcome this disadvantage. Nevertheless, the haplotype and candidate gene analyses reported here reveal promising alleles that function in stripe rust resistance and provide potential targets for further functional analysis and inclusion in future wheat resistance breeding.
Materials and methods
Phenotypic evaluation of stripe rust infection
The association mapping panel used in this study comprised 411 breeding lines from CIMMYT and ICARDA (Table S1). An independent diversity panel of 1045 wheat accessions from a global collection, a set of 63 common wheat resequencing genotypes (Table S1), and a bi‐parent genetic population from a cross of Zhengmai 9023 (ZM9023) × Sunbird 'S' (Snb 'S') were used for validation of the significant MTAs. The wheat lines Avocet S (AvS), Mingxian 169 (MX169) and Xiaoyan 22 (XY22) were used as the susceptible controls.
Evaluations of seedling resistance to stripe rust were conducted under controlled greenhouse conditions. The tested Pst races contained pre‐V26 prevalent races, such as CYR32 and CYR33, and post‐V26 groups collected from different origins, such as V26/Laboratory (V26/Lab), V26/Sichuan (V26/SC), V26/Shaanxi (V26/SX) and V26/Gansu (CYR34). The avirulence/virulence characteristics of the races were reported by Wu et al. (2020). Details of inoculation and disease evaluation were described previously (Wu et al., 2018b). Wheat accessions AvS and Xingzi 9104 (XZ9104) and Pst race V26/Lab were used for gene expression analysis in this study. Xingzi 9104, carrying YrSnb.1, displays resistance at the adult‐plant stage. Flag leaves inoculated with V26/Lab or sterile distilled water (control) at the adult‐plant stage were harvested at 0, 24, 36, 48, 72, 96, 144 and 168 h post‐inoculation (hpi). Time points were selected based on a previous study (Zhang et al., 2012).
Adult‐plant resistance (APR) evaluations were carried out at Yangling in Shaanxi province (over‐wintering region), Tianshui in Gansu province (over‐summering region) and Jiangyou in Sichuan province (over‐wintering region) during three cropping seasons (2016–2017, 2017–2018, 2018–2019). Detailed methods of plant growth, management and evaluation have been published previously (Mu et al., 2019).
Phenotypic data analyses
For each environment, the arithmetic mean of phenotypic observations was used as the phenotypic data. Genotype (411 cultivars and breeding lines) and environment (three years in three locations) were treated as random effects in a linear mixed model to estimate the best linear unbiased predictions (BLUPs) using the lme4 package in the R 3.5.3 program (Bates et al., 2014). For each trait, each single environment phenotypic data set and BLUP data set were used for analysis of variance (ANOVA). Since there was no replication in this study, it was not possible to estimate the genotype by environment interaction. The broad‐sense heritability (H 2) estimates for IT and DS were calculated across nine test environments using the lme4 package with the formula H 2 = V G/(V G + V E), where V G and V E represent the genotypic and environmental variances, respectively. Pearson's correlation coefficients (r) of pairwise environments were computed using the Hmisc package to determine the consistency of stripe rust responses a different environments.
Genotyping, SNP filtering and population structure analysis
Wheat leaf samples including 411 breeding lines and 1045 accessions were collected, and DNA was extracted using an extraction kit (Invitrogen™, Thermo Fisher, Waltham, USA) following the manufacturer's instructions. Genotyping was performed using the wheat 660K genotyping assay by Beijing CapitalBio Technology Company (http://www.capitalbiotech.com). SNP genotype calling and allele clustering were processed with the polyploid version of Affymetrix Genotyping Console™ (GTC) software. To ensure the quality pretreatment of genotyping data, SNP markers with minor allele frequencies (MAF) < 0.05, missing data >10%, or Hardy–Weinberg Equilibrium (HWE) > 0.01 were excluded from further analysis. The most up‐to‐date physical positions of the SNPs were obtained from the Triticeae Multi‐omics Center website (http://202.194.139.32/). Polymorphism information content (PIC) and expected heterozygosity (He), representing two genetic diversity parameters, were calculated using a self‐written programme in Perl. PIC and He values were calculated for each SNP marker and each chromosome based on the formulas described in Botstein et al. (1980) and Nei (1978), respectively.
Population structure was assessed using STRUCTURE software v2.3.4 with unlinked markers (r 2 = 0). The model was applied without the use of prior population information, and the most likely number of subpopulations was determined using a previously described method (Earl and VonHoldt, 2012). Principal components analysis (PCA) of the population was performed using the software GCTA (Yang et al., 2011). The p‐distance was used to construct NJ phylogenetic trees with 1000 bootstrap replicates using the software MEGA‐CC (Kumar et al., 2012). The identity‐by‐state (IBS) relative K‐matrix was calculated between pairs of accessions using PLINK (Purcell et al., 2007). Heat maps of kinship were generated on the basis of the K‐matrix using the pheatmap v1.0.8 R package. Genome‐wide linkage disequilibrium (LD) analysis for the A, B, and D genomes was performed using the software PLINK. LD estimation and LD decay analysis were performed as described in Yu et al. (2020). A locally weighted polynomial regression (LOESS) curve was drawn to fit the data using second‐degree locally weighted scatter plot smoothing in the R program. The confidence interval of quantitative trait loci (QTL) was defined based on the intersection of the fitted LOESS curve with LD r 2 = 0.1.
Genome‐wide association analyses
GWAS was conducted using a univariate linear mixed model with GEMMA software (Zhou and Stephens, 2012). The P‐value threshold was calculated using a modified Bonferroni correction (Genetic type 1 Error Calculator, version 0.2) with a suggested threshold of P = 1/Ne (Ne = effective SNP number) (Li et al., 2012). Our results showed that the suggested P‐value thresholds ranged from 1.36 × 10−4 to 9.90 × 10−4 for each chromosome (Table 1), and thus, we considered the mean value 3.23 × 10−4 as the criterion for genome‐wide significance in this study. Significant markers from the GWAS result were visualized using a Manhattan plot, and important P‐value distributions were visualized by a quantile‐quantile plot (Q‐Q plot), both drawn by the qqman package in R 3.0.3 (http://www.r‐project.org/). The phenotypic variance explained (R 2) by significant SNPs was evaluated using GCTA software.
Comparisons with previously published Yr genes and QTL
To date, more than 150 permanently or temporarily designated Yr genes and over 300 QTL have been described across 21 wheat chromosomes, and most of these are listed in the Catalogue of Gene Symbols for Wheat or summarized in integrated genetic maps (Chen and Kang, 2017). To determine the relationships between significant loci identified in the GWAS and previously reported Yr genes/QTL, we compared the physical locations of these loci based on the Chinese Spring reference genome coupled with integrated genetic maps (Cui et al., 2017; Maccaferri et al., 2015). For previously reported stripe rust resistance genes/QTL, the closest flanking markers were used to generate the confidence intervals reported. Whether the loci identified in the GWAS were novel depended on the interval of the haplotype block.
Revalidation of marker–trait associations, haplotype estimation and association analysis of the STPK gene
To validate the stability and accuracy of significant SNPs located within major QTL regions identified in the first diversity panel, the associated SNPs were retested in the extended independent validation panel and in a bi‐parental genetic population. The second panel was phenotyped across multiple environments in field trials and also genotyped with the wheat 660K SNP array. Univariate ANOVA was used to analyse MTAs in the R package. The local LD patterns were visualized on the basis of the LD squared‐allele frequency correlation (r 2) estimates between markers using the software HAPLOVIEW. Haplotype blocks were identified based on LD, and the effect of each haplotype allele was calculated using the lmer function in R software. In addition, 63 common wheat accessions with resequencing data were also used for association analysis in the candidate region to validate causal genes (Cheng et al., 2019).
Association analysis of the STPK gene (TraesCS2B01G513100) was conducted on 64 representative wheat accessions and six near‐isogenic lines (NILs) derived from the heterozygous inbred families (HIFs) from a ZM9023 × Snb "S" cross. The STPK gene coding regions (including introns) were amplified and sequenced. These sequences were assembled using DNAMAN and aligned using AliView (Larsson, 2014). Nucleotide polymorphisms, including SNPs and indels, were identified (MAF ≥ 0.05) among these genotypes, and their association with the YR responses and pairwise LD were calculated with the software PLINK.
Identification of candidate genes by nucleotide polymorphism analysis
High‐confidence genes located within the LD block around significant SNPs were used for candidate gene analysis based on IWGSC RefSeq v1.0 with gene annotations (IWGSC, 2018). Based on the estimated functional importance of each nucleotide polymorphism as described by Yano et al. (2016), all the polymorphisms in the candidate region were classified into five groups, referred to as G1–5. G1 contained significant MTAs in the GWAS (−log10 P ≥ the threshold value in this chromosome) that putatively caused amino acid conversion. G2 harboured significant MTAs in the 5′ flanking sequences (≤2 kb from the first ATG), which were considered to be promoter regions. G3 included significant MTAs within introns or 3′ non‐coding sequences. G4 contained significant MTAs outside coding regions, and G5 contained polymorphisms but not significant MTAs.
Preliminary verification of causal genes by expression and mutation analysis
Expression data (transcripts per million, TPM) for the potential causal genes from previously mapped RNA‐seq samples were downloaded from the Triticeae Multi‐omics Center website (http://202.194.139.32/) (Ramírez‐González et al., 2018). Total RNA extraction and cDNA synthesis from AvS and XZ9104 samples were performed following Liu et al. (2019). Quantitative real‐time PCR (qRT‐PCR) primer sequences for the twelve candidate genes in the 612.9 kb region are provided in Table S12. Wheat TaActin (AB181991.1) was used as an internal reference for normalization, and transcript abundance estimates were based on three technical replicates each of three biological replicates per each genotype.
In addition, the function of candidate genes was also verified in a durum wheat mutant pool. The mutants induced by ethyl methanesulfonate (EMS) from durum wheat cultivar Kronos have been sequenced using exome capture sequencing and contribute to the analysis of gene variations corresponding to phenotype (Henry et al., 2014). The lines Kronos1064, Kronos2338, Kronos3557, Kronos3545, Kronos2969, Kronos2619, Kronos3312 and Kronos3186 were kindly provided by Drs. Jiajie Wu and Fei Ni, Shandong Agricultural University. The stripe rust responses of these mutants were evaluated at the adult‐plant stage under controlled greenhouse conditions.
Author contributions
JHW conducted the experiments, analysed the data and wrote the manuscript. RY, HYW, CEZ, SH, HXJ, XJN, QLW and SJL participated in field experiments and contributed to the molecular biology experiments. SZY and QDZ assisted in analysing the data. SWN, RPS and SB revised the manuscript. ZSK, DJH and QDZ conceived and directed the project and revised the manuscript.
Conflict of Interest
The authors declare no conflicts of interest and all experiments comply with the current laws of China.
Supporting information
Figure S1 Circos diagram showing genome‐wide physical distributions of filtered single nucleotide polymorphisms (SNPs) from the wheat 660K SNP array and average nucleotide diversity in each chromosome
Figure S2 Genome‐wide average linkage disequilibrium (LD) decay over physical distances
Figure S3 Box plot distributions of seedling infection type (IT) for six Pst races
Figure S4 Phenotypic distribution of the stripe rust responses of different wheat accessions
Figure S5 Regression plot, histograms and Pearson correlation coefficients (P = 0.01) for infection type (IT) and disease severity (DS) among different environments (YL, Yangling; TS, Tianshui; JY, Jingyou; 17, 18, 19 represent the 2016–2017, 2017–2018, and 2018–2019 cropping seasons, respectively)
Figure S6 Genome‐wide association analysis results for stripe rust infection types across ten tested environments including (a) 2017‐Jiangyou, (b) 2017‐Tianshui, (c) 2017‐Yanlging, (d) 2018‐Jiangyou, (e) 2018‐Tianshui, (f) 2018‐Yanlging, (g) 2019‐Jiangyou, (h) 2019‐Tianshui, (i) 2019‐Yanlging, and (j) BLUP
Figure S7 Chromosome positions of QTL identified in this study relative to previously mapped stripe rust resistance genes/QTL
Figure S8 Sequence alignment of TraesCS2B01G513100, TraesCS2B01G513000, and TraesCS2B01G512900 complete gene sequences to those alleles in other subgenomes
Figure S9 The relative expression levels of all high‐confidence genes in the candidate region in two cultivars (AvS and XZ9104) with extremely opposite YR phenotypes using qRT‐PCR
Figure S10 The expression profile of TraesCS2B01G513100 in different tissues and organs of wheat
Table S1 Information and stripe rust responses for the 411 spring wheat lines included in this genome‐wide association study diversity panel, the 1045 accessions in the validation set, and the 63 resequenced samples.
Table S2 Information on 36,324 polymorphic haplotypes (represented by SNP probes) derived from the 660K SNP array.
Table S3 Functional location and type of substitution (synonymous and non‐synonymous) within the coding sequence for single nucleotide polymorphisms (SNPs) present in the diversity panel of spring wheat lines.
Table S4 Summary of the genetic diversity in the subgenomes and chromosomes of the 1045 included wheat accessions.
Table S5 Estimates of variance components and heritability of phenotypic responses (infection type, IT; and disease severity, DS) to Puccinia striiformis f. sp. tritici (Pst) under multiple environments in the 411 spring wheat accessions included in this study.
Table S6 Significant SNP–trait associations.
Table S7 Analysis of the estimated functional importance of each nucleotide polymorphism in candidate regions on chromosome 7D and 2B.
Table S8 The genotype information of heterozygous inbred families (HIFs) from Zhengmai 9023 (ZM9023)/Sunbird 'S' (Snb 'S') on chromosome 2B and near‐isogenic lines (±Yr18) in Avocet S background on chromosome 7D.
Table S9 DNA variations derived from resequencing data in the candidate genomic region (612.9 kb) of YrSnb.1‐2BL.
Table S10 Information on Kronos mutants.
Table S11 Variations in the TraesCS2B01G513100 genomic region using PCR‐based sequencing.
Table S12 The molecular markers or primers used in this study.
Acknowledgements
The authors are grateful to TopEdit LLC for language editing and proofreading during the preparation of this manuscript. We would like to thank Drs. Guangwei Li, Yue Liu, Shengwei Ma, Li Tai and Meng Wang for their valuable advice on data analysis and participation in molecular biology experiment and figure generation. The Kronos EMS mutant lines were developed by Dr. Jorge Dubcovsky at UC Davis and we thank Drs. Jiajie Wu and Fei Ni for providing the mutant lines of Kronos. We also thank Prof. Jizeng Jia for providing 670 genotyping data of wheat 660K SNP array in the validation panel. This study was financially supported by National Science Foundation for Young Scientists in China (Grant nos. 31901494, 31901869 and 31701421), International Cooperation and Exchange of the National Natural Science Foundation of China (Grant no. 31961143019 & 31561143005), National Natural Science Foundation of China (Grant no. 31971890), China Postdoctoral Science Foundation funding (2019M653769), Open Project Program of the State Key Laboratory of Crop Biology (2019KF01), National '111 plan' (Grant no. BP0719026) and Natural Science Basic Research Plan in Shaanxi Province of China (Grant no. 2019JCW‐18).
Wu, J. , Yu, R. , Wang, H. , Zhou, C. , Huang, S. , Jiao, H. , Yu, S. , Nie, X. , Wang, Q. , Liu, S. , Weining, S. , Singh, R. P. , Bhavani, S. , Kang, Z. , Han, D. and Zeng, Q. (2021) A large‐scale genomic association analysis identifies the candidate causal genes conferring stripe rust resistance under multiple field environments. Plant Biotechnol. J., 10.1111/pbi.13452
Contributor Information
Zhensheng Kang, Email: kangzs@nwafu.edu.cn.
Dejun Han, Email: handj@nwafu.edu.cn.
Qingdong Zeng, Email: zengqd@nwafu.edu.cn.
References
- Arora, S. , Steuernagel, B. , Gaurav, K. , Chandramohan, S. , Long, Y. , Matny, O. , Johnson, R. et al. (2019) Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 37, 139–143. [DOI] [PubMed] [Google Scholar]
- Bates, D. , Maechler, M. , Bolker, B. and Walker, S. (2014) lme4: linear mixed‐effects models using Eigen and S4. pp 1–23.
- Bazakos, C. , Hanemian, M. , Trontin, C. , Jimenez‐Gomez, J.M. and Loudet, O. (2017) New strategies and tools in quantitative genetics: how to go from the phenotype to the genotype. Annu. Rev. Plant Biol. 68, 435–455. [DOI] [PubMed] [Google Scholar]
- Botstein, D. , White, R.L. , Skolnick, M. and Davis, R.W. (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331. [PMC free article] [PubMed] [Google Scholar]
- Brown, J.K. (2015) Durable resistance of crops to disease: a Darwinian perspective. Annu. Rev. Phytopathol. 53, 513–539. [DOI] [PubMed] [Google Scholar]
- Cao, A. , Xing, L. , Wang, X. , Yang, X. , Wang, W. , Sun, Y. , Qian, C. et al. (2011) Serine/threonine kinase gene Stpk‐V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc. Natl Acad Sci USA 108, 7727–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X.M. (2013) High‐temperature adult‐plant resistance, key for sustainable control of stripe rust. Am. J. Plant Sci. 04, 608–627. [Google Scholar]
- Chen, X.M. (2014) Integration of cultivar resistance and fungicide application for control of wheat stripe rust. Can. J. Plant Path. 36, 311–326. [Google Scholar]
- Chen, X.M. and Kang, Z.S. , eds. (2017) Stripe Rust. Dordrecht: Springer Netherlands. [Google Scholar]
- Chen, J. , Zhang, F. , Zhao, C. , Lv, G. , Sun, C. , Pan, Y. , Guo, X. et al. (2019) Genome‐wide association study of six quality traits reveals the association of the TaRPP13L1 gene with flour colour in Chinese bread wheat. Plant Biotechnol. J. 17, 2106–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , Liu, J. , Wen, J. , Nie, X. , Xu, L. , Chen, N. , Li, Z. et al. (2019) Frequent intra‐ and inter‐species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol. 20, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, F. , Zhang, N. , Fan, X. , Zhang, W. , Zhao, C. , Yang, L. , Pan, R. et al. (2017) Utilization of a Wheat660K SNP array‐derived high‐density genetic map for high‐resolution mapping of a major QTL for kernel number. Sci. Rep. 7, 3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubcovsky, J. and Dvorak, J. (2007) Genome plasticity a key factor in the success of polyploid wheat under domestication. Science, 316, 1862–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl, D.A. and VonHoldt, B.M. (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. [Google Scholar]
- Fu, D. , Uauy, C. , Distelfeld, A. , Blechl, A. , Epstein, L. , Chen, X. , Sela, H. et al. (2009) A kinase‐START gene confers temperature‐dependent resistance to wheat stripe rust. Science 323, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z. , Liu, G. , Röder, M.S. , Reif, J.C. , Ganal, M.W. and Schnurbusch, T. (2018) Genome‐wide association analyses of plant growth traits during the stem elongation phase in wheat. Plant Biotechnol. J. 16, 2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, C. , Wang, L. , Ge, H. , Dong, Y. and Zhang, X. (2011) Genetic diversity and linkage disequilibrium in Chinese bread wheat (Triticum aestivum L.) revealed by SSR markers. PLoS One 6, e17279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, I.M. , Nagalakshmi, U. , Lieberman, M.C. , Ngo, K.J. , Krasileva, K.V. , Vasquez‐Gross, H. , Akhunova, A. et al. (2014) Efficient genome‐wide detection and cataloging of EMS‐induced mutations using exome capture and next‐generation sequencing. Plant Cell, 26, 1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Zhao, Y. , Wei, X. , Li, C. , Wang, A. , Zhao, Q. , Li, W. et al. (2011) Genome‐wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 44, 32–39. [DOI] [PubMed] [Google Scholar]
- IWGSC . (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, r7191. [DOI] [PubMed] [Google Scholar]
- Juliana, P. , Poland, J. , Huerta‐Espino, J. , Shrestha, S. , Crossa, J. , Crespo‐Herrera, L. , Toledo, F.H. et al. (2019) Improving grain yield, stress resilience and quality of bread wheat using large‐scale genomics. Nat. Genet. 51, 1530–1539. [DOI] [PubMed] [Google Scholar]
- Kang, H.M. , Zaitlen, N.A. , Wade, C.M. , Kirby, A. , Heckerman, D. , Daly, M.J. and Eskin, E. (2008) Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourelis, J. and van der Hoorn, R.A.L. (2018) Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell, 30, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krattinger, S.G. , Jordan, D.R. , Mace, E.S. , Raghavan, C. , Luo, M. , Keller, B. & Lagudah, E.S. (2013) Recent emergence of the wheat Lr34 multi‐pathogen resistance: insights from haplotype analysis in wheat, rice, sorghum and Aegilops tauschii. Theor Appl Genet 126, 663–672. [DOI] [PubMed] [Google Scholar]
- Krattinger, S.G. , Lagudah, E.S. , Spielmeyer, W. , Singh, R.P. , Huerta‐Espino, J. , McFadden, H. , Bossolini, E. et al. (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323, 1360–1363. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Peterson, D. and Tamura, K. (2012) MEGA‐CC: computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 28, 2685–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, A. (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Yeung, J.M.Y. , Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Mao, X. , Wang, J. , Chang, X. , Reynolds, M. and Jing, R. (2019) Genetic dissection of drought and heat‐responsive agronomic traits in wheat. Plant, Cell Environ. 42, 2540–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipka, A.E. , Kandianis, C.B. , Hudson, M.E. , Yu, J. , Drnevich, J. and Gore, P.J.B.A. (2015) From association to prediction: statistical methods for the dissection and selection of complex traits in plants. Curr. Opin. Plant Biol. 24, 110–118. [DOI] [PubMed] [Google Scholar]
- Liu, P. , Guo, J. , Zhang, R. , Zhao, J. , Liu, C. , Qi, T. , Duan, Y. et al. (2019) TaCIPK10 interacts with and phosphorylates TaNH2 to activate wheat defense responses to stripe rust. Plant Biotechnol. J. 17, 956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. , Zhao, Y. , Beier, S. , Jiang, Y. , Thorwarth, P.H. , Longin C.F., Ganal M. et al. (2020) Exome association analysis sheds light onto leaf rust (Puccinia triticina) resistance genes currently used in wheat breeding (Triticum aestivum L.). Plant Biotechnol. J. 18, 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri, M. , Zhang, J. , Bulli, P. , Abate, Z. , Chao, S. , Cantu, D. , Bossolini, E. et al. (2015) A genome‐wide association study of resistance to stripe rust (Puccinia striiformis f. sp. tritici) in a worldwide collection of hexaploid spring wheat (Triticum aestivum L.). G3 5:449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, R. , Mu, J. , Han, D. and Kang, Z. (2018) Wheat stripe rust resistance gene Yr24/Yr26: A retrospective review. Crop J. 6, 321–329. [Google Scholar]
- Moore, J.W. , Herrera‐Foessel, S. , Lan, C. , Schnippenkoetter, W. , Ayliffe, M. , Huerta‐Espino, J. , Lillemo, M. et al. (2015) A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat. Genet. 47, 1494–1498. [DOI] [PubMed] [Google Scholar]
- Mu, J. , Wang, Q. , Wu, J. , Zeng, Q. , Huang, S. , Liu, S. , Yu, S. et al. (2019) Identification of sources of resistance in geographically diverse wheat accessions to stripe rust pathogen in China. Crop Prot. 122, 1–8. [Google Scholar]
- Nei, M. (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, R. , Wiesner‐Hanks, T. , Wisser, R. and Balint‐Kurti, P. (2018) Navigating complexity to breed disease‐resistant crops. Nat. Rev. Genet. 19, 21–33. [DOI] [PubMed] [Google Scholar]
- Niks, R.E. , Qi, X. and Marcel, T.C. (2015) Quantitative resistance to biotrophic filamentous plant pathogens: concepts, misconceptions, and mechanisms. Annu. Rev. Phytopathol. 53, 445–470. [DOI] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A.R. , Bender, D. , Maller, J. et al. (2007) PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal, V.R. , Rao, S.R. and Raina, S.N. (2016) Molecular breeding for sustainable crop improvement, volume 2. Cham: Springer. [Google Scholar]
- Ramírez‐González, R.H. , Borrill, P. , Lang, D. , Harrington, S.A. , Brinton, J. , Venturini, L. , Davey, M. et al.(2018) The transcriptional landscape of polyploid wheat. Science 361, r6089. [DOI] [PubMed] [Google Scholar]
- Rasheed, A. , Hao, Y. , Xia, X. , Khan, A. , Xu, Y. , Varshney, R.K. and He, Z. (2017) Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Molecular Plant 10, 1047–1064. [DOI] [PubMed] [Google Scholar]
- Sánchez‐Martín, J. and Keller, B. (2019) Contribution of recent technological advances to future resistance breeding. Theor. Appl. Genet. 132, 713–732. [DOI] [PubMed] [Google Scholar]
- Schaid, D.J. , Chen, W. and Larson, N.B. (2018) From genome‐wide associations to candidate causal variants by statistical fine‐mapping. Nat. Rev. Genet. 19, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St.Clair, D.A. (2010) Quantitative disease resistance and quantitative resistance loci in breeding. Annu. Rev. Phytopathol. 48, 247–268. [DOI] [PubMed] [Google Scholar]
- Sun, C. , Dong, Z. , Zhao, L. , Ren, Y. , Zhang, N. and Chen, F. (2020) The wheat 660K SNP array demonstrates great potential for marker‐assisted selection in polyploid wheat. Plant Biotechnol. J. 18, 1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Wang, Q. , Liu, S. , Huang, S. , Mu, J. , Zeng, Q. , Huang, L. et al. (2017) Saturation mapping of a major effect QTL for stripe rust resistance on wheat chromosome 2B in cultivar Napo 63 using SNP genotyping arrays. Front. Plant Sci. 8, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Huang, S. , Zeng, Q. , Liu, S. , Wang, Q. , Mu, J. , Yu, S. et al. (2018a) Comparative genome‐wide mapping versus extreme pool‐genotyping and development of diagnostic SNP markers linked to QTL for adult plant resistance to stripe rust in common wheat. Theor. Appl. Genet. 131, 1777–1792. [DOI] [PubMed] [Google Scholar]
- Wu, J. , Liu, S. , Wang, Q. , Zeng, Q. , Mu, J. , Huang, S. , Yu, S. et al. (2018b) Rapid identification of an adult plant stripe rust resistance gene in hexaploid wheat by high‐throughput SNP array genotyping of pooled extremes. Theor. Appl. Genet. 131, 43–58. [DOI] [PubMed] [Google Scholar]
- Wu, J. , Wang, X. , Chen, N. , Yu, R. , Yu, S. , Wang, Q. , Huang, S. et al. (2020) Association analysis identifies new loci for resistance to Chinese Yr26‐virulent races of the stripe rust pathogen in a diverse panel of wheat germplasm. Plant Dis. 104, 1751–1762. [DOI] [PubMed] [Google Scholar]
- Xu, Y. , Li, P. , Zou, C. , Lu, Y. , Xie, C. , Zhang, X. , Prasanna, B.M. et al. (2017) Enhancing genetic gain in the era of molecular breeding. J. Exp. Bot. 68, 2641–2666. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Lee, S.H. , Goddard, M.E. and Visscher, P.M. (2011) GCTA: a tool for genome‐wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, K. , Yamamoto, E. , Aya, K. , Takeuchi, H. , Lo, P. , Hu, L. , Yamasaki, M. et al. (2016) Genome‐wide association study using whole‐genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927–934. [DOI] [PubMed] [Google Scholar]
- Yu, J. , Pressoir, G. , Briggs, W.H. , Vroh Bi, I. , Yamasaki, M. , Doebley, J.F. , McMullen, M.D. et al. (2006) A unified mixed‐model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. [DOI] [PubMed] [Google Scholar]
- Yu, S. , Wu, J. , Wang, M. , Shi, W. , Xia, G. , Jia, J. , Kang, Z. et al. (2020) Haplotype variations in QTL for salt tolerance in Chinese wheat accessions identified by marker‐based and pedigree‐based kinship analyses. Crop J. 10.1016/j.cj.2020.03.007 [DOI] [Google Scholar]
- Zeng, Q. , Wu, J. , Liu, S. , Huang, S. , Wang, Q. , Mu, J. , Yu, S. et al. (2019) A major QTL co‐localized on chromosome 6BL and its epistatic interaction for enhanced wheat stripe rust resistance. Theor. Appl. Genet. 132, 1409–1424. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Wang, C. , Cheng, Y. , Chen, X. , Han, Q. , Huang, L. , Wei, G. et al. (2012) Histological and cytological characterization of adult plant resistance to wheat stripe rust. Plant Cell Rep. 31, 2121–2137. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Huang, J. , Wang, Z. , Jing, S. , Wang, Y. , Ouyang, Y. , Cai, B. et al. (2016) Allelic diversity in an NLR gene BPH9 enables rice to combat planthopper variation. Proc. Natl Acad. Sci. USA, 113, 12850–12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. and Stephens, M. (2012) Genome‐wide efficient mixed‐model analysis for association studies. Nat. Genet. 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, Q.S. ed. (2003) Chinese Wheat improvement and pedigree analysis. Beijing: China Agriculture Press. [In Chinese with English summary]. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Circos diagram showing genome‐wide physical distributions of filtered single nucleotide polymorphisms (SNPs) from the wheat 660K SNP array and average nucleotide diversity in each chromosome
Figure S2 Genome‐wide average linkage disequilibrium (LD) decay over physical distances
Figure S3 Box plot distributions of seedling infection type (IT) for six Pst races
Figure S4 Phenotypic distribution of the stripe rust responses of different wheat accessions
Figure S5 Regression plot, histograms and Pearson correlation coefficients (P = 0.01) for infection type (IT) and disease severity (DS) among different environments (YL, Yangling; TS, Tianshui; JY, Jingyou; 17, 18, 19 represent the 2016–2017, 2017–2018, and 2018–2019 cropping seasons, respectively)
Figure S6 Genome‐wide association analysis results for stripe rust infection types across ten tested environments including (a) 2017‐Jiangyou, (b) 2017‐Tianshui, (c) 2017‐Yanlging, (d) 2018‐Jiangyou, (e) 2018‐Tianshui, (f) 2018‐Yanlging, (g) 2019‐Jiangyou, (h) 2019‐Tianshui, (i) 2019‐Yanlging, and (j) BLUP
Figure S7 Chromosome positions of QTL identified in this study relative to previously mapped stripe rust resistance genes/QTL
Figure S8 Sequence alignment of TraesCS2B01G513100, TraesCS2B01G513000, and TraesCS2B01G512900 complete gene sequences to those alleles in other subgenomes
Figure S9 The relative expression levels of all high‐confidence genes in the candidate region in two cultivars (AvS and XZ9104) with extremely opposite YR phenotypes using qRT‐PCR
Figure S10 The expression profile of TraesCS2B01G513100 in different tissues and organs of wheat
Table S1 Information and stripe rust responses for the 411 spring wheat lines included in this genome‐wide association study diversity panel, the 1045 accessions in the validation set, and the 63 resequenced samples.
Table S2 Information on 36,324 polymorphic haplotypes (represented by SNP probes) derived from the 660K SNP array.
Table S3 Functional location and type of substitution (synonymous and non‐synonymous) within the coding sequence for single nucleotide polymorphisms (SNPs) present in the diversity panel of spring wheat lines.
Table S4 Summary of the genetic diversity in the subgenomes and chromosomes of the 1045 included wheat accessions.
Table S5 Estimates of variance components and heritability of phenotypic responses (infection type, IT; and disease severity, DS) to Puccinia striiformis f. sp. tritici (Pst) under multiple environments in the 411 spring wheat accessions included in this study.
Table S6 Significant SNP–trait associations.
Table S7 Analysis of the estimated functional importance of each nucleotide polymorphism in candidate regions on chromosome 7D and 2B.
Table S8 The genotype information of heterozygous inbred families (HIFs) from Zhengmai 9023 (ZM9023)/Sunbird 'S' (Snb 'S') on chromosome 2B and near‐isogenic lines (±Yr18) in Avocet S background on chromosome 7D.
Table S9 DNA variations derived from resequencing data in the candidate genomic region (612.9 kb) of YrSnb.1‐2BL.
Table S10 Information on Kronos mutants.
Table S11 Variations in the TraesCS2B01G513100 genomic region using PCR‐based sequencing.
Table S12 The molecular markers or primers used in this study.