

Summary
Nonalcoholic fatty liver disease (NAFLD) is currently the most prevalent form of liver disease worldwide. This term encompasses a spectrum of pathologies, from benign hepatic steatosis to non-alcoholic steatohepatitis, which have, to date, been challenging to model in the laboratory setting. Here, we present a human pluripotent stem cell (hPSC)-derived model of hepatic steatosis, which overcomes inherent challenges of current models and provides insights into the metabolic rewiring associated with steatosis. Following induction of macrovesicular steatosis in hepatocyte-like cells using lactate, pyruvate, and octanoate (LPO), respirometry and transcriptomic analyses revealed compromised electron transport chain activity. 13C isotopic tracing studies revealed enhanced TCA cycle anaplerosis, with concomitant development of a compensatory purine nucleotide cycle shunt leading to excess generation of fumarate. This model of hepatic steatosis is reproducible, scalable, and overcomes the challenges of studying mitochondrial metabolism in currently available models.
Subject areas: Endocrine System Physiology, Stem Cells Research, Transcriptomics
Graphical Abstract
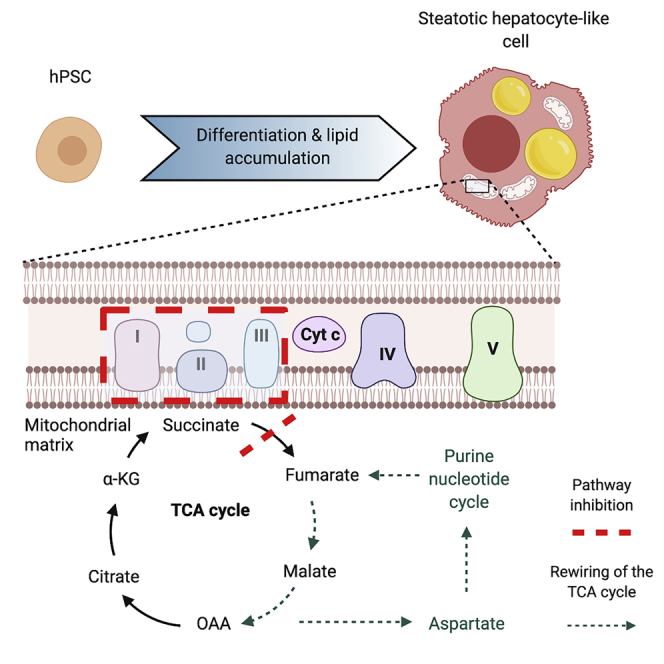
Highlights
-
•
LPO-exposed hPSC-derived hepatocyte-like cells resemble human hepatic steatosis
-
•
Steatosis is associated with decreased mitochondrial maximal respiration
-
•
Purine nucleotide cycle activity increases in steatotic hepatocytes
-
•
Excess fumarate production is associated with induction of macrovesicular steatosis
Endocrine System Physiology; Stem Cells Research; Transcriptomics
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common form of hepatic disease and is strongly associated with obesity and type 2 diabetes (WHO, 2006). In the earliest stage of NAFLD, triglyceride (TG) accumulation in hepatocytes leads to the development of hepatic steatosis (Valenti et al., 2016). This is characterized by the development of macrovesicular steatosis, with TG storage in large lipid droplets (Wang and Yu, 2016). Although steatosis is largely benign, it can progress to nonalcoholic steatohepatitis (NASH), which, in turn, increases the risk of developing cirrhosis and hepatocellular carcinoma (Asrih and Jornayvaz, 2015). However, the mechanism(s) underlying this progression have yet to be determined. At present, there are no specific therapeutics available to reverse or treat NAFLD, and the only effective intervention is through the reduction of obesity through diet and exercise or following bariatric surgery (Laursen et al., 2019).
In order to understand the pathophysiology of the NAFLD disease spectrum, studies have been undertaken in rodent models, in vitro culture systems, and human liver biopsy samples. Dietary intervention or genetic manipulation leading to the development of obesity and/or hepatic steatosis in rodents often fails to fully recapitulate the NAFLD phenotype. For example, although the leptin-deficient Ob/Ob mouse develops extreme obesity and metabolic abnormalities, the profound abnormalities in circulating leptin mean that this model is not representative of human disease in the general population (Anstee and Goldin, 2006; Rotundo et al., 2018). Likewise, dietary interventions mimic specific stages of NAFLD, such as steatosis or inflammation, but do not reflect the disease progression that occurs in humans (Oligschlaeger and Shiri-Sverdlov, 2020). A common dietary intervention is the methyl-donor-deficient (MDD) diet, which recapitulates the inflammatory responses associated with NASH but not the other elements of the metabolic syndrome (Lyall et al., 2017; Rinella and Green, 2004). In vitro approaches commonly utilize immortalized cell lines derived from hepatocellular carcinoma; however, malignant transformation is known to induce profound changes in metabolic phenotype (Huang et al., 2013; Pavlova and Thompson, 2016), limiting their utility for modeling NAFLD metabolism. Using whole liver tissue from rodent models or humans to study metabolism in distinct cell populations necessitates disaggregation and sorting, a process that induces oxidative stress, potentially confounding analyses (Llufrio et al., 2018). Analysis of primary cells may also be confounded by the cell sorting process (Llufrio et al., 2018).
In terms of mechanisms, studies in animal models and humans have shown that NAFLD is associated with mitochondrial dysfunction including altered electron transport chain (ETC) activity (Koliaki et al., 2015; Rector et al., 2010), altered tricarboxylic acid (TCA) cycle activity and anaplerosis (Satapati et al., 2012; Sunny et al., 2011), induced oxidative metabolism, with a proportional increase in oxidative stress (Satapati et al., 2015) and changes in mitochondrial respiration (Koliaki et al., 2015). In these studies, the presence of multiple cell types in tissue samples may confound the results, and this is further complicated by the changes in cell proportions that may occur in disease states.
Here, we present a human-relevant model of hepatic steatosis that can be used for high-resolution analysis of metabolic function and that may provide novel insights into human NAFLD. We hypothesized that nutrient excess in NAFLD leads to increases in ETC activity due to increased availability of respiratory substrates. We aimed to determine the impact of lipid accumulation on mitochondrial respiration specifically in hepatocytes, to avoid the confounding effects associated with bulk tissue analysis.
Results
Lactate, pyruvate, and octanoate treatment induces macrovesicular steatosis in hepatocyte-like cells
We and others have previously reported that the high-energy substrate cocktail LPO promotes intracellular lipid accumulation in pluripotent stem-cell-derived hepatocyte-like cells (HLCs) (Lyall et al., 2018) (schematic of this process is shown in Figure 1A) and human hepatocyte or hepatoblastoma cell lines (Gilchrist et al., 2010; Lockman et al., 2012, 2016). However, it was not known whether LPO promotes lipid droplet biogenesis or increased accumulation of lipids within existing droplets. Stem-cell-derived HLCs were differentiated and characterized by measuring mRNA levels of a marker of pluripotency (NANOG) and markers of hepatocytes (Albumin; ALB and hepatocyte nuclear factor-4-alpha; HNF4A) (Wang et al., 2017) (Figure 1B). CYP3A4 activity was present in control HLCs and those challenged with LPO, demonstrating that they retained hepatocyte function (Figure 1C). Analysis of gene expression using RT-qPCR showed increased expression of transcripts that are typically associated with intracellular lipid droplet membranes: perilipin 2 (PLIN2), PLIN4, and PLIN5, suggesting an increase in lipid droplet size (Figures 1D and 1F). This was supported by analysis of lipid accumulation using high content imaging, which demonstrated that 48 h of LPO exposure was not associated with a change in the number of intracellular lipid droplets (Figure 1G) but led to a ~2-fold increase in their size (Figure 1H), consistent with the development of macrovesicular steatosis.
Figure 1.

LPO treatment induces development of macrovesicular lipid droplets, similar to benign steatosis in humans
(A) Schematic overview showing differentiation process and induction of lipid accumulation.
(B) H9 hPSCs differentiate to HLCs, losing pluripotency as shown by diminished expression of the pluripotency marker NANOG during the differentiation process. Concomitantly, these cells begin expressing the hepatocyte markers albumin (ALB) and hepatocyte nuclear factor-4-alpha (HNF4A) (n = 3 biological replicates/group).
(C) Differentiated HLCs display robust CYP3A4 activity, which is not diminished following challenge with LPO.
(D–F) Perilipin 2 (PLIN2), PLIN4, and PLIN5 expression increases in response to LPO treatment (n = 3 biological replicates per group).
(G) Number of lipid droplets do not increase,
(H) But intracellular lipid droplets increase in size (n = 32 biological replicates/group). Data were analyzed using two-tailed Student t test and expressed as mean ± SD, ∗p < 0.05, ∗∗∗∗p < 0.0001.
LPO treatment induces intracellular lipid accumulation and transcriptomic alterations in key mitochondrial respiratory pathways
Induction of steatosis in HLCs was associated with widespread transcriptomic changes (Figure 2A). Differential gene expression analysis identified 853 downregulated and 826 upregulated genes (log2 fold change cut-off >1.5) in LPO exposed cells compared with controls. A selection of candidate genes was validated by RT-qPCR (Figure S1), based on genes that we previously found to be dysregulated in association with steatosis (Lyall et al., 2018). RNA-seq analysis revealed altered expression of a number of genes previously described as having functional roles in the progression of NAFLD, including PLIN2, PPARGC1A, CYP7A1, and HMGCS2 (Figure 2B). Mapping genes with a log2 fold change >1.5 to the KEGG pathway database identified a number of enriched pathways (Figures 2C and 1D), including those related to steroid hormone biosynthesis (18 genes) and ascorbate and aldarate metabolism (7 genes) (Table S4). There was extensive downregulation of the histone structural units H1, H2A/B, H3, and H4 and UDP-glucuronosyltransferases, which were enriched in multiple pathways. Specifically comparing transcriptomic data with the KEGG terms “TCA cycle” and “oxidative phosphorylation” revealed marked gene expression changes within these pathways (Figures 2E and 2F) including in the majority of genes encoding enzymes that catalyze metabolite interconversion (Table S5). Analysis of the oxidative phosphorylation pathway revealed an overall downregulation of transcription of genes encoding components of respiratory complexes I (ND1, ND2, ND4L, NDUFS2, NDUFV1, NDUFA10, NDUFB2 NDUFA2, NDUFB10) and IV (COX1, COX4I1, COX8A, COX6B1), as well as ATP synthase (ATP6V1A, ATP5MF, ATP6V0D1, ATP6V1E1, ATP6, APT5MC2, ATP5MC1) (Table S6).
Figure 2.

Steatosis in hepatocytes is associated with transcriptional rewiring reflecting human NAFLD
(A) Heatmap analysis of transcriptional changes associated with macrovesicular steatosis.
(B) Expression of key NAFLD-associated genes is disrupted in steatotic HLCs.
(C–D) Pathway enrichment analysis reveals a number of disrupted pathways associated with macrovesicular steatosis.
(E–F) Analysis of KEGG TCA cycle and oxidative phosphorylation pathways reveals extensive disruption of expression of key genes. Data in (A) were analyzed using a two-tailed Student's t test. Data in (B) were analyzed by two-way ANOVA with Sidak post-hoc testing. n = 3 biological replicates per group. Data are expressed as mean ± SD.
Macrovesicular steatosis in hepatocytes is associated with electron transport chain dysfunction
It has been suggested that NAFLD-associated macrovesicular steatosis results in impairment of mitochondrial respiration (Koliaki et al., 2015), and we therefore proceeded to analyze this in steatotic HLCs (Figure 3A). Basal oxygen consumption rate (OCR), representing combined mitochondrial and non-mitochondrial oxygen consumption, was unchanged following LPO exposure (Figure 3B). In addition, oligomycin A, a complex V inhibitor decreased OCR equally effectively in control and treatment group. Firstly, this indicated that there were no changes in ATP-linked respiration in response to macrovesicular steatosis (Figure 3C). Secondly, when comparing oligomycin-A-induced alterations in OCR with those following addition of rotenone/antimycin A, we could detect no changes in proton leak between groups (Figure 3D). The addition of the ETC uncoupler FCCP revealed a decrease in maximal respiration in the steatotic HLCs, suggesting mild ETC dysfunction (Figure 3E). Using OCR measurements following FCCP treatment, and comparing with the basal OCR, we calculated that there was a decrease in reserve capacity in the LPO-treated cells, compared with controls (Figure 3F). Subsequently, complex I and III were targeted with rotenone/antimycin A, to completely inhibit oxidative phosphorylation, which reduced OCR to a similar level in both groups, suggesting no difference in non-mitochondrial sources of OCR between the control and steatotic HLCs.
Figure 3.

LPO treatment results in decreased mitochondrial maximal respiration and reserve capacity, suggestive of respiratory complex impairment.
HLCs were injected sequentially from ports A–C with 4 μM oligomycin, 0.5–1 μM FCCP, and 1 μM antimycin combined with 200 nM rotenone.
(A) Raw trace of OCR comparing cells with or without LPO treatment; (B) basal respiration (C) ATP-linked respiration and (D) proton leak were unchanged in LPO-treated HLCs. In contrast, maximal respiration (E) and reserve capacity (F) were both diminished following LPO treatment (n = 18 and 16 biological replicates in the control and treatment group, respectively, from two plates). (G) High-content microscopy revealed no changes in fluorescence of mitochondrial in response to treatment (n = 4 and 6 biological replicates in the control and treatment group, respectively). Representative images of mitochondrial content in control and LPO-treated groups 10x magnification. Blue staining = NucBlue, red staining = MitoTracker Deep Red. Scale bar: 25 μm (H). There were no changes in abundance of the mitochondrial proteins succinate dehydrogenase subunit (SDHA) or pyruvate dehydrogenase α1 and α2 subunits (PDH) (n = 6 biological replicates/group). (I) There were no significant changes in the ratio of mitochondrial to genomic DNA, when comparing mitochondrial region 2 (MT2); mitochondrial region 3 (MT3); or genomic region beta-globin (HBB), indicating no alterations in the quantity of mitochondria in response to treatment (n = 4 biological replicates/group). Levels of citrate synthase activity appears unaltered by LPO treatment (n = 3 biological replicates/group). Data were analyzed using two-tailed Student t test for parametric data, or Mann-Whitney U test (D, I) for non-parametric data, and expressed as mean ± SD.
Induction of macrovesicular steatosis is not associated with altered mitochondrial number or mitochondrial integrity
We wanted to analyze whether changes in ETC function, as demonstrated by decreased cellular maximal respiration and reserve capacity, were due to altered numbers of mitochondria. Because these were the only respiratory measurements to change, we hypothesized that this was not the case. To test this, we used a high-content microscopy-based approach to measure intracellular mitochondrial fluorescence and observed no changes following exposure to LPO (Figure 3G), indicating no change in quantity. We then measured protein levels of succinate dehydrogenase subunit A (SDHA) and pyruvate dehydrogenase (PDH) α1 and α2 subunits (Figure 3H; representative blot Figure S2), which localize to the mitochondria. Protein levels of SDHA and PDH did not change in response to intracellular lipid accumulation. Furthermore, there were no changes in mitochondrial or nuclear DNA (Figure S3) and no change in the ratio of nuclear to mitochondrial DNA (Figure 3I), suggesting that mitochondrial number is not altered by LPO-induced macrovesicular steatosis. To analyze whether the observed changes in OCR could be due to diminished mitochondrial integrity rather than ETC dysfunction, we measured citrate synthase (CS) activity. This has previously been described as a measure of mitochondrial integrity (Boutagy et al., 2015; Short et al., 2005). Our findings suggest that LPO treatment has no marked impact on CS activity (Figure 3J), suggesting that mitochondria remain intact following intracellular lipid accumulation. Taken together, these data demonstrate that mitochondrial number was not altered in response to macrovesicular steatosis. This suggests that the altered respiratory complex activity may arise due to reactive oxygen species (ROS)-mediated dysfunction, rather than changes in protein content, although this requires further investigation.
Induction of an NAFLD-like phenotype in HLCs is associated with increased pyruvate carboxylase activity
To investigate TCA cycle dynamics in HLCs, lactate in the LPO cocktail was replaced with 13C3-lactate. Lactate can enter the TCA cycle via mitochondrial pyruvate metabolism, through either pyruvate carboxylase (PC) or pyruvate dehydrogenase (PDH). Alternatively, pyruvate can be transported into the cytosol and used in gluconeogenesis (Figure 4A). LPO exposure was associated with increases in steady state levels of pyruvate, aspartate, and citrate, but there were no changes in metabolites associated with the gluconeogenesis pathway (Figure 4B). Although pyruvate was added as part of the LPO cocktail, increased pyruvate generation also occurred as a result of increased lactate dehydrogenase activity (Figure 4C). Although we were unable to directly measure oxaloacetate levels, we used aspartate as a surrogate and found increased m+3 labeling, demonstrating direct synthesis of aspartate from pyruvate (Figure 4D). Furthermore, isotopomer labeling of citrate indicated increased incorporation from pyruvate-derived oxaloacetate and decreased conversion of labeled acetyl-CoA by PDH (Figure 4E). An alternative pathway for pyruvate is conversion to alanine, and although there were no changes in steady state alanine levels, there were moderate, but significant, changes in the incorporation of 13C (Figure 4F), indicating overall transamination of pyruvate toward alanine. Despite there being no increase in steady state levels of gluconeogenesis-associated metabolites, the increased m+2 labeling of 3-PG (Figure 4G), serine (Figure 4H), and glycine (Figure 4I) suggests increased flux through the gluconeogenesis pathway.
Figure 4.

Macrovesicular steatosis is associated with increased PC activity, leading to preferential anaplerosis of pyruvate into the TCA cycle
(A) Schematic outlining conversion of lactate to pyruvate and routes by which this can be converted. Black circles denote carbon atoms.
(B) Steady state measurements of pyruvate and metabolites that pyruvate can be converted into. Isotopomer labeling patterns of pyruvate (C), aspartate (D), and citrate (E) show anaplerosis into the TCA cycle. There is minimal conversion of pyruvate to alanine (F–G), but increased flux of the m+2 isotopomer through metabolites related to gluconeogenesis, with sustained conversion to 3-PG, serine, and glycine (H–K). For NMR data, labels are: 1 = 13C labeling; 0 = no 13C labeling. All GC-MS data consisted of 10 biological replicates and 2 technical replicates. Isotopomer data were calculated by multiplying MID (multiple ion detection) by normalized total ion count. For NMR data (G and K) n = 4 biological replicates/group. Data were analyzed by two-way ANOVA with Sidak post-hoc testing, or (I–J) two-tailed Student's t test. Data are expressed as mean ± SD.
Steatosis in HLCs is associated with increased TCA cycle anaplerosis
Isotopic labeling of TCA metabolites can produce a number of different isotopomers, depending on the directionality of metabolite synthesis (Figure 5A). In steatotic HLCs, we observed increases in steady state levels of the TCA cycle metabolites αKG, fumarate, and malate (Figure 5B). In contrast, there were no changes in the steady state levels of succinate and no evidence for increased metabolism of glutamate. To further analyze alterations in TCA cycle dynamics we measured 13C incorporation into metabolites. Although there was increased 13C incorporation into PC-derived citrate (Figure 4E), this was not the case for αKG (Figure 5C). The decreased levels of 13C incorporation into glutamate suggests that this does not result from increased cataplerosis through glutamate (Figure 5D). The increased expression of OGDHL and decreased expression of SUCG1 (Table S5) suggest that steatosis may be associated with metabolic reprogramming within the mitochondrial matrix supported by altered protein synthesis. Although decreased incorporation of 13C into succinate was observed (Figure 5E), a parallel increase in 13C incorporation into fumarate, malate, and aspartate was also apparent (Figures 5F and 5G). Because the overall directionality of the malate dehydrogenase and fumarate hydratase reactions can vary depending on the metabolic environment (Dasika et al., 2015; Tyrakis et al., 2017), increased incorporation of 13C into both malate and fumarate indicate increased overall flux of carbons into these metabolites from the oxaloacetate pool. Taken together, this suggests dysregulated TCA cycle flux in steatotic cells, which may be associated with changes in SDH activity (Figure 5H). The increased incorporation of 13C into oxaloacetate (using aspartate as a surrogate) demonstrates increased PC activity, driving the conversion of pyruvate to oxaloacetate and suggests possible disruption of associated metabolic cycles, including the malate-aspartate shuttle (MAS) and the purine nucleotide cycle (PNC).
Figure 5.

Macrovesicular steatosis in HLCs results in truncation of the TCA cycle, inhibiting conversion of succinate to fumarate.
Paradoxically, this is associated with increased accumulation of fumarate.
(A) Schematic outlining the TCA cycle and possible 13C labeled isotopomers from a single cycle.
(B) Steady state measurements of TCA cycle-associated metabolites that could be measured by GC-MS.
(C) PC-derived αKG is unchanged in the presence of steatosis, and PDH-derived αKG is reduced.
(D–E) Labeling patterns of αKG are not due to increased cataplerosis through glutamate, as both PC- and PDH-derived glutamate are reduced in response to steatosis.
(F) Furthermore, reduced 13C labeling of succinate indicates inefficient conversion from αKG.
(G–H) Despite reduced labeling of succinate, both fumarate and malate show increased PC-derived label incorporation.
(I) Proposed model of preferential anaplerosis of pyruvate into the TCA cycle, with concomitant truncation, preventing conversion of succinate to fumarate. For NMR data, labels are: 1 = 13C labeling; 0 = no 13C labeling. All GC-MS data consisted of 10 biological replicates and 2 technical replicates. Isotopomer data were calculated by multiplying MID (multiple ion detection) by normalized total ion count. For NMR data (E) n = 4 biological replicates/group. Data were analyzed by two-way ANOVA with Sidak post-hoc testing and are expressed as mean ± SD.
Increased PNC and MAS activity drives NAFLD-associated fumarate accumulation
Next, we wanted to determine the source of fumarate accumulation in steatotic HLCs. Transcriptomic analysis identified dysregulation of multiple genes associated with the MAS (Figure 6A) and PNC (Figure 6B), indicating that these pathways may be involved in fumarate accumulation. To investigate this further, HLCs incubated with 13C3-lactate-labeled LPO were co-incubated with either 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranosyl 5′-monophosphate (AICAR) or O-(Carboxymethyl)hydroxylamine hemihydrochloride (AOA) to inhibit the PNC or MAS, respectively. Addition of AICAR increased only the steady state levels of αKG, and neither AICAR nor AOA had a significant impact on the synthesis of pyruvate, citrate, succinate, or malate (Figure 6C). However, both inhibitors significantly reduced steady state levels of fumarate and aspartate. Although addition of AICAR did not impact on the synthesis of PDH-derived citrate (Figure 6D), it did result in increased accumulation of PDH-derived 13C in αKG (Figure 6E), suggesting impaired conversion to succinyl-CoA or increased anaplerosis from glutamate. AICAR was also able to partially restore the effects of steatosis on succinate levels (Figure 6F), resulting in small but significant increases in PC- and PDH-derived isotopomers. The most profound effect of AICAR was on fumarate, with each isotopomer reduced to levels below those observed in the control group (Figure 6G), suggesting that in steatotic HLCs, fumarate accumulation is primarily driven by the PNC. Following AOA treatment, we observed a moderate decrease in PC-derived 13C incorporation into fumarate, demonstrating a small contribution from the MAS. AICAR had a moderate impact on incorporation of PC-derived incorporation of 13C into malate, whereas AOA impacted on both PC- and PDH-derived 13C incorporation, suggesting limited contribution of the MAS and PNC to malate accumulation (Figure 6H). Both AICAR and AOA reduced incorporation of 13C into aspartate (Figure 6I). These data show that the metabolic consequences of LPO treatment, likely driven by increased TCA cycle anaplerosis through OAA, include an increase in both MAS and PNC activity (Figure 6J).
Figure 6.

In the presence of TCA cycle truncation, the purine nucleotide cycle and malate-aspartate shuttle fuel fumarate accumulation
(A and B) Transcriptomic analysis showed increased expression of malate-aspartate shuttle (MAS) and purine nucleotide cycle (PNC) transcripts, indicating perturbed activity.
(C) Inhibition of the PNC and MAS reversed steatosis-induced accumulation of fumarate and aspartate but not malate.
(D–G) (D) PNC inhibition moderately impacted PC-derived citrate 13C labeling but not (E) αKG or (F) succinate. (G) In contrast, PNC inhibition profoundly reduced incorporation of 13C into fumarate. MAS inhibition also limited PC-derived fumarate accumulation, but to a lesser extent.
(H) PNC and MAS expression also inhibited generation of PC-derived malate but to a much smaller extent than fumarate.
(I) PNC and MAS inhibition also resulted in a significant reduction in the generation of PC-derived aspartate.
(J) Schematic outlining of the proposed pathways by which fumarate accumulation occurs in response to steatosis.
(K) Inhibition of fumarate through the PNC reduced LPO-induced macrovesicular steatosis, which was partially restored through addition of exogenous monomethyl fumarate. For NMR data, labels are: 1 = 13C labeling; 0 = no 13C labeling. AICAR = PNC inhibitor; AOA = MAS inhibitor. All control and LPO group GC-MS data consisted of 10 biological replicates and 2 technical replicates, as shown in Figure 3. GC-MS LPO + AICAR and LPO + AOA groups consisted of six biological replicates/group. Isotopomer data were calculated by multiplying MID by normalized total ion count. For lipid droplet analysis (K), the LPO group is as shown in Figure 1. For the LPO + AICAR and the LPO + AICAR + Fumarate group, n = 31 and 28 biological replicates/group, respectively. Data were analyzed by two-way ANOVA with Sidak post-hoc testing and are expressed as mean ± SD.
Inhibition of fumarate accumulation inhibits lipid droplet hypertrophy in LPO-treated HLCs
To determine whether manipulation of fumarate levels affected the development of macrovesicular steatosis, we performed HCA microscopy on HLCs. Cells were treated with LPO in the presence or absence of AICAR. Cells that received AICAR did not develop macrovesicular steatosis (Figure 6K). These results were corroborated by the addition of exogenous fumarate to steatotic HLCs treated with AICAR, which resulted in the development of larger lipid droplets.
Fumarate accumulation is not associated with widescale alterations of 5hmC in protein-coding regions in steatotic HLCs
One potential consequence of fumarate accumulation in steatotic HLCs is inhibition of αKG-dependent dioxygenase enzyme activity. Given our previous data showing altered 5hmC in a mouse model of NAFLD and the potential importance of the TET enzymes in hepatocellular carcinoma (Lyall et al., 2020; Thomson et al., 2016), we used a DIP-seq approach to measure changes in 5hmC across the genome. Within each group, samples were highly correlated, suggesting that the 5hmC patterns are stable between replicate samples (Figure 7A). We identified 3,294 differentially hydroxymethylated regions (DHRs) between LPO exposed and control cells (>2-fold change, see methods), with the majority of changes located in intragenic or intronic regions (Figure 7B). 5hmC in gene body regions may functionally relate to mRNA transcription (Thomson et al., 2016), and we therefore generated heatmaps representing mean changes across the gene body, with cluster 1 showing subtle increases in 5hmC within the TSS region (Figure 7C). Analysis of genic 5hmC patterns revealed that a number of promoter regions displayed changes in 5hmC levels following LPO exposure (Figure 7D). Integration of DHR and RNA-seq data for promoter regions upstream of the transcriptional start site revealed 12 promoter regions with differential enrichment of 5hmC (>2-fold) and where mRNA expression was altered >0.5-fold. Linear regression of these regions identified a moderate but significant negative relationship between 5hmC enrichment and mRNA expression (Figure S4; Table S7). Of these genes 7 (EPHX3, ERO1B, CSGALNACT1, DOC2A, COL6A1, CASP1 and TMEM88) were previously shown to be dysregulated in the pathogenesis of NAFLD or progression to cirrhosis (Atanasovska et al., 2017; Cazanave et al., 2017; Parafati et al., 2018; Revill et al., 2013; Wilson and Kumar, 2018). Conversely, to our knowledge, there are no described associations between NAFLD and the remaining genes in this list (DDN, OGFRL1, OCIAD2, ABAT, CCDC150). Our findings may indicate a role for 5hmC in the regulation of these genes in NAFLD pathogenesis.
Figure 7.

Macrovesicular steatosis in HLCs does not correlate with wide-scale changes in 5hmC enrichment
(A) Correlation heatmap of control versus LPO groups, following hmeDIP sequencing, clustered by Euclidean distance.
(B) Proportion of DHRs associated with different regions of the genome, showing the majority located in intragenic and intronic regions.
(C) Heatmap of DHRs, with an FDR of <0.05 and k-means clustering.
(D) Sliding window analysis of the transcriptional start site (TSS), gene body, and transcriptional end site (TES) shows minimal changes between control and LPO groups. For both control and LPO, n = 3 biological replicates/group.
Discussion
NAFLD is a challenging disease to study in humans. This is due to the difficulty of obtaining tissues that are not compromised by confounding conditions, making high-resolution analysis of transcriptional and metabolic rewiring challenging. In this study we sought to explore the utility of a human-HLC-based model for the study of transcriptomic, epigenomic, and metabolomic dysfunction associated with hepatic steatosis.
Using a combination of high content microscopy and gene expression analyses, we showed that treatment of HLCs with LPO induces intracellular lipid accumulation, recapitulating the macrovesicular steatosis observed in NAFLD (Figure 1G). This was in parallel with changes in the expression of genes associated with lipid droplet size (Figures 1D–1F) and is in agreement with findings from human studies (Fujii et al., 2009; Sahini and Borlak, 2016).
The disruption of the expression of genes in multiple pathways related to metabolism confirms the findings of a number of previous studies in humans and mice (Collison et al., 2009; Hardwick et al., 2013; Kolwankar et al., 2007; Liu et al., 2016; Nikolaou et al., 2019; Schiöth et al., 2016; Suppli et al., 2019; Yamaguchi and Murata, 2013). Consistent with the altered expression of multiple subunits of respiratory complexes I, IV, and ATPase, respirometry-based analyses revealed decreased maximal mitochondrial respiration and spare capacity (Figures 3E and 3F). Previous data suggest that NALFD associates with inhibition of respiratory activity in complexes I–III in hepatocytes (Koliaki et al., 2015). Our transcriptomic and metabolomic data suggest that this inhibition most likely occurs in complexes I and II. In terms of mechanisms, octanoic acid inhibits complex I–III in rat liver (Scaini et al., 2012) and excess fumarate may also inhibit complex II activity, although manipulation of fumarate did not alter activity of this complex, suggesting that this is not the case.
We then questioned whether changes in maximal respiration arose from increased biogenesis or from changes in mitochondrial integrity. Our findings suggest that mitochondrial biogenesis was not affected by LPO treatment (Figure 3), because maximal respiration was the only parameter of respiration that changed, and the ratio of nuclear to mitochondrial DNA, as well as HCA microscopy analysis of mitochondrial mass were not altered. In addition, the finding that CS activity was not adversely affected suggests that mitochondrial integrity was not compromised, although this requires further study. These findings are consistent with human studies of NAFLD that show no changes in mitochondrial mass in steatotic liver tissue (Koliaki et al., 2015). In contrast, liver tissue from patients with NASH contains greater numbers of mitochondria and a decrease in maximal respiration, indicating that a transition in mitochondrial function occurs between these two disease states (Koliaki et al., 2015). The reasons for this are unclear; differences in maximal respiration between NAFLD and NASH could reflect impairment of the ETC and/or changes in baseline mitochondrial respiration. In NASH, the decrease in maximal respiration may be a consequence of increased mitochondrial fragmentation and concurrent decreases in mitophagy (Knott et al., 2008; Koliaki et al., 2015). The differences between studies may also reflect the isolation of mitochondria from the multiple cell types present in whole tissues, which may be altered as a consequence of the disease process. We suggest that the model of hepatic steatosis presented here may mirror the transition between NAFLD and NASH, with respiratory dysfunction in the absence of mitochondrial biogenesis. Future experiments, with longer exposure to LPO and/or the incorporation of non-parenchymal cells into the model, may shed light on this.
In agreement with studies in mouse models, and indirect studies in humans, NAFLD was associated with increased hepatic TCA cycle activity (Figure 5B) (Satapati et al., 2012; Sunny et al., 2011). We identified substantial disruption of TCA cycle and oxidative phosphorylation pathways indicating compromised energy metabolism in steatotic HLCs. As TCA cycle flux generates NADH and FADH2, which transfer electrons to the electron transport chain (ETC), alterations in TCA cycling may have a direct impact on respiration and oxidative stress. In order to delineate detailed information on TCA cycle flux dynamics we utilized stable isotopic tracing with 13C3-lactate, which provides greater metabolite labeling than other substrates (Hui et al., 2017). In the liver, flux of substrates into the TCA cycle predominantly occurs via PC (Lardy et al., 1965), and there is evidence suggesting that PC activity increases in NAFLD (Sunny et al., 2011) and contributes to increased rates of gluconeogenesis through conversion of oxaloacetate to phosphoenolpyruvate (Satapati et al., 2012). Our data support the assertion that PC activity increases in steatotic HLCs and that pyruvate is predominantly utilized to sustain TCA cycle activity. Although we observed sustained synthesis of gluconeogenesis-associated metabolites in steatotic HLCs, there was no increase in gluconeogenesis detected (Figure 4). Increased incorporation of only two 13C atoms into each of these gluconeogenesis-associated metabolites may suggest that pyruvate-derived acetyl-CoA is being oxidized to OAA via citrate as a means to enter the gluconeogenic pathway.
Although increased flux of substrates into the TCA cycle leads to higher levels of TCA cycle activity (Sunny et al., 2011), our data suggest that changes in the TCA cycle are more nuanced than was previously suggested, with enhanced anaplerosis from pyruvate/lactate into the TCA cycle and inhibition of the conversion of succinate to fumarate in steatotic cells. This raised the question of what leads to the diminished incorporation of 13C into succinate. The decreased cataplerosis through glutamate suggests that this could be due to perturbation of TCA cycle equilibrium, leading to altered interconversion of succinyl-CoA and succinate, which may, in turn, lead to the generation of an increased pool of succinyl-CoA. This is supported by the observation of decreases in PC-derived glutamate and succinate, despite no changes in PC-derived αKG. Because interconversion between succinyl-CoA and succinate is in near equilibrium and readily reversible (Lynn and Guynn, 1978), we propose that this effect could be a result of an alteration in the equilibrium of this reaction, with enhanced succinyl-CoA synthesis, which could be elicited through branched-chain amino acid catabolism.
Despite truncation of the TCA cycle, steatotic HLCs rewire their metabolic circuitry to compensate for this, generating increased levels of fumarate through the PNC, and to a lesser extent the MAS. A similar metabolic bypass has been reported in cardiac ischemia (Chouchani et al., 2014), as well as in human and mouse in vitro models of tumorigenesis (Tyrakis et al., 2017). In ischemic reperfusion injury, increased PNC activity results in fumarate overflow, driving reversal of succinate dehydrogenase activity and accumulation of succinate (Chouchani et al., 2014). We did not observe the same phenomenon here, but rather found evidence for inhibition of succinate dehydrogenase activity in steatotic HLCs. Although fumarate can be generated from reversal of fumarate hydratase (FH) activity (Chouchani et al., 2014), we were unable to directly manipulate the activity of this enzyme in order to assess its contribution to the fumarate pool in steatotic HLCs. However, as FH operates at equilibrium (Ajalla Aleixo et al., 2019) it is possible that PNC-fueled fumarate accumulation prevents reverse catalytic activity. Defects in complex I can lead to a reduction in the levels of NAD+ (Porcelli et al., 2010) and regeneration of fumarate via the PNC may be one mechanism of increasing the NADH pool and maintaining the hydrogen ion gradient of the ETC. Our data suggest that fumarate is important for the development of macrovesicular steatosis; inhibition of the PNC prevented development of macrovesicular lipid droplets, and this was partially restored following the addition of exogenous fumarate (Figure 6K). This correlates with findings in oligodendrocytes and CD8+ T cells, in which exposure to exogenous fumarate resulted in perturbed lipid metabolism (Bhargava et al., 2019; Huang et al., 2015), although the mechanism by which this occurs is unknown.
Fumarate accumulation can impact the activity of the αKG-dependent dioxygenase enzyme family, including the ten-eleven translocation (TET) enzymes, which are involved in the conversion of 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC) and subsequent steps in the DNA demethylation pathway (Tahiliani et al., 2009). We have previously shown that the TET enzymes play an important role in the development of hepatocellular carcinoma and that 5hmC enrichment is reversibly altered at specific loci in response to hepatic fat accumulation (Lyall et al., 2020; Thomson et al., 2016). 5hmC is a stable epigenetic modification and studies suggest that 5hmC enrichment at transcriptional start sites (TSSs) associates with transcriptional repression (Wu et al., 2011). We did not observe wide-scale changes in 5hmC enrichment, and 5hmC changes at specific loci did not, in general, correlate with transcriptional changes in steatotic cells, except at a limited number of promoter regions.
This human-cell-based model displays many of the transcriptional and metabolic characteristics associated with human hepatic steatosis, and we propose it as a useful addition to the field. Although there are a number of current in vitro approaches for the study of steatosis, these often use HCC cell lines, which have perturbed metabolic function arising from the process of malignant transformation and immortalization (Huang et al., 2013; Pavlova and Thompson, 2016). This may limit their utility for studying steatosis-associated metabolic changes in non-malignant cells. Furthermore, culturing of HCC cell lines is associated with loss of 5hmC, making them unsuitable for studying the interplay between mitochondrial metabolism and TET-mediated modulation of the epigenome (Chen et al., 2013; Hernandez-Vargas et al., 2010; Liu et al., 2013; Thomson et al., 2016; Xu et al., 2011). As an alternative, primary mouse and human hepatocytes can be used to model hepatic steatosis, but these cells have a limited lifespan, cannot be passaged or expanded in vitro, and human cells are often isolated from fundamentally abnormal, e.g. fatty livers (Hirsova et al., 2016; Lyall et al., 2018). The process of cell sorting may also confound metabolomic, transcriptomic, and epigenomic analyses in cell lines (Llufrio et al., 2018), and the necessity for both tissue disaggregation and cell sorting, which are needed to enable the study downstream effects in individual cell types from whole tissue biopsies, can make the interpretation of results even more challenging (Llufrio et al., 2018). The model system presented here overcomes many of these challenges, as HLCs display a similar metabolic phenotype to human hepatocytes and cell sorting is not required (Wang et al., 2017). Furthermore, these cells retain 5hmC in patterns similar to those observed in humans (Ivanov et al., 2013), making them well suited to studying the interplay between mitochondrial metabolism and the epigenome.
The system, allowing the study of transcriptomic, epigenomic, metabolomic, and proteomic effects in a single cell type, can be adapted for the analysis of effects induced by a variety of alternative agents that may induce steatosis. In addition, this model system can be scaled up to model liver injury (Lucendo-Villarin et al., 2020a; Szkolnicka et al., 2014), genetically modified to study gene function (Wang et al., 2019), and moved into 3D culture for longer-term experimentation (Lucendo-Villarin et al., 2020b). These approaches will be valuable to further determine which genes link metabolic disruption and steatosis and for high-throughput drug screening to identify targets for preclinical analysis.
In conclusion, we propose that steatotic HLCs represent a new and attractive tool to develop a deeper understanding of steatosis-associated biology and for the development of new therapeutics.
Limitations of the study
One limitation of this study is the use of AICAR to block PNC activity and limit fumarate accumulation (Chouchani et al., 2014). AICAR is also an AMP-activated protein kinase (AMPK) agonist (Zhao and Saltiel, 2020). AMPK is a regulator of cellular energy homeostasis, and its activation by Metformin, a commonly used treatment for type 2 diabetes, inhibits gluconeogenesis in the liver (Zhou et al., 2001), which may explain why we did not observe increased activity in this pathway. AICAR may have a number of additional effects, notably through downstream pathways, which make the interpretation of its effects problematic, and further studies exploring the effects of overexpression and/or silencing of metabolic enzymes using siRNA on lipid accumulation and the metabolome will be useful to delineate the precise pathways involved. Similarly, there is some evidence that AOA may exert effects upstream of the MAS, leading to inhibition of gluconeogenesis (Subramanian et al., 2007), supporting the need for additional experiments to dissect the precise effect of this inhibitor in the model presented here. Octanoic acid increases oxidative stress through ROS production (Scaini et al., 2012), so future studies should additionally measure oxidative stress and ROS levels in steatotic HLCs. Further studies are required to determine if the mitochondrial dysfunction is due to defects in individual respiratory complexes or changes in a combination of complexes and whether other aspects of mitochondrial respiration may become compromised with longer LPO exposure. Finally, LPO may induce ER stress (Lockman et al., 2016), which may influence mitochondrial oxygen consumption and contribute to the development of NAFLD (Liu and Green, 2019; Wang et al., 2011).
Resource availability
Lead contact
David C Hay: Center for Regenerative Medicine, University of Edinburgh, Institute for Regeneration and Repair, Edinburgh BioQuarter, 5 Little France Crescent, Edinburgh, EH16 4UU. davehay@talktalk.net.
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for transcriptomic sequencing data reported in this paper is GEO: GSE138052. The accession number for 5hmC sequencing data reported in this paper is GEO: GSE144955. Original raw GC-MS data have been deposited to Mendeley Data: https://data.mendeley.com/datasets/gd5k5psvf4/1; https://doi.org/10.17632/gd5k5psvf4.1.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
MCS was supported by a British Heart Foundation PhD studentship (FS/16/54/32730) and the British Heart Foundation Centre of Research Excellence. SW-Z was funded by the Deutsche Forschungsgemeinschaft (SFB960). This work was supported in part by the Wellcome Trust [grant number 208400/Z/17/Z], and we thank HWB-NMR at the University of Birmingham for providing open access to their Wellcome Trust-funded NMR equipment. RNC and NMM were supported by a Wellcome Trust New Investigator Award to NMM (100981/Z/13/Z). MJL was supported by a Wellcome Trust PhD Fellowship as part of the Edinburgh Clinical Academic Track scheme (102839/Z/13/Z). RRM is supported by the Medical Research Council and by grants from the BBSRC. Research in RRM's lab leading to these results is partly funded by the Innovative Medicine Initiative Joint Undertaking (IMI JU) under grant agreement number 115001 (MARCAR project: URL: http://www.imi-marcar.eu/). DCH, JMR, and BLV were supported with awards from the MRC Doctoral Training Partnership (MR/K501293/1) and the Chief Scientist Office (TCS/16/37). AJD was funded by the British Heart Foundation Centre of Research Excellence, University of Edinburgh. Our thanks go to the Wellcome Trust Clinical Research Facility Genetics Core, Western General Hospital, Edinburgh, UK. We thank Will Cawthorn for discussions about mitochondrial quantification. Graphical abstract and Figure 1A were created using the BioRender platform.
Author contributions
MCS was involved in conceptualization, methodology, validation, formal analysis, investigation, data curation, writing (original draft preparation, reviewing, and editing) and visualization. BLV, JMR, MJL, PDW, and AT were involved in methodology, validation, formal analysis, investigation, and writing (reviewing and editing). SW-Z and JPT were involved in software, formal analyses, data curation, and writing (reviewing and editing). RRM, CL, and DAT were involved in conceptualization, methodology, validation, formal analysis, investigation, data curation, provision of resources, writing (reviewing and editing), project administration, funding acquisition, and supervision. DCH and AJD were involved in conceptualization, methodology, validation, formal analysis, provision of resources, writing (reviewing and editing), project administration, funding acquisition, and supervision.
Declaration of interests
Professor David Hay is a founder, shareholder, and director in Stemnovate Limited. All other authors declare that they have no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101931.
Contributor Information
David C. Hay, Email: davehay@talktalk.net.
Amanda J. Drake, Email: mandy.drake@ed.ac.uk.
Supplemental information
References
- Ajalla Aleixo M.A., Rangel V.L., Rustiguel J.K., de Pádua R.A.P., Nonato M.C. Structural, biochemical and biophysical characterization of recombinant human fumarate hydratase. FEBS J. 2019;286:1925–1940. doi: 10.1111/febs.14782. [DOI] [PubMed] [Google Scholar]
- Anstee Q.M., Goldin R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrih M., Jornayvaz F.R. Metabolic syndrome and nonalcoholic fatty liver disease: is insulin resistance the link? Mol. Cell. Endocrinol. 2015;418:55–65. doi: 10.1016/j.mce.2015.02.018. [DOI] [PubMed] [Google Scholar]
- Atanasovska B., Rensen S.S., van der Sijde M.R., Marsman G., Kumar V., Jonkers I., Withoff S., Shiri-Sverdlov R., Greve J.W.M., Faber K.N. A liver-specific long noncoding RNA with a role in cell viability is elevated in human nonalcoholic steatohepatitis. Hepatology. 2017;66:794–808. doi: 10.1002/hep.29034. [DOI] [PubMed] [Google Scholar]
- Bhargava P., Fitzgerald K.C., Venkata S.L.V., Smith M.D., Kornberg M.D., Mowry E.M., Haughey N.J., Calabresi P.A. Dimethyl fumarate treatment induces lipid metabolism alterations that are linked to immunological changes. Ann. Clin. Transl. Neurol. 2019;6:33–45. doi: 10.1002/acn3.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutagy N.E., Pyne E., Rogers G.W., Ali M., Hulver M.W., Frisard M.I. Isolation of mitochondria from minimal quantities of mouse skeletal muscle for high throughput microplate respiratory measurements. J. Vis. Exp. 2015;2015:53217. doi: 10.3791/53217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazanave S., Podtelezhnikov A., Jensen K., Seneshaw M., Kumar D.P., Min H.K., Santhekadur P.K., Banini B., Mauro A.G., Oseini A.M. The transcriptomic signature of disease development and progression of nonalcoholic fatty liver disease. Sci. Rep. 2017;7:17193. doi: 10.1038/s41598-017-17370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.L., Shen F., Huang W., Qi J.H., Wang Y., Feng Y.Q., Liu S.M., Yuan B.F. Quantification of 5-methylcytosine and 5-hydroxymethylcytosine in genomic DNA from hepatocellular carcinoma tissues by capillary hydrophilic-interaction liquid chromatography/quadrupole TOF mass spectrometry. Clin. Chem. 2013;59:824–832. doi: 10.1373/clinchem.2012.193938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E.T., Pell V.R., Gaude E., Aksentijević D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N.J., Smith A.C. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison K.S., Maqbool Z., Saleh S.M., Inglis A., Makhoul N.J., Bakheet R., Al-Johi M., Al-Rabiah R., Zaidi M.Z., Al-Mohanna F.A. Effect of dietary monosodium glutamate on trans fat-induced nonalcoholic fatty liver disease. J. Lipid Res. 2009;50:1521–1537. doi: 10.1194/jlr.M800418-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasika S.K., Vinnakota K.C., Beard D.A. Determination of the catalytic mechanism for mitochondrial malate dehydrogenase. Biophys. J. 2015;108:408–419. doi: 10.1016/j.bpj.2014.11.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H., Ikura Y., Arimoto J., Sugioka K., Iezzoni J.C., Park S.H., Naruko T., Itabe H., Kawada N., Caldwell S.H. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J. Atheroscler. Thromb. 2009;16:893–901. doi: 10.5551/jat.2055. [DOI] [PubMed] [Google Scholar]
- Gilchrist E.S., Lockman K., Pryde A., Cowan P., Lee P., Plevris J.N. PWE-002 Cell growth is impaired in a cellular model of non-alcoholic fatty liver disease, but is unaffected by simple steatosis. Gut. 2010;59:A85–A86. [Google Scholar]
- Hardwick R.N., Ferreira D.W., More V.R., Lake A.D., Lu Z., Manautou J.E., Slitt A.L., Cherrington N.J. Altered UDP-glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver diseases. Drug Metab. Dispos. 2013;41:554–561. doi: 10.1124/dmd.112.048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Vargas H., Lambert M.P., Le Calvez-Kelm F., Gouysse G., McKay-Chopin S., Tavtigian S.V., Scoazec J.Y., Herceg Z. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One. 2010;5:e9749. doi: 10.1371/journal.pone.0009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsova P., Ibrahim S.H., Krishnan A., Verma V.K., Bronk S.F., Werneburg N.W., Charlton M.R., Shah V.H., Malhi H., Gores G.J. Lipid-Induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016;150:956–967. doi: 10.1053/j.gastro.2015.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Taraboletti A., Shriver L.P. Dimethyl fumarate modulates antioxidant and lipid metabolism in oligodendrocytes. Redox Biol. 2015;5:169–175. doi: 10.1016/j.redox.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Tan Y., Yin P., Ye G., Gao P., Lu X., Wang H., Xu G. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013;73:4992–5002. doi: 10.1158/0008-5472.CAN-13-0308. [DOI] [PubMed] [Google Scholar]
- Hui S., Ghergurovich J.M., Morscher R.J., Jang C., Teng X., Lu W., Esparza L.A., Reya T., Zhan L., Yanxiang Guo J. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov M., Kals M., Kacevska M., Barragan I., Kasuga K., Rane A., Metspalu A., Milani L., Ingelman-Sundberg M. Ontogeny, distribution and potential roles of 5-hydroxymethylcytosine in human liver function. Genome Biol. 2013;14:R83. doi: 10.1186/gb-2013-14-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott A.B., Perkins G., Schwarzenbacher R., Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliaki C., Szendroedi J., Kaul K., Jelenik T., Nowotny P., Jankowiak F., Herder C., Carstensen M., Krausch M., Knoefel W.T. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Kolwankar D., Vuppalanchi R., Ethell B., Jones D.R., Wrighton S.A., Hall S.D., Chalasani N. Association between nonalcoholic hepatic steatosis and hepatic cytochrome P-450 3A activity. Clin. Gastroenterol. Hepatol. 2007;5:388–393. doi: 10.1016/j.cgh.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Lardy H.A., Paetkau V., Walter P. Paths of carbon in gluconeogenesis and lipogenesis: the role of mitochondria in supplying precursors of phosphoenolpyruvate. Proc. Natl. Acad. Sci. U. S. A. 1965;53:1410–1415. doi: 10.1073/pnas.53.6.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen T.L., Hagemann C.A., Wei C., Kazankov K., Thomsen K.L., Knop F.K., Grønbæk H. Bariatric surgery in patients with non-alcoholic fatty liver disease - from pathophysiology to clinical effects. World J. Hepatol. 2019;11:138–249. doi: 10.4254/wjh.v11.i2.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Green R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019;3:55–64. doi: 10.1016/j.livres.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Liu L., Chen X., Shen J., Shan J., Xu Y., Yang Z., Wu L., Xia F., Bie P. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One. 2013;8:e62828. doi: 10.1371/journal.pone.0062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Pathak P., Boehme S., Chiang J.Y.L. Cholesterol 7α-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J. Lipid Res. 2016;57:1831–1844. doi: 10.1194/jlr.M069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llufrio E.M., Wang L., Naser F.J., Patti G.J. Sorting cells alters their redox state and cellular metabolome. Redox Biol. 2018;16:381–387. doi: 10.1016/j.redox.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman K.A., Baren J.P., Pemberton C.J., Baghdadi H., Burgess K.E., Plevris-Papaioannou N., Lee P., Howie F., Beckett G., Pryde A. Oxidative stress rather than triglyceride accumulation is a determinant of mitochondrial dysfunction in in vitro models of hepatic cellular steatosis. Liver Int. 2012;32:1079–1092. doi: 10.1111/j.1478-3231.2012.02775.x. [DOI] [PubMed] [Google Scholar]
- Lockman K.A., Htun V., Sinha R., Treskes P., Nelson L.J., Martin S.F., Rogers S.M., Le Bihan T., Hayes P.C., Plevris J.N. Proteomic profiling of cellular steatosis with concomitant oxidative stress in vitro. Lipids Health Dis. 2016;15:114. doi: 10.1186/s12944-016-0283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucendo-Villarin B., Nell P., Hellwig B., Filis P., Feuerborn D., O’Shaughnessy P.J., Godoy P., Rahnenführer J., Hengstler J.G., Cherianidou A. Genome-wide expression changes induced by bisphenol A, F and S in human stem cell derived hepatocyte-like cells. EXCLI J. 2020;19:1459–1476. doi: 10.17179/excli2020-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucendo-Villarin B., Meseguer-Ripolles J., Drew J., Fischer L., Ma W.S.E., Flint O., Simpson K., Machesky L., Mountford J., Hay D. Development of a cost effective automated platform to produce human liver spheroids for basic and applied research. Biofabrication. 2020;13:015009. doi: 10.1088/1758-5090/abbdb2. [DOI] [PubMed] [Google Scholar]
- Lyall M.J., Cartier J., Richards J.A., Cobice D., Thomson J.P., Meehan R.R., Anderton S.M., Drake A.J. Methyl donor deficient diets cause distinct alterations in lipid metabolism but are poorly representative of human NAFLD. Wellcome Open Res. 2017;2:67. doi: 10.12688/wellcomeopenres.12199.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall M.J., Cartier J., Thomson J.P., Cameron K., Meseguer-Ripolles J., O’Duibhir E., Szkolnicka D., Villarin B.L., Wang Y., Blanco G.R. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. B Biol. Sci. 2018;373:20170362. doi: 10.1098/rstb.2017.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall M.J., Thomson J.P., Cartier J., Ottaviano R., Kendall T.J., Meehan R.R., Drake A.J. Non-alcoholic fatty liver disease (NAFLD) is associated with dynamic changes in DNA hydroxymethylation. Epigenetics. 2020;15:61–71. doi: 10.1080/15592294.2019.1649527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn R., Guynn R.W. Equilibrium constants under physiological conditions for the reactions of succinyl coenzyme A synthetase and the hydrolysis of succinyl coenzyme A to coenzyme A and succinate. J. Biol. Chem. 1978;253:2546–2553. [PubMed] [Google Scholar]
- Nikolaou N., Gathercole L.L., Marchand L., Althari S., Dempster N.J., Green C.J., van de Bunt M., McNeil C., Arvaniti A., Hughes B.A. AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease. Metabolism. 2019;99:67–80. doi: 10.1016/j.metabol.2019.153947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oligschlaeger Y., Shiri-Sverdlov R. NAFLD preclinical models: more than a handful, less of a concern? Biomedicines. 2020;8:28. doi: 10.3390/biomedicines8020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parafati M., Kirby R.J., Khorasanizadeh S., Rastinejad F., Malany S. A nonalcoholic fatty liver disease model in human induced pluripotent stem cell-derived hepatocytes, created by endoplasmic reticulum stress-induced steatosis. DMM Dis. Model. Mech. 2018;11:dmm033530. doi: 10.1242/dmm.033530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli A.M., Ghelli A., Ceccarelli C., Lang M., Cenacchi G., Capristo M., Pennisi L.F., Morra I., Ciccarelli E., Melcarne A. The genetic and metabolic signature of oncocytic transformation implicates HIF1α destabilization. Hum. Mol. Genet. 2010;19:1019–1032. doi: 10.1093/hmg/ddp566. [DOI] [PubMed] [Google Scholar]
- Rector R.S., Thyfault J.P., Uptergrove G.M., Morris E.M., Naples S.P., Borengasser S.J., Mikus C.R., Laye M.J., Laughlin M.H., Booth F.W. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010;52:727–736. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revill K., Wang T., Lachenmayer A., Kojima K., Harrington A., Li J., Hoshida Y., Llovet J.M., Powers S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145:1424–1435.e1-25. doi: 10.1053/j.gastro.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella M.E., Green R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Rotundo L., Persaud A., Feurdean M., Ahlawat S., Kim H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: a population-based study. Clin. Mol. Hepatol. 2018;24:392–401. doi: 10.3350/cmh.2018.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahini N., Borlak J. Genomics of human fatty liver disease reveal mechanistically linked lipid droplet–associated gene regulations in bland steatosis and nonalcoholic steatohepatitis. Transl. Res. 2016;177:41–69. doi: 10.1016/j.trsl.2016.06.003. [DOI] [PubMed] [Google Scholar]
- Satapati S., Sunny N.E., Kucejova B., Fu X., He T.T., Méndez-Lucas A., Shelton J.M., Perales J.C., Browning J.D., Burgess S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satapati S., Kucejova B., Duarte J.A.G., Fletcher J.A., Reynolds L., Sunny N.E., He T., Arya Nair L., Livingston K., Fu X. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Invest. 2015;125:4447–4462. doi: 10.1172/JCI82204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaini G., Simon K.R., Tonin A.M., Busanello E.N.B., Moura A.P., Ferreira G.C., Wajner M., Streck E.L., Schuck P.F. Toxicity of octanoate and decanoate in rat peripheral tissues: evidence of bioenergetic dysfunction and oxidative damage induction in liver and skeletal muscle. Mol. Cell. Biochem. 2012;361:329–335. doi: 10.1007/s11010-011-1119-4. [DOI] [PubMed] [Google Scholar]
- Schiöth H.B., Boström A., Murphy S.K., Erhart W., Hampe J., Moylan C., Mwinyi J. A targeted analysis reveals relevant shifts in the methylation and transcription of genes responsible for bile acid homeostasis and drug metabolism in non-alcoholic fatty liver disease. BMC Genomics. 2016;17:462. doi: 10.1186/s12864-016-2814-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short K.R., Bigelow M.L., Kahl J., Singh R., Coenen-Schimke J., Raghavakaimal S., Nair K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian R.K., Kasumov T., Yang L., Cendrowski A., David F., Minkler P.E., Hoppel C.L., Anderson V.A., Brunengraber H. John Wiley & Sons, Ltd); 2007. Reassessment of the Mechanisms by Which Aminooxyacetate (AOA) Inhibits Gluconeogenesis (GNG) from Lactate. [Google Scholar]
- Sunny N.E., Parks E.J., Browning J.D., Burgess S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppli M.P., Rigbolt K.T.G., Veidal S.S., Heebøl S., Lykke Eriksen P., Demant M., Bagger J.I., Nielsen J.C., Oró D., Thrane S.W. Hepatic transcriptome signatures in patients with varying degrees of non-alcoholic fatty liver disease compared to healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019;316:G462–G472. doi: 10.1152/ajpgi.00358.2018. [DOI] [PubMed] [Google Scholar]
- Szkolnicka D., Farnworth S.L., Lucendo-Villarin B., Storck C., Zhou W., Iredale J.P., Flint O., Hay D.C. Accurate prediction of drug-induced liver injury using stem cell-derived populations. Stem Cells Transl. Med. 2014;3:141–148. doi: 10.5966/sctm.2013-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (80-. ) 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J.P., Ottaviano R., Unterberger E.B., Lempiainen H., Muller A., Terranova R., Illingworth R.S., Webb S., Kerr A.R.W., Lyall M.J. Loss of tet1-associated5-hydroxymethylcytosine is concomitant with aberrant promoter hypermethylation in liver cancer. Cancer Res. 2016;76:3097–3108. doi: 10.1158/0008-5472.CAN-15-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrakis P.A., Yurkovich M.E., Sciacovelli M., Papachristou E.K., Bridges H.R., Gaude E., Schreiner A., D’Santos C., Hirst J., Hernandez-Fernaud J. Fumarate hydratase loss causes combined respiratory chain defects. Cell Rep. 2017;21:1036–1047. doi: 10.1016/j.celrep.2017.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti L., Bugianesi E., Pajvani U., Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int. 2016;36:1563–1579. doi: 10.1111/liv.13185. [DOI] [PubMed] [Google Scholar]
- Wang L., Yu S. Pathology of non-alcoholic fatty liver disease. Int. J. Dig. Dis. 2016;2:85–89. [Google Scholar]
- Wang X., Eno C.O., Altman B.J., Zhu Y., Zhao G., Olberding K.E., Rathmell J.C., Li C. ER stress modulates cellular metabolism. Biochem. J. 2011;435:285–296. doi: 10.1042/BJ20101864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Alhaque S., Cameron K., Meseguer-Ripolles J., Lucendo-Villarin B., Rashidi H., Hay D.C. Defined and scalable generation of hepatocyte-like cells from human pluripotent stem cells. J. Vis. Exp. 2017:55355. doi: 10.3791/55355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Tatham M.H., Schmidt-Heck W., Swann C., Singh-Dolt K., Meseguer-Ripolles J., Lucendo-Villarin B., Kunath T., Rudd T.R., Smith A.J.H. Multiomics analyses of HNF4α protein domain function during human pluripotent stem cell differentiation. IScience. 2019;16:206–217. doi: 10.1016/j.isci.2019.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . WHO Media Center; 2006. Obesity and Overweight; pp. 1–3. [Google Scholar]
- Wilson C.H., Kumar S. Caspases in metabolic disease and their therapeutic potential. Cell Death Differ. 2018;25:1010–1024. doi: 10.1038/s41418-018-0111-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., D’Alessio A.C., Ito S., Wang Z., Cui K., Zhao K., Sun Y.E., Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Wu F., Tan L., Kong L., Xiong L., Deng J., Barbera A.J., Zheng L., Zhang H., Huang S. Genome-wide Regulation of 5hmC, 5mC, and gene expression by tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M., Murata T. Involvement of regucalcin in lipid metabolism and diabetes. Metabolism. 2013;62:1045–1051. doi: 10.1016/j.metabol.2013.01.023. [DOI] [PubMed] [Google Scholar]
- Zhao P., Saltiel A.R. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J. Biol. Chem. 2020;295:12279–12289. doi: 10.1074/jbc.REV120.011356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for transcriptomic sequencing data reported in this paper is GEO: GSE138052. The accession number for 5hmC sequencing data reported in this paper is GEO: GSE144955. Original raw GC-MS data have been deposited to Mendeley Data: https://data.mendeley.com/datasets/gd5k5psvf4/1; https://doi.org/10.17632/gd5k5psvf4.1.