

Engineering liquid-liquid phase separation into synthetic and optogenetic transcription factors increases gene expression.
Abstract
Light-inducible gene switches represent a key strategy for the precise manipulation of cellular events in fundamental and applied research. However, the performance of widely used gene switches is limited due to low tissue penetrance and possible phototoxicity of the light stimulus. To overcome these limitations, we engineer optogenetic synthetic transcription factors to undergo liquid-liquid phase separation in close spatial proximity to promoters. Phase separation of constitutive and optogenetic synthetic transcription factors was achieved by incorporation of intrinsically disordered regions. Supported by a quantitative mathematical model, we demonstrate that engineered transcription factor droplets form at target promoters and increase gene expression up to fivefold. This increase in performance was observed in multiple mammalian cells lines as well as in mice following in situ transfection. The results of this work suggest that the introduction of intrinsically disordered domains is a simple yet effective means to boost synthetic transcription factor activity.
INTRODUCTION
Optogenetic switches to control signaling and gene expression in mammalian cells and mammals have revolutionized our understanding of cell fate and function and paved the avenue for precision interventions in gene- and cell-based therapies (1–6). Optogenetic switches are constructed by functionally fusing plant- or bacteria-derived photoreceptors to cellular effector molecules, the activity of which can subsequently be triggered by light. This allows the control of cellular events with unmatched spatial and temporal resolution, reversibility, and dose dependence. Despite excellent functionality in cultivated cells, the performance of optogenetic switches is limited in tissue cultures and living animals by the low penetrance of the inducing light and further by phototoxicity when higher light doses are administered (7). This is especially the case for switches responsive to blue light, the class of switches which is most widely used (6). To overcome these limitations, we here devise a likely generically applicable strategy of increasing the potency of transcription factors (TFs). The engineered optogenetic tools described in this study already show a higher activity at lower light doses or an increased activity at the same light dose compared with conventional switches. We achieve this advance by driving optogenetic TFs to undergo liquid-liquid phase separation (LLPS) and to form TF droplets around their target promoters.
Eukaryotic cells are highly compartmentalized into specific organelles, which are either membrane enclosed or membrane-less. The recent discovery that membrane-less organelles are formed by LLPS has triggered an avalanche of discoveries that have greatly improved our understanding of cell biology (8). We now understand that LLPS forms chemically distinct cellular compartments with diverse biological functions including adaptive responses to changes in temperature and pH (9), regulation of cellular metabolism (10), local enrichment of molecules to activate cell signaling (11), nucleation of the cytoskeleton (12), acceleration of biochemical reactions (13), selective localization of mRNA (14), and sorting at the nuclear pore (15). These and other functional implications of LLPS in cell biology are discussed in recent reviews (16–18).
Biomolecular condensates are particularly common in eukaryotic cell nuclei, therefore also called the “liquid nucleome” (19, 20). Recently, a computational model suggested a possible link between high expression levels from super-enhancers and LLPS of TFs (21). Since then, a substantial body of biological evidence has confirmed this notion. For example, nuclear transcription factories contain clusters of crowded RNA polymerase II (Pol II) (22), and in mouse embryonic stem cells, Pol II was shown to form stable transcriptional condensates together with Mediator (23). In addition, single-molecule imaging revealed that eukaryotic TFs form high-density interaction hubs with Mediator and Pol II via LLPS in the promoter region (24). Condensate formation was linked to intrinsically disordered regions (IDRs) of transcriptional coactivators Med1 and BRD4 (25), as well as the C-terminal domain of Pol II (26).
In this work, we develop the concept of forming coacervates from synthetic TFs (DropletTFs) that bind to and trigger transcription from synthetic target promoters. We demonstrate that DropletTFs trigger strongly increased transcription compared with non–coacervate-forming factors. Generalizing these findings, we design TFs that form coacervates in response to blue or red light. These OptoDropletTFs combine the transcription-increasing effect of DropletTFs with the excellent control modalities of optogenetic gene switches. We anticipate that the concept developed here will have broad implications not only in boosting synthetic gene switches but also in the fundamental understanding of how cells modulate endogenous transcription rates via coacervate formation.
RESULTS
In a first approach, we capitalized on the TetOff system for mammalian transgene expression (27). For this purpose, we designed the TF consisting of TetR-VP16-NLS-eYFP (TF-), where TetR constitutes the DNA binding domain, VP16 the trans-activating domain (TAD), and NLS (a nuclear localization signal). The fluorescent reporter protein eYFP (enhanced yellow fluorescent protein) enables the monitoring of transfection efficiencies and normalization of expression levels in transiently transfected cells. To engineer coacervate formation into the transcription factor TF-, we additionally incorporated the N-terminal IDR of human oncogene FUS (FUSn; amino acids 1 to 214), forming TF + FUS (TetR-VP16-NLS-FUSn-eYFP). FUSn mainly consists of amino acids Gly (24.9%), Ser (23.9%), Gln (20.2%), Tyr (12.7%), Pro (5.2%), and Thr (4.7%) and can drive LLPS, likely due to π-π, CH-π, OH-π, and NH-π interactions (28).
As reporter for transcriptional activation, we used the secreted alkaline phosphatase (SEAP) (29) under the control of a tetracycline response element (TRE) consisting of varying numbers of tetO operators and a minimal promoter PhCMVmin. Upon cotransfection of TF- with the reporter, the TF binds the TRE and induces SEAP expression (Fig. 1A, left). In contrast, we hypothesized that for cotransfection of TF + FUS and the reporter, FUSn would trigger LLPS and the formation of a condensate on the TRE, which, in turn, would lead to a higher local concentration of VP16 and increase reporter expression (Fig. 1A, right).
Fig. 1. Design of droplet transcription factors (DropletTF).
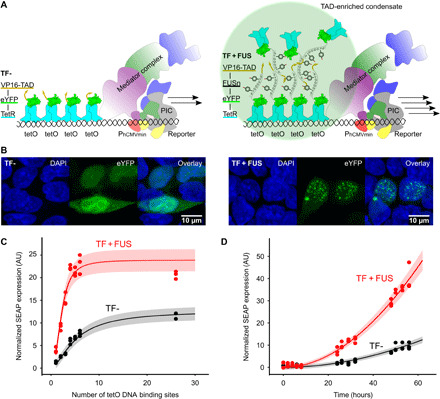
(A) Left: The conventional synthetic TF (TF-, TetR-eYFP-VP16) binds as a homodimer to tetO operators and recruits the preinitiation complex (PIC), initiating the expression of a downstream gene. Right: DropletTF (TF + FUS, TetR-eYFP-FUSn-VP16). FUS addition triggers the formation of coacervates at the tetO operator sites. The locally increased VP16 density increases promoter activity and downstream gene expression. (B) Distribution of TF- and TF + FUS. Human embryonic kidney (HEK)–293 cells were transfected with TF- or TF + FUS constructs and analyzed by fluorescence microscopy. (C) Effects of TF- and TF + FUS on transgene expression. tetOn-based SEAP reporters (n = 1 to 6, 26) were cotransfected with TF- and TF + FUS constructs. The stoichiometry of the expression vectors was adjusted to achieve approximately equal expression levels of TF- and TF + FUS. After 48 hours, SEAP production was quantified and TF expression levels were determined by flow cytometric detection of eYFP. (D) Temporal dynamics of SEAP production induced by TF- and TF + FUS and a tetO4 reporter. SEAP production was quantified at the indicated time points. (C and D) SEAP production was normalized to eYFP fluorescence. Curves represent the model fit of the data. Error bands are estimated with a model with a constant and relative Gaussian error. AU, arbitrary unit.
To test the IDR-dependent formation of coacervates described above, we first studied the effect of IDR insertion on protein localization via fluorescence microscopy. As expected, a diffuse nuclear distribution was observed for TF-, while cells transfected with TF + FUS showed a high number of protein droplets, reflecting FUSn-driven LLPS (Fig. 1B). As it is known that fusion proteins of different composition are produced at varying amounts in different cells (30), we adjusted the amount of transfected DNA to obtain comparable TF- and TF + FUS levels as judged from TF quantification via eYFP fluorescence. We then measured the transactivation potential of TF- and TF + FUS by transfecting the respective constructs together with SEAP reporters comprising 1, 2, 3, 4, 5, 6, or 26 tetO repeats.
We observed a marked difference in normalized SEAP production levels (Fig. 1C), with TF + FUS resulting in an up to 4.7-fold increase compared with TF- (for tetO2). In both cases, reporter production was saturated at a high number of tetO repeats. For raw data of SEAP and TF production, see also Supplementary Materials and Methods and fig. S1 (A and B).
To obtain quantitative insights into the effect of coacervate formation, we next developed a quantitative mathematical model based on ordinary differential equations (ODEs). We used the measured protein production data to parameterize the model. The set of ODEs on which the model is based are
| (1) |
| (2) |
These equations describe the changes in the molecule concentrations of SEAP protein and SEAPmRNA. The existence of tetO in the cells leads to transcription of SEAPmRNA with the rate . This process becomes saturated for a high concentration of tetO. For these high tetO concentrations, the SEAPmRNA levels approach . The saturation behavior at intermediate tetO concentrations is described by the parameter , which indicates the tetO level where half of the maximal SEAPmRNA production rate is reached. Thus, the two parameters jointly describe the tetO-induced transcription of SEAPmRNA and its saturation behavior. The cooperativity of this process is modeled with the Hill coefficient h*.
SEAPmRNA is degraded with a constant rate kdeg, SEAP. SEAP is then translated from SEAPmRNA with the rate ktransl, SEAP. Because of the high stability of the SEAP protein [t1/2 = 502 hours; (31)], SEAP degradation is not considered in the model.
The two complexes TF- and TF + FUS are modeled by equations of the same structure; however, some parameters are assumed to be different to account for the differences in the behavior of the two complexes. Asterisks indicate these parameters. A more detailed derivation of the model and its equations is provided in the Supplementary Materials, Modeling, section S1.
The model was calibrated using a maximum likelihood approach based on the data in Fig. 1C (fig. S1A), as well as a time course experiment of SEAP production for up to 56 hours after transfection in Fig. 1D (fig. S1B). The model is able to describe the saturation effect of the tetO dose-response data as well as the time course data. It furthermore is able to quantify the differences between the TF- and the TF + FUS system by simultaneously estimating the differing parameters for both settings. A detailed description of the maximum likelihood approach to parametrize the models is provided in the Supplementary Materials, Modeling, section S3.
For TF + FUS, the maximum specific transcription rate ktransk is 1.9 times higher than for TF-. Similarly, Km for TF + FUS is 2.4-fold lower compared with TF-, indicating that saturation is reached for lower TF concentrations for the coacervate-forming variant. The Hill coefficients h of the TF- and TF + FUS systems are equal within their uncertainties, suggesting that the cooperativity with regard to tetO-binding sites is not linked to the droplet formation (see table S1 for the full set of parameter values).
Encouraged by the initial results for the Tet system–based gene switch, we next applied our concept to increase expression levels of an optogenetic gene switch to combine the advantages of both, increased expression due to coacervate formation and excellent control opportunities due to optical stimulation. To this aim, we capitalized on the optoDroplet concept (32), in which IDRs, such as FUSn, DDX4n, or hnRNAPA1c, were fused to a fluorescent protein and the blue light receptor Cryptochrome 2 (Cry2). In darkness, proteins were diffusely localized; however, upon blue light illumination, Cry2 oligomerized, thus initiating LLPS and the formation of coacervates. We based our switch on the BLInCR system (33) using CIBn-TetR as DNA binding domain and a fusion protein of Cry2(PHR)-eYFP-NLS-VP16 as base construct for the activation domain, in the following termed OptoTF-. In this construct, we inserted between the eYFP and NLS-VP16 three different IDRs, DDX4n [OptoTF+DDX(1–236)], FUSn [OptoTF + FUS(1–214)], or hnRNPA1c [OptoTF+RNP(186–320)] (32).
The resulting split TF is inactive in darkness; however, upon blue light illumination (465 nm), simultaneous dimerization of CIBn and Cry2(PHR) as well as homo-oligomerization of Cry2(PHR) are induced (34). In the case of OptoTF-, this effect already leads to recruitment of VP16-TAD to the promoter (Fig. 2A, left). For OptoTF + IDR constructs, illumination leads to localized induction of LLPS. We therefore hypothesized that DNA-bound optoDroplets should again enrich VP16 relative to OptoTF- and lead to increased reporter gene expression (Fig. 2A, right). When transfecting these constructs together with a tetO13-PhCMVmin-mCherry reporter and cultivating the cells in the dark, the OptoTF- and OptoTF + IDR constructs were diffusely distributed in the cell nuclei and no mCherry reporter expression was observed. However, upon 24-hour constant blue light illumination (465 nm, 5 μmol m−2 s−1), all OptoTFs formed aggregates (no IDR: 24.2%; DDX4: 64.6%; FUS: 56.2%; RNP: 54.5% of all cells), and in all cases, the reporter mCherry was expressed (Fig. 2B and fig. S2A). We further quantified droplet formation at the single-cell level, indicating that IDR insertion leads to a 12.8-, 10.9-, and 8.4-fold increase of the mean aggregate count for FUS, DDX4, and RNP toward OptoTF-, respectively (fig. S2A). To further estimate the propensity of different IDR constructs to form coacervates, we determined the integrated fluorescence of all droplets per single cell where TF + FUS showed the highest propensity [4.4 relative fluorescence unit (RFU)], followed by TF + DDX (3.3 RFU) and TF + RNP (2.7 RFU) (fig. S2B). For this reason, we performed the following comprehensive characterization with the TF + FUS construct.
Fig. 2. Design of droplet optogenetic transcription factors (OptoTF).

(A) Left: Conventional optogenetic TFs. CIBn-TetR is continuously bound to tetO. Upon blue light illumination, Cry2-eYFP-VP16 (OptoTF-) is recruited to the promoter via heterodimerization of Cry2 and CIBn. Homomultimerization of Cry2 leads to further accumulation of the TAD VP16, resulting in high expression of the reporter gene. Right: Droplet optogenetic TFs (Cry2-eYFP-IDR-VP16, OptoTF + IDR). Blue light triggers coacervate formation of OptoTF + IDR due to binding to CIBn-TetR and homo-oligomerization of Cry2. The resulting locally increased TAD concentration further increases reporter gene expression. (B) Light-responsive distribution of OptoTF- and different OptoTF + IDRs in HEK-293 cells. OptoTF- or OptoTF + IDR with three different IDRs (DDX4, FUS, and hnRNPA1) was cotransfected with a tetO13-based mCherry reporter and analyzed by fluorescence microscopy after 24-hour cultivation in the dark (left) or under blue light (465 nm, 5 μmol m−2 s−1). (C) Mobile fractions and half-recovery times of OptoTF + FUS droplets determined by fluorescence recovery after photobleaching (FRAP). OptoTF + FUS, CIBn-TetR, and a tetO7-based reporter were cotransfected into HEK-293 cells. Cells were illuminated with blue light (465 nm, 5 μmol m−2 s−1) for either 10 min or 24 hours before measurement. Both groups were compared by a two-tailed Welch’s t test.
First, we confirmed that the observed aggregates were liquid as neighboring droplets coalesced to bigger droplets over time (fig. S3A). In addition, we performed fluorescence recovery after photobleaching (FRAP) experiments by bleaching the YFP fluorescence of aggregates after 10-min or 24-hour blue light illumination of the cells (Fig. 2C and fig. S3, B to D). We observed mobile fractions (10 min: 62 ± 8%, 24 hours: 52 ± 8%) and half recovery times (10 min: 90 ± 30 s, 24 hours: 135 ± 34 s) both in the same range as previously observed for OptoFUS-based droplets (32). These data suggest that our observed aggregates are mainly liquid-phase droplets. The increased half recovery time and the decreased mobile fraction upon 24-hour blue light illumination are likely caused by an aging of the droplets similar to the decrease in the mobile fraction for an increased supersaturation depth (32). Then, we verified that OptoTF + FUS droplets form on-targeted DNA regions. For this analysis, we used U2OS 2-6-3 cells, which carry 200 cassettes each containing 256 × lacO and 96 × tetO operators in the euchromatin of chromosome 1 (35). We labeled the genomic locus with an mCherry-lacI construct binding to lacO and then observed the localization of OptoTF + FUS via confocal fluorescence microscopy (fig. S4A). In the dark, eYFP fluorescence was diffusely localized. Upon blue light illumination, we observed CIBn-TetR–dependent recruitment of large coacervates to the lacO/tetO genomic locus (fig. S4B, left). To verify that we have similar colocalization in the plasmid-based system, we cloned a 256 × lacO cassette into a 6 × tetO-SEAP reporter plasmid. Upon cotransfection of this reporter with CIBn-TetR and OptoTF + FUS, we again observed colocalization of mCherry and eYFP, demonstrating that OptoTF + FUS droplets formed at transfected reporter constructs (fig. S4B, right). We then went on to test the influence of LLPS on transgene activation in the optogenetic system, resulting in a 4.1-fold increase in SEAP production for OptoTF + FUS relative to the OptoTF- construct (Fig. 3A). We wondered if this difference might be an effect of a greater spatial distance between the Cry2(PHR) domain and VP16 due to the IDR in the middle. To exclude this possibility, we rearranged the OptoTF + FUS constructs to carry the IDR at the N terminus. In this arrangement, FUS resulted in a 3.5-fold increase in SEAP production. Dark controls confirmed the low leakiness of the optogenetic gene switch (Fig. 3A). To further underline that the observed increase was due to LLPS and not due to altered protein domain interactions, we also performed the experiments with OptoTF constructs carrying mCherry instead of eYFP and tested an exchange of the TAD from VP16 to E2F4. In every case, we observed a significant increase in SEAP reporter expression for OptoTF + FUS (fig. S5).
Fig. 3. Effects of FUSn insertion into OptoTF constructs on gene expression levels.

(A) Effect of the IDR integration site. FUSn was integrated into OptoTF- either between eYFP and VP16 or at the N terminus of Cry2. The constructs were cotransfected into HEK-293 cells together with a tetO7-based SEAP reporter and CIBn-TetR. Cells were cultivated in the dark or under blue light (465 nm, 5 μmol m−2 s−1) for 48 hours before quantifying SEAP production. (B) Gene expression mediated by OptoTF constructs. The experiment was performed as described in (A) except that reporters with different numbers of tetO repeats (1 to 6, 26) were used. The model fit to the data is represented by the curves, while the shaded error bands are estimated with an error model with a constant and relative Gaussian error. (C) OptoTF-mediated expression kinetics. OptoTF + FUS and OptoTF- were cotransfected together with a tetO4-based SEAP reporter and CIBn-TetR. Cells were cultivated in the dark or under blue light, and SEAP production was quantified at the indicated time points. The model fit to the data is represented by the curves, while the shaded error bands are estimated with an error model with a constant and relative Gaussian error.
The recurrent and pronounced effect of FUSn addition might also be caused by FUSn acting as TAD itself. In sarcomas, it has been described that fusion of the N-terminal domain of RNA binding protein FUS/TLS to a C-terminal DNA binding domain (e.g., DDIT3/CHOP) can form potent TFs (36). To exclude this possibility, we deleted the VP16-TAD from the OptoTF/FUS construct. The finding that we did not observe blue light–dependent SEAP production for the ΔVP16 construct suggests that FUSn alone is not sufficient to result in measurable SEAP output (fig. S6). Together, these findings suggest that LLPS-mediated formation of optogenetic TF droplets (DropletTFs) strongly increases expression from specific target promoters.
Furthermore, we tested the OptoTF + FUS–induced gene expression for reversibility (fig. S7). We measured SEAP production of samples that were returned to dark after 6- or 24-hour blue light illumination in comparison to samples that were continuously illuminated. We found that after 18 or 24 hours in darkness, gene expression can be completely terminated for samples previously illuminated for 6 hours, but only partially for samples that were illuminated for 24 hours, respectively (fig. S7, A and B). This suggests slower dissolution of the OptoTF + FUS droplets after prolonged illumination. To asses this effect more closely, we determined the half-life period of droplet disassembly via microscopy. Droplets that formed at a 10-min blue light illumination were dissolved exponentially with t1/2 = 6.8 min, while droplets that were illuminated for 24 hours had a half-life of t1/2 = 58 min and did not dissolve completely (fig. S7, C to F). This suggests an aging effect leading to irreversible aggregates, as previously reported for optoDroplets (32).
To quantitatively characterize the OptoTF system, we extended the mathematical model to incorporate the optogenetic switch. To this aim, we modeled the light dependence by introducing an additional state describing the concentration of the VP16 fusions in their active and inactive states for both the optogenetic and the previously modeled TFs. Thus, the sets of ODEs are
| (3) |
| (4) |
| (5) |
| (6) |
VP16 is activated from its inactive state VP16inactive to its active state VP16active with the rate kon, VP16 by the blue light intensity Iblue. A constant dark revision of this process with the rate koff, VP16 exists, too. These two processes are only relevant for the optogenetic conditions; thus, the Boolean variable bopto deactivates them for the light-insensitive TF- and TF + FUS conditions. The tetO-induced transcription and saturation behavior is similar to Eq. 1; however, VP16 in its active conformation is now also actively influencing the transcription. The cooperativity of the VP16 transcription is modeled with the Hill coefficient hVP16. The set of condition-specific parameters described by , , and h* now incorporates both the two previous conditions TF- and TF + FUS as well as the two additional conditions OptoTF- and OptoTF + FUS. Last, the SEAP translation is modeled with the constant rate ktransl, SEAP. A full derivation of the extended model is given in the Supplementary Materials, Modeling, section S2.
To parameterize the model, we measured the dose-response curve with tetO counts of 1, 2, 3, 4, 5, 6, and 26 (Fig. 3B) and a time course experiment for up to 32 hours after start of blue light illumination (Fig. 3C). Raw data are provided in fig. S8. The extended model is able to quantitatively explain the experimental data and to quantify the differences between the OptoTF and the initial TF system. Identified parameters were highly comparable, reinforcing the notion that the FUS-dependent effect holds true between different model systems of synthetic gene switches. Furthermore, a direct comparison of the DropletTF and OptoDropletTF systems allows to discern a Cry2-dependent increase in gene expression (fig. S9). The effect is in line with our expectation, that Cry2 homo-oligomerization recruits additional TADs to the promoter.
We next compared the TF and OptoTF results with and without FUS and showed that the incorporation of the IDR sequence triggers in both cases a strong increase in reporter expression especially at low tetO copy numbers (Fig. 4A). This confirms our initial hypothesis that coacervate formation yields in stronger gene expression (up to 4.9-fold increase for tetO2), an effect that becomes less pronounced when many transactivators can bind via strongly increased tetO repeat numbers (2.0-fold change at tetO26). This finding indicates that promoter complexity can be strongly reduced (i.e., lowering the number of operator copies) when invigorating the TFs’ multiplicity via coacervate formation. We further applied the quantitative mathematical model to analyze the impact of the light dose on reporter gene expression in the OptoTF systems. To this aim, we incorporated the light intensity into the model based on our previous work (37). Analyzing this model predicts that at the same light dose, the OptoTF + FUS system should reach higher gene expression levels than the OptoTF- variant. This would allow reducing the light dose while still achieving high gene expression, an experimental advantage allowing the mitigation of the phototoxic effects of energy-rich blue light. We tested this model prediction by comparing the performance of the OptoTF- and OptoTF + FUS systems at increasing intensities of blue light (Fig. 4B).
Fig. 4. Difference in gene expression levels for transcription factors with and without FUS.
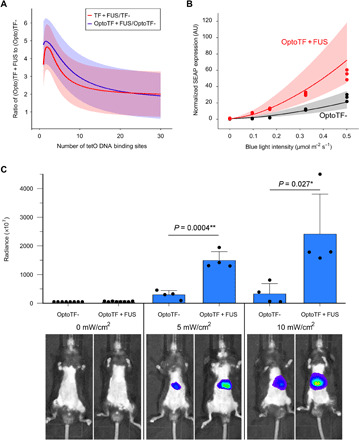
(A) Ratio of the gene expression levels for TF + FUS/TF- (red) and OptoTF + FUS/OptoTF- (blue). Curves correspond to the ratio of the calibrated model trajectories for TF- and TF + FUS (Fig. 1C) and OptoTF- and OptoTF + FUS (Fig. 3B). (B) Light dose–dependent reporter gene expression. HEK-293 cells were cotransfected with OptoTF + FUS, CIBn-TetR, and a tetO7-based SEAP reporter and cultivated for 48 hours with the indicated blue light intensities. SEAP production was quantified. Curves represent the model prediction. Uncertainties (shaded bands) were calculated using the prediction profile likelihood method. (C) Differential reporter gene expression in mice. CIBn-TetR, OptoTF- or OptoTF + FUS, and a tetO7-Luciferase reporter were coadministered via hydrodynamic tail vein injection. Mice were either kept in darkness or exposed to blue light pulses for 11 hours (460 nm, 2-min pulses with the indicated intensity). For in vivo bioluminescence imaging, luciferin was injected intraperitoneally. Top: Mean bioluminescent radiance (p s−1 cm−2 sr−1) ± SEM, n = 4. P values were calculated with Student’s t test *P < 0.05, **P < 0.01. Bottom: Representative images for each condition (photo credit: Deqiang Kong, East China Normal University).
The experimental data are in accordance with the model predictions within its uncertainties. This agreement indicates that coacervate formation of optically inducible TFs can be used to reduce the light dose on biological samples. Furthermore, the agreement underlines the predictive potential of the quantitative mathematical model for model-guided design of desired transgene expression scenarios. This holds especially true because the experimental conditions of the validation dataset are fundamentally different from the data used in the calibration of the model.
To further evaluate the general applicability of TF coacervate formation for increased transgene expression, we reengineered a previously described red light–responsive gene expression system (38). To this end, we inserted the FUSn(1–214) domain between the Arabidopsis red light receptor PhyB and the VP16 transactivation domain (fig. S10A). We cotransfected cells with this construct together with an expression cassette for the phytochrome-interacting factor PIF6(1–100) fused to TetR and with a tetO7-PhCMVmin–containing SEAP reporter. Under red light, PhyB and PIF6(1–100) dimerize and thus reconstitute a functional TF that is able to activate tetO-containing target promoters. However, under far-red light, PhyB and PIF dissociate, thus resulting in transcriptional deactivation. When comparing the performance of the FUS-containing system with the original system, we observed a 6.5-fold increase in maximal SEAP production (fig. S10B), suggesting that the concept of increasing transcriptional performance by LLPS might be broadly applicable.
On the basis of the findings above that coacervate formation of optogenetic synthetic TFs increases transgene expression, we evaluated whether this approach could be applied in mice. Light-mediated gene expression in mammals is complicated by the low tissue penetration of inducing light, a limitation that is especially prominent for light with short wavelength such as in the blue spectrum. To evaluate whether OptoDropletTFs could alleviate this limitation, we implemented the OptoTF- and OptoTF + FUS systems together with a tetO7-driven luciferase reporter in mice via hydrodynamic tail vein injection. With this method, plasmid DNA is primarily taken up into liver cells due to high hydrostatic pressure (39). The mice were externally illuminated (fig. S11) at increasing light intensities, and luciferase production was quantified by whole-body bioluminescence imaging. In the experiments using OptoTF + FUS, a fivefold increase in bioluminescence radiance was observed compared with OptoTF- configurations (Fig. 4C and fig. S12). This demonstrates that the OptoDropletTF approach is as well functional in vivo and can be used to overcome limitations linked to the poor tissue penetration of blue light.
Biomolecular condensates and membrane-less organelles have extensively been studied for more than a decade, and first biotechnological applications are beginning to emerge. For example, artificial organelles were already used to recruit and subsequently release cargo molecules upon a trigger (40), while optogenetically controlled phase separation was used for metabolic channeling, enabling a sixfold increase in product formation of a two-step metabolic pathway (41). Furthermore, membrane-less organelles have also been used to increase the efficiency of noncanonical amino acid integration into engineered proteins (42).
Simultaneously, our growing understanding of the role of coacervates in gene expression opens previously unidentified and exciting avenues to apply these principles in synthetic gene switches. On the basis of our work, a next step could be to apply phase-separating TFs to endogenous genomic loci and to transform weak enhancers into synthetic super-enhancers. In this context, it is also advantageous that the IDR-dependent increase in gene expression allows the reduction of the number of DNA binding sites and thus the complexity of gene targeting. It might also be interesting to further explore the interchange between FUSn and the transcription machinery by modifying the amino acid composition and liquid properties of the coacervates, e.g., by changing the number of tyrosine and glycine residues (43).
While the practical application of biomolecular condensates and synthetic membrane-less organelles in synthetic gene expression systems is still in its early days, we expect that this work contributes to substantial advances and that IDR incorporation into TFs will unlock new functionalities and applications.
MATERIALS AND METHODS
DNA cloning and production
All used gene constructs were created via Gibson assembly (44) or AQUA cloning (45). The growth of Escherichia coli was performed at 30°C for DNA constructs containing sequence repeats (tetO and lacO) to avoid recombination and at 37°C for all other constructs. Before use, sequences were verified by Sanger sequencing. For details of the vectors developed and used in this work, see table S1.
Mammalian cell culture and transfection
Human embryonic kidney cells (HEK-293, ATCC CRL-1573) and U2OS 2-6-3 cells (35) were cultivated at 37°C and 5% CO2 in DMEM (Dulbecco’s modified Eagle’s medium) complete medium [DMEM PAN, P04-03550; supplemented with 10% fetal calf serum (FCS, PAN, P30-3602) and 1% penicillin-streptomycin (PAN, P06-07100)] and passaged every 2 to 3 days upon reaching ~80% confluency. For experiments with SEAP reporter and flow cytometry readout, cells were cultivated and transfected in 24-well plates (1.9 cm2 surface area, Corning 3524). Trypsinized cells were adjusted to a density of 150,000 cells/ml in DMEM complete medium followed by addition of 500 μl to each well. Cells were then grown for ~24 hours until reaching a confluency of ~30%. Cells were transfected using a polyethyleneimine (PEI)–based method as described before (46). For each 24-well, a total of 750 ng of DNA, 2.5 μl of PEI solution (1 mg/ml PEI in H2O, pH 7), and 50 μl of OptiMEM (Invitrogen) were mixed, incubated at room temperature for 15 min, and added dropwise to cells. For each condition to be measured, biological triplicates were carried out. Detailed information on transfection mixture compositions is listed in table S2. Cells were subsequently grown for the indicated times until harvest of supernatant and/or cells. In the case of optogenetic experiments, experimental steps after transfection and with living cells were carried out under red safelight (660 nm). Cells were first grown in the dark for 24 hours and then transferred into light boxes, followed by either blue light exposure (465 nm, 5 μmol m−2 s−1) or not (dark control). For preparation of fixed cell samples for imaging, HEK-293 or U2OS 2-6-3 cells were cultivated in 24-well plates. Before cell seeding, high-precision cover slips (Roth, LH23.1) were added to empty wells and coated for 1 hour with 500 μl of Rat Tail Collagen I (Thermo Fisher Scientific, A1048301) at a concentration of 20 μg/ml in DMEM complete medium. Subsequently, wells were washed twice with 500 μl of Dulbecco’s phosphate-buffered saline (DPBS) and used for cell cultivation and transfection as described above. For live cell imaging, cells were grown in 35-mm μ-Dishes (Ibidi, 81156).
SEAP readout and calculation
To measure SEAP reporter production, for each experimental condition, 200 μl of cell supernatant was harvested at indicated times. If analysis was not carried out directly (e.g., in the case of time course experiments), samples were stored at −20°C until further use. Supernatants were then transferred into V-bottom 96-well plates. Plates were sealed with adhesive tape, incubated at 65°C for 30 min to inactivate heat-labile phosphatases, and centrifuged for 5 min at 300g to remove cell debris. Next, 80 μl of each sample was added to 100 μl 2× SEAP buffer [21% (v:v) diethanolamine, 20 mM l-homoarginine, 1 mM MgCl2, (pH 9.8)] in transparent flat-bottom 96-well plates. Directly before measurement, 20 μl of para-nitrophenylphosphate solution (120 mM) was added, and conversion to para-nitrophenol was measured at room temperature via absorbance measurement at 405 nm at time intervals of 1 min. SEAP activity was calculated as described before (31). For the quantitative mathematical model, SEAP values were normalized to the amount of eYFP-labeled TF constructs to account for variations of TF expression levels. To that end, SEAP values were divided by the integrated eYFP fluorescence (RFU) of the ungated cell population.
Flow cytometry readout and gating
To determine expression rates of different TFs—TF-, TF + FUS, OptoTF-, and OptoTF + IDR—in parallel to SEAP experiments, we routinely measured single-cell eYFP fluorescence via flow cytometry. After harvest of cell supernatants, the remaining medium was removed and HEK-293 cells were treated with 100 μl of Trypsin-EDTA. After ~3 min, 100 μl of DMEM complete medium was added to each well, and detached cells were gently resuspended and then transferred into a round bottom 96-well plates. Cells were spun down at 300g for 3 min, washed twice with DBPS, and lastly resuspended in 300 μl of DPBS + 10% FCS. Cells were then placed on ice and covered with aluminum foil until analysis. Single-cell fluorescence was measured with an Attune NxT Flow Cytometer with autosampler (Thermo Fisher Scientific). eYFP was excited with a 488-nm laser and detected using a 530/30-nm emission filter. Obtained data were gated for singlets with the FlowJo_V10 software. For comparison of TF expression, we explicitly did not select the subpopulation of YFP-expressing cells but included the entire population of singlets. That way, we obtain a measure of eYFP expression in the whole population, comparable to, e.g., Western blot analysis.
Imaging and image analysis
For imaging of OptoTF constructs, fixed cell samples on coverslips were used. Under red safelight, the medium of the wells was first removed and then carefully replaced with 500 μl of prewarmed 4% methanol-free formaldehyde solution (Science Services, E15714). After 15 min of incubation at room temperature, fixed cells were washed twice with 500 μl of DPBS. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 0.1 μg/ml) for 15 min. Cells were then again washed twice and mounted on microscope slides in 4 μl of Mowiol mounting medium [2.4 g of Mowiol, 6 g of glycerol, 6 ml of H2O, 12 ml of tris/HCl (pH 8.5)]. Coverslips were subsequently fixed with nail polish. Cell samples were imaged with an inverted wide-field Axiovert 200 microscope equipped with a Colibri light-emitting diode (LED) illumination source (excitation DAPI: 400 nm, excitation eYFP: 505 nm) and a 63× objective. To capture optoDroplets within the entire cell volumes, 12 focal planes with a distance of 1 μm were acquired. To obtain >1000 cells per condition, regions of 20 image tiles were imaged and stitched with the ZEN2.6 pro (Zeiss) software. Next, three-dimensional z-stacks were reduced to two-dimensional images via maximum intensity projection in Fiji (47). Cell nuclei and aggregates of eYFP were segmented, counted, and analyzed with custom-written ImageJ macros. Live cell imaging to observe droplet fusion was performed with an inverted Zeiss Axiovert 200 microscope, equipped with a confocal scan head LSM 510 Meta using a 63× water immersion objective and a 514-nm laser for excitation of YFP. FRAP experiments were performed with a Zeiss LSM 880 laser scanning confocal microscope using a 63× Plan-Apochromat oil objective (NA 1.4). For both experiments, cells were always kept at 37°C and 5% CO2 in a stage top Tokai Hit incubator during imaging. For the FRAP experiments, a zoom factor of 6 was used, and YFP was excited with a 514-nm laser (0.7% power) and detected at 526 to 597 nm. Before and after bleaching, z-stacks (nine slices, slice distance: 0.6 μm) were acquired every 5 s. In each FRAP experiment, three droplets were bleached each with a spot of ~0.8-μm diameter and 100% laser (514 nm) intensity (total bleaching time: ~1.5 s). During the whole FRAP experiment, cells were illuminated with blue light with an external LED (465 nm, 5 μmol m−2 s−1). For analysis, the droplets were tracked manually in the x, y, and z directions, and the mean intensity of each droplets was measured for each time point (using a circular mask with constant diameter). Afterward, the acquired intensities were corrected for photobleaching and normalized to the pre- and postbleaching intensities. FRAP parameters were calculated by fitting the indicated exponential recovery equation to the recovery data.
OptoTF-mediated transgene expression in mice
Wild-type C57BL/6 mice [6 weeks old, male, East China Normal University (ECNU) Laboratory Animal Center] were randomly divided into groups. The mice were hydrodynamically injected with plasmid DNA encoding the OptoTF- [pNS1000 (PhCMV-CIBN-TetR-pA), 12 μg; pNS1001 (PhCMV-Cry2-eYFP-VP16-pA), 12 μg; pAF101(P7×tetO-hCMVmin-Luciferase-pA), 300 μg] or the OptoTF + FUS [pNS1000, 12 μg; pNS026 (PhCMV-Cry2-eYFP-FUS-VP16-pA), 12 μg; pAF101, 300 μg] with a total of 324 μg of plasmids in 2 ml (10% of the body weight in grams) of Ringer’s solution (147 mM NaCl, 4 mM KCl, and 1.13 mM CaCl2) within 8 s via the tail vein for each mouse. Eight hours after plasmid injection, the mice were exposed to blue light pulses for 11 hours [460-nm LED (Shenzhen Kiwi Lighting Co. Ltd.); 5 or 10 mW·cm−2; 2 min on, 2 min off, alternating]. The mouse illumination device is depicted in fig. S11. Control mice were kept in the dark. For in vivo bioluminescence imaging, each mouse was intraperitoneally injected with 100 mM luciferin substrate solution (SYNCHEM; CAS no. 115144-35-9) under ether anesthesia. Five minutes after luciferin injection, bioluminescence images of the mice were obtained using the IVIS Lumina II In Vivo Imaging System (Perkin Elmer, USA). Radiance (p s−1 cm−2 sr−1) values were calculated for region of interest using Living Image 4.3.1 software.
Ethics
The experiments involving mice were approved by the ECNU Animal Care and Use Committee and in direct accordance with the Ministry of Science and Technology of the People’s Republic of China on Animal Care guidelines. The protocol (protocol ID: m20200501) was approved by the ECNU Animal Care and Use Committee. All mice were euthanized after the termination of the experiments.
Supplementary Material
Acknowledgments
We thank all members of the Weber laboratory for the helpful comments. We also thank P. Heun and V. Lazou for providing us plasmids pLacO and pIB_mCherry_LacI_v5. We are especially grateful to A. Rademacher and K. Rippe, who provided plasmids Cry(PHR)-YFP-YP16 and CIBn-TetR along with U2OS 2-6-3 cells. We also want to thank D. Spector for agreeing to our use of the U2OS 2-6-3 cell line developed in laboratory. We are very grateful to M. Klenzendorf for excellent technical assistance and to L. Qiao for excellent assistance in the animal work. We would like to thank the Faculty of Biology technical workshop for construction of the illumination devices. We acknowledge the excellent scientific and technical assistance of the Signalling Factory Core Facility staff of the Albert-Ludwigs-University Freiburg for help on flow cytometry. We thank the staff of the Life Imaging Center (LIC) in the Center for Biological Systems Analysis (ZBSA) of the Albert-Ludwigs-University Freiburg for help with microscopy resources, and the excellent support in image recording and analysis. Funding: This work was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) grant WE 4733/7-1, under Germany’s Excellence Strategy (EXC-2189; project ID: 390939984) and under the Excellence Initiative of the German Federal and State Governments (EXC-294 and GSC-4), and in part by the Ministry for Science, Research and Arts of the State of Baden-Württemberg. J.T. and F.-G.W. were supported by the German Federal Ministry for Education and Research (grant no. 031L0048). This work was also partially supported by the grants from the National Key R&D Program of China, Synthetic Biology Research (no. 2019YFA0904500), the National Natural Science Foundation of China (NSFC; nos. 31971346 and 31861143016), and the Science and Technology Commission of Shanghai Municipality (no. 18JC1411000) to H.Y. Author contributions: W.W. and N.S. conceived the project. W.W., N.S., H.Y., F.-G.W., and J.T. designed the experiments. N.S., F.-G.W., D.K., A.A.M.F., and M.H. performed the experimental work. N.S., F.-G.W., A.A.M.F., M.H., J.T., H.Y., and W.W. analyzed the results and wrote the manuscript. All authors edited and approved the manuscript. Competing interest: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Raw data (images, FACS data, SEAP measurements) are available from the authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd3568/DC1
REFERENCES AND NOTES
- 1.Mansouri M., Strittmatter T., Fussenegger M., Light-controlled mammalian cells and their therapeutic applications in synthetic biology. Adv. Sci. (Weinh) 6, 1800952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartmann J., Krueger D., De Renzis S., Using optogenetics to tackle systems-level questions of multicellular morphogenesis. Curr. Opin. Cell Biol. 66, 19–27 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Krueger D., Izquierdo E., Viswanathan R., Hartmann J., Cartes C. P., de Renzis S., Principles and applications of optogenetics in developmental biology. Development 146, dev175067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye H., Baba M. D.-E., Peng R.-W., Fussenegger M., A synthetic optogenetic transcription device enhances blood-glucose homeostasis in mice. Science 332, 1565–1568 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Kolar K., Weber W., Synthetic biological approaches to optogenetically control cell signaling. Curr. Opin. Biotechnol. 47, 112–119 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Kolar K., Knobloch C., Stork H., Žnidarič M., Weber W., OptoBase: A web platform for molecular optogenetics. ACS Synth. Biol. 7, 1825–1828 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Kaberniuk A. A., Shemetov A. A., Verkhusha V. V., A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat. Methods 13, 591–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brangwynne C. P., Eckmann C. R., Courson D. S., Rybarska A., Hoege C., Gharakhani J., Jülicher F., Hyman A. A., Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Riback J. A., Katanski C. D., Kear-Scott J. L., Pilipenko E. V., Rojek A. E., Sosnick T. R., Drummond D. A., Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168, 1028–1040.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prouteau M., Loewith R., Regulation of cellular metabolism through phase separation of enzymes. Biomolecules. 8, 160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su X., Ditlev J. A., Hui E., Xing W., Banjade S., Okrut J., King D. S., Taunton J., Rosen M. K., Vale R. D., Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Case L. B., Zhang X., Ditlev J. A., Rosen M. K., Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science 363, 1093–1097 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheu-Gruttadauria J., MacRae I. J., Phase transitions in the assembly and function of human miRISC. Cell 173, 946–957.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langdon E. M., Gladfelter A. S., A new lens for RNA localization: Liquid-liquid phase separation. Annu. Rev. Microbiol. 72, 255–271 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Schmidt H. B., Görlich D., Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem. Sci. 41, 46–61 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Holehouse A. S., Pappu R. V., Functional implications of intracellular phase transitions. Biochemistry 57, 2415–2423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin Y., Brangwynne C. P., Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strom A. R., Brangwynne C. P., The liquid nucleome–phase transitions in the nucleus at a glance. J. Cell Sci. 132, jcs235093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feric M., Vaidya N., Harmon T. S., Mitrea D. M., Zhu L., Richardson T. M., Kriwacki R. W., Pappu R. V., Brangwynne C. P., Coexisting liquid phases underlie nucleolar subcompartments. Cell 165, 1686–1697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hnisz D., Shrinivas K., Young R. A., Chakraborty A. K., Sharp P. A., A phase separation model for transcriptional control. Cell 169, 13–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cisse I. I., Izeddin I., Causse S. Z., Boudarene L., Senecal A., Muresan L., Dugast-Darzacq C., Hajj B., Dahan M., Darzacq X., Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341, 664–667 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Cho W.-K., Spille J.-H., Hecht M., Lee C., Li C., Grube V., Cisse I. I., Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boija A., Klein I. A., Sabari B. R., Dall’Agnese A., Coffey E. L., Zamudio A. V., Li C. H., Shrinivas K., Manteiga J. C., Hannett N. M., Abraham B. J., Afeyan L. K., Guo Y. E., Rimel J. K., Fant C. B., Schuijers J., Lee T. I., Taatjes D. J., Young R. A., Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabari B. R., Dall’Agnese A., Boija A., Klein I. A., Coffey E. L., Shrinivas K., Abraham B. J., Hannett N. M., Zamudio A. V., Manteiga J. C., Li C. H., Guo Y. E., Day D. S., Schuijers J., Vasile E., Malik S., Hnisz D., Lee T. I., Cisse I. I., Roeder R. G., Sharp P. A., Chakraborty A. K., Young R. A., Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boehning M., Dugast-Darzacq C., Rankovic M., Hansen A. S., Yu T., Marie-Nelly H., McSwiggen D. T., Kokic G., Dailey G. M., Cramer P., Darzacq X., Zweckstetter M., RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 25, 833–840 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Gossen M., Bujard H., Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin Y., Currie S. L., Rosen M. K., Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 292, 19110–19120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berger J., Hauber J., Hauber R., Geiger R., Cullen B. R., Secreted placental alkaline phosphatase: A powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 66, 1–10 (1988). [DOI] [PubMed] [Google Scholar]
- 30.Sitton G., Hansgate A., Srienc F., Transient gene expression in CHO cells monitored with automated flow cytometry. Cytotechnology 52, 13–24 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlatter S., Rimann M., Kelm J., Fussenegger M., SAMY, a novel mammalian reporter gene derived from Bacillus stearothermophilus α-amylase. Gene 282, 19–31 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Shin Y., Berry J., Pannucci N., Haataja M. P., Toettcher J. E., Brangwynne C. P., Spatiotemporal control of intracellular phase transitions using light-activated optoDroplets. Cell 168, 159–171.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rademacher A., Erdel F., Trojanowski J., Schumacher S., Rippe K., Real-time observation of light-controlled transcription in living cells. J. Cell Sci. 130, 4213–4224 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Che D. L., Duan L., Zhang K., Cui B., The dual characteristics of light-induced cryptochrome 2, homo-oligomerization and heterodimerization, for optogenetic manipulation in mammalian cells. ACS Synth. Biol. 4, 1124–1135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janicki S. M., Tsukamoto T., Salghetti S. E., Tansey W. P., Sachidanandam R., Prasanth K. V., Ried T., Shav-Tal Y., Bertrand E., Singer R. H., Spector D. L., From silencing to gene expression: Real-time analysis in single cells. Cell 116, 683–698 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riggi N., Cironi L., Suvà M. L., Stamenkovic I., Sarcomas: Genetics, signalling, and cellular origins. Part 1: The fellowship of TET. J. Pathol. 213, 4–20 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Müller K., Engesser R., Timmer J., Zurbriggen M. D., Weber W., Orthogonal optogenetic triple-gene control in mammalian cells. ACS Synth. Biol. 3, 796–801 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Müller K., Engesser R., Metzger S., Schulz S., Kämpf M. M., Busacker M., Steinberg T., Tomakidi P., Ehrbar M., Nagy F., Timmer J., Zubriggen M. D., Weber W., A red/far-red light-responsive bi-stable toggle switch to control gene expression in mammalian cells. Nucleic Acids Res. 41, e77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu F., Song Y. K., Liu D., Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6, 1258–1266 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Schuster B. S., Reed E. H., Parthasarathy R., Jahnke C. N., Caldwell R. M., Bermudez J. G., Ramage H., Good M. C., Hammer D. A., Controllable protein phase separation and modular recruitment to form responsive membraneless organelles. Nat. Commun. 9, 2985 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao E. M., Suek N., Wilson M. Z., Dine E., Pannucci N. L., Gitai Z., Avalos J. L., Toettcher J. E., Light-based control of metabolic flux through assembly of synthetic organelles. Nat. Chem. Biol. 15, 589–597 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reinkemeier C. D., Girona G. E., Lemke E. A., Designer membraneless organelles enable codon reassignment of selected mRNAs in eukaryotes. Science 363, eaaw2644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J., Choi J.-M., Holehouse A. S., Lee H. O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D., Poser I., Pappu R. V., Alberti S., Hyman A. A., A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison III C. A., Smith H. O., Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Beyer H. M., Gonschorek P., Samodelov S. L., Meier M., Weber W., Zurbriggen M. D., AQUA cloning: A versatile and simple enzyme-free cloning approach. PLOS ONE 10, e0137652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Müller K., Engesser R., Schulz S., Steinberg T., Tomakidi P., Weber C. C., Ulm R., Timmer J., Zurbriggen M. D., Weber W., Multi-chromatic control of mammalian gene expression and signaling. Nucleic Acids Res. 41, e124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raue A., Steiert B., Schelker M., Kreutz C., Maiwald T., Hass H., Vanlier J., Tönsing C., Adlung L., Engesser R., Mader W., Heinemann T., Hasenauer J., Schilling M., Höfer T., Klipp E., Theis F., Klingmüller U., Schöberl B., Timmer J., Data2Dynamics: A modeling environment tailored to parameter estimation in dynamical system. Bioinformatics 31, 3558–3560 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Hindmarsh A. C., Brown P. N., Grant K. E., Lee S. L., Serban R., Shumaker D. E., Woodward C. S., SUNDIALS: Suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. 31, 363–396 (2005). [Google Scholar]
- 50.Coleman T. F., Li Y., An interior trust region approach for nonlinear minimization subject to bounds. SIAM J. Optim. 6, 418–445 (1996). [Google Scholar]
- 51.Tucker C. L., Manipulating cellular processes using optical control of protein–protein interactions. Prog. Brain Res. 196, 95–117 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Kreutz C., Raue A., Timmer J., Likelihood based observability analysis and confidence intervals for predictions of dynamic models. BMC Syst. Biol. 6, 120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raue A., Kreutz C., Maiwald T., Bachmann J., Schilling M., Klingmüller U., Timmer J., Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics 25, 1923–1929 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Beyer H. M., Engesser R., Hörner M., Koschmieder J., Beyer P., Timmer J., Zurbriggen M. D., Weber W., Synthetic biology makes polymer materials count. Adv. Mater. 30, e1800472 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Hinkley D., Predictive likelihood. Ann. Statist. 7, 718–728 (1979). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabd3568/DC1