

In this study, Han et al. demonstrate that JNK signaling in adipocytes causes an increased circulating concentration of the hepatokine fibroblast growth factor 21 (FGF21) that regulates systemic metabolism. This regulatory loop represents a novel signaling paradigm that connects autocrine and endocrine signaling modes of the same hormone in different tissues.
Keywords: autocrine, endocrine, FGF21, JNK, organ cross-talk
Abstract
The cJun NH2-terminal kinase (JNK) signaling pathway is activated by metabolic stress and promotes the development of metabolic syndrome, including hyperglycemia, hyperlipidemia, and insulin resistance. This integrated physiological response involves cross-talk between different organs. Here we demonstrate that JNK signaling in adipocytes causes an increased circulating concentration of the hepatokine fibroblast growth factor 21 (FGF21) that regulates systemic metabolism. The mechanism of organ crosstalk is mediated by a feed-forward regulatory loop caused by JNK-regulated FGF21 autocrine signaling in adipocytes that promotes increased expression of the adipokine adiponectin and subsequent hepatic expression of the hormone FGF21. The mechanism of organ cross-talk places circulating adiponectin downstream of autocrine FGF21 expressed by adipocytes and upstream of endocrine FGF21 expressed by hepatocytes. This regulatory loop represents a novel signaling paradigm that connects autocrine and endocrine signaling modes of the same hormone in different tissues.
Fibroblast growth factor 21 (FGF21) is secreted by hepatocytes and circulates in the blood (Fisher and Maratos-Flier 2016; BonDurant and Potthoff 2018; Kliewer and Mangelsdorf 2019). This liver-derived hormone (hepatokine) functions by binding receptors expressed by target tissues formed by one of several FGF receptors together with the coreceptor β-Klotho. It is established that FGF21 plays an important role in the regulation of metabolism during the initial phase of the starvation response by promoting hepatic fatty acid oxidation and ketone body formation (Fisher and Maratos-Flier 2016; Kliewer and Mangelsdorf 2019). The central nervous system represents a major target of FGF21 signaling by acting on receptors present in the suprachiasmatic nucleus of the hypothalamus and the dorsal vagal complex of the hindbrain (Bookout et al. 2013). These central actions of FGF21 include suppression of physical activity and altered circadian behavior (Owen et al. 2014), food choice preference (sugar and alcohol) (Schumann et al. 2016; Talukdar et al. 2016; von Holstein-Rathlou et al. 2016; Søberg et al. 2017), water consumption (Song et al. 2018), protection from lipid-induced liver and muscle insulin resistance (Camporez et al. 2013), and increased sympathetic outflow from the brainstem that promotes adipose tissue thermogenesis and reduced body mass (Owen et al. 2014; Douris et al. 2015).
Hepatic Fgf21 gene expression is increased in response to a wide array of nutritional stresses (Fisher and Maratos-Flier 2016; BonDurant and Potthoff 2018; Kliewer and Mangelsdorf 2019). Thus, Fgf21 gene expression is increased by the nuclear hormone receptor PPARα in response to starvation or consumption of a ketogenic diet (Badman et al. 2007; Inagaki et al. 2007), by the transcription factor ChREBP in response to simple sugars (Fisher et al. 2017; Iroz et al. 2017), by the transcription factor ATF2 in response to cold exposure (Hondares et al. 2011), and by the integrated stress response pathway mediated by eIF2α/ATF4 and IRE1α/XBP1 in response to ER stress, amino acid starvation, and mitochondrial dysfunction (Salminen et al. 2017).
The liver is the major source of the endocrine FGF21 that circulates in the blood and regulates energy expenditure, glucose/lipid metabolism, and insulin sensitivity (Markan et al. 2014; Vernia et al. 2016a), but muscle can act also as an endocrine source of FGF21 in response to mitochondrial myopathy (Tyynismaa et al. 2010). Interestingly, pancreatic acinar cells secrete exocrine FGF21 that functions to maintain acinar cell proteostasis (Coate et al. 2017). In contrast, adipocyte FGF21 functions primarily in an autocrine/paracrine manner in response to feeding/fasting cycles (Dutchak et al. 2012) and cold exposure (Chartoumpekis et al. 2011; Hondares et al. 2011; Fisher et al. 2012). These autocrine actions of FGF21 may be mediated, in part, by increased expression of the coactivator PGC1α and the expression of thermogenic genes (Fisher et al. 2012), increased PPARγ activity and insulin sensitization (Dutchak et al. 2012), and increased expression of adipokines (Holland et al. 2013; Lin et al. 2013; Huang et al. 2017). The absence of an endocrine function for adipocyte FGF21 may reflect expression levels and the very short half-life of FGF21 in the blood (Fisher and Maratos-Flier 2016; BonDurant and Potthoff 2018; Kliewer and Mangelsdorf 2019).
The responsiveness of FGF21 to nutritional stress (BonDurant and Potthoff 2018) is shared by the cJun NH2-terminal kinase (JNK) signaling pathway (Sabio and Davis 2010). Indeed, JNK acts to suppress PPARα-induced hepatic Fgf21 gene expression and promotes systemic insulin resistance by reducing the amount of FGF21 circulating in the blood (Vernia et al. 2014, 2016a). The observed insulin resistance is characterized by decreased endocrine signaling by FGF21 and reduced sympathetic tone, fatty acid β-oxidation, energy expenditure, hyperglycemia, and hyperlipidemia (Owen et al. 2014; Douris et al. 2015). This role of hepatic JNK to regulate endocrine expression of hepatic FGF21 raises questions concerning the potential function of JNK-regulated FGF21 expression by adipocytes. We report that autocrine signaling by adipocyte FGF21 triggers a feed-forward regulatory loop that promotes endocrine FGF21 signaling by the liver. This mechanism represents a novel signaling paradigm that connects autocrine and endocrine signaling modes of the same hormone in different tissues.
Results
JNK in adipose tissue promotes systemic diet-induced insulin resistance
We established mice with deficiency of JNK1 plus JNK2 in adipocytes (Adipoq-Cre+/− Mapk8LoxP/LoxP Mapk9LoxP/LoxP) and Control mice (Adipoq-Cre+/−). Genotype analysis of adipose tissue depots demonstrated Mapk8 and Mapk9 gene ablation in inguinal fat (IngF), epididymal fat (EpiF), retroperitoneal fat (RetF), brown adipose tissue (BAT), and isolated adipocyte fraction (ADF), but not in liver, muscle, or the adipose tissue stromal vascular fraction (SVF) (Supplemental Fig. S1A). Immunoblot analysis demonstrated that JNK expression was decreased in the adipocyte fraction, but not in the stromal vascular fraction, of epididymal adipose tissue of JNK-deficient (ADKO) mice compared with Control (ADWT) mice (Supplemental Fig. S1B). These studies demonstrate an adipocyte-selective defect in JNK expression in ADKO mice.
The mice were fed a chow diet (ND) or a high fat diet (HFD) for 16 wk. No difference in total body mass between the ADWT mice and ADKO mice was detected (Fig. 1A,B). However, JNK deficiency caused a reduction in adipose tissue mass in HFD-fed mice (Supplemental Fig. S1C). Histological examination of white fat demonstrated that JNK deficiency in adipocytes did not cause major changes in adipose tissue morphology in ND-fed mice (Fig. 1D). In contrast, adipocyte size was reduced in the subcutaneous IngF and BAT of ADKO mice compared with ADWT mice when fed a HFD (Fig. 1D; Supplemental Fig. S1D,E). Moreover, hepatic steatosis of HFD-fed ADWT mice was suppressed in HFD-fed ADKO mice (Fig. 1D; Supplemental Fig. S1F).
Figure 1.

HFD-induced metabolic syndrome is suppressed in mice with JNK deficiency in adipocytes. (A) Whole body mass of ND-fed and HFD-fed (16 wk) ADWT and ADKO mice was measured (mean ± SEM; n = 8∼9). (B) The fat mass and lean mass of ND-fed and HFD-fed (16 wk) mice were measured using 1H-MRS (mean ± SEM; n = 8∼9). (C) The blood glucose concentration of ND-fed and HFD-fed (16 wk) mice was measured (mean ± SEM; n = 8∼9). (***) P < 0.001. (D) Sections prepared from ND-fed and HFD-fed (16 wk) ADWT and ADKO mice were stained with H&E. Scale bar, 100 µm. (E–G) Tolerance tests for glucose (GTT), pyruvate (PTT), and insulin (ITT) were performed using ND-fed and HFD-fed (16 wk) mice (mean ± SEM; n = 7∼9). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (H) ND-fed and HFD-fed (16 wk) mice were treated with insulin (15 min). Immunoblot analysis was performed with extracts prepared from adipose tissue (IngF and EpiF), liver, and gastrocnemius muscle using antibodies to phospho-AKT, AKT, and αTubulin.
Gene expression analysis demonstrated increased expression of markers for macrophage infiltration (Adgre1) in IngF of HFD-fed ADKO mice compared with ADWT mice. Expression of markers for both M1-like (Cd11c) and M2-like (Mgl1 and Mrc1) polarization were detected in IngF of ADKO and ADWT mice (Supplemental Fig. S2A,B). In contrast, similar expression levels of marker genes for extracellular matrix remodeling (including Col1α1, Mmp2, Mmp9, and Tgfβ1) were detected in IngF of ADKO and ADKO mice (Supplemental Fig. S2C).
We found that consumption of a HFD caused hyperglycemia in ADWT mice, but not in ADKO mice (Fig. 1C). Glucose tolerance tests demonstrated that feeding a HFD caused glucose intolerance of ADWT mice, but not ADKO mice (Fig. 1E; Supplemental Fig. S1G). Similarly, pyruvate tolerance tests demonstrated that HFD consumption promoted greater increases in plasma glucose concentrations in ADWT mice compared with ADKO mice during a pyruvate tolerance test (Fig. 1F; Supplemental Fig. S1H). Moreover, insulin tolerance tests demonstrated that the HFD feeding caused whole body insulin resistance in ADWT mice, but not in ADKO mice (Fig. 1G; Supplemental Fig. S1I). Collectively, these data indicate that HFD promotes the development of insulin resistance in ADWT mice, but not in ADKO mice.
To examine the impact of JNK deficiency on adipose insulin signaling, we examined the activation of the AKT signaling pathway by immunoblot analysis using antibodies to AKT, pThr308-AKT, and pSer473-AKT. Similar insulin-stimulated activation of AKT was detected in IngF and EpiF of ND-fed ADWT mice and ADKO mice (Fig. 1H). This AKT activation in adipose tissue was suppressed in HFD-fed ADWT mice, but insulin-stimulated AKT activation was detected in HFD-fed ADKO mice (Fig. 1H). Similarly, insulin-stimulated AKT activation was detected in the gastrocnemius muscle of HFD-fed ADKO mice (Fig. 1H). These data indicate that JNK in adipocytes impairs insulin signaling in HFD-fed mice in adipose tissue and other tissues.
Studies of the circulating concentration of insulin demonstrated that feeding a HFD caused hyperinsulinemia in ADWT mice, but not in ADKO mice (Fig. 2A). This reduction in hyperinsulinemia was associated with suppression of islet hypertrophy in the pancreas (Fig. 2B) and increased glucose-stimulated insulin secretion (Fig. 2C,D). Moreover, the HFD-induced increase in the blood concentration of IL6 and leptin in ADWT mice was not detected in ADKO mice (Fig. 2A).
Figure 2.

Adipocyte JNK promotes insulin resistance in HFD-fed mice. (A) The blood concentration of insulin, IL6, and leptin was measured by ELISA using ND-fed and HFD-fed (16 wk) ADWT and ADKO mice (mean ± SEM; n = 7∼8). (*) P < 0.05. (B) Pancreatic sections prepared from ND-fed and HFD-fed (16 wk) ADWT and ADKO mice were stained with DAPI and antibodies to insulin and glucagon. Scale bar, 150 µm. Islet area was measured (mean ± SEM; n = 6∼8). (***) P < 0.001. (C,D) Glucose-stimulated insulin secretion (GSIS) was measured using ND-fed and HFD-fed (16 wk) ADWT and ADKO mice (mean ± SEM; n = 7∼8). (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (E–K) Hyperinsulinemic-euglycemic clamp studies were performed using ND-fed and HFD-fed (8 wk) ADWT and ADKO mice to measure glucose infusion rate, glycogen synthesis, nonesterified fatty acid (NEFA) suppression, whole body glycerol turnover, whole body palmitate turnover, whole body glucose uptake, and glucose uptake by EpiF and gastrocnemius muscle (mean ± SEM; n = 5∼7). (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
Our analysis of ADWT and ADKO mice indicates that JNK deficiency in adipocytes suppresses the development of several hallmarks of insulin resistance in HFD-fed mice (Fig. 1). To confirm this conclusion, we performed a hyperinsulinemic-euglycemic clamp study (Fig. 2E–K). The glucose infusion rate (a measure of whole body insulin sensitivity) of ND-fed ADWT and ADKO mice was similar. Consumption of a HFD by ADWT mice markedly reduced the glucose infusion rate, consistent with the development of insulin resistance (Fig. 2E,F). However, the glucose infusion rate of HFD-fed ADKO mice was significantly higher than HFD-fed ADWT mice (Fig. 2E,F). These data demonstrate that adipocyte JNK deficiency did not alter insulin sensitivity in ND-fed mice, but adipocyte JNK deficiency strongly promoted insulin sensitivity in HFD-fed mice. This increase in insulin sensitivity was associated with increased glycogen plus lipid synthesis (Fig. 2G) and reduced lipolysis, as monitored by a reduction in circulating non-esterified fatty acids (NEFA) (Fig. 2H) and reduced turnover of both glycerol and palmitate (Fig. 2I,J). Moreover, HFD-fed ADKO mice exhibited increased whole body glucose uptake and increased glucose uptake by adipose tissue and muscle compared with HFD-fed ADWT mice (Fig. 2K). These data demonstrate that JNK deficiency in adipocytes caused systemic protection against the development of insulin resistance in HFD-fed mice.
JNK deficiency in adipocytes causes increased energy expenditure
Metabolic cage analysis of gas exchange demonstrated that JNK deficiency in adipocytes caused increased VO2, VCO2, and energy expenditure (EE) in HFD-fed mice, but changes in the respiratory exchange ratio (RER) were not detected (Fig. 3A; Supplemental Fig. S2D). This increased energy expenditure may contribute to the improved metabolic phenotype of HFD-fed ADKO mice compared with HFD-fed ADWT mice (Figs. 1, 2). However, this increased energy expenditure would be expected to cause decreased body mass, which was not observed (Fig. 1A,B). It is most likely that the body mass is the consequence of a composite phenotype caused by a balance between the increased energy expenditure (Fig. 3A) and increased food consumption by HFD-fed ADKO mice compared with HFD-fed ADWT mice (Supplemental Fig. S2E). The hyperleptinemia in HFD-fed ADWT mice, but not in HFD-fed ADKO mice (Fig. 2A), may contribute to this difference in feeding behavior and contribute to the improved metabolic profile of HFD-fed ADKO mice compared with HFD-fed ADWT mice (Zhao et al. 2020).
Figure 3.

JNK in adipocytes suppresses adipose tissue thermogenesis. (A) ND-fed and HFD-fed (6 wk) ADWT and ADKO mice were examined using metabolic cages to measure VO2, VCO2, respiratory exchange ratio (RER), and energy expenditure (EE) (mean ± SEM; n = 8). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (B) Surface temperature was measured using infrared photography and core body temperature was measured using an implanted probe by telemetry (mean ± SEM; n = 6∼12). (*) P < 0.05; (**) P < 0.01. (C) IngF sections of HFD-fed ADWT and ADKO mice (16 wk) were stained with DAPI and an antibody to UCP1. The mean fluorescence intensity was measured (mean ± SEM; n = 5). (*) P < 0.05. (D) RNA isolated from the IngF of ND-fed and HFD-fed (16 wk) ADWT and ADKO mice was examined using TaqMan assays to measure mRNA expression and presented as a heat map (n = 6∼8). (E) The core body temperature of ND-fed ADWT and ADKO mice at 21°C (day 0) and at 6°C was measured using implanted probes by telemetry (mean ± SEM; n = 10∼12). (*) P < 0.05; (**) P < 0.01. (F) IngF sections of ND-fed ADWT and ADKO mice housed at 21°C and 6°C (21 d) were stained with H&E. Representative sections are presented. Scale bar, 100 µm. (G) RNA isolated from the IngF tissue of ADWT and ADKO mice housed at 6°C (21 d) was examined using TaqMan assays to measure mRNA expression and presented as a heat map (n = 6∼8).
An expected consequence of increased energy expenditure is a change in body temperature. We therefore measured core body temperature using telemetry with an implanted probe and surface temperature by infrared photography. These studies demonstrated that the body temperature of ADKO mice was increased compared with ADWT mice (Fig. 3B). These data indicate that JNK deficiency in adipocytes may cause increased adipose tissue thermogenesis. Indeed, studies of IngF demonstrated increased expression of a thermogenic gene signature and increased expression of the thermogenic protein UCP1 (Fig. 3C,D) and an increase in PPAR-dependent gene expression (Fig. 3D).
Increased thermogenesis would be expected to provide protection against reduced environmental temperature. We therefore tested ND-fed ADWT and ADKO mice with a chronic cold challenge (6°C) and measured core body temperature. The ADKO mice maintained a significantly higher body temperature than ADWT mice during 21 d of cold exposure (Fig. 3E). Analysis of IngF tissue sections demonstrated morphological evidence of adipose tissue browning following cold exposure, including the presence of small multilocular adipocytes (Fig. 3F). The cold-induced browning of IngF in ADKO mice was greater than ADWT mice based on adipose tissue morphology (Fig. 3F) and expression of a browning gene signature (Fig. 3G).
Isothermal housing suppresses the metabolic phenotype of mice with JNK deficiency in adipocytes
Increased thermogenesis may contribute to the improved metabolic phenotype of HFD-fed ADKO mice compared with HFD-fed ADWT mice. To test this hypothesis, we housed mice under thermoneutral conditions (30°C). We found that when housed at thermoneutrality, JNK deficiency in adipocytes caused no significant change in HFD-induced obesity (Supplemental Fig. S3A), hyperglycemia (Supplemental Fig. S3B), hyperinsulinemia (Supplemental Fig. S3C) or the increased circulating concentrations of IL6 and leptin (Supplemental Fig. S3C). The isothermal housing also suppressed the effects of JNK deficiency in adipocytes on intolerance to glucose and insulin (Supplemental Fig. S3D,E).
Analysis of inguinal adipose tissue sections demonstrated morphological evidence of reduced adipose tissue browning, including the presence of large unilocular adipocytes, in both of ADWT and ADKO mice (Supplemental Fig. S3F). Indeed, JNK deficiency caused only modest changes in the expression of a browning gene signature in the inguinal adipose tissue of mice housed under thermoneutral conditions (Supplemental Fig. S3G) compared with mice at 21°C (Fig. 3D).
Together, these data demonstrate that the improved metabolic phenotype of HFD-fed ADKO mice compared with HFD-fed ADWT mice is caused, in part, by increased thermogenesis caused by JNK deficiency in adipocytes.
JNK deficiency in adipocytes causes an increase in the circulating concentration of FGF21
We have previously reported that JNK deficiency in hepatocytes causes increased energy expenditure by promoting hepatic Fgf21 mRNA expression and increasing the circulating concentration of FGF21 in the blood (Vernia et al. 2014, 2016a). We therefore investigated whether JNK deficiency in adipocytes might also cause an increase in the circulating concentration of FGF21. Indeed, HFD-fed ADKO mice exhibited increased Fgf21 mRNA in adipose tissue and an increased blood concentration of FGF21 compared with HFD-fed ADWT mice (Fig. 4A,B). These changes were also detected at thermoneutrality (Supplemental Fig. S3H). The increased adipocyte Fgf21 mRNA expression was associated with increased receptor expression in HFD-fed mice (Fgfr1 and Klb) (Fig. 4C,D; Supplemental Fig. S3I).
Figure 4.

FGF21 mediates the metabolic actions of JNK signaling in adipocytes. (A) The blood concentration of FGF21 in ND-fed and HFD-fed (16 wk) ADWT, ADKO, and ADKOΔFgf21 mice was measured by ELISA (mean ± SEM; n = 6∼8). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (B–D) The expression of Fgf21, Fgfr1, and Klb by IngF and liver was measured by TaqMan assays (mean ± SEM; n = 6∼8). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (E) The fasted blood glucose concentration was measured (mean ± SEM; n = 6∼8). (**) P < 0.01. (F) Surface temperature was measured using infrared photography (mean ± SEM; n = 8∼14). (**) P < 0.01; (***) P < 0.001.
To test whether FGF21 contributed to the phenotype of ADKO mice, we established Adipoq-Cre+/− Mapk8LoxP/LoxP Mapk9LoxP/LoxP Fgf21LoxP/LoxP (ADKOΔFgf21) mice with a deficiency of JNK1/2 plus FGF21 in adipocytes (Supplemental Fig. S4A). We found that the effect of JNK deficiency in adipocytes to both increase the circulating concentration of FGF21 and adipocyte Fgf21 mRNA expression was suppressed by adipocyte Fgf21 gene ablation (Fig. 4A,B). Functional analysis demonstrated that adipocyte Fgf21 gene ablation in ADKO mice prevented HFD-induced hyperglycemia (Fig. 4E) and suppressed thermogenesis (Fig. 4F). These data demonstrate that JNK acts to suppress Fgf21 expression by adipocytes.
JNK in adipocytes regulates hepatic FGF21 expression
The finding that adipocyte Fgf21 mRNA expression regulates the circulating concentration of FGF21 (Fig. 4) was unexpected because it has been previously established that no FGF21 can be detected in the blood of mice with Fgf21 gene ablation in hepatocytes (Markan et al. 2014; Vernia et al. 2016a). The simplest explanation for this observation is that adipocyte FGF21 promotes expression of hepatic FGF21. To test this hypothesis, we examined hepatic Fgf21 expression in ADWT mice, ADKO mice, and ADKOΔFgf21 mice. This analysis demonstrated that JNK deficiency in adipocytes caused increased expression of hepatic Fgf21 mRNA expression in HFD-fed ADKO mice compared with HFD-fed ADWT mice (Fig. 4B) and that this increase in hepatic Fgf21 mRNA expression was suppressed by Fgf21 gene ablation in adipocytes (Fig. 4B). These data demonstrate the presence of a feed-forward loop that is initiated by JNK-regulated Fgf21 expression in adipocytes that promotes Fgf21 expression by hepatocytes to increase the circulating concentration of FGF21 in the blood.
To test the role of hepatic FGF21, we ablated the Fgf21 gene in the hepatocytes of ADWT mice, ADKO mice, and ADKOΔFgf21 mice by tail vein administration of AAV8-TBG-Cre (Supplemental Fig. S5A). FGF21 receptor expression (Fgfr1 and Klb mRNA) was detected in adipose tissue and the liver (Fig. 5A,B). Ablation of the hepatocyte Fgf21 gene prevented the increased circulating FGF21 concentration detected in mice with JNK deficiency in adipocytes (Fig. 5C,D). This observation confirms that the circulating FGF21 in mice with JNK deficiency in adipocytes is secreted by the liver.
Figure 5.

Hepatic FGF21 is required for the metabolic actions of JNK signaling in adipocytes. (A–C) ADWT, ADKO, and ADKOΔFgf21 mice were treated with AAV8-TBG-Cre and fed a HFD (16 wk). The expression of Fgf21, Fgfr1, and Klb by IngF and liver was measured by TaqMan assays (mean ± SEM; n = 5∼8). (*) P < 0.05; (**) P < 0.01. (D) The blood concentration of FGF21 was measured by ELISA (mean ± SEM; n = 6∼7). (**) P < 0.01; (***) P < 0.001. (E–G) Tolerance tests for glucose (GTT), pyruvate (PTT), and insulin (ITT) were performed (mean ± SEM; n = 6∼7). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (H) The fasted blood glucose concentration was measured (mean ± SEM; n = 6∼7). (**) P < 0.01. (I) The blood concentration of insulin was measured by ELISA (mean ± SEM; n = 6∼7). (*) P < 0.05; (**) P < 0.01. (J) Sections prepared from IngF and liver were stained with H&E. Scale bar, 100 µm. (K) RNA isolated from IngF was examined using TaqMan assays to measure mRNA expression and presented as a heat map (n = 6∼8).
We found that hepatic Fgf21 gene ablation did not affect body mass (Supplemental Fig. S5B,C). Nevertheless, the improved tolerance to glucose and pyruvate caused by JNK deficiency in adipocytes was suppressed by hepatic Fgf21 gene ablation in HFD-fed mice (Fig. 5E,F; Supplemental Fig. S5D,E). Similarly, the improved insulin tolerance caused by adipocyte JNK deficiency was reversed by hepatic Fgf21 gene ablation in HFD-fed mice (Fig. 5G; Supplemental S5F). We also found that hepatic Fgf21 gene ablation suppressed the effect of adipocyte JNK deficiency to reduce HFD-induced hyperglycemia (Fig. 5H) and hyperinsulinemia (Fig. 5I). Hepatic Fgf21 gene ablation also suppressed the effect of adipocyte JNK deficiency to decrease hepatic steatosis (Fig. 5J) and promote adipocyte browning (Fig. 5J,K). These data indicate that hepatic FGF21 is important for the metabolic actions of JNK in adipocytes.
Adipose signaling to the liver increases FGF21 circulating in the blood
Adipose signaling to the liver is physiologically important and may be mediated by several different mechanisms (Titchenell et al. 2017; Petersen and Shulman 2018; Funcke and Scherer 2019). For example, it has been proposed that adipose tissue lipolysis generates fatty acids that can activate the hepatic PPARα nuclear hormone receptor and increase FGF21 expression by the liver (Chen et al. 2011). Since ADKO mice display reduced adipose tissue lipolysis compared with ADWT mice (Fig. 2H–J), it appears unlikely that this mechanism contributes to adipose tissue cross-talk to the liver in ADKO mice. A second potential mechanism is represented by the adipokine IL6 (Sabio et al. 2008; Stanford et al. 2013) that circulates in the blood of HFD-fed ADWT mice and is suppressed in HFD-fed ADKO mice (Fig. 2A). This mechanism also appears unlikely because it has recently been demonstrated that adipocyte-derived IL6 does not cause changes in systemic insulin resistance (Whitham et al. 2019; Han et al. 2020). We also considered a third potential mechanism of adipose to liver cross-talk that involves exosome-mediated microRNA delivery from adipose tissue to the liver to regulate hepatic Fgf21 expression (Thomou et al. 2017). Indeed, we found reduced adipocyte expression of microRNAs that may target Fgf21 mRNA (miR-99a, miR-99b-5p, miR-99b-3p, and miR-100) in adipose tissue of ADKO mice compared with ADWT mice (Supplemental Fig. S4B). This reduction in adipocyte microRNA expression was not detected in ADKOΔFgf21 mice (Supplemental Fig. S4B); this finding implicates FGF21 in the suppression of these microRNAs in adipose tissue. However, no significant difference in the hepatic expression of these microRNAs between ADKO and ADWT mice was detected (Supplemental Fig. S4B). These microRNAs therefore do not mediate the effects of adipocyte JNK deficiency on hepatic Fgf21 mRNA expression. A different mechanism must therefore account for adipose tissue cross-talk to the liver in ADKO mice.
It is established that FGF21 increases adiponectin expression by activating PPARγ (Holland et al. 2013; Lin et al. 2013) and that JNK acts to suppress adiponectin expression (Manieri et al. 2019). Indeed, treatment of mice with recombinant FGF21 caused increased Adipoq and Fgf21 gene expression by adipose tissue (Supplemental Fig. S6A). We therefore examined Adipoq mRNA expression and the circulating concentration of adiponectin in ADWT and ADKO mice. We found that mice with JNK deficiency in adipocytes expressed increased amounts of Adipoq mRNA and exhibited a higher concentration of circulating adiponectin in the blood (Fig. 6A,B). These changes were disrupted by ablation of the Fgf21 gene in adipocytes (Fig. 6A,B). In contrast, ablation of the Fgf21 gene in hepatocytes using AAV8-TBG-Cre did not prevent the effect of adipocyte JNK deficiency to increase adipose tissue expression of Adipoq mRNA and the circulating concentration of adiponectin (Fig. 6C,D). These data indicate that autocrine/paracrine signaling by adipocyte FGF21 promotes the expression of adiponectin.
Figure 6.

Endocrine FGF21 secreted by the liver is required for the metabolic actions of JNK signaling in adipocytes. (A,B) ADWT, ADKO, and ADKOΔFgf21 mice were fed a HFD (16 wk). The circulating concentration of adiponectin was measured by ELISA (mean ± SEM; n = 7∼8). (**) P < 0.01. The expression of Adipoq mRNA by IngF was measured by TaqMan assay (mean ± SEM; n = 7). (*) P < 0.05; (**) P < 0.01. (C,D) Mice were treated with AAV8-TBG-Cre and fed a HFD (16 wk). The circulating concentration of adiponectin was measured by ELISA (mean ± SEM; n = 6). (*) P < 0.05; (**) P < 0.01. The expression of Adipoq mRNA by IngF was measured by TaqMan assay (mean ± SEM; n = 6∼7). (**) P < 0.01. (E) The blood concentration of FGF21 in ND-fed and HFD-fed (16 wk) ADWTΔAdq and ADKOΔAdq mice was measured by ELISA (mean ± SEM; n = 6∼10). (F–H) The expression of Fgf21, Fgfr1, and Klb by IngF and liver was measured by TaqMan assays (mean ± SEM; n = 6∼8). (**) P < 0.01. (I) Whole body mass of ND-fed and HFD-fed (16 wk) mice was measured (mean ± SEM; n = 6∼11). (J,K) Tolerance tests for glucose (GTT) and insulin (ITT) were performed using ND-fed and HFD-fed (16-wk) mice (mean ± SEM; n = 8∼11). (L) The fasted blood glucose concentration was measured (mean ± SEM; n = 8∼11). (M) Sections prepared from IngF and liver were stained with H&E. Scale bar, 100 µm.
Adiponectin may mediate cross-talk between adipose tissue and the liver (Straub and Scherer 2019). We found that treatment with adiponectin caused increased Fgf21 gene expression in cell culture (Supplemental Fig. S6B). To test whether adiponectin contributes to the phenotype of ADKO mice, we established Adipoq-Cre+/− Mapk8+/+ Mapk9+/+ Adipoq−/− (ADWTΔAdq) mice with adiponectin deficiency and Adipoq-Cre+/− Mapk8LoxP/LoxP Mapk9LoxP/LoxP Adipoq−/− (ADKOΔAdq) mice with deficiency of adiponectin plus JNK1/2 in adipocytes. No adiponectin in the blood of ADWTΔAdq and ADKOΔAdq mice was detected. We found that adipocyte JNK deficiency in adiponectin-deficient mice did not increase hepatic Fgf21 mRNA expression or the circulating concentration of FGF21 (Fig. 6E,F). FGF21 receptor expression (Fgfr1 and Klb mRNA) was detected in adipose tissue and the liver (Fig. 6G,H). Moreover, adipocyte JNK deficiency did not alter body mass (Fig. 6I), resistance to cold stress (Supplemental Fig. S7), tolerance to glucose or insulin (Fig. 6J,K; Supplemental Fig. S8A,B), hyperglycemia (Fig. 6L), adipocyte morphology or hepatic steatosis (Fig. 6M) in adiponectin-deficient mice. These data indicate that adiponectin is required for the effect of adipocyte JNK deficiency to increase hepatic Fgf21 mRNA expression and the circulating concentration of FGF21.
Endocrine signaling by hepatic FGF21 does not require adiponectin
It is established that adiponectin contributes to the response of mice to treatment with recombinant FGF21 (Holland et al. 2013; Lin et al. 2013). We therefore tested whether adiponectin is required for the endocrine function of endogenous FGF21. We have reported that JNK deficiency in hepatocytes causes increased hepatic Fgf21 mRNA expression and an increased circulating concentration of FGF21 (Vernia et al. 2014). Moreover, hepatic FGF21 expression is required for the effects of hepatocyte JNK deficiency on metabolism (Vernia et al. 2016a). To test the role of adiponectin, we established Alb-Cre+/− Mapk8+/+ Mapk9+/+ Adipoq−/− (AlbWTΔAdq) mice with adiponectin deficiency and Alb-Cre+/− Mapk8LoxP/LoxP Mapk9LoxP/LoxP Adipoq−/− (AlbKOΔAdq) mice with deficiency of adiponectin plus JNK1/2 in hepatocytes. No adiponectin was detected in the blood of AlbWTΔAdq and AlbKOΔAdq mice.
We found that adiponectin deficiency did not prevent the effect of hepatocyte JNK deficiency to increase hepatic Fgf21 mRNA expression or the circulating concentration of FGF21 (Fig. 7A,B). These data demonstrate that adiponectin is not required for hepatic FGF21 expression. Moreover, FGF21 receptor expression (Fgfr1 and Klb mRNA) was detected in adipose tissue and the liver (Fig. 7C,D).
Figure 7.

Adiponectin is not required for endocrine signaling by FGF21. (A) AlbWTΔAdq and AlbKOΔAdq mice were fed a HFD (16 wk). The blood concentration of FGF21 was measured by ELISA (mean ± SEM; n = 7∼8). (*) P < 0.05; (***) P < 0.001. (B–D) The expression of Fgf21, Fgfr1, and Klb by IngF and liver was measured by TaqMan assays (mean ± SEM; n = 7∼8). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (E,F) The body weight was measured (mean ± SEM; n = 7∼10). Fat mass and lean mass were measured by 1H-MRS analysis (mean ± SEM; n = 7∼10). (**) P < 0.01. (G) The fasted blood glucose concentration was measured (mean ± SEM; n = 7∼10). (***) P < 0.001. (H–J) Tolerance tests for glucose (GTT), pyruvate (PTT), and insulin (ITT) were performed using ND-fed and HFD-fed (16-wk) mice (mean ± SEM; n = 7∼10). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (K) Core body temperature was measured using an implanted probe by telemetry (mean ± SEM; n = 7∼8). (**) P < 0.01. (L) Sections prepared from IngF and liver were stained with H&E. Scale bar, 100 µm.
To test whether adiponectin is required for hepatic FGF21 function, we characterized the metabolic phenotype of HFD-fed AlbWTΔAdq and AlbKOΔAdq mice. We found that JNK deficiency in hepatocytes decreased body fat mass (Fig. 7E,F), suppressed HFD-induced hyperglycemia (Fig. 7G), improved tolerance to glucose, pyruvate, and insulin in HFD-fed mice (Fig. 7H–J; Supplemental Fig. S8C–E), increased core body temperature in HFD-fed mice (Fig. 7K), reduced hepatic steatosis and increased adipose browning (Fig. 7L). Together, these data indicate that adiponectin is not required for the biological actions of endocrine FGF21. It is likely that these actions of FGF21 are mediated, in part, by a central mechanism, including an increase in sympathetic nervous system outflow (Fisher and Maratos-Flier 2016; BonDurant and Potthoff 2018; Kliewer and Mangelsdorf 2019).
Discussion
A major target of endocrine FGF21 signaling is the central nervous system, including the suprachiasmatic nucleus of the hypothalamus and the dorsal vagal complex of the hindbrain (Bookout et al. 2013). FGF21 causes behavioral changes (Bookout et al. 2013) and increases sympathetic outflow from the brain (Owen et al. 2014; Douris et al. 2015). This sympathetic outflow causes increased energy expenditure by stimulation of brown adipose tissue and promotion of white adipose tissue “browning,” increased glucose uptake, reduced blood glucose and lipid concentrations, and increased insulin sensitivity (Camporez et al. 2013; Fisher and Maratos-Flier 2016; BonDurant and Potthoff 2018; Kliewer and Mangelsdorf 2019). The importance of FGF21 signaling in the brain was established by examination of mice with neuron-specific deficiency of the obligate coreceptor β-Klotho. These studies demonstrated that neuronal FGF21 signaling is essential for the chronic metabolic actions of FGF21 (Owen et al. 2014; Lan et al. 2017). Similar studies of mice with β-Klotho deficiency in adipocytes demonstrated that FGF21 signaling in adipose tissue can mediate acute metabolic actions of FGF21, including increased adipose tissue glucose uptake (BonDurant et al. 2017; Lan et al. 2017). In contrast, no requirement for hepatic FGF21 signaling was detected (Lan et al. 2017). The physiological response to FGF21 is therefore mediated primarily by the central nervous system, but adipocytes contribute to FGF21 metabolic responses.
The liver is the major source of the endocrine FGF21 that circulates in the blood (Markan et al. 2014; Vernia et al. 2016a). Adipocyte FGF21 functions primarily in an autocrine/paracrine manner in response to feeding/fasting cycles (Dutchak et al. 2012) and cold exposure (Chartoumpekis et al. 2011; Hondares et al. 2011; Fisher et al. 2012). These autocrine actions of FGF21 may be mediated, in part, by increased expression of the coactivator PGC1α (Fisher et al. 2012) and increased PPARγ activity (Dutchak et al. 2012). Functions of adipocyte FGF21 signaling include increased glucose uptake and insulin sensitivity (BonDurant et al. 2017; Lan et al. 2017), and increased expression of adipokines (Holland et al. 2013; Lin et al. 2013; Huang et al. 2017).
Role of FGF21 in the metabolic stress response to JNK activation
The JNK signaling pathway is activated by nutritional stress and is associated with the development of hallmarks of metabolic syndrome (Davis 2000; Sabio and Davis 2010). The major effects of JNK on obesity are mediated by hypothalamic signaling to regulate energy expenditure by JNK1/2 (Belgardt et al. 2010; Sabio et al. 2010a; Vernia et al. 2013) and satiety by JNK3 (Vernia et al. 2016b). Insulin resistance requires the actions of JNK in the innate immune system (Han et al. 2013) and also peripheral insulin target tissues (liver, muscle, and adipose tissue) (Sabio et al. 2008, 2010b; Sabio and Davis 2010; Vernia et al. 2014; Perry et al. 2015). Studies of hepatic JNK signaling demonstrate that JNK suppresses PPARα-mediated Fgf21 gene expression and endocrine secretion of FGF21 (Vernia et al. 2014). Ablation of the Fgf21 gene in hepatocytes is sufficient to suppress the effects of JNK deficiency in hepatocytes on resistance to the development of metabolic syndrome (Vernia et al. 2016a). This observation demonstrates that FGF21 represents an important mediator of hepatic JNK signaling that can systemically regulate metabolism.
Here we report that JNK signaling in adipocytes can also systemically regulate metabolism by regulating hepatic FGF21 expression. The hormone FGF21 therefore plays a key role in the metabolic actions of JNK. This hormonal function of hepatic FGF21 regulates systemic metabolism (Fisher and Maratos-Flier 2016; BonDurant and Potthoff 2018; Kliewer and Mangelsdorf 2019). An important focus for future studies will be to identify FGF21-independent mechanisms that also contribute to metabolic stress signaling by JNK in adipocytes.
Cross-talk between adipose tissue and the liver
Substantial evidence links adipose tissue to the regulation of hepatic metabolism (Titchenell et al. 2017; Petersen and Shulman 2018; Funcke and Scherer 2019). For example, insulin-regulated adipose tissue lipolysis contributes to hepatic glucose production (Perry et al. 2015; Titchenell et al. 2016). Our study of JNK signaling in adipocytes provides another example of adipose tissue cross-talk with the liver.
We show that reduced JNK signaling in adipocytes promotes FGF21 expression and increases autocrine/paracrine FGF21 signaling. PPARγ activation by FGF21 in adipocytes increases Adipoq mRNA expression and increases the concentration of adiponectin circulating in the blood (Holland et al. 2013; Lin et al. 2013). Subsequent adiponectin binding to hepatic receptors (AdipoR2) causes PPARα activation in hepatocytes (Yamauchi et al. 2007; Ishtiaq et al. 2019) that may promote Fgf21 mRNA expression and endocrine secretion of FGF21 (Badman et al. 2007; Inagaki et al. 2007). This mechanism indicates that autocrine/paracrine signaling by adipocyte FGF21 triggers a feed-forward loop that promotes endocrine FGF21 signaling by the liver (Fig. 8). This signaling paradigm provides new insight into the complexity of adipose tissue cross-talk with the liver.
Figure 8.
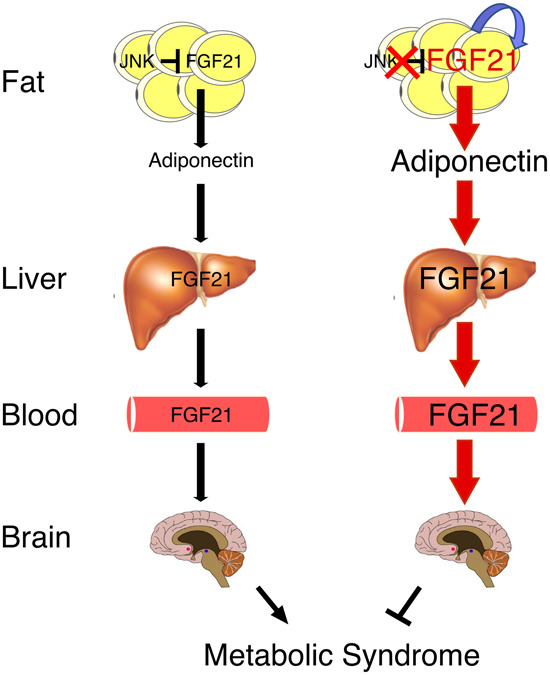
Schematic illustration of organ cross-talk by autocrine FGF21 signaling by adipocytes and endocrine signaling by the liver.
Adiponectin plays an important role in organ cross-talk by mediating a connection between autocrine/paracrine FGF21 signaling by adipocytes and endocrine FGF21 signaling by hepatocytes. Adiponectin functions downstream from adipocyte FGF21 and upstream of hepatic FGF21. Consequently, endocrine signaling by hepatic FGF21 is independent of adiponectin. Adiponectin is therefore a key mediator of autocrines/paracrine signaling by adipocyte FGF21 because adiponectin can promote endocrine expression of FGF21 by the liver.
Conclusions
This study identifies a mechanism for organ cross-talk that is initiated by JNK-regulated FGF21 expression in adipose tissue. Autocrine/paracrine signaling by adipocyte FGF21 increases the circulating concentration of adiponectin that promotes expression of the hormone FGF21 by the liver. This feed-forward regulatory loop represents a novel signaling paradigm that connects autocrine and endocrine signaling modes of the same hormone in different tissues.
Materials and methods
Resources
Key resources are listed in Supplemental Table S1.
Mice
C57BL/6J mice (RRID: IMSR_JAX:000664), B6;FVB-Tg(Adipoq-Cre)1Evdr mice (RRID: IMSR_JAX:028020) (Eguchi et al. 2011), and B6.Cg-Speer6-ps1Tg(alb-cre)21Mgn/J mice (RRID: IMSR_ JAX:003574) (Postic et al. 1999) were obtained from The Jackson Laboratory. We have previously described Adipoq−/− mice (Nawrocki et al. 2006), Fgf21LoxP/LoxP mice (Vernia et al. 2016a), Mapk8LoxP/LoxP mice (Das et al. 2007), and Mapk9LoxP/LoxP mice (Han et al. 2013). The mice were backcrossed to the C57BL/6J strain (10 generations) and housed in a specific pathogen-free facility accredited by the American Association for Laboratory Animal Care using laminar flow cages at 21°C. Mice were housed at thermoneutrality (30°C) in the vivarium using rodent incubators (Powers Scientific, Inc. model RIT33SD). Cold exposure was performed by reducing the temperature 2°C/day from ambient to the maintenance temperature (6°C) using rodent incubators in the vivarium. Male mice (age 8 wk) were fed a chow diet (Purina IsoPro 3000) or a high-fat diet (Bio-Serv S3282) (Han et al. 2013). Whole-body fat and lean mass were noninvasively measured by 1H-MRS (Echo Medical Systems). The studies were approved by the Animal Care and Use Committees of the University of Massachusetts Medical School, Yale Medical School, and the University of Cincinnati.
Genotype analysis
Genomic DNA was genotyped using a PCR-based method. The amplimers 5′-GGACCCCTGAACTTGCTTCAC-3′, 5′-TTCAATTCCCAGCACCCACAGTAA-3′, and 5′-GGCTACCCGTGATATTGCTGAAGAG-3′ were used to detected the AdipoqWT (214-bp) and AdipoqΔ (520-bp) alleles. Cre+ (450 bp) was detected using amplimers 5′- TTACTGACCGTACACCAAATTTGCCTGC-3′ and 5′-CCTGGCAGCGATCGCTATTTTCCATGAGTG-3′. The amplimers 5′-CCTGACTCTTCCTGAATC-3′ and 5′-GAGCCCAAAATGGTG-3′ were used to detect the Fgf21WT (216-bp) and Fgf21LoxP (288-bp) alleles. The amplimers 5′-GCCAAGTGTCAGAACTG-3′ and 5′-CCGTGGTACCAGGAATGC-3′ were used to detect the Fgf21LoxP (1211-bp) and Fgf21Δ (298-bp) alleles. The amplimers 5′- CCTCAGGAAGAAAGGGCTTATTTC-3′ and 5′-GAACCACTGTTCCAATTTCCATCC-3′ detected the Mapk8WT allele (1550 bp), the Mapk8LoxP allele (1095 bp), and the Mapk8Δ allele (395 bp). The amplimers 5′-GTTTTGTAAAGGGAGCCGAC-3′ and 5′-CCTGACTACTGAGCCTGGTTTCTC-3′ were used to detect the Mapk9WT allele (224 bp) and the Mapk9LoxP allele (264 bp). The amplimers 5′-GGAATGTTTGGTCCTTTAG-3′, 5′-GCTATTCAGAGTTAAGTG-3′, and 5′-TTCATTCTAAGCTCAGACTC-3′ were used to detect the Mapk9LoxP allele (560 bp) and the Mapk9Δ allele (400 bp). The amplimers were obtained from Eurofins Genomics LLC.
AAV transduction studies
High-titer AAV8-TBG-Cre stocks were prepared using plasmid AAV.TBG.PI.Cre.rBG (Addgene plasmid 107787; RRID: Addgene_107787) by the University of Massachusetts Medical School Viral Vector Core. Mice were injected intravenously with 1.5 × 1011 genome copies of AAV8 vector per mouse.
RNA analysis
The expression of mRNA and microRNA were examined by quantitative PCR analysis using a Quantstudio PCR machine (Thermo Fisher Scientific) and TaqMan probes (Supplemental Table S2). Fgf21 mRNA expression was measured using Thermo Fisher TaqMan probes (Supplemental Table S2) in studies of Fgf21+/+ cells and mice. Studies of Fgf21 mRNA expression in mice with floxed Fgf21 alleles were performed using Universal ProbeLibrary Probe 67 (Roche, Cat# 4688651001) and amplimers 5′-AGATGGAGCTCTCTATGGATCG-3′ and 5′-GGGCTTCAGACTGGTACACAT-3′ (Eurofins Genomics LLC). Normalized relative mRNA and microRNA expression was performed by measurement of the amount of 18S RNA and U6 RNA, respectively, in each sample.
Insulin treatment
Intraperitoneal injection of mice with saline (control) or 1 U/kg insulin (15 min) was performed. Tissue extracts were prepared using Triton lysis buffer (20 mM Tris at pH 7.4, 1% Triton X-100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 µg/mL aprotinin plus leupeptin).
Immunoblot analysis
Extracts (20–50 µg of protein) were examined by protein immunoblot analysis by probing with antibodies to AKT (Cell Signaling Technology 9272; RRID: AB_329827), pSer473 AKT (Cell Signaling Technology 9271; RRID: AB_329825), pThr308 AKT (Cell Signaling Technology 4056; RRID: AB_331163), JNK1/2 (BD Biosciences 554285, RRID: AB_395344), GAPDH (Santa Cruz Biotechnology sc-25778, RRID: AB_10167668), and αTubulin (clone B-5-1-2; Millipore-Sigma T5168; :RRID: 477579). IRDye 680LT-conjugated donkey anti-mouse immunoglobulin G (IgG) antibody (LI-COR Biosciences 926-68022, RRID: AB_10715072) and IRDye 800CW conjugated-goat anti-rabbit IgG (LI-COR Biosciences 926-32211, RRID: AB_621843) were used to detect and quantitate immune complexes with the Odyssey infrared imaging system (LI-COR Biosciences).
Measurement of blood glucose and adipokine concentration
Blood glucose was measured with an Ascensia Breeze 2 glucometer (Bayer). Insulin, IL6, and leptin in plasma were measured by multiplexed ELISA (Millipore Sigma MMHMAG-44K) using a Luminex 200 machine (Millipore Sigma). The plasma concentration of FGF21 (Millipore Sigma EZRMFGF21-26K) and adiponectin (Millipore Sigma EZMADP-60K) was measured by ELISA.
Measurement of hepatic triglyceride
Liver triglyceride was measured using a kit (Millipore Sigma TR0100).
Hyperinsulinemic-euglycemic clamp studies
Basal infusion and hyperinsulinemic-euglycemic clamp studies were performed after an overnight fast to assess whole-body, hepatic, and adipose tissue insulin sensitivity (Perry et al. 2015).
Glucose, pyruvate, and insulin tolerance tests
Glucose, pyruvate, and insulin tolerance tests were performed using methods described previously (Sabio et al. 2008).
Glucose-induced insulin release
Glucose-stimulated insulin secretion (GSIS) measurements were performed by intraperitoneal injection of glucose (2 g/kg) and measurement of blood insulin concentration at 0, 5, and 15 min.
Body temperature
Biocompatible and sterile microchip transponders (IPTT-300 extended accuracy calibration; Bio Medic Data Systems) were implanted and core body temperature was measured by telemetry. Thermal imaging was performed using an infrared thermal camera (FLIR-T62101) and analyzed with FLIR Tools software.
Metabolic cages
The analysis was performed by the Mouse Metabolic Phenotyping Center at the University of Cincinnati. The mice were housed under controlled temperature and lighting with free accesses to food and water. Respiratory exchange ratio and energy exchange ratio were measured using metabolic cages.
Analysis of tissue sections
Histology was performed using tissue fixed in 10% formalin for 24 h, dehydrated, and embedded in paraffin. Sections (5 µm) were cut and stained using hematoxylin and eosin (American Master Tech Scientific). Adipocyte size and number were assessed using H&E-stained sections with Image J software. Immunohistochemistry was performed using deparaffinized tissue sections that were incubated in blocking buffer for 1 h (1% BSA, 10% normal goat serum in PBS) and permeabilized with 0.1% Triton X-100. The sections were incubated in blocking buffer containing primary antibodies to insulin (Agilent A0564, RRID: AB_10013624), glucagon (Abcam ab10988, RRID: AB_2917642), and UCP1 (Abcam ab10983, RRID: AB_2241462). The insulin antibody was detected using Alexa-Fluor 488 conjugated-goat anti-guinea pig IgG (H + L) antibody (Thermo Fisher Scientific A-11073, RRID: AB_2534117), the glucagon antibody was detected using Alexa Fluor 546 conjugated-donkey anti-mouse IgG (H + L) antibody (Thermo Fisher Scientific A-10036, RRID: AB_2534012), and the UCP1 antibody was detected using Alexa Fluor 488-conjugated goat anti-rabbit IgG (H + L) antibody (Thermo Fisher Scientific A-11034, RRID: AB_2576217). DNA was detected by staining with DAPI (Thermo Fisher Scientific D3571, RRID: AB_2307445). Fluorescence was visualized using a Leica TCS SP2 confocal microscope equipped with a 405-nm diode laser. Islet area was determined by dividing total β-cell area (marked by staining with insulin antibodies) by the pancreatic area per section.
Adipocyte and stromal vascular cell isolation
The stromal vascular cell fraction (SVF) of white fat (EpiF) was isolated (Herrero et al. 2010). Briefly, EpiF was excised and minced into 10 mL of KRB solution (12.5 mM HEPES at pH 7.4, 120 mM NaCl, 6 mM KCl, 1.2 mM MgSO4, 1 mM CaCl2, 2% BSA, 2.5 mM glucose). Collagenase II (1 mg/mL; Millipore Sigma C6885) and DNase I (0.2 mg/mL; Millipore Sigma DN-25) were added and the tissue was incubated at 37°C with shaking (40 min). Larger particles were removed using a 100-µm nylon sieve and the filtrates were centrifuged at 1000 rpm (3 min). Floating adipocytes were washed twice with phosphate-buffered saline (PBS) prior to RNA isolation. Nonadipocyte layers (SVF) were collected, centrifuged at 3000 rpm (5 min), and washed with PBS.
Statistical analysis
Data are presented as the mean and standard error. Statistical analysis was performed using GraphPad Prism version 7 (GraphPad Software). ANOVA with Bonferroni's test was used to determine significance with an assumed confidence interval of 95%. Two-tailed, unpaired t-test with Welch's correction was used for pairwise comparisons. Statistical significance was defined as P < 0.05.
Acknowledgments
We thank Aafreen Syed, Armanda Roy, Shanzah Chaudhry, Julie Cavanaugh-Kyros, Ali Nasiri, and Xiaoxian Ma for expert technical assistance, and Kathy Gemme for administrative assistance. These studies were supported by American Heart Association grant 19CDA34660270 to M.S.H., Sao Paulo Research Foundation grant 2018/04956-5 to J.-P.C., and National Institutes of Health grants R01 DK107220 and R01 DK112698 to R.J.D., R00 CA215315 to R.J.P., R01 DK55758 and P01 AG051459 to P.E.S., and R01 DK116774, R01 DK114793, and P30 DK045735 to G.I.S.
Author contributions: M.S.H. and R.J.D. designed the study. M.S.H., R.J.P., and J.-P.C. performed experiments. M.S.H., R.J.P., P.E.S., G.I.S., G.G., and R.J.D. analyzed data and wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.344556.120.
Freely available online through the Genes & Development Open Access option.
References
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. 2007. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5: 426–437. 10.1016/j.cmet.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, et al. 2010. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci 107: 6028–6033. 10.1073/pnas.1001796107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BonDurant LD, Potthoff MJ. 2018. Fibroblast growth factor 21: a versatile regulator of metabolic homeostasis. Annu Rev Nutr 38: 173–196. 10.1146/annurev-nutr-071816-064800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR, Walsh SA, Ornitz DM, Potthoff MJ. 2017. FGF21 regulates metabolism through adipose-dependent and -independent mechanisms. Cell Metab 25: 935–944.e4. 10.1016/j.cmet.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, Ding X, Elmquist JK, Takahashi JS, Mangelsdorf DJ, et al. 2013. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med 19: 1147–1152. 10.1038/nm.3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camporez JP, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, Zhang D, Kahn M, Samuel VT, Jurczak MJ, et al. 2013. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 154: 3099–3109. 10.1210/en.2013-1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, Papavassiliou AG. 2011. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med 17: 736–740. 10.2119/molmed.2011.00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hoo RL, Konishi M, Itoh N, Lee PC, Ye HY, Lam KS, Xu A. 2011. Growth hormone induces hepatic production of fibroblast growth factor 21 through a mechanism dependent on lipolysis in adipocytes. J Biol Chem 286: 34559–34566. 10.1074/jbc.M111.285965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate KC, Hernandez G, Thorne CA, Sun S, Le TDV, Vale K, Kliewer SA, Mangelsdorf DJ. 2017. FGF21 is an exocrine pancreas secretagogue. Cell Metab 25: 472–480. 10.1016/j.cmet.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Jiang F, Sluss HK, Zhang C, Shokat KM, Flavell RA, Davis RJ. 2007. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci 104: 15759–15764. 10.1073/pnas.0707782104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252. 10.1016/S0092-8674(00)00116-1 [DOI] [PubMed] [Google Scholar]
- Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, Zarebidaki E, Adams AC, Kharitonenkov A, Flier JS, et al. 2015. Central fibroblast growth factor 21 browns white Fat via sympathetic action in male mice. Endocrinology 156: 2470–2481. 10.1210/en.2014-2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, Kliewer SA. 2012. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 148: 556–567. 10.1016/j.cell.2011.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. 2011. Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13: 249–259. 10.1016/j.cmet.2011.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Maratos-Flier E. 2016. Understanding the physiology of FGF21. Annu Rev Physiol 78: 223–241. 10.1146/annurev-physiol-021115-105339 [DOI] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al. 2012. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 26: 271–281. 10.1101/gad.177857.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kim M, Doridot L, Cunniff JC, Parker TS, Levine DM, Hellerstein MK, Hudgins LC, Maratos-Flier E, Herman MA. 2017. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab 6: 14–21. 10.1016/j.molmet.2016.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funcke JB, Scherer PE. 2019. Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res 60: 1648–1684. 10.1194/jlr.R094060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. 2013. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–222. 10.1126/science.1227568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, White A, Perry RJ, Camporez JP, Hidalgo J, Shulman GI, Davis RJ. 2020. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci 117: 2751–2760. 10.1073/pnas.1920004117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero L, Shapiro H, Nayer A, Lee J, Shoelson SE. 2010. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc Natl Acad Sci 107: 240–245. 10.1073/pnas.0905310107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, et al. 2013. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab 17: 790–797. 10.1016/j.cmet.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, Villarroya F. 2011. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J Biol Chem 286: 12983–12990. 10.1074/jbc.M110.215889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Zhong L, Lee JTH, Zhang J, Wu D, Geng L, Wang Y, Wong CM, Xu A. 2017. The FGF21-CCL11 axis mediates beiging of white adipose tissues by coupling sympathetic nervous system to type 2 immunity. Cell Metab 26: 493–508.e4. 10.1016/j.cmet.2017.08.003 [DOI] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, et al. 2007. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab 5: 415–425. 10.1016/j.cmet.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Iroz A, Montagner A, Benhamed F, Levavasseur F, Polizzi A, Anthony E, Régnier M, Fouché E, Lukowicz C, Cauzac M, et al. 2017. A specific ChREBP and PPARα cross-talk Is required for the glucose-mediated FGF21 response. Cell Rep 21: 403–416. 10.1016/j.celrep.2017.09.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishtiaq SM, Rashid H, Hussain Z, Arshad MI, Khan JA. 2019. Adiponectin and PPAR: a setup for intricate crosstalk between obesity and non-alcoholic fatty liver disease. Rev Endocr Metab Disord 20: 253–261. 10.1007/s11154-019-09510-2 [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Mangelsdorf DJ. 2019. A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab 29: 246–253. 10.1016/j.cmet.2019.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T, Morgan DA, Rahmouni K, Sonoda J, Fu X, Burgess SC, Holland WL, Kliewer SA, Mangelsdorf DJ. 2017. FGF19, FGF21, and an FGFR1/β-Klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab 26: 709–718.e3. 10.1016/j.cmet.2017.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, Itoh N, Wang Y, Bornstein SR, Xu A, et al. 2013. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab 17: 779–789. 10.1016/j.cmet.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Manieri E, Herrera-Melle L, Mora A, Tomás-Loba A, Leiva-Vega L, Fernández DI, Rodríguez E, Morán L, Hernández-Cosido L, Torres JL, et al. 2019. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J Exp Med 216: 1108–1119. 10.1084/jem.20181288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, Mohammadi M, Potthoff MJ. 2014. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63: 4057–4063. 10.2337/db14-0595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, et al. 2006. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor γ agonists. J Biol Chem 281: 2654–2660. 10.1074/jbc.M505311200 [DOI] [PubMed] [Google Scholar]
- Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, Kliewer SA, Mangelsdorf DJ. 2014. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab 20: 670–677. 10.1016/j.cmet.2014.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. 2015. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160: 745–758. 10.1016/j.cell.2015.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Shulman GI. 2018. Mechanisms of insulin action and insulin resistance. Physiol Rev 98: 2133–2223. 10.1152/physrev.00063.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274: 305–315. 10.1074/jbc.274.1.305 [DOI] [PubMed] [Google Scholar]
- Sabio G, Davis RJ. 2010. Cjun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci 35: 490–496. 10.1016/j.tibs.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. 2008. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322: 1539–1543. 10.1126/science.1160794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK, et al. 2010a. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev 24: 256–264. 10.1101/gad.1878510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. 2010b. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol 30: 106–115. 10.1128/MCB.01162-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Kauppinen A. 2017. Integrated stress response stimulates FGF21 expression: systemic enhancer of longevity. Cell Signal 40: 10–21. 10.1016/j.cellsig.2017.08.009 [DOI] [PubMed] [Google Scholar]
- Schumann G, Liu C, O'Reilly P, Gao H, Song P, Xu B, Ruggeri B, Amin N, Jia T, Preis S, et al. 2016. KLB is associated with alcohol drinking, and its gene product β-Klotho is necessary for FGF21 regulation of alcohol preference. Proc Natl Acad Sci 113: 14372–14377. 10.1073/pnas.1611243113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søberg S, Sandholt CH, Jespersen NZ, Toft U, Madsen AL, von Holstein-Rathlou S, Grevengoed TJ, Christensen KB, Bredie WLP, Potthoff MJ, et al. 2017. FGF21 is a sugar-induced hormone associated with sweet intake and preference in humans. Cell Metab 25: 1045–1053.e6. 10.1016/j.cmet.2017.04.009 [DOI] [PubMed] [Google Scholar]
- Song P, Zechner C, Hernandez G, Cánovas J, Xie Y, Sondhi V, Wagner M, Stadlbauer V, Horvath A, Leber B, et al. 2018. The hormone FGF21 stimulates water drinking in response to ketogenic diet and alcohol. Cell Metab 27: 1338–1347.e4. 10.1016/j.cmet.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng YH, et al. 2013. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest 123: 215–223. 10.1172/JCI62308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub LG, Scherer PE. 2019. Metabolic messengers: adiponectin. Nat Metab 1: 334–339. 10.1038/s42255-019-0041-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Owen BM, Song P, Hernandez G, Zhang Y, Zhou Y, Scott WT, Paratala B, Turner T, Smith A, et al. 2016. FGF21 regulates sweet and alcohol preference. Cell Metab 23: 344–349. 10.1016/j.cmet.2015.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, et al. 2017. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542: 450–455. 10.1038/nature21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD, et al. 2016. Direct hepatocyte insulin signaling Is required for lipogenesis but Is dispensable for the suppression of glucose production. Cell Metab 23: 1154–1166. 10.1016/j.cmet.2016.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Lazar MA, Birnbaum MJ. 2017. Unraveling the regulation of hepatic metabolism by insulin. Trends Endocrinol Metab 28: 497–505. 10.1016/j.tem.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkilä S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola-Mjøsund KE, Tulkki V, et al. 2010. Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet 19: 3948–3958. 10.1093/hmg/ddq310 [DOI] [PubMed] [Google Scholar]
- Vernia S, Cavanagh-Kyros J, Barrett T, Jung DY, Kim JK, Davis RJ. 2013. Diet-induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes Dev 27: 2345–2355. 10.1101/gad.223800.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, Kim JK, Xu J, Shulha HP, Garber M, et al. 2014. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab 20: 512–525. 10.1016/j.cmet.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh-Kyros J, Barrett T, Tournier C, Davis RJ. 2016a. Fibroblast growth factor 21 mediates glycemic regulation by hepatic JNK. Cell Rep 14: 2273–2280. 10.1016/j.celrep.2016.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Morel C, Madara JC, Cavanagh-Kyros J, Barrett T, Chase K, Kennedy NJ, Jung DY, Kim JK, Aronin N, et al. 2016b. Excitatory transmission onto AgRP neurons is regulated by cJun NH2-terminal kinase 3 in response to metabolic stress. Elife 5: e10031 10.7554/eLife.10031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Holstein-Rathlou S, BonDurant LD, Peltekian L, Naber MC, Yin TC, Claflin KE, Urizar AI, Madsen AN, Ratner C, Holst B, et al. 2016. FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab 23: 335–343. 10.1016/j.cmet.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham M, Pal M, Petzold T, Hjorth M, Egan CL, Brunner JS, Estevez E, Iliades P, Zivanovic B, Reibe S, et al. 2019. Adipocyte-specific deletion of IL-6 does not attenuate obesity-induced weight gain or glucose intolerance in mice. Am J Physiol Endocrinol Metab 317: E597–E604. 10.1152/ajpendo.00206.2019 [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, et al. 2007. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 13: 332–339. 10.1038/nm1557 [DOI] [PubMed] [Google Scholar]
- Zhao S, Kusminski CM, Elmquist JK, Scherer PE. 2020. Leptin: less is more. Diabetes 69: 823–829. 10.2337/dbi19-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]