

Abstract
Mathematical models are routinely calibrated to experimental data, with goals ranging from building predictive models to quantifying parameters that cannot be measured. Whether or not reliable parameter estimates are obtainable from the available data can easily be overlooked. Such issues of parameter identifiability have important ramifications for both the predictive power of a model, and the mechanistic insight that can be obtained. Identifiability analysis is well-established for deterministic, ordinary differential equation (ODE) models, but there are no commonly adopted methods for analysing identifiability in stochastic models. We provide an accessible introduction to identifiability analysis and demonstrate how existing ideas for analysis of ODE models can be applied to stochastic differential equation (SDE) models through four practical case studies. To assess structural identifiability, we study ODEs that describe the statistical moments of the stochastic process using open-source software tools. Using practically motivated synthetic data and Markov chain Monte Carlo methods, we assess parameter identifiability in the context of available data. Our analysis shows that SDE models can often extract more information about parameters than deterministic descriptions. All code used to perform the analysis is available on Github.
Keywords: stochasticity, identifiability, stochastic differential equations, moment dynamics, noise, Markov chain Monte Carlo
1. Introduction
Stochastic mathematical models are rapidly becoming an essential tool for interpreting biological phenomena [1–7]. These models are necessitated, in part, by increasing experimental interest in capturing finer-scale time-series observations [8–12] as well as spatial information [13–18] rather than coarse-scale deterministic trends (figure 1). As computational inference techniques for stochastic models have improved [22–26], a fundamental question that often remains overlooked is whether or not model parameters can be confidently estimated from the available data. Drug development, for example, often relies on the quantification of cell growth rates from a proliferation assay (figure 1a–d) [27]. If a mean-field model is applied to interpret the most frequently reported observation—cell count data—only the net growth rate is identifiable, not the proliferation and death rates [28,29]. Establishing the identifiability of model parameters is critical as predictions, and parameter estimates, from a non-identifiable model may be unreliable [30–33], with further analysis required to quantify prediction uncertainty in non-identifiable models [34–36]. Identifiability should always, therefore, be established before parameter estimation is attempted. Such identifiability analysis is well-established for deterministic ordinary differential equation (ODE) models [31,37–44], but there is a scarcity of methods available for the stochastic models that are becoming increasingly important.
Figure 1.

(a–d) Cell proliferation and death observed in vitro over 36 h in a proliferation assay [19]. Each snapshot has a field-of-view of 1440 × 1440 μm and the location of each cell is indicated with a yellow marker. (e) Data from the early stages of the coronavirus pandemic comprising the observed number of (i) infected individuals, deaths, and (ii) daily new case count in Australia during 2020 [20]. (f) Continuous glucose monitoring data from a single individual over three consecutive days [21].
Stochasticity is fundamental to many processes [2,45–51]. Diabetic patients, for example, rely on the rapid interpretation of highly volatile blood glucose measurements to determine insulin input (figure 1f ) [52,53]. Data from the COVID-19 pandemic [1] is also volatile (figure 1e), and inferences of epidemic data must often be drawn from a single, stochastic, time series. Finally, for systems at equilibrium in the mean-field, such as ion-channel data, models that account for system noise are required to establish parameters [54,55]. Stochastic differential equation (SDE) models of the Itô form are widely applied in systems biology to describe stochastic phenomena [56–59]. SDE models can describe intrinsic noise in, for example, gene expression [2,9,26] or a bio-chemical reaction network [60]; extrinsic noise describing volatility in the environment [51,56,61,62]; and model approximations and unknown effects in so-called grey-box models [63,64]. Explicitly modelling this variability in biological systems can often capture more information about a process than a deterministic model is able to [65–68]. Furthermore, SDE models can account for the correlations inherent to time-series data and account for noise that might otherwise obscure parameters. We demonstrate how to establish parameter identifiability for SDE models that encode information about the intrinsic noise of the process [65]. Our focus is on SDE state-space models that can be formulated through the chemical Langevin equation (CLE), although our analysis is applicable to any SDE of the Itô form. While simulation [69], inference [70,71], and identifiability analysis [72] can, for many stochastic systems, be conducted for discrete Markov models, SDE approximations can offer a significant computational advantage and have a long and extensive history of use in the systems biology literature. Furthermore, use of reflected SDEs [73] can guarantee good agreement with their discrete counterparts at boundaries [73,74].
A prerequisite for parameter estimation is that model parameters be structurally identifiable [31,37–39,75–77]. Structural identifiability refers to the question of whether a parameter can be identified given an infinite amount of noise-free data. A state-space model is said to be structurally identifiable if distinct values of the parameters imply distinct observed model outputs (or in the case of a stochastic model, distinct observed output distributions [78]), and vice versa [79–81]. Techniques such as differential algebra [41,82,83] and transfer function approaches [37,38] can establish structural identifiability in ODE models. These approaches are also used to establish identifiable relationships between parameters [38,84]—for example, the net growth rate in a proliferation assay—which can aid model design and model reduction [84–86]. Many of these techniques have accessible implementations in symbolic computation packages [41,87–90], meaning structural identifiability analysis does not require a detailed understanding of the, often complex, underlying mathematical analysis [41].
When experimental data are considered, a more useful question is that of practical identifiability or estimability [31,81,87,91]. That is, can parameters in the model be accurately estimated given a finite amount of noisy experimental data? This kind of analysis is routinely used in the field of experimental design to assess the nature of data required to adequately identify biophysical parameters [32,54,58,92–94]. Practical identifiability is established in conjunction with an inference technique, such as profile or maximum likelihood [94–97] or Markov chain Monte Carlo (MCMC) [32,54]. These techniques provide information about the flatness (or otherwise) of the likelihood function—in the Bayesian case, the posterior distribution—that describes knowledge about the parameters after the experimental data is taken into consideration. For deterministic and simple stochastic models, this information can be obtained directly from the Fisher information matrix [97,98]. Compared with structural identifiability, which is a property of the model, practical identifiability is more nuanced and additionally dependent upon prior knowledge; the experimental data; and consequentially, the experiment itself [32,87]. For example, should the model and data provide no more information about a parameter than that already established in previous studies, the parameter may be classified as practically non-identifiable from the data and model in question. For this reason, we take a Bayesian approach to parameter estimation and encode existing knowledge about the parameters in a prior distribution. Therefore, we classify a model parameter as practically non-identifiable if it cannot be uniquely established within a level of confidence not already established from prior knowledge [31,32]. This question of practical identifiability has not yet been demonstrated for SDE models in systems biology.
Computational inference for stochastic models is a significant challenge [25]. Unlike approaches to parameter estimation for deterministic models, the likelihood function for a realistic stochastic model is, generally, intractable [25]. Techniques based on approximations, such as a linear noise approximation [97] or approximate Bayesian computation [23,99–103], are available for SDEs but are, naturally, approximations. Pseudo-marginal methods [104,105], developed relatively recently, are computationally costly, but provide an unbiased estimate of the true likelihood function for partially observed time series described by nonlinear stochastic models. In this review, we use a pseudo-marginal MCMC approach, where we estimate the likelihood with a particle filter, which we refer to as particle MCMC [106–108]. There are many excellent articles and reviews of inference for stochastic models in systems biology [23,25,108,109], so we do not focus on the details of our implementation here. Despite the established importance of identifiability, it is all too common in parts of the inference literature to draw the standard assumption that the model parameters are identifiable: we note that all the aforementioned review articles make no mention of identifiability. The computational cost of inference for stochastic models, in itself, motivates us to consider identifiability. For example, identifiability can guide model selection: if both a deterministic and stochastic description of a process are practically non-identifiable, the cheaper deterministic model may, in some cases, be adequate for parameter estimation. Where structural non-identifiability is detected, practical non-identifiability necessarily follows and does not need to be established separately.
The focus of this review is to provide an accessible guide to establishing identifiability in SDE models in biology. To do this, we analyse identifiability in SDE descriptions of four case study models, shown in figure 2. The simplest model we consider is a birth–death process (figure 2a) that is routinely used to describe cell proliferation and death in a range of in vitro and in vivo biological systems, such as that shown in figure 1a. We demonstrate that, from cell count-data, the cell proliferation and death rates are structurally non-identifiable for a routinely employed ODE model, but can be identified for an SDE model. Next, we consider two multi-state models where only partial observations of the system are available. First, a two-pool model (figure 2b) that can describe, for example, the decay of human cholesterol while it transfers between two organs [38,110]. We assume that data from the two-pool model comprises several time-series observations of the substance concentration in a single pool. Second, an epidemic model (figure 2c) [111–113] describes individuals infected owing to interactions between susceptible and infectious individuals. We model a testing procedure such that unknown proportions of the number of infectious and recovered individuals are observed, and inferences are drawn from a single time series. The last model we consider is a nonlinear SDE model for insulin regulation by β-cells (figure 2d) [114,115]. This type of model can describe the volatility associated with data from a continuous glucose monitoring device (figure 1f) [21]. The equivalent ODE description of the β-insulin-glucose circuit is not structurally or practically identifiable [116], and we demonstrate how the analysis for the ODE description can inform a parameter transformation to aid identifiability analysis for the SDE model.
Figure 2.

We demonstrate identifiability in an SDE CLE description of four models: (a) a birth–death process; (b) a two-pool model; (c) an epidemic model; and (d) a β-insulin-glucose circuit. The coloured boxes indicate the observed quantity, which is coupled to a noisy observation process.
We demonstrate two main approaches to assess identifiability in SDE models. First, we assess structural identifiability through a surrogate model, taken to be a system of ODEs that describe the time-evolution of the statistical moments of the SDE [117–121]. This allows us to apply the established open-source structural identifiability software package DAISY (written for the freeware REDUCE software) to the SDE models through the moment equations. We repeat this analysis in the more recent open-source software package GenSSI2 [122,123], written for MATLAB, which can be more efficient for nonlinear systems. We interpret these results as a proxy for identifiability of the SDE model itself. While this approach is not always conclusive, it can provide a rapid preliminary screening tool and allows direct comparison of identifiability for an SDE model, which contains information about the mean, variance and higher moments; to identifiability for a corresponding ODE model that is typically assumed to describe an approximation of the mean. We only apply this approach where an exact system of moment equations can be derived, which occurs when the reaction rates are polynomial. For more complex stochastic models containing terms such as Hill functions, as found in the β-insulin-glucose circuit model, an exact system of moment equations cannot be derived, we do not apply the moment dynamics approach in this case. Second, we assess practical identifiability using the full SDE models through MCMC [32,54], first demonstrating how practical identifiability can be cheaply established from a naive proposal kernel. To compute credible intervals for each parameter, and visualize potential correlations between parameters, we produce results using a tuned proposal kernel where we can be more certain of convergence.
The outline of this review is as follows. In §2, we establish the types of SDE models and observation processes that we consider, and then outline the techniques used to generate synthetic data. Following this, in §2.2, we summarize moment closure techniques for SDEs and describe how we implement the software tools DAISY and GenSSI to assess for structural identifiability. Next, in §2.3, we provide a brief overview of our implementation of the particle MCMC algorithm. Full details of particle MCMC for SDE models can be found in the existing literature [108,109] and as supporting material. In §3, we use these tools to assess identifiability using an SDE description of four models. In §§4 and 5, we discuss our results and provide an outlook on the future of identifiability for stochastic models in biology. Specifically, we discuss alternative approaches, including those based upon approximations, to perform practical identifiability analysis in light of the computational challenges of working with SDE models. To aid in the accessibility of the techniques we review, we provide our MCMC code in the form of a module (https://github.com/ap-browning/SDE-Identifiability) for the open-source, high-performance Julia programming language [124].
2. Mathematical techniques
In this section, we outline the mathematical and statistical techniques we use to perform identifiability analysis. Full details of all algorithms used are provided as electronic supplementary material.
2.1. Stochastic models in biology
We consider Itô SDE state space models of the form
| 2.1 |
and
| 2.2 |
Here, the state is described by , is a Q-dimensional Wiener process with independent components; α( · ) maps to an N-dimensional vector; and σ( · ) maps to an N × Q matrix. The observables, , are connected to the state variables according to an observation process with probability density function g(Yt|Xt, t; θ). We consider several forms of observation function, including partial observations of the state with both additive and multiplicative Gaussian noise with unknown variance . In equations (2.1) and (2.2), θ is a vector of unknown parameters to be determined through inference. In this review, all variables and parameters are dimensionless.
The focus of this review is on Itô SDE models that are formulated through the CLE description of a system of bio-chemical reactions [60,125,126]. Therefore, additional information about rate parameters is encoded in the noise of the process. The first three models we consider (figure 2a–c) can be expressed directly as a network of reactions. As the β-insulin-glucose circuit model (figure 2d) involves state variables modelled as concentrations, not individual counts, we derive a stochastic description from the CLE but scale the noise term in proportion to the concentration of each species.
In summary, a bio-chemical reaction network comprises N species, X1, X2, …, XN, that interact through Q reactions [69,127,128]. The population of each species is given by . By the law of mass action [60,129], each reaction occurs with a rate described by a propensity function, ak(Xt, t; θ), which is equal to the product of the reactants and the rate constant. The net effect of the kth reaction is described by the stoichiometry νk such that, should reaction k occur in [t, t + dt),
| 2.3 |
For bio-chemical reaction networks without an explicit time-dependent input, the propensity functions will be independent of t and the system can be simulated exactly using an event-driven stochastic simulation algorithm (SSA) [5,129–131]. The principle behind an exact SSA is that reactions can be modelled by an inhomogeneous Poisson process. The time interval between reactions, Δt, is exponentially distributed such that
| 2.4 |
A single reaction occurs at each time-step; the kth reaction occurs with probability proportional to ak(Xt; θ). A typical implementation of the SSA first samples a time-step using equation (2.4); then samples the next event to occur; and finally updates the state. Full details of our implementation of an SSA are given as supporting material, and the reader is directed to [127] for a comprehensive review of simulation algorithms for bio-chemical reaction networks. We generate synthetic data for the first three models, for which the propensity functions are independent of t, using the SSA. In figure 3a–c, we show 100 realizations of the SSA for the birth–death process, two-pool model and epidemic model, respectively.
Figure 3.

(a–d) One hundred example realizations of each model, produced using: (a–c) the SSA; and (d) the SDE. (e–h) Synthetic data used for practical identifiability analysis. Synthetic data comprise noisy observations of the (e) full and (f–h) partial state. In (e,f,h), experimental replicates used simultaneously for parameterization are shown semi-transparent, with the first replicate fully opaque. For the epidemic model, both short-time (opaque) and long-time (semi-transparent) data are considered separately. In both cases of the epidemic model, an unknown proportion of the number of infected individuals (green), and the recovered individuals (black), is observed. In (d,h), the β cell concentration, βt (and the measured concentration Y1,t) is shown on the right axis.
When the population of each species is large and reactions sufficiently frequent, the dynamics of a bio-chemical reaction network can be approximated using the CLE [28,125,127]. Such an approximation is widely applied in systems biology [132,133], and it is often necessary as the SSA quickly becomes computationally expensive as the populations become large and reactions are frequent enough [134]. The CLE is an Itô SDE of the form
| 2.5 |
Here, Wt = (W1,t, W2,t, …, WQ,t) is a Q-dimensional Wiener process with independent components. In this study, we derive an SDE description for each model using the CLE, and we calibrate this SDE to the synthetic data to approximate the parameters in each model. For the first three models, where data are generated using the SSA, not the SDE, this means that identifiability analysis is conducted in such a way that model misspecification could potentially arise. This pragmatically mirrors experimental data, where any model (including an ODE and SDE description) is an approximation. The forward simulation for each SDE is approximated using the Euler–Maruyama algorithm [135], where we apply reflected SDEs to ensure positivity [73]. Full details of the numerical algorithm are given as electronic supplementary material.
2.2. Moment dynamics
To enable the application of established methods for structural identifiability analysis to SDE models, we formulate a system of ODEs that describe the statistical moments of the random variable . We denote as a raw moment of Xt, such that [118–120,126]
| 2.6 |
where 〈 · 〉 indicates the expectation taken with respect to the probability measure of the random variable Xt. Here, is the order of the moment. For example, the first-order moments correspond to the mean of each dimension of Xt, the second-order moments relate to the variances and covariances, and so forth.
We apply the software packages DAISY [41] and GenSSI2 [123] to establish structural identifiability of the resultant system of moment equations. The software package takes a system of ODEs describing the state equations—in our case, the moment equations—in addition to an explicit algebraic relationship between the observables and the state. We, therefore, provide the moments of the observables, Yt, in the noise-free limit, which we denote
| 2.7 |
In many cases, the observation distribution, g(Yt|Xt, t; θ), will depend upon the unknown parameters, θ, if, for example, an unknown proportion of the state is observed. This is captured in the structural identifiability analysis as the equations derived for the observed moments, n, may depend on θ. We provide well-commented input and output obtained using DAISY on Github as supporting material.
An expression for the time derivative of each moment can be found using Itôs lemma (electronic supplementary material). When each component of σTσ is an analytic function, which occurs when all the propensity functions in the bio-chemical reaction network are also analytic functions, we obtain [136]
| 2.8 |
where H( · ) denotes the N × N Hessian matrix of its argument and . In the case that N = 1, equation (2.8) reduces to
where α and σ are now scalar functions.
When each component of α and σTσ are polynomials in Xt, the expectation in equation (2.8) can be carried through to replace powers of Xt with appropriate moments. This, in general, provides an infinite system of ODEs that exactly describe the time evolution of the moments. In practice, we consider a finite system of moments, up to and including moments of order J. We express this now finite system of ODEs as
| 2.9 |
where m≤J(t) is a vector containing all the moments up to, and including, order J; and m>J(t) is a vector containing all moments of order J + 1 and above. In the case that f≤J( · ) depends only on moments up to order J, the system is said to be closed at order J. That is, the infinite system of equations can be truncated at order J and solved directly to obtain an exact solution for the moments. This is the case if α and σTσ are linear in Xt, which occurs in SDEs derived from the CLE if each propensity is linear in Xt, as is the case for the first two models we consider (figure 2a,b).
For more complicated models, including the epidemic model (figure 2c), the system will not, in general, be closed. We must, therefore, apply a moment closure approximation to express moments of order higher than J in terms of lower-order moments [48]. Moment closures typically make an a priori assumption about the distribution of the random variable Xt. For example, assuming components of Xt are independent or normally distributed is a common approach. In this review, we consider three common moment closures: (i) a mean-field closure [119]; (ii) a pairwise closure [119]; and (iii) a Gaussian closure [118].
The mean-field closure, we consider makes the approximation
| 2.10 |
This closure is derived from the assumption that components of Xt are weakly correlated [119] and is also referred to as the covariance closure [137]. In the case, a closure is drawn at J = 1, the mean-field closure often corresponds to an ODE description of the process. For our analysis of the epidemic model, we find that the mean-field closure behaves poorly, suggesting that an assumption that the components of Xt are independent may not be appropriate (electronic supplementary material, figure S1).
While the mean-field closure is commonly drawn at order J = 1, it is more common for the pair-approximation closure to be applied for second and higher-order closures [119]. The pair-approximation closure assumes that a third-order moment can be expressed as
| 2.11 |
The Gaussian closure approximates higher-order moments to match those of the normal distribution, and gives a closure in terms of the mean and covariances. Higher-order moments can be approximated with [118,138]
| 2.12 |
Here, denotes a central moment; Cov(Xj,tXk,t) denotes the covariance between Xj,t and Xk,t; and Is are the sets formed by partitioning the set into unordered pairs, where s is the number of sets. The raw moments, can then be solved from the expressions for the central moments obtained from equation (2.12). For a practical example of the Gaussian closure, see [118].
Other choices of moment closure are routinely used in systems biology, such as those based upon a multivariate lognormal distribution [118] or a derivative matching scheme [139]. However, more complex closures add further complexity to the moment equations, which is a significant computational disadvantage for automated assessment of structural identifiability in software packages such as DAISY and GenSSI2. Furthermore, an approximate system of moment equations (which must then also be closed) could be obtained by applying a series expansion approximation, or an approximation similar to the mean-field closure, to systems containing non-polynomial analytic functions; this is the case for the fourth model we consider (figure 2d). We do not consider the moment dynamics approach for such non-polynomial models in this review as there are difficulties in determining how approximations of different types of nonlinearities impinge on identifiability. This is beyond the scope of this review.
2.3. Inference with Markov chain Monte Carlo
We take a Bayesian approach to parameter estimation to update our knowledge about the parameters, θ, from a set of observations, , using the likelihood function, , such that [140]
| 2.13 |
Here, p(θ) is the prior distribution, and represents our knowledge of θ before consideration of the observations . The prior distribution may encode information from, for example, previous experiments, established knowledge, or physical restrictions on the parameters. In the context of practical identifiability, our goal is to significantly increase our understanding of θ from our prior knowledge. We specify p(θ) to be a truncated uniform distribution: all parameters within a specified region of realistic parameter values (the support) are considered equally likely [32]. An advantage of a uniform prior in the context of identifiability is that the posterior corresponds to the truncated likelihood function, and, therefore, high density regions of the posterior correspond to regions of high likelihood. Furthermore, should an improper, unbounded uniform prior be considered, the posterior will be directly proportional to the likelihood. Thus, our methodology can also be applied to assess parameter identifiability using a purely likelihood-based approach.
We use an MCMC technique, based on the Metropolis–Hastings algorithm, to sample from the posterior distribution [140–143]. The principle behind MCMC in Bayesian inference is to construct a Markov chain, {θm}m≥0, with a stationary distribution equal to . We make a standard choice to initiate the chain from a prior sample, θ0 ∼ p(θ). At each iteration of the algorithm, a new state is proposed, θ* ∼ q(θ*|θm), where q is termed the proposal kernel. The proposal is accepted, θm+1 ← θ*, with probability
| 2.14 |
else the proposal is rejected, θm+1 ← θm. Full details of our implementation are provided as electronic supplementary material. In this review, we use a multivariate normal proposal so that q(θm|θ*) = q(θ*|θm). An interpretation of the Metropolis choice of acceptance probability, equation (2.14), where the proposal is normal and, therefore, symmetric, is that proposals which increase the posterior density are always accepted, whereas proposals that decrease the posterior density are accepted with some reduced probability [32].
We refer to the first set of MCMC chains for each problem as pilot chains [144]. The proposal distribution for each pilot chain is set to be a multivariate normal distribution with independent components and variances equal to one-tenth the corresponding prior variance for each parameter, a typical choice. We always produce four pilot chains, each of 10 000 iterations, which we find to be sufficient to indicate identifiability for our models. These pilot chains are then used to tune the MCMC proposal kernel [145]. We then produce four tuned chains, which can be reliably used to estimate credible intervals and other features of the posterior distribution. The proposal distribution for each tuned chain is chosen to be multivariate normal, with covariance given by [144]
| 2.15 |
Here, is the number of unknown parameters, and is the covariance matrix for the pooled samples from the four pilot chains (a total of 28 000 samples after 3000 samples are discarded as burn-in from each pilot chain). To assess convergence, we calculate the commonly used [146] and neff (effective sample size) [140] diagnostics. In summary, measures the ratio of between-chain and within-chain variance; and neff measures the effective number of independent samples drawn from the posterior. To draw reliable inferences, Gelman et al. [140] suggest ensuring that . Full details of these convergence statistics are available in [140].
The primary challenge with performing inference for SDE models, with time-series data, is computing the likelihood function. In this review, we consider synthetic data from E independent experiments, each with NE time-series observations. The data are denoted
| 2.16 |
and correspond to the likelihood function
| 2.17 |
In most cases, the likelihood for noisy time-series data modelled by an SDE will be intractable [108]. This contrasts with data modelled by a deterministic model, which are typically assumed to be independent and normally distributed about the model output [32]. Likelihood free methods, such as ABC [23,102] and pseudo-marginal approaches [105], are routinely used in systems biology to calibrate complex stochastic models to experimental data by approximating equation (2.17). In this study, we apply a pseudo-marginal approach based on a bootstrap particle filter to approximate the likelihood and calibrate each SDE model to synthetic experimental data [108]. In summary, the bootstrap particle filter approximates equation (2.17) by
| 2.18 |
Here, the observation probability density, g (equation (2.2)), is averaged over R samples from the SDE, to approximate the likelihood. The bootstrap particle filter then resamples from the set of weighted samples, , at each time-step to form the starting locations for each SDE sample to sample forward to tn+1. This process is repeated for each independent experiment, and the result is an unbiased Monte Carlo estimate of the likelihood function, , that replaces in the Metropolis acceptance probability (equation (2.14)). Full details of the particle MCMC algorithm, including an implementation for an ODE model used in one case study, are provided as electronic supplementary material, and for further information the reader is directed to [108,109].
3. Case studies
Using the moment equations and MCMC, we provide a practical guide for assessing parameter identifiability in SDE CLE models through four case studies. We generate synthetic data for each model using the SSA when the propensity functions are time-independent (the birth–death process, two-pool model and epidemic model), and the corresponding CLE when the propensity functions are time-dependent (the β-insulin-glucose circuit). In practice, we would first assess practical identifiability using the experimental data available. However, working with synthetic data provides the means to evaluate the effect of different experiment designs, and observation protocols, on practical identifiability. Our focus is on data comprising partial observations of the process that realistically captures potential experimental data.
3.1. Birth–death process
The first model we consider is a birth–death process (figure 2a). The birth–death processes can describe, for example, the growth of a well-mixed cell population where individuals proliferate and die according to rates θ1 and θ2, respectively. We consider practical identifiability for synthetic data comprising noisy measurements of the cell count at 10 equally spaced times in 10 identically prepared experiments. Such data are typical for in vitro cell proliferation experiments [27,147], an example of which is shown in figure 1a–d.
3.1.1. Model formulation and moment equations
The birth–death process can be expressed as the bio-chemical reaction network
with stoichiometries ν1 = 1 and ν2 = −1; and propensities a1(Xt) = θ1Xt and a2(Xt) = θ2Xt. Here, we denote Xt as the number of individuals in the population. The observed number of individuals, Yt, is described by the noise model
| 3.1 |
Here, we consider a noise process that scales with the total population, that is, multiplicative Gaussian noise. We show 100 realizations of the SSA for the birth–death process in figure 3a, and the synthetic data used for practical identifiability analysis in figure 3e. The data are generated using the initial condition X0 = 50 and target parameter values θ1 = 0.2, θ2 = 0.1 and σerr = 0.05. Here, σerr ≪ 1, which ensures that Yt remains positive.
The CLE for the birth–death process is
| 3.2 |
and the first- and second-order moment equations are
| 3.3 |
The moment equations for the SDE description of the birth–death model above are identical to the moment equations for the discrete Markov model that we simulate using the SSA [148]. The moments of the observable (in the noise-free limit) are given to second order by n1 = m1 and n2 = m2. As α( · ) and σ2( · ) are linear in Xt, the moment equations of the birth–death process are closed at every order and so equations (3.3) are exact. Furthermore, we note that the common mean-field model for the birth–death process,
| 3.4 |
corresponds to the first moment, and describes the average behaviour of Xt. The solution to equation (3.4) is
| 3.5 |
Here, the population, , undergoes exponential growth with a net-growth rate of θ1 − θ2. Therefore, intuitively, it is not possible to identify θ1 and θ2 if only average growth behaviour is observed [28].
3.1.2. Structural identifiability
We first assess structural identifiability of the moment equations in DAISY [41]. If only the first moment, n1, is observed, the system is structurally non-identifiable, meaning the model parameters cannot be uniquely estimated with any amount of data. However, the system becomes structurally identifiable if n2 is also observed. As the moment, equations are closed at every order, and therefore exact, this analysis indicates that the ODE model (equation (3.4), corresponding to the first moment equation) is structurally non-identifiable, while the SDE model is structurally identifiable.
These structural identifiability results can be intuitively understood through re-parameterization [43]. The first moment equation (or the ODE model) can be re-parameterized with , where is the sole parameter in the model. Therefore, for a fixed , all values on the line produce indistinguishable behaviour in the first moment, m1, and hence in the observation, n1. On the other hand, when re-parameterized the second moment equation contains a second, linearly independent, parameter . For the birth–death process, the second moment provides enough additional information to uniquely identify both parameters θ1 and θ2, provided enough data is available. Thus, the birth–death process is structurally identifiable from the first two moments.
3.1.3. Practical identifiability
We assess practical identifiability of the parameter vector θ = (θ1, θ2, σerr) for the ODE and SDE models using MCMC. We place independent uniform priors on each parameter so that and . If prior knowledge about the population (i.e. the cell line) is available, perhaps based upon previously conducted experiments, this can be incorporated into the analysis through an informative prior. For example, upper bounds that define reasonable values for biological parameters are routinely applied in this context [94].
In figure 4a–i, we show MCMC results for the birth–death process using the ODE model. Based on the structural identifiability results, we expect the likelihood (and for a uniform prior, the posterior density) to be constant along the identifiable parameter combination , and we see this in figure 4d. These results also suggest that, should one of θ1 or θ2 be known (for example, if the cells are treated with an anti-proliferative drug that enforces θ1 = 0 [149]) the other be identifiable. However, lower and upper bounds for θ1 and θ2, respectively, are able to be established as a direct consequence of the prior assumption that all parameters are strictly positive. Examination of univariate credible intervals, shown in table 1, reveals that each parameter cannot individually be identified within 3–4 orders of magnitude, a hallmark of non-identifiability [32]. We note that σerr is practically identifiable (figure 4i, 95% credible interval: (0.1448, 0.1907)) from the ODE model, however it will always be overestimated as the observation model for the ODE model must also account for the intrinsic noise of the process.
Figure 4.

MCMC results for (a–i) an ODE and (j–r) an SDE description of the birth–death process. (a,c,f) and (j,l,o) show trace plots for the ODE and SDE models, respectively. Kernel density estimates of the posterior for each parameter (b,e,i) and (k,n,r), and bivariate scatter plots ((d,g,h) and (m,p,q)), are produced by thinning the MCMC chains by using every 100th sample from four independent MCMC chains, after burn-in.
Table 1.
This shows 95% credible intervals (CrI), and diagnostics, for the parameter estimates for the birth–death process. (Credible intervals are approximated using the MCMC quantiles after burn-in.)
| ODE |
SDE |
||||||
|---|---|---|---|---|---|---|---|
| true | 95% CrI | Seff | 95% CrI | Seff | |||
| θ1 | 0.2 | (0.1276,0.5891) | 1.00056 | 2292 | (0.1609,0.4059) | 1.01068 | 104 |
| θ2 | 0.1 | (0.0130,0.4744) | 1.00056 | 2300 | (0.0477,0.3016) | 1.01107 | 107 |
| σerr | 0.05 | (0.1448,0.1907) | 1.00242 | 2254 | (0.0270,0.0667) | 1.00079 | 364 |
We repeat the analysis for the SDE model, results of which are shown in figure 4j–r. For the prior support chosen, both θ1 and θ2 are practically identifiable, as seen in figure 4k,n. Furthermore, 95% credible intervals identify each parameter within a single order of magnitude (table 1). While structural identifiability analysis revealed that the SDE model is identifiable in the limit of infinite, noise-free data, it is not necessarily so for data with a realistic signal-to-noise ratio, characterized by the noise model parameter σerr. In our case, if prior knowledge provided an upper bound for θ1 and θ2 at, for example, 0.3, conclusions of practical identifiability may be analogous to those of the ODE model. We see this in table 1, where the upper bounds of the credible intervals for θ1 and θ2 extend beyond 0.3. This is also evident from both the bivariate scatter plot (figure 4m) and MCMC trace plots (figure 4j,l), where posterior samples above 0.3 are regularly drawn for both θ1 and θ2. As the SDE explicitly accounts for intrinsic noise, σerr is identifiable with estimates close to the true value, in contrast to results from the ODE model.
3.2. Two-pool model
Next, we consider partial observations of a process governed by a two-pool model, describing the decay of a substance that is able to transfer between two pools (figure 2b). Identifiability of a two-pool model was first examined in the fundamental study of Bellman & Åström [37] as they introduced the concept of structural identifiability. The model can represent, for example, human cholesterol distribution dispersed through two pools (for example, two organs), where measurements are taken from a tracer in the first pool [110]. Bellman [37] and later Cobelli [38] show that, for an ODE model, the pool transfer and decay rates are not structurally identifiable. We consider practical identifiability for synthetic data comprising noisy measurements of the first pool at 10 equally spaced time points in five identically prepared experiments. Although measurements of the second pool are not taken, we assume, for demonstration purposes, that the initial concentration in each pool is zero before a known amount is introduced to the first pool, thus the full initial condition is known. In practice, the initial condition may also depend on a set of unknown parameters, and we focus on this with the epidemic model.
3.2.1. Model formulation and moment equations
The two-pool model can be expressed as the bio-chemical reaction network
with stoichiometries ν1 = ( − 1,0)T, ν2 = (0, −1)T, ν3 = ( − 1,1)T and ν4 = (1, −1)T; and propensities a1(Xt) = θ1X1, a2(Xt) = θ2X2, a3(Xt) = θ3X1 and a4(Xt) = θ4X2. Here, we denote Xt = (X1,t,X2,t)T as the concentration of cholesterol in the first and second pools, respectively. The observed concentration, Yt, is described by the noise model
| 3.6 |
in which we consider that the data are subject to measurement error in the form of additive Gaussian noise [9,106,150]. We show 100 realizations of the SSA for the two-pool model in figure 3b, and the synthetic data used for practical identifiability analysis in figure 3f. The data are generated using the initial condition X0 = (100, 0)T and target parameter values θ1 = 0.1, θ2 = 0.2, θ3 = 0.2, θ4 = 0.5 and σerr = 2. Here, we note that σerr ≪ Xt (figure 3c), which ensures Yt > 0.
The CLE for the two-pool model is
| 3.7 |
and the moment equations are given to second order by
| 3.8 |
The moments of the observed cholesterol concentration are given in the noise-free limit by n1 = m10 and n2 = m20. As with the birth–death process, all elements of α and σ( · )Tσ( · ) are linear in Xt, so the moment equations are closed at every order and, therefore, exact.
3.2.2. Structural identifiability
The two-pool model provides an archetypical example of structural non-identifiability in an ODE model [37,38]. Unless a restriction is placed on one of the parameters (for example, if decay of the substance can only occur from the first pool so θ2 = 0), the model parameters are structurally non-identifiable: many parameter combinations give identical behaviour in the ODE model. Therefore, the model parameters cannot be uniquely determined from any amount of noise-free experimental data if observations are made from only the first pool.
We assess structural identifiability of an SDE description of the two-pool SDE model using DAISY with the system of moment equations up to second order (equation (3.8)). While the ODE model is structurally non-identifiable, the SDE model is structurally locally identifiable: the parameters identifiable, but non-uniquely. For the parameter values considered, which are rates and, by definition, positive, DAISY indicates that the parameters are uniquely identifiable. Therefore, in the limit of infinite, noise-free data, the model parameters can be determined from an SDE description of the two-pool model.
3.2.3. Practical identifiability
To assess practical identifiability of the two-pool model, we apply MCMC to infer θ = (θ1, θ2, θ3, θ4, σerr). Initially, independent uniform priors are chosen such that , , , , and . The support of each prior is chosen to cover a range of magnitudes over the target parameter values. Results from four independent pilot chains, each initiated at a random sample from the prior, are shown in figure 5a–f . In figure 5a, we see that the log-likelihood estimate rapidly stabilizes, indicating that the chain has moved to a high-likelihood region of the parameter space. Results for σerr and θ3 also rapidly stabilize, indicating that these parameters are practically identifiable [32]. Results for the remaining three kinetic rate parameters in figure 5c,d,f indicate that θ1, θ2 and θ4 are practically non-identifiable. In particular, chains for θ1 and θ2 spend a non-negligible time near zero, indicating that the model may be indistinguishable (using the available data) from a model where removal only occurs from a single pool.
Figure 5.

Pilot MCMC trace plots, and log-likelihood estimates, of four chains for the two pool SDE model with (a–f) untransformed parameters; and (g–l) transformed parameters. Priors for each parameter are uniform with support corresponding to the respective axis limits. The target parameters, used to generate synthetic data, are indicated (black dashed line).
We next repeat the analysis using MCMC to infer . Inferring the logarithm of rate parameters will provide more detailed information about the magnitude of rate parameters potentially close to zero [108]. This transformation provides an excellent example of why even a uniform prior is informative, as a uniform prior placed on the linear-scale is not uniform on the log-scale. A uniform prior on the linear-scale makes parameters of a smaller magnitude less likely than a larger magnitude. The priors are again chosen to be independent and uniform (on the log-scale), such that for all i and as before. The support of each prior is chosen, again, to cover a range of magnitudes above and below that of the target parameter values. Results in figure 5k confirm that θ3 is practically identifiable, while θ2 and θ4 are practically non-identifiable. From results in figure 5l, we term θ4 one-sided identifiable: the parameter has an identifiable lower bound, and is distinguishable from zero.
To visualize correlations between inferred parameters, we tune the proposal kernel (equation (2.15)) and run the MCMC algorithm for 30 000 iterations, results are shown in figure 6 and table 2. If only the univariate marginal distributions are considered, all parameters except for θ4 may be classified as practically identifiable. However, our analysis shows that θ1 and θ2 are distinguishable only within a large range of magnitudes. A strong correlation is seen between θ1 and θ2, indicating that the total substance exit rate, θ1 + θ2, may be practically identifiable. If one of θ1 or θ2 were known in advance, perhaps based on past experimental knowledge, the other may become practically identifiable. Furthermore, results from the tuned chains verify that θ3 is practically identifiable (95% credible interval (0.1356, 0.4857)) and θ4 is distinguishable from zero.
Figure 6.

Tuned MCMC results for the two-pool model with a parameters on the linear-scale. The left-most column shows an MCMC trace from a single chain. Kernel density estimates of the marginal posterior for each parameter and bivariate scatter plots are produced using every 300th sample from four independent MCMC chains, after burn-in. The autocorrelation function (ACF) for a single chain is shown in (c), indicating that every 300th sample is approximately independent.
Table 2.
This shows 95% credible intervals, and diagnostics, for the parameter estimates (on the linear-scale) for the two-pool model. (Credible intervals are approximated using the MCMC quantiles after burn-in.)
| true | 95% CrI | Seff | ||
|---|---|---|---|---|
| θ1 | 0.1 | (0.0042,0.1503) | 1.0024 | 510 |
| θ2 | 0.2 | (0.0307,1.0699) | 1.0014 | 456 |
| θ3 | 0.2 | (0.1356,0.4857) | 1.0023 | 515 |
| θ4 | 0.5 | (0.4372,1.9585) | 1.0004 | 741 |
| σerr | 2.0 | (0.5715,2.8773) | 1.0089 | 409 |
3.3. Epidemic model
Here, we consider a four-compartment epidemic model—the SEIR model [111–113,151] (figure 2c). In this model, susceptible individuals, S, are infected due to interactions with infectious individuals, I, and undergo an unknown period of time during which they have been exposed, E, but are not themselves infectious. Infectious individuals either recover or are removed from the total population, R. A noisy unknown proportion, ξ, with mean μobs, of the number of infectious and recovered individuals is monitored. This captures a testing regime where not all infectious or recovered individuals are tested. We supplement these results by considering a scenario where the same unknown proportion of the exposed individuals is also monitored during the early part of the epidemic.
The kind of data available for the epidemic model differs significantly from that for the experiment-based models we have considered thus far: we are interested in a practical identifiability problem where data from only a single time-series is available, which mirrors data available from an actual epidemic [152]. We first consider practical identifiability using data from the early part of the epidemic, before the number of cases is observed to decrease. Next, these results are compared to a case where data further through the course of the epidemic is considered (figure 3g). Initially, 10 infected individuals and 10 recovered individuals are detected. For simplicity, we assume there is no noise in these initial observations, so the number of infected and recovered individuals is given by 10/μobs. An unknown number of individuals, E0, are initially exposed. In our analysis, we assume that E0 is not of direct interest, and we class it a nuisance parameter.
3.3.1. Model formulation and moment equations
The SEIR model can be represented by the following bio-chemical reactions:
with stoichiometries ν1 = (−1, 1, 0, 0)T, ν2 = (0, −1, 1, 0)T and ν3 = (0, 0, −1, 1)T; and propensities a1(Xt) = θ1 St It, a2(Xt) = θ2 Et and a3(Xt) = θ3 It. Here, we denote Xt = (St, Et, It, Rt)T as the number of individuals in each compartment. Two observations are made:
| 3.9 |
and
| 3.10 |
Here, Y1,t and Y2,t describe the observed number of infected individuals and recovered individuals, respectively. We further assume that μobs, the average observed proportion; and σerr, the observation error, are unknown and must be estimated. We show 100 realizations of the SSA for the epidemic model in figure 3c, and synthetic data used for practical identifiability analysis in figure 3g. The data are generated using the initial condition X0 = (500 − E0, E0, 10/μobs, 10/μobs)T and target parameter values θ1 = 0.01, θ2 = 0.2, θ3 = 0.1, E0 = 20, μobs = 0.5 and σerr = 0.05. Here, we note that σerr ≪ μobs, ensuring that Y1,t and Y2,t remain positive.
The moment equations differ from the previous two models considered, in that they are not closed. Therefore, the first-order moment equations are not equivalent to those for the corresponding ODE model [33], unless a mean-field closure is drawn at first order. To make progress, we close the moment equations after second order to form an approximate system of moment equations for the first two moments. We give the system of 14 moment equations, under all three moment closures considered, as supporting material. The moments of the observation variables are given in the noise-free limit by
| 3.11 |
In the electronic supplementary material, we produce numerical solutions to the moment equations for the epidemic model for each closure considered (electronic supplementary material, figure S1). All closures predict visually identical behaviour at first order, and the pair-approximation and Gaussian closures are in agreement at second order. For the target parameters, we consider the mean-field closure does not agree at second order with the more advanced closures. Whereas a numerical solution to the moment equations for the pair-approximation and Gaussian closures is readily obtainable from a standard solver in Julia [153], the mean-field closure required a positivity-preserving Patankar-type method [154] to avoid blow up.
3.3.2. Structural identifiability
We assess structural identifiability of the approximate system of moment equations in DAISY and GenSSI2, results are shown in table 3. The ODE model, equivalent to a mean-field closure (equation (2.10)) drawn after the first moment, is structurally non-identifiable. The second-order systems, for all closures, are structurally identifiable (table 3). As the second-order systems are approximate, this analysis is not conclusive for the SDE. However, we can conclude that if the mean and variance of the epidemic model (the first two moments) are modelled using the system of moment equations, and data are available accordingly, the parameters are identifiable in the limit of infinite, noise-free data. We highlight the computational cost in DAISY of introducing complexity into the moment equations through the closure methods. The pairwise closure, equation (2.11), which introduces a quotient, and the Gaussian closure, equation (2.12), which introduces a cubic, take significantly longer using DAISY to assess than the mean-field closure, equation (2.10), yet give the same result. However, unlike MCMC, we note that structural identifiability results are deterministic, and independent of user choices such as prior, number of particles, and generated or real synthetic data.
Table 3.
Structural identifiability of the partially observed SEIR model assessed in DAISY and GenSSI2. (Structural identifiability of the SDE is assessed using each closure method for third and higher-order moments. Note that the ODE model is equivalent to the SDE model with a mean-field closure for second- and higher-order moments. Runtimes correspond to a 3.7GHz quad-core i7 desktop machine running Windows 10.)
| model | structural identifiability | runtime (DAISY) | runtime (GenSSI2) |
|---|---|---|---|
| ODE | non-identifiable | 5 s | 5 s |
| SDE (mean-field closure) | identifiable | 5 min | 2 s |
| SDE (pairwise closure) | identifiable | 16 h | 2 s |
| SDE (Gaussian closure) | identifiable | 7 h | 2 s |
3.3.3. Practical identifiability
We assess practical identifiability of the epidemic model using MCMC to infer θ = (θ1, θ2, θ3, E0, μobs, σerr). Independent uniform priors are placed on each parameter so that , , , , and . Results are shown in figure 7, where we initiate each chain at the same location for all forms of data we consider.
Figure 7.

(a–u) Pilot MCMC trace plots, and log-likelihood estimate, of four chains for the epidemic model. We consider data comprising noisy observations of an unknown proportion of the number of infected and recovered individuals during the early part of the epidemic (first column) and throughout the epidemic (second column). We supplement these results by considering the case where we are able to observe the same unknown proportion of the number of exposed individuals during the early part of the epidemic (third column). Priors for each parameter are uniform with support corresponding to the respective axis limits. The target parameter set, used to generate synthetic data, are indicated (black dashed line).
First, we assess identifiability when only early-time data is available. The log-likelihood estimate rapidly stabilizes (figure 7a), indicating that the chains have moved to a high-likelihood region of the parameter space [32]. Results for θ3, the recovery rate, also stabilize, indicating that θ3 is structurally identifiable. Eventually, we see the estimate for θ2 stabilizes in all chains, however, they underestimate the target value, although proposals equal to and greater than the target value θ2 = 0.2 are occasionally accepted. To compensate, the estimate of θ1 stabilizes, and covers a region an order of magnitude greater than the target (θ1 = 0.01). Therefore, although θ1 is practically identifiable to a large, but finite, range of values, we classify θ1 as non-identifiable from the short-time data. Estimates for E0 and μobs in figure 7m,p do not stabilize, and are practically non-identifiable.
Next, we consider a scenario where long-time data are available, such that the number of infected individuals is observed to eventually decrease. The log-likelihood estimate (figure 7b) and chains for all parameters, except E0, are observed to stabilize, indicating that all parameters of interest are now practically identifiable. We supplement these results by considering a third scenario, where only early-time data are available, but the same unknown proportion of the number of exposed individuals is also monitored. As with the long-time data, all parameters of interest are now practically identifiable.
We perform a posterior predictive check [140] of the epidemic model to compare the model prediction—which accounts for parameter uncertainty, intrinsic noise and observation error—to the synthetic data used for inference. We discard the first 3000 samples from each pilot chain as burn-in, and resample 10 000 parameter combinations for each data type considered. Results in figure 8 show that, in all cases, the model predictions are in agreement with the full time-course (although, we note, the long-time data are only used to calibrate parameters in figure 8b). Results in figure 8a, for the short-time data, highlight how practical non-identifiability affects model predictions. These results predict an epidemic size at t = 30 is noticeably wider and higher than those for the data types where θ1 is practically identifiable. Furthermore, the lower 95% credible interval for the observed number of infected individuals reduces much faster than that predicted by the other data types.
Figure 8.

Posterior predictive distribution for the epidemic model using (a) short-time data; (b) long-time data; and (c) short-time data where observations are also made of the number of exposed individuals. In (a,c), the dashed line indicates the last observation point used for inference. The first 3000 samples from each pilot chain is discarded as burn-in. We resample 10 000 parameter combinations (with replacement) and solve the SDE model to estimate posterior predictive intervals (PIs). Shown are 50% (darker) and 95% (lighter) prediction intervals computed from the quantiles of the posterior predictive distribution.
3.4. β-insulin-glucose circuit
Finally, we consider a nonlinear model of glucose homeostasis, the β-insulin-glucose circuit [114,115] (figure 2d). Parameterizing mathematical models of glucose homeostasis is important for the development of patient-specific insulin delivery for type 1 diabetics [52]. Time-series data of blood glucose concentration is available from continuous glucose monitoring sensors, a critical component of type 1 diabetes management [21,53], an example of which is shown in figure 1f. The model describes the regulation of blood plasma glucose by insulin secreted by pancreatic β cells. Glucose is introduced into the system through a base production plus a meal intake, u(t), and decays linearly according to the insulin concentration. Insulin is secreted by β cells at a rate given by a nonlinear Hill function [115]. β cells are produced and decay in a non-response to the glucose concentration. We consider identifiability for synthetic data comprising noisy measurements of the β cell and glucose concentrations, but not the insulin concentration. The data consist of five independent experiments, each comprising 15 time-series observations following a meal intake. We only consider inference for two biophysical parameters: θ1, the insulin secretion rate; and θ2, the insulin sensitivity. The nonlinearities in the model mean that the moment equation approach is not available, and inference using MCMC is computationally expensive. We demonstrate how structural identifiability analysis of the corresponding ODE system [155] can guide analysis of the SDE system and alleviate some of the computational challenges.
3.4.1. Model formulation
We consider a stochastic analogue of the model presented by Karin et al. [115]. Denoting Xt = (βt, It, Gt)T as the concentrations of β cells, insulin and glucose, respectively, the propensity functions and corresponding stoichiometries are given by
where
and
Because βt, It and Gt denote the concentrations of each substance, and not the population counts, we scale the diffusion term in the CLE to represent the relative concentrations of each substance [73]. Denoting , NI and NG the relative concentration of β cells, insulin and glucose, respectively, we write
| 3.12 |
Two observations are made:
and
such that Y1,t and Y2,t are the observed β cell and glucose concentrations, respectively. We show 100 realizations of the SSA for the β-insulin-glucose circuit in figure 3d, and the synthetic data used for practical identifiability analysis in figure 3h. The data are generated using the initial condition X0 = (322, 10, 5)T with fixed parameters, μ+ = 0.021/(24 × 60), μ− = 0.025/(24 × 60), η = 7.85, γ = 0.3, u0 = 1/30, c = 10−3, , NI = NG = 20, and target parameters θ1 = 0.03, θ2 = 0.0005 and σerr = 1 [115]. Here, we note σerr ≪ βt, Gt (figure 3d), which ensures that Y1,t and Y2,t remain positive.
3.4.2. Parameter transform
Villaverde et al. [156] study structural identifiability of the corresponding ODE model using differential geometry. In the ODE model, θ1 and θ2 are structurally non-identifiable, unless the insulin concentration is also observed or one of these two parameters is known. We demonstrate this using MCMC in figure 9a, where the marginal posterior for (θ1,θ2) covers a hyperbolic region of the parameter space of equal posterior density. In the ODE model, the product θ1θ2 is structurally identifiable. To demonstrate this, we perform MCMC on the ODE model with transformed variables and , results shown in figure 9b. These results also show how inefficient a naive MCMC proposal can be when correlations between posterior parameters are nonlinear. Structural identifiability analysis [156] indicates that the hyperbolic region defined by (for a fixed ) produces indistinguishable behaviour, corresponding to a flat posterior when a uniform prior is applied. Despite this, the tail regions in figure 9a are rarely sampled, which could give the impression that the parameters are practically identifiable.
Figure 9.

Kernel density of the bivariate marginal posterior distribution of the biophysical parameters in the β-insulin-glucose circuit, using the ODE and 100 000 pilot MCMC iterations (the first 3000 are discarded as burn-in). (a) The posterior for the untransformed parameters, (θ1, θ2) shows non-identifiability. (b) The posterior for the transformed parameters, , demonstrates that is identifiable, but is not.
As the propensity functions for the β-insulin-glucose circuit model contain non-polynomial functions, we cannot produce an exact expression for the moment equations. Therefore, we only study practical identifiability using MCMC, and do not consider structural identifiability of the SDE for the β-insulin-glucose circuit using the moment equations. Motivated by the structural identifiability analysis of the ODE model, we use MCMC to infer , where we only consider the transformed variables and .
3.4.3. Practical identifiability
We show MCMC results from four pilot chains in figure 10. The log-likelihood estimate rapidly stabilizes (figure 10a), as do results for and σerr (figure 10b,d). As with the ODE model, is practically identifiable, but is not. To visualize possible correlations between inferred parameters, we tune the proposal kernel (equation (2.15)) and run the MCMC algorithm for 10 000 iterations, results shown in figure 11. The univariate marginal distributions, and MCMC trace plots, show that (95% credible interval: (1.34, 1.67) × 10−5) and σerr (95% credible interval: (0.812, 1.049)) are practically identifiable, whereas is not (95% credible interval: (8.21, 97.79)). No large correlations are seen between the parameters (), and θ2 is clearly practically non-identifiable as samples cover the entire range of the prior.
Figure 10.
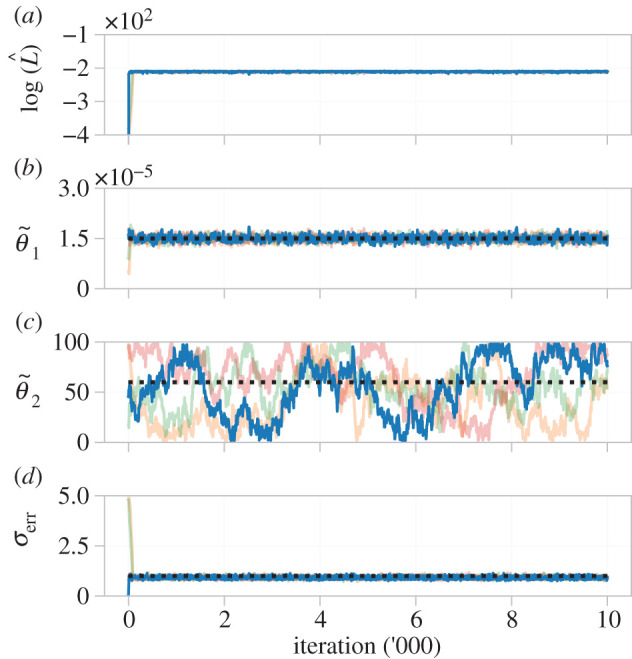
Pilot MCMC trace plots, and log-likelihood estimate, of four chains for the β-insulin-glucose circuit in the transformed parameter space. The likelihood quickly stabilizes, but estimates for do not, indicating practical non-identifiability. Priors for each parameter are uniform with support corresponding to the respective axis limits. The target parameter set, used to generate synthetic data, are indicated (black dashed line).
Figure 11.

(a–j) Tuned MCMC results for the β-insulin-glucose circuit in the transformed parameter space. The left-most column shows an MCMC trace from a single chain. Kernel density estimates of the marginal posterior for each parameter and bivariate scatter plots are produced using every 100th sample from four independent MCMC chains, after burn-in. The autocorrelation function (ACF) for a single chain is shown in (c), indicating that every 20th sample is approximately independent.
4. Discussion
Mathematical models are routinely calibrated to experimental data, with goals ranging from building a predictive model to quantifying biophysical parameters that cannot be directly measured. Much of the usefulness of calibrated models hinges on an assumption that model parameters are identifiable. Heavily over-parameterized models, with large numbers of practically non-identifiable parameters, are often referred to as sloppy in the systems biology literature [76,157–160]. Worryingly, these issues of parameter identifiability can often go undetected: models with non-identifiable parameters can still match experimental data (figure 8), but may have poor predictive power and provide little or no mechanistic insight [31]. Identifiability analysis is well-developed for deterministic ODE models, but there is little guidance in the literature to conducting such analysis for the stochastic models that are often vital for interpreting complex experimental data. In this review, we demonstrate how existing techniques can be applied to assess both structural and practical identifiability of SDE models in biology.
4.1. Moment dynamics approach
We demonstrate how existing ODE identifiability techniques can be applied directly to stochastic problems by formulating a system of moment equations. In the birth–death process and the two-pool model, the derived moment equations are closed and, therefore, exactly describe the time-evolution of the moments of the SDE. In these two case studies, we find that the moment equations are structurally identifiable. This implies structural identifiability of the corresponding SDE model, and parameters can be uniquely estimated in the limit of infinite, noise-free data. For an SDE model, this implies an infinite number of observation-noise free trajectories of the SDE, since the variability, which relates to higher-order moments, contains information. While we find that the two-pool model of cholesterol distribution is not practically identifiable, establishing structural identifiability is useful as it suggests to the practitioner that the observation process (i.e. observe cholesterol in the first pool) is sufficient, in principle, to fully parameterize the model.
For the epidemic model, the moment equations are not closed, so we study structural identifiability through an approximate system of second-order moment equations. The idea of studying identifiability through an approximate system was first suggested by Pohjanpalo [79], who studies identifiability of ODE systems through a power series expansion. The closed system of moment equations suggest the epidemic SDE model could be structurally identifiable, and these results agree, in our case, with practical identifiability detected using MCMC. More research is needed to establish how identifiability is affected when closing, or truncating, a system of moment equations. For example, if information required to identify model parameters is contained in third or higher-order moments, results suggesting that a model is practically non-identifiable from a second order closure will not be indicative of non-identifiability in the SDE model. Furthermore, if structural identifiability differs between moment closures, such a preliminary screening tool needs to be interpreted with caution. If this were the case, a conclusion of structural identifiability is indicative of the model under a particular closure. Recent work suggests that a finite number of moments often contain the information required to identify parameters [161], even for a bimodal distribution and if a closure is applied [162].
Owing to the computational constraints placed on analysing structural identifiability of non-polynomial ODE models, we do not attempt to apply the moment dynamics approach to the stochastic β-insulin-glucose circuit model. However, for many models, a mean-field closure corresponds to an ODE description of the system, and studying identifiability of this ODE model can aid practical identifiability analysis of the corresponding SDE. In our case, the corresponding ODE model is structurally non-identifiable owing to a hyperbolic relationship between the two parameters of interest: for a fixed θ1θ2, model outputs are indistinguishable [156]. The question of whether an SDE description can provide enough information to practically identify θ1 and θ2 can be answered through MCMC, however simple variants of MCMC can struggle when correlations between parameters are strong and nonlinear. Therefore, we work in a transformed parameter space where, for the ODE model, is identifiable but is not (figure 9). This analysis provides a better sense of whether the SDE model captures enough information to identify the parameters, and provides more robust results that are less dependent upon choices made in the MCMC algorithm.
4.2. Particle Markov chain Monte Carlo
We demonstrate practical identifiability by calibrating each model to synthetic data using particle MCMC. We observe the MCMC chains to stabilize in a region of high posterior density, after which time transitions produce samples from the posterior distribution [32]. By visualizing MCMC trace plots, we see that estimates of practically identifiable parameters also stabilize, but those of practically non-identifiable parameters do not. These results also demonstrate that, although estimates made of practically identifiable parameters are precise (that is, within a reasonable level of confidence), they are not necessarily accurate. For example, in figure 7g, the rate at which exposed individuals become infectious is practically identifiable, but it is underestimated compared to the target value, which could hint at model misspecification.
Given that particle MCMC is computationally expensive, our implementation of a standard technique to detect identifiability from pilot chains carries several advantages. First, pilot chains are regularly generated in the early stages of many inference procedures to establish efficient proposal kernels. Practical identifiability can, therefore, be established as part of an existing workflow. Second, more sophisticated methods are by their very nature more difficult to implement and dependent on practitioner choices, which could obscure results and require more algorithmic experimentation. In comparison, we take an automated approach: aside from the model and choice of prior, the procedure to perform MCMC for each model we consider is identical. Once identifiability is established, the computational cost of MCMC can be alleviated to some extent by adopting a more efficient inference technique. For example, adaptive MCMC [163], sequential Monte Carlo [164], multi-level methods [165–167], sub-sampling techniques [168] and model-based proposal methods [169] provide significant performance improvements over the standard technique we employ. Furthermore, we expect applying higher-order SDE simulation algorithms, such as a Runge–Kutta method [170], or considering graphical processing unit approaches to particle MCMC [171], to improve performance.
As we calibrate to synthetic data for the purpose of a didactic demonstration, we take a pragmatic approach by treating the true values as unknown. Hence, we initiate each chain as a random sample from the prior distribution. This involves a burn-in phase before the MCMC chain settles in an area of high posterior density. For computationally expensive models, such as those found in the cardiac modelling literature [172], synthetic data can be used with pilot chains initiated at the target values. If models have already been calibrated to experimental data using, for example, maximum-likelihood estimation, the chain can be initiated at the calibrated values. MCMC then, relatively cheaply, provides information about the posterior distribution about this point, akin to the Fisher information for models where it can be calculated [44].
MCMC can be applied to detect identifiability for any stochastic model provided an approximation to the likelihood is available. Recent developments to particle MCMC have seen its adoption for more complicated SDE models, such as SDE mixed effects models [173]. For systems with relatively small populations, it may be more appropriate to work directly with an SSA with, for example, a tau-leap method [57,125]. Alternative approximations to the likelihood, such as those employed by ABC, may be necessary if model complexity requires; for example, should the model include spatial effects [18]. A major drawback of ABC in the context of identifiability is that one must typically decide a priori which features of the model and data to match. Common applications of ABC for SDE models match the mean and variance of system [109] or the mean square error between simulations and data [174]. Estimating the likelihood directly, as particle MCMC does, is advantageous when assessing identifiability as it is not clear a priori which features of the data and model are significant. For example, some systems might contain the information required for identifiability in higher-order moments or auto-correlations between time-series observations. If ABC is used, a variant that preserves features of the model distribution might be desirable [175].
4.3. Modelling noise
In contrast to many studies of identifiability analysis for ODEs, we do not pre-specify parameters in the observation distribution. In a deterministic modelling framework, it is common to assume that all the variability in the data is uncorrelated and sourced from the observation process [44,94,176]. Therefore, for an ODE model, the observation parameters can be reliably estimated using the pooled sample variance. For inference on the birth–death ODE model (figure 4), we see that, because the observation variance must now also account for intrinsic noise, the identified value of σerr is significantly larger than the target value. For an ODE model with additive homoscedastic Gaussian noise, the posterior mode (in the case of an improper uniform prior), maximum-likelihood estimate and least-squares estimate are identical and are independent of the choice of the observed variance. For an SDE model, this is not the case as the intrinsic component of the noise is also modelled implicitly. Therefore, pre-specifying the observation variance could lead to biased estimates and obscure parameter identifiability. We account for this by treating the observation distribution variance as a nuisance parameter that we infer using MCMC, finding it to be identifiable in every case-study considered.
We have focussed our analysis on SDEs derived through the CLE, where the intrinsic noise can provide more information about the process. However, for large populations, the information contained in higher-order moments dissipates: to leading order, 〈X2〉 → 〈X〉2 as X → ∞. We see this in the epidemic model (figure 3c), where the variance is small compared to the scale of the mean. This loss of information in higher-order moments will not be detected by structural identifiability analysis of the moment equations, which is independent of the relative sizes of each moment. As populations become large, the information tends towards that obtained from the equivalent ODE system: this is the assumption behind many mean-field models. There are, however, many other models that contain sources of variability in their own right. For example, Mummert & Otunuga [62] study identifiability of an epidemic model where the infection rate varies according to a white noise process. Other external effects, such as seasonal effects, are often incorporated into epidemic models [177,178]. In other systems, extrinsic noise describing, for example, the environment, forms a core part of the process and is described by an SDE independent of the population size [51]. Grey-box models use a diffusion term to characterize uncertain physiological effects [64] that could obscure inference, rather than contain information. Making high-level assumptions about which noise process contains information can help with some of the computational challenges by formulating hybrid models containing a mixture of ODEs and SDEs. Particle MCMC carries across, trivially, to any Itô SDE, and the moment equation approach can be applied provided a system of moments be constructed. We have not considered identifiability of SDE models containing non-diffusion noise, such as coloured noise or jump noise. These models lend themselves to different inference techniques, such as forms of rejection sampling [179].
4.4. Approaches to computational challenges
The primary computational cost of working with SDE models stems from the need to simulate a suite of trajectories at each iteration of the particle MCMC algorithm. This cost increases not only with the dimensionality of the problem (as for deterministic models) but also with the amount of data, because the number of particles required for an unbiased likelihood estimate increases with the sample size [28,105]. We see this, in particular, when conducting practical identifiability analysis of the two-pool and β-insulin-glucose models. These issues have important ramifications for identifiability, as it may not always be feasible to increase the amount of experimental data to rectify practical non-identifiability. Working with a surrogate model, such as a system of moment equations, can help alleviate some of these challenges. For example, establishing structural identifiability—which is requisite for parameter estimation [31]—indicates that the computational investment is worthwhile. Furthermore, such surrogate models can also form a computationally efficient alternative for assessing practical identifiability and performing inference [97,162,180], while still capturing more information than a purely deterministic description. The linear noise approximation (LNA) [181] captures information about the mean and variance of a process through a linear SDE. Although the LNA neglects higher-order behaviour, a tractable likelihood function allows practical identifiability analysis directly through the Fisher information matrix (in the frequentist context) or through likelihood-based MCMC techniques (in the Bayesian context). As there is an extensive body of work analysing inference and identifiability using the LNA [28,97,182], we do not consider this approach here. However, conclusions of practical identifiability analysis drawn through MCMC are independent of whether the likelihood is calculated analytically (as through the LNA) or estimated through the particle filter approach.
A large class of high-dimensional stochastic models lend themselves to structural identifiability analysis through moment equations. For example, CLE descriptions of multi-state ion-channel systems [73] and cascades with many bimolecular reactions [183], can be analysed in terms of a surrogate model using moment equations. This approach can be used because the propensity functions are often polynomial. However, the systems of moment equations are often infinite and require a moment closure approximation to facilitate this analysis. When a moment closure assumption is required, we find that newer software, such as GenSSI2 in Matlab, is computationally advantageous over DAISY (table 3). Furthermore, for larger systems, it may not be necessary to consider a full system of second-order moments: a closed system that neglects covariances is also a potentially useful surrogate model.
Many stochastic problems are both computationally expensive to assess using particle MCMC and may not directly permit moment dynamics analysis. The β-insulin-glucose model comprises eight reactions and takes approximately 30 h to perform 10 000 iterations of a pilot chain. (Runtimes for all results are available on Github: https://github.com/ap-browning/SDE-Identifiability.) These issues are magnified by the quantity of data we consider in this example: five time series of 15 observations each. Non-polynomial propensity functions mean that an exact expression for the moment equations cannot be derived, so it may not be possible to pre-detect structural non-identifiability through the moment dynamics approach. Fortunately, other approaches, such as those that use polynomial chaos [184], Gaussian processes [185], or the LNA [97] can provide alternative means of deriving surrogate models. This kind of approximation is already routine in the field of uncertainty quantification, which has deep connections to identifiability [186,187]. In the future, many of these ideas could allow tractable structural and practical identifiability analysis of large systems of SDEs and, by extension, analysis of spatial problems described by stochastic partial differential equations.
The computational cost of MCMC, in particular for stochastic models with many parameters, has spurred the development of alternatives to explore and exploit thegeometry of the likelihood near parameter estimates. The concept of information geometry [159,188] generalizes Fisher information and can be applied to detect identifiability through local information [189], and improve the performance of MCMC algorithms [190]. For SDE models, in particular, variational Bayesian techniques provide an efficient alternative to MCMC for parameter estimation [191]. In many cases, mathematical models are calibrated to experimental data to establish the value of a biophysical parameter, not to fully parameterise a model. Profile likelihood [96,192] is widely applied to assess identifiability in ODE models by maximizing out parameters that are not of direct interest to reduce the dimensionality of the analysis. Because the bootstrap particle filter that we employ estimates the likelihood function, profile likelihood could be applied to SDE problems.
5. Conclusion
It is essential to consider identifiability when performing inference. Yet, there is a scarcity of methods available for assessing identifiability of the stochastic models that are becoming increasingly important. We have provided, through this review, an introduction to identifiability and a guide for performing identifiability analysis of SDE models in systems biology. By formulating a system of moment equations, we show how existing techniques for structural identifiability analysis of ODE models can be applied directly to SDE models [31,37,38,155]. Through synthetic data and particle MCMC, we have demonstrated how to establish practical identifiability for SDE models from data [32,54].
The analysis we demonstrate is critical for tailoring model complexity to the available data [31]. When a structurally identifiable model is found to be practically non-identifiable, identifiability analysis can guide experiment design to discern the quality and quantity of data required to estimate model parameters [193]. On the other hand, models found to be structurally non-identifiable should be re-parameterized, reduced in complexity, or changed [91,194]. Moving from an ODE to an SDE model can often provide enough information to render an otherwise structurally non-identifiable parameter identifiable: we demonstrate this with the birth–death process. As increasing computing power facilitates inference of complex stochastic models, we expect identifiability to become ever more relevant.
Supplementary Material
Acknowledgements
The authors thank Oliver Maclaren for helpful discussions, and the three anonymous referees for their comments.
Data accessibility
This study contains no experimental data. Code used to produce the numerical results is available as a Julia module on GitHub at github.com/ap-browning/SDE-Identifiability.
Authors' contributions
All authors designed the research; A.P.B. performed the research and wrote the manuscript; A.P.B. and D.J.W. implemented the computational algorithms. All authors provided direction, feedback and gave approval for final publication.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the Australian Research Council (DP200100177) and the Air Force Office of Scientific Research(BAA-AFRL-AFOSR-20160007). A.P.B. acknowledges funding from the Australian Centre for Excellence in Mathematical and Statistic Frontiers International Mobility Program. R.E.B. acknowledges funding from the Leverhulme Trust for a Leverhulme Research Fellowship, the BBSRCvia BB/R00816/1 and the Royal Society for a Wolfson Research Merit Award.
References
- 1.Abkowitz JL, Catlin SN, Guttorp P. 1996. Evidence that hematopoiesis may be a stochastic process in vivo. Nat. Med. 2, 190–197. ( 10.1038/nm0296-190) [DOI] [PubMed] [Google Scholar]
- 2.Elowitz MB, Leibler S. 2000. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338. ( 10.1038/35002125) [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson DJ. 2009. Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet. 10, 122–133. ( 10.1038/nrg2509) [DOI] [PubMed] [Google Scholar]
- 4.Neuert G, Munsky B, Tan RZ, Teytelman L, Khammash M, Oudenaarden Av. 2013. Systematic identification of signal-activated stochastic gene regulation. Science 339, 584–587. ( 10.1126/science.1231456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Székely T, Burrage K. 2014. Stochastic simulation in systems biology. Comput. Struct. Biotechnol. J. 12, 14–25. ( 10.1016/j.csbj.2014.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang HW, KhudaBukhsh WR, Koeppl H, Rempała GA. 2019. Quasi-steady-state approximations derived from the stochastic model of enzyme kinetics. Bull. Math. Biol. 81, 1303–1336. ( 10.1007/s11538-019-00574-4) [DOI] [PubMed] [Google Scholar]
- 7.Xu B, Kang HW, Jilkine A. 2019. Comparison of deterministic and stochastic regime in a model for Cdc42 oscillations in fission yeast. Bull. Math. Biol. 81, 1268–1302. ( 10.1007/s11538-019-00573-5) [DOI] [PubMed] [Google Scholar]
- 8.Swameye I, Muller TG, Timmer J, Sandra O, Klingmuller U. 2003. Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by databased modeling. Proc. Natl Acad. Sci. USA 100, 1028–1033. ( 10.1073/pnas.0237333100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heron EA, Finkenstädt B, Rand DA. 2007. Bayesian inference for dynamic transcriptional regulation; the Hes1 system as a case study. Bioinformatics 23, 2596–2603. ( 10.1093/bioinformatics/btm367) [DOI] [PubMed] [Google Scholar]
- 10.Locke JCW, Elowitz MB. 2009. Using movies to analyse gene circuit dynamics in single cells. Nat. Rev. Microbiol. 7, 383–392. ( 10.1038/nrmicro2056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young JW, Locke JCW, Altinok A, Rosenfeld N, Bacarian T, Swain PS, Mjolsness E, Elowitz MB. 2011. Measuring single-cell gene expression dynamics in bacteria using fluorescence time-lapse microscopy. Nat. Protoc. 7, 80–88. ( 10.1038/nprot.2011.432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho H, Rockne RC. 2019. Mathematical modeling with single-cell sequencing data. bioRxiv 710640 ( 10.1101/710640) [DOI] [Google Scholar]
- 13.Ritchie K, Shan XY, Kondo J, Iwasawa K, Fujiwara T, Kusumi A. 2005. Detection of non-Brownian diffusion in the cell membrane in single molecule tracking. Biophys. J. 88, 2266–2277. ( 10.1529/biophysj.104.054106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isaacson SA. 2008. Relationship between the reaction–diffusion master equation and particle tracking models. J. Phys. A: Math. Theor. 41, 065003 ( 10.1088/1751-8113/41/6/065003) [DOI] [Google Scholar]
- 15.Rienzo CD, Piazza V, Gratton E, Beltram F, Cardarelli F. 2014. Probing short-range protein Brownian motion in the cytoplasm of living cells. Nat. Commun. 5, 5891 ( 10.1038/ncomms6891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnoerr D, Grima R, Sanguinetti G. 2016. Cox process representation and inference for stochastic reaction–diffusion processes. Nat. Commun. 7, 11729 ( 10.1038/ncomms11729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brückner DB, Ronceray P, Broedersz CP. 2020. Inferring the dynamics of underdamped stochastic systems. Phys. Rev. Lett. 125, 058103 ( 10.1103/physrevlett.125.058103) [DOI] [PubMed] [Google Scholar]
- 18.Browning AP, Jin W, Plank MJ, Simpson MJ. 2020. Identifying density-dependent interactions in collective cell behaviour. J. R. Soc. Interface 17, 20200143 ( 10.1098/rsif.2020.0143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Browning AP, McCue SW, Binny RN, Plank MJ, Shah ET, Simpson MJ. 2018. Inferring parameters for a lattice-free model of cell migration and proliferation using experimental data. J. Theor. Biol. 437, 251–260. ( 10.1016/j.jtbi.2017.10.032) [DOI] [PubMed] [Google Scholar]
- 20.University Center for Systems Science and Engineering (CSSE) at Johns Hopkins. 2020 COVID-19 data repository. See https://github.com/CSSEGISandData/COVID-19. (accessed 7 July 2020).
- 21.Vigers T, Chan CL, Snell-Bergeon J, Bjornstad P, Zeitler PS, Forlenza G, Pyle L. 2019. cgmanalysis: an R package for descriptive analysis of continuous glucose monitor data. PLoS ONE 14, e0216851 ( 10.1371/journal.pone.0216851) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golightly A, Wilkinson DJ. 2006. Bayesian sequential inference for stochastic kinetic biochemical network models. J. Comput. Biol. 13, 838–851. ( 10.1089/cmb.2006.13.838) [DOI] [PubMed] [Google Scholar]
- 23.Toni T, Welch D, Strelkowa N, Ipsen A, Stumpf MPH. 2009. Approximate Bayesian computation scheme for parameter inference and model selection in dynamical systems. J. R. Soc. Interface 6, 187–202. ( 10.1098/rsif.2008.0172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liepe J, Kirk P, Filippi S, Toni T, Barnes CP, Stumpf MPH. 2014. A framework for parameter estimation and model selection from experimental data in systems biology using approximate Bayesian computation. Nat. Protoc. 9, 439–456. ( 10.1038/nprot.2014.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnoerr D, Sanguinetti G, Grima R. 2017. Approximation and inference methods for stochastic biochemical kinetics—a tutorial review. J. Phys. A: Math. Theor. 50, 093001 ( 10.1088/1751-8121/aa54d9) [DOI] [Google Scholar]
- 26.Bressloff PC. 2017. Stochastic switching in biology: from genotype to phenotype. J. Phys. A: Math. Theor. 50, 133001 ( 10.1088/1751-8121/aa5db4) [DOI] [Google Scholar]
- 27.Bosco DB, Kenworthy R, Zorio DAR, Sang QXA. 2015. Human mesenchymal stem cells are resistant to paclitaxel by adopting a non-proliferative fibroblastic state. PLoS ONE 10, e0128511 ( 10.1371/journal.pone.0128511) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilkinson DJ. 2012. Stochastic modelling for systems biology, 2nd edn Boca Raton, FL: CRC Press. [Google Scholar]
- 29.Liao S, Vejchodský T, Erban R. 2015. Tensor methods for parameter estimation and bifurcation analysis of stochastic reaction networks. J. R. Soc. Interface 12, 20150233 ( 10.1098/rsif.2015.0233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP. 2007. Universally sloppy parameter sensitivities in systems biology models. PLoS Comput. Biol. 3, e189 ( 10.1371/journal.pcbi.0030189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmüller U, Timmer J. 2009. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics 25, 1923–1929. ( 10.1093/bioinformatics/btp358) [DOI] [PubMed] [Google Scholar]
- 32.Hines KE, Middendorf TR, Aldrich RW. 2014. Determination of parameter identifiability in nonlinear biophysical models: a Bayesian approach. J. Gen. Physiol. 143, 401–416. ( 10.1085/jgp.201311116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roosa K, Chowell G. 2019. Assessing parameter identifiability in compartmental dynamic models using a computational approach: application to infectious disease transmission models. Theor. Biol. Med. Modell. 16, 1 ( 10.1186/s12976-018-0097-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kreutz C, Raue A, Timmer J. 2012. Likelihood based observability analysis and confidence intervals for predictions of dynamic models. BMC Syst. Biol. 6, 120 ( 10.1186/1752-0509-6-120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cedersund G. 2016. Prediction uncertainty estimation despite unidentifiability: an overview of recent developments. In Uncertainty in Biology (eds L Geris, D Gomez-Cabrero), pp. 449–466. Cham, Switzerland: Springer International Publishing. ( 10.1007/978-3-319-21296-8_17). [DOI]
- 36.Villaverde AF, Raimúndez E, Hasenauer J, Banga JR. 2019. A comparison of methods for quantifying prediction uncertainty in systems biology. IFAC-PapersOnLine 52, 45–51. ( 10.1016/j.ifacol.2019.12.234) [DOI] [Google Scholar]
- 37.Bellman R, Åström K. 1970. On structural identifiability. Math. Biosci. 7, 329–339. ( 10.1016/0025-5564(70)90132-X) [DOI] [Google Scholar]
- 38.Cobelli C, DiStefano JJ. 1980. Parameter and structural identifiability concepts and ambiguities: a critical review and analysis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 239, 7–24. ( 10.1152/ajpregu.1980.239.1.r7) [DOI] [PubMed] [Google Scholar]
- 39.Walter E. 1987. Identifiability of parametric models. London, UK: Elsevier Science & Technology; ( 10.1016/C2013-0-03836-4) [DOI] [Google Scholar]
- 40.Jaqaman K, Danuser G. 2006. Linking data to models: data regression. Nat. Rev. Mol. Cell Biol. 7, 813–819. ( 10.1038/nrm2030) [DOI] [PubMed] [Google Scholar]
- 41.Bellu G, Saccomani MP, Audoly S, D’Angiò L. 2007. DAISY: A new software tool to test global identifiability of biological and physiological systems. Comput. Methods Programs Biomed. 88, 52–61. ( 10.1016/j.cmpb.2007.07.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miao H, Xia X, Perelson AS, Wu H. 2011. On identifiability of nonlinear ODE models and applications in viral dynamics. SIAM Rev. 53, 3–39. ( 10.1137/090757009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eisenberg MC, Hayashi MA. 2014. Determining identifiable parameter combinations using subset profiling. Math. Biosci. 256, 116–126. ( 10.1016/j.mbs.2014.08.008) [DOI] [PubMed] [Google Scholar]
- 44.Daly AC, Gavaghan D, Cooper J, Tavener S. 2018. Inference-based assessment of parameter identifiability in nonlinear biological models. J. R. Soc. Interface 15, 20180318 ( 10.1098/rsif.2018.0318) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raj A, Oudenaarden Av. 2008. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. ( 10.1016/j.cell.2008.09.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balázsi G, van Oudenaarden A, Collins J. 2011. Cellular decision making and biological noise: from microbes to mammals. Cell 144, 910–925. ( 10.1016/j.cell.2011.01.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bar-Joseph Z, Gitter A, Simon I. 2012. Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 13, 552–564. ( 10.1038/nrg3244) [DOI] [PubMed] [Google Scholar]
- 48.Ruess J, Lygeros J. 2015. Moment-based methods for parameter inference and experiment design for stochastic biochemical reaction networks. ACM Trans. Model. Comput. Simul. 25, 8 ( 10.1145/2688906) [DOI] [Google Scholar]
- 49.Soltani M, Vargas-Garcia CA, Antunes D, Singh A. 2016. Intercellular variability in protein levels from stochastic expression and noisy cell cycle processes. PLoS Comput. Biol. 12, e1004972 ( 10.1371/journal.pcbi.1004972) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith S, Grima R. 2018. Single-cell variability in multicellular organisms. Nat. Commun. 9, 345 ( 10.1038/s41467-017-02710-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Browning AP, Sharp JA, Mapder T, Baker CM, Burrage K, Simpson MJ. In press. Persistence as an optimal hedging strategy. Biophys. J. ( 10.1016/j.bpj.2020.11.2260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hovorka R, Canonico V, Chassin LJ, Haueter U, Massi-Benedetti M, Federici MO, Pieber TR, Schaller HC, Schaupp L, Vering T, Wilinska ME. 2004. Nonlinear model predictive control of glucose concentration in subjects with type 1 diabetes. Physiol. Meas. 25, 905-–920. ( 10.1088/0967-3334/25/4/010) [DOI] [PubMed] [Google Scholar]
- 53.Facchinetti A. 2016. Continuous glucose monitoring sensors: past, present and future algorithmic challenges. Sensors 16, 1–12. ( 10.3390/s16122093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siekmann I, Sneyd J, Crampin E. 2012. MCMC can detect nonidentifiable models. Biophys. J. 103, 2275–2286. ( 10.1016/j.bpj.2012.10.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi B, Rempala GA, Kim JK. 2017. Beyond the Michaelis-Menten equation: accurate and efficient estimation of enzyme kinetic parameters. Sci. Rep. 7, 17018 ( 10.1038/s41598-017-17072-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turelli M. 1977. Random environments and stochastic calculus. Theor. Popul. Biol. 12, 140–178. ( 10.1016/0040-5809(77)90040-5) [DOI] [PubMed] [Google Scholar]
- 57.Turner TE, Schnell S, Burrage K. 2004. Stochastic approaches for modelling in vivo reactions. Comput. Biol. Chem. 28, 165–178. ( 10.1016/j.compbiolchem.2004.05.001) [DOI] [PubMed] [Google Scholar]
- 58.Ruess J, Milias-Argeitis A, Lygeros J. 2013. Designing experiments to understand the variability in biochemical reaction networks. J. R. Soc. Interface 10, 20130588 ( 10.1098/rsif.2013.0588) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parsons TL, Lambert A, Day T, Gandon S. 2018. Pathogen evolution in finite populations: slow and steady spreads the best. J. R. Soc. Interface 15, 20180135 ( 10.1098/rsif.2018.0135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gillespie DT. 2000. The chemical Langevin equation. J. Chem. Phys. 113, 297–306. ( 10.1063/1.481811) [DOI] [Google Scholar]
- 61.Hidalgo J, Pigolotti S, Muñoz MA. 2015. Stochasticity enhances the gaining of bet-hedging strategies in contact-process-like dynamics. Phys. Rev. E 91, 032114 ( 10.1103/physreve.91.032114) [DOI] [PubMed] [Google Scholar]
- 62.Mummert A, Otunuga OM. 2019. Parameter identification for a stochastic SEIRS epidemic model: case study influenza. J. Math. Biol. 79, 705–729. ( 10.1007/s00285-019-01374-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kristensen NR, Madsen H, Jørgensen SB. 2004. Parameter estimation in stochastic grey-box models. Automatica 40, 225–237. ( 10.1016/j.automatica.2003.10.001) [DOI] [Google Scholar]
- 64.Duun-Henriksen AK, Schmidt S, Røge RM, Møller JB, Nørgaard K, Jørgensen JB, Madsen H. 2013. Model identification using stochastic differential equation grey-box models in diabetes. J. Diabetes Sci. Technol. 7, 431–440. ( 10.1177/193229681300700220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Munsky B, Trinh B, Khammash M. 2009. Listening to the noise: random fluctuations reveal gene network parameters. Mol. Syst. Biol. 5, 318 ( 10.1038/msb.2009.75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leander J, Lundh T, Jirstrand M. 2014. Stochastic differential equations as a tool to regularize the parameter estimation problem for continuous time dynamical systems given discrete time measurements. Math. Biosci. 251, 54–62. ( 10.1016/j.mbs.2014.03.001) [DOI] [PubMed] [Google Scholar]
- 67.Enciso G, Kim J. 2019. Embracing noise in chemical reaction networks. Bull. Math. Biol. 81, 1261–1267. ( 10.1007/s11538-019-00575-3) [DOI] [PubMed] [Google Scholar]
- 68.Enciso G, Erban R, Kim J. 2020 Identifiability of stochastically modelled reaction networks. arXiv 2006.02272. (http://arxiv.org/abs/2006.02272. )
- 69.Warne DJ, Baker RE, Simpson MJ. 2019. Simulation and inference algorithms for stochastic biochemical reaction networks: from basic concepts to state-of-the-art. J. R. Soc. Interface 16, 20180943 ( 10.1098/rsif.2018.0943) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boys RJ, Wilkinson DJ, Kirkwood TBL. 2008. Bayesian inference for a discretely observed stochastic kinetic model. Stat. Comput. 18, 125–135. ( 10.1007/s11222-007-9043-x) [DOI] [Google Scholar]
- 71.Ruttor A, Opper M. 2009. Efficient statistical inference for stochastic reaction processes. Phys. Rev. Lett. 103, 230601 ( 10.1103/physrevlett.103.230601) [DOI] [PubMed] [Google Scholar]
- 72.Simpson MJ, Browning AP, Drovandi C, Carr EJ, Maclaren OJ, Baker RE. 2020 doi: 10.1098/rspa.2021.0214. Profile likelihood analysis for a stochastic model of diffusion in heterogeneous media. arXiv 2011.03638. (http://arxiv.org/abs/2011.03638. ) [DOI] [PMC free article] [PubMed]
- 73.Dangerfield CE, Kay D, Burrage K. 2012. Modeling ion channel dynamics through reflected stochastic differential equations. Phys. Rev. E 85, 051907 ( 10.1103/physreve.85.051907) [DOI] [PubMed] [Google Scholar]
- 74.Dangerfield CE, Kay D, Burrage K. 2011. Comparison of continuous and discrete stochastic ion channel models. 2011 Annual Int. Conf. of the IEEE Engineering in Medicine and Biology Society 2011 , 704–707. ( 10.1109/iembs.2011.6090159). [DOI]
- 75.Hengl S, Kreutz C, Timmer J, Maiwald T. 2007. Data-based identifiability analysis of non-linear dynamical models. Bioinformatics 23, 2612–2618. ( 10.1093/bioinformatics/btm382) [DOI] [PubMed] [Google Scholar]
- 76.Chis OT, Banga JR, Balsa-Canto E. 2011. Structural identifiability of systems biology models: a critical comparison of methods. PLoS ONE 6, e27755 ( 10.1371/journal.pone.0027755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janzén DLI, Bergenholm L, Jirstrand M, Parkinson J, Yates J, Evans ND, Chappell MJ. 2016. Parameter identifiability of fundamental pharmacodynamic models. Front. Physiol. 7, 590 ( 10.3389/fphys.2016.00590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reiersøl O. 1950. Identifiability of a linear relation between variables which are subject to error. Econometrica 18, 375 ( 10.2307/1907835) [DOI] [Google Scholar]
- 79.Pohjanpalo H. 1978. System identifiability based on the power series expansion of the solution. Math. Biosci. 41, 21–33. ( 10.1016/0025-5564(78)90063-9) [DOI] [Google Scholar]
- 80.White LJ, Evans ND, Lam TJGM, Schukken YH, Medley GF, Godfrey KR, Chappell MJ. 2001. The structural identifiability and parameter estimation of a multispecies model for the transmission of mastitis in dairy cows. Math. Biosci. 174, 77–90. ( 10.1016/s0025-5564(01)00080-3) [DOI] [PubMed] [Google Scholar]
- 81.Maclaren OJ, Nicholson R. 2019 What can be estimated? Identifiability, estimability, causal inference and ill-posed inverse problems. arXiv 1904.02826. (http://arxiv.org/abs/1904.02826. )
- 82.Margaria G, Riccomagno E, Chappell MJ, Wynn HP. 2001. Differential algebra methods for the study of the structural identifiability of rational function state-space models in the biosciences. Math. Biosci. 174, 1–26. ( 10.1016/s0025-5564(01)00079-7) [DOI] [PubMed] [Google Scholar]
- 83.Saccomani MP, Audoly S, D’Angiò L. 2003. Parameter identifiability of nonlinear systems: the role of initial conditions. Automatica 39, 619–632. ( 10.1016/S0005-1098(02)00302-3) [DOI] [Google Scholar]
- 84.Brouwer AF, Eisenberg MC. 2018 The underlying connections between identifiability, active subspaces, and parameter space dimension reduction. arXiv 1802.05641. (http://arxiv.org/abs/1802.05641. )
- 85.Jacquez JA, Greif P. 1985. Numerical parameter identifiability and estimability: integrating identifiability, estimability, and optimal sampling design. Math. Biosci. 77, 201–227. ( 10.1016/0025-5564(85)90098-7) [DOI] [Google Scholar]
- 86.Saccomani MP, Thomaseth K. 2018. The union between structural and practical identifiability makes strength in reducing oncological model complexity: a case study. Complexity 2018, 1–10. ( 10.1155/2018/2380650) [DOI] [Google Scholar]
- 87.Raue A, Karlsson J, Saccomani MP, Jirstrand M, Timmer J. 2014. Comparison of approaches for parameter identifiability analysis of biological systems. Bioinformatics 30, 1440–1448. ( 10.1093/bioinformatics/btu006) [DOI] [PubMed] [Google Scholar]
- 88.Meshkat N, Kuo CEz, DiStefano J. 2014. On finding and using identifiable parameter combinations in nonlinear dynamic systems biology models and COMBOS: a novel web implementation. PLoS ONE 9, e110261 ( 10.1371/journal.pone.0110261) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Villaverde AF, Barreiro A, Papachristodoulou A. 2016. Structural identifiability of dynamic systems biology models. PLoS Comput. Biol. 12, e1005153 ( 10.1371/journal.pcbi.1005153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hong H, Ovchinnikov A, Pogudin G, Yap C. 2019. SIAN: software for structural identifiability analysis of ODE models. Bioinformatics 35, 2873–2874. ( 10.1093/bioinformatics/bty1069) [DOI] [PubMed] [Google Scholar]
- 91.Brouwer AF, Meza R, Eisenberg MC. 2017. Parameter estimation for multistage clonal expansion models from cancer incidence data: a practical identifiability analysis. PLoS Comput. Biol. 13, e1005431 ( 10.1371/journal.pcbi.1005431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnston ST, Ross JV, Binder BJ, McElwain DLS, Haridas P, Simpson MJ. 2016. Quantifying the effect of experimental design choices for in vitro scratch assays. J. Theor. Biol. 400, 19–31. ( 10.1016/j.jtbi.2016.04.012) [DOI] [PubMed] [Google Scholar]
- 93.Warne DJ, Baker RE, Simpson MJ. 2017. Optimal quantification of contact inhibition in cell populations. Biophys. J. 113, 1920–1924. ( 10.1016/j.bpj.2017.09.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simpson MJ, Baker RE, Vittadello ST, Maclaren OJ. 2020. Practical parameter identifiability for spatio-temporal models of cell invasion. J. R. Soc. Interface 17, 20200055 ( 10.1098/rsif.2020.0055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lehmann EL, Fienberg S, Casella G. 1998. Theory of point estimation, 2nd edn Secaucus, NJ: Springer; ( 10.1007/b98854). [DOI] [Google Scholar]
- 96.Murphy SA, Vaart AWVD. 2000. On profile likelihood. J. Am. Stat. Assoc. 95, 449–465. ( 10.1080/01621459.2000.10474219) [DOI] [Google Scholar]
- 97.Komorowski M, Costa MJ, Rand DA, Stumpf MPH. 2011. Sensitivity, robustness, and identifiability in stochastic chemical kinetics models. Proc. Natl Acad. Sci. USA 108, 8645–8650. ( 10.1073/pnas.1015814108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gunawan R, Cao Y, Petzold L, Doyle FJ. 2005. Sensitivity analysis of discrete stochastic systems. Biophys. J. 88, 2530–2540. ( 10.1529/biophysj.104.053405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tavaré S, Balding DJ, Griffiths RC, Donnelly P. 1997. Inferring coalescence times from DNA sequence data. Genetics 145, 505–518. (doi:pmid:9071603) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW. 1999. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol. Biol. Evol. 16, 1791–1798. ( 10.1093/oxfordjournals.molbev.a026091) [DOI] [PubMed] [Google Scholar]
- 101.Beaumont MA, Zhang W, Balding DJ. 2002. Approximate Bayesian computation in population genetics. Genetics 162, 2025–2035. (doi:pmid:12524368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sunnåker M, Busetto AG, Numminen E, Corander J, Foll M, Dessimoz C. 2013. Approximate Bayesian computation. PLoS Comput. Biol. 9, e1002803 ( 10.1371/journal.pcbi.1002803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wilkinson RD. 2013. Approximate Bayesian computation (ABC) gives exact results under the assumption of model error. Stat. Appl. Genet. Mol. Biol. 12, 129–141. ( 10.1515/sagmb-2013-0010) [DOI] [PubMed] [Google Scholar]
- 104.Beaumont MA. 2003. Estimation of population growth or decline in genetically monitored populations. Genetics 164, 1139–1160. (doi:pmid:12871921) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Andrieu C, Roberts GO. 2009. The pseudo-marginal approach for efficient Monte Carlo computations. Ann. Stat. 37, 697–725. ( 10.1214/07-aos574) [DOI] [Google Scholar]
- 106.Golightly A, Wilkinson DJ. 2008. Bayesian inference for nonlinear multivariate diffusion models observed with error. Comput. Stat. Data Anal. 52, 1674–1693. ( 10.1016/j.csda.2007.05.019) [DOI] [Google Scholar]
- 107.Andrieu C, Doucet A, Holenstein R. 2010. Particle Markov chain Monte Carlo methods. J. R. Stat. Soc.: Ser. B 72, 269–342. ( 10.1111/j.1467-9868.2009.00736.x) [DOI] [Google Scholar]
- 108.Golightly A, Wilkinson DJ. 2011. Bayesian parameter inference for stochastic biochemical network models using particle Markov chain Monte Carlo. Interface Focus 1, 807–820. ( 10.1098/rsfs.2011.0047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Warne DJ, Baker RE, Simpson MJ. 2020. A practical guide to pseudo-marginal methods for computational inference in systems biology. J. Theor. Biol. 496, 110255 ( 10.1016/j.jtbi.2020.110255) [DOI] [PubMed] [Google Scholar]
- 110.Nestel PJ, Whyte HM, Goodman DS. 1969. Distribution and turnover of cholesterol in humans. J. Clin. Invest. 48, 982–991. ( 10.1172/jci106079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kermack WO, McKendrick AG. 1927. A contribution to the mathematical theory of epidemics. Proc. R. Soc. Lond. A 115, 700–721. ( 10.1098/rspa.1927.0118) [DOI] [Google Scholar]
- 112.Tuncer N, Le TT. 2018. Structural and practical identifiability analysis of outbreak models. Math. Biosci. 299, 1–18. ( 10.1016/j.mbs.2018.02.004) [DOI] [PubMed] [Google Scholar]
- 113.Alahmadi A. et al. 2020. Influencing public health policy with data-informed mathematical models of infectious diseases: recent developments and new challenges. Epidemics 32, 100393 ( 10.1016/j.epidem.2020.100393) [DOI] [PubMed] [Google Scholar]
- 114.Topp B, Promislow K, Devries G, Miura RM, Finegood DT. 2000. A model of β-cell mass, insulin, and glucose kinetics: pathways to diabetes. J. Theor. Biol. 206, 605–619. ( 10.1006/jtbi.2000.2150) [DOI] [PubMed] [Google Scholar]
- 115.Karin O, Swisa A, Glaser B, Dor Y, Alon U. 2016. Dynamical compensation in physiological circuits. Mol. Syst. Biol. 12, 886 ( 10.15252/msb.20167216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Villaverde AF, Tsiantis N, Banga JR. 2019. Full observability and estimation of unknown inputs, states and parameters of nonlinear biological models. J. R. Soc. Interface 16, 20190043 ( 10.1098/rsif.2019.0043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Engblom S. 2006. Computing the moments of high dimensional solutions of the master equation. Appl. Math. Comput. 180, 498–515. ( 10.1016/j.amc.2005.12.032) [DOI] [Google Scholar]
- 118.Lakatos E, Ale A, Kirk PDW, Stumpf MPH. 2015. Multivariate moment closure techniques for stochastic kinetic models. J. Chem. Phys. 143, 094107 ( 10.1063/1.4929837) [DOI] [PubMed] [Google Scholar]
- 119.Kuehn C. 2016. Moment closure - a brief review. In Control of self-organizing nonlinear systems (eds E Schöll, SHL Klapp, P Hövel), pp. 253–271. Cham, Switzerland: Springer. ( 10.1007/978-3-319-28028-8). [DOI]
- 120.Fan S, Geissmann Q, Lakatos E, Lukauskas S, Ale A, Babtie AC, Kirk PDW, Stumpf MPH. 2016. MEANS: python package for moment expansion approximation, iNference and simulation. Bioinformatics 32, 2863–2865. ( 10.1093/bioinformatics/btw229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brouwer AF, Meza R, Eisenberg MC. 2017. A systematic approach to determining the identifiability of multistage carcinogenesis models. Risk Anal. 37, 1375–1387. ( 10.1111/risa.12684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chiş O, Banga JR, Balsa-Canto E. 2011. GenSSI: a software toolbox for structural identifiability analysis of biological models. Bioinformatics 27, 2610–2611. ( 10.1093/bioinformatics/btr431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ligon TS, Fröhlich F, Chiş OT, Banga JR, Balsa-Canto E, Hasenauer J. 2017. GenSSI 2.0: multi-experiment structural identifiability analysis of SBML models. Bioinformatics 34, 1421–1423. ( 10.1093/bioinformatics/btx735) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bezanson J, Edelman A, Karpinski S, Shah VB. 2017. Julia: a fresh approach to numerical computing. SIAM Rev. 59, 65–98. ( 10.1137/141000671) [DOI] [Google Scholar]
- 125.Gillespie DT. 2001. Approximate accelerated stochastic simulation of chemically reacting systems. J. Chem. Phys. 115, 1716–1733. ( 10.1063/1.1378322) [DOI] [Google Scholar]
- 126.Schnoerr D, Sanguinetti G, Grima R. 2014. The complex chemical Langevin equation. J. Chem. Phys. 141, 024103 ( 10.1063/1.4885345) [DOI] [PubMed] [Google Scholar]
- 127.Higham DJ. 2008. Modeling and simulating chemical reactions. SIAM Rev. 50, 347–368. ( 10.1137/060666457) [DOI] [Google Scholar]
- 128.Erban R, Chapman SJ. 2009. Stochastic modelling of reaction–diffusion processes: algorithms for bimolecular reactions. Phys. Biol. 6, 046001 ( 10.1088/1478-3975/6/4/046001) [DOI] [PubMed] [Google Scholar]
- 129.Gillespie DT. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361. ( 10.1021/j100540a008) [DOI] [Google Scholar]
- 130.Kurtz TG. 1972. The relationship between stochastic and deterministic models for chemical reactions. J. Chem. Phys. 57, 2976–2978. ( 10.1063/1.1678692) [DOI] [Google Scholar]
- 131.Gibson MA, Bruck J. 2000. Efficient exact stochastic simulation of chemical systems with many species and many channels. J. Phys. Chem. A 104, 1876–1889. ( 10.1021/jp993732q) [DOI] [Google Scholar]
- 132.Rao CV, Wolf DM, Arkin AP. 2002. Control, exploitation and tolerance of intracellular noise. Nature 420, 231–237. ( 10.1038/nature01258) [DOI] [PubMed] [Google Scholar]
- 133.Samad HE, Khammash M, Petzold L, Gillespie D. 2005. Stochastic modelling of gene regulatory networks. Int. J. Robust Nonlinear Cont. 15, 691–711. ( 10.1002/rnc.1018) [DOI] [Google Scholar]
- 134.Golightly A, Wilkinson DJ. 2005. Bayesian inference for stochastic kinetic models using a diffusion approximation. Biometrics 61, 781–788. ( 10.1111/j.1541-0420.2005.00345.x) [DOI] [PubMed] [Google Scholar]
- 135.Maruyama G. 1955. Continuous Markov processes and stochastic equations. Rendiconti del Circolo Matematico di Palermo 4, 48 ( 10.1007/bf02846028) [DOI] [Google Scholar]
- 136.Socha L. 2008. Linearization methods for stochastic dynamic systems. Lecture Notes in Physics Berlin, Heidelberg, Germany: Springer. [Google Scholar]
- 137.Hausken K, Moxnes JF. 2010. A closure approximation technique for epidemic models. Math. Comput. Modell. Dyn. Syst. 16, 555–574. ( 10.1080/13873954.2010.496149) [DOI] [Google Scholar]
- 138.Isserlis L. 1918. On a formula for the product-moment coefficient of any order of a normal frequency distribution in any number of variables. Biometrika 12, 134 ( 10.2307/2331932) [DOI] [Google Scholar]
- 139.Singh A, Hespanha JP. 2007. A derivative matching approach to moment closure for the stochastic logistic model. Bull. Math. Biol. 69, 1909–1925. ( 10.1007/s11538-007-9198-9) [DOI] [PubMed] [Google Scholar]
- 140.Gelman A, Carlin JB, Stern HS, Dunson DB, Vehtari A, Rubin DB. 2014. Bayesian data analysis, 3rd edn Boca Raton, FL: CRC Press. [Google Scholar]
- 141.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21, 1087–1092. ( 10.1063/1.1699114) [DOI] [Google Scholar]
- 142.Hastings WK. 1970. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 57, 97–109. ( 10.1093/biomet/57.1.97) [DOI] [Google Scholar]
- 143.Geyer CJ. 1992. Practical Markov chain Monte Carlo. Stat. Sci. 7, 473–483. ( 10.1214/ss/1177011137) [DOI] [Google Scholar]
- 144.Roberts GO, Rosenthal JS. 2001. Optimal scaling for various Metropolis-Hastings algorithms. Stat. Sci. 16, 351–367. ( 10.1214/ss/1015346320) [DOI] [Google Scholar]
- 145.Gelman A, Rubin DB. 1992. Inference from iterative simulation using multiple sequences. Stat. Sci. 7, 457–472. ( 10.1214/ss/1177011136) [DOI] [Google Scholar]
- 146.Brooks SP, Gelman A. 1998. General methods for monitoring convergence of iterative simulations. J. Comput. Graph. Stat. 7, 434 ( 10.2307/1390675) [DOI] [Google Scholar]
- 147.Johnston ST, Shah ET, Chopin LK, McElwain DLS, Simpson MJ. 2015. Estimating cell diffusivity and cell proliferation rate by interpreting IncuCyte ZOOM assay data using the Fisher-Kolmogorov model. BMC Syst. Biol. 9, 38 ( 10.1186/s12918-015-0182-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Allen LJS. 2011. An introduction to stochastic processes with applications to biology. Boca Raton, FL: Chapman & Hall/CRC Press. [Google Scholar]
- 149.Matsiaka OM, Baker RE, Shah ET, Simpson MJ. 2019. Mechanistic and experimental models of cell migration reveal the importance of cell-to-cell pushing in cell invasion. Biomed. Phys. Eng. Express 5, 045009 ( 10.1088/2057-1976/ab1b01) [DOI] [Google Scholar]
- 150.Poovathingal SK, Gunawan R. 2010. Global parameter estimation methods for stochastic biochemical systems. BMC Bioinf. 11, 414–414 ( 10.1186/1471-2105-11-414) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dargatz C. 2008. A diffusion approximation for an epidemic model. Sonderforschungsbereich 386, 517. [Google Scholar]
- 152.Warne DJ, Ebert A, Drovandi C, Hu W, Mira A, Mengersen K. In press. Hindsight is 2020 vision: a characterisation of the global response to the COVID-19 pandemic. BMC Pub. Health ( 10.1186/s12889-020-09972-Z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rackauckas C, Nie Q. 2016. DifferentialEquations.jl – a performant and feature-rich ecosystem for solving differential equations in Julia. J. Open Res. Softw. 5, 15 ( 10.5334/jors.151) [DOI] [Google Scholar]
- 154.Burchard H, Deleersnijder E, Meister A. 2003. A high-order conservative Patankar-type discretisation for stiff systems of production–destruction equations. Appl. Numer. Math. 47, 1–30. ( 10.1016/s0168-9274(03)00101-6) [DOI] [Google Scholar]
- 155.Villaverde AF, Banga JR. 2014. Reverse engineering and identification in systems biology: strategies, perspectives and challenges. J. R. Soc. Interface 11, 20130505 ( 10.1098/rsif.2013.0505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Villaverde AF. 2019. Observability and structural identifiability of nonlinear biological systems. Complexity 2019, 1–12. ( 10.1155/2019/8497093) [DOI] [Google Scholar]
- 157.Brown KS, Sethna JP. 2003. Statistical mechanical approaches to models with many poorly known parameters. Phys. Rev. E 68, 021904 ( 10.1103/physreve.68.021904) [DOI] [PubMed] [Google Scholar]
- 158.Erguler K, Stumpf MPH. 2011. Practical limits for reverse engineering of dynamical systems: a statistical analysis of sensitivity and parameter inferability in systems biology models. Mol. Biosyst. 7, 1593–1602. ( 10.1039/c0mb00107d) [DOI] [PubMed] [Google Scholar]
- 159.Transtrum MK, Machta BB, Brown KS, Daniels BC, Myers CR, Sethna JP. 2015. Perspective: sloppiness and emergent theories in physics, biology, and beyond. J. Chem. Phys. 143, 010901 ( 10.1063/1.4923066) [DOI] [PubMed] [Google Scholar]
- 160.Dufresne E, Harrington HA, Raman DV. 2018. The geometry of sloppiness. J. Algebr. Stat. 9, 30–68. ( 10.18409/jas.v9i1.64) [DOI] [Google Scholar]
- 161.Smadbeck P, Kaznessis YN. 2013. A closure scheme for chemical master equations. Proc. Natl Acad. Sci. USA 110, 14261–14265. ( 10.1073/pnas.1306481110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zechner C, Ruess J, Krenn P, Pelet S, Peter M, Lygeros J, Koeppl H. 2012. Moment-based inference predicts bimodality in transient gene expression. Proc. Natl Acad. Sci. USA 109, 8340–8345. ( 10.1073/pnas.1200161109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Roberts GO, Rosenthal JS. 2009. Examples of adaptive MCMC. J. Comput. Graph. Stat. 18, 349–367. ( 10.1198/jcgs.2009.06134) [DOI] [Google Scholar]
- 164.Moral PD, Doucet A, Jasra A. 2006. Sequential Monte Carlo samplers. J. R. Stat. Soc.: Ser. B 68, 411–436. ( 10.1111/j.1467-9868.2006.00553.x) [DOI] [Google Scholar]
- 165.Giles MB. 2008. Multilevel Monte Carlo path simulation. Oper. Res. 56, 607–617. ( 10.1287/opre.1070.0496) [DOI] [Google Scholar]
- 166.Jasra A, Kamatani K, Law KJH, Zhou Y. 2017. Multilevel particle filters. SIAM J. Numer. Anal. 55, 3068–3096. ( 10.1137/17m1111553) [DOI] [Google Scholar]
- 167.Warne DJ, Baker RE, Simpson MJ. 2018. Multilevel rejection sampling for approximate Bayesian computation. Comput. Stat. Data Anal. 124, 71–86. ( 10.1016/j.csda.2018.02.009) [DOI] [Google Scholar]
- 168.Quiroz M, Kohn R, Villani M, Tran MN. 2019. Speeding up MCMC by efficient data subsampling. J. Am. Stat. Assoc. 114, 831–843. ( 10.1080/01621459.2018.1448827) [DOI] [Google Scholar]
- 169.Pooley CM, Bishop SC, Marion G. 2015. Using model-based proposals for fast parameter inference on discrete state space, continuous-time Markov processes. J. R. Soc. Interface 12, 20150225 ( 10.1098/rsif.2015.0225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Burrage K, Burrage P. 1996. High strong order explicit Runge-Kutta methods for stochastic ordinary differential equations. Appl. Numer. Math. 22, 81–101. ( 10.1016/S0168-9274(96)00027-X) [DOI] [Google Scholar]
- 171.Mingas G, Bottolo L, Bouganis CS. 2017. Particle MCMC algorithms and architectures for accelerating inference in state-space models. Int. J. Approx. Reason. 83, 413–433. ( 10.1016/j.ijar.2016.10.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee YS, Liu OZ, Hwang HS, Knollmann BC, Sobie EA. 2013. Parameter sensitivity analysis of stochastic models provides insights into cardiac calcium sparks. Biophys. J. 104, 1142–1150. ( 10.1016/j.bpj.2012.12.055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Botha I, Kohn R, Drovandi C. In press. Particle methods for stochastic differential equation mixed effects models. Bayesian Anal. ( 10.1214/20-ba1216) [DOI] [Google Scholar]
- 174.Picchini U. 2012. Inference for SDE models via approximate Bayesian computation. J. Comput. Graph. Stat. 23, 1080–1100. ( 10.1080/10618600.2013.866048) [DOI] [Google Scholar]
- 175.Buckwar E, Tamborrino M, Tubikanec I. 2020. Spectral density-based and measure-preserving ABC for partially observed diffusion processes. An illustration on Hamiltonian SDEs. Stat. Comput. 30, 627–648. ( 10.1007/s11222-019-09909-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liepe J, Filippi S, Komorowski M, Stumpf MPH. 2013. Maximizing the information content of experiments in systems biology. PLoS Comput. Biol. 9, e1002888 ( 10.1371/journal.pcbi.1002888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Evans ND, White LJ, Chapman MJ, Godfrey KR, Chappell MJ. 2005. The structural identifiability of the susceptible infected recovered model with seasonal forcing. Math. Biosci. 194, 175–197. ( 10.1016/j.mbs.2004.10.011) [DOI] [PubMed] [Google Scholar]
- 178.Chapman JD, Evans ND. 2008. The structural identifiability of SIR type epidemic models with incomplete immunity and birth targeted vaccination. IFAC Proc. Volumes 41, 9075–9080. ( 10.3182/20080706-5-kr-1001.01532) [DOI] [Google Scholar]
- 179.Beskos A, Papaspiliopoulos O, Roberts GO. 2006. Retrospective exact simulation of diffusion sample paths with applications. Bernoulli 12, 1077–1098. ( 10.3150/bj/1165269151) [DOI] [Google Scholar]
- 180.Fröhlich F, Thomas P, Kazeroonian A, Theis FJ, Grima R, Hasenauer J. 2016. Inference for stochastic chemical kinetics using moment equations and system size expansion. PLoS Comput. Biol. 12, e1005030 ( 10.1371/journal.pcbi.1005030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Elf J, Ehrenberg M. 2003. Fast evaluation of fluctuations in biochemical networks with the linear noise approximation. Genome Res. 13, 2475–2484. ( 10.1101/gr.1196503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fearnhead P, Giagos V, Sherlock C. 2014. Inference for reaction networks using the linear noise approximation. Biometrics 70, 457–466. ( 10.1111/biom.12152) [DOI] [PubMed] [Google Scholar]
- 183.Plotnikov A, Zehorai E, Procaccia S, Seger R. 2011. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochimica et Biophysica Acta (BBA) - Mole. Cell Res. 1813, 1619–1633. ( 10.1016/j.bbamcr.2010.12.012) [DOI] [PubMed] [Google Scholar]
- 184.Xiu D, Karniadakis GE. 2002. The Wiener-Askey polynomial chaos for stochastic differential equations. SIAM J. Sci. Comput. 24, 619–644. ( 10.1137/s1064827501387826) [DOI] [Google Scholar]
- 185.Archambeau C, Cornford D, Opper M, Shawe-Taylor JS. 2007. Gaussian process approximations of stochastic differential equations. Proc. Mach. Learn. Res. 1, 1–16. [Google Scholar]
- 186.Mirams GR, Pathmanathan P, Gray RA, Challenor P, Clayton RH. 2016. Uncertainty and variability in computational and mathematical models of cardiac physiology. J. Physiol. 594, 6833–6847. ( 10.1113/jp271671) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kaintura A, Dhaene T, Spina D. 2018. Review of polynomial chaos-based methods for uncertainty quantification in modern integrated circuits. Electronics 7, 30 ( 10.3390/electronics7030030) [DOI] [Google Scholar]
- 188.Ran ZY, Hu BG. 2017. Parameter identifiability in statistical machine learning: a review. Neural Comput. 29, 1151–1203. ( 10.1162/neco_a_00947) [DOI] [PubMed] [Google Scholar]
- 189.Lill D, Timmer J, Kaschek D. 2019. Local Riemannian geometry of model manifolds and its implications for practical parameter identifiability. PLoS ONE 14, e0217837 ( 10.1371/journal.pone.0217837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Livingstone S, Girolami M. 2014. Information-geometric Markov chain Monte Carlo methods using diffusions. Entropy 16, 3074–3102. ( 10.3390/e16063074) [DOI] [Google Scholar]
- 191.Archambeau C, Opper M, Shen Y, Cornford D, Shawe-Taylor JS. 2008. Variational inference for diffusion processes. In Advances in neural information processing systems (eds JC Platt, D Koller, Y Singer, ST Roweis), vol. 20, pp. 17–24. New York, NY: Curran Associates, Inc.
- 192.Raue A, Kreutz C, Theis FJ, Timmer J. 2013. Joining forces of Bayesian and frequentist methodology: a study for inference in the presence of non-identifiability. Phil. Trans. R. Soc. A 371, 20110544 ( 10.1098/rsta.2011.0544) [DOI] [PubMed] [Google Scholar]
- 193.Faller D, Klingmüller U, Timmer J. 2003. Simulation methods for optimal experimental design in systems biology. Simulation 79, 717–725. ( 10.1177/0037549703040937) [DOI] [Google Scholar]
- 194.Walter E, Lecourtier Y. 1981. Unidentifiable compartmental models: what to do? Math. Biosci. 56, 1–25. ( 10.1016/0025-5564(81)90025-0) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study contains no experimental data. Code used to produce the numerical results is available as a Julia module on GitHub at github.com/ap-browning/SDE-Identifiability.