

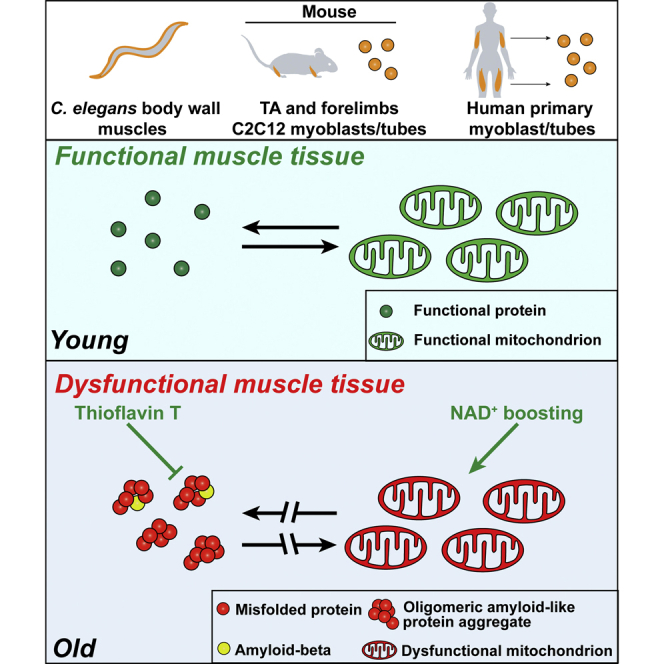
Summary
Aging is characterized by loss of proteostasis and mitochondrial homeostasis. Here, we provide bioinformatic evidence of dysregulation of mitochondrial and proteostasis pathways in muscle aging and diseases. Moreover, we show accumulation of amyloid-like deposits and mitochondrial dysfunction during natural aging in the body wall muscle of C. elegans, in human primary myotubes, and in mouse skeletal muscle, partially phenocopying inclusion body myositis (IBM). Importantly, NAD+ homeostasis is critical to control age-associated muscle amyloidosis. Treatment of either aged N2 worms, a nematode model of amyloid-beta muscle proteotoxicity, human aged myotubes, or old mice with the NAD+ boosters nicotinamide riboside (NR) and olaparib (AZD) increases mitochondrial function and muscle homeostasis while attenuating amyloid accumulation. Hence, our data reveal that age-related amyloidosis is a contributing factor to mitochondrial dysfunction and that both are features of the aging muscle that can be ameliorated by NAD+ metabolism-enhancing approaches, warranting further clinical studies.
Keywords: aging, amyloidosis, inclusion body myositis, NAD+, mitochondria, skeletal muscle, proteostasis, amyloid-beta, nicotinamide riboside, olaparib
Graphical Abstract
Highlights
-
•
Amyloidosis and mitochondrial dysfunction typify muscle aging and disease across species
-
•
NAD+ homeostasis is required to maintain proteostasis in nematodes and mammalian cells
-
•
Reducing age-associated amyloidosis improves healthspan and mitochondrial function
-
•
Late-life NAD+ boosting reduces amyloidosis and mitochondrial dysfunction during aging
Romani et al. demonstrate that aging is typified by muscle amyloidosis in a cross-species conserved manner and that late-life treatments aimed at restoring NAD+ levels and mitochondrial homeostasis also increase cellular and organismal proteostasis, therefore beneficially impacting on health- and lifespan in a pleiotropic fashion.
Introduction
Aging is a complex, multifactorial, biological process typified by a decline of many cellular functions, including mitochondrial dysfunction and collapse of proteostasis, which ultimately impact on organismal homeostasis (Labbadia and Morimoto, 2015; Riera et al., 2016). Proteostasis includes all cellular processes required to maintain homeostasis of the cellular proteome. These involve regulation of protein synthesis, folding and assembly, and degradation of proteins or aggregates (D’Amico et al., 2017).
Due to the fact that mitochondrial function and proteostasis are essential to ensure cellular homeostasis, are functionally interconnected, and decline in aging (Riera et al., 2016), it is not surprising that mitochondrial dysfunction and abnormal proteostasis are involved in chronic age-associated neuromuscular proteinopathies, such as Alzheimer’s disease (AD) (Irvine et al., 2008), and inclusion body myositis (IBM), a debilitating age-associated muscle disease (Askanas and Engel, 2011; Joshi et al., 2014). Although affecting different organs, AD and IBM are both protein aggregation diseases characterized by the accumulation of amyloid protein deposits (Benveniste et al., 2015). IBM is the most common muscle proteinopathy affecting the elderly; however, it is generally considered a rare disorder, with its overall prevalence still under debate (Needham et al., 2008). Skeletal muscle decay instead is one of the most prominent features of aging, characterized by loss of muscle mass and function (Goodpaster et al., 2006) and by a decline in mitochondrial function (Short et al., 2005). In addition, muscle aging is also typified by dysfunctional proteostasis pathways, including altered ubiquitin-proteasome system (UPS) activity (Husom et al., 2004) and defective autophagy (Wohlgemuth et al., 2010). Currently, the mechanism underlying the collapse of proteostasis in the aging muscle is not fully elucidated, and it is furthermore unclear whether amyloid deposition, a hallmark of IBM, is also at play in the aging muscle.
Here, we report that, during natural aging, muscle tissues accumulate amyloid-like deposits, a process which is evolutionary conserved in C. elegans, in mouse and human muscle cells and tissues, with molecular features recapitulating some aspects of IBM. Moreover, we also discovered the reversible nature of these deposits, which can be reduced by interventions aimed at restoring mitochondrial homeostasis, such as by enhancing nicotinamide adenine dinucleotide (NAD+) metabolism, even at the onset of aging. Importantly, we show that reduction of the accumulation of amyloid-like deposits in aging is sufficient to improve muscle mitochondrial homeostasis.
Results
Gene signatures of proteostasis and mitochondrial function are altered in muscle aging and diseases
To analyze whether transcriptional signatures associated with protein homeostasis and aggregation and with mitochondria are altered in muscle during aging or diseases, we analyzed transcriptome data from a collection of human muscle datasets of aging; genetic muscle diseases, such as muscle dystrophies; and muscle proteinopathies, such as IBM, polymyositis, and amyotrophic lateral sclerosis (ALS) (Table S1). Gene sets related to mitochondrial respiration and tricarboxylic acid (TCA) cycle were markedly downregulated in the majority of the aging and disease conditions (Figure 1A; Table S1), in line with the consensus in the field (Joshi et al., 2014; Ryu et al., 2016; Short et al., 2005). Conversely, gene sets related to protein degradation, such as the UPS, and chaperone-mediated protein folding were overall upregulated across the different muscle diseases and in aging (Figure 1A). Interestingly, amyloidosis and protein-misfolding-related gene sets were among the most consistently induced in all the muscle aging and disease conditions analyzed (Figure 1A). This indicates a high similarity between aging- and disease-associated alterations of proteostasis pathways, as highlighted in the volcano plots of selected IBM and aging datasets (Figures 1B and 1C; Table S2).
Figure 1.

Mitochondrial and proteostasis transcript signatures are perturbed in aging and muscle disease
(A) Heatmap analysis of expression datasets from human muscles during aging and a panel of different muscle diseases. The mitochondrial category of gene sets includes sets related to mitochondrial respiration and processes, although proteostasis includes gene sets related to the UPS, amyloid-related processes, and aggregation. See Table S1.
(B and C) Volcano plot representations of an IBM (B; GSE39454, control, n = 5; IBM, n = 10) and a human muscle aging (C; GSE9676, young, n = 14; old, n = 16) dataset, with the mitochondrial and proteostasis gene sets highlighted in green and blue, respectively. See Table S2.
(D) Heatmap of correlations of mitochondrial and proteostasis gene sets with age in the GTEx skeletal muscle expression datasets (male, n = 316; female, n = 175). The datasets used for this analysis are listed in Table S3.
(E) Heatmap analysis of selected gene expression datasets from mouse muscles during aging and selected muscle diseases. The datasets used for this analysis are listed in Table S1.
(F) Module-module association analysis of amyloid-beta formation module (GO:0034205) in human and mouse, using 12 and 11 muscle datasets, respectively (Li et al., 2019). The threshold of significant module-module connection is indicated by the red dashed line. MMAS, module-module association score.
We then used the human Genotype-Tissue Expression (GTEx) muscle datasets (Figure 1D; Table S3) to confirm our results generated in disease- and age-related muscle expression datasets. We separated the GTEx data based on gender and then the age of the subjects was correlated against the expression of genes. We then determined the gene sets correlating with age by using gene set enrichment analysis (GSEA) on the resulting correlation coefficients for all genes (Sergushichev, 2016). Mitochondrial and proteostasis signatures were also altered in an opposite fashion in GTEx. In agreement with the human data, we observed repression of mitochondrial pathways, mirrored by the induction of protein folding and degradation but also of aggregation and amyloid formation and regulation, in muscle expression data of mouse models of aging, proteinopathies, and dystrophy (Figure 1E; Table S1). Finally, module-module association analysis performed on 12 human muscle datasets using the genetic toolkit GeneBridge (Li et al., 2019) indicated that the amyloid-beta (Aβ) formation module has strong negative associations with respiratory electron transport and mitochondrial translation modules (Figure 1F, top), further pointing at an opposite link between mitochondrial function and amyloid pathways. Module-module association in the mouse, using 11 mouse muscle datasets, also showed similar negative associations (Figure 1F, bottom).
These analyses hence suggest that there is an overall decline of mitochondrial function paired with perturbed cellular proteostasis in skeletal muscle in established proteinopathies, such as IBM, in aging, as well as in other chronic muscle conditions, including muscular dystrophies. These shared alterations, suggestive of increased protein misfolding and amyloidosis hence may represent a major common hallmark of muscle aging and disease.
Muscle aging is characterized by accumulation of amyloid-like deposits across species
Similarly to what is observed in brain tissues affected by AD, IBM is characterized by the accumulation of well-known aggregation-prone proteins, such as Aβ peptides (Schmidt et al., 2008). One common feature of aggregation-prone proteins or peptides is their propensity to form oligomeric aggregates, which share a similar conformational status typical of amyloid aggregates (Kayed et al., 2003; Sontag et al., 2012). However, whether amyloid-based protein deposits might occur in muscle also during physiological aging across different species is yet to be defined.
Based on these premises and our bioinformatic analysis pointing to the induction of amyloid-related pathways in aging and disease (Figures 1A–1E), we explored whether aging indeed correlated with the accumulation of amyloid-like aggregates in the muscle. First, we analyzed primary myoblasts obtained from 2 healthy young (18 and 25 years old) and 2 aged donors (73 and 82 years old) and from 2 IBM patients (61 years, heterozygote for a VCP mutation found in another IBM patient, Watts et al., 2004, and 73 years, sporadic case) for the presence of these aggregates, using Proteostat-based stainings (Shen et al., 2011; Figures 2A and S1A). The Proteostat dye is reactive toward intracellular aggresomes and related inclusion bodies as well as amyloid-like aggregates (Laor et al., 2019; Navarro and Ventura, 2014). Cells from aged donors showed a marked enrichment for the Proteostat signal compared to young donors (Figures 2A and S1A). These results were corroborated by the increased Proteostat signal in primary cells from IBM patients (Figures 2A and S1A). This observation is in line with our bioinformatics analysis which also indicate the induction of inclusion-bodies-related gene sets (Figures 1A, 1D, and 1E). Accordingly, protein aggregates were also detected in the murine muscle cell line C2C12, when overexpressing the amyloidogenic Swedish mutation (K595N/M596L) of human amyloid precursor protein (APPSwe) (Sorrentino et al., 2017; Zheng et al., 2011; Figures 2B and S1B–S1D), along with an altered mitochondrial homeostasis typified by reduced mitochondrial membrane potential (Figure 2C) and increased presence of circular mitochondria, suggestive of altered mitochondrial dynamics (Kowaltowski et al., 2019), and by impaired mitochondrial respiration (Figures S1E and S1F). We further validated our observations using the A11 anti-oligomer antibody, which is reactive toward protein aggregates of amyloidogenic nature (conformational epitope) and can be easily implemented in dot blotting assays (Condello et al., 2015; Upadhaya et al., 2012). APPSwe-expressing C2C12 accumulated amyloid deposits (2.06 relative signal compared with empty vector cells), in spite of an equal total protein amount (Figures S2A and S2B; Table S4), although negative control dot blots, performed in the presence only of the secondary antibody, did not detect aspecific signals (Figure S2C), confirming the presence of amyloid-like deposits in the APPSwe-expressing myotubes. To validate our hypothesis also in vivo, we tested muscle tissues from young (3 months) and old (2 years) C57BL/6J mice for presence of amyloid-like aggregates, via A11-immunostaining of tibialis anterioris (TA) (Figures 2D and S1G). Aged muscle displayed a marked accumulation of amyloid oligomers, and importantly, A11-positive aggregates were not detected in interstitial space (Figure S1G), in line with intracellular presence of oligomeric aggregates seen also in IBM patients (Askanas and Engel, 2011). Additionally, we also investigated protein lysates from forelimb muscles (Figures S2D–S2F; Table S4) and TA muscle of young (11 weeks), adult (30 weeks), and old (2 years or 104 weeks) mice (Figures S2G and S2H; Table S4). Both muscle groups revealed a specific, strong enrichment for the A11 signal in the old animals (104-week-old animals compared with 11-week-old mice had 1.39 increase in A11 signal in TA muscle and 6.08 increase in forelimbs). Altogether, these findings provide evidence for the occurrence of amyloid-like deposits in the aging mammalian muscle.
Figure 2.

Accumulation of amyloid-like deposits in aging muscle across species
(A and B) Representative confocal images of primary human muscle cells from young, aged, and IBM donors (scale bar, 50 μm; A; representative images of n = 2 individuals per group) or of control and APPSwe-expressing C2C12 myotubes (scale bar, 10 μm) stained with the Proteostat fluorescent dye (B; n = 3 biological replicates per group).
(C) Microscopy images of tetramethylrhodamine methyl ester (TMRM) staining of C2C12 ev, C2C12 ev treated with trifluoromethoxy carbonylcyanide phenylhydrazonen (FCCP) (10 μM; internal control for membrane depolarization), and C2C12 APPSwe (C2C12 ev + Veh, n = 17; C2C12 ev + FCCP, n = 5; C2C12 APPSwe + Veh, n = 20 biological replicates) and relative quantification. Scale bar, 200 μm. ∗∗∗∗p < 0.0001.
(D) Representative images and corresponding quantification of immunostainings of A11-positive deposits in tibialis anterior (TA) muscles of young (3 months) or aged (24 months) male C57BL/6J mice (n = 12 animals). Scale bar, 50 μm. ∗∗∗p < 0.001 (relative % area).
(E) Dot blot of protein lysates from young (day 1) and aged N2 worms (day 11; n = 3 biological replicates per group) and from GMC101 worms, fed with bacteria containing either an empty vector (e.v.) or the atfs-1 RNAi. Dot blots were stained for the presence of oligomers using the A11 antibody. Relative quantification of the blot intensities is reported in Table S4.
(F) Confocal images of TMRM staining of young (day 1) and aged (day 11) N2 worms (day 1, n = 8; day 11, n = 5 worms) and relative quantification. Scale bar, 100 μm. ∗∗∗p < 0.001.
(G and H) Loss of healthy mitochondrial morphology in SJ4103 worms (G) and muscle fiber structural decay in the RAW1596 (myo-3p::GFP) C. elegans strain (H) between day 1 and day 11 of aging.
(I) A11 dot blotting of N2 (day 6) worms treated with pnc-1 RNAi (n = 2 biological replicates per group).
Relative quantification of the blot intensities is reported in Table S4. See also Figures S1 and S2. For all the individual p values, see Table S6 (Excel data source Figure 2). Values in the figure are mean ± S.E.M.
Recently, the A11 antibody was reported to detect aging-associated aggregates also in C. elegans muscles (Alavez et al., 2011). We therefore performed A11 dot blot analyses on whole-worm protein lysates from adult (day 1) and old (day 11) wild-type N2 worms (as summarized in Figure S2I). Lysates from aged N2 showed a marked A11 enrichment compared to young worms (Figure 2E; 6.08 relative signal compared with young worms), and this signal was specifically dependent on the presence of the A11 antibody (Figures S2J and S2K; Table S4). In addition, we performed A11 dot blotting assay on the inducible muscle Aβ proteotoxicity model, GMC101 (McColl et al., 2012; day 1), whose proteostasis is hampered by the impairment of mitochondrial homeostasis induced by the RNAi depletion of the mitochondrial unfolded protein response (UPRmt) master regulator atfs-1 (Sorrentino et al., 2017; Figure 2E). The amyloid signal, already detectable in GMC101 worms in basal conditions (Figure 2E: ev), was further enhanced by atfs-1 silencing. Aging in N2 worms also resulted in reduced mitochondrial membrane potential (Figure 2F); decreased levels of mitochondrial proteins, including the UPRmt effector, HSP-6, and certain oxidative phosphorylation (OXPHOS) components (Figure S2L); alteration of mitochondrial networks, with the accumulation of large and circular mitochondria (Figure 2G; D’Amico et al., 2019); and in the decay of muscle structural integrity (Figure 2H).
Aging is also known to be linked to a decline in NAD+ levels in muscle tissue in lower organisms and mammals (Gomes et al., 2013; Mouchiroud et al., 2013). In line with this premise and to investigate whether NAD+ homeostasis is required to maintain proteostasis, we fed C. elegans with bacteria expressing an RNAi targeting pnc-1, a key enzyme in NAD+ salvage in the nematode muscle (Vrablik et al., 2009, 2011). Genetic attenuation of pnc-1 function and NAD+ salvage exacerbated the accumulation of amyloid-like aggregates (1.98 relative signal compared with ev worms; Figures 2I and S2M). Moreover, addition of paraquat, an approach that was shown to deplete NAD+ levels and to reduce lifespan in C. elegans (Mouchiroud et al., 2013), also accelerated the loss of proteostasis in day 4 adult worms (Figures S2N and S2O). Similarly, pharmacological blocking NAD+ salvage using the nicotinamide phosphoribosyltransferase (NAMPT) inhibitor, FK866, in APPSwe-expressing C2C12 myoblast (Hasmann and Schemainda, 2003; Oakey et al., 2019) also robustly induced protein aggregation in these amyloid-producing cells (Figure S2P), in line with that observed in nematodes. Collectively, these data confirm that the processes of amyloidosis and aging-associated loss of proteostasis are evolutionary conserved and directly linked to NAD+ homeostasis. These data furthermore suggest that these processes could be beneficially influenced by mitochondrial or NAD+-modulating interventions (Figures 2E–2I and S2M–S2P).
Late-life treatment with NAD+ boosters attenuates amyloid formation and aging phenotypes in C. elegans
Given the reversal of mitochondrial dysfunction by life-extending metabolic interventions during aging (Houtkooper et al., 2013; Mouchiroud et al., 2013) and proteotoxic damage (Schöndorf et al., 2018; Sorrentino et al., 2017), the reduced Aβ aggregation and functional improvement in the context of AD following NAD+ replenishment (Gong et al., 2013; Sorrentino et al., 2017), and the increased amyloid-like deposition following NAD+ depletion (Figure 2I), we then tested the impact of the well-known NAD+ boosters nicotinamide riboside (NR) and the poly(ADP-ribose)polymerase (PARP) inhibitor olaparib (AZD) on aging-associated amyloid formation in C. elegans. Rather than starting treatment from the embryonic stage, as commonly performed (Fang et al., 2014; Mouchiroud et al., 2013; Ryu et al., 2016), we only provided the compounds in late adulthood, allowing to bypass all the early effects due to the impact of NAD+ boosting on development and to only investigate aging-related events (Figure S3A). Treating N2 worms with NR (1 mM) starting at day 4 of adulthood (Figure S3A) induced the expression of the mitochondrial stress response (MSR) signature genes (Sorrentino et al., 2017) in day 11 old worms (Figure 3A), similar to what we previously observed in N2 or in the Aβ aggregation strain GMC101 treated from the egg stage (Sorrentino et al., 2017). Additionally, late-life treatment with either NR (1 mM) or olaparib (AZD; 300 nM) had a profound effect on proteostasis in aged worms, with amyloid-like deposits levels being reduced by both compounds (0.33 and 0.08 relative signal, respectively, compared with untreated worms; Figures 3B, S3B, and S3C; Table S4). Importantly, the NR-mediated reduction of A11-positive aggregates deposition required both atfs-1 (Nargund et al., 2012, 2015) and dct-1 (Palikaras et al., 2015; Figure 3B). This confirmed the key contribution of both the UPRmt and mitophagy pathways of the MSR to cellular proteostasis in line with our previous work on Aβ proteotoxicity (Sorrentino et al., 2017). Late-life NAD+ boosting interventions also restored mitochondrial membrane potential and muscle mitochondrial morphology in aged animals (Figures 3C, 3D, S3D, and S3E) and increased mitochondrial DNA content (Figure S3F), indicative of increased mitochondrial biogenesis. This furthermore improved fitness, measured as spontaneous movement (Mouchiroud et al., 2016; Figures 3E and S3G), muscle integrity (Figures 3F and S3H), and percentage of paralysis and death at day 18 (Figure S3I).
Figure 3.

Boosting NAD+ levels late in life reduces amyloid-like deposits and improves mitochondrial function and fitness in aged C. elegans
(A) Transcript analysis of the mitochondrial stress response (MSR) signature (n = 5 biological replicates) in aged N2 worms treated with nicotinamide riboside (NR) (1 mM) from day 4 until day 11 of adulthood.
(B) A11 dot blotting of aged (day 11) N2 worms treated as indicated in Figure S3A, with NR (1 mM) and fed with either ev, atfs-1, or dct-1 RNAi (n = 3 biological replicates per group). Relative quantification of the blot intensities is reported in Table S4.
(C) Confocal images of TMRM staining of aged (day 11) N2 worms treated as indicated in Figure S3A, with NR (1 mM; vehicle, n = 11; NR, n = 14 worms) and relative quantification. Scale bar, 100 μm.
(D) Representative confocal images of mitochondrial networks and corresponding morphology analyses, including mitochondria outline and circularity assessment (in which 1 represents a perfect circle and 0 a line) in young (day 1) and aged (day 11) SJ4103 (myo-3p::mt-GFP) worms treated with NR (1 mM) as in (A) (n = 20 per group). Scale bar, 10 μm.
(E) Spontaneous mobility and average mobility of N2 worms treated with vehicle or NR (1 mM) as in (A) (vehicle, n = 55; NR, n = 49 worms). Overall differences between conditions were assessed by two-way ANOVA (interaction and average mobility); differences between conditions at individual time points were assessed using post hoc Sidak’s multiple comparison test.
(F) Confocal images of GFP-labeled muscle fibers in young (day 1) and aged (day 11) RAW1596 (myo-3p::GFP) worms treated with NR (1 mM) as in (A) (n = 20 per group). Scale bar, 10 μm. See STAR methods for further details.
Values in the figure are mean ± SEM. ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001. Differences for two groups were assessed using two-tailed t tests (95% confidence interval) in (A), (C), and (D). All experiments were performed independently at least twice. AU, arbitrary units. See also Figures S2G–S2I and S3. For all the individual p values, see the Table S6 (Excel data source Figure 3).
Given the beneficial effects on proteostasis observed following boosting of mitochondrial stress responses and function, we decided to test also whether prevention of amyloid-like oligomer accumulation could have a favorable impact on muscle and mitochondrial homeostasis. To test this, we took advantage of the amyloid-binding compound thioflavin T (ThT); this dye, used to detect protein aggregates, was previously shown to reduce proteotoxicity in C. elegans during aging and in models of human proteotoxic disease (Alavez et al., 2011). The reduction in protein aggregates after ThT was concomitant with a preservation of fitness in nematodes, which is also in line with our results using NAD+ boosters, showing a simultaneous reduction of amyloid formation and increased fitness in old treated worms (Figures 3B–3F and S3B–S3H). N2 worms treated with ThT displayed a marked reduction of A11-positive protein aggregates (Figure 4A), in agreement with previous data (Alavez et al., 2011). Importantly, we also observed that ThT treatment robustly promoted mitochondrial homeostasis in aging, as shown by higher mitochondrial membrane potential (Figure 4B), increased mitochondrial content (Figure 4C), and normalized muscle mitochondrial dynamics (Figure 4D), to a similar extent to what was observed following administration of NAD+ boosters (Figures 3C, 3D, and S3D–S3F). Collectively, these data indicate that age-associated, amyloid-like deposits likely contribute to the mitochondrial dysfunction observed in aged nematodes muscle tissue and that reducing amyloid formation can be advantageous for mitochondrial homeostasis too (Figure 4).
Figure 4.

Thioflavin T prevents amyloid-like accumulation and improves mitochondrial homeostasis in aging
(A) A11 dot blotting of aged (day 11) N2 worms treated with ThT (50 μM; n = 3 biological replicates per group). Relative quantification of the blot intensities is reported in Table S4.
(B) Confocal images of TMRM staining of aged (day 11) N2 worms treated with ThT (50 μM; vehicle, n = 11; ThT, n = 11 worms) and relative quantification. Scale bar, 100 μm.
(C) mitochondrial +DNA/nuclear DNA ratio in aged (day 11) N2 worms treated with ThT (5 or 50 μM; n = 12 worms per group).
(D) Representative confocal images of mitochondrial networks and corresponding morphology analyses, including mitochondria outline and circularity assessment (in which 1 represents a perfect circle and 0 a line), in aged (day 11) SJ4103 (myo-3p::mt-GFP) worms treated with ThT (5 μM; vehicle, n = 9; ThT, n = 8 worms). Scale bar, 10 μm.
Values in the figure are mean ± SEM. ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. For all the individual p values, see the Table S6 (Excel data source Figure 4).
Finally, we verified whether such late-life NAD+ boosting interventions impact mitochondrial and protein homeostasis in a corrective, rather than preventive, fashion in a severe model of inducible amyloidosis in the worm. To that end, we treated the GMC101 strain during adulthood (starting from day 1) with NR and AZD only after the induction of Aβ aggregation, achieved by shifting these worms from 20°C to 25°C at the L4 stage (McColl et al., 2012; Sorrentino et al., 2017; Figure S4A). Onset of amyloidosis in the GMC101 strain reduced worm fitness (Figure S4B) and mitochondrial membrane potential (Figure S4C) in line with previous work (Sorrentino et al., 2017). Moreover, amyloid aggregates accumulation in the AUW15 strain, generated from the cross of GMC101 with the SJ4103 strain expressing mitochondrial GFP in the body wall muscle, also resulted in a marked alteration of mitochondrial morphology when cultured at amyloid-promoting temperature (Figure S4D) or when compared to the strain AUW14, derived from a cross between SJ4103 and the control strain CL2122 (Sorrentino et al., 2017; Figures 5B, vehicle, and S4F, vehicle). As observed for aged N2 worms, both NR and AZD normalized the mitochondrial membrane potential (Figures 5A and S4E) and dynamics in the GMC101 worms (Figures 5B and S4F) but also reduced protein aggregation (0.18 [NR] and 0.1 [AZD] relative signal compared with untreated worms; Figure 5C; Table S4) and muscle damage (Figure 5D), whereas they improved fitness and decreased death at day 8 of adulthood (Figures 5E, S4G, and S4H).
Figure 5.

NAD+ boosting after the induction of proteotoxic stress in muscle of Aβ-expressing worms reduces amyloid deposits and improves mitochondria and healthspan
(A) Confocal images of TMRM staining of aged (day 11) N2 worms treated ThT (50 μM; vehicle, n = 5; NR, n = 8 worms) and relative quantification. Scale bar, 100 μm.
(B) Representative confocal images of mitochondrial networks at day 2 and corresponding morphology analyses, including mitochondria outline and circularity assessment (in which 1 represents a perfect circle and 0 a line), in control AUW14 and Aβ1–42-expressing AUW15 worms treated with NR (1 mM) and AZD (300 nM; n = 20 per group). Scale bar, 10 μm.
(C) A11 dot blotting of day 2 GMC101 worms treated as indicated in Figure S4A, with NR (1 mM) and AZD (300 nM; n = 3 biological replicates per group). Relative quantification of the blot intensities is reported in Table S4.
(D) Confocal images of phalloidin-stained muscle fibers on day 2 in CL2122 (control) and GMC101 worms treated as indicated in Figure S4A (n = 8 worms per group). Scale bar, 10 μm.
(E) Spontaneous mobility and average mobility of GMC101 treated with vehicle, NR (1 mM), or AZD (300 nM) as in Figure S4A (vehicle, n = 38; NR, n = 37 worms; AZD, n = 36 worms). Overall differences between conditions were assessed by two-way ANOVA (average mobility); differences between conditions at individual time points were assessed by post hoc Sidak’s multiple comparison test.
See STAR methods for further details. Values in the figure are mean ± SEM. ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001. Differences for two groups were assessed using two-tailed t tests (95% confidence interval) in (A), (B) (individual time points), (F), and (G). All experiments were performed independently at least twice. For all the individual p values, see the Table S6 (Excel data source Figure 5).
NAD+ boosting reduces amyloid formation and improves mitochondrial parameters during aging in mammals
To translate our findings in C. elegans to mammals, we also treated primary human myotubes obtained from aged subjects and IBM patients with NR and AZD and assessed the presence of amyloid-like proteins by Proteostat staining (Figures 6A, 6B, S5A, and S5B). Importantly, both treatments decreased the amount of Proteostat-positive signal in cells derived from aged or IBM donors (Figures 6A, 6B, S5A, and S5B). Protein aggregates were also reduced in C2C12-APPSwe-expressing myotubes following these interventions, both when scored by Proteostat-based staining (Figures 6C and S5C) and A11 dot blotting (0.4 [NR] and 0.15 [AZD] relative signal compared with untreated cells; Figures S5D and S5E; Table S4). Importantly, co-treating cells with a global integrative stress response (ISR) inhibitor (ISRIB) (Sidrauski et al., 2015), attenuated the reduction in aggregates seen upon NAD+ boosting (Figure S5F). This suggests the involvement of the ISR in resolving amyloid proteotoxic stress in mammalian cells, supporting our worm data (Figure 3B). Reduction of amyloid-like deposits was mirrored by an improvement of the mitochondrial membrane potential (Figure 6D), increased mitochondrial respiration (Figure S5G), and a more reticular mitochondrial network (Figure S5H). Together, the data in primary cells and C2C12 cells with Aβ-aggregates confirm that NAD+ boosting, in addition to its established impact on mitochondrial function, also improves proteostasis and attenuates amyloid formation in mammalian cells.
Figure 6.

Boosting NAD+ levels reduces amyloid-like deposits in muscle cells and in vivo in aged mice
(A–C) Representative confocal images of primary human muscle cells from an aged and an IBM donor (scale bar, 50 μm; A and B) or APPSwe-expressing C2C12 myotubes (scale bar, 10 μm; C) treated with NR (3 mM) and stained with the Proteostat fluorescent dye. See also Figures S6A–S6C for quantification.
(D) Microscopy images of TMRM staining of C2C12 APPSwe treated with NR (3 mM; n = 10) and relative quantification. Scale bar, 200 μm.
(E) Representative images and corresponding quantification of immunostainings of A11-positive protein deposits in TA muscles of young or aged male C57BL/6J mice, fed for 8 weeks with chow diet (CD) or chow diet supplemented with NR (400 mg/kg/day; young, n = 12 animals; young+NR, n = 7 animals; old, n = 12 animals; old+NR, n = 11 animals). Scale bar, 50 μm. ∗∗∗p < 0.001 (relative % area).
(F) MSR transcript analysis of forelimbs muscles of aged male mice C57BL/6J following NR treatment (n = 5 animals per group).
(G) Immunoblot of the OXPHOS proteins, MTCO1 and SDHB, from forelimb muscles of the animals in (E) and Figure S4J (n = 3 animals per group).
See STAR methods for further details. Values in the figure are mean ± SEM. ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. Differences for two groups were assessed using two-tailed t tests (95% confidence interval). For all the individual p values, see the Table S6 (Excel data source Figure 6).
Given that NR treatment in vivo beneficially impacts on mitochondrial and muscle homeostasis (including running performance), healthspan, and lifespan in adult (Cantó et al., 2012) and aged mice (Zhang et al., 2016) and reduces brain amyloid plaques in AD mice (Sorrentino et al., 2017), we assessed the effects of NR on age-associated muscle amyloid-like deposits in vivo. Dietary NR supplementation (8 weeks) consistently reduced protein deposits in TA and forelimbs muscles of old C57BL/6J mice as evidenced by A11 immunohistochemistry (Figure 6E) and dot blotting (0.45 relative signal compared to untreated animals; Figure S5I; Table S4). In agreement with our previous work (Sorrentino et al., 2017), the improved proteostasis after NR treatment was reflected by the increased expression of MSR signature genes in both young and old forelimbs tissues (Figures 6F and S5J). The expression of the OXPHOS proteins, MTCO1 and SDHB, which were decreased in aged forelimbs, was restored by NR treatment to levels similar to the ones observed in young animals (Figure 6G).
To better characterize the composition of muscular age-associated amyloid-like aggregates, and given the fact that APP processing-related pathways appeared to be enriched in aging in skeletal muscle in our bioinformatic analysis (Figure 1), we verified whether APP or peptides derived from APP processing are part of the A11-positive deposits in vivo. We took advantage of the anti-Aβ 4G8 antibody, reactive also to murine endogenous APP, its cleavage products, and to Aβ (Teich et al., 2013), and stained 2 types of muscles. Similar to that observed with the A11 antibody, histology analysis of TA muscles revealed increased signals for 4G8 reactive proteins in aged animals, compared to young, which were reduced following NR treatment (Figure S6A). Accordingly, accumulation of the 4G8-dependent signal was also reduced in lysates from the forelimbs from aged animals treated with NR (Figure S6B). Although we cannot conclude which other proteins are part of these protein accumulations in aging, our data suggest that these amyloid-like deposits accumulating in aging muscles also contain APP, APP byproducts, and possibly Aβ and show that NR treatment decreases the amount of these A11- and 4G8-positive aggregates.
Discussion
Aging is characterized by a collapse of cellular proteostasis and mitochondrial homeostasis, which in the neuromuscular system is often associated with degenerative disorders, such as AD and IBM, characterized by detrimental protein aggregation (Askanas and Engel, 2011; Joshi et al., 2014; Labbadia and Morimoto, 2015; Riera et al., 2016). Here, we provided bioinformatic and experimental evidence for the accumulation of amyloid-like deposits and mitochondrial dysfunction during muscle aging across different species, ranging from C. elegans to human primary myotubes and mouse skeletal muscle. Interestingly, muscle aging phenocopies some key molecular features of IBM. These findings therefore indicate that muscle aging involves evolutionary conserved processes whose alteration results not only in mitochondrial dysfunction but also in the onset of amyloidosis across the tested species. Importantly, our data indicate that prevention of amyloidosis in aging is sufficient to correct age-associated mitochondrial dysfunction, underlining the contribution of these protein aggregates to the general process of muscle aging (Figure 4). Although it is known that protein aggregation occurs not only in these disorders but can also be present during healthy aging in several species (Ayyadevara et al., 2016; Basisty et al., 2018; Walther et al., 2015), the major types of protein aggregates occurring in physiological aging in different organs and tissues are yet to be defined. Recent work in C. elegans has shown that certain proteins form age-dependent insoluble aggregates with amyloid-like structure resembling that of protein aggregates observed in amyloidogenic diseases (Huang et al., 2019); these observations are in line with our A11 dot blotting data in whole protein lysates from old N2 worms (Figure 2D). Although the identity of the proteins forming the A11-positive accumulations we observed both in aged worms and mammals in muscle tissues remains to be determined, our data in mouse using the anti-Aβ 4G8 antibody suggest that APP, APP byproducts, and possibly Aβ could compose in part the observed aggregates (Figures S6A and S6B). Interestingly, marked accumulation of 4G8-positive deposits was also detected in brain tissues from aged (24 months) animals and again decreased following NR supplementation (Figure S6C). This last observation is in line with recent work showing increased Proteostat dye reactivity in different brain regions of 24-month-old mice (Vonk et al., 2020); therefore, we speculate that similar amyloid-like protein aggregates to those we observed in aged muscles may characterize aging across different tissues. This knowledge will aid to identify novel aging biomarkers and potentially to better understand the aging process.
Our bioinformatic analyses also showed that the mitochondrial and proteostasis alterations observed in aging and IBM also occur in other muscle diseases, such as in various types of myositis and dystrophies (Figure 1A). Although these conditions are known to present a profound mitochondrial dysfunction (Alhatou et al., 2004; Gong et al., 2013; Temiz et al., 2009), to the best of our knowledge, they have not been investigated in depth for the occurrence of alterations in proteostasis that underpin protein aggregation or amyloidosis.
Mitochondrial dysfunction in aging and disease, and Aβ aggregation in the brain during AD, can be reversed by NAD+ replenishment approaches, such as dietary supplementation with NR and treatment with PARP inhibitors, such as olaparib (AZD) (Gong et al., 2013; Mouchiroud et al., 2013; Sorrentino et al., 2017). Importantly, our current work now shows that direct targeting and reduction of amyloid aggregates using ThT in aged worms (Figure 4) restores mitochondrial homeostasis. In addition, we also have demonstrated a critical role of NAD+ homeostasis to control amyloid aggregates levels (Figures 2I and S2M–S2P). Conversely, NAD+ boosting interventions also improve proteostasis in aging muscle, promoting the reduction of age-associated amyloid-like deposits. Additionally, and in line with our previous work (Sorrentino et al., 2017), we show that the reversibility of these aggregates in C. elegans requires the MSR pathways of UPRmt and mitophagy and, in Aβ-producing C2C12 cells, a functional ISR (Figures 3B and S5F). Of note, NR and olaparib treatment decreased Proteostat levels also in primary cells from patients with IBM (Figure 6B), an established example of muscle amyloidosis pathology (Irvine et al., 2008). The results in these cells are also in line with the data, obtained using A11 antibody, from the GMC101 worms (Figure 5), which express the human Aβ isoform 1–42 mostly in the muscle and develop age progressive paralysis, muscle degeneration, and amyloid deposition (McColl et al., 2012). This worm strain should hence be considered as a general model of Aβ disease that mimics the proteotoxic phenotypes of both AD and IBM. Therefore, our findings in IBM cells and GMC101 worms, supported also by the compound-dependent effects observed in APPSwe-expressing C2C12 (Figure 6C), strongly suggest a therapeutic potential for NAD+ enhancing strategies in treating or delaying the progression of IBM and other amyloidogenic proteinopathies, such as AD (Sorrentino et al., 2017).
Collectively, our results in C. elegans, mice, and humans support the notion that natural aging is typified by muscle amyloidosis and, very importantly, that late-life treatments aimed at restoring metabolic and mitochondrial homeostasis also increase cellular and organismal proteostasis, therefore beneficially impacting on health- and lifespan in a more pleiotropic fashion than that reported so far.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HSP-6 | Abmart | Cat# X-P11141-N, RRID:AB_2885192 |
| ATP-5 | Abcam | Cat# ab110413, RRID:AB_2629281 |
| UCR-1 | Abcam | Cat# ab110413, RRID:AB_2629281 |
| CTC-1 | Abcam | Cat# ab110413, RRID:AB_2629281 |
| GAPDH (14C10) | Cell Signaling | Cat# 2118, RRID:AB_561053 |
| APP | Abcam | Cat# ab32136, RRID:AB_2289606 |
| SDHB | Abcam | Cat# ab110413, RRID:AB_2629281 |
| MTCO1 | Abcam | Cat# ab14705, RRID:AB_2084810 |
| 4G8 | Biolegend | Cat# 800701, RRID:AB_2564633 |
| anti-Oligomer (A11) | Thermo Scientific | Cat# AHB0052, RRID:AB_2536236 |
| Laminin | Sigma | Cat# L9393, RRID:AB_477163 |
| Bacterial and virus strains | ||
| atfs-1 RNAi | Vidal library | 11070-E7; ZC376.7 |
| dct-1 RNAi | Ahringer library | X-4G11; C14F5.1 |
| Chemicals, peptides, and recombinant proteins | ||
| Nicotinamide riboside triflate (NR) | Novalix | http://www.novalix-pharma.com/ |
| Olaparib (AZD2281) | Glentham Life Sciences | 763113-22-0 |
| Thioflavin T (ThT) | Sigma Aldrich | T3516-5G |
| Rhodamine Phalloidin | Invitrogen | 219920-04-4 |
| TMRM | Invitrogen | T668 |
| DAPI | Invitrogen | D1306 |
| DAKO mounting medium | DAKO | S3023 |
| Hoechst | Invitrogen | C10340 |
| Paraquat | Sigma Aldrich | 856177 |
| Lipofectamine 2000 | Thermo | 11668019 |
| TRIzol | Life technologies | 15596026 |
| FK866 | Sigma Aldrich | F8557-5MG |
| Critical commercial assays | ||
| Proteostat® Aggresome Detection kit | Enzo life sciences | ENZ-51035-K100 |
| LightCycler 480 SYBR Green I Master reagent | Roche Life Science | 04887352001 |
| Experimental models: cell lines | ||
| Human primary myoblasts | Hospices Civils de Lyon | N/A |
| C2C12 ev or APPSwe | Generated in house | N/A |
| Experimental models: organisms/strains | ||
| C. elegans N2 | Caenorhabditis Genetics Center | WormBase ID: WBStrain00000001 |
| C. elegans SJ4103 (myo-3::GFP(mit)) | Caenorhabditis Genetics Center | WormBase ID: WBStrain00034069 |
| C. elegans GMC101 (unc-54p::A-beta-1-42::unc-54 3′-UTR + mtl-2p::GFP) | Caenorhabditis Genetics Center | WormBase ID: WBStrain00007866 |
| C. elegans CL2122 ((pPD30.38) unc-54(vector) + (pCL26) mtl-2::GFP) | Caenorhabditis Genetics Center | WormBase ID: WBStrain00005101 |
| C. elegans RW1596 (myo-3p::GFP) | Caenorhabditis Genetics Center | WormBase ID: WBStrain00033508 |
| C. elegans AUW15: (cross GMC101+SJ4103) | Generated in house | N/A |
| C. elegans AUW14: (cross CL2122+SJ4103) | Generated in house | N/A |
| Mouse C57BL/6JRj | Janvier Labs | https://www.janvier-labs.com/en/fiche_produit/aged_c57bl-6jrj_mouse/ |
| Oligonucleotides | ||
| Primers for qPCR analysis of C. elegans samples, see Table “list of primers used in C.elegans” | This paper | N/A |
| Primers for qPCR analysis of mouse samples, see Table “list of primers used in Mus musculus” | This paper | N/A |
| Recombinant DNA | ||
| Plasmid pCAX APPSwe/Ind | Addgene | #30145 |
| Software and algorithms | ||
| Fiji | Schindelin et al., 2012 | https://imagej.net/Fiji |
| ImageJ | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| GeneBridge | Li et al., 2019 | https://www.systems-genetics.org/mmad |
| Movement Tracker software | Mouchiroud et al., 2016 | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Johan Auwerx (admin.auwerx@epfl.ch).
Materials availability
The worm strains generated in this study are available upon request from the corresponding author.
Data and code availability
All data generated during this study are included in this published article (and its Supplemental information). Source Data for all the individual P values are provided in Table S6 - Excel data source table.
The datasets supporting the current study have been included in Tables S1, S2, S3, and S4.
The published article includes all datasets generated using GeneBridge: https://www.systems-genetics.org/mmad.
Experimental model and subject details
Animal experiments and ethical approval
Male 24 months old C57BL/6JRj mice were purchased from Janvier Labs. Mice were fed with pellets containing vehicle or NR (400 mg/kg/day) for 8 weeks. The pellets were prepared by mixing powdered chow diet (2016S, Harlan Laboratories) with water or with NR dissolved in water. Pellets were dried under a laminar flow hood for 48 h. Mice were housed in groups of two to four animals per cage and randomized to 7–8 animals per experimental group according to their body weight. All experiments were performed in compliance with all relevant ethical regulations and authorized by the local animal experimentation committee (Commission cantonale pour l’expérimentation animale) of the Canton de Vaud under license 2890.
Cell culture
The C2C12 cell line expressing the APP Swedish K670N/M671L double mutation (APPSwe) was generated in house. The pCAX APPSwe/Ind plasmid was purchased from Addgene (Plasmid #30145) and the human APPSwe/Ind gene was cloned into pLX304 lentiviral vector (Addgene, Plasmid #25890) by gateway cloning. The newly generated pLX304-APPSwe/Ind vector and lentiviral plasmid packaging mix (pCMV-dR8.2 with pCMV-VSV-G) were transfected into HEK293T cells by Lipofectamine 2000 (Thermo, #11668019). 48 hours after transfection, the cell culture medium was collected as a source of viral particles. Then C2C12 myoblasts were infected with lentivirus-containing medium with polybrene for 24 hours. Cells were selected in 5 μg/ml blasticidin (Sigma-Aldrich, #15205). The C2C12 control and APPSwe-expressing myoblast cells were cultured in high glucose DMEM (GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO), penicillin-streptomycin (1x, GIBCO) at 37°C under 5% CO2. After cells reach confluence, differentiation was initiated by replacing 10% FBS to 2% horse serum. The differentiation media were changed every two days.
Human primary myoblasts lines were purchased from “Hospices Civils de Lyon.” VCP IBM myoblasts were derived from a patient with heterozygote variation in the VCP gene: c.475C > T (p.Arg159Cys), previously associated with IBM in another study (Watts et al., 2004). The IBM diagnosis in the patients was validated by the “Hospices Civils de Lyon” through scores for inflammation, p62-positive aggregates in fibers and vacuoles, mitochondrial dysfunction, and indicative clinical signs. Cells were grown in DMEM/F-10, supplemented with 12% FBS (GIBCO) and penicillin–streptomycin (1 × , GIBCO). Cells were cultured at 37°C in a 5% CO2 atmosphere and tested for mycoplasma using Mycoprobe (CUL001B, R&D systems).
C. elegans strains and RNAi experiments
C. elegans strains were cultured at 20°C on nematode growth medium (NGM) agar plates seeded with E. coli strain OP50 unless stated otherwise. Strains used in this study were the wild-type Bristol N2, SJ4103 (myo-3::GFP(mit)), GMC101 (unc-54p::A-beta-1-42::unc-54 3′-UTR + mtl-2p::GFP) (McColl et al., 2012), CL2122 ((pPD30.38) unc-54(vector) + (pCL26) mtl-2::GFP) and RW1596 (myo-3p::GFP). Strains were provided by the Caenorhabditis Genetics Center (University of Minnesota). AUW14: (cross CL2122+SJ4103) and AUW15: (cross GMC101+SJ4103) strains were generated in house. Briefly, for the generation of these lines, SJ4103 males were generated after exposure of L4 worms to 30°C for 5.30 h, and allowed to mate with L4 hermaphrodites from CL2122 or GMC101 mutant strains. The derived progeny was selected for homozygosis of SJ4103 by the presence of GFP in body wall muscle and for homozygosis of CL2122 and GMC101 by the intestinal GFP marker for a few generations. For RNAi experiments, worms were exposed to atfs-1 and dct-1 RNAi starting from egg stage to ensure robust knock down of the investigated genes.
Method details
Bioinformatics analysis
Human or mouse gene expression data from muscle aging or various muscle diseases were obtained from GEO under the respective accession identifiers (Table S1). To identify the enriched gene sets across the different conditions, we performed gene set enrichment analysis (GSEA) (Subramanian et al., 2005) using the fgsea package (Sergushichev, 2016). Specifically, genes were ranked based on the fold change in muscle aging or muscle diseases against controls, and enriched gene sets were determined by enrichment analysis.
For the analysis using GTEx datasets (GTEx Consortium, 2013), the skeletal muscle RNA-seq data and covariates were obtained from the GTEx Portal (https://gtexportal.org) under version v7. The expression residuals after accounting for the known and hidden covariates were used for further analysis. To identify the enriched gene sets correlated with age of the sample donors, we ranked the genes based on their Pearson correlation coefficient against age and performed GSEA to determine the enriched gene sets.
The modules associated to the amyloid beta formation (GO:0034205) module were determined using muscle datasets with over 80 samples through GeneBridge at https://www.systems-genetics.org/mmad (Li et al., 2019).
Imaging and image processing
Confocal images were acquired with Zeiss LSM 700 Upright confocal microscope (Carl Zeiss AG) under non-saturating exposure conditions. The worms were immobilized with 7.5 mM solution of tetramisole hydrochloride (Sigma-Aldrich) in M9 and mounted on 6% agarose pads on glass slides. For immunostaining, cells were fixed with 1 × Formal-Fixx (Thermo Scientific) for 15 min. After 15 min permeabilization with 0.1% Triton X-100, cells were blocked in PBS supplemented with 5% FBS for 1 h and immuno-stained overnight at 4°C the anti-TOM20 antibody or stained using Proteostat. For aggregate detection with this dye, cells were fixed with 4% formaldehyde for 30 min at 21°C. Then cells were washed with PBS, followed by permeabilization for 30 min on ice using permeabilizing solution. Next, the cells were washed twice in PBS and incubated with Proteostat® dye for 30 min at 21°C. Cells were then washed with PBS, covered with glass coverslip, and observed by fluorescence microscopy. Proteostat® Aggresome Detection kit (ENZ-51035-K100) was purchased from Enzo Life Sciences, Inc and all components were prepared according to manufacturer’s instruction. For cell immunostaining procedures, the secondary antibody was coupled to an Alexa 488 fluorochrome (Thermo Scientific) and nuclei were stained with DAPI (Invitrogen, D1306). Cell slides were mounted with DAKO mounting medium (DAKO, S3023) and analyzed with a Zeiss LSM 700 confocal microscope. Image processing was performed with the Fiji software (https://imagej.nih.gov/ij; version 1.47b).
Mitochondrial circularity
To quantify mitochondrial morphology, we used an ImageJ macro publicly available for download from the ImageJ Wiki site (https://imagejdocu.tudor.lu/plugin/morphology/mitochondrial_morphology_macro_plug-in/start) and previously validated(Dagda et al., 2009). Briefly, the color channel of cells stained with TOM20 or worms expressing mito-GFP was extracted to grayscale, inverted to show mitochondria-specific fluorescence as black pixels, and thresholded to optimally resolve individual mitochondria. The macro traces mitochondrial outlines using “analyze particles.”
Gene expression analyses in C. elegans and mice
Total RNA was extracted from mouse tissues using TriPure RNA isolation reagent (Roche) according to the product manual. To analyze N2 worms at day 11 of adulthood a total of approximately 3,000 worms per condition, divided into three biological replicates, was recovered in M9 buffer from NGM plates and lysed in the TriPure RNA reagent. Each experiment was repeated twice. Total RNA was transcribed to cDNA using the QuantiTect Reverse Transcription Kit (QIAGEN). Expression of selected genes was analyzed using the LightCycler480 system (Roche) and LightCycler 480 SYBR Green I Master reagent (Roche). For worms two housekeeping genes were used to normalize the expression data, actin (act-1) and peroxisomal membrane protein 3 (pmp-3); for mice, the β2-microglobulin (B2m) gene was used as housekeeping reference.
Pharmacological treatment of C. elegans
Nicotinamide riboside triflate (NR) was custom synthesized by Novalix (http://www.novalix-pharma.com/) and dissolved in water, and used at a final concentration of 1 mM. Olaparib (AZD2281) was purchased from Sigma Aldrich dissolved in DMSO to experimental concentrations of 300 nM. Thioflavin T (ThT) was purchased from Sigma-Aldrich (reference T3516-5G) dissolved in methanol and used at final concentrations of 5 and 50 uM (provided from L4 stage). Paraquat was purchased from Sigma-Aldrich (reference 856177), dissolved in water and used at final concentration of 4mM. Compounds were added just before pouring the plates. To ensure a permanent exposure to the compound, plates were changed twice a week.
Worm phenotypic assays
Mobility
C. elegans movement analysis started from day 6 (N2) or day 1 (GMC101) of adulthood, using the Movement Tracker software (Mouchiroud et al., 2016). The experiments were repeated at least twice.
Paralysis and death score
45 to 60 worms per condition were manually scored for paralysis after poking, as described (McColl et al., 2012; Sorrentino et al., 2017). Worms that were unable to respond to any repeated stimulation were scored as dead. Results are representative of data obtained from at least two independent experiments.
TMRM staining
Worms were incubated 24h on NGM agar plates containing TMRM (5uM) added to the medium before pouring the plates. Before imaging, worms were washed 3 times with M9 and incubated 30 minutes on regular NGM agar plates in the dark.
Phalloidin staining
A population of 100 L4 worms was incubated for 24 h at 25°C. The worms were then washed in M9 and frozen in liquid nitrogen. Immediately after, worms were lyophilized using a centrifugal evaporator and permeabilized using acetone. 2 U of phalloidin (Thermo Scientific) was resuspended in 20 μl of a buffer containing: Na-phosphate pH 7.5 (final concentration 0.2 mM), MgCl2 (final concentration 1 mM), SDS (final concentration 0.004%) and dH2O to volume. The worms were incubated for 1 h in the dark and then washed twice in PBS.
Real-time PCR for mitochondrial:nuclear DNA ratio
Relative quantification of the mtDNA copy number in worms was performed by real-time PCR. Relative values for mtce-26 and act-3 were compared within each sample to generate a ratio representing the relative level of mitochondrial DNA per nuclear genome. The results obtained were confirmed with a second mitochondrial gene, nd-1. The average of at least two technical repeats was used for each biological data point. Each experiment was performed on at least ten independent biological samples.
Cell culture and treatments
Differentiated C2C12 cells were treated with 1uM AZD or 3mM NR in differentiation medium for 24 hours after 4 days differentiation. Human primary cells were treated with AZD or NR after 14 days myotubes differentiation. Then myotubes were collected or fixated for analysis.
For TMRM staining, cells were exposed to medium containing TMRM (100 nM final) and Hoechst (5ug/mL) for 30 minutes at 37°C, then washed 3 times with PBS and imaged. Proteostat staining was performed using the manufacturer protocol.
Mitochondrial oxygen consumption
Mitochondrial oxygen consumption rate (OCR) was evaluated using Seahorse analyzer (XF96, Agilent Technologies). OCR was measured using 1 μM of oligomycin to inhibit ATP synthase, 2 μM of the protonophore Carbonyl cyanide p-trifluoromethoxyphenyl hydrazone (FCCP) to uncouple mitochondrial oxidative phosphorylation, and 1 μM rotenone/antimycin to block mitochondrial respiration and determine non‐mitochondrial OCR. ATP‐linked respiration was calculated by subtracting the uncoupled (after the addition of oligomycin) from the basal OCR. Maximum respiratory capacity was determined after FCCP addition, and spare capacity was determined by subtracting basal from the FCCP‐induced OCR. All Seahorse measurements were normalized by protein quantified by Bradford assay.
Western and dot blot analysis
C.elegans were lysed by sonication with TBS buffer containing protease and phosphatase inhibitors (Roche), and analyzed by western and dot blot. The concentration of extracted protein was determined using the Bio-Rad Protein Assay. Membranes were blocked in 10% milk for 2h or overnight. Homogeneous loading was monitored by ponceau red. Antibody detection reactions for all immunoblot experiments were developed by enhanced chemiluminescence (Advansta) and imaged using the c300 imaging system (Azure Biosystems). Each immunoblot experiment was repeated at least twice using at least three biological replicates each containing approximately 1,000 worms.
C2C12 cell lysates were prepared for immunoblotting as for C.elegans. Each experiment was repeated at least twice using 3 biological replicates.
For mouse tissues, frozen forelimbs samples were lysed by mechanical homogenization with RIPA buffer containing protease and phosphatase inhibitors for western blot analysis. Lysates were prepared and analyzed by dot blot as described for worms.
Histology
TA muscles and brains were harvested from anaesthetized mice and immediately frozen in Tissue-TEK® OCT compound (PST). 8-μm cryosections were fixed with 4% paraformaldehyde. For immunostainings, heat activated antigen retrieval was performed in pH 6.0 citrate buffer for 10min at 95°C. After washing with PBS-0.1% tween 20 (PBST), the sections were blocked with 10% affinipure Fab goat anti mouse IgG (Jackson Immunoresearch) in PBS for 60min and PBST containing 2% BSA and 5% goat serum for 30min at room temperature. The primary anti-Oligomer A11 (Thermo Scientific, AHB0052) or anti-Aβ 4G8 (Biolegend, 800701) or laminin (Sigma, L9393) antibodies were then applied over night at 4°C. Subsequently, the slides were washed in PBST and incubated with appropriate secondary antibodies and labeling dyes. For immunofluorescence, secondary antibodies were coupled to Alexa 488 or Alexa-568 fluorochromes (Life technology), and nuclei were stained with DAPI (Invitrogen, D1306). After washing in PBST, tissue sections were mounted with Dako mounting medium (Dako, S3023). Images were acquired using Leica DMI 4000 (Leica Microsystems) or Olympus Slide Scanner VS120 (Olympus) at the same exposure time. Quantitative analysis of the immunofluorescence data was carried out by histogram analysis of the fluorescence intensity at each pixel across the images using ImageJ (Fiji; National Institutes of Health). Appropriate thresholding was employed to all the images of each single experiment to eliminate background signal in the images before histogram analysis. Fluorescence intensity and signal positive areas were calculated using the integrated “analyse particles” tool of the Fiji software.
Quantification and statistical analysis
No statistical methods were used to predetermine sample size. Differences between two groups were assessed using two-tailed t tests. Differences between more than two groups were assessed by using two-way ANOVA. Prism 6 (GraphPad Software, Inc.) was used for all statistical analyses. Variability in all plots and graphs is presented as the s.e.m. All p < 0.05 were considered to be significant. ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001 unless stated otherwise. All mouse experiments were performed once. Animals that showed signs of severity, predefined by the animal authorizations were euthanized. These animals, together with those who died spontaneously during the experiments, were excluded from the analysis. These criteria were established before starting the experiments. For motility, fitness, and death scoring in C. elegans, sample size was estimated based on the known variability of the assay. All experiments were done non-blinded and repeated at least twice.
Acknowledgments
We are grateful to the Caenorhabditis Genetics Center for providing worm strains and to the “Hospices Civils de Lyon” for providing samples of and information on IBM patients. H.L. is supported by the China Scholarship Council. C.-M.O. is supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education (2016R1A6A3A04010466 to C.-M.O.). T.L. is supported by FAPESP (2019/11171-7). The research project in the J.A. lab was supported by the EPFL, the European Research Council (ERC-AdG-787702), the Swiss National Science Foundation (SNSF) (310030B_160318 and 31003A_179435), the National Research Foundation of Korea (GRL 2017K1A1A2013124), the Fondation Suisse de Recherche sur les Maladies Musculaires (FSRMM), and the Fondation Marcel Levaillant.
Author contributions
M.R., V.S., and J.A. conceived and designed the project. M.R., V.S., and C.-M.O. performed the experiments. M.R. and V.S. independently replicated worm experiments. C.-M.O. and T.L. generated the C2C12 APPSwe-expressing cell line. M.R., C.-M.O., and T.L. performed the cell experiments. H.Z. performed the in vivo mouse experiments, and H.L. performed the bioinformatics analyses. M.S. provided advice. J.A., C.-M.O., and M.S. contributed to the funding of the project. M.R., V.S., and J.A. wrote the manuscript, which was edited by all co-authors.
Declaration of interests
The authors declare no competing interests.
Published: January 19, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108660.
Supplemental information
References
- Alavez S., Vantipalli M.C., Zucker D.J.S., Klang I.M., Lithgow G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature. 2011;472:226–229. doi: 10.1038/nature09873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhatou M.I., Sladky J.T., Bagasra O., Glass J.D. Mitochondrial abnormalities in dermatomyositis: characteristic pattern of neuropathology. J. Mol. Histol. 2004;35:615–619. doi: 10.1007/s10735-004-2194-6. [DOI] [PubMed] [Google Scholar]
- Askanas V., Engel W.K. Sporadic inclusion-body myositis: conformational multifactorial ageing-related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-β42 oligomers and phosphorylated tau. Presse Med. 2011;40:e219–e235. doi: 10.1016/j.lpm.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Ayyadevara S., Balasubramaniam M., Suri P., Mackintosh S.G., Tackett A.J., Sullivan D.H., Shmookler Reis R.J., Dennis R.A. Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging (Albany NY) 2016;8:3486–3497. doi: 10.18632/aging.101141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basisty N.B., Liu Y., Reynolds J., Karunadharma P.P., Dai D.-F., Fredrickson J., Beyer R.P., MacCoss M.J., Rabinovitch P.S. Stable isotope labeling reveals novel insights into ubiquitin-mediated protein aggregation with age, calorie restriction, and rapamycin treatment. J. Gerontol. A Biol. Sci. Med. Sci. 2018;73:561–570. doi: 10.1093/gerona/glx047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste O., Stenzel W., Hilton-Jones D., Sandri M., Boyer O., van Engelen B.G.M. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 2015;129:611–624. doi: 10.1007/s00401-015-1384-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C., Houtkooper R.H., Pirinen E., Youn D.Y., Oosterveer M.H., Cen Y., Fernandez-Marcos P.J., Yamamoto H., Andreux P.A., Cettour-Rose P. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C., Yuan P., Schain A., Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015;6:6176. doi: 10.1038/ncomms7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico D., Sorrentino V., Auwerx J. Cytosolic proteostasis networks of the mitochondrial stress response. Trends Biochem. Sci. 2017;42:712–725. doi: 10.1016/j.tibs.2017.05.002. [DOI] [PubMed] [Google Scholar]
- D’Amico D., Mottis A., Potenza F., Sorrentino V., Li H., Romani M., Lemos V., Schoonjans K., Zamboni N., Knott G. The RNA-binding protein PUM2 impairs mitochondrial dynamics and mitophagy during aging. Mol. Cell. 2019;73:775–787.e10. doi: 10.1016/j.molcel.2018.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda R.K., Cherra S.J., 3rd, Kulich S.M., Tandon A., Park D., Chu C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang E.F., Scheibye-Knudsen M., Brace L.E., Kassahun H., SenGupta T., Nilsen H., Mitchell J.R., Croteau D.L., Bohr V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A.P., Price N.L., Ling A.J.Y., Moslehi J.J., Montgomery M.K., Rajman L., White J.P., Teodoro J.S., Wrann C.D., Hubbard B.P. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B., Pan Y., Vempati P., Zhao W., Knable L., Ho L., Wang J., Sastre M., Ono K., Sauve A.A., Pasinetti G.M. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging. 2013;34:1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodpaster B.H., Park S.W., Harris T.B., Kritchevsky S.B., Nevitt M., Schwartz A.V., Simonsick E.M., Tylavsky F.A., Visser M., Newman A.B. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J. Gerontol. A Biol. Sci. Med. Sci. 2006;61:1059–1064. doi: 10.1093/gerona/61.10.1059. [DOI] [PubMed] [Google Scholar]
- GTEx Consortium The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmann M., Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Houtkooper R.H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R.W., Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wagner-Valladolid S., Stephens A.D., Jung R., Poudel C., Sinnige T., Lechler M.C., Schlörit N., Lu M., Laine R.F. Intrinsically aggregation-prone proteins form amyloid-like aggregates and contribute to tissue aging in Caenorhabditis elegans. eLife. 2019;8:e43059. doi: 10.7554/eLife.43059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husom A.D., Peters E.A., Kolling E.A., Fugere N.A., Thompson L.V., Ferrington D.A. Altered proteasome function and subunit composition in aged muscle. Arch. Biochem. Biophys. 2004;421:67–76. doi: 10.1016/j.abb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Irvine G.B., El-Agnaf O.M., Shankar G.M., Walsh D.M. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P.R., Vetterke M., Hauburger A., Tacik P., Stoltenburg G., Hanisch F. Functional relevance of mitochondrial abnormalities in sporadic inclusion body myositis. J. Clin. Neurosci. 2014;21:1959–1963. doi: 10.1016/j.jocn.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kowaltowski A.J., Menezes-Filho S.L., Assali E.A., Gonçalves I.G., Cabral-Costa J.V., Abreu P., Miller N., Nolasco P., Laurindo F.R.M., Bruni-Cardoso A., Shirihai O.S. Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. FASEB J. 2019;33:13176–13188. doi: 10.1096/fj.201901136R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J., Morimoto R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laor D., Sade D., Shaham-Niv S., Zaguri D., Gartner M., Basavalingappa V., Raveh A., Pichinuk E., Engel H., Iwasaki K. Fibril formation and therapeutic targeting of amyloid-like structures in a yeast model of adenine accumulation. Nat. Commun. 2019;10:62. doi: 10.1038/s41467-018-07966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Rukina D., David F.P.A., Li T.Y., Oh C.-M., Gao A.W., Katsyuba E., Bou Sleiman M., Komljenovic A., Huang Q. Identifying gene function and module connections by the integration of multispecies expression compendia. Genome Res. 2019;29:2034–2045. doi: 10.1101/gr.251983.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl G., Roberts B.R., Pukala T.L., Kenche V.B., Roberts C.M., Link C.D., Ryan T.M., Masters C.L., Barnham K.J., Bush A.I., Cherny R.A. Utility of an improved model of amyloid-beta (Aβ1−42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 2012;7:57. doi: 10.1186/1750-1326-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L., Houtkooper R.H., Moullan N., Katsyuba E., Ryu D., Cantó C., Mottis A., Jo Y.-S., Viswanathan M., Schoonjans K. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L., Sorrentino V., Williams E.G., Cornaglia M., Frochaux M.V., Lin T., Nicolet-Dit-Félix A.A., Krishnamani G., Ouhmad T., Gijs M.A.M. The movement tracker: a flexible system for automated movement analysis in invertebrate model organisms. Curr. Protoc. Neurosci. 2016;77 doi: 10.1002/cpns.17. 8.37.1–8.37.21. [DOI] [PubMed] [Google Scholar]
- Nargund A.M., Pellegrino M.W., Fiorese C.J., Baker B.M., Haynes C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund A.M., Fiorese C.J., Pellegrino M.W., Deng P., Haynes C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt) Mol. Cell. 2015;58:123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro S., Ventura S. Fluorescent dye ProteoStat to detect and discriminate intracellular amyloid-like aggregates in Escherichia coli. Biotechnol. J. 2014;9:1259–1266. doi: 10.1002/biot.201400291. [DOI] [PubMed] [Google Scholar]
- Needham M., Corbett A., Day T., Christiansen F., Fabian V., Mastaglia F.L. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J. Clin. Neurosci. 2008;15:1350–1353. doi: 10.1016/j.jocn.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Oakey L.A., Fletcher R.S., Elhassan Y.S., Cartwright D.M., Doig C.L., Garten A., Thakker A., Maddocks O.D.K., Zhang T., Tennant D.A. Metabolic tracing reveals novel adaptations to skeletal muscle cell energy production pathways in response to NAD + depletion. Wellcome Open Res. 2019;3:147. doi: 10.12688/wellcomeopenres.14898.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K., Lionaki E., Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–528. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- Riera C.E., Merkwirth C., De Magalhaes Filho C.D., Dillin A. Signaling networks determining life span. Annu. Rev. Biochem. 2016;85:35–64. doi: 10.1146/annurev-biochem-060815-014451. [DOI] [PubMed] [Google Scholar]
- Ryu D., Zhang H., Ropelle E.R., Sorrentino V., Mázala D.A.G., Mouchiroud L., Marshall P.L., Campbell M.D., Ali A.S., Knowels G.M. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J., Barthel K., Wrede A., Salajegheh M., Bähr M., Dalakas M.C. Interrelation of inflammation and APP in sIBM: IL-1 β induces accumulation of β-amyloid in skeletal muscle. Brain. 2008;131:1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöndorf D.C., Ivanyuk D., Baden P., Sanchez-Martinez A., De Cicco S., Yu C., Giunta I., Schwarz L.K., Di Napoli G., Panagiotakopoulou V. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 2018;23:2976–2988. doi: 10.1016/j.celrep.2018.05.009. [DOI] [PubMed] [Google Scholar]
- Sergushichev A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv. 2016:060012. [Google Scholar]
- Shen D., Coleman J., Chan E., Nicholson T.P., Dai L., Sheppard P.W., Patton W.F. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem. Biophys. 2011;60:173–185. doi: 10.1007/s12013-010-9138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short K.R., Bigelow M.L., Kahl J., Singh R., Coenen-Schimke J., Raghavakaimal S., Nair K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C., McGeachy A.M., Ingolia N.T., Walter P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife. 2015;4:e05033. doi: 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E.M., Lotz G.P., Yang G., Sontag C.J., Cummings B.J., Glabe C.G., Muchowski P.J., Thompson L.M. Detection of mutant Huntingtin aggregation conformers and modulation of SDS-soluble fibrillar oligomers by small molecules. J. Huntingtons Dis. 2012;1:119–132. doi: 10.3233/JHD-2012-129004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino V., Romani M., Mouchiroud L., Beck J.S., Zhang H., D’Amico D., Moullan N., Potenza F., Schmid A.W., Rietsch S. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teich A.F., Patel M., Arancio O. A reliable way to detect endogenous murine β-amyloid. PLoS ONE. 2013;8:e55647. doi: 10.1371/journal.pone.0055647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiz P., Weihl C.C., Pestronk A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J. Neurol. Sci. 2009;278:25–29. doi: 10.1016/j.jns.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Upadhaya A.R., Lungrin I., Yamaguchi H., Fändrich M., Thal D.R. High-molecular weight Aβ oligomers and protofibrils are the predominant Aβ species in the native soluble protein fraction of the AD brain. J. Cell. Mol. Med. 2012;16:287–295. doi: 10.1111/j.1582-4934.2011.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk W.I.M., Rainbolt T.K., Dolan P.T., Webb A.E., Brunet A., Frydman J. Differentiation drives widespread rewiring of the neural stem cell chaperone network. Mol. Cell. 2020;78:329–345.e9. doi: 10.1016/j.molcel.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrablik T.L., Huang L., Lange S.E., Hanna-Rose W. Nicotinamidase modulation of NAD+ biosynthesis and nicotinamide levels separately affect reproductive development and cell survival in C. elegans. Development. 2009;136:3637–3646. doi: 10.1242/dev.028431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrablik T.L., Wang W., Upadhyay A., Hanna-Rose W. Muscle type-specific responses to NAD+ salvage biosynthesis promote muscle function in Caenorhabditis elegans. Dev. Biol. 2011;349:387–394. doi: 10.1016/j.ydbio.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther D.M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P., Morimoto R.I., Dobson C.M., Vendruscolo M., Mann M., Hartl F.U. Widespread proteome remodeling and aggregation in aging C. elegans. Cell. 2015;161:919–932. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts G.D.J., Wymer J., Kovach M.J., Mehta S.G., Mumm S., Darvish D., Pestronk A., Whyte M.P., Kimonis V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth S.E., Seo A.Y., Marzetti E., Lees H.A., Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010;45:138–148. doi: 10.1016/j.exger.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Ryu D., Wu Y., Gariani K., Wang X., Luan P., D’Amico D., Ropelle E.R., Lutolf M.P., Aebersold R. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- Zheng L., Terman A., Hallbeck M., Dehvari N., Cowburn R.F., Benedikz E., Kågedal K., Cedazo-Minguez A., Marcusson J. Macroautophagy-generated increase of lysosomal amyloid β-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy. 2011;7:1528–1545. doi: 10.4161/auto.7.12.18051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated during this study are included in this published article (and its Supplemental information). Source Data for all the individual P values are provided in Table S6 - Excel data source table.
The datasets supporting the current study have been included in Tables S1, S2, S3, and S4.
The published article includes all datasets generated using GeneBridge: https://www.systems-genetics.org/mmad.